

Abstract
DNA nanostructures (DNs) can be designed in a controlled and programmable manner, and these structures are increasingly used in a variety of biomedical applications, such as the delivery of therapeutic agents. When exposed to biological liquids, most nanomaterials become covered by a protein corona, which in turn modulates their cellular uptake and the biological response they elicit. However, the interplay between living cells and designed DNs are still not well established. Namely, there are very limited studies that assess protein corona impact on DN biological activity. Here, we analyzed the uptake of functionalized DNs in three distinct hepatic cell lines. Our analysis indicates that cellular uptake is linearly dependent on the cell size. Further, we show that the protein corona determines the endolysosomal vesicle escape efficiency of DNs coated with an endosome escape peptide. Our study offers an important basis for future optimization of DNs as delivery systems for various biomedical applications.
Keywords: nanotechnology, DNA nanotechnology, cellular uptake, protein corona, bionano interactions, endolysosomal escape
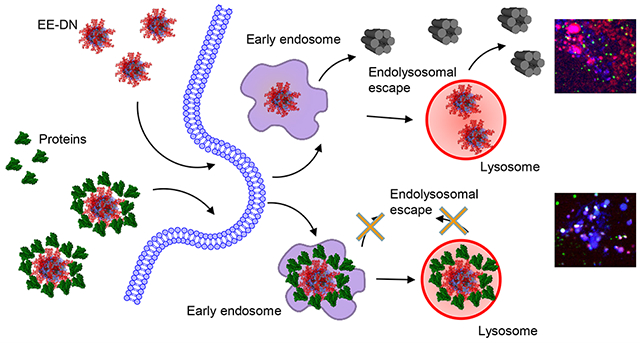
Graphical Abstract
INTRODUCTION
In recent decades, nanoparticle (NP) vehicles have shown substantial potential in different biomedical applications,1–4 including the potential to change the biodistribution and pharmacokinetics of conventional free therapeutics.5,6 As a result, a plethora of NP formulations have been designed for uses like targeted drug delivery, imaging, biosensing, and other various biomedical and therapeutic applications.1–4,7,8 NPs designed as delivery vehicles were expected to solve several key persistent problems (e.g., degradation, poor solubility, toxicity, and incapability to cross biological barriers) of free drugs.3,4,6,9 Different formulations of NPs showed success in preclinical settings and clinical trials, and some NPs received clearance for clinical use.10,11 However, the majority of NP formulations possess very low success rates of clinical translation.9,12–15 Despite isolated success cases, NP formulations are unable to reach maximal targeting effectiveness while concomitantly minimizing off-target effects.16,17 Poor knowledge of the fundamental cellular mechanisms of NP–cell interactions substantially contributes to such shortcomings.15,17–23 To a large extent, our capabilities to accurately produce NPs with tightly controlled size, shape, and surface chemistry are still rather limited.24–26 This in turn challenges the systematic investigation of NP–cell interactions, resulting in poor delivery efficacy.
DNA-based structural nanotechnology27—including polyhedral cages,28,29 bundles,30 or the complex assemblies afforded by DNA origami31—offer unique opportunities to build oligonucleotide nanostructures with tightly controlled size, shape, and surface functionality.32–36 Such remarkable molecular control over DNA nanostructures (DNs) has enabled applications like nanofabrication,37–39 biosensing,40 vehicles for spatiotemporal release of active compounds,41 cell engineering,42–44 and drug delivery.45,46 Importantly, DNs have been recognized as an alternative to conventional NP-based cargos for cellular delivery of various content, including small molecule drugs,45,47 proteins,48 and nucleic acids.49 Foreseeing potential clinical translation of DN-based applications, it is imperative to understand interactions between DNs and living cells in a well-defined and controlled manner. In fact, studies that analyze DN–cell interactions, as well as the ingestion routes and mechanisms of designed DNs are quite limited.50–52 For example, recent advancements were achieved in analyzing how the size and shape of DNs affect their cellular uptake.50,51 Although these reports analyze how different cell lines internalize DNs, no systematic investigation has been undertaken to directly compare the observed effects on closely related cell lines.50–52 Furthermore, from the nanoparticle field it is well established that, upon interaction with biological fluids, NPs form a so-called protein corona.53–56 Importantly, this protein corona affects the physicochemical characteristics of NPs but most importantly may change the overall bioreactivity of the nanoparticles.53–57 To our knowledge, there are no studies assessing the impact of this protein corona on the biological properties and delivery efficiency of functionalized DNs.32 Of note, such studies may open critical insights for the design and optimization of DNs for their successful clinical translation.
Thus, we studied the cellular uptake and fate of functionalized DNs in three closely related cell lines: HepG2, Huh7, and Alexander cells. We performed a comparative analysis of DN uptake in those cell lines and analyzed how the presence of serum proteins affects the desired bioreactivity of functionalized DNs. We also explored the effect of functionalizing the DNs with electrostatic peptide coatings58 and incorporated an endosome escape peptide sequence59 for improving cytosolic delivery of the structures.
RESULTS AND DISCUSSION
In order to probe the effect of serum proteins and DN modification on their cellular interaction, we used a 6-helix bundle (6HB) nanostructure initially designed by Howorka and colleagues30 (Figure 1a). It was found that such bundles directly interact with cell membranes and exhibit selective interaction with distinct cell types.30,60 Additionally, the 6HB may remodel lipid membranes and mediate the formation of nanopores.61,62 However, their ability to facilitate endosomal escape in living cells is unknown, and to our knowledge, no DN has been functionalized with an endosome escape peptide to impart this activity. This structure has the advantage of simplicity (being composed of only six strands), ease of formation through a simple annealing process, and high yield. The bundle is also a rigid and monomeric assembly roughly 7 × 6 nm2 in size. In addition to the bare 6HB nanostructures, we also explored the effect of cationic oligolysine peptide coatings, as initially reported by Shih and co-workers,58 to both stabilize the nanostructures to biological media conditions and to functionalize them with bioactive peptides. Toward this end, we synthesized a (Lys)10 peptide (K10), as well as a peptide flanking K10 with two copies of a sequence termed aurein 1.2, which was found to facilitate endosomal escape (vide infra).59 The K10 and K10-[aurein 1.2]2 (which we termed EE) peptides were obtained by solid-phase peptide synthesis (see the Experimental Section, Figures S1 and S2, and Tables S1 and S2) and used to coat the 6HB nanostructures (Figure 1b,c). As evidenced by native agarose gel electrophoresis, the peptides were able to most effectively coat the nanostructures though electrostatic interactions at a nitrogen/phosphate (N/P) ratio of ~1 (Figure S3). It is worth noting that some aggregation occurs in both K10 and EE structures (Figure S3). However, subsequent atomic force microscopy (AFM) and dynamic laser light scattering (DLS) analysis revealed that this aggregation is very minor (Figure 2a and Figure S4). We used AFM imaging to visualize the 6HB structures (Figure 2a) and saw that samples were primarily monodispersed, with a minimal amount of aggregates. DLS analysis in aqueous solution revealed distinct mean hydrodynamic diameters of about 15, 25, and 28 nm for 6HB, K10, and EE (Figure S4), respectively, where we surmise that the greater diameter for the latter two structures corresponds to the peptide coatings. These data confirmed the theoretically estimated DN sizes (Figure 1).
Figure 1.
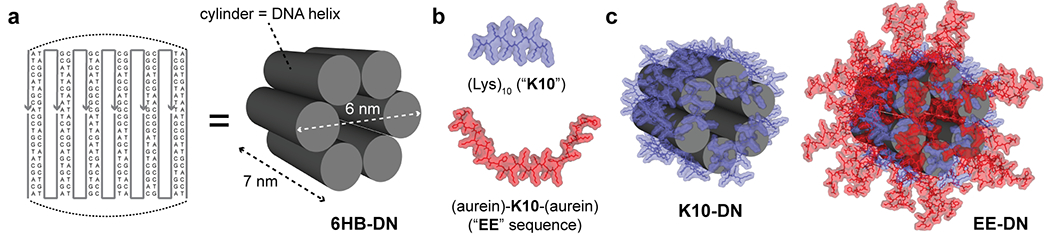
Design and functionalization of DNA nanostructures. (a) Strand diagram showing the six oligonucleotides that comprise the six-helix bundle (6HB) DN. (b) Schematics of the two peptides used for coating DNs in this study: a decalysine (“K10”) peptide in blue and K10 flanked by two copies of the aurein 1.2 endolysosomal escape (“EE”) peptide. (c) Schematics of the DNs coated with either the K10 peptide (left) or an 80:20 mixture of EE/K10 (right).
Figure 2.
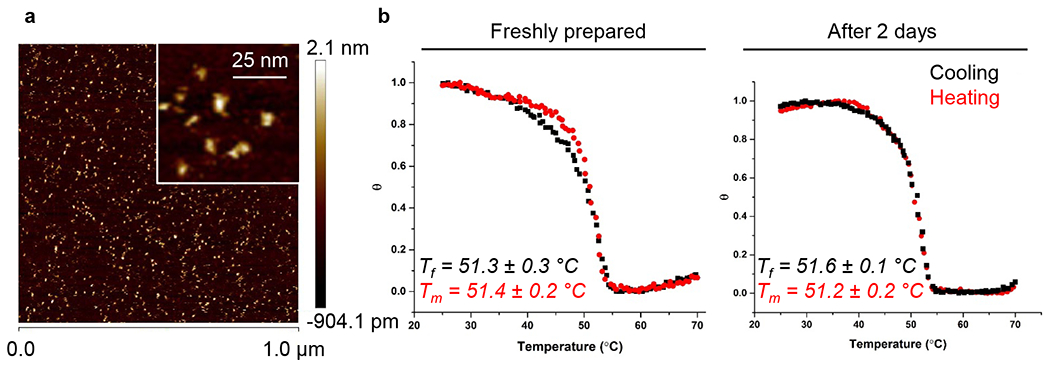
Analysis of the stability of DNA nanostructures. (a) AFM characterization of the 6HB DN. (b) Overlay of the normalized FRET efficiency plots corresponding to cooling (black) and heating cycles (red) that reveals the reversible assembly and disassembly of the structure. 6HB was incubated for either 0 or 48 h in phosphate buffered saline (PBS), followed by determining the temperature of folding (Tf) and the temperature of melting (Tm) via melting profile analysis. Values are representative from three independent repeats.
Multiple studies have shown that various DNA nanostructures remain substantially intact in different physiological media and even within cells for at least 24 h.60,63–67 However, the stability of DNs greatly depends on multiple parameters, e.g., temperature, exposure time, and DN design.68 Therefore, we assessed the structural stability of DNA nanostructures in physiological buffer (PBS). We utilized a previously reported temperature-induced unfolding assay for DNs.60,69 This assay relies on measuring of the transition temperatures (the temperature of folding (Tf) and the temperature of melting (Tm)) by monitoring fluorescence resonance energy transfer (FRET) between two incorporated dyes upon heating. The analysis of Tf and Tm serves as a tool to reveal the local structural changes of DNA nanostructures in detail.60,69 When DNA nanostructures are intact the FRET pairs are held in close contact, leading to a high FRET efficiency.60,69 Conversely, the melting and disassembly of DNA nanostructures result in increased donor–acceptor distances and a subsequent decrease of FRET efficiency.60,69 We designed 6HB structures (Figure S5) containing the FRET reporter dyes (donor, 6-carboxyfluorescein; acceptor, TAMRA). The melting analysis revealed that the transition temperature (Tf and Tm) values were approximately equal in freshly prepared 6HB structures and incubated in PBS for 2 days (Figure 2b and Figures S6 and S7) and were approximately 51 °C. This data suggest that the DNA nanostructures remained stable and assembled under physiological conditions and buffers.
Accumulating evidence suggests that, upon intravenous injection, the majority of nanomaterials are ultimately sequestered by the liver; for a review, see refs 15 and 70. Additionally, nanomaterials have been shown to directly interact with hepatocytes and not only Kupffer cells (liver resident macrophages).70,71 Hence, it is crucial to study the DN properties that might accelerate or obstruct their uptake by hepatocytes. Surprisingly, there is no data on DN–hepatocyte interactions in the current literature. Hepatic cell lines of varying degrees of differentiation have frequently been used to model hepatocyte functions, since primary tissue hepatocytes cannot be readily expanded ex vivo.72,73 Thus, in this study, we assessed DN–cell interactions utilizing three commonly used hepatic cell lines: HepG2, Huh7, and Alexander cells.
To ensure that DNs do not induce any toxic effects on the cells during the experiments, we first analyzed the effects of different DN types on cell viability. Huh7 or HepG2 as well as Alexander cells cultured in medium for 24 h in the presence or absence of 6HB, or the K10- and EE-coated nanostructures, showed no decrease in cell viability (Figure S8a). Additionally, the treatment of all three cell lines with different DN types did not result into any noticeable morphological changes (Figure S8b,c). On the contrary, treatment with ethanol, used as a positive control, led to marked membrane rupture as evident by dye cytosolic accumulation and massive vesicle shedding (Figure S8c). Further, we analyzed the uptake of differently functionalized DNs by three cell lines. To enable tracking of cellular uptake, one strand comprising the DNs was labeled with AlexaFluor-488. First, an end-point uptake study was conducted to examine and compare different DN cellular uptakes in all cell lines, which was characterized qualitatively and quantitatively by high-resolution spinning disc confocal microscopy. Incubation for 24 h with 50 nM of different types of DNs led to a noticeable intracellular accumulation of the nanostructures in all three cell types (Figure 3a). DNs exhibited different internalization behaviors in different cell lines (Figure 3b). Alexander cells showed a significantly higher cellular uptake efficiency of DNs compared to Huh7 and HepG2 (Figure 3b). HepG2 were the least effective in engulfing DNs (Figure 3b). However, we found no significant differences in the uptake of differently functionalized DNs within the same cell line (Figure 3b). All uptake data is presented as corrected total cell fluorescence (CTCF)74 of the full area of interest to average a single cell fluorescence measured by confocal microscopy. CTCF represents the sum of pixel intensity for a single image with the subtraction of average signal per pixel for a background region. The detailed description of CTCF calculations is presented in the Experimental Section. To ensure that the cell membrane-bound DNs were removed from quantification and only internalized DNs were considered in calculations, we performed cell membrane counterstaining and the 3D microscopic image analysis of the particle internalization (Figure 3a). From the orthogonal sections in the x–z planes, one can estimate that measurements in the x–y plane, performed at a z-position of about half height of the cell, enable reliable discrimination of the membrane-associated or intracellular DNs (Figure 3a, white and yellow arrows).
Figure 3.
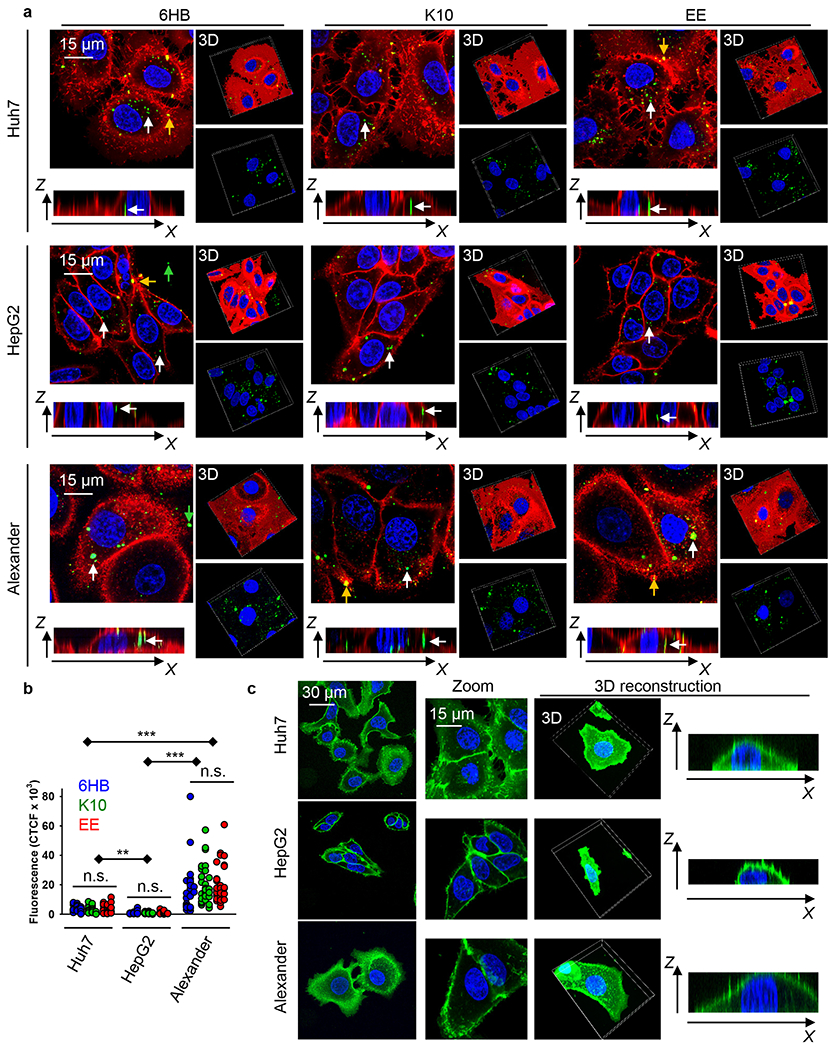
Uptake of different DNs by three distinct hepatic cell lines. (a) Alexander, HepG2, and Huh7 cell lines were treated with a 50 nM concentration of different fluorescently labeled (green fluorescence) DNs for 24 h. After treatment, cells were stained using CellMask Orange (Thermo Fisher Scientific) plasma membrane stain. Stained cells were imaged using spinning disk confocal microscopy IXplore SpinSR (Olympus, Tokyo, Japan). 3D rendering orthogonal projections were done using ImageJ software (NIH). Representative images from three independent experiments are presented. White arrows indicate internalized DNs; yellow arrows show DNs attached to the membrane surface; and green arrows depict extracellular DNs. (b) Quantification of DNs uptake. Cells were treated and imaged as in (a). The intracellular DNs were measured as corrected total cell fluorescence (CTCF) of the full area of interest using ImageJ software (NIH). Data are expressed out of at least three independent experiments (n = 30 cells). (**) P < 0.01 and (***) P < 0.001 denote significant differences. (c) Assessment of cell size and morphology in Huh7, HepG2, and Alexander cells. Cells were stained with CellMask Green (Thermo Fisher Scientific) plasma membrane stain. Nuclei were counterstained with Hoechst 33342 (Thermo Fisher Scientific). Stained cells were imaged using spinning disk confocal microscopy IXplore SpinSR (Olympus, Tokyo, Japan). Representative images out of three independent experiments are presented. 3D rendering orthogonal projections were done using ImageJ software (NIH).
In fact, it is known that even closely related cell lines respond differently to external stimuli.75–77 Specifically, NP uptake may dramatically differ in distinct cell lines of the same lineage.78 Additionally, cell geometry and morphology have been recognized as important factors affecting cell behavior and intracellular trafficking.79 Given these factors, we further analyzed the cellular morphology of HepG2, Huh7, and Alexander cells. Confocal microscopy revealed that cells of different lines are distinct in size and morphology (Figure 3c and Figure S9a). HepG2 showed an elongated shape, whereas Huh7 bore a cuboidal epithelial-like morphology (Figure 3c). Alexander cells had a hexagonal epithelial-like morphology (Figure 3c). Further, using a distinct membrane labeling dye, we confirmed that 24 h treatment with 50 nM of different types of DNs resulted in sufficient intracellular accumulation of the nanostructures in all three cell types (Figures S9b and S10).
Next, we conducted a time-course study (1, 6, and 24 h) to examine and compare DN cellular uptake over time. The analysis of the uptake of three types of DNs by HepG2, Huh7, and Alexander cells by quantitative confocal microscopy revealed that within 1 h all types of DNs were engulfed by all three cell lines (Figure 4a and Figure S11). However, the uptake process did not stop at the 1 h time point and continued up to 24 h of incubation (Figure 4a and Figure S11). Consistent with end-point analysis (Figure 3a), a time-course study (Figure 4a) revealed that Alexander cells showed the highest extent of DN internalization. We showed above that all three cell lines are distinct in size and morphology (Figure 3c). Further, we quantitatively assessed the size differences between cell lines, revealing that the average area of Alexander cells is about 1700 μm2, that of Huh7 is 1100 μm2, and the of HepG2 is 500 μm2 (Figure 4b). It becomes evident that cell size plays a central role to many cellular functions.79,80 Of note, cell size has been identified as a factor that determines the rate of cellular uptake of proteins, endocytic structures,81 and nanomaterials.82
Figure 4.
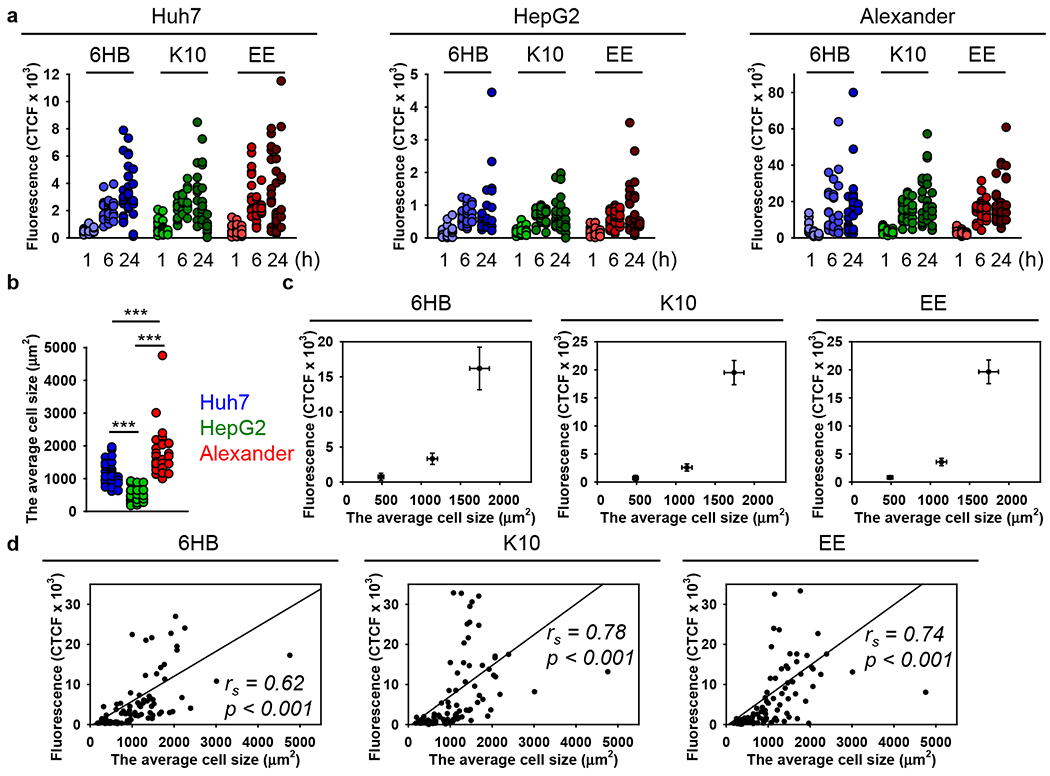
Uptake kinetics assessment of different DNs. (a) Alexander, HepG2, and Huh7 cell lines were treated with a 50 nM concentration of different DNs for 1, 6, and 24 h. After treatment, cells were fixed with 4% paraformaldehyde (VWR) and labeled with CellBrite Blue (Biotium) plasma membrane stain. Stained cells were imaged using spinning disk confocal microscopy IXplore SpinSR (Olympus, Tokyo, Japan). The intracellular DNs were measured as corrected total cell fluorescence (CTCF) of the full area of interest using ImageJ software (NIH). Data are expressed out of at least three independent experiments (n = 28–34 cells). (b) Assessment of cell size in Huh7, HepG2, and Alexander cells. Cells were stained with CellMask Green (Thermo Fisher Scientific) plasma membrane stain. Nuclei were counterstained with Hoechst 33342 (Thermo Fisher Scientific). Stained cells were imaged using spinning disk confocal microscopy IXplore SpinSR (Olympus, Tokyo, Japan). The average cell area was measured using ImageJ software (NIH) and is presented as means of n = 30 cells. (***) p < 0.001 denotes significant differences. (c) Cell-size-dependent DNs uptake. The intracellular DNs presented as CTCF after 24 h treatment with 50 nM concentration of different DNs were plotted versus corresponding cell size. (d) Linear correlation between cell size and DNs uptake. Each black point represents confocal microscopy-measured single-cell DN uptake plotted against corresponding cell size. The uptake is expressed as CTCF after 24 h treatment with 50 nM concentration of different DNs. Correlation coefficients and P values were calculated using SigmaPlot 13 software (Systat Software, Inc.).
Indeed, a number of studies have been conducted to reveal endocytic recognition and engulfment of different DNs by distinct cell types.32,50–52,83–85 In these reports, the primary focus of the research is how either the physiochemical parameters (e.g., mass, shape, size, surface functionalization) of DNs or cell phenotype modulate the average uptake of the nanostructures.32,50–52,83–85 However, little attention has been paid to how the cell size or other cellular characteristics at the single-cell level might affect DN ingestion. Of note, the size of a cell plays a crucial role in determining the rate of cellular uptake of materials. Although the correlation between cell size and uptake appears to be intuitive,78,86–89 it is still not established exactly how the physical parameters of a single cell govern its ability to uptake particles.82 Additionally, for DNA nanostructures, there is still only limited literature that analyzes the correlation of cell size with DNs uptake efficacy.86 The average cell sizes were mapped to the corresponding fluorescence of the different types of internalized DNs (Figure 4c). This analysis revealed a linear increase in the uptake of all three types of DNs with cell size (Figure 4c). Spearman rank order correlation analysis confirmed that the cellular uptake of DNs follows a linear relationship with the cell size (Figure 4d).
To elaborate on the key question of how the presence of serum proteins affects the desired endosome escape of DNs, we first wanted to cross-check whether DNs stay intact in harsh lysosomal conditions. Indeed, the intracellular fate of DNA nanostructures remains elusive and controversial.86 Some evidence suggests that DNs end up in the lysosomes, whereas other studies claim that DNs accumulate in the cytosolic compartments.86 In fact, all three types of DNs were localized in the lysosomal compartments as revealed by confocal imaging (Figure S12). Further, we analyzed the stability of DNs by utilizing FRET analysis. We used the above-mentioned 6HB structures containing the FRET reporter dyes (Figure S5). 6HB structures, labeled with either donor-only or acceptor-only fluorophores, served as negative controls. The Forster distance of the FRET reporter dyes used (6-carboxyfluorescein donor, TAMRA acceptor) is ~5 nm, which allows for the sensitive detection of the changes in FRET efficiency that occur during the structural changes (e.g., assembly/disassembly) of DNs.69 The validation of FRET by fluorescence microscopy revealed that all three cell lines, treated with a 50 nM concentration of 6HB for 24 h, showed high FRET efficiency compared with negative controls (Figure 5). These data indicate that DNs remained largely stable in lysosomal compartments for up to 24 h of incubation.
Figure 5.

FRET microscopy images of Alexander, HepG2, and Huh7 cells treated with 6HB labeled with FRET reporter dyes (6-carboxyfluorescein donor and TAMRA acceptor). Images of the three detection channels (donor, acceptor, and FRET) are shown. The calculated colocalization diagram and colocalized FRET index after the subtraction of spectral bleed-through. Alexander, HepG2, and Huh7 cells were treated with a 50 nM concentration of 6HB labeled with FRET reporter dyes for 24 h. Nuclei were counterstained with Hoechst 33342 (Thermo Fisher Scientific). Confocal images were taken and analyzed for FRET using the “FRET and colocalization analyzer” ImageJ plug-in.115 “Colocalized FRET index” images present the calculated amount of FRET for each pixel in the FRET channel. The ImageJ plug-in color codes the relative FRET efficiency ranging from blue (none FRET efficiency) to red-yellow (high FRET efficiency). The “Colocalization diagram” plots display pixel colocalization as well as color coded FRET efficiency in a 2D plot.
It is worth noting here that DNs are recognized as novel smart delivery platforms for different macromolecules and drugs.32,46,83,90 Generally, the delivery of different biological agents utilizing nanobased vehicles relies on the endocytic pathway as the predominant uptake mechanism.91,92 This process leads to the entrapment of cargo inside the endosome and lysosome, where the contents can be degraded by lysosomal enzymes.91,92 In order to bypass this problem, a number of molecules and other pharmacological agents, which facilitate escape the endolysosomal compartment, have been identified.91 Interestingly, studies that experimentally verify endolysosomal escape are usually conducted utilizing serum-free medium.93–96 Indeed, as is well-known from nanoparticle–cell interactions, the presence of proteins and their adsorption onto a NP surface dramatically affects the resultant biological effects.53–56 Specifically, it has been shown that the protein corona substantially impairs the endolysosomal escape efficiency of different nanomaterials.97,98 Thus, we wanted to probe whether the protein corona has any effect on the escape efficiency of the DN functionalized with an endolysosomal escape enhancer. Bearing in mind that the uptake process continues up to 24 h (Figure 4a and Figure S11), we needed to select an appropriate time point for endolysosomal escape assessment. It is well-established that serum-free cell culturing results in autophagy that dramatically biases endolysosomal interactions. However, short-term starvation up to 6 h of hepatic cells has incremental effect on autophagy, whereas 8 h and longer leads to substantial autophagic flux activation.99–101 Therefore, 6 h represented an optimal time point to monitor endolysosomal escape without concomitant autophagic flux.
Recently a 13-residue peptide, termed aurein 1.2 (GLFDIIKKIAESF), was discovered that enhances endolysosomal escape and improved the cytosolic delivery of proteins it was appended to by up to ~5-fold.59,102 In fact, this peptide can disrupt endolysosomal membranes and in such a way trigger the escape of cargo to cytosol.59,102,103 Importantly, aurein facilitates endolysosomal escape without concomitant disruption of the cell membrane and does not exhibit cytotoxicity.59,102,103 Therefore, we used this peptide to electrostatically coat DNs (Figure 1c) for potential enhancement of endolysosomal escape.
One of the straightforward methods to evaluate endolysosomal escape is to use microscopic imaging of fluorescently labeled materials with localization to endolysosomal compartments.91,104,105 Confocal fluorescence microscopy is indispensable in assessing the colocalization of labeled macromolecular species of interest. However, such analysis might be substantially hampered by undesirable phototoxic effects from laser irradiation, especially during live-cell imaging.106,107 In order to observe undamaged living cells with engulfed DNs and avoid phototoxic effects from imaging, we utilized a novel ultrafast imaging system based on the IXplore SpinSR Olympus spinning disk confocal microscope.107,108 In order to track the peptide coating and the DN separately, we coated the DN with an 80:20 ratio of the EE peptide and K10 labeled with the pH-sensitive dye pHrodo Red, which dramatically increases its fluorescence in acidic pH.109 We estimate that, given the N/P ratio of these coatings and the number of phosphates in the DNA nanostructure, there are ~48 copies of the aurein 1.2 peptide per DN.
DNs with green fluorescence-labeled DNA and LysoTracker Blue DND-22 staining were used to monitor the nanostructures and endo-/lysosomes, respectively, by confocal microscopy. Indeed, aurein 1.2-decorated DNs in serum-free medium were able to escape from the endosomes/lysosomes and be released into the cytoplasm in all three cell lines after 6 h of treatment (Figure 6 and Figure S11). Specifically, a large amount of plain/uncoated DNs (the 6HB) accumulated in endo-/lysosomes at 6 h, while the colocalization of EE-coated DNs with endo-/lysosomes was markedly lower in all three cell lines (Figure 6a,d,g). Additionally, the Pearson’s correlation coefficient of EE-coated DN colocalization with endo-/lysosomes was below 0.5 in all three cell lines (Figure 6b,e,h). These results suggested that a great portion of EE-coated DNs efficiently escaped from endo-/lysosomes. Further, the Pearson’s correlation coefficient of DNA colocalization with pHrodo was below 0.5 for the EE-DNs in all three cell lines (Figure 6c,f,i), suggesting that the peptide coating was probably removed from the DNA construct after endolysosomal escape.
Figure 6.
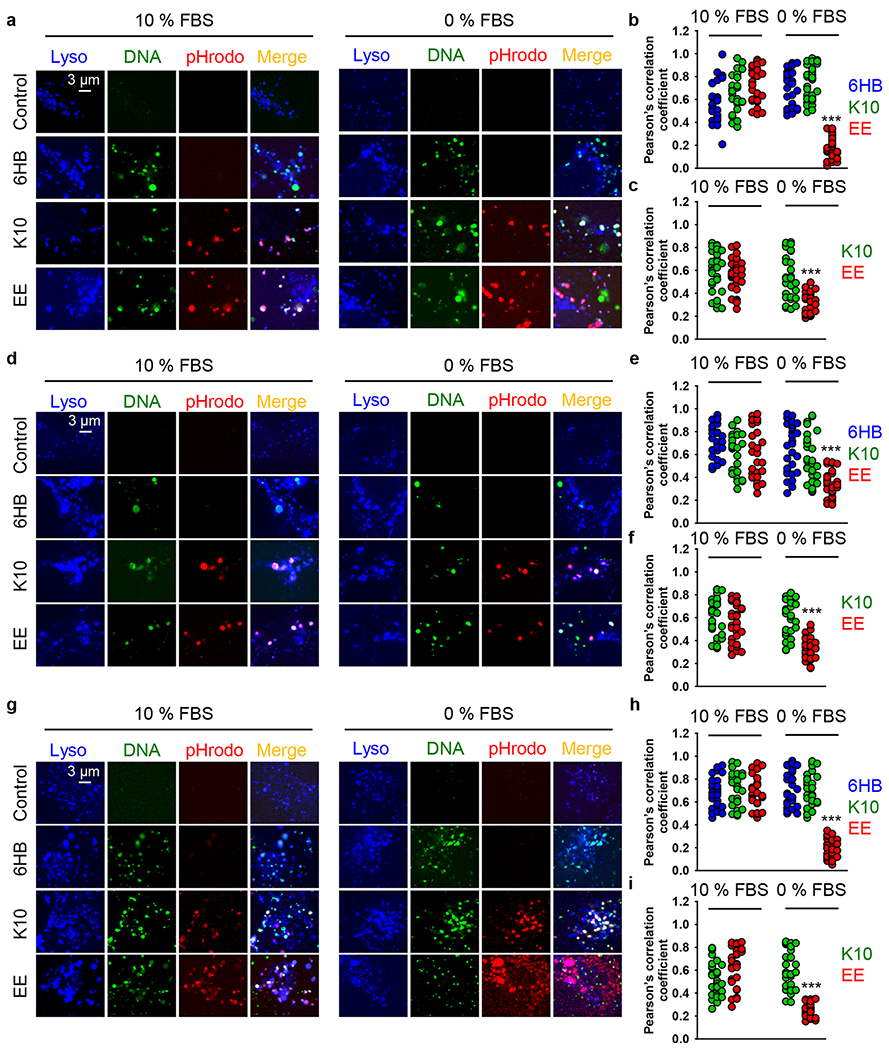
Colocalization analysis of different DNs. (a–c) Huh7 cells were treated with different types of DNs (at 50 nM concentration) for 6 h either in full medium (10% FBS EMEM) or in serum-free medium (0% FBS EMEM). After incubation, cells were labeled with lysosomal marker LysoTracker Blue DND-22 (Thermo Fisher Scientific). Stained cells were imaged using spinning disk confocal microscopy IXplore SpinSR (Olympus, Tokyo, Japan). The Pearson’s correlation coefficient for fluorophore pairs either (b) DNA–Lysosomes or (c) DNA–pHrodo was calculated using the Coloc 2 tool available in ImageJ software (NIH) and is presented as means of n = 30 cells. (***) P < 0.001 denotes significant differences. (d–f) HepG2 cells were treated with different types of DNs (at 50 nM concentration) for 6 h either in full medium (10% FBS EMEM) or in serum-free medium (0% FBS EMEM). After incubation, cells were labeled with lysosomal marker LysoTracker Blue DND-22 (Thermo Fisher Scientific). Stained cells were imaged using spinning disk confocal microscopy IXplore SpinSR (Olympus, Tokyo, Japan). The Pearson’s correlation coefficient for fluorophore pairs eithers (e) DNA–Lysosome or (f) DNA–pHrodo was calculated using the Coloc 2 tool available in ImageJ software (NIH) and is presented as means of n = 30 cells. (***) P < 0.001 denotes significant differences. (g–i) Alexander cells were treated with different types of DNs (at 50 nM concentration) for 6 h either in full medium (10% FBS EMEM) or in serum-free medium (0% FBS EMEM). After incubation cells were labeled with lysosomal marker LysoTracker Blue DND-22 (Thermo Fisher Scientific). Stained cells were imaged using spinning disk confocal microscopy IXplore SpinSR (Olympus, Tokyo, Japan). The Pearson’s correlation coefficient for fluorophore pairs either (h) DNA–Lysosomes or (i) DNA–pHrodo was calculated using the Coloc 2 tool available in ImageJ software (NIH) and is presented as means of n = 30 cells. (***) P < 0.001 denotes significant differences.
By contrast, in the presence of serum the endolysosomal escape of EE-DNs was diminished in all three cell lines (Figure 6a,d,g). In the presence of serum, the Pearson’s correlation coefficient of EE-DN colocalization with endo-/lysosomes was substantially higher than 0.5 in all three cell lines (Figure 6b,e,h). Moreover, there was no statistically significant difference in colocalization with endo-/lysosomes between the plain 6HB-DNs and EE-DNs (Figure 6b,e,h). These results suggest that, in the presence of serum, the EE-DNs stayed in endo-/lysosomal vesicles during the test period. Interestingly, the Pearson’s correlation coefficient of EE-DN DNA colocalization with pHrodo was higher than 0.5 in all three cell lines (Figure 6c,f,i). These results imply that the DNA construct and aurein 1.2 peptide in the EE-DN sample did not dissociate when DNs were added to serum-containing medium.
Aurein 1.2 is a derivative of so-called antimicrobial peptides,59 which penetrate membranes utilizing electrostatic interactions followed by the displacement of lipids and alteration of membrane structure.110 It was postulated that in this way antimicrobial peptides may enhance endolysosomal escape.59,91 To verify the specificity of aurein 1.2 as an endolysosomal escape enhancer, we decorated DNs with the short, highly charged peptide deca-lysine (K10). This peptide was also labeled with pHrodo Red. In fact, in neither serum-free nor serum-containing medium were K10-decorated DNs able to induce any noticeable endolysosomal escape (Figure 6). Additionally, we created DNA bundles decorated with scrambled aurein 1.2 sequence, which was composed of the same amino acids but in a random order not expected to facilitate endolysosomal escape. In fact, those DNA bundles were unable to induce any noticeable endolysosomal escape either in the presence or absence of serum (Figures S13 and S14). Our data support previous findings that the effectiveness of aurein 1.2 is highly dependent on its sequence, and even closely related peptides cannot enhance endolysosomal escape to a similar extent.59
By analogy with NPs, where a protein corona is quickly formed within 1 h upon injection to biological media,53,54 we hypothesized that DNs would follow a similar pattern. Thus, to confirm the protein corona formation, we analyzed protein adsorption to the surface of DNs. We incubated all three types of DN either in serum-containing medium or serum-free buffer for 2 h. After incubation, DNs were collected by centrifugation, washed with PBS buffer, and subjected to 1D sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie staining for the total proteins detached from the structures (Figure 7a). Indeed, DNs incubated in HBSS did not carry any proteins (Figure 7a). By contrast, clear protein bands were eluted from DNs incubated in serum-containing medium (Figure 7a). In fact, SDS-PAGE and Coomassie staining require denaturing conditions for the analysis. Further, we wanted to confirm and monitor protein adsorption while the DNs are immersed in the solution utilizing in situ methodology. Thus, we used fluorescence correlation spectroscopy (FCS), a widely used method enabling precise measurements of the increase in hydrodynamic radius of the particle upon corona formation.111 The increasing particle size resulting from protein adsorption was assessed by measuring the increase in diffusion time.111 In fact, a 60 min incubation of DNs in serum-containing medium led to a statistically significant increase of the diffusion time, reflected by a shift of the autocorrelation curve (Figure 7b,c). Of note, the diffusion time reflects the size of particles; the longer the time the larger the particles,111 but the hydrodynamic radius calculated from FCS measurements can only be taken as an estimate and its value depends on the chosen assumptions. This is why Figure 7b shows less processed results in the form of mean diffusion times. If we estimate the sizes on the basis of our calibration measurements and assumption of ideally spherical particles the diameter of all DNs is 15–28 nm, while the thickness of the protein corona is 2.2 ± 0.8, 1.7 ± 1.1, and 2.1 ± 1.4 nm, for K10, 6HB, and EE, respectively. DNs that were incubated in HBSS buffer showed no signs of the increase of diffusion time and subsequently the size of particles (Figure 7b,c). Thus, FCS data confirmed the results from Coomassie staining (Figure 7a) and imply that, upon incubation of DNs in serum-containing medium, a protein corona is formed.
Figure 7.
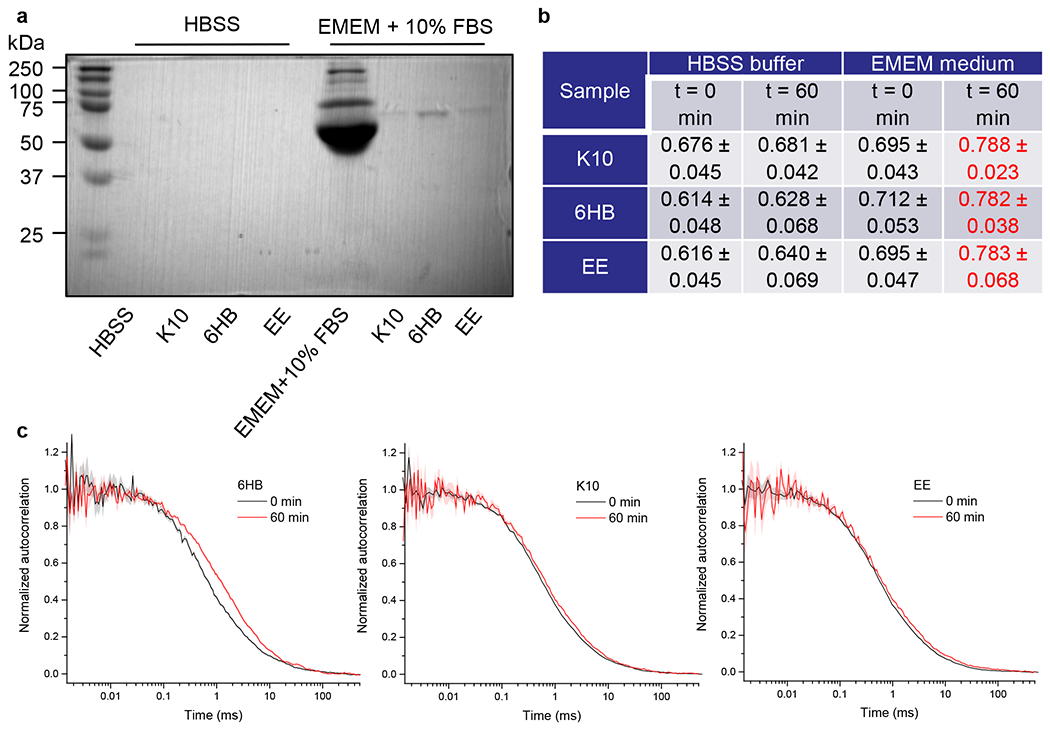
DN–protein interaction. (a) Different types of DNs at concentration 50 nM were incubated either in HBSS or in EMEM medium (ATCC) supplemented with 10% fetal bovine serum (FBS, Thermo Fisher Scientific) for 2 h at 37 °C. The DNs were centrifuged and washed with PBS. Elution and denaturation in sample loading buffer was used to detach proteins associated with the particles. Afterward, proteins were separated by gel electrophoresis. Gels were stained with Coomassie blue (AppliChem). (b and c) Analysis of the protein corona on the particles assessed by Fluorescence Correlation Spectroscopy (FCS). Different types of DNs were incubated either in HBSS, or in EMEM medium (ATCC) supplemented with 10% fetal bovine serum (FBS, Thermo Fisher Scientific), and the mean diffusion time was measured by FCS. (b) Table summarizing diffusion times of different DNs incubated in different buffer conditions in milliseconds. The data are presented as mean ± SE, n = 3. The mean diffusion time is given in milliseconds (ms). (c) Examples of autocorrelation curves obtained for the diffusion of fluorescently labeled particles in EMEM medium supplemented with 10% fetal bovine serum. The measurements were performed immediately after adding the particles to the medium and after 60 min after addition.
CONCLUSION
In summary, in this study, we performed a comparative investigation of the cellular uptake of differently functionalized DNs in distinct but closely related human hepatic cancer cell lines. The three cell types examined (Alexander, HepG2, and Huh7) showed different internalization efficiency. Overall, our study reveals that the extent of DN internalization and the kinetics of uptake may grossly differ between distinct cell lines, even between phenotypically related cells. The analysis clearly shows that the efficiency of DN engulfment by cells is strongly associated with the cell size. Additionally, we demonstrated that modifying DNs with a dense coating of the peptide aurein 1.2 can facilitate endolysosomal escape, which has been a key challenge for the application of DNs in cell delivery studies. We revealed that DNs rapidly form a protein corona when exposed to serum-containing medium and that this protein corona dramatically reduces the endolysosomal escape efficiency of aurein 1.2-decorated DNs.
Foreseeing the use of DNA nanotechnology in therapeutic applications, our presented results provide a foundation and design strategies for the rational optimization of DN-based delivery vehicles.
EXPERIMENTAL SECTION
Materials.
The following fluorescent probes were used. To visualize the plasma membrane in confocal imaging, we used the following plasma membrane stains: CellMask Green (Cat. No. C37608, Thermo Fisher Scientific), CellMask Orange (Cat. No. C10045, Thermo Fisher Scientific), or CellBrite Blue (Cat. No. 30024, Biotium). Nuclei were counterstained with Hoechst 33342 (Cat. No. H3570, Thermo Fisher Scientific). Lysosomes were labeled with lysosomal marker LysoTracker Blue DND-22 (Cat. No. L7525, Thermo Fisher Scientific). The optimal incubation time for each probe was determined experimentally.
Fabrication and Characterization of DNs.
All oligonucleotides were obtained from Integrated DNA Technologies (Coralville, Iowa) and purified using 14% urea-based denaturing polyacrylamide gel electrophoresis (PAGE). One strand was labeled with AlexaFluor-488 for imaging in the agarose gels and in microscopy experiments. Each strand was added to a mixture at 10 μM in 1x tris-acetic acid-EDTA (TAE) buffer with 12.5 mM MgCl2 and annealed from 95 to 4 °C over 2 h. The successful formation of the 6-helix bundle was confirmed using agarose gel electrophoresis. DN size was characterized utilizing a Zetasizer Nano (Malvern Instruments). DNs were dispersed in PBS, pH 7.4.
Atomic Force Microscopy.
AFM images were captured on a Bruker Multimode 8 system with Nanoscope V controller in a ScanAsyst in Fluid mode with ScanAsyst-Fluid+ AFM probes (Bruker, k ~ 0.7 N/m, tip radius <10 nm). Two microliters of sample was deposited on freshly cleaved mica followed by the addition of 48 μL of 1x TAE with 12.5 mM Mg2+ for 2 min. One mM NiCl2 buffer can be used to enhance the adsorption of DNA nanostructures on the mica surface.
DNs Stability Assay with Melting Profile Analysis.
The melting transitions of the DNA nanostructures were assessed using previously published methodology60,69 utilizing a MX3005P real-time thermocycler (Stratagene). The DNs were assembled containing FRET reporter dyes [6-carboxyfluorescein (FAM) donor and TAMRA acceptor] pairs (folded at 1 μM in 1x TAE with 12.5 mM Mg2+). The DNA constructs were diluted into the stated buffer systems to give a final DNA concentration of 0.15 μM (total volume of 300 μL) in eight-well optical tube strips (Agilent, 100 μL per tube). Optical quality sealing tape (Agilent) was placed on top to prevent evaporation. The samples were heated from 25 to 80 °C at a rate of 0.5 °C per min. The efficiency of energy transfer (E) was determined at each temperature according to E(T) = 1 – IDA(T)/ID(T), where IDA and ID are, respectively, the fluorescence intensities of the FRET donor (FAM) in the presence and absence of the FRET acceptor (TAMRA). All experiments were repeated in three replicates to ensure reproducibility. The melting temperature was determined from taking the first derivative of the donor fluorescence profile.
Peptide Synthesis and Characterization.
All peptides were synthesized on a CEM Liberty Blue using a Rink amide resin via standard Fmoc-based solid phase peptide synthesis. Briefly, 0.5 M diisopropylcarbodiimide was used as an activator, 1 M oxyma with 0.1 M diisopropylethylamine was used as an activator base, and 20% piperidine was used as a deprotecting agent. The peptide was cleaved from the resin using a solution of 95% trifluoroacetic acid with 2.5% triisopropylsilane and 2.5% water, followed by ether precipitation. Following pellet suspension, the crude peptide was purified on a reverse phase HPLC instrument (Waters), using a gradient of 0—100% acetonitrile with 0.1% TFA. Pure fractions were identified using MALDI-TOF mass spectrometry (Bruker Microflex) with α-cyano-4-hydroxycinnamic acid as a matrix. The K10-cysteine was labeled with a maleimide-C2-pHrodo Red by addition of the dye (10 equiv) in PBS pH 7.
DN Coating and Characterization.
The DNs (1 μM) were mixed with the desired K10-containing peptide at a 1:1 N/P ratio and incubated at room temperature for a minimum of 2 h. All coated DNs used for cell experiments utilized the pHrodo-labeled K10 at 20 mol % of the total K10 concentration. All coated DNs run on agarose gels utilized the fluorescein labeled K10 at 20 mol % of the total K10 concentration. In order to determine the optimal N/P ratio for complete coating of the DNs, the structures were electrophoresed using 1.5% agarose gels at 65 V for 60 min and imaged using the fluorescein labeled K10.
Cell Culture.
In this study, we utilized well-established cellular models of hepatic cells, namely, the human hepatoblastoma HepG2 cell line (American Type Culture Collection, ATCC) and the human hepatocellular carcinoma cell lines Alexander (PLC/PRF/5, ATCC) and Huh7 (Japanese Collection of Research Bioresources, JCRB). We used standard culturing media composition, i.e., EMEM medium (ATCC) supplemented with 10% fetal bovine serum (FBS, Thermo Fisher Scientific) and 1% penicillin/streptomycin (Thermo Fisher Scientific).8,74,107,112,113 Mycoplasma testing, using the MycoAlert mycoplasma detection kit (LT07-418, Lonza, Basel, Switzerland), was performed routinely. Cells were grown in a humidified 5% CO2 atmosphere at 37 °C. Once per week, fresh cell culture medium was added.8,74,107,112,113
Cell Viability Assay.
The potential toxicity of synthesized DNs was assessed using a well-established alamarBlue viability assay (Thermo Fisher Scientific).8,74,107,112,113 The technique is based on the cleavage of resazurin to resorufin by undamaged live cells. This cleavage leads to an increase of the overall alamarBlue color intensity.8,74,107,112,113 Subsequently, the percentage of metabolically active cells in the culture was calculated on the basis of the absorbance. We assessed cell viability via the alamarBlue assay according to guidelines of the manufacturer and our previously established treatment protocol.8,112 In short, distinct cell lines were grown in 96-well plates at a density of 10 000 cells per well and incubated with different concentrations of DNs for 24 h. Afterward, the alamarBlue reagent was supplemented to each well, and plates were incubated for 2 h at 37 °C. The TECAN microplate reader SpectraFluor Plus (TECAN, Mannedorf, Switzerland) was utilized to detect the absorbance of the alamarBlue reagent at 570 nm. Readings were done in triplicate, with three independent experiments performed for each measurement. Furthermore, we analyzed DN interference with the assay reagent and verified that the nanostructures do not react with alamarBlue (data not shown).
Cellular Uptake Analysis.
We utilized confocal microscopy to assess the cellular uptake of DNs. To analyze intracellular DN distribution, cells were cultured in 6-channel μ-Slides (Ibidi, Martinsried) and treated with different concentrations of fluorescently labeled DNs for 1, 6, and 24 h. Then, cells were fixed with 4% paraformaldehyde (VWR) and stained with CellBrite Blue (Biotium) plasma membrane stain. Labeled cells were visualized using spinning disk confocal microscopy IXplore SpinSR (Olympus, Tokyo, Japan) according to our verified protocols.8,74,107,112,113 For live cell imaging, the cell membrane was labeled with a CellMask Orange (Thermo Fisher Scientific) plasma membrane stain. We performed dual-color imaging of confocal cross sections at about half the cell height for the quantitative assessment of DN intracellular distribution. From the image of the stained membrane, binary masks were extracted that enabled the definition of the membrane-associated regions and the cytosolic space. The corresponding image of the cell membrane was converted into a mask of the cell in all imaged confocal planes. By applying this mask to the relevant image of DNs, the engulfed particles can be discriminated. The intracellular DNs were measured as the corrected total cell fluorescence (CTCF) of the full area of interest, i.e., intracellular region bordered by cell membrane mask. We used a published methodology to define the net average CTCF intensity for each image.114 The CTCF was calculated by following formula: CTCF = integrated density – (area of selected cell × mean fluorescence of background readings). The mean fluorescence of the background was defined as an image area without fluorescent objects. CTCF was determined as the sum of pixel intensity for a single image with the subtracted average signal per pixel for a region selected as the background, according to our previously published methodology.8,74,107,112,113 Image quantifications were performed using ImageJ software (NIH).
Analysis of DN Stability in Cells by FRET Imaging.
An Olympus confocal imaging system (Olympus, Tokyo, Japan), described below, was used for FRET measurements. Cells were grown in 6-channel μ-Slides (Ibidi, Martinsried) and incubated with 6HB-containing FRET reporter dyes [6-carboxyfluorescein (FAM) donor and TAMRA acceptor] at a 50 nM concentration for 24 h. For imaging, the FAM cells were excited with the 488 nm laser and fluorescence was collected with a BA510-550 filter (Olympus), whereas the FRET-signal was detected with a BA575IF filter (Olympus). To image TAMRA, we utilized the 561 nm excitation laser while emission was detected using a BA575IF filter (Olympus). Confocal FRET analysis was performed as described in the “FRET and colocalization analyzer – Users guide”.115 6HBs containing either 6-carboxyfluorescein (FAM) donor or TAMRA acceptor only were used as negative controls.
DN–Protein Interaction.
DNs at 50 nM concentration were incubated either in HBSS, or in EMEM medium (ATCC) supplemented with 10% fetal bovine serum (FBS, Thermo Fisher Scientific) for 2 h at 37 °C. Centrifugation was utilized to collect the particles. To remove any remaining unbound proteins, DNs were washed extensively with PBS. The samples were centrifuged for 15 min at 15 000g followed by pellet resuspension in PBS. The washing with PBS was performed three times to eliminate all the molecules not bound to DNs. According to the literature,116 such methodology has been shown to be effective for the isolation of particle–protein corona complexes. Indeed, the main aim of this work was not the most accurate possible determination of the protein corona composition but rather the demonstration that the formation of protein corona occurs. The process of elution and denaturation in sample loading buffer was used to detach proteins associated with the particles. Afterward, proteins were separated by gel electrophoresis (1D SDS-PAGE). Full cell culture EMEM medium supplemented with 10% fetal bovine serum was utilized as a control. Gels were stained with Coomassie blue (AppliChem).
Protein Corona Analysis Using Fluorescence Correlation Spectroscopy.
To test for the formation of protein corona on the particles their diffusivity was measured using fluorescence correlation spectroscopy (FCS). The method is based on the analysis of the fluorescence intensity fluctuations resulting from a diffusion of diluted fluorescent particles through a small volume (~1 fL) from which the signal is collected. The signal is autocorrelated and fitted to a 3D free diffusion model to get the mean diffusion time, τD (see refs 111 and 117 for details). Under the assumption of reasonably unchanged shape of the particles, this parameter is proportional to the hydrodynamic radius of the particles according to the Stokes–Einstein equation. Therefore, the increase of τD, can be interpreted as an enlargement of the particles, e.g., due to protein corona formation.
FCS data acquisition was carried out by utilizing an inverted confocal fluorescence microscope, Olympus IX71 (Olympus, Hamburg, Germany), equipped with single-photon counting unit MicroTime 200 (PicoQuant, Berlin, Germany). An excitation of 470 nm was achieved with a diode laser (LDH-P-C-470; PicoQuant, Berlin, Germany) operating at 20 MHz. A water immersion objective (1.2 NA, 60×, Olympus) was utilized to visualize a sample. The fluorescence signal was collected through the main dichroic mirror (Z473/635, Chroma, Rockingham, VT), a 50 μm pinhole, and guided to the single photon avalanche diode using 515/50 band-pass filter (Chroma). All FCS data acquisitions were carried out at 25 °C in 8-well μ-Slides (Ibidi, Gräfelfing, Germany). Due to particle adsorption to the glass, we utilized plastic bottom μ-Slides, which showed resistance to adsorption. Atto 488 (Atto-tec, Siegen, Germany) dye was used as a reference for calibration measurements.
The measured data were fitted utilizing a standard 3D diffusion model implemented in Symphotime 64 software (PicoQuant, Berlin, Germany). Fluorescence decay data were used to correct for the noise. On the basis of the intensity histogram, a small fraction of particles with the highest intensity (>99.8% threshold) was excluded from the analysis as possible aggregates. Mean diffusion times obtained from the fitting of the data from three separate experiments were used to calculate the average diffusion time; the weighted-average based on the error of the fitting was used.
High-Resolution Spinning Disk Confocal Microscopy.
In order to be able to reveal clear subcellular details of DNs localization, we utilized a novel IXplore SpinSR Olympus high-resolution imaging system (Olympus, Tokyo, Japan). We utilized 6-channel μ-Slides (Ibidi, Martinsried) for cell seeding. Afterward, cells were treated with different concentrations of fluorescently labeled DNs. Then cells were stained for CellBrite Blue or LysoTracker Blue DND-22. The imaging system consists of the following units: an inverted microscope (IX83; Olympus, Tokyo, Japan) and a spinning disc confocal unit (CSUW1-T2S SD; Yokogawa, Musashino, Japan). Fluorescence data for image reconstruction were collected via either a 100× silicone immersion objective (UPLSAPO100XS NA 1.35 WD 0.2 silicone lens, Olympus, Tokyo, Japan) or a 20× objective (LUCPLFLN20XPH NA 0.45 air lens, Olympus, Tokyo, Japan). The following lasers were used to excite fluorophores: 405 nm laser diode (50 mW), 488 nm laser diode (100 mW), and 561 nm laser diode (100 mW). Confocal images were acquired at a 2048 × 2048-pixel resolution. The fluorescent images were collected by appropriate emission filters (BA420-460; BA575IF; BA510-550; Olympus, Tokyo, Japan) and captured concurrently by two digital CMOS cameras ORCA-Flash4.0 V3 (Hamamatsu, Hamamatsu City, Japan). Fluorescence confocal images were acquired using software cellSens (Olympus, Tokyo, Japan). Quantitative image analysis was performed by selecting randomly ~5–10 visual fields per each sample, using the same setting parameters (i.e., spinning disk speed, laser power, and offset gain). ImageJ software (NIH) was used for image processing, quantification, and 3D reconstruction.
Image Quantification.
To measure cell size, cells were stained with CellMask Green (Thermo Fisher Scientific) plasma membrane stain. Nuclei were counterstained with Hoechst 33342 (Thermo Fisher Scientific). Spinning disk confocal microscopy IXplore SpinSR (Olympus, Tokyo, Japan) was used to acquire images of the labeled cells. The analysis of DN uptake is described above in the Cellular Uptake Analysis section.
To analyze the endolysosomal escape of DNs colocalization analysis was performed. Cells were incubated with different types of DNs (at 50 nM concentration) for 6 h either in full medium (10% FBS EMEM) or in serum-free medium (0% FBS EMEM). After incubation, cells were labeled with lysosomal marker LysoTracker Blue DND-22 (Thermo Fisher Scientific). Stained cells were analyzed using the confocal system described above. Fluorescence images were acquired by software cellSens (Olympus, Tokyo, Japan). In order to quantitatively assess colocalization, we calculated the Pearson correlation coefficient. To robustly analyze overall association between two fluorescent probes, it is well-established to calculate the Pearson correlation coefficient,118,119 which defines pixel-by-pixel correlation by representing mean-normalized to values from −1 (anticorrelation) to +1 (correlation).118,119 The Pearson correlation coefficient for fluorophore pairs (either DNA–lysosomes or DNA–pHrodo) was calculated using the Coloc 2 tool available in ImageJ.120
Statistical Analysis.
Cellular viability was analyzed and represented as mean ± SEM. The ANOVA analysis with subsequent Newman–Keuls test was utilized to assess the statistical significance of differences between the groups. MaxStat Pro 3.6 software (MaxStat Software, Cleverns, Germany) was used to perform all statistical analyses. Differences were considered statistically significant at (*) P < 0.05. Correlation analysis between the cell size and cellular uptake of DNs was done utilizing Spearman rank order correlation. Correlation coefficients and P values were calculated using SigmaPlot 13 software (Systat Software, Inc.).
Fluorescence microscopy analysis (namely, the analysis of cell size and uptake and colocalization of DNA–lysosomes or DNA–pHrodo) was subjected to quantitative assessment in accordance with rigorously defined guidelines.121 Guidance for quantitative confocal microscopy was employed to perform a quantitative assessment in accordance with previous publications.122,123 Quantitative microscopy analysis was carried out using images from three independent experiments. Each microscopy experiment included 10 randomly selected fields from each sample. The determination of sample size was performed in accordance with a previously published statistical methodology.124 Accordingly, the sample size for 95% confidence level and 0.9 statistical power was calculated as n = 30. Therefore, we analyzed at least 30 randomly selected cells for statistically relevant fluorescence microscopy image quantification.
Overall, a statistical methodology published previously in ref 124 was used to determine the sample size, assuming 95% confidence level and 0.9 statistical power.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge the Czech Ministry of Education, Youth and Sports (Project No. LTC19040) and MH CZ - DRO Institute for Clinical and Experimental Medicine – IKEM, IN 00023001. M.H. and P.J. would like to thank the Czech Science Foundation for support via an EXPRO Grant 19-26854X. N.S. acknowledges startup funds from Arizona State University and support by the National Science Foundation (DMR-BMAT CAREER award 1753387). Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under grant number 1DP2GM132931-01. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.1c14401.
Tables of full sequences of DNA strands and sequences of the synthesized peptides along with their expected masses and figures of topology diagram, MALDI-TOF mass spectra, agarose gel electrophoresis, size distribution, AFM characterization, FRET-based monitoring, schematic representation of the 6HB structure, raw fluorescent intensity versus temperature for the heating cycle, derivatives of heating and cooling curves, cytotoxicity of nanoparticles, DIC images, and Pearson’s correlation coefficient (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acsami.1c14401
The authors declare no competing financial interest.
Contributor Information
Barbora Smolková, Department of Optical and Biophysical Systems, Institute of Physics of the Czech Academy of Sciences, Prague 18221, Czech Republic.
Tara MacCulloch, Biodesign Center for Molecular Design and Biomimetics, Arizona State University, Tempe, Arizona 85287, United States; School of Molecular Sciences, Arizona State University, Tempe, Arizona 85287, United States.
Tyler F. Rockwood, Biodesign Center for Molecular Design and Biomimetics, Arizona State University, Tempe, Arizona 85287, United States; School of Molecular Sciences, Arizona State University, Tempe, Arizona 85287, United States
Minghui Liu, Biodesign Center for Molecular Design and Biomimetics, Arizona State University, Tempe, Arizona 85287, United States; School of Molecular Sciences, Arizona State University, Tempe, Arizona 85287, United States.
Skylar J. W. Henry, Biodesign Center for Molecular Design and Biomimetics, Arizona State University, Tempe, Arizona 85287, United States; School of Molecular Sciences, Arizona State University, Tempe, Arizona 85287, United States.
Adam Frtús, Department of Optical and Biophysical Systems, Institute of Physics of the Czech Academy of Sciences, Prague 18221, Czech Republic.
Mariia Uzhytchak, Department of Optical and Biophysical Systems, Institute of Physics of the Czech Academy of Sciences, Prague 18221, Czech Republic.
Mariia Lunova, Department of Optical and Biophysical Systems, Institute of Physics of the Czech Academy of Sciences, Prague 18221, Czech Republic; Institute for Clinical & Experimental Medicine (IKEM), Prague 14021, Czech Republic.
Martin Hof, J. Heyrovský Institute of Physical Chemistry of the Czech Academy of Sciences, Prague 18223, Czech Republic.
Piotr Jurkiewicz, J. Heyrovský Institute of Physical Chemistry of the Czech Academy of Sciences, Prague 18223, Czech Republic.
Alexandr Dejneka, Department of Optical and Biophysical Systems, Institute of Physics of the Czech Academy of Sciences, Prague 18221, Czech Republic.
Nicholas Stephanopoulos, Biodesign Center for Molecular Design and Biomimetics, Arizona State University, Tempe, Arizona 85287, United States; School of Molecular Sciences, Arizona State University, Tempe, Arizona 85287, United States.
Oleg Lunov, Department of Optical and Biophysical Systems, Institute of Physics of the Czech Academy of Sciences, Prague 18221, Czech Republic.
REFERENCES
- (1).Shi JJ; Kantoff PW; Wooster R; Farokhzad OC Cancer Nanomedicine: Progress, Challenges and Opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ahrens ET; Bulte JWM Tracking Immune Cells in Vivo Using Magnetic Resonance Imaging. Nat. Rev. Immunol 2013, 13, 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Lammers T; Ferrari M The Success of Nanomedicine. Nano Today 2020, 31, 100853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Pelaz B; Alexiou C; Alvarez-Puebla RA; Alves F; Andrews AM; Ashraf S; Balogh LP; Ballerini L; Bestetti A; Brendel C; Bosi S; Carril M; Chan WC; Chen C; Chen X; Chen X; Cheng Z; Cui D; Du J; Dullin C; Escudero A; Feliu N; Gao M; George M; Gogotsi Y; Grunweller A; Gu Z; Halas NJ; Hampp N; Hartmann RK; Hersam MC; Hunziker P; Jian J; Jiang X; Jungebluth P; Kadhiresan P; Kataoka K; Khademhosseini A; Kopecek J; Kotov NA; Krug HF; Lee DS; Lehr CM; Leong KW; Liang XJ; Ling Lim M; Liz-Marzan LM; Ma X; Macchiarini P; Meng H; Mohwald H; Mulvaney P; Nel AE; Nie S; Nordlander P; Okano T; Oliveira J; Park TH; Penner RM; Prato M; Puntes V; Rotello VM; Samarakoon A; Schaak RE; Shen Y; Sjoqvist S; Skirtach AG; Soliman MG; Stevens MM; Sung HW; Tang BZ; Tietze R; Udugama BN; VanEpps JS; Weil T; Weiss PS; Willner I; Wu Y; Yang L; Yue Z; Zhang Q; Zhang Q; Zhang XE; Zhao Y; Zhou X; Parak WJ Diverse Applications of Nanomedicine. ACS Nano 2017, 11, 2313–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Allen TM; Cullis PR Drug Delivery Systems: Entering the Mainstream. Science 2004, 303, 1818–22. [DOI] [PubMed] [Google Scholar]
- (6).Blanco E; Shen H; Ferrari M Principles of Nanoparticle Design for Overcoming Biological Barriers to Drug Delivery. Nat. Biotechnol 2015, 33, 941–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Davis ME; Chen Z; Shin DM Nanoparticle Therapeutics: An Emerging Treatment Modality for Cancer. Nat. Rev. Drug Discovery 2008, 7, 771–782. [DOI] [PubMed] [Google Scholar]
- (8).Lunov O; Uzhytchak M; Smolkova B; Lunova M; Jirsa M; Dempsey NM; Dias AL; Bonfim M; Hof M; Jurkiewicz P; Petrenko Y; Kubinova S; Dejneka A Remote Actuation of Apoptosis in Liver Cancer Cells Via Magneto-Mechanical Modulation of Iron Oxide Nanoparticles. Cancers 2019, 11, 1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).van der Meel R; Sulheim E; Shi Y; Kiessling F; Mulder WJM; Lammers T Smart Cancer Nanomedicine. Nat. Nanotechnol 2019, 14, 1007–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ventola CL Progress in Nanomedicine: Approved and Investigational Nanodrugs. P&T. 2017, 42 (12), 742–755. [PMC free article] [PubMed] [Google Scholar]
- (11).Bobo D; Robinson KJ; Islam J; Thurecht KJ; Corrie SR Nanoparticle-Based Medicines: A Review of Fda-Approved Materials and Clinical Trials to Date. Pharm. Res 2016, 33, 2373–87. [DOI] [PubMed] [Google Scholar]
- (12).Park K The Beginning of the End of the Nanomedicine Hype. J. Controlled Release 2019, 305, 221–222. [DOI] [PubMed] [Google Scholar]
- (13).Danhier F To Exploit the Tumor Microenvironment: Since the Epr Effect Fails in the Clinic, What Is the Future of Nanomedicine? J. Controlled Release 2016, 244, 108–121. [DOI] [PubMed] [Google Scholar]
- (14).Venditto VJ; Szoka FC Cancer Nanomedicines: So Many Papers and So Few Drugs! Adv. Drug Delivery Rev 2013, 65, 80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Frtus A; Smolkova B; Uzhytchak M; Lunova M; Jirsa M; Kubinova S; Dejneka A; Lunov O Analyzing the Mechanisms of Iron Oxide Nanoparticles Interactions with Cells: A Road from Failure to Success in Clinical Applications. J. Controlled Release 2020, 328, 59–77. [DOI] [PubMed] [Google Scholar]
- (16).Cheng YH; He CL; Riviere JE; Monteiro-Riviere NA; Lin ZM Meta-Analysis of Nanoparticle Delivery to Tumors Using a Physiologically Based Pharmacokinetic Modeling and Simulation Approach. ACS Nano 2020, 14, 3075–3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wilhelm S; Tavares AJ; Dai Q; Ohta S; Audet J; Dvorak HF; Chan WCW Analysis of Nanoparticle Delivery to Tumours. Nat. Rev. Mater 2016, 1, 16014. [Google Scholar]
- (18).Shi J; Kantoff PW; Wooster R; Farokhzad OC Cancer Nanomedicine: Progress, Challenges and Opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Gause KT; Wheatley AK; Cui J; Yan Y; Kent SJ; Caruso F Immunological Principles Guiding the Rational Design of Particles for Vaccine Delivery. ACS Nano 2017, 11, 54–68. [DOI] [PubMed] [Google Scholar]
- (20).Anchordoquy TJ; Barenholz Y; Boraschi D; Chorny M; Decuzzi P; Dobrovolskaia MA; Farhangrazi ZS; Farrell D; Gabizon A; Ghandehari H; Godin B; La-Beck NM; Ljubimova J; Moghimi SM; Pagliaro L; Park JH; Peer D; Ruoslahti E; Serkova NJ; Simberg D Mechanisms and Barriers in Cancer Nanomedicine: Addressing Challenges, Looking for Solutions. ACS Nano 2017, 11, 12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Qin SY; Zhang AQ; Cheng SX; Rong L; Zhang XZ Drug Self-Delivery Systems for Cancer Therapy. Biomaterials 2017, 112, 234–247. [DOI] [PubMed] [Google Scholar]
- (22).Cheng YH; He C; Riviere JE; Monteiro-Riviere NA; Lin Z Meta-Analysis of Nanoparticle Delivery to Tumors Using a Physiologically Based Pharmacokinetic Modeling and Simulation Approach. ACS Nano 2020, 14, 3075–3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Sindhwani S; Syed AM; Ngai J; Kingston BR; Maiorino L; Rothschild J; MacMillan P; Zhang YW; Rajesh NU; Hoang T; Wu JLY; Wilhelm S; Zilman A; Gadde S; Sulaiman A; Ouyang B; Lin Z; Wang LS; Egeblad M; Chan WCW The Entry of Nanoparticles into Solid Tumours. Nat. Mater 2020, 19, 566–575. [DOI] [PubMed] [Google Scholar]
- (24).Groeneveld E; de Mello Donegá C The Challenge of Colloidal Nanoparticle Synthesis. In Nanoparticles: Workhorses of Nanoscience, 1st ed.; de Mello Donega C, Ed.; Springer, 2014; pp 145–189. [Google Scholar]
- (25).Abedini A; Bakar AA; Larki F; Menon PS; Islam MS; Shaari S Recent Advances in Shape-Controlled Synthesis of Noble Metal Nanoparticles by Radiolysis Route. Nanoscale Res. Lett 2016, 11 (1), 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).da Silva AG; Rodrigues TS; Slater TJ; Lewis EA; Alves RS; Fajardo HV; Balzer R; da Silva AH; de Freitas IC; Oliveira DC; Assaf JM; Probst LF; Haigh SJ; Camargo PH Controlling Size, Morphology, and Surface Composition of Agau Nanodendrites in 15 S for Improved Environmental Catalysis under Low Metal Loadings. ACS Appl. Mater. Interfaces 2015, 7, 25624–32. [DOI] [PubMed] [Google Scholar]
- (27).Seeman NC DNA in a Material World. Nature 2003, 421, 427–31. [DOI] [PubMed] [Google Scholar]
- (28).Goodman RP; Schaap IAT; Tardin CF; Erben CM; Berry RM; Schmidt CF; Turberfield AJ Rapid Chiral Assembly of Rigid DNA Building Blocks for Molecular Nanofabrication. Science 2005, 310, 1661–1665. [DOI] [PubMed] [Google Scholar]
- (29).He Y; Ye T; Su M; Zhang C; Ribbe AE; Jiang W; Mao CD Hierarchical Self-Assembly of DNA into Symmetric Supra-molecular Polyhedra. Nature 2008, 452, 198–201. [DOI] [PubMed] [Google Scholar]
- (30).Burns JR; Seifert A; Fertig N; Howorka S A Biomimetic DNA-Based Channel for the Ligand-Controlled Transport of Charged Molecular Cargo across a Biological Membrane. Nat. Nanotechnol 2016, 11, 152–156. [DOI] [PubMed] [Google Scholar]
- (31).Rothemund PW Folding DNA to Create Nanoscale Shapes and Patterns. Nature 2006, 440, 297–302. [DOI] [PubMed] [Google Scholar]
- (32).Wang PF; Meyer TA; Pan V; Dutta PK; Ke YG The Beauty and Utility of DNA Origami. Chem. 2017, 2, 359–382. [Google Scholar]
- (33).Han D; Pal S; Nangreave J; Deng Z; Liu Y; Yan H DNA Origami with Complex Curvatures in Three-Dimensional Space. Science 2011, 332, 342–6. [DOI] [PubMed] [Google Scholar]
- (34).Benson E; Mohammed A; Gardell J; Masich S; Czeizler E; Orponen P; Hogberg B DNA Rendering of Polyhedral Meshes at the Nanoscale. Nature 2015, 523, 441–444. [DOI] [PubMed] [Google Scholar]
- (35).Zhang F; Jiang S; Wu S; Li Y; Mao C; Liu Y; Yan H Complex Wireframe DNA Origami Nanostructures with Multi-Arm Junction Vertices. Nat. Nanotechnol 2015, 10, 779–84. [DOI] [PubMed] [Google Scholar]
- (36).Veneziano R; Ratanalert S; Zhang K; Zhang F; Yan H; Chiu W; Bathe M Designer Nanoscale DNA Assemblies Programmed from the Top Down. Science 2016, 352, 1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Zhan P; Dutta PK; Wang P; Song G; Dai M; Zhao SX; Wang ZG; Yin P; Zhang W; Ding B; Ke Y Reconfigurable Three-Dimensional Gold Nanorod Plasmonic Nanostructures Organized on DNA Origami Tripod. ACS Nano 2017, 11, 1172–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Knudsen JB; Liu L; Bank Kodal AL; Madsen M; Li Q; Song J; Woehrstein JB; Wickham SF; Strauss MT; Schueder F; Vinther J; Krissanaprasit A; Gudnason D; Smith AA; Ogaki R; Zelikin AN; Besenbacher F; Birkedal V; Yin P; Shih WM; Jungmann R; Dong M; Gothelf KV Routing of Individual Polymers in Designed Patterns. Nat. Nanotechnol 2015, 10, 892–8. [DOI] [PubMed] [Google Scholar]
- (39).Gopinath A; Miyazono E; Faraon A; Rothemund PW Engineering and Mapping Nanocavity Emission Via Precision Placement of DNA Origami. Nature 2016, 535, 401–5. [DOI] [PubMed] [Google Scholar]
- (40).Langecker M; Arnaut V; Martin TG; List J; Renner S; Mayer M; Dietz H; Simmel FC Synthetic Lipid Membrane Channels Formed by Designed DNA Nanostructures. Science 2012, 338, 932–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Veetil AT; Chakraborty K; Xiao K; Minter MR; Sisodia SS; Krishnan Y Cell-Targetable DNA Nanocapsules for Spatiotemporal Release of Caged Bioactive Small Molecules. Nat. Nanotechnol 2017, 12, 1183–1189. [DOI] [PubMed] [Google Scholar]
- (42).Ge Z; Liu J; Guo L; Yao G; Li Q; Wang L; Li J; Fan C Programming Cell-Cell Communications with Engineered Cell Origami Clusters. J. Am. Chem. Soc 2020, 142, 8800–8808. [DOI] [PubMed] [Google Scholar]
- (43).Veneziano R; Moyer TJ; Stone MB; Wamhoff EC; Read BJ; Mukherjee S; Shepherd TR; Das J; Schief WR; Irvine DJ; Bathe M Role of Nanoscale Antigen Organization on B-Cell Activation Probed Using DNA Origami. Nat. Nanotechnol 2020, 15, 716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Akbari E; Mollica MY; Lucas CR; Bushman SM; Patton RA; Shahhosseini M; Song JW; Castro CE Engineering Cell Surface Function with DNA Origami. Adv. Mater 2017, 29, 1703632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Halley PD; Lucas CR; McWilliams EM; Webber MJ; Patton RA; Kural C; Lucas DM; Byrd JC; Castro CE Daunorubicin-Loaded DNA Origami Nanostructures Circumvent Drug-Resistance Mechanisms in a Leukemia Model. Small 2016, 12, 308–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Zhang Q; Jiang Q; Li N; Dai LR; Liu Q; Song LL; Wang JY; Li YQ; Tian J; Ding BQ; Du Y DNA Origami as an in Vivo Drug Delivery Vehicle for Cancer Therapy. ACS Nano 2014, 8, 6633–6643. [DOI] [PubMed] [Google Scholar]
- (47).Yan J; Hu C; Wang P; Zhao B; Ouyang X; Zhou J; Liu R; He D; Fan C; Song S Growth and Origami Folding of DNA on Nanoparticles for High-Efficiency Molecular Transport in Cellular Imaging and Drug Delivery. Angew. Chem., Int. Ed 2015, 54, 2431–5. [DOI] [PubMed] [Google Scholar]
- (48).Douglas SM; Bachelet I; Church GM A Logic-Gated Nanorobot for Targeted Transport of Molecular Payloads. Science 2012, 335, 831–4. [DOI] [PubMed] [Google Scholar]
- (49).Schuller VJ; Heidegger S; Sandholzer N; Nickels PC; Suhartha NA; Endres S; Bourquin C; Liedl T Cellular Immunostimulation by Cpg-Sequence-Coated DNA Origami Structures. ACS Nano 2011, 5, 9696–702. [DOI] [PubMed] [Google Scholar]
- (50).Wang P; Rahman MA; Zhao Z; Weiss K; Zhang C; Chen Z; Hurwitz SJ; Chen ZG; Shin DM; Ke Y Visualization of the Cellular Uptake and Trafficking of DNA Origami Nanostructures in Cancer Cells. J. Am. Chem. Soc 2018, 140, 2478–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Bastings MMC; Anastassacos FM; Ponnuswamy N; Leifer FG; Cuneo G; Lin CX; Ingber DE; Ryu JH; Shih WM Modulation of the Cellular Uptake of DNA Origami through Control over Mass and Shape. Nano Lett. 2018, 18, 3557–3564. [DOI] [PubMed] [Google Scholar]
- (52).Whitehouse WL; Noble JE; Ryadnov MG; Howorka S Cholesterol Anchors Enable Efficient Binding and Intracellular Uptake of DNA Nanostructures. Bioconjugate Chem. 2019, 30, 1836–1844. [DOI] [PubMed] [Google Scholar]
- (53).Del Pino P; Pelaz B; Zhang Q; Maffre P; Nienhaus GU; Parak WJ Protein Corona Formation around Nanoparticles - from the Past to the Future. Mater. Horizons 2014, 1 (3), 301–313. [Google Scholar]
- (54).Ke PC; Lin S; Parak WJ; Davis TP; Caruso F A Decade of the Protein Corona. ACS Nano 2017, 11, 11773–11776. [DOI] [PubMed] [Google Scholar]
- (55).Ge C; Tian J; Zhao Y; Chen C; Zhou R; Chai Z Towards Understanding of Nanoparticle-Protein Corona. Arch. Toxicol 2015, 89, 519–39. [DOI] [PubMed] [Google Scholar]
- (56).Kharazian B; Hadipour NL; Ejtehadi MR Understanding the Nanoparticle-Protein Corona Complexes Using Computational and Experimental Methods. Int. J. Biochem. Cell Biol 2016, 75, 162–174. [DOI] [PubMed] [Google Scholar]
- (57).Saptarshi SR; Duschl A; Lopata AL Interaction of Nanoparticles with Proteins: Relation to Bio-Reactivity of the Nanoparticle. J. Nanobiotechnology 2013, 11, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Ponnuswamy N; Bastings MMC; Nathwani B; Ryu JH; Chou LYT; Vinther M; Li WA; Anastassacos FM; Mooney DJ; Shih WM Oligolysine-Based Coating Protects DNA Nanostructures from Low-Salt Denaturation and Nuclease Degradation. Nat. Commun 2017, 8, 15654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Li M; Tao Y; Shu YL; LaRochelle JR; Steinauer A; Thompson D; Schepartz A; Chen ZY; Liu DR Discovery and Characterization of a Peptide That Enhances Endosomal Escape of Delivered Proteins in Vitro and in Vivo. J. Am. Chem. Soc 2015, 137, 14084–14093. [DOI] [PubMed] [Google Scholar]
- (60).Arulkumaran N; Lanphere C; Gaupp C; Burns JR; Singer M; Howorka S DNA Nanodevices with Selective Immune Cell Interaction and Function. ACS Nano 2021, 15, 4394–4404. [DOI] [PubMed] [Google Scholar]
- (61).Burns JR; Howorka S Defined Bilayer Interactions of DNA Nanopores Revealed with a Nuclease-Based Nanoprobe Strategy. ACS Nano 2018, 12, 3263–3271. [DOI] [PubMed] [Google Scholar]
- (62).Birkholz O; Burns JR; Richter CP; Psathaki OE; Howorka S; Piehler J Multi-Functional DNA Nanostructures That Puncture and Remodel Lipid Membranes into Hybrid Materials. Nat. Commun 2018, 9, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Mei QA; Wei XX; Su FY; Liu Y; Youngbull C; Johnson R; Lindsay S; Yan H; Meldrum D Stability of DNA Origami Nanoarrays in Cell Lysate. Nano Lett. 2011, 11, 1477–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Keum JW; Bermudez H Enhanced Resistance of DNA Nanostructures to Enzymatic Digestion. Chem. Commun 2009, 7036–7038. [DOI] [PubMed] [Google Scholar]
- (65).Massich MD; Giljohann DA; Schmucker AL; Patel PC; Mirkin CA Cellular Response of Polyvalent Oligonucleotide-Gold Nanoparticle Conjugates. ACS Nano 2010, 4, 5641–5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Shen XB; Jiang Q; Wang JY; Dai LR; Zou GZ; Wang ZG; Chen WQ; Jiang W; Ding BQ Visualization of the Intracellular Location and Stability of DNA Origami with a Label-Free Fluorescent Probe. Chem. Commun 2012, 48, 11301–11303. [DOI] [PubMed] [Google Scholar]
- (67).Walsh AS; Yin HF; Erben CM; Wood MJA; Turberfield AJ DNA Cage Delivery to Mammalian Cells. ACS Nano 2011, 5, 5427–5432. [DOI] [PubMed] [Google Scholar]
- (68).Hahn J; Wickham SFJ; Shih WM; Perrault SD Addressing the Instability of DNA Nanostructures in Tissue Culture. ACS Nano 2014, 8, 8765–8775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Wei XX; Nangreave J; Jiang SX; Yan H; Liu Y Mapping the Thermal Behavior of DNA Origami Nanostructures. J. Am. Chem. Soc 2013, 135, 6165–6176. [DOI] [PubMed] [Google Scholar]
- (70).Zhang YN; Poon W; Tavares AJ; McGilvray ID; Chan WCW Nanoparticle-Liver Interactions: Cellular Uptake and Hepatobiliary Elimination. J. Controlled Release 2016, 240, 332–348. [DOI] [PubMed] [Google Scholar]
- (71).Tsoi KM; MacParland SA; Ma XZ; Spetzler VN; Echeverri J; Ouyang B; Fadel SM; Sykes EA; Goldaracena N; Kaths JM; Conneely JB; Alman BA; Selzner M; Ostrowski MA; Adeyi OA; Zilman A; McGilvray ID; Chan WC Mechanism of Hard-Nanomaterial Clearance by the Liver. Nat. Mater 2016, 15, 1212–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Guo L; Dial S; Shi LM; Branham W; Liu J; Fang JL; Green B; Deng H; Kaput J; Ning BT Similarities and Differences in the Expression of Drug-Metabolizing Enzymes between Human Hepatic Cell Lines and Primary Human Hepatocytes. Drug Metab. Dispos 2011, 39, 528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Fernandez-Checa JC; Bagnaninchi P; Ye H; Sancho-Bru P; Falcon-Perez JM; Royo F; Garcia-Ruiz C; Konu O; Miranda J; Lunov O; Dejneka A; Elfick A; McDonald A; Sullivan GJ; Aithal G; Lucena MI; Andrade RJ; Fromenty B; Krannendonk M; Cubero FJ; Nelson LJ Advanced Preclinical Models for Evaluation of Drug Induced Liver Injury - Consensus Statement by the European Drug-Induced Liver Injury Network [Pro-Euro-Dili-Net]. J. Hepatol 2021, 75 (4), 935–959. [DOI] [PubMed] [Google Scholar]
- (74).Lunova M; Prokhorov A; Jirsa M; Hof M; Olzynska A; Jurkiewicz P; Kubinova S; Lunov O; Dejneka A Nanoparticle Core Stability and Surface Functionalization Drive the Mtor Signaling Pathway in Hepatocellular Cell Lines. Sci. Rep 2017, 7, 16049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Ni Z; Wang B; Dai X; Ding W; Yang T; Li X; Lewin S; Xu L; Lian J; He F Hcc Cells with High Levels of Bcl-2 Are Resistant to Abt-737 Via Activation of the Ros-Jnk-Autophagy Pathway. Free Radical Biol. Med 2014, 70, 194–203. [DOI] [PubMed] [Google Scholar]
- (76).Andreu N; Phelan J; de Sessions PF; Cliff JM; Clark TG ; Hibberd ML Primary Macrophages and J774 Cells Respond Differently to Infection with Mycobacterium Tuberculosis. Sci. Rep 2017, 7, 42225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Smolkova B; Lunova M; Lynnyk A; Uzhytchak M; Churpita O; Jirsa M; Kubinova S; Lunov O; Dejneka A Non-Thermal Plasma, as a New Physicochemical Source, to Induce Redox Imbalance and Subsequent Cell Death in Liver Cancer Cell Lines. Cell. Physiol. Biochem 2019, 52 (1), 119–140. [DOI] [PubMed] [Google Scholar]
- (78).Lunov O; Syrovets T; Loos C; Beil J; Delacher M; Tron K; Nienhaus GU; Musyanovych A; Mailander V; Landfester K; Simmet T Differential Uptake of Functionalized Polystyrene Nanoparticles by Human Macrophages and a Monocytic Cell Line. ACS Nano 2011, 5, 1657–69. [DOI] [PubMed] [Google Scholar]
- (79).Bade ND; Kamien RD; Assoian RK; Stebe KJ Curvature and Rho Activation Differentially Control the Alignment of Cells and Stress Fibers. Sci. Adv 2017, 3, No. e1700150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Lunova M; Zablotskii V; Dempsey NM; Devillers T; Jirsa M; Sykova E; Kubinova S; Lunov O; Dejneka A Modulation of Collective Cell Behaviour by Geometrical Constraints. Integr. Biol 2016, 8, 1099–1110. [DOI] [PubMed] [Google Scholar]
- (81).Amodeo AA; Skotheim JM Cell-Size Control. Cold Spring Harbor Perspect. Biol 2016, 8, No. a019083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Khetan J; Shahinuzzaman M; Barua S; Barua D Quantitative Analysis of the Correlation between Cell Size and Cellular Uptake of Particles. Biophys. J 2019, 116, 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Balakrishnan D; Wilkens GD; Heddle JG Delivering DNA Origami to Cells. Nanomedicine 2019, 14, 911–925. [DOI] [PubMed] [Google Scholar]
- (84).Mikkila J; Eskelinen AP; Niemela EH; Linko V; Frilander MJ; Torma P; Kostiainen MA Virus-Encapsulated DNA Origami Nanostructures for Cellular Delivery. Nano Lett. 2014, 14, 2196–200. [DOI] [PubMed] [Google Scholar]
- (85).Liu K; Xu C; Liu J Regulation of Cell Binding and Entry by DNA Origami Mediated Spatial Distribution of Aptamers. J. Mater. Chem. B 2020, 8, 6802–6809. [DOI] [PubMed] [Google Scholar]
- (86).Lee DS; Qian H; Tay CY; Leong DT Cellular Processing and Destinies of Artificial DNA Nanostructures. Chem. Soc. Rev 2016, 45, 4199–4225. [DOI] [PubMed] [Google Scholar]
- (87).Mosquera J; Garcia I; Liz-Marzan LM Cellular Uptake of Nanoparticles Versus Small Molecules: A Matter of Size. Acc. Chem. Res 2018, 51, 2305–2313. [DOI] [PubMed] [Google Scholar]
- (88).Behzadi S; Serpooshan V; Tao W; Hamaly MA; Alkawareek MY; Dreaden EC; Brown D; Alkilany AM; Farokhzad OC; Mahmoudi M Cellular Uptake of Nanoparticles: Journey inside the Cell. Chem. Soc. Rev 2017, 46, 4218–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Rennick JJ; Johnston APR; Parton RG Key Principles and Methods for Studying the Endocytosis of Biological and Nanoparticle Therapeutics. Nat. Nanotechnol 2021, 16, 266–276. [DOI] [PubMed] [Google Scholar]
- (90).Linko V; Ora A; Kostiainen MA DNA Nanostructures as Smart Drug-Delivery Vehicles and Molecular Devices. Trends Biotechnol. 2015, 33, 586–594. [DOI] [PubMed] [Google Scholar]
- (91).Pei D; Buyanova M Overcoming Endosomal Entrapment in Drug Delivery. Bioconjugate Chem. 2019, 30, 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Huotari J; Helenius A Endosome Maturation. EMBO J. 2011, 30, 3481–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Yang X; Fan B; Gao W; Li L; Li T; Sun J; Peng X; Li X; Wang Z; Wang B; Zhang R; Xie J Enhanced Endosomal Escape by Photothermal Activation for Improved Small Interfering Rna Delivery and Antitumor Effect. Int. J. Nanomed 2018, 13, 4333–4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Ahmed S; Fujita S; Matsumura K Enhanced Protein Internalization and Efficient Endosomal Escape Using Polyampholyte-Modified Liposomes and Freeze Concentration. Nanoscale 2016, 8, 15888–901. [DOI] [PubMed] [Google Scholar]
- (95).Gao Y; Li Y; Li Y; Yuan L; Zhou Y; Li J; Zhao L; Zhang C; Li X; Liu Y Psma-Mediated Endosome Escape-Accelerating Polymeric Micelles for Targeted Therapy of Prostate Cancer and the Real Time Tracing of Their Intracellular Trafficking. Nanoscale 2015, 7, 597–612. [DOI] [PubMed] [Google Scholar]
- (96).Huang G; Chen Q; Wu W; Wang J; Chu PK; Bai H; Tang G Reconstructed Chitosan with Alkylamine for Enhanced Gene Delivery by Promoting Endosomal Escape. Carbohydr. Polym 2020, 227, 115339. [DOI] [PubMed] [Google Scholar]
- (97).Zhu DC; Yan HJ; Zhou ZX; Tang JB; Liu XR; Hartmann R; Parak WJ; Feliu N; Shen YQ Detailed Investigation on How the Protein Corona Modulates the Physicochemical Properties and Gene Delivery of Polyethylenimine (Pei) Polyplexes. Biomater. Sci 2018, 6, 1800–1817. [DOI] [PubMed] [Google Scholar]
- (98).Quagliarini E; Di Santo R; Palchetti S; Ferri G; Cardarelli F; Pozzi D; Caracciolo G Effect of Protein Corona on the Transfection Efficiency of Lipid-Coated Graphene Oxide-Based Cell Transfection Reagents. Pharmaceutics 2020, 12, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Zeidler JD; Fernandes-Siqueira LO; Carvalho AS; Cararo-Lopes E; Dias MH; Ketzer LA; Galina A; Da Poian AT Short-Term Starvation Is a Strategy to Unravel the Cellular Capacity of Oxidizing Specific Exogenous/Endogenous Substrates in Mitochondria. J. Biol. Chem 2017, 292, 14176–14187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Yuan HF; Tan BD; Gao SJ Tenovin-6 Impairs Autophagy by Inhibiting Autophagic Flux. Cell Death Dis. 2017, 8, No. e2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Fischer TD; Wang JH; Vlada A; Kim JS; Behrns KE Role of Autophagy in Differential Sensitivity of Hepatocarcinoma Cells to Sorafenib. World J. Hepatol 2014, 6, 752–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Zhang K; Cheng X; Zhao L; Huang M; Tao Y; Zhang H; Rosenholm JM; Zhuang M; Chen Z-Y; Chen B; Shu Y Direct Functional Protein Delivery with a Peptide into Neonatal and Adult Mammalian Inner Ear In vivo. Mol. Ther.–Methods Clin. Dev 2020, 18, 511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Bruce VJ; McNaughton BR Inside Job: Methods for Delivering Proteins to the Interior of Mammalian Cells. Cell Chem. Biol 2017, 24, 924–934. [DOI] [PubMed] [Google Scholar]
- (104).Wensley HJ; Johnston DA; Smith WS; Holmes SE; Flavell SU; Flavell DJ A Flow Cytometric Method to Quantify the Endosomal Escape of a Protein Toxin to the Cytosol of Target Cells. Pharm. Res 2019, 37 (1), 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Martens TF; Remaut K; Demeester J; De Smedt SC; Braeckmans K Intracellular Delivery of Nanomaterials: How to Catch Endosomal Escape in the Act. Nano Today 2014, 9, 344–364. [Google Scholar]
- (106).Artifacts of Light. Nat. Methods 2013, 10, 1135–1135. [Google Scholar]
- (107).Lunova M; Smolkova B; Uzhytchak M; Janouskova KZ; Jirsa M; Egorova D; Kulikov A; Kubinova S; Dejneka A; Lunov O Light-Induced Modulation of the Mitochondrial Respiratory Chain Activity: Possibilities and Limitations. Cell. Mol. Life Sci 2020, 77, 2815–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Hayashi S; Okada Y Ultrafast Superresolution Fluorescence Imaging with Spinning Disk Confocal Microscope Optics. Mol. Biol. Cell 2015, 26, 1743–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Miksa M; Kornura H; Wu RQ; Shah KG; Wang P A Novel Method to Determine the Engulfment of Apoptotic Cells by Macrophages Using Phrodo Succinimidyl Ester. J. Immunol. Methods 2009, 342, 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Zasloff M Antimicrobial Peptides of Multicellular Organisms. Nature 2002, 415, 389–95. [DOI] [PubMed] [Google Scholar]
- (111).Shang L; Nienhaus GU In Situ Characterization of Protein Adsorption onto Nanoparticles by Fluorescence Correlation Spectroscopy. Acc. Chem. Res 2017, 50, 387–395. [DOI] [PubMed] [Google Scholar]
- (112).Uzhytchak M; Smolkova B; Lunova M; Jirsa M; Frtus A; Kubinova S; Dejneka A; Lunov O Iron Oxide Nanoparticle-Induced Autophagic Flux Is Regulated by Interplay between P53-Mtor Axis and Bcl-2 Signaling in Hepatic Cells. Cells 2020, 9, 1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Frtus A; Smolkova B; Uzhytchak M; Lunova M; Jirsa M; Hof M; Jurkiewicz P; Lozinsky VI; Wolfova L; Petrenko Y; Kubinova S; Dejneka A; Lunov O Hepatic Tumor Cell Morphology Plasticity under Physical Constraints in 3d Cultures Driven by Yap-Mtor Axis. Pharmaceuticals 2020, 13, 430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Gavet O; Pines J Progressive Activation of Cyclinb1-Cdk1 Coordinates Entry to Mitosis. Dev. Cell 2010, 18, 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Hachet-Haas M; Converset N; Marchal O; Matthes H; Gioria S; Galzi JL; Lecat S Fret and Colocalization Analyzer - a Method to Validate Measurements of Sensitized Emission Fret Acquired by Confocal Microscopy and Available as an Imagej Plug-In. Microsc. Res. Tech 2006, 69, 941–956. [DOI] [PubMed] [Google Scholar]
- (116).Bohmert L; Voss L; Stock V; Braeuning A; Lampen A; Sieg H Isolation Methods for Particle Protein Corona Complexes from Protein-Rich Matrices. Nanoscale Adv. 2020, 2, 563–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Widengren J; Mets Ü Conceptual Basis of Fluorescence Correlation Spectroscopy and Related Techniques as Tools in Bioscience. Single Molecule Detection in Solution: Methods and Applications, 1st ed.; Wiley-VCH, 2002; pp 69–120. [Google Scholar]
- (118).Manders EM; Stap J; Brakenhoff GJ; van Driel R; Aten JA Dynamics of Three-Dimensional Replication Patterns During the S-Phase, Analysed by Double Labelling of DNA and Confocal Microscopy. J. Cell Sci 1992, 103 (Pt 3), 857–862. [DOI] [PubMed] [Google Scholar]
- (119).Arena ET; Rueden CT; Hiner MC; Wang S; Yuan M; Eliceiri KW Quantitating the Cell: Turning Images into Numbers with Imagej. Wiley Interdiscip. Rev.-Dev. Biol 2017, 6, No. e260. [DOI] [PubMed] [Google Scholar]
- (120). https://imagej.net/plugins/coloc-2.
- (121).Hamilton N Quantification and Its Applications in Fluorescent Microscopy Imaging. Traffic 2009, 10, 951–61. [DOI] [PubMed] [Google Scholar]
- (122).Jonkman J; Brown CM; Wright GD; Anderson KI; North AJ Tutorial: Guidance for Quantitative Confocal Microscopy. Nat. Protoc 2020, 15, 1585–1611. [DOI] [PubMed] [Google Scholar]
- (123).Lee JY; Kitaoka M A Beginner’s Guide to Rigor and Reproducibility in Fluorescence Imaging Experiments. Mol. Biol. Cell 2018, 29, 1519–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Dell RB; Holleran S; Ramakrishnan R Sample Size Determination. ILAR J. 2002, 43, 207–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.