

Abstract
During tendon healing, macrophages are thought to be a key mediator of scar tissue formation, which prevents successful functional restoration of the tendon. However, macrophages are critical for successful tendon healing as they aid in wound debridement, extracellular matrix deposition, and promote fibroblast proliferation. Recent work has sought to better define the multi-faceted functions of macrophages using depletion studies, while other studies have identified a tendon resident macrophage population. To begin to delineate the functions of tendon-resident versus circulation-derived macrophages, we examined the tendon healing phenotype in Chemokine Receptor 2 (CCR2) reporter (CCR2GFP/+), and knockout mice. CCR2 is a chemokine receptor primarily found on the surface of circulating bone marrow-derived monocytes, with CCR2 being an important mediator of macrophage recruitment to wound environments. Surprisingly, CCR2GFP/+ cells were present in the tendon during adult homeostasis, and single-cell RNA sequencing identified these cells as tendon-resident macrophages and T cells. During both homeostasis and healing, CCR2 knockout resulted in a substantial decrease in CCR2GFP+ cells and pan-macrophages. Additionally, loss of CCR2 resulted in reduced numbers of myofibroblasts and impeded functional recovery during late healing. This study highlights the heterogeneity of tendon-resident and recruited immune cells and their contributions following injury, and establishes an important role for CCR2 in modulating both the adult tendon cell environment and tendon healing process.
Keywords: CCR2, fibrosis, macrophage, myofibroblast, tendon healing
1 |. INTRODUCTION
Tendons are dense connective tissues that are primarily composed of type I collagen. Once injured, tendon function is never fully restored. Instead, tendons heal with the formation of a fibrovascular scar, characterized by excessive and disorganized extracellular matrix (ECM) deposition.1,2 While this scar tissue provides tissue continuity and a degree of stability, the mechanical composition of the tendon is compromised, increasing the risk of re-rupture.3 Recent work has suggested that macrophages are a key driver of this scar tissue response both in the tendon1,2,4,5 and other tissues.6,7
While tissue-resident macrophages are thought to be a relatively small population of the tendon cell environment during homeostasis,8 there is a robust increase in extrinsic macrophage infiltration to the tendon after acute injury, which is critical for the initiation of the inflammatory phase of healing.1,4,5,9 Macrophages are a dynamic cell population that interact with tissue-resident cell populations, extrinsic immune cells, and extracellular proteins.6,7,10 Following tissue injury, apoptotic cells release damage-associated molecular patterns (DAMPS) that activate pathways in macrophages that recruit neutrophils, monocytes, and other inflammatory cells to the injured tissue. Macrophages also secrete anti-inflammatory molecules, growth factors, and matrix metalloproteinases that promote tissue repair and matrix degradation. Macrophages are also known to create a pro-fibrotic feedback loop with myofibroblasts, a specialized contractile fibroblast that drives both physiological healing and the transition to fibrosis.11–13 The diverse function of macrophages allows them to successfully evade pathogens, clear wound debris, and deposit collagen in an ever-changing microenvironment. However, failure to clear macrophages following the acute inflammatory phase is associated with a transition to a more chronic, fibrotic phenotype resulting in excessive scar tissue deposition.
In contrast to other tissues, the contributions of macrophages during tendon healing are not well defined. Given the fibrotic healing process that occurs in tendons and the intricate link between macrophages and fibrosis, ascertaining the role of macrophages during tendon healing is therefore critical to identifying potential therapeutic approaches. Recent work has utilized macrophage depletion approaches to better understand the role of macrophages during tendon healing. Macrophage depletion in adult tendons promoted cell proliferation and ECM accumulation but lead to inferior tensile strength.14 Consistent with this, macrophage depletion in neonatal Achilles tendons resulted in impaired functional healing, however, cell proliferation was reduced.9 In addition, extracellular vesicles derived from human mesenchymal stem cells (MSCs) primed macrophages toward an M2 anti-inflammatory phenotype that improved Achilles tendon biomechanics following injury.15 However, there are still important questions regarding the potentially unique roles of tissue-resident vs. circulation-derived macrophages during tendon healing. Therefore, we sought to better understand the contribution of extrinsic macrophages during flexor tendon healing, using Chemokine Receptor 2 (CCR2)-reporter and CCR2 knock-out (CCR2KO) mice. CCR2 is a chemokine receptor primarily found on the surface of bone marrow-derived monocytes circulating in the peripheral blood, with CCR2 thought to mediate the recruitment of macrophages to the wound environment.16–18 While the role of CCR2 during tendon healing has yet to be investigated, it has been studied extensively in a variety of different tissues. For example, CCR2KO kidneys had impaired macrophage and fibroblast accumulation during renal fibrosis19 and CCR2KO liver had reduced macrophage content concomitant with delayed liver fibrosis.20 In the present study, we defined the functional effects of CCR2 inhibition and tracked changes in CCR2+ cell recruitment during tendon healing. We hypothesized that following acute tendon injury, CCR2KO would reduce recruitment of extrinsic macrophages and that diminished macrophage recruitment would subsequently impair myofibroblast differentiation and functional recovery.
2 |. METHODS
2.1 |. Animal ethics
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (Bethesda, MD, USA). All animal procedures were approved by the University Committee on Animal Research (UCAR) at the University of Rochester (UCAR Number: 2014-004E).
2.2 |. CCR2GFP mouse model
CCR2GFP/+ mice were obtained from Jackson Laboratories (strain #027619). These transgenic mice have the green fluorescent protein (GFP) sequence followed by a polyadenylation signal inserted into the chemokine receptor 2 (CCR2) gene.21 In CCR2GFP/+ mice, one copy of CCR2 has been replaced with GFP, however, mice are phenotypically normal and are used as a CCR2 reporter. In CCR2KO mice, GFP has replaced both copies of CCR2, resulting in CCR2 knockout and functional impairment in extrinsic monocyte/macrophage recruitment.21 CCR2WT mice have normal CCR2 expression and do not express GFP. CCR2GFP/+ mice were bred to obtain CCR2GFP/+ reporter mice, knockout mice (CCR2GFP/GFP; CCR2KO), and CCR2WT controls.
2.3 |. Flexor tendon repair
At 10–12 weeks of age, CCR2WT, CCR2GFP/+, and CCR2KO mice underwent complete transection and repair of the flexor digitorum longus (FDL) tendon in the hind-paw as previously described.22 Briefly, mice were given a pre-surgical dose of sustained-release buprenorphine (0.5–1.0 mg/kg). Mice were then anesthetized with ketamine (60 mg/kg) and xylazine (4 mg/kg). Following preparation and sterilization of the surgical site, the FDL was surgically transected in the transverse plane at the myotendinous junction in the calf to prevent early-stage strain-induced rupture of the repair site. The skin was closed with a 5–0 suture. A small incision was then made on the posterior surface of the hind paw, and the FDL tendon was located and completely transected using micro spring scissors. The tendon was then repaired using 8–0 suture and the skin were closed with a 5–0 suture. Following surgery, mice were allowed to resume normal cage activity with regular food intake and water consumption.
2.4 |. Histology and immunofluorescence
Following sacrifice, the hind paws of CCR2WT, CCR2GFP/+, and CCR2KO were harvested for frozen sectioning. Hind paws were harvested at 8-, 14-, 21-, and 28-days post-repair (n = 3–5 per genotype per timepoint). Injured and contralateral hind paws were then fixed in 10% neutral buffer formalin for 24 h at 4°C, decalcified in Webb-Jee 14% EDTA solution for 5 days at 4°C, and processed in 30% sucrose for 24 h at 4°C to cryo-protect the tissue. Samples were then embedded in Cryomatrix (Thermo Fisher Scientific, Waltham, MA, USA) and sectioned into 8 μm sagittal sections using a cryotape-transfer method.23 Sections were mounted on glass slides using 1% chitosan in 0.25% acetic acid. Slides were fixed and probed with antibodies for F4/80 (1:100 Abcam, #ab6640), CX3CR1 (1:200, Abcam, #ab8021), αSMA-Cy3 (1:250, Sigma-Aldrich, #C6198), and fibroblast activation protein (1:500, Abcam, #ab53066) and counterstained with NucBlue Live Cell Stain (Invitrogen, #R37605). Endogenous fluorescence was imaged with a VS120 Virtual Slide Microscope (Olympus, Waltham, MA).
2.5 |. Quantification of fluorescence
Fluorescent images were processed using Visiopharm image analysis software v.6.7.9.2590 (Visiopharm, Horsholm, Denmark). Regions of interest (ROI) were drawn to include the tendon stubs and bridging scar tissue for processing. For CCR2GFP staining, automatic segmentation using a threshold classifier was used to define discrete cell populations based on fluorescent intensity, and data are given as a percentage of all cells within an ROI. For F4/80 and αSMA staining, the area of each fluorescent signal was calculated and data are presented as percent positive staining normalized to total area. An n = 3–5 for CCR2WT, CCR2GFP/+, and CCR2KO mice was used for quantification.
2.6 |. Assessment of gliding function and biomechanical testing
Tendon gliding function was assessed as previously described.24–26 Briefly, injured and uninjured contralateral hindlimbs were harvested at the knee joint, and skin was removed down to the ankle. The end of the tendon was released just proximal to the tarsal tunnel without disrupting the skin or the ankle. The tendon end was secured using cyanoacrylate between two pieces of tape. The tibia was then secured in an alligator clip and loaded incrementally with small weights ranging from 0–19 g. Digital images were taken at each weight. The flexion angle of the metatarsophalangeal (MTP) joint was measured from these images. The MTP flexion angle corresponds to the degrees of flexion upon application of the 19 g weight. Uninjured tendons undergo complete flexion of the digits at 19 g. Gliding resistance is based on the changes in the MTP flexion angle of the range of the applied weights. Higher gliding resistance is indicative of impaired gliding function and the formation of adhesive scar tissue between the tendon and surrounding tissue.
Following gliding testing, the tibia was removed at the ankle and the toes and proximal section of the FDL in the tape were secured in opposing ends of custom grips on an Instron 8841 DynaMight™ axial servo-hydraulic testing system (Instron Corporation, Norwood, MA). The tendons were loaded at a rate of 30 mm/min until failure to test tensile strength. Force-displacement curves were plotted to determine maximum tensile force and stiffness. An n = 9–12 per genotype per time point was analyzed for both injured and contralateral tendons.
2.7 |. Statistical analysis
Quantitative data were analyzed in GraphPad Prism and are presented as means ± standard deviation. Normality was assessed using the Shapiro–Wilk normality test for all data. A student’s t test was used to assess differences in the number of CCR2GFP cells between genotypes for uninjured data. A two-way Analysis of Variance (ANOVA) with Sidak’s multiple comparisons test was used to determine the significance between genotypes at any given timepoint for injured quantification data and functional analysis (gliding and biomechanics). GraphPad Prism was used to detect statistical outlier data points (ROUT method, Q value = 1%). One outlier was identified in the D28 CCR2GFP + cell quantification data set and was removed from the analysis. For all experiments, n = 1 represents one mouse. For all outcome measures, p ≤.05 was considered significant. Significance is noted in all figures with the following conventions: *p ≤.05, **p ≤.01, ***p ≤.001, ****p ≤.0001.
3 |. RESULTS
3.1 |. CCR2 identifies tendon-resident cell populations
Given that CCR2+ cells are typically considered to be recruited from the systemic circulation, we first determined whether any CCR2+ cells were present during adult flexor digitorum longus (FDL) tendon homeostasis using CCR2GFP/+ reporter mice. Surprisingly, CCR2+ cells accounted for 7.77 ± 5.372% of the total resident tendon cell population in CCR2GFP/+ mice (Figure 1A,B). However, in CCR2KO tendons, only 1.73 ± 1.33% of the cells were GFP+ (Figure 1A,B). Collectively, these data suggest that CCR2 is required for the incorporation of most CCR2GFP cells into the native tendon, but that there are also CCR2-independent mechanisms responsible for the presence of CCR2+ cells during adult tendon homeostasis.
FIGURE 1.

CCR2 identifies tendon-resident macrophages and T cells. (A) To assess the effects of CCR2 deficiency during tendon homeostasis, uninjured flexor digitorum longus tendons from 10–12 week old CCR2GFP/+ and CCR2KO mice were probed for GFP (CCR2GFP)) and F4/80. Relative to the CCR2GFP/+ control, CCR2KO tendons have a reduced presence of CCR2GFP+ cells (green arrows), F4/80+ macrophages (red arrows), double-positive cells (yellow arrows), and CCR2GFP+ F4/80− cells (green arrows). (B) Quantification of % CCR2GFP+ cells and (C) % area of F4/80+ macrophages in uninjured CCR2GFP/+ and CCR2KO tendons. (D) UMAP of cells from uninjured C57BL/6J tendons, with cells expressing CCR2 highlighted in purple and clustering in the macrophage and T cell clusters, and a heatmap of genes expressed by CCR2+ cells in the macrophage and T-cell clusters. (E) To examine the relationship between CCR2 and CXRCR1, CCR2GFP/+ reporter mice were stained for GFP (CCR2GFP), F4/80, and CX3CR1. Immunofluorescence exhibits 5 cell populations: CCR2GFP+ CX3CR1+ F4/80+ macrophages (orange arrows), CCR2GFP+ CX3CR1− F4/80+ (yellow arrows), CCR2GFP− CX3CR1+ F4/80+ macrophages (pink arrows) and CCR2GFP− CX3CR1− F4/80+ macrophages (red arrows) and CCR2GFP− CX3CR1+ F4/80− cells (white arrows). The nuclear live cell stain is blue. Tendon is outlined in a white dotted line. Scale bars, 20 and 50 μM. N = 5 per genotype. Student’s t test was used to assess statistical significance. *p ≤ .05.
We next examined the relationship between tendon resident CCR2GFP cells and the pan-macrophage marker F4/80. The majority of CCR2GFP+ cells were F4/80+ (Figure 1A,C; yellow arrows); however, there were also CCR2GFP- F4/80+ macrophages (Figure 1A; red arrows). With the loss of CCR2, both of these populations are reduced, relative to CCR2GFP/+ controls (Figure 1A; yellow arrows). We also observed a significant 77.75% reduction in overall F4/80 surface-area staining in CCR2KO tendons (Figure 1C; p = .0451) compared to the CCR2GFP/+ tendons. CCR2KO tendons had an average of 8.92 ± 3.83% F4/80 area staining, compared to 28.07 ± 12.23% F4/80 area staining in the CCR2GFP/+ tendons (Figure 1A,C; red arrows). We also identified a CCR2GFP+ F4/80− resident cell population (Figure 1A; green arrow). Interestingly, these reductions in the uninjured CCR2KO tendon are restricted to the mid-substance of the tendon, with the remaining CCR2GFP+ cells and F4/80 macrophages in the epitenon region. Taken together, these data indicate that CCR2 identifies a subset of tendon-resident macrophages in addition to a non-macrophage cell population in uninjured flexor tendons, and that loss of CCR2 decreases the presence of both populations.
To better understand the identity of the CCR2+ population in the adult tendon, we analyzed single-cell RNA sequencing data from uninjured C57BL/6J FDL tendons (full study to be published elsewhere). Consistent with our immunofluorescence staining, CCR2 expression was largely restricted to the tendon-resident macrophage population (Figure 1D). Interestingly, we also found that CCR2 was expressed by a tendon-resident T cell population (Figure 1D). While we initially hypothesized that CCR2 regulates the recruitment of circulation-derived monocytes/macrophages, these substantial alterations in the resident tendon cell environment in CCR2KO tendons identify a potential role for CCR2 in mediating the composition of the tendon intrinsic cell environment as well.
A tendon resident macrophage population has been previously defined as CX3CR1+.8 Thus, we stained uninjured CCR2GFP/+ adult tendons for CX3CR1 and F4/80 to assess the relationship between these populations during homeostasis. Based on this panel, six different populations or cell states were identified: CCR2+ F4/80+ macrophages (Figure 1E; yellow arrows), CCR2+ CX3CR1+ F4/80+ macrophages (Figure 1E; orange arrows), CCR2− CX3CR1+ F4/80+ macrophages (Figure 1E; pink arrows), CCR2−CX3CR1−F4/80+ macrophages (Figure 1E; red arrows), CCR2− CX3CR1+ F4/80− non macrophages (Figure 1E, white arrows), and a triple-negative population. While tissue-resident macrophages only comprise about ~28% (Figure 1B) of the overall tendon cell environment, these data provide evidence of heterogeneity within the resident macrophage population.
3.2 |. CCR2 deficiency disrupts macrophage presence during tendon healing
Next, we assessed the presence of CCR2GFP+ cells during early and late flexor tendon healing, as well as the impact of CCR2KO on the cellular environment. At 8 days post-repair, CCR2GFP cells comprised 50.57 ± 7.65% of the total cell population in CCR2GFP/+ control mice (Figure 2A,B) CCR2KO reduced this population to 10.16 ± 5.66% (Figure 2A,B, p = .0020). This significant decrease continued through late healing, with CCR2GFP cells accounting for 45.26 ± 7.65% of all cells in CCR2GFP/+ mice, while this population was reduced to 2.53 ± 0.64% in CCR2KO mice at D28 (Figure 2A,B, p = .0005). Loss of CCR2GFP cells led to a significant reduction in F4/80+ area, relative to controls during early (D8 CCR2GFP/+: 33.43 ± 11.82%; D8 CCR2KO: 7.38 ± 4.84%, p = .0072) and late healing (D28 CCR2GFP/+: 62.26 ± 17.53%; D28 CCR2KO: 19.26 ± 11.37%, p < .0001). (Figure 2A,C). Consistent with our homeostasis data, CCR2KO healing tendons had reduced numbers of CCR2GFP+ F4/80+ macrophages (Figure 2D, yellow arrows), CCR2GFP− F4/80+ macrophages (Figure 2D, red arrows) and CCR2GFP+ F4/80− cells (Figure 2D, green arrows). Taken together, these data suggest that CCR2KO hinders the presence of CCR2GFP+ cells and macrophages during early and late tendon healing.
FIGURE 2.

Blunted CCR2 recruitment reduces macrophage presence during early and late healing. (A) To determine the effect of CCR2 deficiency during flexor tendon healing, immunofluorescence of GFP (CCR2GFP) and F4/80 at 8- and 28-days post-repair were stained in CCR2GFP/+ and CCR2KO healing tendons. Tendon stubs are outlined in a dotted white line. (B) Quantification of % of CCR2GFP and (C) % area of F4/80+ macrophages at 8- and 28-days post-repair in CCR2GFP/+ and CCR2KO healing tendons. A two-way ANOVA with Sidak’s multiple comparisons test was used to determine statistical significance. *p ≤ .05, **p ≤ .01, ***p ≤ .001. (D) Co-immunofluorescence of GFP (CCR2GFP) and F4/80 at 8 and 28 post-repair. The nuclear live cell stain is blue. Scale bars, 50 and 20 μM.
3.3 |. CCR2 deficiency does not affect tendon biomechanics during homeostasis
To determine the functional effects of CCR2KO on flexor tendon homeostasis, contralateral hindlimbs from CCR2WT and CCR2KO mice were harvested at 14-, 21-, 28-, and 35-days post-repair for assessment of gliding function and biomechanical analysis. A two-way ANOVA with Sidak’s multiple comparisons test between genotypes and timepoints for the contralateral revealed no significant differences in gliding (gliding resistance and metatarsophalangeal flexion angle) and biomechanical properties (maximum load at failure and stiffness), as such contralateral data from each time-point is presented in aggregate (Figure 3).
FIGURE 3.
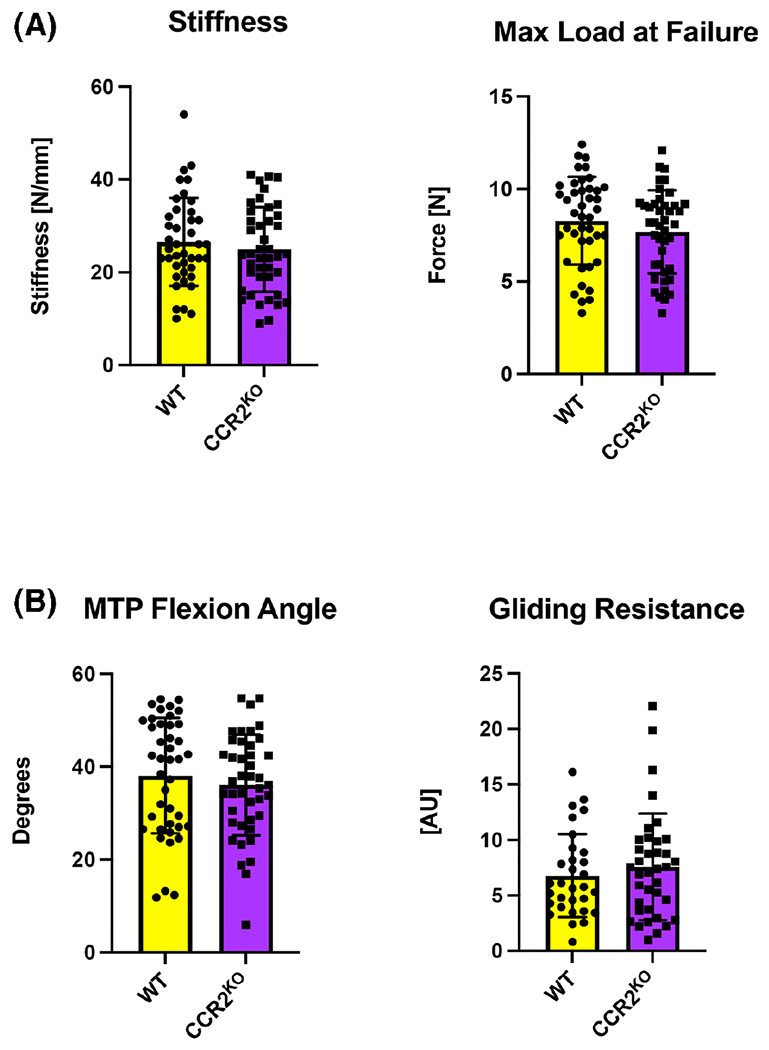
CCR2 deficiency does not affect flexor tendon biomechanics during homeostasis. (A) Measurement of biomechanical properties (stiffness and maximum load at failure) and (B) gliding properties (gliding resistance and metatarsophalangeal joint flexion angle) in uninjured WT and CCR2KO tendons. Student’s t test was used to assess statistical significance. A two-way ANOVA with Sidak’s multiple comparisons test between genotypes and timepoints for the contralateral revealed no significant differences in gliding and biomechanical properties, as such contralateral data (n = 9–12 per genotype) from each time-point is presented in aggregate. Student’s t test was used to assess statistical significance.
3.4 |. Reduced CCR2+ cell recruitment impairs late tendon healing but does not alter the range of motion
To determine the functional effects of CCR2 deficiency during the flexor healing process, CCR2WT and CCR2KO hindlimbs were harvested between 14 and 35 days post-repair for gliding function and biomechanical analysis. No significant differences in biomechanical properties were observed at D14 and D21 post-repair between CCR2WT and CCR2KO mice. However, at D28, significant decreases in both maximum load at failure (CCR2WT: 2.41 ± 0.87 N, CCR2KO: 1.29 ± 0.64 N, p = .0076) and stiffness (CCR2WT: 8.19 ± 4.27 N/mm, CCR2KO: 4.39 ± 2.26 N/mm, p = .0448) were observed in CCR2KO tendons compared to the control (Figure 4A). By D35 the impairments in maximum load (CCR2WT 3.18 ± 1.04 N, CCR2KO: 2.54 ± 0.56 N, p = .4181) and stiffness (CCR2WT: 8.20 ± 2.97 N/mm, CCR2KO: 6.91 ± 2.07 N/mm, p = .9605) in CCR2KO mice were resolved (Figure 4A). Taken together, these data suggest that CCR2KO alters the timing and rate of reacquisition of mechanical properties during tendon healing.
FIGURE 4.

CCR2KO impedes late flexor tendon healing. Measurement of (A) biomechanical (stiffness and maximum load at failure) and (B) gliding properties (gliding resistance and metatarsophalangeal joint flexion angle) at 14-, 21-, 28-, and 35-days post-repair in WT and CCR2KO tendons. N = 9–12 per genotype/timepoint. Two-way ANOVA with Sidak’s multiple comparisons test was used to assess statistical significance. *p ≤ .05, **p ≤ .01.
Because macrophages are implicated in modulating scar tissue formation, we also examined the effects of CCR2KO on post-operative tendon range of motion and gliding resistance (Figure 4B). No significant differences in MTP flexion angle or gliding resistance were observed between CCR2WT and CCR2KO tendons at any time point between 14 and 35 days.
3.5 |. CCR2 deficiency reduces myofibroblast content during tendon healing
Because macrophages can influence fibroblast activation and engage in a pro-fibrotic feedback loop with myofibroblasts,11,27 we assessed the effects of CCR2 deficiency on the myofibroblast population during healing. At D14 there was an observable decrease in αSMA staining in CCR2KO tendon repairs at, however, quantitative analyses identified a trending, non-significant increase in percent αSMA+ area (Figure 5B,C; p = .057). At D28 there was some variation in the %+ αSMA+ area in CCR2WT tendons, and the decrease in % αSMA+ area in CCR2KO repairs was not significantly different than CCR2WT (Figure 5B,C; p = .1151). A key step in myofibroblast differentiation is the activation of tissue resident fibroblasts.28 To determine whether CCR2KO may impact this initial step in differentiation, we stained for fibroblast activation protein (FAP) at D14. Indeed, CCR2KO tendons had reduced FAP staining relative to CCR2WT, although this trending difference did not reach statistical significance upon quantification of the FAP+ area (Figure 5D,E, p = .0791). Collectively, these data suggest that CCR2KO may impair tendon cell activation and subsequent differentiation to myofibroblasts.
FIGURE 5.

CCR2 deficiency transiently reduces myofibroblast presence. (A) Schematic of fibrotic tendon healing outlining tendon stub and bridging scar tissue. The orange box indicates the location of immunofluorescent images. (B) Immunofluorescence staining of αSMA+ myofibroblasts in WT and CCR2KO flexor tendon stub ends at 14- and 28-days post-repair to define changes in myofibroblasts during CCR2 deficient healing. (C) Quantification of % αSMA+ area in WT and CCR2KO healing tendon at 14- and 28-days post-repair. Student’s t test was used to assess statistical significance. (D) Immunofluorescence staining of fibroblast activation protein (FAP) in WT and CCR2KO tendon stub ends at 14 days post-repair. (E) Quantification of % area of Fibroblast Activation Protein (FAP) at 14 days post-repair in WT and CCR2KO healing tendons Nuclear live cell stain is blue. Scale bars, 50 μM. N = 3–4 per genotype/timepoint.
4 |. DISCUSSION
In the present study, we examined the role of CCR2 in modulating the flexor tendon cell environment during homeostasis and the healing process and tested the hypothesis that loss of CCR2 would be sufficient to obstruct the recruitment of extrinsic CCR2+ macrophages. In contrast to this hypothesis, we found that CCR2 also marks tendon-intrinsic cell populations including resident macrophage and T cell populations and that CCR2KO decreases the presence of tendon-resident macrophages during homeostasis. Moreover, CCR2KO decreases the overall macrophage response to the tendon injury. Interestingly, we also found that there is CCR2-independent incorporation of macrophages into the tendon during both homeostasis and healing as we observed CCR2GFP+ cells present even in CCR2KO mice. In addition to the diminution of the macrophage response, we also observed a reduction in both fibroblast activation and αSMA+ myofibroblast presence during flexor tendon healing. Collectively, these data identify CCR2 as a tendon-resident macrophage and T cell marker, and demonstrate that CCR2KO results in transient impairments during later tendon healing.
Given that CCR2 is predominantly considered a regulator of the release of monocytes from the bone marrow and the recruitment of monocytes to tissues during injury, we were particularly interested in this population during tendon healing. However, we first demonstrated the presence of a CCR2+ tendon resident population, consistent with similar findings in the brain,29 heart,30 and liver.31 While the mechanisms that dictate CCR2+ cell incorporation into homeostatic tissue are unclear, there is some evidence that circulating monocytes replenish tissue resident macrophages during post-natal development via CCR2-mediated mechanisms.32,33 Interestingly, while there was a reduction in CCR2GFP cells in CCR2KO tendons, a complete reduction in this population was not observed, suggesting CCR2-independent mechanisms of cell incorporation as well. The continued presence of CCR2GFP cells in the CCR2KO tendons is consistent with other types of injury using this model,31,34,35 in which CCR2KO results in ~80% reduction in CCR2 cells, likely because the remaining cells are being recruited in a CCR2-independent manner.36
The relationship between CCR2+ cells and other known macrophage populations in the tendon during both homeostasis and healing is important to further unravel the complex cell environment that regulates the maintenance of tendon health and physiological tendon healing.
For example, CX3CR1+ cells have been proposed to be a macrophage-like tendon cell population, or “tenophage,” that regulates proliferative and fibrotic responses and has vasculoprotective functions.8 Here, we show that while there is some overlap between CCR2+ and CX3CR1+ cells, there are distinctly separate CCR2+ and CX3CR1+ populations, further demonstrating the molecular complexity of the resident tendon cell environment. Finally, the finding of both CCR2+ and CCR2− tissue resident macrophages will be an important area of future work as cardiac resident CCR2+ macrophages express higher levels of inflammatory cytokines and act as critical drivers of monocyte and neutrophil recruitment, while CCR2− macrophages express several growth factors and enhance macrophage proliferation.30 Additionally, recent single-cell sequencing data has defined three distinct murine resident macrophage subpopulations across several organs, emphasizing resident macrophage heterogeneity.37
Consistent with our single-cell RNA sequencing data, CCR2 expression has also been reported in regulatory T cells38 and Th17 cells.39 Regulatory T cells (Tregs) are crucial to the maintenance of tissue homeostasis by preventing autoimmunity and controlling excessive inflammation.40 CCR2−/− T cells promoted accumulation of Tregs and decreased levels of Th17 cells,41 supporting an important role for CCR2 in modulating the T cell response. Recent work has only begun to appreciate the role of T cells in the tendon.42,43 Moreover, while a lack of B and T cells did not disrupt tendon development,5 adoptive transfer of neonatal Tregs into adult hosts improved Achilles tendon functional recovery and polarized macrophages into an anti-inflammatory profile.44 Collectively, these data support an important role for T cells in the healing process and merit future work to better define the role of these cells in the healing process.
Given the potential for macrophages and myofibroblasts to engage in a pro-fibrotic feedback loop in many tissues,11,27 we were particularly interested in the impact of CCR2KO on the myofibroblast environment. In response to macrophage-derived cytokines, fibroblasts undergo an activation process and subsequent myofibroblast differentiation. Moreover, myofibroblast activity can sustain macrophage function, creating an effective pro-fibrotic loop. Prior work has shown that CCR2KO reduces myofibroblast differentiation in injured kidneys.19 Consistent with these data and given the role of macrophages in wound debridement and myofibroblasts in ECM remodeling, reductions in these cell populations and decreased fibroblast activation are a potential culprits of the functional deficit we see during late tendon healing (D28). However, it is not entirely clear why CCR2KO results in only transient mechanical deficits, such that by D35 CCR2KO and CCR2GFP/+ tendons are not significantly different. One potential mechanism may relate to different myofibroblasts dynamics during healing. While CCR2KO tendons healed with qualitative reductions in myofibroblast content at D14, by D28 these differences had resolved so it is possible that restoration of the myofibroblast environment by D28 is sufficient to drive functional restoration by D35. In addition, it remains unknown whether it is the reduction in tissue resident or circulating CCR2+ cells that leads to decreased fibroblast activation and myofibroblast content, and subsequent delays in functional recovery in CCR2KO tendon repairs.
While this study clearly demonstrates that CCR2KO alters the composition of the basal flexor tendon cell environment and the flexor tendon healing process, there are a few limitations that must be considered. This CCR2KO model results in global and sustained deletion of CCR2, and thus global and sustained alterations in macrophage recruitment. Given that macrophages are a highly plastic cell type that can change in response to their microenvironment, depletion of macrophages likely results in altered responsiveness of other cell populations. Future work using temporally-controlled CCR2 inhibition will be important to define the function of these cells during different phases of the healing process. Furthermore, the lack of specific markers for tendon-resident and extrinsic macrophages complicates our ability to delineate the role of these cells during healing. Given the clear presence of CCR2+ macrophages and T cells in the adult tendon during homeostasis, we have defined these as tendon resident cells. However, it is not clear when these cells are incorporated into the tendon during development or post-natal growth, and we cannot determine whether these are indeed yolk sac-derived cells.45 Finally, these studies were conducted in young-adult mice and we focused our analyses on the flexor tendon. The impact of CCR2 deficiency may be different in middle-aged or aged animals due to an overall decline in tendon cell density,46–50 and potential changes in both the tendon-intrinsic and extrinsic response to injury. Moreover, while the scar-mediated process is conserved between different tendons,51 it is important to understand how the loss of CCR2 may alter the basal tendon cell environment and healing process in anatomically distinct tendons such as the Achilles tendon or tendons of the rotator cuff.
Taken together, our results suggest that CCR2 identifies tendon resident macrophage and T cell populations and that CCR2KO alters the composition of the basal tendon-resident cell environment. In addition, hindered recruitment of CCR2+ cells decreases the macrophage response to tendon injury as hypothesized, with decreased macrophage response during tendon healing leading to decreased myofibroblast content and a subsequent transient slowing of the reacquisition of mechanical properties after tendon injury. As such, these data suggest that a combination of altering the tissue resident and extrinsic macrophage response can drive impaired tendon healing; however, parsing out the relative contributions of these populations, as well as their time-dependent functions will be critical to informing the therapeutic target selection moving forward.
ACKNOWLEDGMENTS
We would like to thank the Histology, Biochemistry and Molecular Imaging (HBMI), the Biomechanics, Biomaterials and Multimodal Tissue Imaging (BBMTI), and the Multiphoton Imaging Cores for technical assistance. This work was supported in part by NIH/NIAMS R01 AR077527 and R01AR073169 (to AEL), NIH/NIAMS T32 AR076950, and NIH/NIAMS K99 AR080757 (to AECN). The HBMI and BBMTI Cores are supported by NIH/NIAMS P30AR069655.
Abbreviations:
- CCR2
Chemokine Receptor 2
- CCR2−/−
CCR2 knockout mice
- FAP
Fibroblast Activation Protein
- FDL
Flexor Digitorum longus
- GFP
Green Fluorescent Protein
- αSMA
alpha-smooth muscle actin
Footnotes
DISCLOSURES
The authors declare no conflict of interest.
DATA AVAILABILITY STATEMENT
The data that support the findings are available in the methods of this article.
REFERENCES
- 1.Nichols AEC, Best KT, Loiselle AE. The cellular basis of fibrotic tendon healing: challenges and opportunities. Transl Res. 2019;209:156–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ackerman JE, Best KT, Muscat SN, Loiselle AE. Metabolic regulation of tendon inflammation and healing following injury. Curr Rheumatol Rep. 2021;23:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leong NL, Kator JL, Clemens TL, James A, Enamoto-Iwamoto M, Jiang J. Tendon and ligament healing and current approaches to tendon and ligament regeneration. J Orthop Res. 2020;38:7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sunwoo JY, Eliasberg CD, Carballo CB, Rodeo SA. The role of the macrophage in tendinopathy and tendon healing. J Orthop Res. 2020;38:1666–1675. [DOI] [PubMed] [Google Scholar]
- 5.Crosio G, Huang AH. Innate and adaptive immune system cells implicated in tendon healing and disease. Eur Cell Mater. 2022;43:39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shapouri-Moghaddam A, Mohammadian S, Vazini H, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233:6425–6440. [DOI] [PubMed] [Google Scholar]
- 7.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lehner C, Spitzer G, Gehwolf R, et al. Tenophages: a novel macrophage-like tendon cell population expressing CX3CL1 and CX3CR1. Dis Model Mech. 2019;12:dmm041384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Howell KL, Kaji DA, Li TM, et al. Macrophage depletion impairs neonatal tendon regeneration. FASEB J. 2021;35:e21618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watanabe S, Alexander M, Misharin AV, Budinger GRS. The role of macrophages in the resolution of inflammation. J Clin Invest. 2019;129:2619–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pakshir P, Hinz B. The big five in fibrosis: macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol. 2018;68-69:81–93. [DOI] [PubMed] [Google Scholar]
- 12.Vierhout M, Ayoub A, Naiel S, et al. Monocyte and macrophage derived myofibroblasts: is it fate? A review of the current evidence. Wound Repair Regen. 2021;29:548–562. [DOI] [PubMed] [Google Scholar]
- 13.Pakshir P, Noskovicova N, Lodyga M, et al. The myofibroblast at a glance. J Cell Sci. 2020;133:jcs227900. [DOI] [PubMed] [Google Scholar]
- 14.de la Durantaye M, Piette AB, van Rooijen N, Frenette J. Macrophage depletion reduces cell proliferation and extracellular matrix accumulation but increases the ultimate tensile strength of injured Achilles tendons. J Orthop Res. 2014;32:279–285. [DOI] [PubMed] [Google Scholar]
- 15.Chamberlain CS, Clements AEB, Kink JA, et al. Extracellular vesicle-educated macrophages promote early Achilles tendon healing. Stem Cells. 2019;37:652–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chu HX, Arumugam TV, Gelderblom M, Magnus T, Drummond GR, Sobey CG. Role of CCR2 in inflammatory conditions of the central nervous system. J Cereb Blood Flow Metab. 2014;34:1425–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boniakowski AE, Kimball AS, Joshi A, et al. Murine macrophage chemokine receptor CCR2 plays a crucial role in macrophage recruitment and regulated inflammation in wound healing. Eur J Immunol. 2018;48:1445–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blanc RS, Kallenbach JG, Bachman JF, Mitchell A, Paris ND, Chakkalakal JV. Inhibition of inflammatory CCR2 signaling promotes aged muscle regeneration and strength recovery after injury. Nat Commun. 2020;11:4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xia Y, Entman ML, Wang Y. CCR2 regulates the uptake of bone marrow-derived fibroblasts in renal fibrosis. PLoS One. 2013;8:e77493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitchell C, Couton D, Couty JP, et al. Dual role of CCR2 in the constitution and the resolution of liver fibrosis in mice. Am J Pathol. 2009;174:1766–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Satpathy AT, Briseno CG, Lee JS, et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat Immunol. 2013;14:937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ackerman JE, Loiselle AE. Murine flexor tendon injury and repair surgery. J Vis Exp. 2016;115:e54433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dyment NA, Jiang X, Chen L, et al. High-throughput, multi-image cryohistology of mineralized tissues. J Vis Exp. 2016;115:54468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hasslund S, Jacobson JA, Dadali T, et al. Adhesions in a murine flexor tendon graft model: autograft versus allograft reconstruction. J Orthop Res. 2008;26:824–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ackerman JE, Nichols AE, Studentsova V, Best KT, Knapp E, Loiselle AE. Cell non-autonomous functions of S100a4 drive fibrotic tendon healing. Elife. 2019;8:e45342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Best KT, Korcari A, Mora KE, et al. Scleraxis-lineage cell depletion improves tendon healing and disrupts adult tendon homeostasis. eLife. 2021;10:e62203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuster R, Rockel JS, Kapoor M, Hinz B. The inflammatory speech of fibroblasts. Immunol Rev. 2021;302:126–146. [DOI] [PubMed] [Google Scholar]
- 28.Kis K, Liu X, Hagood JS. Myofibroblast differentiation and survival in fibrotic disease. Expert Rev Mol Med. 2011;13:e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Komiya H, Takeuchi H, Ogawa Y, et al. CCR2 is localized in microglia and neurons, as well as infiltrating monocytes, in the lumbar spinal cord of ALS mice. Mol Brain. 2020;13:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bajpai G, Bredemeyer A, Li W, et al. Tissue resident CCR2− and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ Res. 2019;124:263–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seki E, de Minicis S, Inokuchi S, et al. CCR2 promotes hepatic fibrosis in mice. Hepatology. 2009;50:185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bain CC, Bravo-Blas A, Scott CL, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol. 2014;15:929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Epelman S, Lavine KJ, Beaudin AE, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40:91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pedragosa J, Miró-Mur F, Otxoa-de-Amezaga A, et al. CCR2 deficiency in monocytes impairs angiogenesis and functional recovery after ischemic stroke in mice. J Cereb Blood Flow Metab. 2020;40:S98–s116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuroda N, Masuya M, Tawara I, et al. Infiltrating CCR2(+) monocytes and their progenies, fibrocytes, contribute to colon fibrosis by inhibiting collagen degradation through the production of TIMP-1. Sci Rep. 2019;9:8568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi C, Jia T, Mendez-Ferrer S, et al. Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll-like receptor ligands. Immunity. 2011;34:590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dick SA, Wong A, Hamidzada H, et al. Three tissue resident macrophage subsets coexist across organs with conserved origins and life cycles. Sci Immunol. 2022;7:eabf7777. [DOI] [PubMed] [Google Scholar]
- 38.Bruhl H, Cihak J, Schneider MA, et al. Dual role of CCR2 during initiation and progression of collagen-induced arthritis: evidence for regulatory activity of CCR2+ T cells. J Immunol. 2004;172:890–898. [DOI] [PubMed] [Google Scholar]
- 39.Kara EE, McKenzie DR, Bastow CR, et al. CCR2 defines in vivo development and homing of IL-23-driven GM-CSF-producing Th17 cells. Nat Commun. 2015;6:8644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zeng Q, Sun X, Xiao L, Xie Z, Bettini M, Deng T. A unique population: adipose-resident regulatory T cells. Front Immunol. 2018;9:2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bakos E, Thaiss CA, Kramer MP, et al. CCR2 regulates the immune response by modulating the interconversion and function of effector and regulatory T cells. J Immunol. 2017;198: 4659–4671. [DOI] [PubMed] [Google Scholar]
- 42.Garcia-Melchor E, Cafaro G, MacDonald L, et al. Novel self-amplificatory loop between T cells and tenocytes as a driver of chronicity in tendon disease. Ann Rheum Dis. 2021;80:1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Watad A, Rowe H, Russell T, et al. Normal human enthesis harbours conventional CD4+ and CD8+ T cells with regulatory features and inducible IL-17A and TNF expression. Ann Rheum Dis. 2020;79:1044–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arvind V, Howell K, Huang AH. Reprogramming adult tendon healing using regenerative neonatal regulatory T cells. bioRxiv. 2021. 10.1101/2021.05.12.443424 [DOI] [Google Scholar]
- 45.Gomez Perdiguero E, Klapproth K, Schulz C, et al. Tissue-resident macrophages originate from yolk-sac-derived erythromyeloid progenitors. Nature. 2015;518:547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Korcari A, Przybelski SJ, Gingery A, Loiselle AE. Connect Tissue Res. 2022;1–13.Published online July 28, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sugiyama Y, Naito K, Goto K, et al. Effect of aging on the tendon structure and tendon-associated gene expression in mouse foot flexor tendon. Biomed Rep. 2019;10:238–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stanley RL, Fleck RA, Becker DL, Goodship AE, Ralphs JR, Patterson-Kane JC. Gap junction protein expression and cellularity: comparison of immature and adult equine digital tendons. J Anat. 2007;211:325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yan Z, Yin H, Brochhausen C, Pfeifer CG, Alt V, Docheva D. Aged tendon stem/progenitor cells are less competent to form 3D tendon organoids due to cell autonomous and matrix production deficits. Front Bioeng Biotechnol. 2020;8:406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Freedman BR, Knecht RS, Tinguely Y, Eskibozkurt GE, Wang CS, Mooney DJ. Aging and matrix viscoelasticity affect multi-scale tendon properties and tendon derived cell behavior. Acta Biomater. 2022;143:63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Galatz LM, Gerstenfeld L, Heber-Katz E, Rodeo SA. Tendon regeneration and scar formation: the concept of scarless healing. J Orthop Res. 2015;33:823–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings are available in the methods of this article.