

Abstract
Homozygous familial hypercholesterolemia (HoFH) is a rare genetic disorder, and a genetic analysis is important to make a definitive diagnosis. A comprehensive genetic analysis using next generation sequencing (NGS) and whole exome sequencing (WES) is feasible. However, the application of NGS in the assessment of genomic structural variations is generally limited, and a substantial number of control samples are needed for such assessments. Thus, NGS alone is unlikely to detect genomic structural variations in a “singleton.” We present the case of a patient with compound HeFH (heterozygous FH), whose causative mutations in the LDLR gene could not be identified by WES, necessitating the application of the multiplex ligation-dependent probe amplification (MLPA) technique.
Keywords: familial hypercholesterolemia, multiplex ligation-dependent probe amplification, whole exome sequencing
Introduction
Familial hypercholesterolemia (FH; OMIM no. 143890) is a relatively common monogenic hyper low-density lipoprotein (LDL) cholesterolemia, the frequency of which is estimated to be 1 in 250 to 300 general population. This condition is associated with premature death due to atherosclerotic cardiovascular disease (ASCVD) (1,2). Homozygous FH (HoFH), including compound heterozygous FH (HeFH) and double HeFH, is a rare genetic disorder that is clinically characterized by xanthoma tuberosum in infancy, supravalvular aortic stenosis (SVAS), and premature ASCVD associated with extremely elevated levels of LDL cholesterol (LDL-C) (3-5). In addition, there are patients with oligogenic FH who present deleterious mutations in the FH gene coding for the LDL receptor (LDLR), the apolipoprotein B gene, the proprotein convertase subtilisin/kexin type 9 (PCSK9), as well as in some “accessory” genes, such as the ATP-binding cassette sub-family G member (ABCG) 5 and ABCG8 gene (6). Moreover, there are patients with “severe HeFH” who exhibit a more severe phenotype in comparison to those with typical HeFH (7). Accordingly, there are substantial overlaps between LDL-C and the phenotype of HoFH and HeFH, which result in different prognoses according to their genetic backgrounds. Furthermore, to achieve an accurate diagnosis, a comprehensive genetic analysis using next generation sequencing (NGS) has emerged as an efficient tool in cases of FH (8,9). Among several comprehensive genetic analysis methods, whole exome sequencing (WES) is widely used as an established method. This technique has also been used to clarify the genetic background of FH (10). However, NGS is generally limited in terms of the assessment of genomic structural variations, including large deletions, duplications, copy-number variants, insertions, inversions, and translocations. We herein present the case of a patient with compound HeFH who exhibited typical clinical features of HoFH and whose causative mutations in the LDLR gene could not be identified through WES alone, which necessitated the application of a multiplex ligation-dependent probe amplification (MLPA) technique to identify structural genetic variations.
Case Report
A 41-year-old woman with a history of chest pain and dyspnea on exertion for 1 month accompanied by extreme hyper LDL-cholesterolemia was referred to our hospital for further examination of her lipid and atherosclerotic cardiovascular diseases. She was born from a non-consanguineous marriage. She had a xanthoma tuberosum in her wrist joint at 3 years of age, which was resected. She received statins from 16 years of age for severe hypercholesterolemia (≧800 mg/dL); however, she discontinued the medications due to the risk of fetal malformation during her first pregnancy at 31 years of age. Moreover, she had a family history of hyperlipidemia as well as sudden cardiac death on the paternal and maternal sides of her family (Fig. 1, Table 1).
Figure 1.

A pedigree chart of the present case. Pin dots indicate affected members carrying the Cys302Gly/Arg303Trp mutations and black symbols indicate affected members carrying an exon 6 large deletion. Question marks indicate members with unknown genotype. “I,” “II,” “III,” and “IV” indicate the generation of this family. Squares indicate men, circles indicate women. Asterisks indicate members clinically diagnosed with hypercholesterolemia.
Table 1.
Genotype and Lipid Profile of the Proband and Her Family Members.
| Patient | Genotype | Age | Sex | T-Cho | TG | HDL-C | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Proband | c.904T>G/c.907C>T, exon 6 del, LDLR | 41 | female | 523 | 122 | 43 | ||||||
| II-1* | c.904T>G/c.907C>T, LDLR | 75 | male | 205 | 140 | 49 | ||||||
| II-14 | exon 6 del, LDLR | 68 | female | 226 | 137 | 91 | ||||||
| III-4* | exon 6 del, LDLR | 49 | female | 197 | 490 | 84 | ||||||
| III-6 | exon 6 del, LDLR | 46 | female | 323 | 54 | 88 | ||||||
| III-12 | exon 6 del, LDLR | 45 | male | 256 | 77 | 63 | ||||||
| III-14 | 41 | male | 205 | 336 | 51 | |||||||
| IV-5 | c.904T>G/c.907C>T, LDLR | 10 | male | 312 | 73 | 81 | ||||||
| IV-6 | exon 6 del, LDLR | 7 | female | 254 | 42 | 51 |
* Asterisk indicates the patients under lipid lowering therapy.
T-Cho: total cholesterol, TG: triglycerides, HDL-C: High-density lipoprotein cholesterol
A physical examination revealed xanthelasma and tendinous xanthoma on her finger extensor muscle and her Achilles' tendons (Fig. 2). An ejection systolic grade IV/VI murmur was heard on the right second intercostal space radiating toward the jugular. The plasma lipid profile showed severe primary hypercholesterolemia, even under statin treatment (Table 2). Although the leaflets of her aortic valve were almost intact with preserved mobility (Fig. 3A, B, Supplementary material 1), severe left ventricular outflow tract stenosis was detected, with a peak gradient of 87 mmHg on transthoracic echocardiography (Fig. 3C). The patient had mild concentric left ventricular hypertrophy (10 mm) and a narrow annulus followed by SVAS (Fig. 3D). Cardiac computed tomography demonstrated severe calcification and a hypoplastic sinus of Valsalva due to SVAS (Fig. 4). Although coronary angiography showed slight irregularity of both the left main trunk and right coronary ostium, there was no significant stenotic lesion in her coronary arteries (Fig. 5A, B). Cardiac catheterization examination revealed SVAS with severe calcification and a pressure gradient of 75 mmHg between the left ventricle and aortic root (Fig. 5C, D).
Figure 2.

Xanthelasma palpebrarum (A), xanthoma tendinosum on the finger’s extensor muscle (B), Achilles’ tendons (C), and radiograph of the left Achilles’ tendon (D).
Table 2.
Lipid and Apolipoprotein Profile of the Proband under Statin Therapy at the First Visit.
| Total cholesterol | 423 | mg/dL | Apolipoprotein A-I | 104 | mg/dL | |||
| TG | 52 | mg/dL | Apolipoprotein A-II | 19.1 | mg/dL | |||
| HDL-cholesterol | 46 | mg/dL | Apolipoprotein B | 222 | mg/dL | |||
| LDL-cholesterol | 354 | mg/dL | Apolipoprotein C-II | 1.1 | mg/dL | |||
| F-cholesterol | 132 | mg/dL | Apolipoprotein C-III | 5.7 | mg/dL | |||
| Phospholipids | 313 | mg/dL | Apolipoprotein E | 5.3 | mg/dL | |||
| Free fatty acids | 0.07 | mEq/L | Lipoprotein (a) | 55.1 | mg/dL | |||
| PG | 105 | mg/dL | Free triiodothyronine | 2.49 | pg/mL | |||
| HbA1c | 5.4 | % | Free thyroxine | 1.15 | ng/dL | |||
| Total protein | 7.2 | g/dL | Thyroid stimulating hormone | 1 | ng/dL | |||
| Albumin | 3.9 | g/dL | Urine albumin | (-) |
TG: triglycerides, HDL: high-density lipoprotein, LDL: low-density lipoprotein, PG: plasma glucose, Hb: hemoglobin
Figure 3.

The transesophageal echocardiogram shows that the leaflets of the patient’s aortic valve had preserved mobility in the parasternal long axis view (A; end systolic) and in the parasternal short axis view (B; end systolic). Severe left ventricular outflow tract stenosis with a peak gradient of 87 mmHg is revealed by transthoracic echocardiography using continuous wave Doppler (C). Transesophageal echocardiography shows left ventricular outflow tract stenosis in the parasternal long axis view (D; end diastolic, AA=19 mm, STJ=23 mm, Ao=27 mm). LA: left atrium, LV: left ventricle, Ao: aorta, RA: right atrium, AA: aortic annulus, STJ: sinotubular junction
Figure 4.
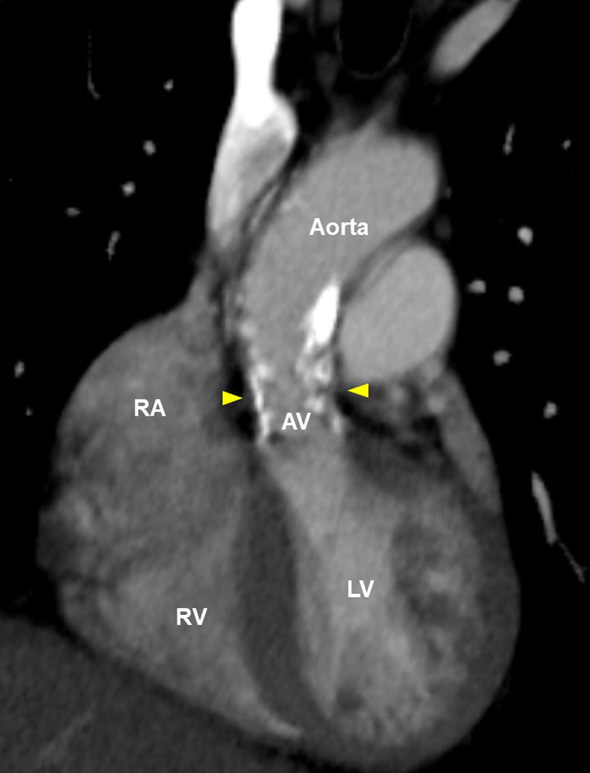
Computed tomographic findings of the patient’s aortic valve and ascending aorta. Arrowheads indicate the site of supravalvular aortic stenosis. AV: aortic valve, LV: left ventricle, RA: right atrium, RV: right ventricle
Figure 5.

Coronary angiography and aortography. Coronary angiography of the right coronary artery (A) and left coronary artery (B) demonstrates slight irregularity of both the left main trunk and right coronary ostium. Aortography of the aortic root shows supravalvular aortic stenosis with severe calcification (C; LAO: left anterior oblique, D; RAO: right anterior oblique). Arrowheads indicate the site of supravalvular aortic stenosis. LCA: left coronary artery, RCA: right coronary artery
Accordingly, her symptoms could be only be accounted for by SVAS, and aortic valve replacement surgery (Medtronic, ATS, 18 mm) with aortic root angioplasty was performed. Postoperative echocardiography showed significant hemodynamic improvement.
Her clinical diagnosis was tentatively estimated as HoFH because of her severe hyper LDL-cholesterolemia, which was refractory to statins, and her severe phenotype, which included xanthoma and SVAS. We attempted to determine her genetic background in order to explain the extreme disease manifestations.
Exome Sequencing and Bioinformatic Analysis
At first, we performed WES only for the proband, and subsequently performed bioinformatic filtering as previously described (5). This study was approved by the Research Ethics Committee of Kanazawa University (Kanazawa, Japan), and written informed consent was obtained from the patient and her family members. All procedures were conducted in accordance with the Helsinki Declaration of 2008. Four independent filters were applied after standard variant quality control to successfully discover the causal variants. Variants were filtered out as follows: 1) benign variants predicted by SnpEff; 2) minor allele frequency >1% in the Asian population; 3) variants found in two healthy subjects; 4) unknown variants that could be causal mutations of FH.
As a result, we identified two point-mutations with amino acid substitutions in the LDLR gene (NM_000527.4:c.904 T>G, c.907 C>T) on chromosome 19. We confirmed these two point-mutations in exon 7 using Sanger sequencing. Initially, we recognized that the two adjacent missense-mutations were derived from each parent. However, we later confirmed both mutations in her father and older brother, but not in her maternal relatives, including those exhibiting hyper LDL cholesterolemia. Consequently, these observations led to the conclusion that the proband may have another mutation (i.e., a structural genetic variation, that was undetectable through WES in a singleton). Accordingly, we investigated the presence of such structural genetic variations in LDLR using the MLPA technique and discovered a large maternal deletion in exon 6 (Supplementary material 2). Finally, our patient was diagnosed with compound HeFH with three different LDLR mutations.
Cholesterol lowering therapy
The patient was sequentially treated with rosuvastatin, ezetimibe, and colestimide, which reduced her LDL-C level from 429 to 253 mg/dL (Fig. 6). LDL apheresis therapy (once every 4 weeks) was also introduced because of the insufficient reduction of LDL-C (11). In addition to these therapies, the patient was treated with evolocumab, a PCSK9 inhibitor (420 mg/biweekly), because her average LDL cholesterol level during LDL apheresis therapy was over 100 mg/dL. As expected, the final therapeutic regimen substantially reduced her average LDL cholesterol level (to <70 mg/dL).
Figure 6.

The clinical course of the patient. LDL: low-density lipoprotein
Discussion
We reported a rare case of compound HeFH with xanthoma tuberosum in infancy. SAVS is also recognized as a typical clinical manifestation of HoFH. A study using magnetic resonance imaging showed that 41% of patients with HoFH exhibiting SVAS (12) required aortic root enlargement and aortic valve replacement (mechanical prosthetic heart valve), even while under aggressive medical therapy (4).
HoFH exhibiting double deleterious mutations in the FH gene is a rare genetic condition, the prevalence of which is currently considered to be approximately 1 in 160,000-360,000 in many countries, including Japan (11,13).
When we encounter a patient with severe hypercholesterolemia, it is important to differentiate HoFH from HeFH. Although most available cholesterol lowering medicines are effective for reducing LDL-C in HeFH (half of their LDLR are intact), they could not effectively reduce the LDL-C level in patients with HoFH without an intact LDLR (14). Thus, most patients with HoFH require LDL-apheresis therapy and/or microsomal triglyceride transfer protein inhibitor treatment, neither of which requires LDLR to reduce LDL-C. In the present case, we firstly attempted to determine the patient's genetic background using WES. Two adjacent missense mutations in the LDLR gene could be revealed, however, these mutations originated from her father, and they were also found in some paternal relatives with the HeFH phenotype. However, no genetic mutations responsible for the elevation of LDL-C could be found among the patient's maternal relatives by WES. Although WES can detect mutations across the exome, enabling us to establish a differential diagnosis, its application is not sufficient for the detection of structural genetic variations such as the large deletion reported in our patient. One possible way to identify such structural variations is through NGS data with consideration of the depth of coverage; however, this method typically requires hundreds of “controls” data-points. On the other hand, the MLPA technique could be used to detect structural variants with higher sensitivity (15,16). It is noteworthy that up to approximately 10% of the genetic background of FH showed such structural genetic variations (15).
In conclusion, we suggest the use of the MLPA method along with WES for the detection of structural genetic variations to establish a quick diagnosis of FH, as significant numbers of patients with FH show structural genetic variations.
The authors state that they have no Conflict of Interest (COI).
Financial Support
A scientific research grant from the Ministry of Education, Science, and Culture of Japan (No. 19K17552), Ministry of Health, Labor and Welfare Sciences Research Grant for Research on Rare and Intractable Diseases, and Japanese Circulation Society (project for genome analysis in cardiovascular diseases).
Supplementary Materials
These videos represents still images, Figure 3B and 3D.
The MLPA technique revealed a large deletion in exon 6 of maternal family members.
{kind=link}
References
- 1. Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: The Metabolic and Molecular Bases of Inherited Disease. Vol 2. 8th ed. Scriver CR, Beaudet AL, Sly WS, Valle D, Eds. McGraw-Hill, New York, 2001: 2863-2913. [Google Scholar]
- 2. Okada H, Tada H, Hayashi K, et al. Aortic root calcification score as an independent factor for predicting major adverse cardiac events in familial hypercholesterolemia. J Atheroscler Thromb 25: 634-642, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Awan Z, Alrasadi K, Francis GA, et al. Vascular calcifications in homozygote familial hypercholesterolemia. Arterioscler Thromb Vasc Biol 28: 777-785, 2008. [DOI] [PubMed] [Google Scholar]
- 4. Kakuta T, Fujita T, Fukushima S, et al. Two cases of surgical management of supravalvular aortic stenosis in familial hypercholesterolemia. Ann Thorac Surg 105: e171-e174, 2018. [DOI] [PubMed] [Google Scholar]
- 5. Tada H, Kawashiri MA, Nomura A, et al. Oligogenic familial hypercholesterolemia, LDL cholesterol, and coronary artery disease. J Clin Lipidol 12: 1436-1444, 2018. [DOI] [PubMed] [Google Scholar]
- 6. Santos RD, Gidding SS, Hegele RA, et al. Defining severe familial hypercholesterolaemia and the implications for clinical management: a consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Lancet Diabetes Endocrinol 4: 850-861, 2016. [DOI] [PubMed] [Google Scholar]
- 7. Futema M, Plagnol V, Li K, et al. Whole exome sequencing of familial hypercholesterolaemia patients negative for LDLR/APOB/PCSK9 mutations. J Med Genet 51: 537-544, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Iacocca MA, Wang J, Dron JS, et al. Use of next-generation sequencing to detect LDLR gene copy number variation in familial hypercholesterolemia. J Lipid Res 58: 2202-2209, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tada H, Hosomichi K, Okada H, et al. A de novo mutation of the LDL receptor gene as the cause of familial hypercholesterolemia identified using whole exome sequencing. Clin Chim Acta 453: 194-196, 2016. [DOI] [PubMed] [Google Scholar]
- 10. Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J 34: 3478-3490, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Makino H, Koezuka R, Tamanaha T, et al. Familial hypercholesterolemia and lipoprotein apheresis. J Atheroscler Thromb 26: 679-687, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Summers RM, Andrasko-Bourgeois J, Feuerstein IM, et al. Evaluation of the aortic root by MRI - insights from patients with homozygous familial hypercholesterolemia. Circulation 98: 509-518, 1998. [DOI] [PubMed] [Google Scholar]
- 13. Nohara A, Tada H, Ogura M, et al. Homozygous familial hypercholesterolemia. J Atheroscler Thromb 28: 665-678, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Santos RD, Stein EA, Hovingh GK, et al. Long-term evolocumab in patients with familial hypercholesterolemia. J Am Coll Cardiol 75: 565-574, 2020. [DOI] [PubMed] [Google Scholar]
- 15. Wang J, Ban MR, Hegele RA. Multiplex ligation-dependent probe amplification of LDLR enhances molecular diagnosis of familial hypercholesterolemia. J Lipid Res 46: 366-372, 2005. [DOI] [PubMed] [Google Scholar]
- 16. Mu W, Li B, Wu S, et al. Detection of structural variation using target captured next-generation sequencing data for genetic diagnostic testing. Genet Med 21: 1603-1610, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
These videos represents still images, Figure 3B and 3D.
The MLPA technique revealed a large deletion in exon 6 of maternal family members.