

This systematic review and meta-analysis investigates the molecular diagnostic yield of exome sequencing and chromosomal microarray in cerebral palsy.
Key Points
Question
What is the molecular diagnostic yield of exome sequencing and chromosomal microarray for cerebral palsy (CP)?
Findings
Through a systematic review and meta-analysis of 15 exome-sequencing and 5 chromosomal microarray CP study cohorts comprising 2419 individuals from 11 articles and 294 individuals from 5 articles, respectively, the molecular diagnostic yield of these technologies was found to be 23% and 5%, respectively.
Meaning
For individuals with cryptogenic CP, these data suggest the use of exome sequencing followed by chromosomal microarray (if whole-genome sequencing is not accessible) to identify molecular disorders for this group of patients.
Abstract
Importance
There are many known acquired risk factors for cerebral palsy (CP), but in some cases, CP is evident without risk factors (cryptogenic CP). Early CP cohort studies report a wide range of diagnostic yields for sequence variants assessed by exome sequencing (ES) and copy number variants (CNVs) assessed by chromosomal microarray (CMA).
Objective
To synthesize the emerging CP genetics literature and address the question of what percentage of individuals with CP have a genetic disorder via ES and CMA.
Data Sources
Searched articles were indexed by PubMed with relevant queries pertaining to CP and ES/CMA (query date, March 15, 2022).
Study Selection
Inclusion criteria were as follows: primary research study, case series with 10 or more nonrelated individuals, CP diagnosis, and ES and/or CMA data used for genetic evaluation. Nonblinded review was performed.
Data Extraction and Synthesis
Preferred Reporting Items for Systematic Reviews and Meta-analyses guidelines were used for assessing data quality and validity. Data were extracted by a single observer.
Main Outcomes and Measures
A separate meta-analysis was performed for each modality (ES, CMA). The primary outcome was proportion/molecular diagnostic yield (number of patients with a discovered genetic disorder divided by the total number of patients in the cohort), evaluated via meta-analysis of single proportions using random-effects logistic regression. A subgroup meta-analysis was conducted, using risk factor classification as a subgroup. A forest plot was used to display diagnostic yields of individual studies.
Results
In the meta-analysis of ES yield in CP, the overall diagnostic yield of ES among the cohorts (15 study cohorts comprising 2419 individuals from 11 articles) was 23% (95% CI, 15%-34%). The diagnostic yield across cryptogenic CP cohorts was 35% (95% CI, 27%-45%), compared with 7% (95% CI, 4%-12%) across cohorts with known risk factors (noncryptogenic CP). In the meta-analysis of CMA yield in CP, the diagnostic yield of CMA among the cohorts (5 study cohorts comprising 294 individuals from 5 articles) was 5% (95% CI, 2%-12%).
Conclusions and Relevance
Results of this systematic review and meta-analysis suggest that for individuals with cryptogenic CP, ES followed by CMA to identify molecular disorders may be warranted.
Introduction
Cerebral palsy (CP) is a neurodevelopmental disorder (NDD) that affects motor development and functioning, resulting in abnormalities in movement, coordination, tone, reflexes, posture, and/or balance.1 CP is a clinical diagnosis, independent of etiology. Known acquired risk factors for CP include prematurity,2 hypoxic ischemic encephalopathy,3 maternal and fetal infection,4,5,6 and perinatal stroke.7 The term cryptogenic CP describes cases without compelling risk factors.8
A substantial portion of individuals with CP, particularly cryptogenic CP, may have an underlying genetic disorder. For example, in a review of inborn errors of metabolism presenting with CP-like symptoms, researchers identified 54 treatable and 43 nontreatable inborn errors of metabolism that could mimic or present as CP.9 In 1 study10 of 115 children with CP, approximately 5% of the affected individuals had a pathogenic chromosomal copy number variant (CNV) detected with chromosomal microarray (CMA). In another study11 involving exome sequencing (ES) to identify monogenic causes of CP, 14% of individuals had a potentially causative genetic alteration, including variants in candidate genes. These studies have involved heterogeneous cohorts, with variable methods applied to ascertain cases, assess phenotypes, and classify CP and its subtypes.
Despite these data, genetic testing for CP is inconsistently used in clinical practice. Historically, CP has been associated with acquired injury. There is also variability in the diagnosis of CP in the setting of a known genetic etiology,12 possibly fueled by conventional thinking that genetic disorders are progressive, thus precluding the diagnosis of CP.13 Furthermore, guidelines for genetic testing in this population, such as those from the American Academy of Neurology,14 are not modernized. To address the gap in knowledge regarding the percentage of individuals with CP who may have a genetic disorder, we conducted a systematic review and meta-analysis evaluating the molecular diagnostic yield of identifying a genetic disorder in patients with CP using ES and CMA. We selected these 2 modalities because of their use as first-tier tests in the etiologic evaluation of patients with NDDs more broadly.15,16
Methods
Eligibility, Search Strategy, and Selection Process
This study did not require a registered research protocol or statement of approval by an ethical standards committee, given that no human participants/animals were involved, and the study was a meta-analysis of already published literature. We searched articles indexed by PubMed (title, abstract, and keyword search via https://pubmed.ncbi.nlm.nih.gov/) with relevant queries pertaining to CP and ES, as well as CP and CMA (Figure 1). We reviewed the abstracts and full texts of the resulting articles, applying inclusion/exclusion criteria. Inclusion criteria included primary research study, case series with 10 or more nonrelated individuals, CP diagnosis, and ES and/or CMA data used for genetic evaluation. Exclusion criteria included non-English articles; erratum to another article; commentary articles; review articles (unless there was a previously unreported cohort embedded in the review); an animal, in vitro/in vivo, biomarker, or other biological study; case reports/case series with fewer than 10 nonrelated participants; study population not of interest (eg, hereditary spastic paraplegia) or with a focus on a neurodevelopmental disorder other than CP; focus on a specific biochemical defect or specific genetic variant/disorder; no evaluation of interest (eg, no use of CMA or ES; focus on whole-genome sequencing); no determinable outcome of interest (eg, no determinable diagnostic yield); unclear pathogenicity of reported variants; and cohort analyzed in subsequent study. This study followed the Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA) reporting guidelines.
Figure 1. Articles Included and Excluded in the Meta-analysis of Exome Sequencing and Chromosomal Microarray for Cerebral Palsy.

A, Search queries for the articles included and excluded in exome sequencing for cerebral palsy include "cerebral palsy" AND "exome sequencing" OR "next-generation sequencing.” B, Search queries included and excluded in chromosomal microarray for cerebral palsy include “copy number” OR “microarray” OR “deletion” OR “duplication” OR “chromosomal rearrangement."
Articles had to explicitly state that the patient population was diagnosed with CP. We designated whether the diagnosis of CP was using existing consensus criteria1 if there was explicit reference to these criteria or a comparable description. We excluded articles citing an alternative diagnosis (such as hereditary spasticity paraplegia) or referencing progressive symptoms. We focused on microarray-based CNV analysis and did not include the results of exome-based CNV analysis, given variability in usage and technical application of this technique. We defined pathogenic or likely pathogenic as (1) explicit designation of “pathogenic” or “likely pathogenic” in accordance with American College of Medical Genetics and Genomics criteria17 (as interpreted at the time of the study), and/or (2) explicit designation of “pathogenic,” “likely pathogenic,” or “predicted deleterious” variants in known disease genes, with the assertion that the variants were causative/diagnostic of a genetic disorder. For some studies, if these designations were unclear, we communicated with the study authors to ascertain pathogenicity assertions of reported variants.
Data Collection and Data Items
For articles included in the meta-analysis, we determined the number of participants, involvement of trios or mixed trios/nontrios in the analysis, participant characteristics (CP vs specific motor subtype, use of standard criteria for CP diagnosis, risk factor classification), testing modality (ES, CMA), and the number of participants diagnosed with a genetic disorder due to pathogenic/likely pathogenic variant(s) in a known human disease gene consistent with the inheritance pattern of that disorder. We classified included cohorts as belonging to 1 of 3 risk factor categories: (1) cryptogenic CP (absence of known acquired risk factors for CP), (2) noncryptogenic CP (presence of known acquired risk factors for CP), or (3) mixture of cryptogenic and noncryptogenic CP. In studies with cohorts suspected of having a genetic cause (eg, patients with positive family history, normal brain magnetic resonance imaging, severe clinical features despite absence of perinatal complications, and/or isolated hypotonia), we designated the cohort as cryptogenic CP. In studies with cohorts that referred to a clinical laboratory for genetic testing, we assumed that the individuals had cryptogenic CP because clinical practice has not included genetic testing for patients with CP with known acquired risk factors. We did not include variants in candidate genes (as designated at the time of the study or those not yet implicated in human disease) in the number of participants with a genetic disorder.
In articles investigating phenotypes in addition to CP, we included only those individuals clearly delineated as having a CP diagnosis. In articles that reported separate findings for both cryptogenic and noncryptogenic CP, we differentiated between each cohort/subgroup in the meta-analysis. If we determined that a subset of participants was part of a separately published study, we counted those individuals only once in the meta-analysis. If we were unsure about the pathogenicity of variants in some individuals but clear about the pathogenicity of variants in others, we only counted those patients for whom we were clear about variant pathogenicity. For ES studies that performed CNV analysis from ES data, we included as part of the number of patients with a genetic disorder only those individuals with single-nucleotide variants and small insertion-deletions. If a single study performed ES and CMA, we included the study in both meta-analyses.
Statistical Analysis
We performed a separate meta-analysis for each testing modality (ES, CMA). The primary outcome was proportion/molecular diagnostic yield (ie, the number of patients with a discovered genetic disorder divided by the total number of patients in the cohort) evaluated via meta-analysis of single proportions using random-effects logistic regression. We conducted subgroup meta-analysis, using risk factor classification as a subgroup. We used a forest plot to display diagnostic yields of individual studies.
To adjust for effects of multiple covariates on the diagnostic yield, we performed meta-regression using random-effects logistic regression modeling. In the meta-regression, the covariates were year of article publication, use of trios, and risk factor classification (dichotomized into cryptogenic vs noncryptogenic/mixture of cryptogenic and noncryptogenic). We performed a Fisher exact test to compare prevalence of participant characteristics between the ES study cohorts and the CMA study cohorts. There were no missing data from among the data items we collected. We used R, version 4.1.2 (R Project for Statistical Computing), including the metaprop function in the meta package, version 5.2-0, and the rma.glmm function in the metafor package, version 3.0-2.
Results
ES
The PubMed query for studies on ES for CP yielded 94 articles. Among these, we excluded 83 articles and included 15 study cohorts comprising 2419 individuals from 11 articles (Figure 1A and eTable 5 in the Supplement).11,18,19,20,21,22,23,24,25,26,27 One article18 investigated 2 separate cohorts, a clinical laboratory cohort (which we deemed cryptogenic CP) and a health care system-based cohort (which we deemed mixture of cryptogenic and noncryptogenic CP). Two studies19,20 evaluated diagnostic yield stratified by cryptogenic and noncryptogenic CP as we have defined it. In 1 article,21 we communicated directly with the study authors to determine which subset of the cohort had cryptogenic CP vs noncryptogenic CP and to determine pathogenicity of reported variants. For these 4 studies, we considered each cohort/subgroup as a separate entry in the meta-analysis. One study28 conducted CMA analysis on trios, as well as ES on CNV-positive trios, but we were unable to ascertain pathogenicity of reported variants from ES; we did, however, include this study in the CMA meta-analysis. Reasons for exclusion of remaining articles are presented in eTable 5 in the Supplement.
Characteristics of the 15 included study cohorts from 11 articles are shown in the Table,11,18,19,20,21,22,23,24,25,26,27 with additional details in eTable 1 in the Supplement. Forty percent of the cohorts (6 of 15) involved trio testing for all participants. Ninety-three percent of the cohorts (14 of 15) involved CP without differentiation between motor subtypes, whereas 1 study25 examined dystonic CP only. Sixty percent of the study cohorts (9 of 15) involved patients with CP whose diagnosis was based on standard criteria. Finally, 53% of the study cohorts (8 of 15) examined cryptogenic CP, 20% (3 of 15) examined noncryptogenic CP, and 27% (4 of 15) examined a mixture of cryptogenic and noncryptogenic CP. Among these 15 study cohorts,11,18,19,20,21,22,23,24,25,26,27 the diagnostic yields of ES varied from 6% to 55%. This wide range likely reflects the heterogeneity of the individual cohorts and makes it difficult to the assess clinical utility of ES for CP from individual studies alone. Thus, we sought to perform a meta-analysis quantifying overall diagnostic yield while accounting for clinical factors which may mediate this yield.
Table. Characteristics of Study Cohorts Included in the Meta-analysis of Exome Sequencing (ES) and Chromosomal Microarray (CMA) for Cerebral Palsy (CP).
| Study and cohort | Analysis using trios or mixed trios/nontrios | Testing modality | Focus on CP or specific CP motor subtype | CP diagnosis based on standard criteria | CP risk factor classification |
|---|---|---|---|---|---|
| ES for CP | |||||
| McMichael et al,11 2015 | Trios | ES | CP | Yes | Mixture of cryptogenic and noncryptogenic |
| Takezawa et al,23 2018 | Trios | ES, CMA | CP | Not specified | Cryptogenic |
| Jin et al,21 2020 | Trios | ES | CP | Yes | Cryptogenic |
| Jin et al,21 2020 | Trios | ES | CP | Yes | Noncryptogenic |
| Zech et al,25 2020 | Trios and nontrios | ES, CNV analysis | Dystonic CP | Not specified | Mixture of cryptogenic and noncryptogenic |
| Rosello et al,27 2021 | Trios | ES, CMA | CP | Yes | Cryptogenic |
| Moreno-De-Luca et al,18 2021 (clinical lab cohort) | Trios and nontrios | ES, CNV analysis | CP | Not specified | Cryptogenic |
| Moreno-De-Luca et al,18 2021 (health care–based cohort) | Trios and nontrios | ES, CNV analysis | CP | Not specified | Mixture of cryptogenic and noncryptogenic |
| May et al,20 2021 (no risk factor cohort) | Trios and nontrios | ES | CP | Not specified | Cryptogenic |
| May et al,20 2021 (risk factor cohort) | Trios and nontrios | ES | CP | Not specified | Non-cryptogenic |
| Yechieli et al,22 2021 | Trios | ES, CMA | CP | Yes | Cryptogenic |
| Nejabat et al,24 2021 | Trios and nontrios | ES | CP | Yes | Cryptogenic |
| Mei et al,26 2022 | Trios and nontrios | ES, CNV analysis | CP | Yes | Mixture of cryptogenic and noncryptogenic |
| Chopra et al,19 2022 (cryptogenic CP cohort) | Trios and nontrios | ES | CP | Yes | Cryptogenic |
| Chopra et al,19 2022 (non-cryptogenic CP cohort) | Trios and nontrios | ES | CP | Yes | Noncryptogenic |
| CMA for CP | |||||
| Oskoui et al,10 2015 | Trios | CMA | CP | Not specified | Mixture of cryptogenic and noncryptogenic |
| Zarrei et al,28 2018 | Trios | CMA plus ES in n = 23 trios with CNV findings | Hemiplegic CP | Not specified | Mixture of cryptogenic and noncryptogenic |
| Takezawa et al,23 2018 | Trios | ES, CMA | CP | Not specified | Cryptogenic |
| Rosello et al,27 2021 | Trios | ES, CMA | CP | Yes | Cryptogenic |
| Yechieli et al,22 2021 | Trios | ES, CMA | CP | Yes | Cryptogenic |
Abbreviation: CNV, copy number variant.
The overall diagnostic yield of ES among all 15 study cohorts was 23% based on random-effects model (95% CI, 15%-34%) (Figure 2A). The 15 study cohorts comprised a total of 2419 patients, 667 of whom were identified with a genetic diagnosis, including 6 with more than 1 genetic disorder. Patients had pathogenic or likely pathogenic variants in 297 unique genes (Figure 3 and eTable 2 in the Supplement). Details about the numerator and denominators used in the analysis that may deviate from the original study are shown in eTable 1 in the Supplement. Subgroup analysis showed that cryptogenic CP was associated with a higher diagnostic yield (35%; 95% CI, 27%-45%) compared with noncryptogenic CP (7%; 95% CI, 4%-12%) and mixed cohorts (18%; 95% CI, 7%-38%) (Figure 2A).
Figure 2. Forest Plot Showing Overall Diagnostic Yield Stratified by Cerebral Palsy (CP) Risk Factor Classification.

Overall diagnostic yield of exome sequencing (A) and chromosomal microarray (B).
Figure 3. Genetic Disorders Identified by Exome Sequencing That Occurred in More Than 5 Individuals Across Study Cohorts.

Meta-regression for the ES cohorts showed that diagnostic yield was significantly related to CP risk factor classification (cryptogenic vs noncryptogenic/mixture of cryptogenic and noncryptogenic; estimate, 1.19; 95% CI, 0.26-2.13; P = .01) but not publication year or use of trios vs mixed trios/nontrios.
CMA
The PubMed query for studies on CMA for CP yielded 122 articles. Among these, we excluded 117 articles and included 5 articles (Figure 1B and eTable 6 in the Supplement).10,22,23,27,28 We excluded 1 study29 due to inability to ascertain pathogenicity of variants. Although this study noted potentially pathogenic variants, the basis for this designation was comparison of CNV calls in patients compared with population controls. The study noted that 10 of 50 patients had a total of 14 CNVs potentially relevant to CP; however, 11 of these 14 CNVs were inherited from an unaffected parent. We excluded another article8 because patients from the study were reanalyzed and reported in a subsequent article.22 Reasons for exclusion of remaining articles are presented in eTable 6 in the Supplement.
Characteristics of the 5 included study cohorts from 5 articles10,22,23,27,28 are shown in the Table, with corroborative details shown in eTable 3 in the Supplement. With respect to analysis with trios vs mixed trios/nontrios, CP vs specific motor subtypes, CP diagnosis based on standard criteria, and CP risk factor classification, the only characteristic that showed a statistically significant difference in prevalence in the CMA analysis cohorts vs the ES cohorts was use of trios vs mixed trios/nontrios (Fisher exact test, P = .03). Among these 5 studies, the diagnostic yields of CMA varied from 0% to 18%. Similar to our rationale for meta-analysis of ES for CP, we sought out to perform meta-analysis quantifying overall diagnostic yield of CMA for CP while accounting for clinical factors which may mediate this yield.
Out of these 5 study cohorts, the diagnostic yield was 5% based on random-effects model (95% CI, 2%-12%) (Figure 2B). Across the 18 of 294 patients with pathogenic or likely pathogenic CNVs, there were 21 CNVs altogether, as some individuals had multiple CNVs (eTable 4 in the Supplement). We did not perform a meta-regression for the CMA cohorts due to the overall small number of patients with a molecular diagnosis.
Discussion
In this systematic review and meta-analysis, the diagnostic yields of ES and CMA in CP were 23% (95% CI, 15%-34%) and 5% (95% CI, 2%-12%), respectively. These results suggest that a substantial portion of individuals with CP may have a genetic disorder, countering conventional thinking that CP is exclusively due to acquired brain injury. The first descriptions of CP arose in 1861 by William Little, who described children with spastic cerebral palsy and linked their presentation to birth complications/perinatal asphyxia.30 Though the conceptualization of CP has evolved over time, in current clinical practice, once an individual receives a diagnosis of CP, clinicians sometimes assume an underlying cause (specifically birth injury, whether supported or not) without undertaking efforts to fully investigate affected individuals.31 We underscore the importance of attempting to delineate a precise cause in any individual with CP,32 which has far-reaching implications. Although individual studies have reported genetic causes of CP, the novelty of our work lies in the fact that we have carefully evaluated the phenotypic features of each individual cohort, particularly whether the CP is cryptogenic vs noncryptogenic. This allows better generalizability of these findings into clinical practice.
For CP, the higher yield of ES compared with CMA parallels similar findings from other NDDs, including autism spectrum disorder (ASD), intellectual disability,15 and epilepsy.33,34 One meta-analysis of 30 ES studies highlighted an overall yield of 36% for ASD and/or intellectual disability.15 In contrast, the yield of CMA for global developmental delay/intellectual disability, ASD, and/or multiple congenital anomalies is 15% to 20%.35 Another meta-analysis found that the diagnostic yield of ES for epilepsy was 24%, whereas the diagnostic yield of CMA was 9%.33
In this analysis, ES studies involving cryptogenic CP demonstrated higher diagnostic yields of genetic testing than did studies involving heterogeneous CP populations (ie, mixture of cryptogenic and noncryptogenic CP). It is worth noting that there were only 3 study cohorts with noncryptogenic CP. Although mendelian etiology discoverable by ES may be less common in noncryptogenic CP cohorts, there may be genetic variations that confer vulnerability to perinatal insult in this population (eg, COL4A1-related risk of stroke).19 Further studies are needed on genetic testing in patients with noncryptogenic CP.
Unsurprisingly, 2 cohorts in the ES meta-analysis with among the highest diagnostic yields were enriched for patients with a suspected genetic cause. One of the highest diagnostic yields (53%) came from a study cohort investigating patients with CP born at full-term gestation without specific abnormalities on brain magnetic resonance imaging.23 Another example of a high diagnostic yield (52%) came from an Iranian study cohort24 investigating patients presenting with atypical CP with specific factors suggesting a genetic cause. Notably, this cohort had many autosomal recessive disorders due to ascertainment in a population with high rates of consanguineous unions. In the CMA meta-analysis, 1 study cohort22 showed a substantially higher diagnostic yield (18%) compared with the other study cohorts. Factors which may play a role include its exclusive focus on cryptogenic CP and high rates of dysmorphism (31%) and congenital anomalies (29%).
In this ES meta-analysis, the most common molecular diagnosis was CTNNB1-related disorder (MIM # 615075), accounting for 23 of 667 patients with genetic disorders (3%). Pathogenic variants in CTNNB1 are associated with variable presentations of intellectual disability and spastic diplegia, among other features.36,37 The second most-frequent molecular diagnosis in the ES meta-analysis was SPAST-related disorder (MIM # 182601). SPAST-related disorder is typically characterized by slowly progressive lower-extremity spasticity and weakness, with variable age of onset, ranging from infancy to adulthood.38 There is recognition of divergent phenotypes for an increasing number of genes. In fact, for SPAST-related disorder specifically, some patients have a stable clinical trajectory lasting decades.38,39 A molecular diagnosis should prompt a careful reevaluation of the phenotype, but if an individual with a diagnosis of CP continues to have a nonprogressive course, the label of CP may still be applicable.40
We did not include the results of CNV analysis from ES, given the small numbers of studies that included CNV analysis from ES, as well as variability in application of this technique from 1 laboratory to the next. Among the studies we included in the ES meta-analysis, there were 3 that performed CNV analysis with clear designation of pathogenicity of reported variants. In the dystonic CP cohort comprising 76 patients in our analysis, in addition to the 34 patients with a molecular diagnosis, there were 2 patients with a molecular diagnosis involving a CNV, which would have added 2.6% to the diagnostic yield of ES.25 In the 2 cohorts reported by Moreno-De-Luca et al,18 CNV analysis identified molecular diagnoses in 19 of 1345 patients (1.4%) and 5 of 181 patients (2.7%). Finally, Corbett et al41 performed CNV analysis from ES on 186 individuals (including 98 who were part of a previously published ES cohort that we included in our ES meta-analysis)11 and found that 7 of 186 patients (3.7%) had a pathogenic or likely pathogenic CNV. Mei et al26 performed CNV analysis from ES, but it was difficult to ascertain the pathogenicity of the reported CNVs. Altogether, these findings suggest that CNV analysis from ES may enhance the diagnostic yield in the CP population by an additional 1% to 3%, consistent with analytical estimates.42
Evidence-based guidelines for other neurodevelopmental disorders (ASD, global developmental delay/intellectual disability, epilepsy) endorse ES as a first-line diagnostic test given the higher diagnostic yield of ES compared with CMA.15,16,34 Due to the small numbers of study cohorts in this meta-analysis, we were not able to make definitive recommendations for choice of genetic testing for CP in general. However, the data we have aggregated here support consideration of a genetic etiology, particularly for individuals presenting with cryptogenic CP. If clinical and radiologic evaluation does not suggest a specific diagnosis, we suggest that trio ES serve as a first-line genetic test for cryptogenic CP (if whole-genome sequencing is not readily available), given the superior diagnostic yield of ES over CMA for potential genetic disorders. Increasingly, CNVs can be detected from ES data, and CMA or CNV analysis should be pursued if ES is unrevealing for a child with cryptogenic CP. Although we are unable to make definitive recommendations pertaining to genetic testing for noncryptogenic CP, further research will provide the data needed to assess its utility.
Whole-genome sequencing is ideally suited for capturing single-nucleotide variants, small insertions-deletions, and CNVs. Thus, whole-genome sequencing is able to capture all variants detected by ES plus structural variants and nonexonic sequence variants. Studies pertaining to other disorders populations have shown that whole-genome sequencing can marginally improve the diagnostic yield over ES in developmental and epileptic encephalopathies43 as well clinically heterogeneous cohorts of patients with suspected genetic disorders.44,45 The main limitation of whole-genome sequencing is that it is not yet commonly available for clinical use.
With respect to use of trios in sequencing, the studies we evaluated involved either trios or mixtures of trios/nontrios (as opposed to singletons). Thus, we do not have the data to definitively prove that trio ES has a superior diagnostic yield than singleton ES for CP. However, multiple studies have shown that trio ES has a higher diagnostic rate than singleton ES across a wide range of genetic disorders, as summarized previously.46
Limitations
Our study had some limitations. First, we evaluated the yield of genetic testing in detecting a genetic disorder but not necessarily one implicated in CP specifically. There is a need to systematically evaluate gene-disease associations of genes implicated in CP, similar to efforts by Clinical Genome Resource for other NDDs.47 Second, we did not evaluate the diagnostic yield of whole-genome sequencing, given that this technology is still emerging in clinical practice; only 1 published study has evaluated its yield in CP.48 That study evaluated the diagnostic yield of whole-genome sequencing in 150 patients with CP, showing that 24.7% of the individuals had a pathogenic/likely pathogenic variant implicated in CP (20% with single nucleotide or insertion-deletion variants and 4.7% with CNVs).48 These numbers are overall consistent with the numbers reported in our meta-analysis.
Conclusions
In summary, results of this systematic review and meta-analysis suggest that a substantial percentage of individuals with CP may have a genetic diagnosis. For individuals with cryptogenic CP or noncryptogenic CP with red flags concerning for a genetic disorder,19 if clinical/radiologic assessment does not suggest a specific cause, we propose using trio ES as a first-line sequencing test followed by CMA (if whole-genome sequencing is not available at the onset) (Figure 4). We also recommend periodic clinical reevaluation and reanalysis of whole-genome sequencing or ES data for those with no molecular diagnosis identified. For noncryptogenic CP, additional data are needed to assess the utility of genetic testing.
Figure 4. Proposed Framework for the Genetic Evaluation of an Individual With Cerebral Palsy (CP).
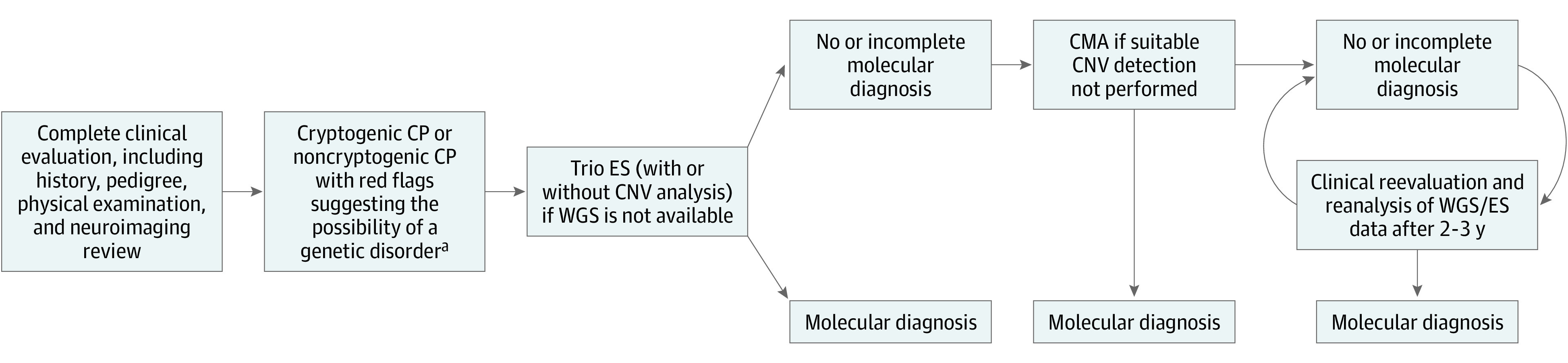
aRed flags raising the possibility of a genetic disorder include: absence of perinatal risk factors; consanguinity; more than 1 affected family member; progressive or regressive course (not initially apparent but eventually apparent after the diagnosis of CP); presence of congenital anomalies; dysmorphic features; normal brain magnetic resonance imaging (MRI) findings; unexplained biochemical/metabolic disturbances; and mismatch between perinatal history, brain MRI, and motor phenotype/severity (such as prematurity at 36 weeks with minimal neonatal intensive care course yet severe spasticity and intellectual disability; globus pallidus signal changes yet spastic diplegia phenotype). Diagnosis may lead to further action, such as enrollment in research registries and/or clinical trials, genetic counseling, and specific management changes. CMA indicates chromosomal microarray; CNV, copy number variant; ES, exome sequencing; WGS, whole-genome sequencing.
eTable 1. Detailed Characteristics of Study Cohorts Included in the Meta-analysis of ES for CP
eTable 2. List of Pathogenic/Likely Pathogenic Variants Among the Studies Included in the Meta-analysis of ES for CP
eTable 3. Detailed Characteristics of Study Cohorts Included in the Meta-analysis of CMA for CP
eTable 4. List of Pathogenic/Likely Pathogenic CNVs Among the Studies Included in the Meta-analysis of CMA for CP
eTable 5. Articles Excluded From the Meta-analysis of ES for CP
eTable 6. Articles Excluded From the Meta-analysis of CMA for CP
References
- 1.Rosenbaum P, Paneth N, Leviton A, et al. A report: the definition and classification of cerebral palsy April 2006. Dev Med Child Neurol Suppl. 2007;109:8-14. [PubMed] [Google Scholar]
- 2.Sukhov A, Wu Y, Xing G, Smith LH, Gilbert WM. Risk factors associated with cerebral palsy in preterm infants. J Matern Fetal Neonatal Med. 2012;25(1):53-57. doi: 10.3109/14767058.2011.564689 [DOI] [PubMed] [Google Scholar]
- 3.Pappas A, Korzeniewski SJ. Long-term cognitive outcomes of birth asphyxia and the contribution of identified perinatal asphyxia to cerebral palsy. Clin Perinatol. 2016;43(3):559-572. doi: 10.1016/j.clp.2016.04.012 [DOI] [PubMed] [Google Scholar]
- 4.Smithers-Sheedy H, Raynes-Greenow C, Badawi N, et al. Congenital cytomegalovirus among children with cerebral palsy. J Pediatr. 2017;181:267-271.e1. doi: 10.1016/j.jpeds.2016.10.024 [DOI] [PubMed] [Google Scholar]
- 5.Ahlin K, Himmelmann K, Hagberg G, et al. Cerebral palsy and perinatal infection in children born at term. Obstet Gynecol. 2013;122(1):41-49. doi: 10.1097/AOG.0b013e318297f37f [DOI] [PubMed] [Google Scholar]
- 6.Bear JJ, Wu YW. Maternal infections during pregnancy and cerebral palsy in the child. Pediatr Neurol. 2016;57:74-79. doi: 10.1016/j.pediatrneurol.2015.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raju TNK. Ischemic perinatal stroke: challenge and opportunities. Int J Stroke. 2008;3(3):169-172. doi: 10.1111/j.1747-4949.2008.00205.x [DOI] [PubMed] [Google Scholar]
- 8.Segel R, Ben-Pazi H, Zeligson S, et al. Copy number variations in cryptogenic cerebral palsy. Neurology. 2015;84(16):1660-1668. doi: 10.1212/WNL.0000000000001494 [DOI] [PubMed] [Google Scholar]
- 9.Leach EL, Shevell M, Bowden K, Stockler-Ipsiroglu S, van Karnebeek CD. Treatable inborn errors of metabolism presenting as cerebral palsy mimics: systematic literature review. Orphanet J Rare Dis. 2014;9:197. doi: 10.1186/s13023-014-0197-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oskoui M, Gazzellone MJ, Thiruvahindrapuram B, et al. Clinically relevant copy number variations detected in cerebral palsy. Nat Commun. 2015;6:7949. doi: 10.1038/ncomms8949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McMichael G, Bainbridge MN, Haan E, et al. Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol Psychiatry. 2015;20(2):176-182. doi: 10.1038/mp.2014.189 [DOI] [PubMed] [Google Scholar]
- 12.Aravamuthan BR, Fehlings D, Shetty S, et al. Variability in cerebral palsy diagnosis. Pediatrics. 2021;147(2):e2020010066. doi: 10.1542/peds.2020-010066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MacLennan AH, Lewis S, Moreno-De-Luca A, et al. Genetic or other causation should not change the clinical diagnosis of cerebral palsy. J Child Neurol. 2019;34(8):472-476. doi: 10.1177/0883073819840449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ashwal S, Russman BS, Blasco PA, et al. ; Quality Standards Subcommittee of the American Academy of Neurology; Practice Committee of the Child Neurology Society . Practice parameter: diagnostic assessment of the child with cerebral palsy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2004;62(6):851-863. doi: 10.1212/01.WNL.0000117981.35364.1B [DOI] [PubMed] [Google Scholar]
- 15.Srivastava S, Love-Nichols JA, Dies KA, et al. ; NDD Exome Scoping Review Work Group . Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med. 2019;21(11):2413-2421. doi: 10.1038/s41436-019-0554-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manickam K, McClain MR, Demmer LA, et al. ; ACMG Board of Directors . Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;23(11):2029-2037. doi: 10.1038/s41436-021-01242-6 [DOI] [PubMed] [Google Scholar]
- 17.Richards S, Aziz N, Bale S, et al. ; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moreno-De-Luca A, Millan F, Pesacreta DR, et al. Molecular diagnostic yield of exome sequencing in patients with cerebral palsy. JAMA. 2021;325(5):467-475. doi: 10.1001/jama.2020.26148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chopra M, Gable DL, Love-Nichols J, et al. Mendelian etiologies identified with whole-exome sequencing in cerebral palsy. Ann Clin Transl Neurol. 2022;9(2):193-205. doi: 10.1002/acn3.51506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.May HJ, Fasheun JA, Bain JM, et al. ; New York Presbyterian Hospital/Columbia University Irving Medical Center Genomics Team . Genetic testing in individuals with cerebral palsy. Dev Med Child Neurol. 2021;63(12):1448-1455. doi: 10.1111/dmcn.14948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin SC, Lewis SA, Bakhtiari S, et al. Mutations disrupting neuritogenesis genes confer risk for cerebral palsy. Nat Genet. 2020;52(10):1046-1056. doi: 10.1038/s41588-020-0695-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yechieli M, Gulsuner S, Ben-Pazi H, et al. Diagnostic yield of chromosomal microarray and trio whole-exome sequencing in cryptogenic cerebral palsy. J Med Genet. 2022;59(8):759-767. doi: 10.1136/jmedgenet-2021-107884 [DOI] [PubMed] [Google Scholar]
- 23.Takezawa Y, Kikuchi A, Haginoya K, et al. Genomic analysis identifies masqueraders of full-term cerebral palsy. Ann Clin Transl Neurol. 2018;5(5):538-551. doi: 10.1002/acn3.551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nejabat M, Inaloo S, Sheshdeh AT, et al. Genetic testing in various neurodevelopmental disorders which manifest as cerebral palsy: a case study from Iran. Front Pediatr. 2021;9:734946. doi: 10.3389/fped.2021.734946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zech M, Jech R, Boesch S, et al. Monogenic variants in dystonia: an exome-wide sequencing study. Lancet Neurol. 2020;19(11):908-918. doi: 10.1016/S1474-4422(20)30312-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mei H, Yang L, Xiao T, et al. Genetic spectrum identified by exome sequencing in a Chinese pediatric cerebral palsy cohort. J Pediatr. 2022;242:206-212.e6. doi: 10.1016/j.jpeds.2021.11.019 [DOI] [PubMed] [Google Scholar]
- 27.Rosello M, Caro-Llopis A, Orellana C, et al. Hidden etiology of cerebral palsy: genetic and clinical heterogeneity and efficient diagnosis by next-generation sequencing. Pediatr Res. 2021;90(2):284-288. doi: 10.1038/s41390-020-01250-3 [DOI] [PubMed] [Google Scholar]
- 28.Zarrei M, Fehlings DL, Mawjee K, et al. De novo and rare inherited copy-number variations in the hemiplegic form of cerebral palsy. Genet Med. 2018;20(2):172-180. doi: 10.1038/gim.2017.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McMichael G, Girirajan S, Moreno-De-Luca A, et al. Rare copy number variation in cerebral palsy. Eur J Hum Genet. 2014;22(1):40-45. doi: 10.1038/ejhg.2013.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Little WJ. On the influence of abnormal parturition, difficult labours, premature birth, and asphyxia neonatorum, on the mental and physical condition of the child, especially in relation to deformities. Clin Orthop Relat Res. 1966;46(46):7-22. doi: 10.1097/00003086-196600460-00002 [DOI] [PubMed] [Google Scholar]
- 31.Moreno-De-Luca A, Ledbetter DH, Martin CL. Genomic insights into the causes and classification of cerebral palsies. Lancet Neurol. 2012;11(3):283-292. doi: 10.1016/S1474-4422(11)70287-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aravamuthan BR, Shusterman M, Green Snyder L, Lemmon ME, Bain JM, Gross P; For Simons Searchlight; Cerebral Palsy Research Network . Diagnostic preferences include discussion of etiology for adults with cerebral palsy and their caregivers. Dev Med Child Neurol. 2022;64(6):723-733. Published online January 29, 2022. doi: 10.1111/dmcn.15164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sheidley BR, Malinowski J, Bergner AL, et al. Genetic testing for the epilepsies: a systematic review. Epilepsia. 2022;63(2):375-387. doi: 10.1111/epi.17141 [DOI] [PubMed] [Google Scholar]
- 34.Sánchez Fernández I, Loddenkemper T, Gaínza-Lein M, Sheidley BR, Poduri A. Diagnostic yield of genetic tests in epilepsy: a meta-analysis and cost-effectiveness study. Neurology. 2019;92(5):e418-e428. doi: 10.1212/WNL.0000000000006850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749-764. doi: 10.1016/j.ajhg.2010.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kharbanda M, Pilz DT, Tomkins S, et al. ; DDD Study . Clinical features associated with CTNNB1 de novo loss of function mutations in ten individuals. Eur J Med Genet. 2017;60(2):130-135. doi: 10.1016/j.ejmg.2016.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rossetti LZ, Bekheirnia MR, Lewis AM, et al. ; Undiagnosed Diseases Network . Missense variants in CTNNB1 can be associated with vitreoretinopathy-Seven new cases of CTNNB1-associated neurodevelopmental disorder including a previously unreported retinal phenotype. Mol Genet Genomic Med. 2021;9(1):e1542. doi: 10.1002/mgg3.1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parodi L, Fenu S, Barbier M, et al. ; SPATAX network . Spastic paraplegia due to SPAST mutations is modified by the underlying mutation and sex. Brain. 2018;141(12):3331-3342. doi: 10.1093/brain/awy285 [DOI] [PubMed] [Google Scholar]
- 39.Schüle R, Wiethoff S, Martus P, et al. Hereditary spastic paraplegia: clinicogenetic lessons from 608 patients. Ann Neurol. 2016;79(4):646-658. doi: 10.1002/ana.24611 [DOI] [PubMed] [Google Scholar]
- 40.Lewis SA, Shetty S, Wilson BA, et al. Insights from genetic studies of cerebral palsy. Front Neurol. 2021;11:625428. doi: 10.3389/fneur.2020.625428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Corbett MA, van Eyk CL, Webber DL, et al. Pathogenic copy number variants that affect gene expression contribute to genomic burden in cerebral palsy. NPJ Genom Med. 2018;3:33. doi: 10.1038/s41525-018-0073-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marchuk DS, Crooks K, Strande N, et al. Increasing the diagnostic yield of exome sequencing by copy number variant analysis. PLoS One. 2018;13(12):e0209185. doi: 10.1371/journal.pone.0209185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palmer EE, Sachdev R, Macintosh R, et al. Diagnostic yield of whole-genome sequencing after nondiagnostic exome sequencing or gene panel in developmental and epileptic encephalopathies. Neurology. 2021;96(13):e1770-e1782. doi: 10.1212/WNL.0000000000011655 [DOI] [PubMed] [Google Scholar]
- 44.Lionel AC, Costain G, Monfared N, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med. 2018;20(4):435-443. doi: 10.1038/gim.2017.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alfares A, Aloraini T, Subaie LA, et al. Whole-genome sequencing offers additional but limited clinical utility compared with reanalysis of whole-exome sequencing. Genet Med. 2018;20(11):1328-1333. doi: 10.1038/gim.2018.41 [DOI] [PubMed] [Google Scholar]
- 46.Tan TY, Lunke S, Chong B, et al. A head-to-head evaluation of the diagnostic efficacy and costs of trio vs singleton exome-sequencing analysis. Eur J Hum Genet. 2019;27(12):1791-1799. doi: 10.1038/s41431-019-0471-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Helbig I, Riggs ER, Barry CA, et al. The ClinGen Epilepsy Gene Curation Expert Panel—bridging the divide between clinical domain knowledge and formal gene curation criteria. Hum Mutat. 2018;39(11):1476-1484. doi: 10.1002/humu.23632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Eyk CL, Webber DL, Minoche AE, et al. Yield of clinically reportable genetic variants in unselected cerebral palsy by whole-genome sequencing. NPJ Genom Med. 2021;6(1):74. doi: 10.1038/s41525-021-00238-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eTable 1. Detailed Characteristics of Study Cohorts Included in the Meta-analysis of ES for CP
eTable 2. List of Pathogenic/Likely Pathogenic Variants Among the Studies Included in the Meta-analysis of ES for CP
eTable 3. Detailed Characteristics of Study Cohorts Included in the Meta-analysis of CMA for CP
eTable 4. List of Pathogenic/Likely Pathogenic CNVs Among the Studies Included in the Meta-analysis of CMA for CP
eTable 5. Articles Excluded From the Meta-analysis of ES for CP
eTable 6. Articles Excluded From the Meta-analysis of CMA for CP