

Abstract
The tumor microenvironment (TME) is considered to be one of the vital mediators of tumor progression. Extracellular matrix (ECM), infiltrating immune cells, and stromal cells collectively constitute the complex ecosystem with varied biochemical and biophysical properties. The development of liver cancer is strongly tied with fibrosis and cirrhosis that alters the microenvironmental landscape, especially ECM composition. Enhanced deposition and cross‐linking of type I collagen are frequently detected in patients with liver cancer and have been shown to facilitate tumor growth and metastasis by epithelial‐to‐mesenchymal transition. However, information on the effect of collagen enrichment on drug resistance is lacking. Thus, the present study has comprehensively illustrated phenotypical and mechanistic changes in an in vitro mimicry of collagen‐enriched TME and revealed that collagen enrichment could induce 5‐fluorouracil (5FU) and sorafenib resistance in liver cancer cells through hypoxia‐induced up‐regulation of lysyl oxidase‐like 2 (LOXL2). LOXL2, an enzyme that facilitates collagen cross‐linking, enhances cell adhesion‐mediated drug resistance by activating the integrin alpha 5 (ITGA5)/focal adhesion kinase (FAK)/phosphoinositide 3‐kinase (PI3K)/rho‐associated kinase 1 (ROCK1) signaling axis. Conclusion: We demonstrated that inhibition of LOXL2 in a collagen‐enriched microenvironment synergistically promotes the efficacy of sorafenib and 5FU through deterioration of focal adhesion signaling. These findings have clinical implications for developing LOXL2‐targeted strategies in patients with chemoresistant liver cancer and especially for those patients with advanced fibrosis and cirrhosis.
In this study, we revealed the collagen‐mediated chemoresistance via activation of HIF1A/LOXL2/ITGA5/FAK/PI3K/ROCK1 signaling and showed that LOXL2 is a promising therapeutic target to enhance the efficacy of 5FU and sorafenib in resistant liver cancer patients, especially those with advanced fibrosis and cirrhosis.
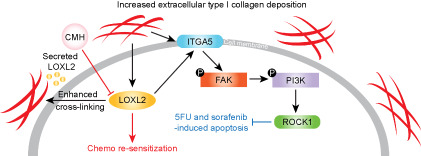
INTRODUCTION
The microenvironment of liver cancer is a highly complicated ecosystem due to the chronic progression of hepatitis B virus (HBV)‐induced fibrosis and cirrhosis. More than 80% of patients with liver cancer are diagnosed with advanced fibrosis and cirrhosis that alters the locoregional extracellular matrix (ECM).[ 1 ] Type I collagen is the major component of the ECM in the fibrotic and cirrhotic liver that contributes to malignant transformation and tumor progression. An excessive number of fibroblasts and hepatic stellate cells in the liver cancer microenvironment are primary cellular sources of collagens, but tumor cells are also found to be capable of secreting type I collagen for ECM remodeling.[ 2 , 3 ] Enhanced type I collagen deposition, cross‐linking, and reduced ECM turnover alter the biochemical and biophysical properties of the liver microenvironment through locoregional stiffening and integrin transduction, which further activates malignancy‐associated signaling pathways and cultivates tumor‐specific characteristics, including uncontrollable growth, therapeutic resistance, and distant metastasis. Ultrasound elastography, such as FibroScan, has demonstrated that liver stiffness measurements are a reliable predictor of liver cancer incidence and growth.[ 4 ] Accumulating evidence suggests that type I collagen facilitates the formation of a premetastatic niche. Thus, it is logical that type I collagen enrichment and tumor–ECM interaction in the liver cancer microenvironment might facilitate tumor progression.
The influence of collagen enrichment on tumor progression has been extensively investigated in respect to metastasis through epithelial–mesenchymal transition (EMT), whereas its function on anti‐apoptosis during therapeutic intervention remains unexplored. The efficacy of chemotherapy and targeted therapy shrinks to attenuate liver cancer progression in patients with advanced fibrosis and cirrhosis.[ 5 , 6 ] Not only does type I collagen accumulation change the liver architecture but it also creates a physical barrier that impedes drug distribution and penetration. Indeed, liver cancer remains one of the most therapeutically resistant cancers owing to the complexity of the locoregional ecosystem. 5‐Fluorouracil (5FU), a well‐established chemotherapeutic agent, and sorafenib, a broad‐spectrum kinase inhibitor, provide modest first‐line treatment efficacy, where approximately 50% of patients do not respond or rapidly develop chemoresistance.[ 7 ] Disruption of the tumor‐promoting microenvironment might serve as a feasible adjuvant strategy to improve chemotherapeutic efficacy because modulation of ECM homeostasis directly influences tumor cell survival. While the late stages of liver fibrosis and cirrhosis are irreversible, microenvironmental regulation on several key ECM components and enzymes remains clinically feasible. Hence, developing new strategies to target the collagen‐enriched liver cancer microenvironment might stop tumor progression and resensitize resistant tumors to chemotherapy and targeted therapy.
Although previous studies have demonstrated that collagen accumulation protects tumor cells from apoptosis in pancreatic ductal adenocarcinoma and small cell lung cancer,[ 8 , 9 ] how type I collagen enrichment affects therapeutic outcomes in liver cancer and the molecular mechanism of collagen‐mediated downstream signaling remains poorly deciphered.[ 10 ] Thus, we have applied an integrated approach to comprehensively investigate whether type I collagen contributes to the development of chemoresistance in liver cancer. Our study has identified and characterized that collagen accumulation in the liver cancer microenvironment can influence the clinical efficacy of chemotherapeutic agents 5FU and sorafenib through enhancing cellular focal adhesion. Cell adhesion mediated drug resistance (CAM‐DR) has been shown to play a vital role in protecting tumor cells from programmed death,[ 11 , 12 ] and many drugs targeting adhesion‐associated signaling have generated promising clinical outcomes.[ 13 ] Through whole transcriptome sequencing, we reveal that lysyl oxidase‐like 2 (LOXL2) from the ECM remodeler family is one of the major hypoxia inducible factor 1A (HIF1A)‐induced molecular drivers of 5FU and sorafenib chemoresistance in the collagen‐enriched liver cancer microenvironment through a focal adhesion‐dependent mechanism. Inhibition of LOXL2 using short hairpin RNA (shRNA) and a LOXL2‐selective inhibitor enhances in vitro and in vivo sensitivity to 5FU and sorafenib through down‐regulating the integrin alpha 5 (ITGA5)/focal adhesion kinase (FAK)/phosphoinositide 3‐kinase (PI3K)/rho‐associated kinase 1 (ROCK1) signaling axis. Altogether, these findings suggest that inhibition of LOXL2 might disrupt the development of CAM‐DR and synergistically increase the efficacy of 5FU and sorafenib in patients with chemoresistant liver cancer, especially for those with advanced fibrosis and cirrhosis.
MATERIALS AND METHODS
Bioinformatics analysis of transcriptome sequencing data
Transcriptome sequencing data of patients with liver cancer were downloaded from The Cancer Genome Atlas‐Liver Hepatocellular Carcinoma (TCGA‐LIHC) and Gene Expression Omnibus (GEO) with accession numbers GSE109211 and GSE28702. The transcriptome sequencing data were subsequently normalized and analyzed using EdgeR (version 3.13).
Establishment of an in vitro collagen‐enriched mimicry and cell culture
Type I collagen solution (100 μg/ml) from rat tail (Sigma) diluted in phosphate‐buffered saline (PBS) was coated on six‐well plates (1 ml/well) and incubated at 37°C overnight. The next day, the type I collagen solution was removed and the plates were air dried for at least 2 h. Subsequently, the plates were washed with serum‐free Dulbecco's modified Eagle's medium (DMEM; Cytiva) twice and were ready for cell seeding. Cell lines used in this study included one immortalized hepatocyte cell line (Miha) and three liver cancer cell lines (HepG2, SNU‐878, and H4M). Miha, HepG2, and SNU‐878 were obtained from the Institute of Virology, Chinese Academy of Medical Sciences (Beijing, China), and H4M cell lines were previously established in our laboratory.[ 14 ] All cell lines were cultured with DMEM + 10% fetal bovine serum (Gibco) + 1% penicillin and streptomycin (Gibco).
Immunofluorescence staining
After the 48‐h culture on collagen‐coated and noncoated plates, cells were washed twice with PBS. Subsequently, cells were fixed with 4% paraformaldehyde (Chemcruz) for 30 min, permeablized with 0.01% Triton X‐100 (Sigma) at room temperature for 10 min, and blocked with 3% bovine serum albumin (BSA; Gibco) at room temperature for 30 min. Cells were incubated with primary antibodies diluted in 0.1% BSA at 4°C overnight. The next day, cells were incubated with secondary antibodies (1:400) and Alexa Fluor 555 phalloidin (1:250; Thermo Fisher) diluted in 0.1% BSA at room temperature for 1 h. In the end, the cell nucleus was stained by 4′,6‐diamidino‐2‐phenylindole (1:1000) diluted in PBS. Fluorescence images were taken with a Zeiss 980. All antibodies used for immunofluorescence staining are listed in Table S1.
Microfluidics‐based single‐cell stiffness quantification
Quantification of whole‐cell stiffness was realized by implementing a previously developed microfluidic cytometer.[ 15 ] Such microfluidic cytometers can be fabricated by following standard soft lithography procedures. Cell samples were injected into the cytometer by using compressive air flow with a proper driving pressure (i.e., 100 Pa). Because exit widths of confining microchannels in the cytometer are much smaller than the cell diameter, injected cells can be captured at different penetrating positions in the microchannels. After capturing micrographs of cell‐penetrating positions, cell elasticity E can be calculated with measured cell diameter and penetration length by using the equation[ 16 ]
| (1) |
where F drag is the drag force on the particle, D c [= (D 2 – W deform 2)½ – D – L deform] is the diameter of the contact area between the cell body and each side of the microchannel walls; Φ and Θ are the correction factors as functions of the deformation level ξ (= 1 – W deform/D) for the hyperelastic properties, calculated by
| (2) |
Western blotting
Protein lysates were extracted from cells using the Pierce radio immunoprecipitation assay buffer (Thermo Fisher) with 1% protease inhibitor. Protein lysates were quantified using the bicinchoninic acid method (Biorad). Quantified protein lysates were diluted in protein loading buffer (Biorad) and denatured at 95°C for 10 min. The protein samples were then resolved on sodium dodecyl sulfate–
polyacrylamide gel electrophoresis, transferred onto a polyvinylidene difluoride membrane (Millipore), and then blocked with 5% BSA in Tris‐buffered saline‐Tween 20 (TBS‐T; Sigma) for 1 h at room temperature. The blocked membrane was incubated with primary antibodies in 5% BSA in TBS‐T at 4°C overnight. All antibodies used for western blotting are listed in Table S1. After washing with TBS‐T, the membrane was incubated for 1 h with horseradish peroxidase‐conjugated secondary antibody (1:3000; Sigma). A complex of primary and secondary antibody‐labeled proteins was detected by an enhanced chemiluminescence system (Biorad) followed by manual exposure using the Hypercassette Autoradiography Cassette (Cytiva Amersham).
In vitro chemoresistance assay
Collagen‐residing/parental/vector control (VEC)/LOXL2‐overexpressed (OE), short hairpin negative control (shNC)/LOXL2‐sh1/sh2 cells were treated with varied concentrations of 5FU and sorafenib for 24 and 48 h, respectively. Clinically used 5FU and sorafenib were obtained from the Sun Yat‐sen University Cancer Center. Cell viability was quantified using the XTT Proliferation Assay Kit II (Roche Diagnostics) with a measurement wavelength of 460 nm and a reference wavelength of 620 nm. Apoptotic cells were stained with phycoerythrin (PE)‐annexin V (early apoptosis) and allophycocyanin (APC)–7‐aminoactinomycin D (7AAD) (late apoptosis), using the apoptosis kit (BD Biosciences). Flow cytometry was performed using BD Canton II, and the flow data were analyzed by FlowJo (version 11).
Signaling pathway enrichment analysis
A total of 375 genes that were co‐up‐regulated in HepG2 and Miha cells were input into the Gene Set Enrichment Analysis (GSEA) molecular signatures database (http://www.gsea‐msigdb.org/gsea/msigdb/annotate.jsp). We computed the gene overlaps in H: hallmark gene sets and CP:Kyoto Encyclopedia of Genes and Genomes (KEGG): KEGG gene sets. In total, we identified the top 10 significantly enriched pathways (false discovery rate [FDR] q ≤ 0.01) in collagen‐residing HepG2 and Miha cells, ordered by −log10 FDR.
Quantitative reverse‐transcription polymerase chain reaction
Total RNA was extracted using TRIZOL reagent (Takara), and complementary DNA (cDNA) was synthesized by reverse transcription using the reverse transcription kit (Takara). We mixed 0.5 ng cDNA with quantitative reverse‐transcription polymerase chain reaction (qRT‐PCR) primers (BGI Genomics) and SYRB Green (Takara) in a 10‐μl volume and analyzed this with a LightCycler 480 II real‐time PCR detector (Roche). qRT‐PCR primers used in this study are listed in Table S1.
Plasmid construction and lentiviral transduction
Full‐length human LOXL2 cDNA was cloned into the pCDH‐CMV‐MCS‐EF1α‐Puro plasmid. Two shRNA specifically targeting LOXL2 were designed and cloned into the psi‐LVRU6P plasmid. We transfected 293FT cells with the functional plasmids with three packaging plasmids, including Gag, Vsvg, and Rev, using Lipofectamine 2000 (Invitrogen). After 72 h of transfection, lentivirus‐containing supernatants were collected. HepG2 and Miha cells were transduced with lentivirus containing the LOXL2‐OE plasmid or an empty pCDH‐CMV‐MCS‐EF1α (VEC) plasmid. SNU‐878 cells were transduced with lentivirus containing a negative control shRNA (shNC) or two LOXL2‐targeted shRNAs (LOXL2‐sh1 and LOXL2‐sh2). We used 2–4 mM puromycin (Sigma) to select for stably transduced cells. shRNA target sequences used in this study are listed in Table S1.
Dual‐luciferase reporter assay
We cloned 2 kb of the promoter sequence of human LOXL2 into the pGL3‐enhancer plasmid. Full‐length human HIF1A cDNA was cloned into the pCDH‐CMV‐MCS‐EF1α plasmid. The luciferase reporter constructs (LOXL2/NC luciferase plasmid, full‐length HIF1A/VEC plasmid, and Renilla plasmid) were transfected into 293FT cells using Lipofectamine 2000 (Invitrogen). After 48 h of transfection, the activity of firefly and Renilla luciferase were detected using the Dual‐Luciferase Reporter Assay System (E1910; Progema). Firefly luminescence was normalized to the Renilla signals.
Enzyme‐linked immunosorbent assay
Culture mediums from different groups were collected after 48 h of cell seeding. A LOXL2 standard curve was calibrated using LOXL2 standard solutions (Abcam) with concentrations of 5000, 2500, 1250, 625, 312.5, 156.25, and 0 pg/ml empty controls. The concentrations of secreted LOXL2 in different cell groups were quantified based on the human LOXL2 enzyme immunosorbent assay (ELISA) kit (Abcam), as per the manufacturer's instructions.
Mouse model
Subcutaneous injection of HepG2 and SNU‐878 cells was performed in 4‐week‐old female BALB/cAnN‐nu (nude) mice. Because LOXL2 overexpression and knockdown influenced in vivo tumorigenicity of liver cancer cells, we first euthanized the mice and collected subcutaneous tumors. The tumors were cut into 2–3‐mm3 chunks using a sterile razor blade and transplanted into new 4‐week‐old nude mice. When the transplanted tumors became visible (2 weeks), 5FU (20 mg/kg) was given through intraperitoneal injection twice per week and sorafenib (10 mg/kg) was given through intragastric administration twice per week. For the in vivo experiment testing the synergistic efficacy of the LOXL2 inhibitor, 5FU (20 mg/kg) and sorafenib (10 mg/kg) combined with (2‐chloropyridin‐4‐yl)methanamine hydrochloride (CMH) (10 mg/kg) was given through intraperitoneal/intragastric administration twice per week. Tumor volume was estimated using the formula tumor volume = (length × width2)/2. All animal experiments were conducted and approved by the Committee on the Use of Live Animals in Teaching and Research at the University of Hong Kong (HKU). Mice were maintained in a pathogen‐free animal facility at the Center for Comparative Medicine Research of HKU.
Statistical analysis
The results are represented as mean ± SD, as indicated in the figure legends. All statistical analyses were performed in GraphPad Prism (version 9) software and SPSS (version 23; IBM) using the two‐sided Student t test for comparisons between two groups, and two‐way analysis of variance (ANOVA) for comparing the change in tumor volume among different treatment groups in the in vivo chemoresistance experiments. p ≤ 0.05 was considered statistically significant. Clinical correlations between collagen type I, alpha 1 (COL1A1)/LOXL2 and liver fibrosis were evaluated by the chi‐square test in SPSS. Gene correlation coefficients (r) were determined by Pearson's correlation analysis in GraphPad Prism. The prognostic association of LOXL2 in overall survival and progression‐free survival was performed using the Kaplan‐Meier method, and statistical significance was calculated by the log‐rank test in SPSS. All experiments were independently repeated at least 3 times.
RESULTS
Collagen enrichment in the liver cancer microenvironment and its association to EMT and therapeutic resistance to 5FU and sorafenib
The development of liver cancer is a chronic process associated with the liver injury caused by viral infection, alcohol, and lipid uptake. One of the major characteristics of liver damage is abnormal collagen deposition in the locoregional microenvironment; this deposition further facilitates the malignant transformation of normal hepatocytes into tumor cells and develops a therapeutically resistant niche (Figure 1A). Although collagen‐associated mechanisms in proliferation and metastasis have been well elucidated, there is a lack of sufficient investigations on how the extracellular deposition of collagen contributes to the development of therapeutic resistance.
FIGURE 1.
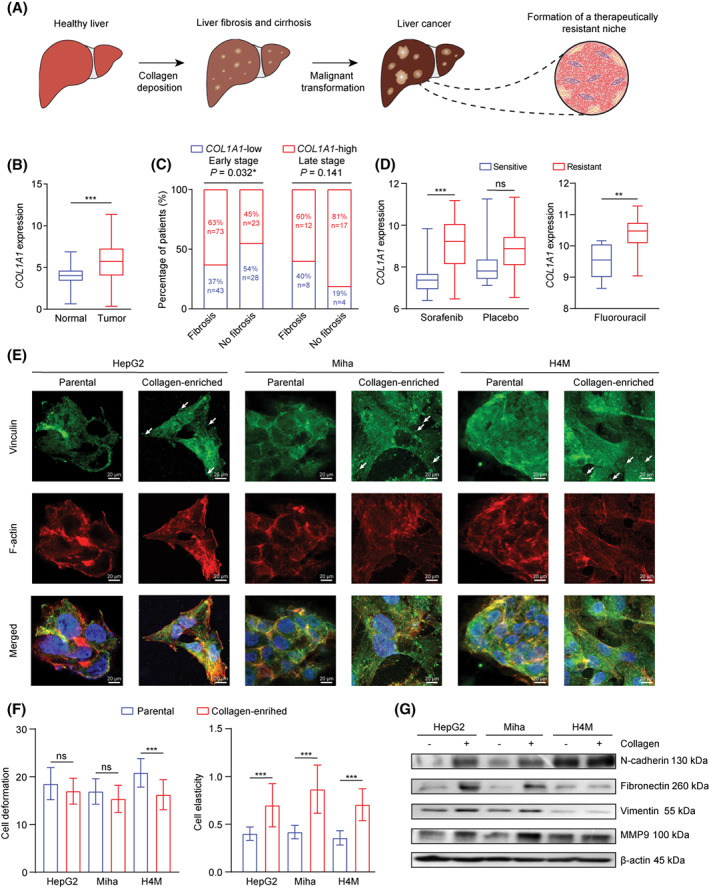
Collagen accumulation in the liver cancer microenvironment and its association with chemoresistance and EMT. (A) Graphical abstract of a three‐stage malignant transformation from a healthy liver to liver cancer. (B) COL1A1 expression in normal individuals (n = 50) and patients with liver cancer (n = 369) from TCGA. (C) Correlation between COL1A1 expression and liver fibrosis in early tumor stage (stage I and II) and late tumor stage (stage III and IV). (D) COL1A1 expression in patients treated with sorafenib (sensitive n = 21, resistant n = 46), placebo (sensitive n = 21, resistant n = 51), and 5FU (sensitive n = 8, resistant n = 15) from GSE109211 and GSE28702. (E) Immunofluorescence staining showed vinculin (green) and F‐actin (red) expression in parental and collagen‐residing HepG2, Miha, and H4M cells. White arrows indicate focal adhesion sites on cells. Scale bar, 20 μm. (F) Cell stiffness qualification was evaluated in parental and collagen‐residing cells based on cell deformation and elasticity in a microfluidic chamber. (G) Western blot result of epithelial and mesenchymal markers expressed in parental and collagen‐residing cells. (B,D,F) *p < 0.05, **p < 0.01, ***p < 0.001. 5FU, 5‐fluorouracil; COL1A1, collagen type I, alpha 1; EMT, epithelial–mesenchymal transition; GSE, gene expression data series; MMP9, matrix metalloproteinase 9; ns, not significant; TCGA, The Cancer Genome Atlas
Type I collagen is one of the most prevalent ECM components in the fibrotic and cancerous liver microenvironment. COL1A1 expression was associated with fibrosis and significantly up‐regulated in patients with liver cancer compared to normal counterparts (Figure 1B,C). Additionally, higher COL1A1 expression was found in patients with sorafenib‐ and 5FU‐resistant liver cancer (Figure 1D) but not in patients treated with placebo, suggesting that synthesis of type I collagen in the liver cancer microenvironment might influence the chemotherapy outcome. Therefore, inhibition of type I collagen‐associated downstream alterations might synergistically improve therapeutic efficacy and lead to a better prognosis in patients with liver cancer. Thus, we established an in vitro mimicry of the collagen‐enriched microenvironment to investigate collagen‐mediated chemoresistance from a molecular perspective. Liver cancer cell lines HepG2, H4M, and an immortalized hepatocyte cell line (Miha) grown on the collagen‐coated plates exhibited an obvious transition from epithelial‐like into mesenchymal‐like morphology (Figure S1A,E). We found that the number of focal adhesion sites was significantly increased in collagen‐residing cells (Figure 1E). Although studies have sufficiently reported that collagen enrichment induces EMT, we further validated the phenomenon based on microfluidics‐based single‐cell stiffness quantification and western blot analysis. Consistent with reports,[ 17 , 18 ] cells grown on a collagen‐enriched microenvironment had higher intracellular stiffness (Figure 1F) and showed higher expression of mesenchymal markers, including N‐cadherin, fibronectin, vimentin, and matrix metalloproteinase 9 (MMP9) (Figure 1G).
To corroborate that locoregional collagen enrichment induced 5FU and sorafenib resistance in liver cancer cells, we conducted the XTT proliferation assay and flow cytometry to evaluate growth inhibition and induced apoptosis during 5FU and sorafenib treatment. The inhibitory effect of 5FU and sorafenib on cell proliferation was significantly decreased in the collagen‐residing HepG2 and Miha cells (Figure 2A), suggesting that collagen enrichment overcame chemotherapy‐induced growth arrest. Following treatment with 5FU and sorafenib, collagen‐residing cells had fewer early and late apoptotic cells (Figure 2B,C), exhibiting stronger resistance to chemotherapy‐induced cell apoptosis. Thus, locoregional collagen enrichment can significantly influence in vitro cell growth and viability under chemotherapeutic treatment independent of drug penetration efficiency.
FIGURE 2.
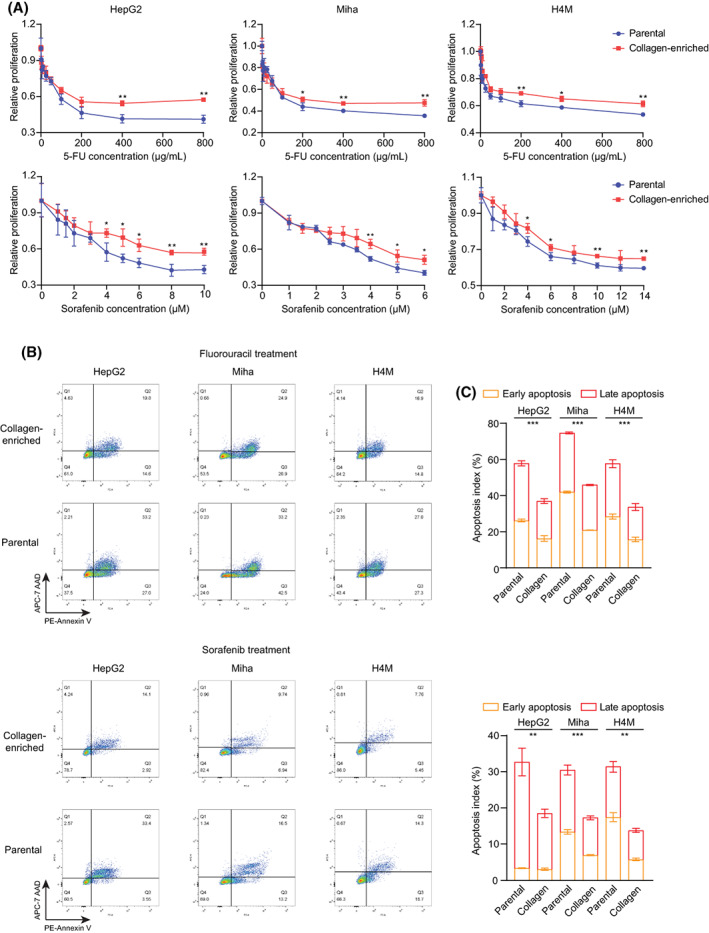
Collagen accumulation induced 5FU and sorafenib resistance in liver cancer cells. (A) Relative proliferation (normalized by control cell proliferation) of parental and collagen‐residing cells under 5FU and sorafenib treatment was quantified by the XTT assay. (B) Flow cytometry results of cell apoptosis determined by PE‐annexin V (early apoptosis) and APC‐7AAD (late apoptosis) staining. Gating was determined based on cells without 5FU and sorafenib treatment. (C) Quantified apoptosis indexes in parental and collagen‐residing cells. *p < 0.05, **p < 0.01, ***p < 0.001. 5FU, 5‐fluorouracil; 7AAD, 7‐aminoactinomycin D; APC, allophycocyanin; PE, phycoerythrin
Collagen enrichment up‐regulated chemoresistance‐related gene LOXL2
To delineate the molecular mechanism that induced collagen‐mediated 5FU and sorafenib resistance, we performed whole transcriptome sequencing on collagen‐residing/parental HepG2 and Miha cells and identified 689 and 387 significantly up‐regulated genes (log2 fold change [FC], ≥1; FDR, ≤0.001) in HepG2 and Miha groups, respectively (Figure 3A). We further selected 375 significantly co‐up‐regulated genes in both HepG2 and Miha groups for Hallmark and KEGG pathway enrichment analysis (Figure 3A,B). Tumor necrosis factor alpha (TNF‐α) signaling through nuclear factor kappa B (NF‐κB), EMT, hypoxia, focal adhesion, and ECM receptor interaction was significantly up‐regulated in collagen‐residing cells. Increased molecular activities in these pathways demonstrated that locoregional type I collagen accumulation cultivated mesenchymal characteristics through enhancing cell–substrate interactions and ECM remodeling.
FIGURE 3.
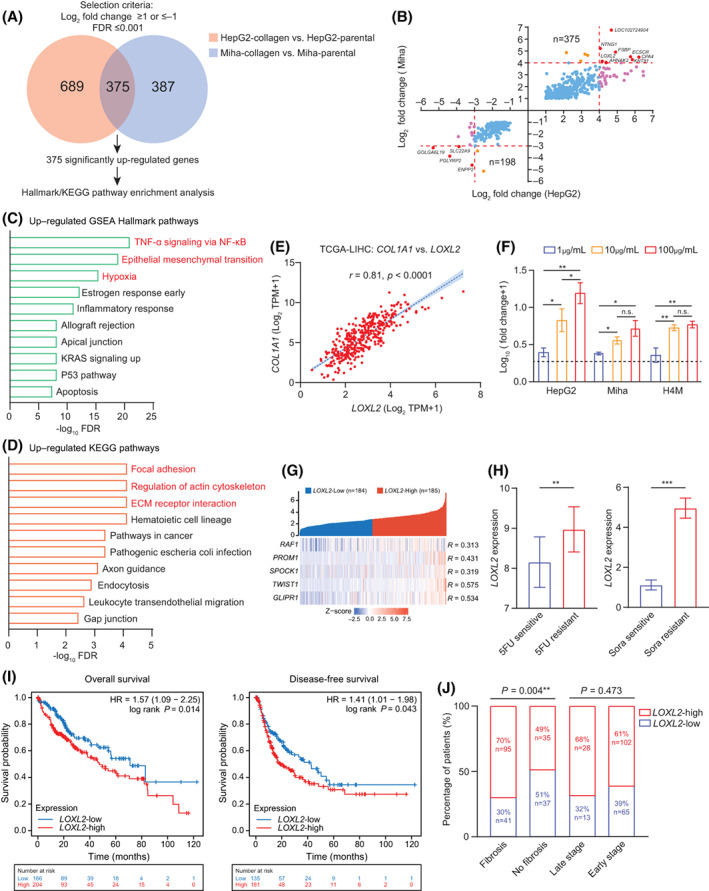
Collagen accumulation up‐regulated an ECM remodeler, LOXL2, in liver cancer cells. (A) Venn diagram shows 375 overlapped genes in 689 up‐regulated genes in collagen‐residing HepG2 cells and 387 up‐regulated genes in collagen‐residing Miha cells. (B) Transcriptome analysis identified 375 co‐up‐regulated and 198 co‐down‐regulated genes, including eight significantly co‐up‐regulated genes (LOXL2, AHNAK2, KRT81, CPA4, ECSCR, FSBP, NTNG1, and LOC102724904) and four significantly co‐down‐regulated genes (SLC22A9, PGLYRP2, ENPP2, and GOLGA6L19). (C) Top10 hallmark pathways enriched in collagen‐residing cells, ordered by –log10 FDR. (D) Top10 KEGG pathways enriched in collagen‐residing cells, ordered by –log10 FDR. (E) Pearson's correlation between LOXL2 and COL1A1 in TCGA‐LIHC (n = 369). (F) Fold change of LOXL2 expression in cells cultured on different densities of collagen, normalized by the LOXL2 expression of parental cells cultured on a noncollagen‐coated plate. (G) Pearson's correlation between LOXL2 and the five chemoresistant gene signature in TCGA‐LIHC (n = 369). (H) LOXL2 expression in patients treated with sorafenib (sensitive n = 21, resistant n = 46) and 5FU (sensitive n = 8, resistant n = 15) from GSE109211 and GSE28702. (I) Prognostic value of LOXL2 expression in overall survival and disease‐free survival of patients with liver cancer (n = 369). (J) Correlation between LOXL2 expression and liver fibrosis and the tumor stage. *p < 0.05, **p < 0.01, ***p < 0.001. AHNAK2, AHNAK nucleoprotein 2; COL1A1, collagen type I, alpha 1; ECM, extracellular matrix; CPA4, carboxypeptidase A4; ECSCR, endothelial cell surface expressed chemotaxis and apoptosis regulator; ENPP2, ectonucleotide pyrophosphatase/phosphodiesterase 2; FDR, false discovery rate; FSBP, fibrinogen silencer binding protein; GLIPR1, GLI pathogenesis related 1; GOLGA6L19, golgin A6 family like 19; GSE, gene expression data series; HR, hazard ratio; KEGG, Kyoto Encyclopedia of Genes and Genomes; KRAS, KRAS proto‐oncogene, guanosine triphosphatase; KRT81, keratin 81; LIHC, liver hepatocellular carcinoma; LOC102724904, methylenetetrahydrofolate dehydrogenase (NADP+ Dependent) 1 like pseudogene; LOXL2, lysyl oxidase‐like 2; NF‐κB, nuclear factor kappa B; n.s., not significant; NTNG1, netrin G1; PGLYRP2, peptidoglycan recognition protein 2; PROM1, prominin 1; RAF1, raf‐1 proto‐oncogene, serine/threonine kinase; SLC22A9, solute carrier family 22 member 9; SPARC, secreted protein acidic and cysteine rich (also known as osteonectin); SPOCK1, cwcv‐ and kazal‐like domains proteoglycan 1; TCGA, The Cancer Genome Atlas; TNF, tumor necrosis factor; TPM, transcript per million; TWIST1, twist family bHLH transcription factor 1
Shown by the transcriptome analysis, LOXL2 is the only highly co‐up‐regulated gene (log2 FC, ≥4; FDR, ≤0.001 in both HepG2 and Miha groups) associated with EMT and the cell–ECM interaction in liver cancer[ 19 , 20 ] (Figure 3B–D). LOXL2 expression was highly correlated to COL1A1 in the TCGA cohort, and collagen density was proportional to the LOXL2 expression of collagen‐residing cells, suggesting that LOXL2 is directly influenced by type I collagen accumulation (Figure 3E,F). LOX and LOXL2 have shown association with hepatitis transactivator protein X (HBX)‐induced collagen fiber stiffening, and enhanced ECM stiffness in turn up‐regulated LOXL2 expression, forming a positive regulatory loop between collagen deposition, cross‐linking, and LOXL2 up‐regulation.[ 21 , 22 ] LOXL2‐mediated ECM remodeling further contributed to establish a fertile ecosystem for intrahepatic and distal tumor invasion through recruitment and activation of VECs and myeloid cell lineages in a Snail family transcriptional repressor (SNAIL)/fructose‐bisphosphatase 1 and FAK/mitogen‐activated protein kinase extracellular signal‐regulated kinase/extracellular signal‐regulated kinase‐dependent mechanism.[ 22 , 23 , 24 ] Nevertheless, the therapeutic effects of LOXL2 on chemoresistance in liver cancer are not explicit, even though some of the lysyl oxidase family members, including LOX, LOXL1, LOXL3, and LOXL4, have been shown to contribute to chemoresistance to platinum, gemcitabine, and doxorubicin or provide prosurvival signaling in cancer cells.[ 25 , 26 , 27 , 28 ] Transcriptome data showed that the expression of other LOX genes did not alter in the collagen‐enriched microenvironment, indicating that they might not directly contribute to the collagen‐mediated chemoresistance in liver cancer (Figure S1B). Moreover, LOXL2 was highly correlated to the chemoresistance signature containing genes that had been found associated with 5FU or sorafenib resistance in liver cancer, including raf‐1 proto‐oncogene, serine/threonine kinase (RAF1); CD133; secreted protein acidic and cysteine rich (SPARC, also known as osteonectin), cwcv‐ and kazal‐like domains proteoglycan 1 (SPOCK1), twist family bHLH transcription factor 1 (TWIST1), and GLI pathogenesis related 1 (GLIPR1)[ 29 , 30 , 31 , 32 ] (Figure 3G). Clinically, LOXL2 was up‐regulated in patients with resistance to 5FU and sorafenib (Figure 3H) and associated with worse overall and disease‐free survival (Figure 3I) as well as correlated to liver fibrosis that was independent of tumor stage (Figure 3J). Thus, we deemed that LOXL2 was the vital driver in collagen‐mediated chemoresistance to 5FU and sorafenib in patients with liver cancer.
LOXL2 influenced 5FU and sorafenib efficacy in liver cancer
To further validate the chemoresistant effect of LOXL2 in patients with liver cancer, we established two cell lines with stable LOXL2 overexpression (HepG2‐VEC, HepG2‐OE, Miha‐VEC, and Miha‐OE) and one with knockdown (SUN‐878‐shNC, SNU‐878‐sh1, and SNU‐878‐sh2). Western blotting and qRT‐PCR confirmed the overexpression and knockdown efficiency based on the exogenous expression of LOXL2 in protein and messenger RNA (mRNA) levels (Figure 4A,B). Because LOXL2 is a secreted protein, we further quantified the concentration of LOXL2, using ELISA (Figure 4C). Subsequently, we found that LOXL2 overexpression in HepG2 and Miha cells increased cellular resistance to 5FU‐ and sorafenib‐induced growth arrest and apoptosis (Figure 4D–F) on collagen‐coated plates but did not significantly enhance 5FU and sorafenib chemoresistance cultured on noncollagen‐coated plates (Figure S1C,D). LOXL2 knockdown also sensitized SNU‐878 cells to 5FU and sorafenib treatment (Figure 4F).
FIGURE 4.
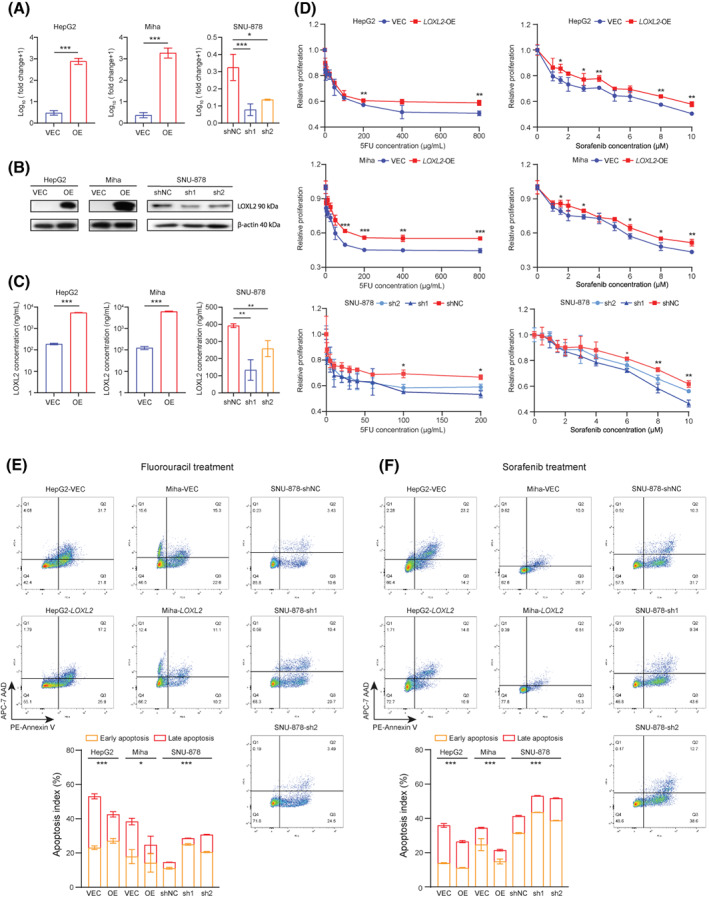
LOXL2 influenced in vitro 5FU and sorafenib resistance in liver cancer cells. (A) Fold change of LOXL2 expression in VEC, LOXL2‐overexpressed, and shNC and LOXL2‐knockdown cells, measured by quantitative reverse‐transcription polymerase chain reaction. (B) LOXL2 expression in VEC, LOXL2‐overexpressed, and shNC and LOXL2‐knockdown cells, determined by western blotting. (C) LOXL2 concentrations in VEC, LOXL2‐overexpressed, and shNC and LOXL2‐knockdown cells, quantified by enzyme‐linked immunosorbent assay. (D) Relative proliferation of VEC, LOXL2‐overexpressed, and shNC and LOXL2‐knockdown cells under 5FU and sorafenib treatment was quantified by the XTT assay. (E) Flow cytometry results and quantified apoptosis indexes of 5FU‐induced cell apoptosis determined by PE‐annexin V (early apoptosis) and APC‐7AAD (late apoptosis) staining. Gating was determined based on cells without 5FU treatment. (F) Flow cytometry results and quantified apoptosis indexes of sorafenib‐induced cell apoptosis determined by PE‐annexin V (early apoptosis) and APC‐7AAD (late apoptosis) staining. Gating was determined based on cells without sorafenib treatment. *p < 0.05, **p < 0.01, ***p < 0.001. 5FU, 5‐fluorouracil; 7AAD, 7‐aminoactinomycin D; APC, allophycocyanin; LOXL2, lysyl oxidase‐like 2; OE, overexpressed; PE, phycoerythrin; shNC, short hairpin negative control; VEC, vector control
Because previous studies have reported that LOXL2 influences cell proliferation,[ 23 ] we established a mouse model with transplant subcutaneous tumor injection to investigate the in vivo effects of LOXL2 on 5FU and sorafenib resistance. Freshly dissected subcutaneous tumors were transplanted into 4‐week BALB/cAnN‐nu mice. After the transplanted tumors became visible (≥4 mm in diameter), the mice were treated with 20 mg/kg 5FU or 10 mg/kg sorafenib twice per week. Despite LOXL2 influencing the tumor growth rate, two‐way ANOVA analysis demonstrated that LOXL2 overexpression induced 5FU and sorafenib resistance in the subcutaneous tumors whereas LOXL2 knockdown synergistically promoted 5FU and sorafenib efficacy in treating tumors (Figure 5A–D). Furthermore, hematoxylin and eosin staining observed more significant apoptotic regions (shown in dashed circles) in LOXL2‐VEC and knockdown tumors, confirming that lower LOXL2 expression in the tumor tissue was associated with decreased chemoresistance (Figure 5A–D). To summarize, we consider that LOXL2 inhibition might be more effective when synergistically used with 5FU and sorafenib in patients with liver cancer, especially those with advanced fibrosis and cirrhosis.
FIGURE 5.
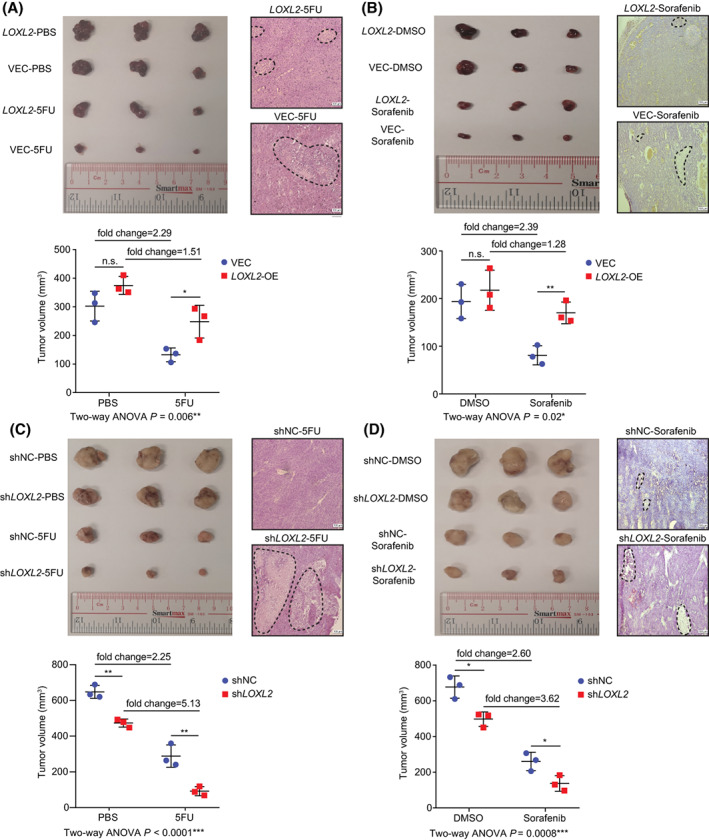
LOXL2 influenced in vivo 5FU and sorafenib resistance in the subcutaneous tumor model. (A) Representative images of HepG2‐VEC (n = 3) and LOXL2‐OE (n = 3) subcutaneous tumors treated with 5FU and phosphate‐buffered saline. H&E‐stained images show representative apoptotic region (in the dashed circle) in the subcutaneous tumors; scale bar, 100 μm. (B) Representative images of HepG2‐VEC (n = 3) and LOXL2‐OE (n = 3) subcutaneous tumors treated with sorafenib and DMSO. H&E‐stained images show representative apoptotic region (in the dashed circle) in the subcutaneous tumors, scale bar = 100 μm. (C) Representative images of SNU‐878‐shNC (n = 3) and SNU‐878‐shLOXL2 (n = 3) subcutaneous tumors treated with 5FU and PBS. H&E‐stained images show representative apoptotic region (in the dashed circle) in the subcutaneous tumors; scale bar, 100 μm. (D) Representative images of SNU‐878‐shNC (n = 3) and SNU‐878‐shLOXL2 (n = 3) subcutaneous tumors treated with sorafenib and DMSO. H&E‐stained images show representative apoptotic region (in the dashed circle) in the subcutaneous tumors; scale bar, 100 μm. *p < 0.05, **p < 0.01, ***p < 0.001. 5FU, 5‐fluorouracil; ANOVA, analysis of variance; DMSO, dimethyl sulfoxide; H&E, hematoxylin and eosin; LOXL2, lysyl oxidase‐like 2; n.s., not significant; OE, overexpressed; PBS, phosphate‐buffered saline; shNC, short hairpin negative control; VEC, vector control
HIF1A‐induced LOXL2 enhanced focal adhesion through ITGA5/FAK/PI3K/ROCK1 signaling
Given the involvement of LOXL2 in ECM remodeling, we hypothesized that LOXL2 could induce integrin activation and enhance cellular adhesion sites. Previous literature also suggested that tumor‐secreted LOXL2 could promote tumor survival and metastasis through focal adhesion‐associated signaling. To validate the hypothesis, we performed GSEA on transcriptome data from 369 patients with liver cancer, divided by median expression of LOXL2. Pathways associated with focal adhesion and tumor–ECM interaction were enriched in patients with high LOXL2 (Figure 6A). Interestingly, we found that the response to oxygen levels (normalized enrichment score [NES], 1.69; FDR, 0.05) in Gene Ontology (GO)‐biological processes (BP) was positively correlated to patients with high LOXL2, indicating that accumulation and stiffening of type I collagen might influence locoregional oxygen distribution and cellular uptake. LOXL2 has been reported to be transcriptional regulated by HIF1A.[ 20 ] Consistently, we found hypoxia signaling was enriched in collagen‐residing cells, and LOXL2 expression was significantly correlated to HIF1A (Figures 3C and 6B). Predicted by JASPER (https://jaspar.genereg.net/), HIF1A can bind to the promoter region of LOXL2 and promote its transcription, which was further validated in dual‐luciferase report assay (Figure 6C), corroborating the LOXL2‐inducing role of HIF1A in the collagen‐enriched microenvironment.
FIGURE 6.
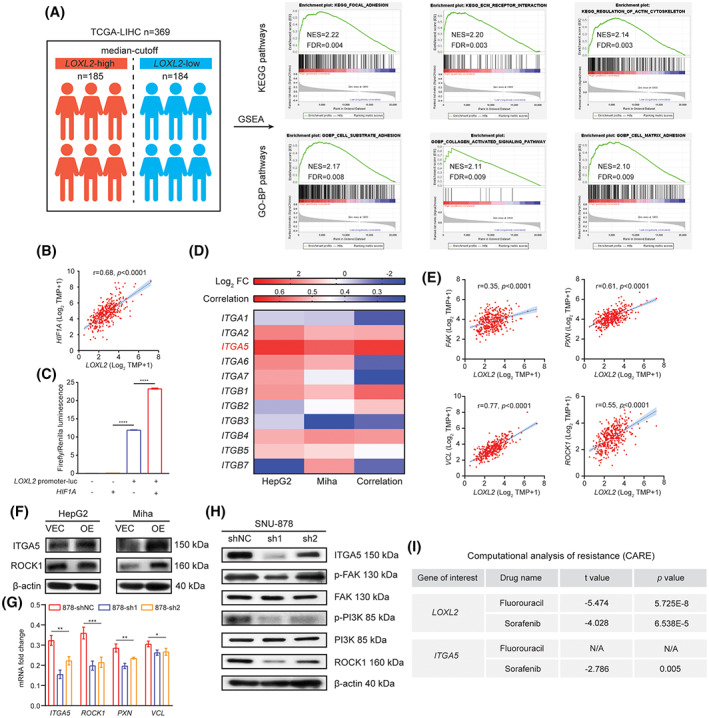
The regulatory circuit of LOXL2 up‐regulation and its downstream factors in the collagen‐enriched microenvironment. (A) A total of 369 patients with liver cancer from TCGA were divided into two groups (n = 185 and n = 184) based on the median expression of LOXL2. GSEA (version 4.1) was used to perform pathway enrichment between the LOXL2‐high and LOXL2‐low groups. The top three enriched pathways with NES ≥2 and FDR ≤0.01 in LOXL2‐high groups are shown. (B) Pearson's correlation between LOXL2 and HIF1A in TCGA‐LIHC (n = 369). (C) Relative firefly luminescence in luciferase‐NC and LOXL2‐promoter‐luciferase‐NC groups with and without HIF1A overexpression, normalized by Renilla luminescence. (D) Log2 fold changes of different integrins in HepG2 and Miha groups based on whole transcriptome sequencing, and Pearson's correlation between LOXL2 and these integrins based on TCGA‐LIHC data (n = 369). (E) Pearson's correlation between LOXL2 and major components in focal adhesion signaling based on TCGA‐LIHC data (n = 369). (F) Changes of ITGA5 and ROCK1 expression between VEC and LOXL2‐overexpressed cells, determined by western blot. (G) Changes of ITGA5, ROCK1, PXN, and VCL expression after LOXL2 knockdown in SNU‐878 cells, measured by quantitative reverse‐transcription polymerase chain reaction. (H) Changes of key proteins in the focal adhesion signaling after LOXL2 knockdown in SNU‐878 cells, determined by western blot. (I) Chemoresistant effects of LOXL2 and its direct downstream effector ITGA5 calculated by the CARE software. *p < 0.05, **p < 0.01, ***p < 0.001. CARE, computational analysis of resistance; ES, enrichment score; FAK, focal adhesion kinase; FC, fold change; FDR, false discovery rate; GO‐BP, Gene Ontology biological processes; GSEA, Gene Set Enrichment Analysis; HIF1A, hypoxia inducible factor 1A; ITGA/B, integrin alpha/beta; KEGG, Kyoto Encyclopedia of Genes and Genomes; LIHC, liver hepatocellular carcinoma; LOXL2, lysyl oxidase‐like 2; mRNA, messenger RNA; NES, normalized enrichment score; OE, overexpressed; p‐, phosphorylated; PI3K, phosphoinositide 3‐kinase; PXN, paxillin; ROCK1, rho‐associated kinase 1; shNC, short hairpin negative control; TCGA, The Cancer Genome Atlas; TPM, transcript per million; VCL, vinculin; VEC, vector control
LOXL2 up‐regulation might enhance focal adhesion signaling, which is a vital mediator in tumor survival, and targeting adhesion‐related molecules has been shown to potently sensitize tumor cells to different therapeutic regimens.[ 33 ] Enhanced focal adhesion is first dependent on integrin activation that transduces biomechanical stimuli sensed from ECM into biochemical stimuli that influence intracellular components. Thus, we examined mRNA changes in varied integrins identified in our transcriptome sequencing result. We found that ITGA5 was significantly up‐regulated in both collagen‐residing HepG2 and Miha cells and was highly correlated to LOXL2 (Figure 6B). The downstream molecules regulated by ITGA5, including FAK, paxillin (PXN), vinculin (VCL), and ROCK1, were also found correlated to LOXL2 (Figure 6C). In LOXL2‐overexpressed cells, protein levels of ITGA5 and ROCK1 were not obviously enhanced (Figure 6D), suggesting that LOXL2 up‐regulation in vitro might not have a large impact on integrin activation because of the locoregional equilibrium between LOXL2 activity and collagen deposition. Nevertheless, in LOXL2‐knockdown cells, the expression of ITGA5 and its downstream signaling molecules were significantly inhibited (Figure 6E,F). LOXL2 knockdown also inhibited the phosphorylation of FAK and its direct downstream factor PI3K (Figure 6F). Based on high throughput drug screening data in the Genomics of Drug Sensitivity in Cancer and the Cancer Cell Line Encyclopedia, the Computational Analysis of Resistance developed by Jiang et al.[ 34 ] has shown that LOXL2 and its direct downstream factor ITGA5 are associated with 5FU and sorafenib resistance (Figure 6G). Here, we validated that the collagen‐enriched liver microenvironment could up‐regulate LOXL2 expression in tumor cells to exert the chemoresistant effect on 5FU and sorafenib treatments through activating the ITGA5/FAK/PI3K/ROCK1 signaling axis.
Therapeutical inhibition of LOXL2 synergistically promoted 5FU and sorafenib efficacy in liver cancer treatment
Unlike the pan‐LOX inhibitor aminopropionitrile (BAPN) that has been widely used to treat liver fibrosis, CMH is a newly synthesized and highly specific LOXL2 inhibitor targeting its catalytic subunit. CMH has been shown to be selective for LOXL2 over LOX.[ 35 ] The use of BAPN in triple‐negative breast cancer could promote the therapeutic efficacy of doxorubicin.[ 26 ] However, it seems that in patients with liver cancer with abnormally high collagen deposition, LOX is not one of the main primary drivers of collagen‐induced chemoresistance (Figure S1B). Thus, the use of BAPN in liver cancer only inhibited proliferation, migration, and invasion of liver cancer cells, whereas no studies have reported the effect of BAPN in chemoresistance in patients with liver cancer. Therefore, in the present study, we used CMH to inhibit LOXL2 secretion and thus decreased locoregional collagen cross‐linking. The half maximal inhibitory concentration of CMH (126 nM) was determined in a previous study.[ 35 ] Consistently, CMH significantly decreased soluble LOXL2 in the culture medium of HepG2, Miha, and H4M cells and synergistically promoted 5FU and sorafenib efficacy in HepG2, Miha, and H4M cells in terms of growth inhibition. Western blotting showed that CMH slightly inhibited the ITGA5/FAK/PI3K/ROCK1 signaling axis in 24 h and significantly inhibited it in 48 h, indicating that a longer time is needed for inhibition of secreted LOXL2 to function in the collagen‐enriched microenvironment. LOXL2 has been shown to be secreted by tumor cells as well as fibroblasts. To further validate that tumor‐secreted LOXL2 in the liver cancer microenvironment is the major source of LOXL2 that influences chemotherapy outcome, we performed transcriptome deconvolution analysis based on TCGA data using xCell (https://xcell.ucsf.edu/).[ 36 ] We found no significant epithelial cell (including malignantly transformed epithelial cells) and fibroblast enrichment in liver cancer compared to patients without liver cancer. However, LOXL2 expression significantly increased in patients with liver cancer, indicating that tumor cells might be the major providers of secreted LOXL2 in the liver cancer microenvironment. We subsequently treated tumor‐bearing mice with 5FU/sorafenib monotherapies and combination therapies of 5FU+CMH and sorafenib+CMH to validate the synergistic effect of CMH. The in vivo result exhibited that tumors treated with the combination therapies significantly decreased in size compared to 5FU and sorafenib monotherapies (Figure 7G,H).
FIGURE 7.
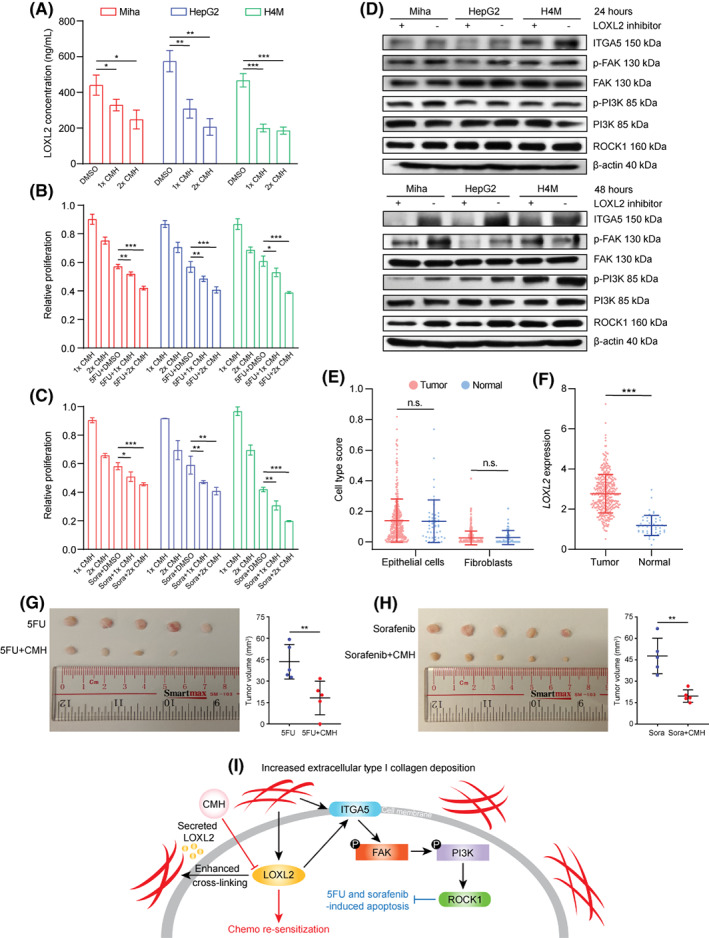
Inhibition of LOXL2 using CMH induced resensitization of 5FU and sorafenib in vitro and in vivo. (A) LOXL2 concentrations in HepG2, Miha, and H4M treated with DMSO, 1×CMH (126 nM), and 2×CMH (252 nM). (B) Relative proliferation of HepG2, Miha, and H4M synergistically treated with 1×CMH, 2×CMH, 5FU+DMSO, 5FU+1×CMH, and 5FU+2×CMH, quantified by the XTT assay. (C) Relative proliferation of HepG2, Miha, and H4M synergistically treated with 1×CMH, 2×CMH, sorafenib+DMSO, sorafenib+1×CMH, and sorafenib+2×CMH, quantified by the XTT assay. (D) Changes in ITGA5/FAK/PI3K/ROCK1 signaling after being treated with 2×CMH for 24 and 48 h. (E) Cell type scores of epithelial cells and fibroblasts in normal individuals (n = 50) and patients with liver cancer (n = 369) from TCGA, estimated by xCell. (F) LOXL2 expression in normal individuals (n = 50) and patients with liver cancer (n = 369) from TCGA. (G) Representative images and quantified volumes of SNU‐878 subcutaneous tumors treated with 5FU (n = 5) and 5FU+CMH (n = 5). (H) Representative images and quantified volumes of SNU‐878 subcutaneous tumors treated with sorafenib (n = 5) and sorafenib+CMH (n = 5). (I) Graphic summary of the molecular mechanism of type I collagen‐LOXL2‐mediated 5FU and sorafenib chemoresistance and proposed therapeutic intervention using CMH. *p < 0.05, **p < 0.01, ***p < 0.001. 5FU, 5‐fluorouracil; CMH, (2‐chloropyridin‐4‐yl) methanamine hydrochloride; DMSO, dimethyl sulfoxide; FAK, focal adhesion kinase; ITGA5, integrin alpha 5; LOXL2, lysyl oxidase‐like 2; n.s., not significant; p‐, phosphorylated; PI3K, phosphoinositide 3‐kinase; ROCK1, rho‐associated kinase 1; TCGA, The Cancer Genome Atlas
DISCUSSION
Liver cancer contributes to 8.3% of cancer death worldwide for its high prevalence and poor prognosis.[ 37 ] HBV infection and dietary factors lead to liver fibrosis and further induce ECM remodeling and malignant transformation of hepatocytes. Although 5FU and sorafenib have been the first‐line treatment option in patients with liver cancer, they can only extend survival time by 3–5 months for patients in late stage.[ 38 ] Particularly, the clinical efficacy of 5FU and sorafenib is worse in patients with severe fibrosis and cirrhosis. Hence, novel synergistic strategies are urgently needed to provide patients with liver cancer a chemoresistant niche. In this study, we found and validated that type I collagen enrichment in the locoregional liver cancer microenvironment caused by fibrosis reduced the efficacy of chemotherapeutic agents 5FU and sorafenib. Based on whole‐transcriptome sequencing of collagen‐residing cells from in vitro collagen mimicry, we revealed that LOXL2 is a vital molecular driver in collagen‐mediated chemoresistance in liver cancer by enhancing the tumor–ECM interplay. Here, we delineated that locoregional collagen accumulation induced hypoxia signaling, which transcriptionally up‐regulated mRNA expression of LOXL2 through HIF1A binding. We subsequently characterized collagen‐LOXL2‐mediated chemoresistance involves activation of the ITGA5/FAK/PI3K/ROCK1 axis, which has been reported associated with a prosurvival function and CAM‐DR in cancers (Figure 7G). Subsequently, we showed that LOXL2‐targeting shRNA and LOXL2‐specific inhibitor CMH could significantly down‐regulate LOXL2 secretion, thus synergistically promote 5FU and sorafenib efficacy, resulting in reversal of chemotherapy resistance in parental and collagen‐residing liver cancer cells.
LOXL2 is an ECM remodeler that is primarily secreted by liver cancer cells to facilitate collagen cross‐linking at cellular focal adhesion sites and promote tumor progression. Furthermore, LOXL2 expression is highly correlated to liver fibrosis and cirrhosis and is currently a promising therapeutic target to treat biliary and nonbiliary fibrosis.[ 39 ] Other studies have demonstrated that LOXL2 is also associated with idiopathic pulmonary fibrosis and primary sclerosing cholangitis.[ 40 , 41 ] Peng et al.[ 42 ] and Park et al.[ 43 ] have shown that LOXL2 promotes cell proliferation, migration, and invasion in gastric and colorectal cancers through FAK/SRC signaling activation, leading to distant metastasis in vivo. LOXL2‐dependent collagen remodeling in liver cancer has also been shown associated with the formation of vasculogenic mimicry and recruitment of bone marrow‐derived cells that provide biophysical stimuli for subsequent tumor settlement and invasion.[ 44 , 45 ] Furthermore, the catalytic domain of LOXL2 participates in elastin and fibronectin remodeling through up‐regulation of MMPs, chemokine (C‐X‐C motif) ligand 12 (CXCL12), and PI3K signaling, pathologically inducing a higher possibility of tumor recurrence and inferior outcome in patients with high‐LOXL2 liver cancer.[ 46 ]
However, in terms of the potential therapeutic effect of LOXL2 on chemotherapy, only a few recent studies have reported that LOXL2 affects the efficacy of 5FU and gemcitabine treatment in colorectal and pancreatic cancers by decreasing ECM deposition to facilitate drug distribution and penetration,[ 47 , 48 ] whereas no studies have comprehensively illustrated the LOXL2‐mediated chemoresistance in liver cancer. In addition to influencing ECM deposition in the tumor microenvironment, LOXL2‐mediated ECM remodeling and collagen cross‐linking can confer 5FU and sorafenib resistance through activating ITGA5 and its downstream effectors that associate with tumor progression, including FAK, PI3K, and ROCK1; this shows how 5FU‐ and sorafenib‐resistant cells evolved from drug‐sensitive cells from a microenvironmental perspective. In many cancer types, the enzymatic activities of LOXL2 can be transcriptionally activated by hypoxia because the promotor region of LOXL2 can be directly bound by HIF1A, suggesting that blocking hypoxia‐associated pathways might serve as an alternative to inhibit LOXL2 activity. In this study, we illustrated the relationship between LOXL2 and CAM‐DR in the collagen‐enriched liver cancer microenvironment and showed that LOXL2 was a clinically effective and feasible target in treating liver fibrosis, making it a promising chemotherapy target in liver cancer.
In summary, we uncovered a molecular mechanism of how the collagen‐enriched liver cancer microenvironment caused by fibrosis influenced the efficacy of 5FU and sorafenib by up‐regulating LOXL2 and its downstream signaling. Notably, LOXL2 enhanced CAM‐DR of liver cancer cells through activating ITGA5/FAK/PI3K/ROCK1 signaling, thus counteracting the apoptotic effect of 5FU and sorafenib. Based on these results, we propose that targeting LOXL2 in patients with liver cancer with severe fibrosis might overcome collagen‐mediated chemoresistance by effectively blocking prosurvival FAK/PI3K signaling. Altogether, our study provides a promising insight into how 5FU and sorafenib chemoresistance is developed from the liver cancer microenvironment and how it can be modulated at a microenvironmental level. Our findings also propose a preclinical strategy for inhibiting a vital molecular driver in CAM‐DR (LOXL2 in patients with liver cancer) to resensitive patients with resistant liver cancer with advanced fibrosis and cirrhosis to a 5FU and sorafenib regimen.
CONFLICT OF INTEREST
Nothing to report.
AUTHOR CONTRIBUTIONS
Xin‐Yuan Guan designed and supervised the study. Lanqi Gong contributed to the study design and wrote the manuscript. Lanqi Gong, Yu Zhang, Yuma Yang, Qian Yan, Jifeng Ren, Jie Luo, and Yuen Chak Tiu performed the experiments. Lanqi Gong and Yu Zhang analyzed the data. Xiaona Fang, Beilei Liu, Raymond Hiu Wai Lam, Ka‐On Lam, and Anne Wing‐Mui Lee contributed to the interpretation of the experimental results and provided important comments. All authors have read and approved the manuscript.
Supporting information
Figure S1 (A) The morphologies of HepG2, Miha, and H4M cells cultured on noncollagen‐coated and collagen‐coated plates. (B) The mRNA fold change of LOX family genes in collagen‐resided/parental HepG2 and Miha cells. (C, D) The relative proliferation index of HepG2‐VEC and HepG2‐LOXL2‐OE cultured on noncollagen‐coated plates after 5FU and sorafenib treatment, measured by the XTT assay
Appendix S1 Supporting Information
Gong L, Zhang Y, Yang Y, Yan Q, Ren J, Luo J, et al. Inhibition of lysyl oxidase‐like 2 overcomes adhesion‐dependent drug resistance in the collagen‐enriched liver cancer microenvironment. Hepatol Commun. 2022;6:3194–3211. 10.1002/hep4.1966
Lanqi Gong, Yu Zhang, and Yuma Yang contributed equally to this work.
Funding informationHong Kong Research Grant Council, Collaborative Research Fund Grants: C7065‐18GF, C7026‐18GF, C4039‐19GF; Theme‐based Research Scheme Grant: T12‐704/16‐R; Research Impact Fund Grants: R4017‐18, R1020‐18F, R7022‐20; National Natural Science Foundation of China, Grants: 81772554 and 82072738; The Shenzhen Peacock Team Project, Grants KQTD20180411185028798
REFERENCES
- 1. Affo S, Yu LX, Schwabe RF. The role of cancer‐associated fibroblasts and fibrosis in liver cancer. Annu Rev Pathol. 2017;12:153–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ma HP, Chang H‐L, Bamodu OA, Yadav VK, Huang T‐Y, Wu ATH, et al. Collagen 1A1 (COL1A1) is a reliable biomarker and putative therapeutic target for hepatocellular carcinogenesis and metastasis. Cancers (Basel). 2019;11:786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fang S, Dai Y, Mei Y, Yang M, Hu L, Yang H, et al. Clinical significance and biological role of cancer‐derived type I collagen in lung and esophageal cancers. Thorac Cancer. 2019;10:277–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sigrist RMS, Liau J, Kaffas AE, Chammas MC, Willmann JK. Ultrasound elastography: review of techniques and clinical applications. Theranostics. 2017;7:1303–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hernandez‐Gea V, Toffanin S, Friedman SL, Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology. 2013;144:512–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Villanueva A, Llovet JM. Targeted therapies for hepatocellular carcinoma. Gastroenterology. 2011;140:1410–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tang W, Chen Z, Zhang W, Cheng Y, Zhang B, Wu F, et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: theoretical basis and therapeutic aspects. Signal Transduct Target Ther. 2020;5:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Armstrong T, Packham G, Murphy LB, Bateman AC, Conti JA, Fine DR, et al. Type I collagen promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Clin Cancer Res. 2004;10:7427–37. [DOI] [PubMed] [Google Scholar]
- 9. Sethi T, Rintoul RC, Moore SM, MacKinnon AC, Salter D, Choo C, et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: a mechanism for small cell lung cancer growth and drug resistance in vivo. Nat Med. 1999;5:662–8. [DOI] [PubMed] [Google Scholar]
- 10. Senthebane DA, Rowe A, Thomford NE, Shipanga H, Munro D, Al Mazeedi MAM, et al. The role of tumor microenvironment in chemoresistance: to survive, keep your enemies closer. Int J Mol Sci. 2017;18:1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM‐DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999;93:1658–67. [PMC free article] [PubMed] [Google Scholar]
- 12. Landowski TH, Olashaw NE, Agrawal D, Dalton WS. Cell adhesion‐mediated drug resistance (CAM‐DR) is associated with activation of NF‐kappa B (RelB/p50) in myeloma cells. Oncogene. 2003;22:2417–21. [DOI] [PubMed] [Google Scholar]
- 13. Schmidmaier R, Baumann P. ANTI‐ADHESION evolves to a promising therapeutic concept in oncology. Curr Med Chem. 2008;15:978–90. [DOI] [PubMed] [Google Scholar]
- 14. Wen JM, Huang JF, Hu L, Wang WS, Zhang M, Sham JST, et al. Establishment and characterization of human metastatic hepatocellular carcinoma cell line. Cancer Genet Cytogenet. 2002;135:91–5. [DOI] [PubMed] [Google Scholar]
- 15. Hu S, Liu G, Chen W, Li X, Lu W, Lam RHW, et al. Multiparametric biomechanical and biochemical phenotypic profiling of single cancer cells using an elasticity microcytometer. Small. 2016;12:2300–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ren J, Li Y, Hu S, Liu Y, Tsao SW, Lau D, et al. Nondestructive quantification of single‐cell nuclear and cytoplasmic mechanical properties based on large whole‐cell deformation. Lab Chip. 2020;20:4175–85. [DOI] [PubMed] [Google Scholar]
- 17. Li J, Lau G, Chen L, Yuan YF, Huang J, Luk JM, et al. Interleukin 23 promotes hepatocellular carcinoma metastasis via NF‐kappa B induced matrix metalloproteinase 9 expression. PLoS ONEj. 2012;7:e46264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luo Q, Kuang D, Zhang B, Song G. Cell stiffness determined by atomic force microscopy and its correlation with cell motility. Biochim Biophys Acta. 2016;1860:1953–60. [DOI] [PubMed] [Google Scholar]
- 19. Wu L, Zhang Y, Zhu Y, Cong Q, Xiang Y, Fu L. The effect of LOXL2 in hepatocellular carcinoma. Mol Med Rep. 2016;14:1923–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wong CC, Tse AP, Huang YP, Zhu YT, Chiu DK, Lai RK, et al. Lysyl oxidase‐like 2 is critical to tumor microenvironment and metastatic niche formation in hepatocellular carcinoma. Hepatology. 2014;60:1645–58. [DOI] [PubMed] [Google Scholar]
- 21. Tse AP, Sze KM, Shea QT, Chiu EY, Tsang FH, Chiu DK, et al. Hepatitis transactivator protein X promotes extracellular matrix modification through HIF/LOX pathway in liver cancer. Oncogenesis. 2018;7:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu S, Zheng Q, Xing X, Dong Y, Wang Y, You Y, et al. Matrix stiffness‐upregulated LOXL2 promotes fibronectin production, MMP9 and CXCL12 expression and BMDCs recruitment to assist pre‐metastatic niche formation. J Exp Clin Cancer Res. 2018;37:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lin HY, Li CJ, Yang YL, Huang YH, Hsiau YT, Chu PY. Roles of lysyl oxidase family members in the tumor microenvironment and progression of liver cancer. Int J Mol Sci. 2020;21:9751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xing X, Wang Y, Zhang X, Gao X, Li M, Wu S, et al. Matrix stiffness‐mediated effects on macrophages polarization and their LOXL2 expression. FEBS J. 2021;288:3465–77. [DOI] [PubMed] [Google Scholar]
- 25. Ye M, Zhou J, Gao Y, Pan S, Zhu X. The prognostic value of the lysyl oxidase family in ovarian cancer. J Clin Lab Anal. 2020;34:e23538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saatci O, Kaymak A, Raza U, Ersan PG, Akbulut O, Banister CE, et al. Targeting lysyl oxidase (LOX) overcomes chemotherapy resistance in triple negative breast cancer. Nat Commun. 2020;11:2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Le Calve B, Griveau A, Vindrieux D, Maréchal R, Wiel C, Svrcek M, et al. Lysyl oxidase family activity promotes resistance of pancreatic ductal adenocarcinoma to chemotherapy by limiting the intratumoral anticancer drug distribution. Oncotarget. 2016;7:32100–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li R, Wang Y, Zhang X, Feng M, Ma J, Li J, et al. Exosome‐mediated secretion of LOXL4 promotes hepatocellular carcinoma cell invasion and metastasis. Mol Cancer. 2019;18:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li Y, Chen L, Chan THM, Liu M, Kong K–L, Qiu J–L, et al. SPOCK1 is regulated by CHD1L and blocks apoptosis and promotes HCC cell invasiveness and metastasis in mice. Gastroenterology. 2013;144:179–91.e4. [DOI] [PubMed] [Google Scholar]
- 30. Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008;27:1749–58. [DOI] [PubMed] [Google Scholar]
- 31. Kim JS, Choi GH, Jung Y, Kim KM, Jang SJ, Yu ES, et al. Downregulation of Raf‐1 kinase inhibitory protein as a sorafenib resistance mechanism in hepatocellular carcinoma cell lines. J Cancer Res Clin Oncol. 2018;144:1487–501. [DOI] [PubMed] [Google Scholar]
- 32. Li R, Wu C, Liang H, Zhao Y, Lin C, Zhang X, et al. Knockdown of TWIST enhances the cytotoxicity of chemotherapeutic drugs in doxorubicin‐resistant HepG2 cells by suppressing MDR1 and EMT. Int J Oncol. 2018;53:1763–73. [DOI] [PubMed] [Google Scholar]
- 33. Aoudjit F, Vuori K. Integrin signaling in cancer cell survival and chemoresistance. Chemother Res Pract. 2012;2012:283181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jiang P, Lee W, Li X, Johnson C, Liu JS, Brown M, et al. Genome‐scale signatures of gene interaction from compound screens predict clinical efficacy of targeted cancer therapies. Cell Syst. 2018;6:343–54.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hutchinson JH, Rowbottom MW, Lonergan D, Darlington J, Prodanovich P, King CD, et al. Small molecule Lysyl oxidase‐like 2 (LOXL2) inhibitors: the identification of an inhibitor selective for LOXL2 over LOX. ACS Med Chem Lett. 2017;8:423–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49. [DOI] [PubMed] [Google Scholar]
- 38. Zhu YJ, Zheng B, Wang HY, Chen L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin. 2017;38:614–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ikenaga N, Peng ZW, Vaid KA, Liu SB, Yoshida S, Sverdlov DY, et al. Selective targeting of lysyl oxidase‐like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut. 2017;66:1697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Aumiller V, Strobel B, Romeike M, Schuler M, Stierstorfer BE, Kreuz S. Comparative analysis of lysyl oxidase (like) family members in pulmonary fibrosis. Sci Rep. 2017;7:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Raghu G, Brown KK, Collard HR, Cottin V, Gibson KF, Kaner RJ, et al. Efficacy of simtuzumab versus placebo in patients with idiopathic pulmonary fibrosis: a randomised, double‐blind, controlled, phase 2 trial. Lancet Respir Med. 2017;5:22–32. [DOI] [PubMed] [Google Scholar]
- 42. Peng L, Ran YL, Hu H, Yu L, Liu Q, Zhou Z, et al. Secreted LOXL2 is a novel therapeutic target that promotes gastric cancer metastasis via the Src/FAK pathway. Carcinogenesis. 2009;30:1660–9. [DOI] [PubMed] [Google Scholar]
- 43. Park PG, Jo SJ, Kim MJ, Kim HJ, Lee JH, Park CK, et al. Role of LOXL2 in the epithelial‐mesenchymal transition and colorectal cancer metastasis. Oncotarget. 2017;8:80325–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shao B, Zhao X, Liu T, Zhang Y, Sun R, Dong X, et al. LOXL2 promotes vasculogenic mimicry and tumour aggressiveness in hepatocellular carcinoma. J Cell Mol Med. 2019;23:1363–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, et al. Hypoxia‐induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wu S, Xing X, Wang Y, Zhang X, Li M, Wang M, et al. The pathological significance of LOXL2 in pre‐metastatic niche formation of HCC and its related molecular mechanism. Eur J Cancer. 2021;147:63–73. [DOI] [PubMed] [Google Scholar]
- 47. Zheng GL, Liu YL, Yan ZX, Xie XY, Xiang Z, Yin L, et al. Elevated LOXL2 expression by LINC01347/miR‐328‐5p axis contributes to 5‐FU chemotherapy resistance of colorectal cancer. Am J Cancer Res. 2021;11:1572–85. [PMC free article] [PubMed] [Google Scholar]
- 48. Ferreira S, Saraiva N, Rijo P, Fernandes AS. LOXL2 inhibitors and breast cancer progression. Antioxidants (Basel). 2021;10:312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (A) The morphologies of HepG2, Miha, and H4M cells cultured on noncollagen‐coated and collagen‐coated plates. (B) The mRNA fold change of LOX family genes in collagen‐resided/parental HepG2 and Miha cells. (C, D) The relative proliferation index of HepG2‐VEC and HepG2‐LOXL2‐OE cultured on noncollagen‐coated plates after 5FU and sorafenib treatment, measured by the XTT assay
Appendix S1 Supporting Information