

Abstract
The endogenous neuropeptide FF (NPFF) and its two cognate G protein-coupled receptors, Neuropeptide FF Receptors 1 and 2 (NPFFR1 and NPFFR2), represent a relatively new target system for many therapeutic applications including pain regulation, modulation of opioid side effects, drug reward, anxiety, cardiovascular conditions and other peripheral effects. Since the cloning of NPFFR1 and NPFFR2 in 2000, significant progress has been made to understand their pharmacological roles and interactions with other receptor systems, notably the opioid receptors. A variety of NPFFR ligands with different mechanisms of action (agonists or antagonists) have been discovered although with limited subtype selectivities. Differential pharmacological effects have been observed for many of these NPFFR ligands, depending on assays/models employed and routes of administration. In this Perspective, we highlight the therapeutic potentials, current knowledge gaps, and the latest updates of the development of peptidic and small molecule NPFFR ligands as tool compounds and therapeutic candidates.
Keywords: Neuropeptide FF, NPFF, opioid, pain, therapeutics development, Structure-Activity Relationship (SAR)
Graphical Abstract
1. Introduction
Neuropeptide FF (NPFF; FLFQPQRF-NH2) is a neuropeptide belonging to the RF-amide family, members of which all share a C-terminal RF-amide motif in vertebrates. NPFF, together with neuropeptide AF (NPAF; AGQGLSSPFWSLAAPQRF-NH2), was identified in 1985 by Yang and colleagues from the isolation of endogenous peptides that displayed immunoreactivity against the cardioexcitatory peptide FMRF-NH2 in bovine brain extract.1 Additional members of the RF-amide neuropeptides were later identified including gonadotropin-inhibitory hormone (GnIH),2, 3 pyroglutamylated RFamide (QRFP),4 prolactin-releasing peptide (PrRP),5 and Kisspeptin.6 The NPFF gene in human, murine, and bovine tissues is highly conserved and encodes precursors for the three neuropeptides NPFF, NPAF, and neuropeptide SF (NPSF; SQAFLFQPQRF-NH2).7
In 2000, two cognate receptors for NPFF were discovered in human and rat brain.8, 9 They were named Neuropeptide FF Receptor 1 (NPFFR1, also known as GPR147) and Neuropeptide FF Receptor 2 (NPFFR2, also known as GPR74), respectively. NPFF binds with high affinities to both receptors (hNPFFR1, Kd = 1.13 nM; hNPFFR2 Kd = 0.37 nM).8
NPFF has long been considered as an endogenous opioid-modulating peptide.1, 10, 11 NPFF modulates opioid-induced antinociception differentially, either blocking or potentiating morphine-induced analgesia, depending on the site of administration.12–15 An increased level of NPFF in the central nervous system (CNS) has been suggested to contribute to the development of tolerance and dependence for opioids.11 The NPFF system has also been shown to play an important role in modulating the effects of other drugs of abuse,11, 16 as well as in physiological processes including insulin release, food intake, memory, blood pressure, electrolyte balance, and neural regeneration.17, 18 Therefore, NPFF as a relatively new target system has attracted increasing attention for many therapeutic applications. In this review, we will first discuss the roles of the NPFF system and its therapeutic potentials, and then summarize NPFFR ligands, either peptides, peptidomimetics, or small molecules, reported to date.
NPFF/NPFFR Distribution and Signaling
NPFF has been found across species including human, rodent, bovine, and guinea pig tissues,7 indicating that NPFF is physiologically important. Distribution of NPFF and its receptors has been described in details in previous reviews.19, 20 Gene expressions of human and rat NPFFRs were found mainly in brain and spinal cord with low peripheral levels except hNPFFR2 which had the highest mRNA level in placenta (Table 1).8 NPFF was found in discrete areas in the rat CNS with the highest levels in the dorsal spinal cord and the posterior lobe of the pituitary gland.19, 21, 22 At tissue level, the distribution of NPFF mRNA is similar to that of NPFF immunoreactivity except in the hypothalamic region.21 However, it should be noted that NPFF mRNA is expressed in cell bodies whereas NPFF antibodies were detected in nerve terminals.19, 21 Radioligand binding with the [125I][Tyr1]NPFF tracer in rat brains confirmed that [125I][Tyr1]NPFF binding was limited to the dorsal portion of the spinal cord; no binding was detected in the membranes of the ventral cord.23 This binding was specific to NPFFRs rather than NPY’s receptors.23 The tracer binding was not affected by chronic morphine treatment across various brain regions and spinal cord.23
Table 1.
Comparisons of distributions of NPFFR1 and NPFFR2 mRNA in human and rat tissues.4
| Tissue | hNPFFR1 | hNPFFR2 | rNPFFR1 | rNPFFR2 |
|---|---|---|---|---|
| Amygdala | 44% | 27% | 57% | 42% |
| Cerebellum | 20% | Trace | 17% | 10% |
| Heart | Trace | Trace | 3% | 82% |
| Hippocampus | 45% | 7% | 20% | 8% |
| Hypothalamus | 21% | 2% | 100% | 84% |
| Kidney | 1% | 1% | 1% | 20% |
| Liver | Trace | Trace | 2% | 3% |
| Lung | 7% | 1% | 4% | 16% |
| Pancreas | Trace | 1% | Trace | Trace |
| Pituitary | 2% | 5% | 24% | 34% |
| Placenta | Trace | 100% | N/A | N/A |
| Spinal cord | 100% | 1% | 24% | 100% |
| Spleen | 8% | 4% | Trace | Trace |
| Stomach | 1% | Trace | Trace | 14% |
| Skeletal muscle | 1% | 1% | Trace | Trace |
| Substantia nigra | 13% | 1% | 49% | 67% |
| Thalamus | 30% | 2% | 3% | 15% |
| Whole brain | 21% | 8% | 21% | 24% |
Note: mRNA levels are expressed as percent of highest expressing tissue: spinal cord for hNPFFR1, placenta for hNPFFR2, hypothalamus for rNPFFR1 and spinal cord for rNPFFR2. N/A: Not Available.
Although distribution of rat and human NPFFR1 and NPFFR2 mRNA is generally similar in the CNS and periphery, there are some notable species differences. The highest level of human NPFFR1 mRNA was found in spinal cord whereas the highest level of human NPFFR2 mRNA was detected in placenta.8 In contrast, the highest level of rat NPFFR1 mRNA was found in hypothalamus, whereas among the tested tissues, the highest level of rat NPFFR2 mRNA was found in spinal cord.8 Rat NPFFR2 mRNA level in placenta was not evaluated in the study.8 Human NPFFR1 mRNA was more abundant in spinal cord, whereas in rat spinal cord only NPFFR2 mRNA was present,8 consistent with the autoradiographic studies.24 While NPFFR2 mRNA was found at high levels in rat heart, little of either NPFFR subtype was found in human heart.8 On the contrary, both NPFFR subtypes were expressed at higher levels in human spleen than in rat spleen.8 Density of each NPFFR subtype in CNS also varies between Sprague-Dawley and Wistar rats and between Swiss and C57BL/6-SV129 mice.25
Several lines of evidence suggest upregulation of NPFF and NPFFR2 gene expression under pathological conditions. While no NPFF-immunoreactivity (NPFF-ir) neuronal cell bodies are found in rat spinal cord under normal conditions, NPFF-ir neurons were found in spinal cord of rats with carrageenan inflammation.26 In agreement to the initial study, NPFF and NPFFR2 mRNA was found to be upregulated by peripheral carrageenan inflammation.7, 27, 28 However, upregulation of spinal cord NPFF mRNA was not observed in neuropathic pain models.7, 28
The signaling of the NPFF system is still not well defined. Both NPFFRs are able to couple to Gi/o proteins when expressed in Chinese hamster ovary (CHO) cells,29 human embryonic kidney (HEK293) cells,8, 9 or human SH-SY5Y neuroblastoma cells.10 NPFFR1 mainly couples to Gαi3 and Gαs proteins, while NPFFR2 couples with Gαi2, Gαi3, Gαo and Gαs proteins in these engineered cells.10, 30 In the mouse neuroblastoma cells endogenously expressing NPFFR2, both NPFF and dNPA (8, Table 2), a selective NPFFR2 agonist increased extracellular signal-regulated kinase (ERK) phosphorylation and stimulate neurite outgrowth.18 NPFF and its analogues which had no effects by themselves, attenuated the opioid-induced inhibition of calcium levels in dorsal root ganglion, dorsal raphe and hypothalamic periventricular nucleus.31–33 However, these effects appear to be modulation of opioid-receptors rather than via direct NPFFR-mediated action on voltage-gated calcium channels.34 dNPA has been reported to increase voltage-dependent potassium outward currents in F-11 DRG cells, a hybridoma derived from rat DRG and mouse neuroblastoma.35 Lastly, NPFF and related RFamides have been reported to directly modulated acid-sensing ion channels (ASICs) which are known to involve in many physiological and pathophysiological processes including pain modulation.36
Table 2.
Biological activities of representative NPFF-derived peptidic probes.
| Ligand | Sequence | Usage | In vitro activities | In vivo activities |
|---|---|---|---|---|
| NPFF (a.k.a. F8Fa, 1) | FLFQPQRF-NH2 | Agonist | hNPFFR1 COS-7 or HEK cells Kd = 1.13 nM4 hNPFFR2 COS-7 or HEK cells Kd = 0.21 – 0.37 nM4, 81 Rat spinal cord Ki = 0.21 – 0.34 nM19, 77, 78, 81 hNPFFR2 EC50 = 3.16 nM81 |
Analgesia1, 10, 11, 36–38, 77, 78 Opioid abuse45, 49, 53 Amphetamine abuse55–57 Cocaine abuse58 Cardiovascular activities32, 59, 60, 62 Gastrointestinal mobility74 |
| [Tyr1]NPFF (a.k.a. Y8Fa, 2) | YLFQPQRF-NH2 | Agonist | Rat spinal cord Ki = 0.20 nM77 | Pain77 Anorectic effect in fasted mice.82 |
| 1DMe (3) | [D-Tyr1,(NMe)Phe3] YLFQPQRF-NH2 | Metabolically stable agonist | hNPFFR1 Ki = 2.0 – 7.9 nM4, 80 hNPFFR2 Ki = 3.2 nM4 rNPFFR1 Ki = 1.6 nM4 rNPFFR2 Ki = 2.0 nM4 Rat spinal cord Ki = 0.07 – 034 nM77, 78, 80, 81 hNPFFR2 EC50 = 2.69 nM81 |
Analgesia77
38, 40, 78, 80 Opioid abuse52 Gastrointestinal mobility75 |
| Nic-1DMe (4) | [D-Tyr1,(NMe)Phe3,Nic-Pro5] YLFQPQRF-NH2 | Metabolically stable agonist | Rat spinal cord Ki = 0.07 nM80 hNPFFR1 Ki = 1.30 nM80 |
Pain80 |
| NPVF (5) | VPNLPQRF-NH2 | NPFFR1 agonist | hNPFFR1 Ki = 0.59 nM23 hNPFFR2 Ki = 23 nM23 |
Cardiovascular activities62 |
| dNPA (6) | [D-Asn1,(NMe)Ala3] NPAFLFQPQRF-NH2 | NPFFR2 agonist | hNPFFR1 Ki = 2.9 nM23 hNPFFR2 Ki = 0.027 nM23 |
Cardiovascular activities62 |
| FMRFamide (7) | FMRF-NH2 | Agonist | Rat spinal cord Ki = 1.8 nM19 hNPFFR2 Ki = 6.6 nM81 hNPFFR2 EC50 = 517 nM81 |
Pain36 Feeding65 Gastrointestinal mobility74 |
| PQRFamide (8) | PQRF-NH2 | Agonist | hNPFFR1 Ki = 40 nM4 Rat spinal cord Ki = 15.5 nM81 hNPFFR2 Ki = 25 nM4 rNPFFR1 Ki = 25 nM4 rNPFFR2 Ki = 25 nM4 hNPFFR2 EC50 = 309 nM81 |
Cardiovascular activities61 |
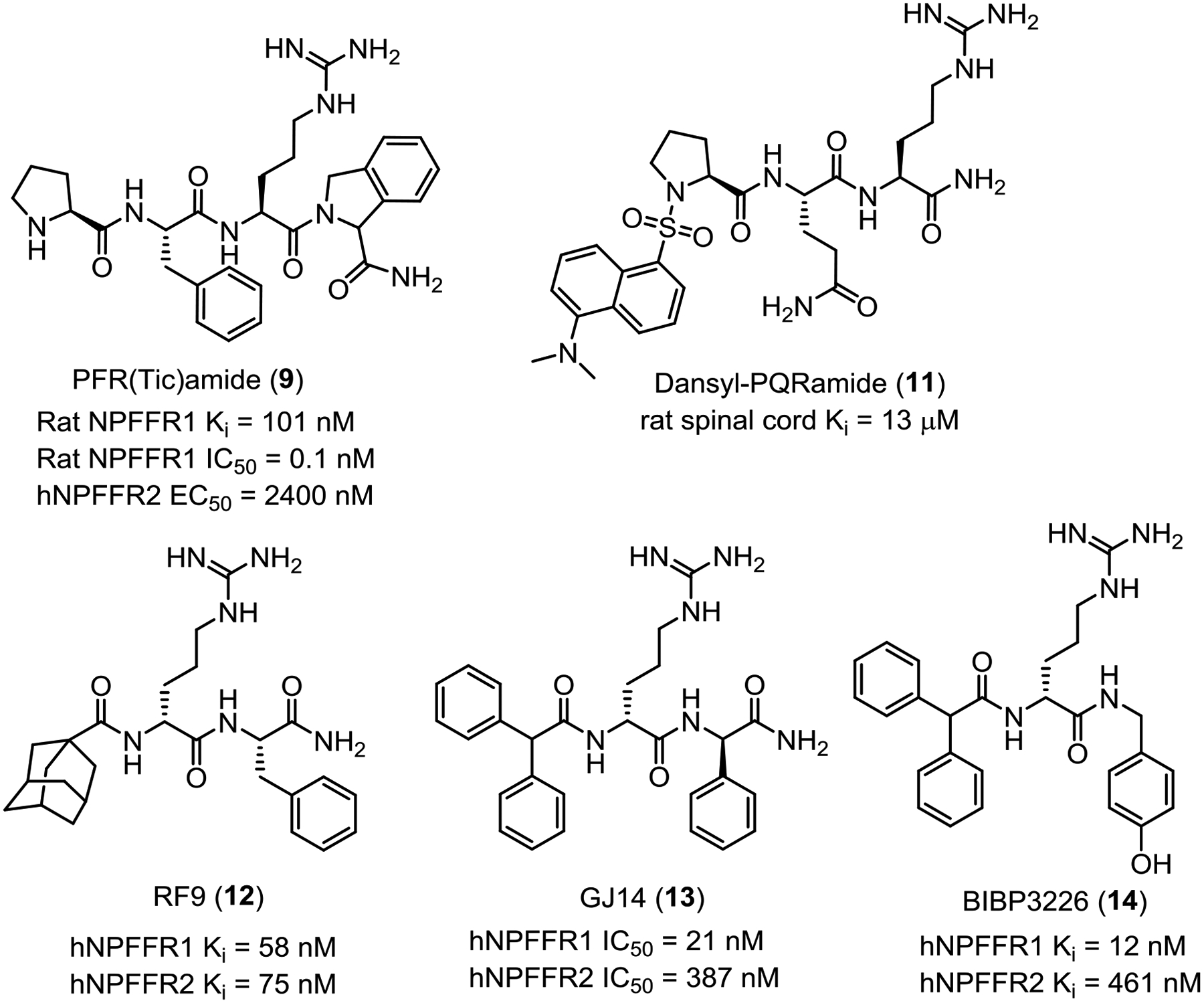
| PFR(Tic)amide (9) | [Tic-Arg3]PFR(Tic)-NH2 (Fig. 1) | NPFFR1 antagonist, NPFFR2 agonist | Rat NPFFR1 Ki = 101 nM56 Rat NPFFR1 IC50 = 0.1 nM56 hNPFFR2 EC50 = 2400 ± 400 nM83 |
Opioid abuse47 Cardiovascular activities61 |
| daY8Ra (10) | [desamino-Tyr1]YFLFQPQR-NH2 | Metabolically stable antagonist | Rat spinal cord Ki = 0.20 ± 0.04 nM50 | Opioid addiction50 |
| Dansyl-PQRNH2 (11) | [dns-Pro1]PQR-NH2 (Fig. 1) | CNS permeable antagonist | Rat spinal cord Ki = 13 μM84 | Opioid addiction47, 84 |
| RF9 (12) | See Fig. 1 | Agonist/ Antagonist | hNPFFR1 Ki = 58 nM32 hNPFFR2 Ki = 75 nM32 |
Pain32 Opioid addiction32 Cardiovascular activities32 |
| GJ14 (13) | See Fig. 1 | Antagonist | hNPFFR1 EC50 = 21 nM72 hNPFFR1 EC50 = 387 nM72 |
Anxiety72 |
Therapeutic applications
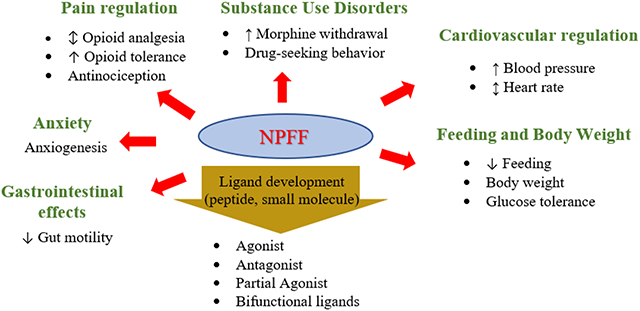
The NPFF system has shown promising therapeutic potential for a number of pathological conditions including pain, substance use disorders, anxiety, mood disorders, cardiovascular and gastrointestinal disorders.
Pain regulation
Initially classified as an endogenous “opioid modulating peptide”,21, 37 NPFF and its receptors have been explored extensively for analgesic development. Consistent with the presence of mRNA of NPFFRs in brain regions regulating pain signals such as the dorsal horn of the spinal cord, the lateral hypothalamus, the spinal trigeminal and thalamic nuclei,8 the potential of the NPFF system as a pain target is strongly supported by the evidence of upregulation of NPFF or its gene in many pain models. NPFF gene is upregulated in the dorsal horn by inflammatory pain.7 In rat spinal cord, gene expression of NPFF and NPFFR2 was upregulated by persistent pain induced by carrageenan inflammation while gene expression of NPVF (7, Table 2) and NPFFR1 was not affected.27 Similarly, activation of spinal NPFF and NPFFR2 was observed during early inflammatory pain.28 Supraspinally, NPFFR2 mRNA was upregulated during acute colonic inflammation and neuropathic pain.28
In animal pain models, NPFF has been reported to elicit both direct antinociceptive and opioid modulatory effects depending on routes of administration and animal pain models.12, 22
In general, the literature suggests that NPFF has no effect in animal models that mimic acute nociception but has analgesic effects in animal models of pathological pain, suggesting that the NPFF system needs to be “primed” by the pain condition first in order for NPFF to be effective against pain processing. For instance, in one study NPFF (i.t. and i.c.v., 10–50 nmol) had no effect in the acute thermal or mechanical nociception.38 Although Gouardères and coworkers reported that i.t. injection of NPFF (0.05–17.5 nmol) produced long-lasting (24–48 h) analgesia in thermal tail flick and mechanical paw pressure pain tests in rats39, this effect was not observed in another study by Kontinen et al using a similar dose range (0.05 – 10 nmol), i.t. administration and the same Sprague-Dawley rat strain.15 In addition, i.c.v. administration of NPFF was initially reported to decrease tail-flick latency in rats.1 but later Oberling et al reported that the analgesic effect of NPFF (i.c.v) in the tail flick test was only observed at night but not during daylight.40 In contrast, a NPFF analogue, (1DMe)NPYF (i.t.), was found effective against both thermal hyperalgesia and mechanical allodynia in a model of carrageenan inflammation and demonstrated antiallodynic effect against cold allodynia in the neuropathic pain model.41 NPFF (i.t. and i.c.v.) was also found to dose-dependently attenuate allodynic response to mechanical stimulation in the inflammatory and neuropathic pain models.38
The role of the NPFF system in modulating opioid analgesia has been well-documented. NPFF (i.c.v.) attenuated morphine-induced tail-flick latency,1, 40 1DMe (i.c.v.) reduced the analgesic effect of opioid agonists at a low dose but increased their analgesia at a high dose.42 Injection of IgG from NPFF antiserum reversed morphine analgesia in morphine-tolerant rats.43
Consistent with the analgesic and opioid modulating effects of NPFF, there is strong evidence of interactions between the NPFF and opioidergic systems.21 NPFF is localized in several brain regions rich in endogenous opioids.44 However, NPFF exhibited no significant binding affinity on opioid receptors. The possibility that NPFF modulated the opioid system by direct interaction with opioid receptors was also ruled out by radioligand binding assays using a tyrosine-substituted NPFF analogue [125I]Y8Fa, which demonstrated that NPFF acted through specific high affinity binding sites distinct from opioid receptors.44–47 In SH-SY5Y neuroblastoma cells engineered to express both NPFFR2 and μ-opioid receptor (MOR), these two receptors appeared to be in close proximity.48 The NPFF agonist 1DMe significantly increased association between these two receptors, reduced MOR internalization, and modified the lateral diffusion of MOR in the cell membrane from a constrained to a free state, suggesting that modulation of the delivery and trafficking of MOR at the cell surface is one of the mechanisms of its opioid modulating actions.48
One interesting and perplexing question in pain and opioid modulation by NPFF and its analogues has been the opposing effects observed with different routes of administration.49 For instance, i.t. injection of NPFF produced analgesia and potentiated opioid effects in rats, whereas i.c.v. administration elicited pronociceptive effects and reversal of morphine analgesia. Similarly, i.t. injection of two metabolically stable NPFF analogues (1DMe and 3DMe) produced antinociceptive effects and enhanced morphine-induced antinociception in the tail flick assay.50 Several high-affinity NPFF analogues displayed both supra-spinal anti-opioid and spinal analgesic activities.47 Although the underlying mechanisms remain elusive, several factors may help explain this discrepancy. Only NPFFR2 is present in the spinal cord in rats, it is likely that the nociceptive effects of i.t. administration of NPFF analogues are produced via NPFFR2 activation, whereas the antinociceptive effects of i.c.v. administration are mediated mainly by NPFFR1, which may play a more prominent role in the CNS of rats.49 These would suggest the development of either NPFFR1 antagonists and/or NPFFR2 agonists as opioid-add on therapies for chronic pain treatment.
Finally, NPFF has been demonstrated to play a role in the development of opioid tolerance, and opioid-induced hyperalgesia. Pharmacological blockade of NPFFR1/2 prevented the development of opioid-induced hyperalgesia and analgesic tolerance.51–53 Blocking NPFFRs with antiserum or antisense43, 54 or NPFFR antagonist RF953 (14, Table 2, Fig. 1) reversed morphine tolerance in rodents. Two hypotheses have been proposed to explain the development of opioid tolerance and hyperalgesia.22 The first theory proposes that opioid tolerance and hyperalgesia are results of opioid receptor desensitization, receptor endocytosis, and degradation upon chronic opioid treatment.55, 56 The second hypothesis suggests that NPFF is an anti-opioid neuropeptide that is released during opioid treatment to counter opioid effects leading to tolerance, dependence and hyperalgesia. The NPFF system remains activated upon cessation of opioid treatment. Therefore, blocking NPFF release reverses opioid tolerance, dependence and hyperalgesia. There is evidence that activation of NPFFR2 resulted in hyperalgesia in mice, which was mediated through the spinal inflammatory mediator CGRP.57 A putative NPFFR1 antagonist and NPFFR2 agonist, (26, Fig. 3) reversed tolerance to morphine’s analgesic effects upon i.p. administration but had no analgesic effect in rats never exposed to morphine.58
Figure 1.
Representative NPFFR peptidomimetics.
Figure 3.
Small molecule NPFFR agonists.
Substance Use Disorders
Substance use disorders represent a large cluster of clinical conditions that include different drug classes and different clinical stages. In this regard, the role of NPFF system has been examined on different drug classes including opioids and stimulants. Because different drugs of abuse have differing pharmacological mechanisms and clinical demonstrations, the results reported in the literature are often complicated and inconsistent. To reconcile the results, the following section will discuss the role of the NPFF system in different drugs of abuse.
The NPFF system seems to play an important role in addiction-related effects of opioids. For example, the NPFF level was elevated in CSF from morphine-dependent rats, but not nondependent rats.59 Activation of NPFFRs by NPFF or its analogues induced withdrawal-like signs or precipitated withdrawal syndrome60–62 while blockade of NPFFRs with a nonselective antagonist dansyl-PQRamide, daY8Ra (13, Table 2) or selective NPFFR1 antagonist AC-262620 (structure not disclosed) prevented naloxone-precipitated morphine withdrawal syndrome.62–64 Similarly, antagonism at both NPFFRs by antiserum or antisense reversed morphine tolerance and dependence in rats and mice43, 54 and blockade of NPFFRs with RF9 (14, Table 2, Fig. 1) prevented the development of morphine tolerance and decreased naltrexone-precipitated withdrawal following chronic morphine treatment.53 Combined, these results consistently suggest that NPFFR antagonists or NPFFR1 selective antagonists have great potential for the treatment of opioid tolerance and dependence.
Strikingly, the role of the NPFF system in the rewarding effects of opioids seems opposite to its role in opioid dependence and withdrawal. NPFF and the NPFFR agonist 1DMe blocked the acquisition of conditioned place preference (CPP) to morphine in C57BL6 mice65–67 while the NPFFR antagonist RF9 increased morphine-induced CPP without producing any rewarding effects by itself.53 Theses results, although preliminary, are particularly interesting as they suggest that NPFF agonists (instead of antagonists) could potentially be useful therapeutics against opioid reward-related effects. More studies are needed to examine whether similar effects can be generalized to the reinforcing effects of opioids using procedures like intravenous self-administration or other related assays (e.g., progressive ratio procedure or demand curve analysis). If confirmed, it would suggest that the use of NPFF ligands for treating opioid use disorder should be carefully tailored toward clinical utility. Thus, NPFF agonists may be useful to control opioid consumption during the early stage of opioid abuse by blocking the positive reinforcing/rewarding effects of opioids, while NPFF antagonists may be useful to reduce opioid dependence and control physical withdrawal signs, which usually occurs at the advanced stage of opioid abuse and are considered driving factors of opioid relapse.
Using neurochemical analyses, Wu and colleagues demonstrated that both NPFFRs were abundantly expressed in the ventral tegmental area (VTA) and lower level in the nucleus accumbens (NAc).67 Immunohistochemical staining revealed that most GAD67-positive neurons showed clear NPFFR expression but only some TH-positive neurons expressed NPFFRs, suggesting that NPFF attenuated morphine reward mainly via its direct action on GABAergic neurons with minor action on VTA dopaminergic neurons.67
The role of the NPFF system in addiction-related effects of stimulants is more complicated. I.c.v. injection of NPFF inhibited the expression of amphetamine70- and cocaine71-induced CPP, which was prevented by the NPFFR antagonist RF9,68 suggesting that NPFF agonist(s) reduces the positive rewarding effects of stimulant drugs. This result is consistent with its role on opioid reward.
However, stimulants such as amphetamine and cocaine induce locomotor sensitization after repeated intermittent treatment and NPFF (i.c.v) potentiated the locomotor sensitization to amphetamine68 but attenuated this effect to cocaine71. It is unclear the cause of this apparenty discrepancy but another study found that the putative NPFFR1 antagonist/NPFFR2 agonist PFR(Tic)amide (11, Table 2, Fig. 1) (i.c.v) blocked amphetamine-induced locomotor sensitization.69. Given that locomotor sensitization is often used as a rodent model related to addiction-correlated behavioral plasticity, these findings stand in contrast to the role of the NPFF system on drug reward-related effects. More studies are needed to decipher whether the role of the NPFF system is assay-dependent, receptor subtype (NPFFR1 vs. NPFFR2)-dependent or both.
There was only one study on the selective NPFFR1 antagonist, AC-262620, on nicotine dependence and it was found that AC-262620 reduced the nicotinic antagonist mecamylamine precipitated withdrawal signs in nicotine-dependent animals.64 This result is consistent with the findings on opioid dependence and further suggest that NPFF antagonists may be the choice for treating drug dependence and withdrawal.
Cardiovascular regulation
As NPFF was originally identified from the isolation of cardioexcitatory peptide FMRF-NH2 immunoreactive peptides1 and is located postsynaptically with respect to vagal afferent fibers in the nucleus tractus solitarius,70 the NPFF system has also been studied for its role in cardiovascular regulation. Studies have shown that activation of the NPFF system leads to increased mean arterial blood pressure (MAP) in rodents. Administration of NPFF via jugular vein and carotid artery catheters elevated MAP in conscious, unrestrained rats.71 This pressor effect was attenuated but not abolished by prior treatment with the antihypertensive drug guanethidine or prazosin, an α1-adrenoreceptor antagonist, suggesting that NPFF plays a role in the regulation and maintenance of blood pressure by both catecholamine-dependent and -independent release mechanisms.71 In agreement to the first study of NPFF on MAP, injection of NPFF and its analogue 1DMe into the commissural nucleus tractus solitarius also produced an increase in MAP which was reversed by prazosin and a significant inhibition of the cardiac component of the baroreceptor reflex.70 I.v. injection of the PFRFamide (NPFFR1 and 2 agonist) or PFR(Tic)amide (11, NPFFR1 antagonist and NPFFR2 agonist) dose-dependently increased or decreased MAP, respectively.72 I.t. administration of NPFF, and its analogues NPVF and dNPA, dose-dependently increased MAP, which was reversed by the NPFFR antagonist RF9, and reduced by pretreatment with the α-adrenoceptor antagonist, phentolamine.73
In contrast to the influence on MAP, effects of the NPFF system on heart rate appear to vary with routes of administration. I.c.v. injection of NPFF and its analogue 1DMe resulted in bradycardia, which was abolished by atropine, a muscarinic receptor antagonist.70 PFRFamide (i.v.) produced a nonsignificant increase on heart rate whereas 11 (i.v.) caused a significant decrease in heart rate.72 However, i.t. administration of NPFF, NPVF (selective NPFFR1 agonist) or dNPA (NPFFR2 agonist) dose-dependently resulted in tachycardia, which was blocked by the β-adrenoceptor antagonist propranolol and the NPFFR antagonist RF9.73 The order of potency of these agonists on MAP was NPFF = NPVF ≥ dNPA and on heart rate was dNPA > NPVF ≥ NPFF, implying that NPFFR1 may play a more prominent role in MAP regulation whereas NPFFR2 contributed more to the control of heart rate.73
Feeding and Body Weight
The NPFF system has also been implicated in the regulation of feeding behaviors depending on the nutritional status. FMRFamide (9, Table 2) was shown to reduce morphine- and food deprivation-induced feeding.74 NPFF (i.c.v.) reduced food intake in food-deprived rats, like the opioid receptor antagonist, naloxone.75 On the other hand, 9 enhanced food intake and increased serotonergic metabolism in ‘cafeteria-fed’ rats (rats with access to foods high in fat and/or sugar), but not normophagic rats, which are classical effects of opioid receptor antagonists such as naltrexone, suggesting a role of the NPFF system in selectively regulating food intake.76
Both NPFFRs have been shown to be involved in the regulation of food intake and body weight. NPFFR2 knockout mice exhibited a stronger bone phenotype and exacerbated obesity when fed a high fat diet due to a failure in activating brown adipose tissue thermogenic response to energy excess, possibly via a hypothalamic NPY-dependent circuitry.77 Interestingly, NPFFR1 displayed a sex-biased role in food intake and metabolic homeostasis. NPFFR1 knockout male mice demonstrated reduced spontaneous food intake and suppressed leptin- and ghrelin-induced feeding. In contrast, food intake was unaltered in NPFFR1 knockout female mice. Ablation of NPFFR1 did not cause any overt alterations in body weight of male mice but increased body weight in female mice associated with increased fat mass. NPFFR1 ablation worsened glucose tolerance and insulin sensitivity caused by high fat diet in male but not in female mice.78 It remains to be determined if reducing the NPFF signaling with NPFFR antagonists produces the same metabolic effects observed with NPFFR knockdown mice.
Anxiety
Both NPFFRs are expressed in subregions of the hypothalamus, including paraventricular nucleus (PVN) upstream of the hypothalamic-pituitary-adrenal (HPA) axis,79, 80 and hyperactivation of HPA axis is highly related to depressive and anxiety-like behaviors.81, 82 Kim and colleagues investigated anxiolytic effect of the NPFFR antagonist GJ14 (15) in mice receiving chronic infusion of RFRP-3, an RFamide-related peptide proposed to play a role in the stress response. As expected, 15 blocked RFRP-3-induced anxiogenic effects and corticosterone release, and stress-induced activation of CRH neurons in the paraventricular nucleus (PVN).83 Consistent with this, NPFFR2 agonists, such as 26 or dNPA, increased corticosteroid and c-Fos protein expression in the hypothalamic PVN levels, inducing an anxiogenic effect in a mouse elevated plus maze model. This anxiogenic effect was also seen with other RFamide peptides such NPAF and NPSF, which are known to activate the HPA axis controlling physiological stress response.84 However, the possibility that the anxiolytic or anxiogenic effects of these NPFFR ligands may be mediated extrahypothalamically should not be discounted.
Gastrointestinal effects
Regarding gastrointestinal effects, NPFF and its analogues elicited opioid-like effects. NPFF, like morphine, delayed colonic bead expulsion time in mice.85 Similarly, both NPFF and 1DMe caused inhibition of intestinal transit in mice, which was not reversed by naloxone.86 The duration of disruption of intestinal migrating myoelectric complexes was significantly reduced by 1DMe.87 Although the effects of NPFFR antagonists on gut motility have not been studied yet, the inhibition of intestinal transit by NPFFR agonists implies that NPFFR antagonists could enhance gut motility, alleviating opioid-induced constipation.
Development of NPFFR ligands
Originally, most of NPFFR ligands were peptides and peptidomimetics designed based on the sequence of NPFF. They were mainly characterized by competitive binding assays in rat spinal cord. Classification of these initial ligands as agonists or antagonists were mostly based on their behavioral effects. Subsequently, advances in the biochemistry field has allowed determination of mechanism of action of NPFFR ligands as agonists or antagonists as cells engineered to overexpress human or rat NPFFR1 or NPFFR2 became available. High throughput screening capability led to the discovery of novel small molecule scaffolds with improved druglike properties.
NPFF-derived peptides and peptidomimetics
Peptidic agonists
The structure-activity relationships (SARs) of NPFF and related RF-amides have been extensively studied.23, 88 To increase stability of NPFF analogues against peptidases in tissues, the first three N-terminal amino acid residues of NPFF were replaced with D-amino acids and/or were N-methylated on the peptide bond. 1DMe (3, Table 2), in which Phe1 and Phe3 were replaced with (D)Tyr1 and [N-Me]Phe3, respectively, emerged as a relatively stable analogue which reduced peptide degradation rate about 68% while maintaining similar binding affinity compared to NPFF.89 In addition, the presence of a phenolic hydroxy group permits easy radiolabeling with 125I, making 1DMe one of the commonly used radiolabeled tracer. Besides [125I][Tyr1]NPFF and [125I]1DMe, [125I]EYF and [125I]YVP24, 29 (4 and 5, Table 2) have also been employed as radiolabeled probes in several studies.51, 90 Similar to 1DMe, Nic-1DMe (6, Table 2) designed with the Pro5 residue capped with a nicotinoyl group to enhance peptidase stability possessed the same binding affinities at both NPFFRs.91 Similar to NPFF, effects of these two NPFF analogues seemed to be dependent on routes of administration. Both i.t. and i.p. administration of 1DMe and 6 potentiated morphine analgesia.91 However, i.c.v. administration of 1DMe reduced morphine analgesia whereas i.c.v. administration of 6 induced a significant potentiation of morphine analgesia.91
The competitive radioligand binding assay against [125I]1DMe tracer in rat spinal cord indicated that truncation of the NPFF sequence from the N-terminus produced only a moderate decrease in affinity up to NPFF(3–8).88 Consistent with this, the last two C-terminal residues (RF) are critical for the NPFFR binding affinity whereas substitution of the first three N-terminal residues (FLF) could be tolerated.92 However, it should be noted that binding affinities were not predictive of their efficacy in reversing morphine-induced analgesia in the tail-flick test even with i.c.v administration. For example, [Tyr1]NPFF had similar binding affinity to rat spinal cord membrane as NPFF, while the former analogue was more effective than NPFF in potentiation of morphine analgesia.88 On the other hand, [Gly5]NPFF had a comparable in vivo effect to [Tyr1]NPFF although [Gly5]NPFF had about six-fold lower binding affinity (Table 3).88 These results suggest that the in vivo effect was greatly influenced by other factors such as metabolic stability.
Table 3.
Binding affinity and relative potency to reverse morphine-induced analgesia of representative NPFF peptidic analogues.77
| Compound | Binding affinity Ki (nM)a | Minimal effective dose (nmol)b |
|---|---|---|
| NPFF | 0.21 ± 0.03 | 22 |
| [Tyr1]NPFF | 0.20 ± 0.04 | 8.8 |
| [Gly5]NPFF | 1.2 ± 0.2 | 8.8 |
Affinity for rat spinal cord membranes using [125I][Tyr1]NPFF tracer.
Mimimal effective dose (i.c.v.) able to significantly reverse morphine-induced analgesia.
Peptidic antagonists
The first putative NPFFR antagonist, daY8Ra (12, Table 2) was designed as an NPFFR antagonist (Ki = 0.20 ± 0.04 nM against rat spinal cord) with the N-terminal extended by a desaminotyrosine to increase stability against aminopeptidase hydrolysis and the C-terminal Phe deleted.63 daY8Ra significantly attenuated opiate abstinence syndromes induced by i.c.v. administration of NPFF.63 Capping the N-terminal of the tripeptide PQR-NH2 with a dansyl group to increase lipophilicity resulting in dansyl-PQR-NH2 (13, Table 2) with improved blood brain barrier permeability, which upon s.c. administration, attenuated opiate dependence in rats infused with morphine chronically.97 Third ventricle administration of 13 dose-dependently antagonized the quasi-morphine abstinence syndrome induced by NPFF (i.c.v.).97 13 was approximately 300 times more stable than NPFF against aminopeptidase.97
The fact that BIBP3226 (16, Fig. 1), an anorexigenic Y1 receptor antagonist that is structurally similar to RFamide, bound to both human and rat NPFFRs led to the discovery of N-capped dipeptide RF9 (Fig. 1) which has been widely used as a nonselective NPFFR antagonist probe to investigate pharmacological effects of the NPFF system in vitro and in vivo.51 Unlike 16, RF9 had no binding affinity for human neuropeptide Y receptor subtype Y1, delta-opioid receptor or three other RFamide receptors including GPR10, GPR103,51 and low binding affinity for GPR54 (KISS1R),98 mu- and kappa-opioid receptors.51 RF9 possessed a Ke = 45 ± 5 nM in shifting the NPFF concentration–effect curve in [35S]GTPγS binding assay to COS cells transiently transfected with hNPFFR2 construct and EC50 = 4.7 ± 1.2 μM in reversing the inhibitory effect of agonist NPVF in CHO cells stably expressing hNPFFR1.51 RF9 (10 μg, i.c.v.) blocked the NPFF-induced mean arterial blood pressure and heart rate in rats without any effects by itself.51 RF9 (0.1 mg/kg, s.c.) injected 30 min before heroin (0.3 mg/kg, s.c.) also prevented heroin-induced delayed hyperalgesia and tolerance.51 However, RF9 did not reverse phosphorylation of MAPK/ERK1/2 or the anorexigenic effect in fasted mice induced by [Tyr1]NPFF as expected for NPFFR antagonists. On the contrary, RF9 itself showed the same anorexigenic effect like [Tyr1]NPFF after both i.c.v. and s.c. administration, thus acting more like an NPFFR agonist in these assay.95 RF9 was also reported to activate GnRH neurons through Kisspeptin receptor (KISS1R).99 A related analogue, 15, was reported as an antagonist in cAMP assays at both NPFFRs (NPFFR1 IC50 = 21 nM; NPFFR2 IC50 = 387 nM) and demonstrated anxiolytic effects.83
Bifunctional ligands
Recently, the development of bi- or multifunctional opioid drugs have emerged as an attractive therapeutic strategy to maintain the powerful antinociceptive effect of opioids without their associated untoward side effects. Several groups have designed bifunctional opioid and NPFF ligands by merging the peptide sequences of an opioid ligand and NPFF as a new class of analgesics (Fig. 2).
Figure 2.
Bifunctional opioid and NPFFR ligands.
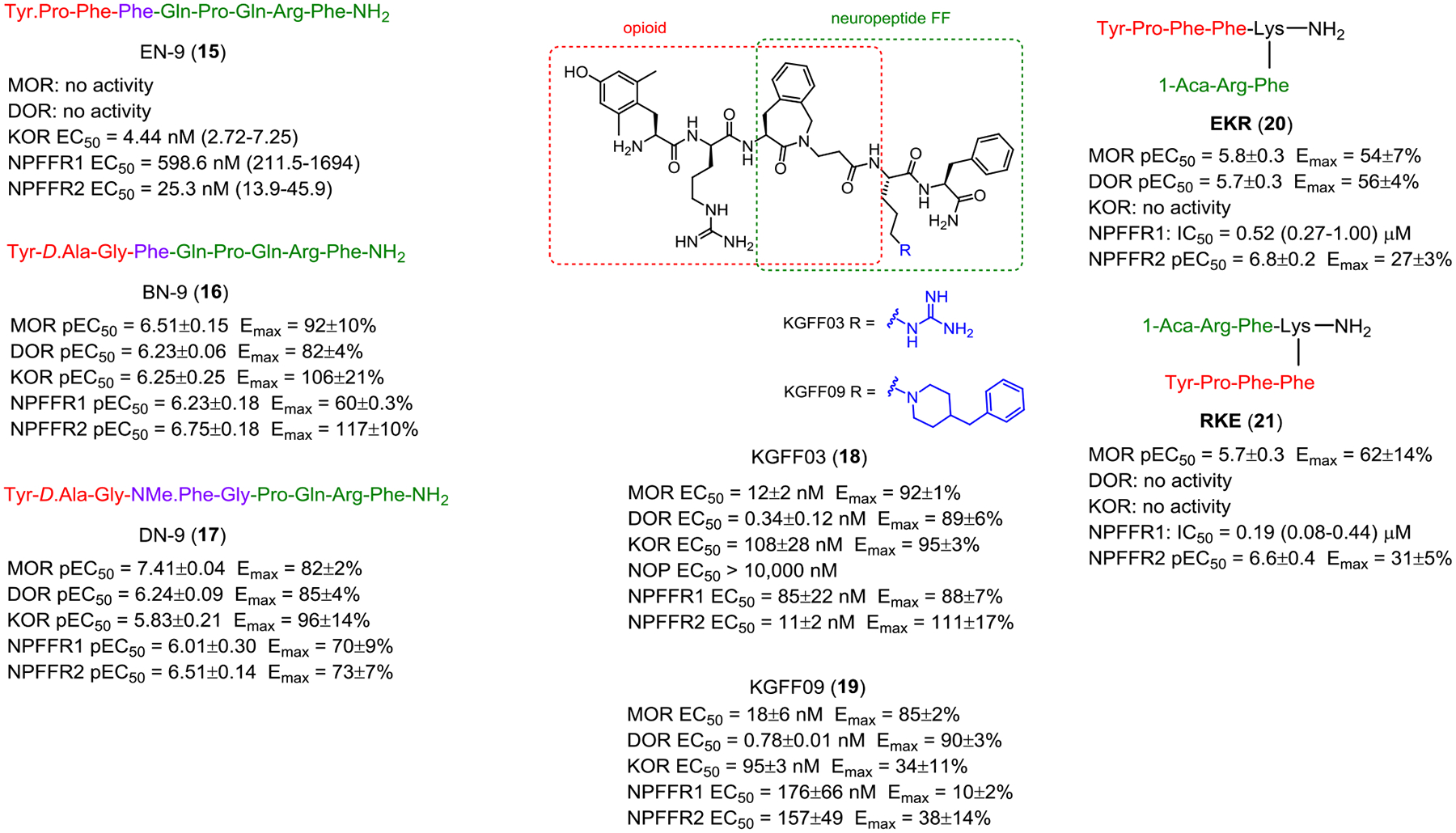
Wang and colleagues reported a chimeric peptide of endomorphin-2 and NPFF (EN-9, 17), a KOR, NPFFR1 and NPFFR2 agonist which produced potent antinociception upon i.c.v., i.v., and s.c. injections.100 the Wang group also designed another chimeric peptide by fusing pharmacophores of the opioid agonist biphalin and NPFF sequence via the common Phe residue.101 BN-9 (18) acted as a full agonist at MOR, DOR, KOR and NPFFR2 and partial agonist at NPFFR1 in the cAMP assays. 18 (i.c.v., i.t. and i.v. administration) produced significant analgesia in the formalin test. Repeated administration of 18 produced analgesia without developing tolerance over 8 days. However, i.c.v. administration of 18 induced CPP and demonstrated reduced inhibitory effect on gastrointestinal transit inhibition. Optimization of the overlapping portion between the opioid and NPFF pharmacophore (i.e. replacing the Phe3-Gln4 with NMe.Phe3-Gly4) resulted in DN-9 (19) with better cell-based potencies and analgesic effects in the formalin pain and complete Freund’s adjuvant (CFA)-induced chronic inflammatory pain models.102 Importantly, 19 did not produce antinociceptive tolerance in the tail-flick and CFA-induced pain models after repeated administration for 6 days. 19 produced weaker constipating effects at antinociceptive doses compared to morphine but significantly inhibited gastrointestinal (GI) transit at high doses.103 These inhibitory effects on GI motility by central and peripheral opioid receptors depend on i.c.v. or i.p. routes of administration.
Drieu la Rochelle and colleagues designed a bifunctional biased μ-opioid agonist-NPFFR antagonist as an analgesic with reduced acute and chronic side effects.104 From the SAR study of the combination of an opioid agonist peptide and neuropeptide FF sequences, the authors identified KGFF03 (H-Dmt-D-Arg-Aba-β-Ala-Arg-PheNH2, 20) as a G-protein biased MOR agonist and NPFFR1/2 agonist and KGFF09 (H-Dmt-D-Arg-Aba-β-Ala-Bpa-PheNH2, 21) as a G-protein biased MOR agonist and NPFFR1/2 antagonist. Acute s.c. administration of 20 and 21 produced dose-dependent, long-lasting antinociception in mice. Moreover, unlike 20, chronic s.c. administration of 21 did not induce hyperalgesia and tolerance in naïve mice and displayed reduced withdrawal syndrome and respiratory depression.
Zhang et al designed two branched peptidomimetics, EKR (22) and RKE (23), by merging the pharmacophores of the opioid peptide endomorphin-2 and the NPFFR antagonist RF9 using a lysine linker.105 In the cAMP assay, both 22 and 23 behaved as multi-functional peptides with MOR agonism, NPFFR1 antagonism and NPFFR2 partial agonism, without any activities at KOR. 22 is an agonist at the DOR whereas 23 has no activity at this receptor. Both compounds completely blocked the NPFFR2-mediated neurite outgrowth of Neuro2A cells. Supraspinal administration of 22 and 23 dose-dependently produced antinociception in both tail-flick and carrageenan-induced inflammatory model. Repeated i.c.v. administration of 22 and 23 produced prolonged antinociceptive effects compared to morphine. Both 22 and 23 delayed the development of antinociceptive tolerance and had no effects on the gastrointestinal motility compared to morphine.
These studies provide a proof of concept that bifunctional opioid-NPFF ligands could lead to novel analgesics with reduced side effects. However, it still requires much work to unravel the complicated relationship between these two receptor systems as both NPFFR agonism or antagonism seems to result in reduced opioid tolerance. It is possible the differential effects are attributed to the opposite actions of each NPFFR subtype as 18 is a partial agonist at NPFFR1 and a NPFFR2 full agonist, 21 is an antagonist at both NPFFRs, and 22 and 23 are NPFFR1 antagonists and NPFFR2 partial agonists.
Small molecule ligands
Small molecule agonists
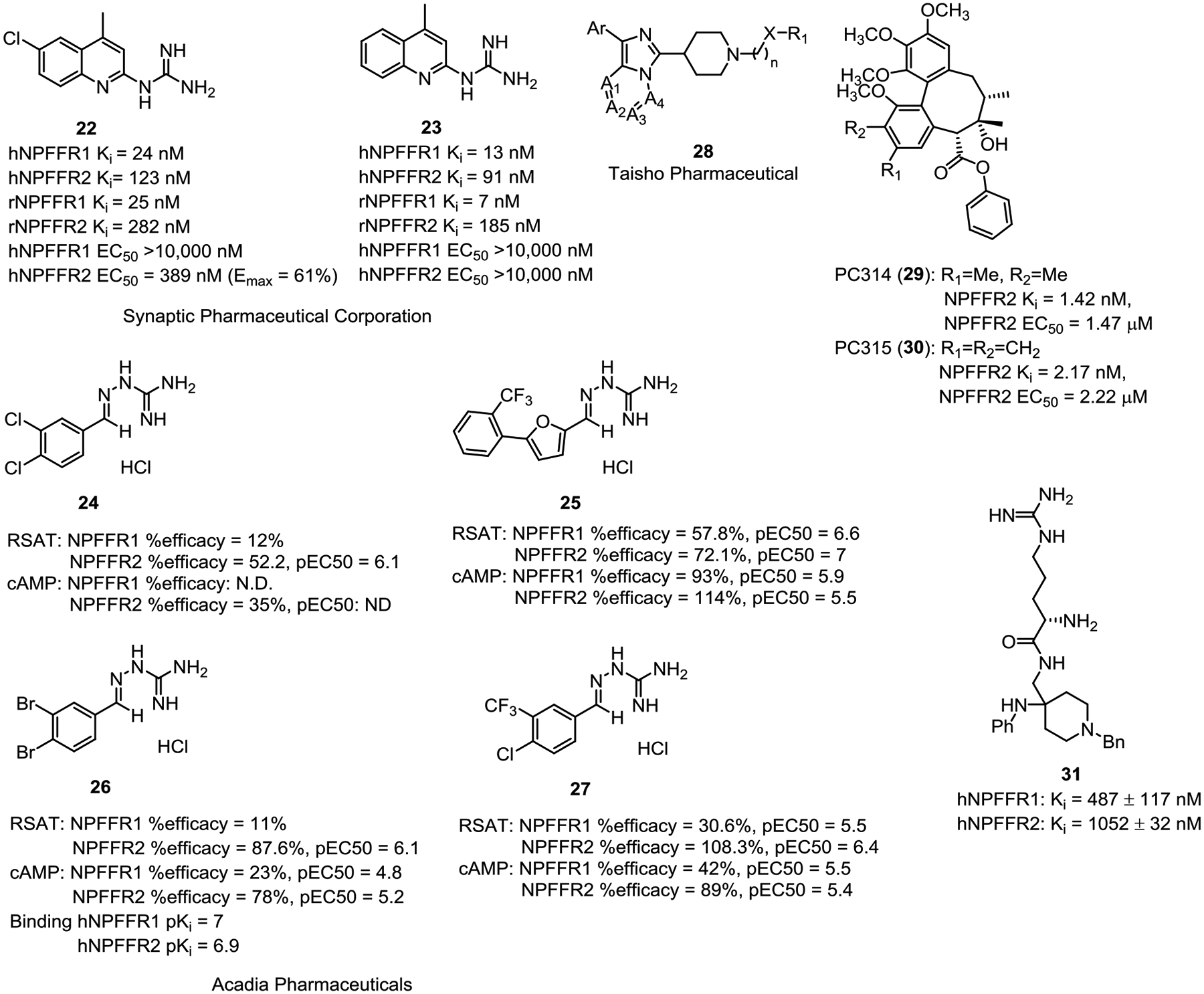
The first small molecule NPFFR agonists were a series of quinazolino- and quinolino-guanidines reported by Synaptic Pharmaceutical Corporation (Fig. 3).106 24 (compound 4005 in the patent) acted as an NPFFR1 antagonist and NPFFR2 agonist in cell based functional assays and demonstrated efficacy to attenuate rhythmic elevations in bladder pressure resulting from distension induced contractions at 1 mg/kg, i.v.. However, this in vivo effect may be nonspecific to NPFFRs as its analogue 25 (3 mg/kg, i.v., compound 4006 in the patent), which was inactive in functional assays, also exhibited efficacy in the same model.
The most extensively investigated small molecule NPFFR agonists to date are a series of aminoguanidine derivatives disclosed by Acadia Pharmaceuticals Inc in a patent.107 28 and 29 (compounds 3093 and 3099 in the patent) were suggested to be selective NPFFR2 agonists as they demonstrated no/low efficacy (Emax) at NPFFR1 (Fig. 3), whereas 26 and 27 (compounds 1045 and 2616 in the patent) showed agonism at both NPFFRs. 28 exhibited comparable binding affinities to hNPFFR1 and hNPFFR2,58 and in addition to agonistic activity at NPFFR2, also acted as NPFFR1 antagonist in the RSAT assay.58 Both 28 and 29 (10 mg/kg, i.p.) are efficacious in reversing inflammatory hyperalgesia and nerve injury-induced allodynia. In contrast, nonselective NPFFR agonists such as 27 increased sensitivity to noxious and non-noxious stimuli,108 which was completely blocked by the NPFFR antagonist dPQR.107 In addition, 28 and 29 demonstrated significant attenuation of formalin-induced flinching in phase II (15–60 min post-formalin injection) but not in phase I (0–15 min post formalin injection), suggesting their potential efficacy in chronic pain including neuropathic and/or inflammatory pain but not in acute pain.107 Compound 28 (10 mg/kg, i.p.) reversed tolerance to morphine analgesia in morphine-tolerant rats in the radiant heat tail flick test, but did not induce an analgesic effect in rats never exposed to morphine.58 26 (i.t.) produced a dose-dependent reversal of thermal hyperalgesia in carrageenan-treated rats but did not have analgesic effect by itself (115.5 μg, i.t.) or alter sensitivity to non-noxious mechanical stimulation.107 27 (10 mg/kg, i.p.) induced thermal hyperalgesia in the 52 °C hot plate test.107 Using homology models of NPFFR1 and NPFFR2, Findeisen and colleagues revealed that the binding pocket of these guanidinium compounds was located at the upper part of transmembrane helices V, VI, VII and the extracellular loop 2.109
Fig. 3 presents small molecule NPFFR agonists from several other groups. Taisho Pharmaceutical Co. Ltd. reported a series of imidazolyl piperidine derivatives (30) as NPFFR2 agonists, but no biological data was disclosed in the patent.110 PC314 (31) and PC315 (32), discovered from a Korean herbal plant extract, were reported to have high binding affinity to NPFFR2 and moderate potency in cAMP assay.111 Rational structural design to preserve the guanidine moiety and lipophilicity of the Phe residue led to 33 (compound 7b in the original paper), which appeared as an NPFFR agonist that had no activities at mu-, delta-, or kappa-opioid receptors up to 5 μM.112
Small molecule antagonists
Figure 4 lists small molecule NPFFR antagonists reported to date. The first few series of guanidine (34 and 35) and indole (36) derivatives were reported in several patents by Actelion Pharmaceuticals Ltd and Kyowa Hakko Kogyo Co. Ltd with no biological data.113–115
Figure 4.
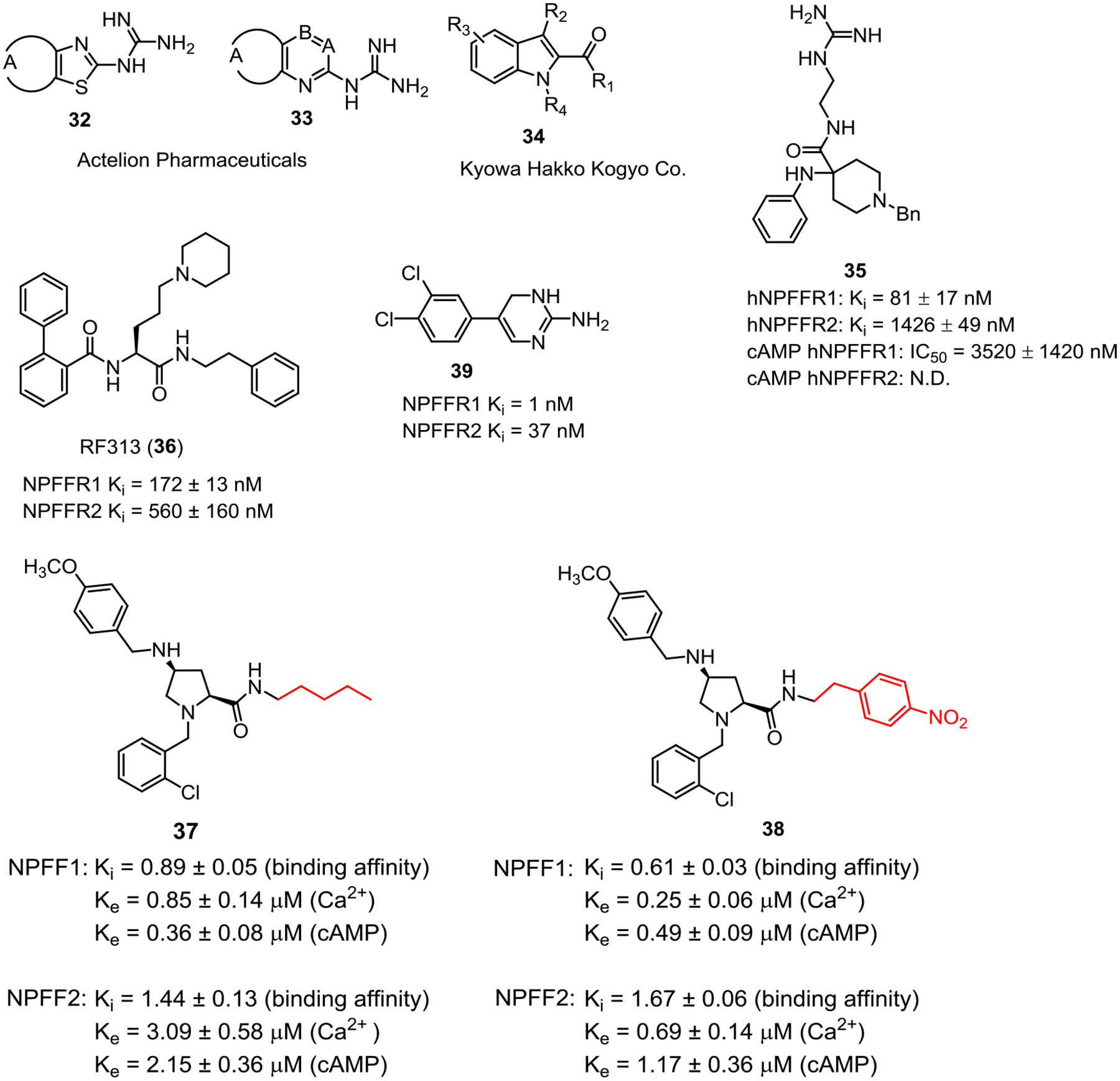
Small molecule NPFFR antagonists.
Using rational structural design, Journigan et al reported a series of benzylpiperidine derivatives containing a guanidine group to mimic the C-terminal dipeptide RF of the NPFF sequence.112 Among this series, 37 (compound 46 in the original paper) appeared as a selective NPFFR1 antagonist with approximate 17-fold selectivity in binding affinities and moderate potency in the cAMP assay at NPFFR1 (IC50 = 3.5 μM). Pretreatment with 37 prevented NPFF-induced hyperalgesia in the mouse warm-water tail-withdrawal test, whereas no effects were seen with administration of 37 alone.
Optimization of RF9 led to RF313 (38) which demonstrated antagonist activity in forskolin-induced cAMP assay in CHO cells expressing human NPFFR1. 38 showed no side effects when administered in mice up to 30 mg/kg s.c. When co-administered with opioids in rodents, 38 improved their analgesic effects and prevented opioid-induced hyperalgesia when administered orally. Unlike RF9, 38 exhibited negligible affinity and no agonist activity toward the Kisspeptin receptor.116, 117
Nguyen and colleagues reported NPFFR antagonists, 39 and 40 (compounds 16 and 33 in the original paper), with a distinct proline scaffold emerging from a high throughput screening campaign using CHO cells stably expressing NPFFRs.52 Both compounds showed antagonist activities at both NPFFRs in calcium mobilization and cAMP assays. They also bind to NPFFRs with submicromolar affinities. Both compounds have good solubility and blood-brain barrier permeability without the liability of being P-glycoprotein substrates. Lastly, they were able to reverse fentanyl-induced hyperalgesia in rats following intraperitoneal administration.52
Hammoud et al performed scaffold hopping by modifying the central core of the aminoguanidine hydrazone first reported by Acadia Pharmaceuticals to arrive at the cyclized guanidine derivative 41 (compound 22e in the original paper) from the lead compound 26.118 These two compounds have no or weak binding affinities to PrRPR (GPR10), Kiss1R (GPR54) and QRFPR (GPR103) (Ki > 5 μM). The functional activities of these compounds were assessed by cAMP assay using HEK293-hNPFFR1/NPFFR2 GloSensor™ stable cell lines. 41 appeared to be a more potent NPFFR1 antagonist than 26 at 1 μM but not at 10 μM. 26 and 41 did not display any agonist or antagonist effect at NPFFR2 up to 10 μM, which is in contrast with the original report showing 26 as a full NPFFR2 agonist with no activity at NPFFR1 in RSAT assay. The inconsistency could be attributed to the different assays used for assessment. In the original report, Acadia Pharmaceuticals tested compounds in the RSAT assay whereas the later study employed the classical cAMP assay. The agonists used could also be a contributing factor as the later study employed RFRP-3 peptide as agonist for the NPFFR1 assay and NPFF as agonist for NPFFR2 assays. Both 26 and 41 were effective in reducing the long lasting fentanyl-induced hyperalgesia in rodents.118
2. Summary and Future Directions
The NPFF system plays an important role in regulating many physiological or pathological processes. While its role in modulation of opioid’s antinociception, tolerance, dependence, and hyperalgesia has emerged as an area of particular interest, manipulation of the NPFF system can be beneficial in the treatment of a host of other disorders including substance use disorders, cardiovascular conditions, anxiety and gastrointestinal disorders. However, like many emerging targets, there are still many knowledge gaps to be filled to fully establish the NPFF system as a promising target for medication development. The pharmacological profiles of NPFFR ligands are complex and not thoroughly understood. One of the most important challenges is discovery of selective receptor subtype agonists and antagonists to elucidate specific roles of each receptor subtype because they seem to exert opposing effects in many pathological conditions. Current NPFFR ligands do not possess the required selectivity and potency, i.e. low nanomolar potency at one of the NPFFR subtypes and absence of activity at the other. This poses a challenge to determine effects of the NPFF system using chemical probes, particularly in the absence of knockout animal models. Therefore, it is important to develop NPFFR ligands with high subtype selectivity either through optimization of existing ligands or identification of new chemical scaffolds. Alternatively, NPFFR1 or NPFFR2 knockout animal models will significantly enhance the knowledge of each NPFFR subtype in contributing to pharmacological and behavioral effects.
A second aspect to address in future research relates to assay variability. This variability makes it difficult to compare NPFFR ligands across different assays, which employ different endpoints, radiolabeled tracers, host cells, receptor origins, or orthosteric agonists. For example, compound 26 from Acadia Pharmaceuticals displayed opposing activity when tested in the RSAT assay and later in the cAMP assay. Further, many ligand’s binding affinities and/or in vitro potencies were measured using different radiolabeled tracers and/or orthosteric agonists for each NPFFR subtype, which creates difficulty in assessing their receptor subtype selectivity. In addition, it has been widely demonstrated that GPCR ligands often display probe-dependent effects. As origins of host cells/membranes play an important factor contributing to variation of potencies and selectivities of compounds, invention of engineered cell systems expressing human NPFFRs has allowed determination of in vitro efficacy of NPFFR ligands in functional assays and classification of ligands as agonists or antagonists, providing better assessment of ligands than initial studies that reported only binding affinities in rat spinal cord. RF9 was originally reported as an NPFFR antagonist, but was later reclassified as a partial agonist in other assays. Similarly, the opposing effects of i.c.v. injections of 1DMe and Nic-1DMe on potentiation of morphine antinociception could be due to differential acting mode (agonist/antagonist) of each ligand, which could better be addressed in functional assays. There is a critical need to assess current and future ligands in functional assays to determine whether they are agonists or antagonists as data shows that ligands can have good binding affinities, but no activities in functional assays.
Although it is indispensable to characterize ligands as agonists and antagonists in cultured cells, binding assays still play an important role to demonstrate direct binding interactions between ligands and receptors. Radiolabeled ligands are essential tools for both in vitro membrane binding assays to assess direct ligand-receptor interaction and binding kinetics as well as autoradiographic studies to evaluate receptor distribution. Issues such as specificity, selectivity, and sensitivity are challenges associated with the design and development of an “ideal” NPFF radioligand. Current NPFF radiolabeled ligands are NPFF-related peptides labeled with Iodine-125 which has a half-life of 60 days, thus requiring frequent costly preparations of these radiotracers on demand. Tritium with a half-life of 12.5 years is a more stable radioactive isotope and allows direct labeling of the native NPFF itself without structure modifications into its analogues. One drawback of tritiated ligands is its lower specific activity compared to 125I-labeled counterparts, thus increasing the background noise. Until small molecule NPFF ligands with high selectivity and potency are developed, radiolabeled peptides remain the only tools to study binding ligand-receptor interactions for the NPFF system.
In drug discovery, compounds are normally first tested in cells engineered to express target receptors, and then tested for therapeutic uses in rodent models before eventually transitioned to humans in clinical studies. This process can be challenging when many target systems display species discrepancies in expression, and may be particularly problematic with the NPFF receptors which differ in abundance by species. NPFFR1 is expressed at a higher level than NPFFR2 in human CNS, while NPFFR2 is more abundant than NPFFR1 in rat brain. NPFFR1 mRNA is more abundant than NPFFR2 in human spinal cord, which is in sharp contrast to the spinal cord of rats where only NPFFR2 is found. Although rat heart expressed high levels of NPFFR2 mRNA, human heart expressed little of either receptor subtype mRNA. Both receptor subtypes are expressed at higher levels in human spleen compared to rats. Therefore, it is entirely possible that NPFFR1 ligands can play a more significant role in humans than in rats because NPFFR1 expression in humans is much higher than in rats. As the first in vivo studies are typically performed in rodents, we recommend determination of binding affinities and activities at both rodent and human NPFFRs to assess species differences to avoid false conclusions about their effects and NPFFR subtype involved.
To aid the design of selective NPFFR receptor subtype ligands, molecular modeling can be employed to facilitate the ligand design process. Although crystal structures of neither NPFFR have been solved yet, homology modeling may shed light on how the two receptors differ structurally, providing clues to guide medicinal chemistry optimization. Site-directed mutagenesis will be useful to confirm in silico prediction of intermolecular interactions. As NPFFRs are known to bind more promiscuously to other neuropeptides,119 allosteric binding site(s) may exist and their identification would provide better clues to develop allosteric modulators that may have better selectivity between the two NPFFR subtypes and/or against other RF-amide receptors.
The most studied therapeutic area of the NPFF system is pain regulation and modulation of opioid actions. Literature supports the potential of NPFFR ligands as analgesics alone or as opioid adjuvants to combat opioid tolerance and hyperalgesia. Although the role of each NPFFR subtypes remains to be ascertained, accumulating evidence implies either NPFFR1 antagonists or NPFFR2 agonists as analgesic therapies. As demonstrated in several studies, antagonism of NPFFRs appears to be a promising approach to treat opioid withdrawal syndromes. Furthermore, NPFFR antagonists have also been implicated to possess therapeutic benefits for the treatment of anxiety and opioid-induced constipation. Currently, there are limited studies, sometimes with conflicting results, on the roles of the NPFF system in the cardiovascular regulation, control of feeding and body weight and the rewarding process of other drugs of abuse such amphetamine/methamphetamine, cocaine, and nicotine. Further studies are required to validate NPFFRs as targets for these therapeutic areas.
In conclusions, the NPFF system represents an attractive drug target for a variety of pathological conditions. In particular, NPFFR ligands emerge as promising opioid adjuvants which may limit the side effects of opioids. Discovery of NPFFR ligands and availability of biochemical assays and behavioral studies has accelerated the progression of scientific knowledge of the pharmacology of the NPFF system. However, the quest to develop NPFFR-acting therapeutic agents still requires much work across medicinal chemistry, pharmacology, and molecular modeling fields to overcome existing limitations of current ligands including low selectivity and poor pharmacokinetic properties.
Acknowledgements
This work was supported by National Institute on Drug Abuse, National Institutes of Health, U.S. (Grants DA045910 to T.N. and DA040693 to Y.Z.) and the Professional Development Award by RTI International to T.N.
Abbreviations
- ASIC
acid-sensing ion channels
- CFA
complete Freund’s adjuvant
- CNS
central nervous system
- CPP
condition placed preference
- DOR
delta-opioid receptor
- ERK
extracellular signal-regulated kinase
- GI
gastrointestinal
- HPA
hypothalamic-pituitary-adrenal
- i.c.v.
intracerebroventricular
- i.p.
intraperitoneal
- i.t.
intrathecal
- i.v.
intravenous
- KOR
kappa-opioid receptor
- MAP
mean arterial pressure
- MOR
mu-opioid receptor
- NAc
nucleus accumbens
- NPFF
Neuropeptide FF
- NPFFR1
Neuropeptide FF Receptor 1
- NPFFR2
Neuropeptide Receptor 2
- s.c.
subcutaneous
- SAR
structure-activity relationship
- RSAT
Receptor Selection and Amplification Technology
- VTA
ventral tegmental area
Biographies
Thuy Nguyen is a research chemist in the Center for Drug Discovery at RTI International working on development of GPCR ligands for neurotherapeutic applications. She received her Ph.D. in medicinal chemistry in 2011 from National University of Singapore. Upon graduation, she worked as Research Fellow at the Experimental Therapeutics Center, A*Star Institute in Singapore focusing on assay development and subsequently held a postdoctoral position performing lead optimization of natural products, peptides, small molecule ligands at the Department of Pharmacy, Virginia Commonwealth University prior to joining RTI International.
Julie Marusich is a research pharmacologist in the Center for Drug Discovery at RTI International where she studies the behavioral pharmacology of substance use disorders. She received her Ph.D. in psychology in 2008 from the University of Florida. She subsequently completed a postdoctoral fellowship in behavioral neuroscience at the University of Kentucky, and then joined RTI International. Her research interests include the behavioral and neurochemical consequences of substances of abuse, with an emphasis on the abuse liability of novel psychoactive substances, and other stimulants of abuse.
Jun-Xu Li is a professor of pharmacology and toxicology at University at Buffalo, the State University of New York. He received his PhD in neuropharmacology in 2005 from Peking University, China. He joined University at Buffalo in 2010 as a tenure-track assistant professor and was promoted to professor in 2020. His research interests include the behavioral neuropharmacology, target validation and drug discovery in the field of pain and drug addiction.
Yanan Zhang is a Senior Research Chemist at the Center for Drug Discovery at Research Triangle Institute (RTI). He received his Ph.D. in organic chemistry from Binghamton University, the State University of New York in 2005 and joined Research Triangle Institute the same year. His current research focuses on medication or probe development targeting G protein-coupled receptors (GPCRs) including cannabinoid, orexin, and neuropeptide FF receptors for therapeutic indications such as pain, addiction, and cognition.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1.Yang HY; Fratta W; Majane EA; Costa E Isolation, sequencing, synthesis, and pharmacological characterization of two brain neuropeptides that modulate the action of morphine. Proc. Natl. Acad. Sci. U. S. A 1985, 82, 7757–7761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsutsui K; Osugi T; Son YL; Ubuka T Review: Structure, function and evolution of gnih. Gen. Comp. Endocrinol 2018, 264, 48–57. [DOI] [PubMed] [Google Scholar]
- 3.Ullah R; Shen Y; Zhou YD; Huang K; Fu JF; Wahab F; Shahab M Expression and actions of gnih and its orthologs in vertebrates: Current status and advanced knowledge. Neuropeptides 2016, 59, 9–20. [DOI] [PubMed] [Google Scholar]
- 4.Leprince J; Bagnol D; Bureau R; Fukusumi S; Granata R; Hinuma S; Larhammar D; Primeaux S; Sopkova-de Oliveiras Santos J; Tsutsui K; Ukena K; Vaudry H The arg-phe-amide peptide 26rfa/glutamine rf-amide peptide and its receptor: Iuphar review 24. Br. J. Pharmacol 2017, 174, 3573–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takayanagi Y; Onaka T Roles of prolactin-releasing peptide and rfamide related peptides in the control of stress and food intake. FEBS J. 2010, 277, 4998–5005. [DOI] [PubMed] [Google Scholar]
- 6.Mills EGA; O’Byrne KT; Comninos AN Kisspeptin as a behavioral hormone. Semin. Reprod. Med 2019, 37, 56–63. [DOI] [PubMed] [Google Scholar]
- 7.Vilim FS; Aarnisalo AA; Nieminen M-L; Lintunen M; Karlstedt K; Kontinen VK; Kalso E; States B; Panula P; Ziff E Gene for pain modulatory neuropeptide npff: Induction in spinal cord by noxious stimuli. Mol. Pharmacol 1999, 55, 804–811. [PubMed] [Google Scholar]
- 8.Bonini JA; Jones KA; Adham N; Forray C; Artymyshyn R; Durkin MM; Smith KE; Tamm JA; Boteju LW; Lakhlani PP; Raddatz R; Yao WJ; Ogozalek KL; Boyle N; Kouranova EV; Quan Y; Vaysse PJ; Wetzel JM; Branchek TA; Gerald C; Borowsky B Identification and characterization of two g protein-coupled receptors for neuropeptide ff. J. Biol. Chem 2000, 275, 39324–39331. [DOI] [PubMed] [Google Scholar]
- 9.Elshourbagy NA; Ames RS; Fitzgerald LR; Foley JJ; Chambers JK; Szekeres PG; Evans NA; Schmidt DB; Buckley PT; Dytko GM; Murdock PR; Milligan G; Groarke DA; Tan KB; Shabon U; Nuthulaganti P; Wang DY; Wilson S; Bergsma DJ; Sarau HM Receptor for the pain modulatory neuropeptides ff and af is an orphan g protein-coupled receptor. J. Biol. Chem 2000, 275, 25965–25971. [DOI] [PubMed] [Google Scholar]
- 10.Mollereau C; Mazarguil H; Zajac JM; Roumy M Neuropeptide ff (npff) analogs functionally antagonize opioid activities in npff2 receptor-transfected sh-sy5y neuroblastoma cells. Mol. Pharmacol 2005, 67, 965–975. [DOI] [PubMed] [Google Scholar]
- 11.Mouledous L; Mollereau C; Zajac JM Opioid-modulating properties of the neuropeptide ff system. Biofactors 2010, 36, 423–429. [DOI] [PubMed] [Google Scholar]
- 12.Roumy M; Zajac JM Neuropeptide ff, pain and analgesia. Eur. J. Pharmacol 1998, 345, 1–11. [DOI] [PubMed] [Google Scholar]
- 13.Panula P; Kalso E; Nieminen M; Kontinen VK; Brandt A; Pertovaara A Neuropeptide ff and modulation of pain. Brain Res. 1999, 848, 191–196. [DOI] [PubMed] [Google Scholar]
- 14.Fang Q; Jiang TN; Li N; Han ZL; Wang R Central administration of neuropeptide ff and related peptides attenuate systemic morphine analgesia in mice. Protein Peptide Lett. 2011, 18, 403–409. [DOI] [PubMed] [Google Scholar]
- 15.Kontinen VK; Kalso EA Differential modulation of alpha 2-adrenergic and mu-opioid spinal antinociception by neuropeptide ff. Peptides 1995, 16, 973–977. [DOI] [PubMed] [Google Scholar]
- 16.Mollereau C; Roumy M; Zajac JM Opioid-modulating peptides: Mechanisms of action. Curr. Top. Med. Chem 2005, 5, 341–355. [DOI] [PubMed] [Google Scholar]
- 17.Simonin F Neuropeptide ff receptors as therapeutic targets. Drug. Future 2006, 31, 603–609. [Google Scholar]
- 18.Yu HP; Zhang N; Zhang T; Wang ZL; Li N; Tang HH; Zhang R; Zhang MN; Xu B; Fang Q; Wang R Activation of npff2 receptor stimulates neurite outgrowth in neuro 2a cells through activation of erk signaling pathway. Peptides 2016, 86, 24–32. [DOI] [PubMed] [Google Scholar]
- 19.Panula P; Aarnisalo AA; Wasowicz K Neuropeptide ff, a mammalian neuropeptide with multiple functions. Prog. Neurobiol 1996, 48, 461–487. [DOI] [PubMed] [Google Scholar]
- 20.Yang HY; Iadarola MJ Modulatory roles of the npff system in pain mechanisms at the spinal level. Peptides 2006, 27, 943–952. [DOI] [PubMed] [Google Scholar]
- 21.Yang HY; Tao T; Iadarola MJ Modulatory role of neuropeptide ff system in nociception and opiate analgesia. Neuropeptides 2008, 42, 1–18. [DOI] [PubMed] [Google Scholar]
- 22.Ayachi S; Simonin F Involvement of mammalian rf-amide peptides and their receptors in the modulation of nociception in rodents. Front. Endocrinol 2014, 5, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Payza K; Akar CA; Yang HY Neuropeptide ff receptors: Structure-activity relationship and effect of morphine. J. Pharmacol. Exp. Ther 1993, 267, 88–94. [PubMed] [Google Scholar]
- 24.Gouarderes C; Quelven I; Mollereau C; Mazarguil H; Rice SQ; Zajac JM Quantitative autoradiographic distribution of npff1 neuropeptide ff receptor in the rat brain and comparison with npff2 receptor by using [125i]yvp and [(125i]eyf as selective radioligands. Neuroscience 2002, 115, 349–361. [DOI] [PubMed] [Google Scholar]
- 25.Gouarderes C; Faura CC; Zajac JM Rodent strain differences in the npff1 and npff2 receptor distribution and density in the central nervous system. Brain Res. 2004, 1014, 61–70. [DOI] [PubMed] [Google Scholar]
- 26.Kontinen VK; Aarnisalo AA; Idanpaan-Heikkila JJ; Panula P; Kalso E Neuropeptide ff in the rat spinal cord during carrageenan inflammation. Peptides 1997, 18, 287–292. [DOI] [PubMed] [Google Scholar]
- 27.Yang HY; Iadarola MJ Activation of spinal neuropeptide ff and the neuropeptide ff receptor 2 during inflammatory hyperalgesia in rats. Neuroscience 2003, 118, 179–187. [DOI] [PubMed] [Google Scholar]
- 28.Nystedt JM; Lemberg K; Lintunen M; Mustonen K; Holma R; Kontinen VK; Kalso E; Panula P Pain- and morphine-associated transcriptional regulation of neuropeptide ff and the g-protein-coupled npff2 receptor gene. Neurobiol. Dis 2004, 16, 254–262. [DOI] [PubMed] [Google Scholar]
- 29.Kotani M; Mollereau C; Detheux M; Le Poul E; Brezillon S; Vakili J; Mazarguil H; Vassart G; Zajac JM; Parmentier M Functional characterization of a human receptor for neuropeptide ff and related peptides. Br. J. Pharmacol 2001, 133, 138–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gouarderes C; Mazarguil H; Mollereau C; Chartrel N; Leprince J; Vaudry H; Zajac JM Functional differences between npff1 and npff2 receptor coupling: High intrinsic activities of rfamide-related peptides on stimulation of [35s]gtpgammas binding. Neuropharmacology 2007, 52, 376–386. [DOI] [PubMed] [Google Scholar]
- 31.Roumy M; Zajac J Neuropeptide ff selectively attenuates the effects of nociceptin on acutely dissociated neurons of the rat dorsal raphe nucleus. Brain Res. 1999, 845, 208–214. [DOI] [PubMed] [Google Scholar]
- 32.Rebeyrolles S; Zajac JM; Roumy M Neuropeptide ff reverses the effect of mu-opioid on ca2+ channels in rat spinal ganglion neurones. Neuroreport 1996, 7, 2979–2981. [DOI] [PubMed] [Google Scholar]
- 33.Roumy M; Garnier M; Zajac JM Neuropeptide ff receptors 1 and 2 exert an anti-opioid activity in acutely dissociated rat dorsal raphe and periventricular hypothalamic neurones. Neurosci. Lett 2003, 348, 159–162. [DOI] [PubMed] [Google Scholar]
- 34.Kersante F; Mollereau C; Zajac JM; Roumy M Anti-opioid activities of npff1 receptors in a sh-sy5y model. Peptides 2006, 27, 980–989. [DOI] [PubMed] [Google Scholar]
- 35.Mollereau C; Roumy M; Zajac JM Neuropeptide ff receptor modulates potassium currents in a dorsal root ganglion cell line. Pharmacol. Rep 2011, 63, 1061–1065. [DOI] [PubMed] [Google Scholar]
- 36.Lingueglia E; Deval E; Lazdunski M Fmrfamide-gated sodium channel and asic channels: A new class of ionotropic receptors for fmrfamide and related peptides. Peptides 2006, 27, 1138–1152. [DOI] [PubMed] [Google Scholar]
- 37.Bihel F Opioid adjuvant strategy: Improving opioid effectiveness. Future Med. Chem 2016, 8, 339–354. [DOI] [PubMed] [Google Scholar]
- 38.Altier N; Dray A; Menard D; Henry JL Neuropeptide ff attenuates allodynia in models of chronic inflammation and neuropathy following intrathecal or intracerebroventricular administration. Eur. J. Pharmacol 2000, 407, 245–255. [DOI] [PubMed] [Google Scholar]
- 39.Gouarderes C; Sutak M; Zajac JM; Jhamandas K Antinociceptive effects of intrathecally administered f8famide and fmrfamide in the rat. Eur. J. Pharmacol 1993, 237, 73–81. [DOI] [PubMed] [Google Scholar]
- 40.Oberling P; Stinus L; Le Moal M; Simonnet G Biphasic effect on nociception and antiopiate activity of the neuropeptide ff (flfqpqrfamide) in the rat. Peptides 1993, 14, 919–924. [DOI] [PubMed] [Google Scholar]
- 41.Xu M; Kontinen VK; Panula P; Kalso E Effects of (1dme)npyf, a synthetic neuropeptide ff analogue, in different pain models. Peptides 1999, 20, 1071–1077. [DOI] [PubMed] [Google Scholar]
- 42.Desprat C; Zajac JM Differential modulation of mu- and delta-opioid antinociception by neuropeptide ff receptors in young mice. Neuropeptides 1997, 31, 1–7. [DOI] [PubMed] [Google Scholar]
- 43.Lake JR; Hammond MV; Shaddox RC; Hunsicker LM; Yang HY; Malin DH Igg from neuropeptide ff antiserum reverses morphine tolerance in the rat. Neurosci. Lett 1991, 132, 29–32. [DOI] [PubMed] [Google Scholar]
- 44.Panula P; Kivipelto L; Nieminen O; Majane EA; Yang HY Neuroanatomy of morphine-modulating peptides. Med. Biol 1987, 65, 127–135. [PubMed] [Google Scholar]
- 45.Allard M; Geoffre S; Legendre P; Vincent JD; Simonnet G Characterization of rat spinal cord receptors to flfqpqrfamide, a mammalian morphine modulating peptide: A binding study. Brain Res. 1989, 500, 169–176. [DOI] [PubMed] [Google Scholar]
- 46.Allard M; Zajac JM; Simonnet G Autoradiographic distribution of receptors to flfqpqrfamide, a morphine-modulating peptide, in rat central nervous system. Neuroscience 1992, 49, 101–116. [DOI] [PubMed] [Google Scholar]
- 47.Gouarderes C; Tafani JA; Zajac JM Affinity of neuropeptide ff analogs to opioid receptors in the rat spinal cord. Peptides 1998, 19, 727–730. [DOI] [PubMed] [Google Scholar]
- 48.Roumy M; Lorenzo C; Mazeres S; Bouchet S; Zajac JM; Mollereau C Physical association between neuropeptide ff and micro-opioid receptors as a possible molecular basis for anti-opioid activity. J. Biol. Chem 2007, 282, 8332–8342. [DOI] [PubMed] [Google Scholar]
- 49.Lameh J; Bertozzi F; Kelly N; Jacobi PM; Nguyen D; Bajpai A; Gaubert G; Olsson R; Gardell LR Neuropeptide ff receptors have opposing modulatory effects on nociception. J. Pharmacol. Exp. Ther 2010, 334, 244–254. [DOI] [PubMed] [Google Scholar]
- 50.Gouarderes C; Jhamandas K; Sutak M; Zajac JM Role of opioid receptors in the spinal antinociceptive effects of neuropeptide ff analogues. Br. J. Pharmacol 1996, 117, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simonin F; Schmitt M; Laulin JP; Laboureyras E; Jhamandas JH; MacTavish D; Matifas A; Mollereau C; Laurent P; Parmentier M; Kieffer BL; Bourguignon JJ; Simonnet G Rf9, a potent and selective neuropeptide ff receptor antagonist, prevents opioid-induced tolerance associated with hyperalgesia. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 466–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nguyen T; Decker AM; Langston TL; Mathews KM; Siemian JN; Li JX; Harris DL; Runyon SP; Zhang Y Discovery of novel proline-based neuropeptide ff receptor antagonists. ACS Chem. Neurosci 2017, 8, 2290–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elhabazi K; Trigo JM; Mollereau C; Mouledous L; Zajac JM; Bihel F; Schmitt M; Bourguignon JJ; Meziane H; Petit-demouliere B; Bockel F; Maldonado R; Simonin F Involvement of neuropeptide ff receptors in neuroadaptive responses to acute and chronic opiate treatments. Br. J. Pharmacol 2012, 165, 424–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gelot A; Frances B; Gicquel S; Zajac JM Antisense oligonucleotides to human sqa-neuropeptide ff decrease morphine tolerance and dependence in mice. Eur. J. Pharmacol 1998, 358, 203–206. [DOI] [PubMed] [Google Scholar]
- 55.Bailey CP; Connor M Opioids: Cellular mechanisms of tolerance and physical dependence. Curr. Opin. Pharmacol 2005, 5, 60–68. [DOI] [PubMed] [Google Scholar]
- 56.Martini L; Whistler JL The role of mu opioid receptor desensitization and endocytosis in morphine tolerance and dependence. Curr. Opin. Neurobiol 2007, 17, 556–564. [DOI] [PubMed] [Google Scholar]
- 57.Lin YT; Liu HL; Day YJ; Chang CC; Hsu PH; Chen JC Activation of npffr2 leads to hyperalgesia through the spinal inflammatory mediator cgrp in mice. Exp. Neurol 2017, 291, 62–73. [DOI] [PubMed] [Google Scholar]
- 58.Malin DH; Henceroth MM; Izygon JJ; Nghiem DM; Moon WD; Anderson AP; Madison CA; Goyarzu P; Ma JN; Burstein ES Reversal of morphine tolerance by a compound with npff receptor subtype-selective actions. Neurosci. Lett 2015, 584, 141–145. [DOI] [PubMed] [Google Scholar]
- 59.Stinus L; Allard M; Gold L; Simonnet G Changes in cns neuropeptide ff-like material, pain sensitivity, and opiate dependence following chronic morphine treatment. Peptides 1995, 16, 1235–1241. [DOI] [PubMed] [Google Scholar]
- 60.Malin DH; Lake JR; Fowler DE; Hammond MV; Brown SL; Leyva JE; Prasco PE; Dougherty TM Fmrf-nh2-like mammalian peptide precipitates opiate-withdrawal syndrome in the rat. Peptides 1990, 11, 277–280. [DOI] [PubMed] [Google Scholar]
- 61.Malin DH; Lake JR; Short PE; Blossman JB; Lawless BA; Schopen CK; Sailer EE; Burgess K; Wilson OB Nicotine abstinence syndrome precipitated by an analog of neuropeptide ff. Pharmacol. Biochem. Behav 1996, 54, 581–585. [DOI] [PubMed] [Google Scholar]
- 62.Tan PP; Chen JC; Li JY; Liang KW; Wong CH; Huang EY Modulation of naloxone-precipitated morphine withdrawal syndromes in rats by neuropeptide ff analogs. Peptides 1999, 20, 1211–1217. [DOI] [PubMed] [Google Scholar]
- 63.Malin DH; Lake JR; Leyva JE; Hammond MV; Rogillio RB; Arcangeli KR; Ludgate K; Moore GM; Payza K Analog of neuropeptide ff attenuates morphine abstinence syndrome. Peptides 1991, 12, 1011–1014. [DOI] [PubMed] [Google Scholar]
- 64.Malin DH; Henceroth MM; Elayoubi J; Campbell JR; Anderson A; Goyarzu P; Izygon J; Madison CA; Ward CP; Burstein ES A subtype-specific neuropeptide ff receptor antagonist attenuates morphine and nicotine withdrawal syndrome in the rat. Neurosci. Lett 2018, 684, 98–103. [DOI] [PubMed] [Google Scholar]
- 65.Marchand S; Betourne A; Marty V; Daumas S; Halley H; Lassalle JM; Zajac JM; Frances B A neuropeptide ff agonist blocks the acquisition of conditioned place preference to morphine in c57bl/6j mice. Peptides 2006, 27, 964–972. [DOI] [PubMed] [Google Scholar]
- 66.Kotlinska J; Pachuta A; Dylag T; Silberring J Neuropeptide ff (npff) reduces the expression of morphine- but not of ethanol-induced conditioned place preference in rats. Peptides 2007, 28, 2235–2242. [DOI] [PubMed] [Google Scholar]
- 67.Wu CH; Tao PL; Huang EY Distribution of neuropeptide ff (npff) receptors in correlation with morphine-induced reward in the rat brain. Peptides 2010, 31, 1374–1382. [DOI] [PubMed] [Google Scholar]
- 68.Kotlinska JH; Gibula-Bruzda E; Koltunowska D; Raoof H; Suder P; Silberring J Modulation of neuropeptide ff (npff) receptors influences the expression of amphetamine-induced conditioned place preference and amphetamine withdrawal anxiety-like behavior in rats. Peptides 2012, 33, 156–163. [DOI] [PubMed] [Google Scholar]
- 69.Chen JC; Lee WH; Chen PC; Tseng CP; Huang EY Rat npff(1) receptor-mediated signaling: Functional comparison of neuropeptide ff (npff), fmrfamide and pfr(tic)amide. Peptides 2006, 27, 1005–1014. [DOI] [PubMed] [Google Scholar]
- 70.Laguzzi R; Nosjean A; Mazarguil H; Allard M Cardiovascular effects induced by the stimulation of neuropeptide ff receptors in the dorsal vagal complex: An autoradiographic and pharmacological study in the rat. Brain Res. 1996, 711, 193–202. [DOI] [PubMed] [Google Scholar]
- 71.Roth BL; Disimone J; Majane EA; Yang HY Elevation of arterial pressure in rats by two new vertebrate peptides flfqpqrf-nh2 and ageglsspfwslaapqrf-nh2 which are immunoreactive to fmrf-nh2 antiserum. Neuropeptides 1987, 10, 37–42. [DOI] [PubMed] [Google Scholar]
- 72.Huang EY; Li JY; Tan PP; Wong CH; Chen JC The cardiovascular effects of pfrfamide and pfr(tic)amide, a possible agonist and antagonist of neuropeptide ff (npff). Peptides 2000, 21, 205–210. [DOI] [PubMed] [Google Scholar]
- 73.Fang Q; Li N; Jiang TN; Liu Q; Li YL; Wang R Pressor and tachycardic responses to intrathecal administration of neuropeptide ff in anesthetized rats. Peptides 2010, 31, 683–688. [DOI] [PubMed] [Google Scholar]
- 74.Kavaliers M; Hirst M; Mathers A Inhibitory influences of fmrfamide on morphine- and deprivation-induced feeding. Neuroendocrinology 1985, 40, 533–535. [DOI] [PubMed] [Google Scholar]
- 75.Murase T; Arima H; Kondo K; Oiso Y Neuropeptide ff reduces food intake in rats. Peptides 1996, 17, 353–354. [DOI] [PubMed] [Google Scholar]
- 76.Robert JJ; Orosco M; Rouch C; Jacquot C; Cohen Y Unexpected responses of the obese “cafeteria” rat to the peptide fmrf-amide. Pharmacol. Biochem. Behav 1989, 34, 341–344. [DOI] [PubMed] [Google Scholar]
- 77.Zhang L; Ip CK; Lee IJ; Qi Y; Reed F; Karl T; Low JK; Enriquez RF; Lee NJ; Baldock PA; Herzog H Diet-induced adaptive thermogenesis requires neuropeptide ff receptor-2 signalling. Nat. Commun 2018, 9, 4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Leon S; Velasco I; Vazquez MJ; Barroso A; Beiroa D; Heras V; Ruiz-Pino F; Manfredi-Lozano M; Romero-Ruiz A; Sanchez-Garrido MA; Dieguez C; Pinilla L; Roa J; Nogueiras R; Tena-Sempere M Sex-biased physiological roles of npff1r, the canonical receptor of rfrp-3, in food intake and metabolic homeostasis revealed by its congenital ablation in mice. Metabolism 2018, 87, 87–97. [DOI] [PubMed] [Google Scholar]
- 79.Kriegsfeld LJ; Gibson EM; Williams WP 3rd; Zhao S; Mason AO; Bentley GE; Tsutsui K The roles of rfamide-related peptide-3 in mammalian reproductive function and behaviour. J. Neuroendocrinol 2010, 22, 692–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pineda R; Garcia-Galiano D; Sanchez-Garrido MA; Romero M; Ruiz-Pino F; Aguilar E; Dijcks FA; Blomenrohr M; Pinilla L; van Noort PI; Tena-Sempere M Characterization of the potent gonadotropin-releasing activity of rf9, a selective antagonist of rf-amide-related peptides and neuropeptide ff receptors: Physiological and pharmacological implications. Endocrinology 2010, 151, 1902–1913. [DOI] [PubMed] [Google Scholar]
- 81.Chalmers DT; Lovenberg TW; De Souza EB Localization of novel corticotropin-releasing factor receptor (crf2) mrna expression to specific subcortical nuclei in rat brain: Comparison with crf1 receptor mrna expression. J. Neurosci 1995, 15, 6340–6350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Post A; Ohl F; Almeida OF; Binder EB; Rucker M; Welt S; Binder E; Holsboer F; Sillaber I Identification of molecules potentially involved in mediating the in vivo actions of the corticotropin-releasing hormone receptor 1 antagonist, nbi30775 (r121919). Psychopharmacology (Berl) 2005, 180, 150–158. [DOI] [PubMed] [Google Scholar]
- 83.Kim JS; Brownjohn PW; Dyer BS; Beltramo M; Walker CS; Hay DL; Painter GF; Tyndall JD; Anderson GM Anxiogenic and stressor effects of the hypothalamic neuropeptide rfrp-3 are overcome by the npffr antagonist gj14. Endocrinology 2015, 156, 4152–4162. [DOI] [PubMed] [Google Scholar]
- 84.Lin YT; Yu YL; Hong WC; Yeh TS; Chen TC; Chen JC Npffr2 activates the hpa axis and induces anxiogenic effects in rodents. Int. J. Mol. Sci 2017, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Raffa RB; Jacoby HI A-18-famide and f-8-famide, endogenous mammalian equivalents of the molluscan neuropeptide fmrfamide (phe-met-arg-phe-nh2), inhibit colonic bead expulsion time in mice. Peptides 1989, 10, 873–875. [DOI] [PubMed] [Google Scholar]
- 86.Gicquel S; Fioramonti J; Bueno L; Zajac JM Effects of f8famide analogs on intestinal transit in mice. Peptides 1993, 14, 749–753. [DOI] [PubMed] [Google Scholar]
- 87.Million M; Fioramonti J; Gicquel S; Zajac JM; Bueno L Comparative action of phe-leu-phe-gln-pro-gln-arg-phe-nh2 analogs on intestinal motility and nociception in rats. J. Pharmacol. Exp. Ther 1993, 265, 96–102. [PubMed] [Google Scholar]
- 88.Gicquel S; Mazarguil H; Desprat C; Allard M; Devillers JP; Simonnet G; Zajac JM Structure-activity study of neuropeptide ff: Contribution of n-terminal regions to affinity and activity. J. Med. Chem 1994, 37, 3477–3481. [DOI] [PubMed] [Google Scholar]
- 89.Gicquel S; Mazarguil H; Allard M; Simonnet G; Zajac JM Analogues of f8famide resistant to degradation, with high affinity and in vivo effects. Eur. J. Pharmacol 1992, 222, 61–67. [DOI] [PubMed] [Google Scholar]
- 90.Gouarderes C; Mollereau C; Tafani JA; Mazarguil H; Zajac JM [(125)i]eyf: A new high affinity radioligand to neuropeptide ff receptors. Peptides 2001, 22, 623–629. [DOI] [PubMed] [Google Scholar]
- 91.Quelven I; Roussin A; Burlet-Schiltz O; Gouarderes C; Tafani JA; Mazarguil H; Zajac JM Dissociation of pharmacological pro- and anti-opioid effects by neuropeptide ff analogs. Eur. J. Pharmacol 2002, 449, 91–98. [DOI] [PubMed] [Google Scholar]
- 92.Mazarguil H; Gouarderes C; Tafani JA; Marcus D; Kotani M; Mollereau C; Roumy M; Zajac JM Structure-activity relationships of neuropeptide ff: Role of c-terminal regions. Peptides 2001, 22, 1471–1478. [DOI] [PubMed] [Google Scholar]
- 93.Chen JC; Li JY; Liang KW; Huang YK Neuropeptide ff potentiates the behavioral sensitization to amphetamine and alters the levels of neurotransmitters in the medial prefrontal cortex. Brain Res. 1999, 816, 220–224. [DOI] [PubMed] [Google Scholar]
- 94.Kotlinska J; Pachuta A; Silberring J Neuropeptide ff (npff) reduces the expression of cocaine-induced conditioned place preference and cocaine-induced sensitization in animals. Peptides 2008, 29, 933–939. [DOI] [PubMed] [Google Scholar]
- 95.Maletinska L; Ticha A; Nagelova V; Spolcova A; Blechova M; Elbert T; Zelezna B Neuropeptide ff analog rf9 is not an antagonist of npff receptor and decreases food intake in mice after its central and peripheral administration. Brain Res. 2013, 1498, 33–40. [DOI] [PubMed] [Google Scholar]
- 96.Engstrom M; Wurster S; Savola JM; Panula P Functional properties of pfr(tic)amide and bibp3226 at human neuropeptide ff2 receptors. Peptides 2003, 24, 1947–1954. [DOI] [PubMed] [Google Scholar]
- 97.Malin DH; Lake JR; Smith DA; Jones JA; Morel J; Claunch AE; Stevens PA; Payza K; Ho KK; Liu J; Ham I; Burgess K Subcutaneous injection of an analog of neuropeptide ff prevents naloxone-precipitated morphine abstinence syndrome. Drug Alcohol Depend. 1995, 40, 37–42. [DOI] [PubMed] [Google Scholar]
- 98.Min L; Leon S; Li H; Pinilla L; Carroll RS; Tena-Sempere M; Kaiser UB Rf9 acts as a kiss1r agonist in vivo and in vitro. Endocrinology 2015, 156, 4639–4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu X; Herbison AE Rf9 excitation of gnrh neurons is dependent upon kiss1r in the adult male and female mouse. Endocrinology 2014, 155, 4915–4924. [DOI] [PubMed] [Google Scholar]
- 100.Wang ZL; Li N; Wang P; Tang HH; Han ZL; Song JJ; Li XH; Yu HP; Zhang T; Zhang R; Xu B; Zhang MN; Fang Q; Wang R Pharmacological characterization of en-9, a novel chimeric peptide of endomorphin-2 and neuropeptide ff that produces potent antinociceptive activity and limited tolerance. Neuropharmacology 2016, 108, 364–372. [DOI] [PubMed] [Google Scholar]
- 101.Li N; Han ZL; Wang ZL; Xing YH; Sun YL; Li XH; Song JJ; Zhang T; Zhang R; Zhang MN; Xu B; Fang Q; Wang R Bn-9, a chimeric peptide with mixed opioid and neuropeptide ff receptor agonistic properties, produces nontolerance-forming antinociception in mice. Br. J. Pharmacol 2016, 173, 1864–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang ZL; Pan JX; Song JJ; Tang HH; Yu HP; Li XH; Li N; Zhang T; Zhang R; Zhang MN; Xu B; Fang Q; Wang R Structure-based optimization of multifunctional agonists for opioid and neuropeptide ff receptors with potent nontolerance forming analgesic activities. J. Med. Chem 2016, 59, 10198–10208. [DOI] [PubMed] [Google Scholar]
- 103.Xu B; Guo Y; Zhang M; Zhang R; Chen D; Zhang Q; Xiao J; Xu K; Li N; Qiu Y; Zhu H; Niu J; Zhang X; Fang Q Central and peripheral modulation of gastrointestinal transit in mice by dn-9, a multifunctional opioid/npff receptor agonist. Neurogastroenterol. Motil 2020, e13848. [DOI] [PubMed] [Google Scholar]
- 104.Drieu la Rochelle A; Guillemyn K; Dumitrascuta M; Martin C; Utard V; Quillet R; Schneider S; Daubeuf F; Willemse T; Mampuys P; Maes BUW; Frossard N; Bihel F; Spetea M; Simonin F; Ballet S A bifunctional-biased mu-opioid agonist-neuropeptide ff receptor antagonist as analgesic with improved acute and chronic side effects. Pain 2018, 159, 1705–1718. [DOI] [PubMed] [Google Scholar]
- 105.Zhang T; Han Z; Shi X; Zhao W; Wang Z; Zhang R; Xu B; Zhang M; Zhang Q; Xiao J; Zhu H; Zheng T; Fang Q Discovery of two novel branched peptidomimetics containing endomorphin-2 and rf9 pharmacophores: Synthesis and neuropharmacological evaluation. Bioorg. Med. Chem 2019, 27, 630–643. [DOI] [PubMed] [Google Scholar]
- 106.Kawakami JK; Konkel MJ; Boteju LW; Wetzel JM; Noble SA; Wan H Quinazolino- and Quinolino-Guanidines as Ligands for the Neuropeptide FF (NPFF) Receptors. WO 200302667 A1, 2003.
- 107.Scully AL; Davis RE; Vanover KE; Gardell LR; Lameh J; Kelly NM; Bertozzi F; Sherbukhin V Treating Neuropathic Pain with Neuropeptide FF Receptor 2 Agonists. WO 2005/031000 A2, 2005.
- 108.Gaubert G; Bertozzi F; Kelly NM; Pawlas J; Scully AL; Nash NR; Gardell LR; Lameh J; Olsson R Discovery of selective nonpeptidergic neuropeptide ff2 receptor agonists. J. Med. Chem 2009, 52, 6511–6514. [DOI] [PubMed] [Google Scholar]
- 109.Findeisen M; Wurker C; Rathmann D; Meier R; Meiler J; Olsson R; Beck-Sickinger AG Selective mode of action of guanidine-containing non-peptides at human npff receptors. J. Med. Chem 2012, 55, 6124–6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nakamura T; Saito S; Yoshinaga M; Ohta H; Kawamura M; Ishizaka T Heterocyclic Compound. WO 2009038012 A1, 2009.
- 111.Do EU; Piao LZ; Choi G; Choi YB; Kang TM; Shin J; Chang YJ; Nam HY; Kim HJ; Kim SI The high throughput screening of neuropeptide ff2 receptor ligands from korean herbal plant extracts. Peptides 2006, 27, 997–1004. [DOI] [PubMed] [Google Scholar]
- 112.Journigan VB; Mesangeau C; Vyas N; Eans SO; Cutler SJ; McLaughlin JP; Mollereau C; McCurdy CR Nonpeptide small molecule agonist and antagonist original leads for neuropeptide ff1 and ff2 receptors. J. Med. Chem 2014, 57, 8903–8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Caroff E; Steger M; Valdenaire O; Fecher A; Breu V; Hilpert K; Fretz H; Giller T Guanidine Derivatives and Use thereof as Neuropeptide FF Receptor Antagonists. WO 2004/083218 A1, 2004.
- 114.Fecher A; Fretz H; Hilpert K; Breu V; Giller T; Valdenaire O Guanidine Derivatives. WO 2005023781 A1, 2005.
- 115.Osakada N; Shinoda K; Kunori S; Shirai T; Toki S; Ichikawa S; Kosaka N; Ichimura M; Shimada J Neuropeptide FF Receptor Antagonist. WO 2004080965 A1, 2004.
- 116.Elhabazi K; Humbert JP; Bertin I; Quillet R; Utard V; Schneider S; Schmitt M; Bourguignon JJ; Laboureyras E; Ben Boujema M; Simonnet G; Ancel C; Simonneaux V; Beltramo M; Bucher B; Sorg T; Meziane H; Schneider E; Petit-Demouliere B; Ilien B; Bihel F; Simonin F Rf313, an orally bioavailable neuropeptide ff receptor antagonist, opposes effects of rf-amide-related peptide-3 and opioid-induced hyperalgesia in rodents. Neuropharmacology 2017, 118, 188–198. [DOI] [PubMed] [Google Scholar]
- 117.Bihel F; Humbert JP; Schneider S; Bertin I; Wagner P; Schmitt M; Laboureyras E; Petit-Demouliere B; Schneider E; Mollereau C; Simonnet G; Simonin F; Bourguignon JJ Development of a peptidomimetic antagonist of neuropeptide ff receptors for the prevention of opioid-induced hyperalgesia. ACS Chem. Neurosci 2015, 6, 438–445. [DOI] [PubMed] [Google Scholar]
- 118.Hammoud H; Elhabazi K; Quillet R; Bertin I; Utard V; Laboureyras E; Bourguignon JJ; Bihel F; Simonnet G; Simonin F; Schmitt M Aminoguanidine hydrazone derivatives as nonpeptide npff1 receptor antagonists reverse opioid induced hyperalgesia. ACS Chem. Neurosci 2018, 9, 2599–2609. [DOI] [PubMed] [Google Scholar]
- 119.Roumeas L; Humbert JP; Schneider S; Doebelin C; Bertin I; Schmitt M; Bourguignon JJ; Simonin F; Bihel F Effects of systematic n-terminus deletions and benzoylations of endogenous rf-amide peptides on npff1r, npff2r, gpr10, gpr54 and gpr103. Peptides 2015, 71, 156–161. [DOI] [PubMed] [Google Scholar]