

Summary
NEU-4438 is a lead for the development of drugs against Trypanosoma brucei, which causes human African trypanosomiasis. Optimized with phenotypic screening, targets of NEU-4438 are unknown. Herein, we present a cell perturbome workflow that compares NEU-4438’s molecular modes of action to those of SCYX-7158 (acoziborole). Following a 6 h perturbation of trypanosomes, NEU-4438 and acoziborole reduced steady-state amounts of 68 and 92 unique proteins, respectively. After analysis of proteomes, hypotheses formulated for modes of action were tested: Acoziborole and NEU-4438 have different modes of action. Whereas NEU-4438 prevented DNA biosynthesis and basal body maturation, acoziborole destabilized CPSF3 and other proteins, inhibited polypeptide translation, and reduced endocytosis of haptoglobin-hemoglobin. These data point to CPSF3-independent modes of action for acoziborole. In case of polypharmacology, the cell-perturbome workflow elucidates modes of action because it is target-agnostic. Finally, the workflow can be used in any cell that is amenable to proteomic and molecular biology experiments.
Subject areas: Microbiology, parasitology, omics, proteomics
Graphical abstract

Highlights
-
•
Cell perturbome proteomics identified polypeptides destabilized after drug addition
-
•
Changes in cellular proteomes induced by drugs were used to develop hypotheses for MOA
-
•
Experimental tests verified hypotheses formulated for NEU-4438 and acoziborole
-
•
Cell perturbomics-based workflow for the prediction of MOA is applicable to all cell types
Microbiology; Parasitology; Omics; Proteomics
Introduction
Human African trypanosomiasis (HAT) is caused by Trypanosoma brucei gambiense and T.b. rhodesiense. Because extensive “antigenic variation” (Bangs, 2018; Ooi and Rudenko, 2017) rules out vaccine development, HAT is managed with chemotherapy. Fexinidazole was approved recently for the treatment of chronic HAT caused by T. b. gambiense (Pelfrene et al., 2019). However, therapies for HAT have limitations that call for continued work to discover drug leads. In the case of fexinidazole, patients’ non-compliance is an issue because of nausea and vomiting. In addition, recrudescence of disease is reported after fexinidazole treatment (Mesu et al., 2018), consistent with emerging trypanosome resistance to the drug. The oxaborole SCYX-7158 (acoziborole) (Nare et al., 2010) is in clinical trials and could replace fexinidazole as the drug of choice for HAT treatment. Nevertheless, given the record of resistance to drugs in protozoans, it may be prudent in a long-term strategy for HAT chemotherapy to include the discovery of leads that have different mode(s) of action than SCYX-7158 (Begolo et al., 2018; Fairlamb et al., 2016; Jones et al., 2015; Steketee et al., 2018; Wall et al., 2018).
Two classic approaches, namely (i) target-based (Kourbeli et al., 2021; Suenaga et al., 2021; Swinney, 2013) and (ii) whole-cell based (i.e., phenotypic) screening are used to find hits for drug development (Ege et al., 2021; Love and McNamara, 2021; Sato, 2020; Swalley, 2020). Modes of action for drugs discovered by target-based approaches are presumed to arise from the engagement of a known target in cells. In many cases, this assumption of a single drug target per cell has been challenged (Cataldi et al., 2004; Chou et al., 2015; Dai et al., 2008; Davis et al., 2011; Dolloff et al., 2011; Dunne et al., 2011; Hafner et al., 2019; Jia et al., 2008; Karaman et al., 2008; Lackey, 2006; Larsson et al., 2012), implying that mechanistic studies focusing on one presumed target may need to be revisited (reviewed in (Mensa-Wilmot, 2021)). In the case of drugs developed by phenotypic screening, as has occurred frequently in anti-trypanosome chemotherapy (Jacobs et al., 2011b; Tarral et al., 2014), targets are not known. As a result, it is not easy to predict modes of action for the drugs. Whereas some anti-trypanosome hits have been used to identify stages of a parasite life cycle where a drug may act, those morphological studies do not identify proteins involved in the disrupted cellular process (Patel et al., 2013; Sullenberger et al., 2017).
Phenotype-driven discovery programs have produced many hits and first-in-class drugs in recent years (Ang et al., 2015; Buckner et al., 2020; Carolino and Winzeler, 2020; Chatelain and Ioset, 2018; Eder et al., 2014; Swinney, 2013). In HAT drug development, the oxaborole SCYX-7158 entered clinical trials without knowledge of a molecular mechanism of action (Jacobs et al., 2011a; Swinney and Lee, 2020). Similarly, NEU-4438 is a lead for HAT whose physiologic target is not known (Bachovchin et al., 2019).
Herein, we use a multi-disciplinary approach to determine modes of NEU-4438 action, and we compare our findings with those for the clinical candidate SCYX-7158 (Eperon et al., 2014; Wring et al., 2014). In many cases, targets of drugs are discovered after approval and it is common for drugs to have multiple cellular targets (i.e., polypharmacology) (Chen et al., 2014; Dowling et al., 2007; Madiraju et al., 2014). Here, we demonstrate that proteome perturbation is a powerful tool for studying polypharmacology as it does not depend on prior knowledge of protein targets.
In the African trypanosome, the biological target of oxaboroles is reputed to be cleavage polyadenylation specificity factor 3 (CPSF3) (Begolo et al., 2018; Wall et al., 2018). However, there is no direct biochemical evidence that the oxaboroles bind CPSF3 (Jones et al., 2015). Furthermore, oxaborole analogs are hits for microorganisms that lack CPSF3 (Bellini et al., 2020; Gupta et al., 2017; Hao et al., 2021; Hernandez et al., 2013; Palencia et al., 2016) (Gupta et al., 2017; Hao et al., 2021; Hernandez et al., 2013; Palencia et al., 2016). We report here that the treatment of trypanosomes with SCYX-7158 causes the loss of 92 proteins including CPSF3. These data lead us to propose a unifying hypothesis for modes of SCYX-7158 action in the African trypanosome that may inform understanding of oxaborole activity in other microorganisms (Bellini et al., 2020; Gupta et al., 2017; Hao et al., 2021; Hernandez et al., 2013; Palencia et al., 2016).
For anti-trypanosome drug discovery, our data highlight differences in molecular modes of action between NEU-4438 and SCYX-7158, in support of furthering the development of NEU-4438 as an anti-trypanosome drug. We present evidence of NEU-4438 efficacy in a mouse model of chronic HAT.
Results
Use of delayed cytocidal concentration in drug modes of action studies
Targets of hits identified by phenotypic screening are unknown (Abo-Rady et al., 2019; Tulloch et al., 2018). Therefore, studies to identify molecules involved in modes of action after lead drug identification can be difficult. Currently, there are no standards for systematically tackling this problem in vitro, and our goal is to devise an experimental protocol that makes it possible to identify proteins involved in drug modes of action.
To make it easy to compare two (or more) drugs in the mode of action studies, it was important to determine how much of each compound to use per experiment. Shunning the use of equimolar concentrations of compounds, we sought concentrations that were normalized for solubility, physicochemical properties, and trypanocidality. For this purpose, we determined DCC25 concentrations of NEU-4438 and SCYX-7158 (Table 1). DCC25 is the concentration of drug which after 6 h treatment of trypanosomes (and after drug wash-off) has no effect on parasite viability but following a 48-h culture cause 25% reduction in trypanosome proliferation. To determine DCC25, trypanosomes were incubated with drug for 6-h and then transferred to drug-free medium for 48-h. The 6-h drug treatment did not harm the membrane integrity of trypanosomes, as propidium iodide did not enter the cells (Figure S1) (Sullenberger et al., 2017). The concentration of drug that caused 25% of cidality under these conditions (DCC25) was calculated from assays with NEU-4438 or SCYX-7158. DCC25 for NEU-4438 was 159 ± 20 nM and that of SCYX-7158 was 489 ± 57 nM (Figure 1, Table 1).
Table 1.
Delayed cytocidal concentrations (DCC) of NEU-4438 and SCYX-7158 (nM)
| Cidality | NEU-4438 | SCYX-7158 |
|---|---|---|
 |
 |
|
| DCC25 (nM) | 159 ± 20 | 489 ± 57 |
| DCC50 (nM) | 334 ± 36 | 619 ± 27 |
| DCC90 (nM) | 1474 ± 116 | 1013 ± 168 |
Figure 1.

Delayed cytocidal concentrations (DCC) of NEU-4438, and SCYX-7158
T.b. brucei (5×105-/mL) culture was treated with serial concentrations of NEU-4438 or SCYX-7158 in 24well plates. After 6 h, cells were washed with and transferred into drug-free HMI-9 medium for 48 h in 384well plates at a starting cell density of 1×104 trypanosomes/mL (total volume 50 μL). After 48 h, cells were lysed and SYBR green dye was added. Fluorescence data obtained were analyzed with non-linear regression plots (GraphPad Prism 9) to determine DCC50 concentrations at 48 h. The mean DCC50 values were obtained from three independent biological experiments each with technical duplicates. DCC25 concentrations (NEU-4438, 150 nM; SCYX-7158, 500 nM) were used for the trypanosome perturbation studies.
Trypanocidal pathways of NEU-4438 and SCYX-7158 are different
Cell death or pro-survival pathways involve multiple proteins that act sequentially in response to extracellular cues that drive proliferation, differentiation, and survival of cells (Goldshmidt et al., 2010; Guzman, 2019; Hao et al., 2016; Hartmann et al., 2018; Ridgley et al., 1999; Win et al., 2018). We hypothesized that NEU-4438 and SCYX-7158 could be trypanocidal by interfering with identical death pathways through interaction with the same drug-binding proteins. To evaluate this hypothesis, we determined the effect of NEU-4438 (334 nM; Table 1) on DCC50 of SCYX-7158 (619 nM; Table 1). As a control, the effect of SCYX-7158 on DCC50 of SCYX-7158 was determined. The presumption is that when two compounds act on the same cell death pathway DCC50 drops when the small molecules are added together to trypanosomes.
In the positive control, SCYX-7158 reduced DCC50 of SCYX-7158 by 0.55 (p = 1.8 × 10−2, T-test) (Figure 2). On the other hand, NEU-4438 raised DCC50 of SCYX-7158 by 1.46 (p = 8.9 × 10−3, T-test) (Figure 2).
Figure 2.

NEU-4438 does not potentiate trypanocidality of SCYX-7158
(A) To determine the effect of NEU-4438 on DCC50 of SCYX-7158, T.b. brucei (5×105/mL) was treated with serial dilutions of SCYX-7158 in a 24-well plate. To each well, either 0.1% DMSO, or DCC50 of SCYX-7158 or NEU-4438 was added, as depicted (panel A) prior to the introduction of SCYX-7158. The plates were incubated for 6 h at 37°C/5% CO2. Cells were washed with and transferred into drug-free HMI-9 medium for 48 h at a starting cell density of 1×104 cells/mL (total volume 50 μL). After 48 h, cells were lysed and fluorescence of SYBR green dye was obtained. GraphPad Prism was used to analyze data and determine DCC25 concentrations.
(B) Error bars denote the mean DCC50 ± SD of SCYX-7158 upon the addition of SCYX-7158 (S + S) or NEU-4438 (S + N) in three independent experiments. A statistical significance of differences in DCC50 was determined with Student’s T-test.
We conclude that NEU-4438 does not enhance the trypanocidality of SCYX-7158; it promotes trypanosome death using pathways that are different from those affected by SCYX-7158.
Perturbomes of NEU-4438 and SCYX-7158 in T. brucei
Having established that NEU-4438 and SCYX-7158 kill T. brucei using different pathways (Figure 2B), we initiated studies to identify specific molecular systems that were affected by each drug. An outline of the steps pursued in this effort is presented in Figure 3. We used DCC25 determined for 5 × 105 trypanosomes/mL for these experiments instead of the alternate approach of using equimolar amounts of drugs.
Figure 3.
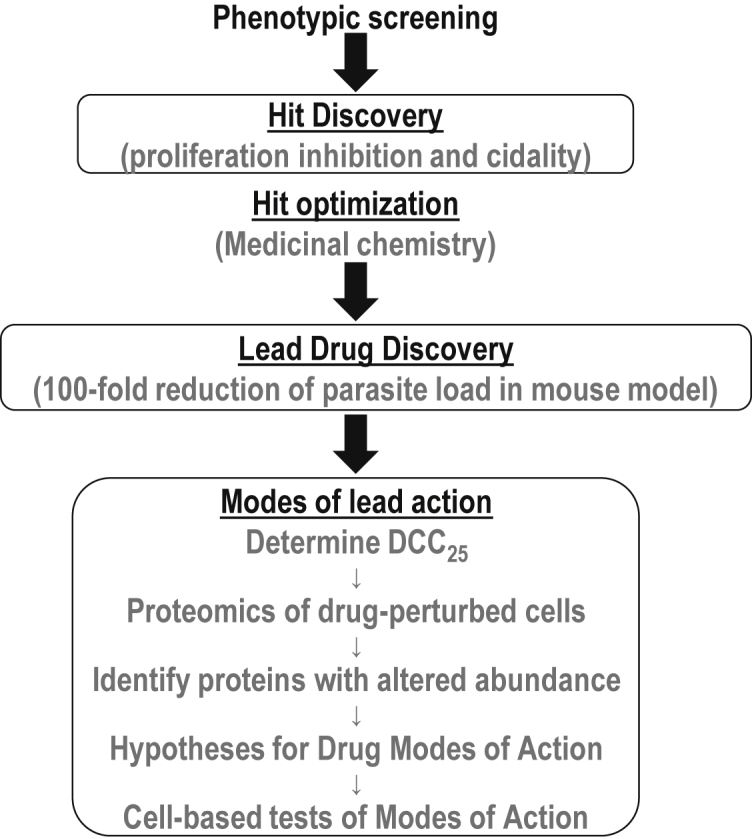
Cell perturbome proteomics workflow to determine modes of molecular action
Perturbation of trypanosomes with NEU-4438 or SCYX-7158 was performed by treating cells with DCC25 equivalents of NEU-4438 or SCYX-7158 for 6 h. Shotgun proteomics was then used to identify polypeptides whose steady-state quantities changed by a factor of two (or more) (Figure 4). SCYX-7158 was used as a control to help rule out proteome changes that might be caused non-specifically by trypanosome treatment with xenobiotics.
Figure 4.

NEU-4438 or SCYX-7158 perturbome in T. brucei
Bloodstream T. brucei labeled with heavy, medium, or light amino acids in HMI-9 for SILAC were treated with SCYX-7158 (500 nM), or NEU-4438 (150 nM) or DMSO (0.1%) for 6 h. Subsequently, trypanosomes from all three samples were combined in 1:1:1: heavy: medium: light, lysed, and separated on SDS-PAGE. Gel fragments were excised, protease digested, desalted, and extracted peptides were identified by LC-MS/MS in three independent experiments. Identified peptides were clustered to match parent proteins and to obtain the sum PEP (Posterior Error Probability). Volcano plots represent proteins with > 2-fold mean abundance ratio for NEU-4438: DMSO (A) or SCYX-7158:DMSO (B) that were identified in at least two out of three experiments. Venn diagrams show the number of proteins with the abundance ratio of ≤0.5 (C) or ≥2 (D) after NEU-4438 or SCYX-7158 treatment compared to DMSO (Tables S1–S6. The source data are available via ProteomeXchange with identifier PXD036393).
NEU-4438 altered the steady-state amount of 94 unique proteins (68 proteins decreased (Table S1), and 26 proteins increased (Table S2) (Figures 4C and 4D). SCYX-7158 affected 129 unique proteins (92 proteins decreased; Table S3), and 37 proteins increased; Table S4) compared to vehicle-treated cells. NEU-4438 and SCYX-7158 perturbomes contained 35 common proteins (30 proteins decreased (Table S5), and 5 proteins increased (Table S6) (Figures 4C and 4D).
To validate the mass spectrometry data, we used western blotting to determine whether a reduction in the amount of peptides for Tb427.10.3700, AMPK-activated protein kinase gamma subunit (AMPKg), induced by NEU-4438 treatment was matched by a reduction in protein level. In the perturbome, NEU-4438 treatment reduced the abundance of AMPKg 0.37-fold compared to DMSO treatment. In western blotting, AMPKg amount was reduced 0.54-fold and 0.86-fold, respectively when trypanosomes were treated with DCC25 and DCC90 of NEU-4438 (Figure 5A). In contrast, quantities of clathrin heavy chain increased after NEU-4438 treatment by 0.19-fold and 0.36-fold for DCC25 and DCC90, respectively (Figure 5B).
Figure 5.

Reduction of AMPK-γ subunit protein after NEU-4438 treatment
Trypanosomes expressing Myc-tagged AMPK-γ regulatory subunit and Myc-tagged clathrin heavy chain (CHC) (5×105/mL) were treated with 0.1% DMSO, DCC25 NEU-4438 or DCC90 NEU-4438. After 6 h incubation, 4×106 or 1×106 cells were pelleted, washed, lysed in SDS-PAGE loading buffer, and electrophoresed on SDS-PAG (12%).
(A) Proteins were transferred to a PVDF membrane and stained with RevertTM 700 total protein stain (LI-COR).
(B) The membrane was blocked with LI-COR® InterceptTM blocking buffer-PBS and incubated with rabbit anti-c-Myc primary antibody. After 1 h, the membrane was washed and incubated with IRDye® 800CW goat anti-rabbit secondary antibody. Images were captured with an LI-COR® Odyssey CLx near-infrared fluorescence imaging system.
(C) Quantitation of IRDye® 800CW fluorescence for myc-tagged protein (in B) normalized for total protein (LI-COR RevertTM. 700 fluorescence from panel A), and statistically analyzed by Student’s T-test.
Hypotheses for modes of drug action from proteome perturbation analysis: NEU-4438 affects DNA synthesis
Hypotheses about possible modes of action for NEU-4438 were developed in two steps. First, we prioritized proteins whose abundance decreased after drug treatment (Table S1) binning them into different biological pathways/functions, based on the homology of domains to proteins in other organisms (Hunter et al., 2009). Second, we reasoned that if NEU-4438 caused the level of a protein to decrease, then in some cases, the drug was triggering the protein equivalent of a genetic “loss-of-function” profile in the trypanosome: a decrease in the function of the protein in question is then expected.
The “NEU-4438 perturbome” (Figure 4 and Table S1) contained proteins whose abundance was reduced 100-fold, including DNA ligase, nucleoside diphosphate kinase, nucleoporin, and Cdc48, several of which have functions related to DNA metabolism (Benz et al., 2017; Madrid et al., 2006; Maric et al., 2017; Srivastava et al., 2018). Because of these observations, we hypothesized that NEU-4438 affected DNA synthesis or degradation, and tested the concept (Figure 6), by quantitating the incorporation of ethynyl deoxyuridine (EdU) into nuclear DNA. We found that that a smaller fraction of NEU-4438 treated trypanosomes incorporated EdU (24.45 ± 3.71%) compared to DMSO (46.49 ± 8.92%) (Student’s T-test, p = 3.5 × 10−2) (Figure 6B). The amount of EdU per nucleus was reduced after NEU-4438 treatment, and the difference in the distribution of data points was statistically significant (Kolmogorov-Smirnov test; p < 1 × 10−4) (Figure 6C).
Figure 6.

NEU-4438 decreases nuclear DNA synthesis
T. b. brucei (5×105/mL) was treated with DMSO (D: 0.1%) or NEU-4438 (N: 150 nM) or SCYX-7158 (S: 500 nM) for 6 h. Cells were washed with drug-free medium and 5×105 cells/mL was labeled with EdU for 1 h. Incorporated EdU was detected with azide-Alexa Fluor-488. For quantitation, at least 100 cells were analyzed from each sample.
(A) Representative images of EdU-positive signal (green) with DAPI (blue) in 0.1% DMSO (top), of NEU-4483-treated (middle), or SCYX-7158-treated cells (bottom). Scale bar = 5 μm.
(B) Quantitation of mean percent trypanosome (n > 100) population that are EdU-positive in nucleus.
(C) Quantitation of nuclear EdU brightness (nEdU). BZ-X800-analyzer software (Keyence) was used to extract EdU signal over the DAPI area. Data are presented as a median with error bars denoting standard deviation(SD) from three biological replicates. Statistical significance of distribution of EdU brightness in different treatment groups was analyzed with a Kolmogorov-Smirnov test.
With SCYX-7158, the difference in the distribution of EdU brightness per nucleus in treated cells was statistically significant (p = 9 × 10−4; Kolmogorov-Smirnov test) compared to control trypanosomes (Figure 6C), and the percentage of the cell population with nuclear EdU after SCYX-7158 treatment (43.28 ± 9.05%) did not differ significantly from that observed after solvent treatment (Student’s T-test, p = 0.68) (Figure 6B). We conclude that SCYX-7158 does not inhibit DNA synthesis in T. brucei.
Basal body maturation is blocked by NEU-4438
Inhibition of nuclear DNA synthesis in NEU-4438-treated trypanosomes might have two explanations. First, the drug interferes with the replisome polymerization of deoxynucleotides on nuclear DNA. Second, NEU-4438 prevents trypanosomes from entering the S-phase, in which case other events are known to occur at that stage of the cell cycle will also fail (Benz et al., 2017). For example, basal body duplication and maturation in the cytoplasm occur in S-phase. To distinguish between these hypotheses, we enumerated basal bodies in drug-treated trypanosomes by tracking TbRP2 with YL1/2 antibody (Andre et al., 2014) (Figure 7A).
Figure 7.

NEU-4438 reduces basal body duplication
T. b. brucei (5 × 105/mL) culture was treated with DMSO (D: 0.1%), NEU-4438 (N: 150 nM) or SCYX-7158 (S: 500 nM) for 6 h. Mature basal bodies were detected with anti-YL1/2 antibody, and nucleic acid was counterstained with DAPI.
(A) Representative image of brightfield, DAPI (blue), YL1/2 signal (red) and merge in 0.1% DMSO (top), NEU-4483 (middle) and SCYX-7158 (bottom) treated cells. Scale bar = 5 μm.
(B) Trypanosomes (n > 100 from each treatment) were binned as one basal body (1BB) and two basal body (2BB) based on number of YL1/2 (red) foci per cell from three biological replicates. Error bars indicate mean ± SD. Statistical significance of differences in population distribution among 1BB and 2BB in drug treated compared to DMSO treated trypanosomes was determined with Pearson’s Chi-square test.
NEU4438 treatment increased the fraction of trypanosomes with one basal body (1BB) while decreasing the proportion of cells with two basal bodies (2BB) (p = 2.8 × 10−2) (Figure 7B). NEU-4438 interference with basal body duplication is consistent with a conclusion that the drug prevents trypanosome entry into S-phase.
SCYX-7158 treatment, in contrast, did not alter the trypanosome cell-type distribution (p = 6.1 × 10−1) (Figure 7B). We conclude that SCYX-7158 does not prevent trypanosome entry into S-phase.
SCYX-7158 reduces protein synthesis
The SCYX-7158 perturbome (Figure 4B and Table S3) contained several proteins associated with proteostasis; pyroglutamyl-peptidase I (PGP) (Tb427.04.2670), E3 ubiquitin-protein ligase (Tb427.06.3780), and SUMO-interacting motif-containing proteins (Tb427.07.4450 NEU-4438 Tb427tmp.160.0400) (Hirano et al., 2003; Vennemann and Hofmann, 2013). To test whether SCYX-7158 affected protein homeostasis, we first tracked the incorporation of a methionine analog homopropargylglycine (HPG) into proteins (Figure S2), and then checked the effect of drugs on that process (Figure 8A).
Figure 8.

SCYX-7158 inhibits protein synthesis
T. b. brucei (5×105/mL) was treated with DMSO (D: 0.1%), or NEU-4438 (N: 150 nM), or SCYX-7158 (S: 500 nM) or cycloheximide (C: 800 nM) for 6 h in HMI-9 medium. Cells were washed and resuspended in the methionine-free medium supplemented with L-homopropargylglycine (HPG) along with respective drugs for 1 h. All samples (2×106 cells) were washed with PBS and fixed in 4% paraformaldehyde. Incorporated of HPG was detected with azide-Alexa Fluor-647.
(A) A flow cytometer was used to detect intensity of fluorescence per cell. Histograms depict fluorescence intensity per cell (n = 15,000) in DMSO, or NEU-4438, or SCYX-7158 or cycloheximide-treated trypanosomes.
(B) Scatter plots represent mean fluorescence intensity of azide-AF-647 from three biological replicates with technical duplicates. Error bars represent SD. Statistical differences in mean AF-647 fluorescence intensity of DMSO-treated trypanosomes were compared to drug-treated cells using Students’ T-test. (See also Figures S2 and S3).
SCYX-7158 (DCC25) reduced protein synthesis 23.6% (p = 1.7 × 10−2) (Figure 8B). Similarly, cycloheximide (DCC25) a control small molecule, inhibited translation by 37.3% (p = 2.16 × 10−2) (Figure 8B). (DCC25 for cycloheximide (786 nM) was determined as shown in Figure S3.) NEU-4438 did not affect synthesis of polypeptides (p = 0.99) (Figure 8B). In other experiments, the degradation of protein was not affected by either NEU-4438 or SCYX-7158 (data not shown).
Endocytosis of haptoglobin-hemoglobin is inhibited by SCYX-7158
Trypanosome treatment with SCYX-7158 reduced the abundance of haptoglobin-hemoglobin (HpHb) receptor (Tb427.06.440), and several proteins associated with endocytosis or intracellular vesicle movement (e.g., trafficking protein particle complex subunit 3 (Tb427.08.5030), synaptojanin (Tb427.07.3490), Arf6 (Tb427.10.4250), vesicular transport protein (Vps51) (Tb427.10.2130)) and dynein light chains (Tb427tmp.211.3770, and Tb427tmp.03.0815) (Figure 4B and Table S3). Haptoglobin-hemoglobin receptor (HpHbR) facilitates the endocytosis of HpHb from vertebrate blood (Vanhollebeke et al., 2008). For these reasons, we tested a hypothesis that SCYX-7158 influenced HpHb uptake in T. brucei.
Endocytosis of HpHb (AlexaFluor 647-labelled) in T. brucei was monitored using flow cytometry (Figure 9A). SCYX-7158 reduced HpHb endocytosis by 7.81% (mean fluorescence (FL) = 135.6 ± 4.36 AU) compared to control trypanosomes (mean FL = 147.15 ± 3.17 AU) (p = 2.5 × 10−2) (Figures 9A and 9B).
Figure 9.

SCYX-7158 inhibits endocytosis of haptoglobin-hemoglobin
T. brucei (5×105 cells/mL) were treated with DMSO (D: 0.1%), or NEU-4438 (N: 150 nM) or SCYX-7158 (S: 500 nM) for 6 h. Cells were washed and resuspended in the serum-free medium (without DMSO or drug). Trypanosomes were incubated with fluorescent cargo, Haptoglobin-hemoglobin (HpHb), or transferrin (Tf) or BSA, for 15 min (37°C). A flow cytometer was used to detect fluorescence intensity per cell. Propidium iodide was used to stain and gate out non-viable cells. Histograms present fluorescence intensity of HpHb-AF647 (A), Tf-AF647 (C), or BSA-AF647 (E), for every cell (n > 10,000 for each cargo) in DMSO or drug-treated samples. Points on the scatter plot represent mean fluorescence intensities of HpHb (B), Tf (D), or BSA (F) from three biological replicates (calculated with FlowJo). Bars indicate mean ± SD. Student’s T-test was used to evaluate a statistical significance of differences in mean intensity of endocytosed ligand in DMSO-treated sample compared to drug-treated samples.
NEU-4438 did not affect HpHb endocytosis in trypanosomes (mean FL = 150.65 AU, (p = 0.54) (Figures 9A and 9B). These data are consistent with selective inhibition of HpHb endocytosis by SCYX-7158.
Both NEU-4438 and SCYX-7158 impede the uptake of transferrin (Tf)
NEU-4438 and SCYX-7158 reduced steady-state amounts of proteins (Figure 4) that affect intracellular vesicle movement in other biological systems, such as a Rab-GTPase-TBC domain-containing protein (Tb427tmp.160.3890), ankyrin repeat protein (Tb427tmp.01.0440) (Givan and Sprague, 1997; Singaraja et al., 2002), and an ARP2/3 complex (Tb427.10.10600) (Table S5). From these data, we postulated that the drugs might affect general endocytosis. We tested the hypothesis by measuring the uptake of fluorophore-labeled BSA or transferrin (Tf) after NEU-4438 or SCYX-7158 treatment of trypanosomes. (BSA was studied as a marker of “bulk-phase” endocytosis.)
Neither SCYX-7158 nor NEU-4438 affected BSA endocytosis (Figures 9C and 9D), indicating that the drugs have no effect on general endocytosis.
Transferrin uptake was affected by the two compounds. NEU-4438 treatment of trypanosomes reduced the uptake of Tf by 37.63% compared to DMSO treatment (p = 1.8 × 10−3) (Figures 9E and 9F). Similarly, SCYX-7158 inhibited Tf endocytosis 28.15% compared to DMSO (p = 8.6 × 10−3) (Figures 9E and 9F). We conclude that both NEU-4438 and SCYX-7158 inhibit endocytosis of Tf in T. brucei.
NEU-4438 reduces trypanosome load in a mouse model of chronic HAT
Most patients with HAT are diagnosed at the chronic stage when trypanosomes are detected in multiple tissues (Trindade et al., 2016). Owing to its excellent potency, aqueous solubility, and metabolic stability, we tested the efficacy of NEU-4438 in a mouse model of chronic HAT. In contrast to haemo-lymphatic HAT (i.e., acute) which causes the death of mice within 7 days of infection (Behera et al., 2014; Thomas et al., 2016), mice infected with T. brucei AnTat1.1 have chronic HAT, present with waves of tissue infection (Capewell et al., 2016; Trindade et al., 2016) and they survive for 60 days before death.
For our study, mice were infected with AnTat1.1 T brucei expressing fire-fly luciferase (McLatchie et al., 2013). On day 0 (not shown), trypanosomes were detected from bioluminescence signals in mice (except animal B). Characteristic of pleomorphic strains of T. brucei [80, 81] infection produced two waves of tissue parasite load within 14 days (Figure S4A). One peak of infection was detected on day 7, and a second wave of trypanosome tissue load was maximal on day 10 (Figure S4).
On day 3, mice treated with NEU-4438 had a 3.92-fold reduction in tissue load (i.e., bioluminescence signal) (Figure S4) (p = 6.8 × 10−3). Tissue infection was undetectable in three out of four mice on day 7 (reduction in median total flux was greater than 100-fold) (Figure S4B). These results confirm NEU-4438 as a lead for drug development against chronic HAT. This dosing regimen was not curative because on day 14 two of four mice had parasites in tissues (Figure S4B). Future pharmacokinetic and pharmacodynamic studies will focus on optimizing the dosing of NEU-4438 to cure chronic HAT.
Discussion
A target-agnostic proteome perturbation strategy to decipher molecular modes of drug action
Drugs for the treatment of infectious diseases kill pathogenic cells either by promoting death pathways or inhibiting pro-survival systems. Mere drug inhibition of protein activity without connection to cell death pathways is unlikely to produce cytotoxic effects. Indeed, stimulation of apoptotic pathways (or suppression of pro-survival pathways) occurs after the engagement of some drugs targets in cells (Nam et al., 2021; O'Hare et al., 2013; Panka et al., 2006; Tang et al., 2018; Venkatesan et al., 2012; Verma et al., 2007). However, it is not routine for investigators interested in drug mechanisms to seek information about cellular events that take place after target engagement. Most work on modes of action involves the alteration of biochemical activities of targets (Futaki, 2021; Lin et al., 2018; Robertson, 2007). For drugs optimized by phenotypic screening, such studies are not possible, so the evaluation of their effect on transcriptomes or genomes contributes to the spectra of possible actions and resistance pathways (Carolino and Winzeler, 2020; Horn, 2021; Tulloch et al., 2018).
Given the increasing contributions of phenotypic screening to infectious and chronic diseases drug discovery, it seems worthwhile to establish general strategies for determining molecular modes of action for drugs that do not rely on prior knowledge of proteins that bind the drug. Proteins mediate many drug actions, so it is logical to focus on them to monitor early cellular responses to drugs (Figure 4). Characterization of drug-induced global proteome perturbation presents an unbiased set of data that could offer a path to understand the comprehensive effects of drugs on a cell. Drug perturbation biology has been performed with other goals in mind (Korkut et al., 2015; Nyman et al., 2020), none to understand molecular modes of drug action.
NEU-4438 modes of action in T. brucei
NEU-4438 was developed in a chemistry campaign that relied on phenotypic assays of anti-trypanosome potency, so its targets are unknown (Bachovchin et al., 2019). In this study, we developed a workflow to study molecular mode(s) of NEU-4438 action in five steps (outlined in Figure 3). First, we determined pharmacologically pertinent drug concentrations for trypanosome perturbation experiments (we used DCC25 determined for 5×105 trypanosomes/mL. DCC25 is normalized for physicochemical and permeability properties of drugs by benchmarking it against trypanocidality at 48 h; it permits comparison of data for different drugs). Second, we analyzed global proteome changes after NEU-4438 perturbation (6 h) of trypanosomes. Third, we used differences in protein quantities to construct hypotheses about the molecular effects of NEU4438 on T. brucei. Fourth, we tested hypotheses of modes of action with molecular biology experiments.
Perturbation proteomics with NEU-4438 revealed a reduction of steady-state amounts of 68 polypeptides after 6 h of drug treatment (Figure 4, Table S1). As most trypanosome proteins have not been studied functionally (Aslett et al., 2010), we worked with those polypeptides with known functions out of necessity. From the analysis of changes in the proteome induced by NEU4438, we hypothesized that the drug affected DNA metabolism and/or endocytosis (Figure 4A, Table S1). In experimental tests of these hypotheses, we established that NEU-4438 inhibited DNA synthesis (Figure 6), basal body maturation (Figure 7), and transferrin endocytosis (Figure 9).
There are additional hypotheses to test, after mining data from the NEU-4438-perturbome, in future studies. For example, enzymes involved in the metabolism of small molecules (e.g., aconitase, dihydroxyacetone phosphate acyltransferase, trans-enoyl CoA isomerase) were reduced (Table S1). Their products can be assayed with metabolomics techniques to determine the effect of NEU-4438 on those pathways (Steketee et al., 2018). In this way analysis of the drug perturbome (Figure 4) can inform the design of metabolomics studies.
NEU-4438 as a candidate anti-human African trypanosomiasis drug lead
Lead compounds that possess different modes of action than the anti-HAT drugs (or clinical candidates) can be backup drugs in case of treatment failure of current clinical candidates or the emergence of drug resistance. Differences in modes of action of NEU-4438 and SCYX-7158 imply that adding the former compound to small molecules in development for HAT chemotherapy is justifiable (Figure S4). Knowledge of modes of molecular action is important for the identification of “physiologic targets” of a drug (Mensa-Wilmot, 2021). So, our future work will include the discovery of NEU-4438-binding proteins whose genetic knockdown (or overexpression) may impede G1/S transition or prevent Tf endocytosis in T. brucei as demonstrated in this work (Meyer and Shapiro, 2021).
A hypothesis for SCYX-7158 modes of action
Perturbation proteomics with SCYX-7158 revealed a reduction of steady-state quantities of 92 polypeptides (Figure 4B, Table S3). From analysis of changes in the proteome we found experimentally that SCYX-7158 inhibited polypeptide synthesis (Figure 8) and prevented endocytosis of HpHb (Figure 9). Our data are consistent with an observation that AN7973, an oxaborole structurally related to SCYX-7158, inhibits protein synthesis (Begolo et al., 2018).
Cleavage polyadenylation specificity factor 3 (CPSF3) has been offered as the biological target of SCYX-7158. In that work, overexpression of CPSF3 restored robust proliferation to SCYX-7158-treated trypanosomes, in support of the hypothesis (Wall et al., 2018). Of note, however, CPSFs were not identified as oxaborole-binding proteins by affinity chromatography (Jones et al., 2015). Our data give us a different perspective on the interpretation of this information. We found that the treatment of trypanosomes with SCYX-7158 reduced the amount of CPSF3 as well as CPSF2 (Tb927.11.230) and 90 other polypeptides (Table S3). To reconcile these three sets of data (above), we propose that SCYX-7158, consistent with data from the biochemical studies, does not bind CPSF3 (Jones et al., 2015). Instead, the drug reduces the stability or synthesis of 92 proteins including CPSF3 (Figure 4; Table S3). Some of the 92 proteins are important for the proliferation of T. brucei (Horn, 2021), so a reduction in quantities diminishes the proliferation of T. brucei. It follows from this perspective that the overexpression of many of these 92 proteins after SCYX-7158 treatment of trypanosomes may, like CPSF3, restore efficient cell division to the drug-treated cells. The data obtained for CPSF3 overexpression (Wall et al., 2018) are “proof of concept” for this hypothesis; overexpression of CPSF3 replenishes lost protein and facilitates proliferation. We note in passing that stability of oxaborole-binding proteins was not perturbed by SCYX-7158 treatment of trypanosomes (Table S7). In future studies, we will test a hypothesis that overexpression of other proteins besides CPSF3 (Table S3) restores robust cell division abilities to SCYX-7158-treated trypanosomes.
A cell perturbome proteomics workflow provides a comprehensive account of modes of drug action, and informs discovery of physiologic drug targets
Modes of action for many clinical drugs remain unsolved. Some drugs developed with target-based approaches have other targets in cells (Dai et al., 2008; Dolloff et al., 2011; Dunne et al., 2011; Hafner et al., 2019; Karaman et al., 2008; Lackey, 2006). This polypharmacology (i.e., one drug with multiple targets) is well-known (Anighoro et al., 2014; Proschak et al., 2019). For example, metformin, an antidiabetic drug, interacts with multiple proteins (Chen et al., 2014; Madiraju et al., 2014; Wang et al., 2014), making it unlikely that all actions of metformin are mediated by a single target. For some clinical candidates, therapeutic effects arise from so-called “off-target” activity (Lin et al., 2019).
In cases of polypharmacology, studies to understand mechanisms of action may be most fruitful when one aims to document all “post-target engagement” effects of the drug. Analysis of a drug perturbome coupled with the development of hypotheses of its modes of action, as we advocate in this work, could contribute to a holistic understanding of the drug’s cellular effects. Similarly, aspirin inhibits multiple proteins (Kaur et al., 2012; Mugge and Silva, 2017; Wang et al., 2015; Yin et al., 1998), so an approach that focuses on events “downstream” of target engagement is likely to be a good path for advancing our understanding of the drug’s molecular effects on cells.
Cells contain tens of thousands of proteins (El-Sayed et al., 2005; Venter et al., 2015), so the view that one drug binds only to a single protein in vivo is debated. Experiments performed with whole proteomes (i.e., cell lysates used for affinity chromatography or photoaffinity labeling or chemoproteomics (Brehmer et al., 2005; Burton et al., 2021; Davis et al., 2011; Jones et al., 2015; Katiyar et al., 2013; Kennedy et al., 2021; Wang et al., 2011; Zuhl et al., 2016)) show that many drugs have other binding partners in addition to proteins for structure-guided optimization (Dai et al., 2008; Dolloff et al., 2011; Dunne et al., 2011; Hafner et al., 2019; Karaman et al., 2008; Lackey, 2006).
As drugs can bind multiple proteins (Bolognesi, 2013; Hafner et al., 2019), and a single binding pocket on a protein can bind more than one ligand (Cerisier et al., 2019), discovery of physiologic drug targets is not a trivial undertaking (Kubota et al., 2019). Many drug-binding proteins are not physiological targets which may be defined as proteins whose genetic disruption yields similar modes of action as the treatment of cells with the drug (Mensa-Wilmot, 2021). Recent efforts to address the topic systematically with multi-disciplinary experimental approaches hold promise (Meyer and Shapiro, 2021; Sanz-Rodriguez et al., 2022).
To identify physiologic targets of a drug, investigators need information on (i) the list of drug-binding proteins, and (ii) a catalog of modes of action of a drug (Figure 10). “Modes of action” correspond to those molecular pathways whose inhibition by a drug compromises cell viability (Bernacchia et al., 2022; Lytton et al., 1993; Ma et al., 2022; Nunes et al., 2022; Pandit et al., 2022; Schafer and Wenzel, 2020) (Figure 10). Two experimental strategies can be used to obtain mode of action data. First, one could identify drug-binding proteins, and from that list predict modes of a drug’s action (Figure 10), as recently demonstrated for CBL0137 (Sanz-Rodriguez et al., 2022; Thomas et al., 2016). Second, one could perturb cells with a drug and after proteome analysis to identify destabilized proteins (i.e., drug perturbome (Table 1), and predict modes of drug action from roles of destabilized proteins (Figure 10). This second strategy can accelerate the determination of modes of action because of not requiring prior knowledge of drug-binding proteins (Figure 10). Data from modes of action studies are compared to molecular effects obtained from the genetic disruption of drug-binding proteins to identify physiologic targets from drug-binding proteins (Jones et al., 2015; Sanz-Rodriguez et al., 2022; Wall et al., 2018) (Figure 10).
Figure 10.

Contributions of data from cell perturbome proteomics and drug binding proteins to the identification of physiologic drug targets
Limitations of the study
First, drug-binding proteins are best identified by affinity-interaction methodologies. Proteins stabilized in the cell perturbome are not necessarily targets of the drug, although some acoziborole binding proteins (Jones et al., 2015) are not destabilized by the drug (Table S7). Second, proteins whose expression levels are very low may require larger numbers of cells to be used for the proteomics work. Finally, many proteins have no known function (so-called “hypothetical proteins” (Berriman et al., 2005)) as they have not been studied experimentally; they cannot be used to develop hypotheses that can be evaluated with chemical biology strategies.
STAR★Methods
Key resources table
| REAGENTS or RESOURCES | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-c-Myc antibody produced in rabbit | Sigma-Aldrich | Cat#C3956 |
| IRDye 800CW Goat anti-Rabbit IgG Secondary Antibody | LI-COR Biosciences | Cat#926-32211 |
| Anti-Tubulin Antibody, clone YL1/2 | EMD Millipore | Cat#MAB1864 |
| Chemicals | ||
| L-Arginine-13C6,15N4 hydrochloride | Sigma-Aldrich | Cat# 608033 |
| L-Arginine-13C6 hydrochloride | Sigma-Aldrich | Cat#643440 |
| L-Lysine-13C6,15N2 hydrochloride | Sigma-Aldrich | Cat#608041 |
| L-Lysine-13C6 hydrochloride | Sigma-Aldrich | Cat#643459 |
| IMDM for SILAC | Thermo Fisher Scientific | Cat#88423 |
| Azide-PEG3-biotin conjugate | Sigma-Aldrich | Cat#762024 |
| EdU, DNA synthesis monitoring probe | abcam | Cat# ab146186 |
| Click-IT™ L-Homopropargylglycine (HPG) | Invitrogen | Cat#C10186 |
| Cycloheximide | Sigma-Aldrich | Cat#C7698 |
| Transferrin From Human Serum, Alexa Fluor™ 647 Conjugate | Invitrogen | Cat#T23366 |
| Haptoglobin (Phenotype 1-1) from human plasma | Sigma-Aldrich | Cat#SRP6507 |
| Albumin from Bovine Serum (BSA), Alexa Fluor™ 647 conjugate | Invitrogen | Cat# A34785 |
| Alexa Fluor™ 647 Protein Labeling Kit | Thermo Fisher Scientific | Cat#A20173 |
| Propidium iodide | Sigma-Aldrich | Cat#P4170 |
| SYBR™ Green I Nucleic Acid Gel Stain 10,000X | Invitrogen | Cat#S7563 |
| D-Luciferin, Sodium Salt | Goldbio | Cat#LUCNA |
| Deposited data | ||
| Drug-perturbome proteomics mass spectrometry | ProteomeXchange Consortium via the PRIDE | PXD036393 |
| Experimental models: Organisms/strain | ||
| Mouse: Swiss-webster | Envigo | Hsd:ND4 |
| Software | ||
| Prism 9.0 | GraphPad | https://www.graphpad.com/ |
Resource availability
Lead contact
Dr. Amrita Sharma, asharm37@kennesaw.edu.
Materials availability
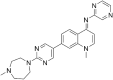
NEU-4438 [(4E)-1-methyl-7-[2-(4-methyl-1,4-diazepan-1-yl) pyrimidin-5-yl]-N-(pyrazin-2-yl)-1,4-dihydroquinolin-4-imine] (Bachovchin et al., 2019) and SCYX-7158 [(4-fluoro-N-(1-hydroxy-3,3-dimethyl-1,3-dihydro-2,1-benzoxaborol-6-yl)-2-(trifluoromethyl)benzamide)] (Jacobs et al., 2011a) were synthesized by Dr. Michael Pollastri (Northeastern University, Boston, MA).
Data and code availability
Data reported in this paper will be shared by the lead contact upon request. Mass spectrometry proteomics data have been deposited at the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2022) partner repository with the dataset identifier PXD036393 and 10.6019/PXD036393. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Trypanosome strains and culture conditions
Bloodstream T. brucei brucei Lister427 was propagated in T-25 vented cap flasks (Corning Inc., Lowell, MA) at 37°C and 5% CO2 with humidity. Trypanosomes were maintained below 106 cell/mL in HMI-9 medium (Hirumi and Hirumi, 1989) supplemented with 9% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA), 9% Serum Plus (SAFC Biosciences, Lenexa, KS), and 1% antibiotic-antimycotic solution (Corning, Corning, NY). Bloodstream T. b. brucei AnTat1.1 was maintained below 5×105 cells/mL in HMI-9 medium supplemented with 10% fetal bovine serum and 10% Serum Plus (Hirumi and Hirumi, 1989).
Bioluminescent T. brucei AnTat1.1 line
Ten micrograms of a linearized (with BamHI and XhoI) construct of pTb-AmLuc DNA (McLatchie et al., 2013) (from Dr. Martin C. Taylor London School of Hygiene and Tropical Medicine, London, United Kingdom) was used for nucleofection. Transgenic T. brucei AnTat1.1 AmLuc clones were selected, cloned, and maintained in HMI-9 medium containing puromycin (0.1 μg/mL) (McLatchie et al., 2013).
Mice and ethical statement
Female Swiss-Webster mice (Mus musculus domesticus), 8-10 weeks old (weighing ∼20-25 g) were purchased for Envigo. All mouse experiments were conducted with the approval of the Institutional Animal Care and Use Committee (IACUC) at the University of Georgia. All experimental protocols involving mice were designed in accordance with IACUC at the University of Georgia.
Mice were acclimatized for a week before commencement of experiments and were maintained at standard feed and water ad libitum.
Method details
Drug stocks and formulations
Drug stocks for in vitro assays were prepared in dimethyl sulfoxide (DMSO). For studies in mice, NEU-4438 was formulated in N-methyl-2-pyrrolidone (NMP) (Sigma Aldrich), 10% of total volume) and 0.2% hydroxypropyl methylcellulose (HPMC) (Sigma Aldrich), 90% of total volume) and dosed orally using 10 mL/kg body weight of mouse.
Delayed cytocidal concentration (DCC) of drugs
Mid-logarithmic growth phase trypanosomes (5×105/mL) were incubated with serial dilutions of NEU-4438 or SCYX-7158 or cycloheximide (1 μL of drug/mL culture medium) for 6 h in 24-well plates at 37°C/5% CO2. Trypanosomes were rinsed with and transferred into drug-free HMI-9 medium for 48 h at the cell density of 1×104 cells/mL (50 μL) in 384-well plates. Trypanosomes were pelleted, lysed and quantitated using SYBR Green I (Invitrogen; Carlsbad, CA) (Faria et al., 2015). T. brucei delayed cytocidal concentration (DCC) values for each drug were obtained after non-linear regression analysis using GraphPad Prism 9.0 (GraphPad Software; La Jolla, CA) from three independent biological replicates (with duplicate samples).
Effect of NEU-4438 on DCC50of SCYX-7158: To T.b. brucei (5×105/mL) in medium containing either DCC50 of SCYX-7158 or DCC50 of NEU-4438, serial dilutions of SCYX-7158 was added in 24-well plates. Plates were incubated for 6 h (37°C/5% CO2) and DCC50 was determined for each condition as summarized above. The data was analyzed for the change in DCC50 of SCYX-7158 after addition of SCYX-7158 or NEU-4438. The experiment was reported three times. Possible significance of differences in DCC50 values was determined with a T-test (Prism 9.0, GraphPad).
In cell perturbation experiments, T. brucei Lister 427 (5×105/mL) was treated with DCC25, a concentration causing 25% delayed cidality, of drug (DCC25 of NEU-448 is 150 nM, and DCC25 of SCYX-7158 is 500 nM) (Table 1) or 0.1% DMSO, a solvent for drugs, at 37°C/5% CO2 for 6 h in HMI-9 medium.
Assessment of cell viability
Trypanosomes (5 ×105 cells/mL) were treated with DMSO (0.1%), or NEU-4438 (150 nM), or SCYX-7158 (500 nM) for 6 h, and pelleted (3000 g for 5 min). Control trypanosomes were treated with digitonin (50 μM) for 15 min. All samples were washed with PBS (1mL) and resuspended in PBS (500 μL) containing propidium iodide (PI) (1.5 μM). Trypanosome fluorescence was determined using Flow Cytometer (Beckman Coulter, CyAn). FlowJo software (FlowJo, LLC, Ashland, OR) was used to gate live cell populations based on size and shape (forward and side scatter) and to quantitate the fluorescence intensity of PI in 20,000 trypanosomes.
Quantitative proteomics of drug perturbed trypanosomes
Trypanosomes were cultured for at least 7 days (∼28 doublings) in HMI-9 medium modified for Stable Isotope Labeling with Amino acids in Cell culture (SILAC) (Guyett et al., 2017) as follows. Iscove’s modified Dulbecco’s medium (IMDM) depleted of Lys and Arg was supplemented either with L-Arg (120 μM) and L-Lys (240 μM) for ‘light’, or with 13C6-L-Arg (120 μM) and 13C6-L-Lys (240 μM) for ‘medium’, or with 13C6, 15N4-L-Arg (120 μM) and 13C6, 15N2-L-Lys (240 μM) for heavy medium. Heavy, medium, or light-labeled trypanosomes (5×105 cells/mL) were treated with SCYX-7158 (500 nM) or NEU-4438 (150 nM) or DMSO. After 6 h, 1×108 cells were harvested from each of the three flasks and combined. The pellet of ∼3×108 trypanosomes was lysed in 2X SDS-PAGE sample buffer (final volume 300 μL), and proteins were resolved by SDS-PAGE (12%) after loading the entire lysate in a single lane. The gel was silver-stained, fragmented into 6-pieces, each of which was separately destained following the manufacturer’s protocol (Thermo Fisher Pierce™ Silver Stain for Mass Spectrometry).
Gel slices were washed with water, 50% acetonitrile/50% water, acetonitrile, ammonium bicarbonate (100 mM), followed by 50% acetonitrile/50% ammonium bicarbonate (100 mM). Proteins were reduced, alkylated and digested as described (Guyett et al., 2017). Eight microliters of desalted peptides were analyzed by LC/ESI MS/MS using a Thermo Scientific Easy-nLC II (Thermo Scientific, Waltham, MA) a nano HPLC system coupled to a Orbitrap Tribrid Fusion mass spectrometer (Thermo Scientific, Waltham, MA). In-line de-salting was accomplished using a reversed-phase column (100 μm × 20 mm) packed with Magic C18AQ (5-μm 200Å resin; Michrom Bioresources, Bruker, Billerica, MA), followed by peptide separation on a reversed-phase column (75 μm × 270 mm) packed with Magic C18AQ (5-μm 100Å resin; Michrom Bioresources, Bruker, Billerica, MA) that was directly mounted on the electrospray ion source. The heated capillary temperature was set to 300°C and a static spray voltage of 2100 V was applied to the electrospray tip. A 90-min gradient from 7% to 35% acetonitrile in 0.1% formic acid (flow rate of 300 nL/min) was used for chromatographic separation. The Orbitrap Fusion instrument was operated in data-dependent mode, switching automatically between MS survey scans in the Orbitrap (AGC target value 500,000, resolution 120,000, and maximum injection time 50 ms) with MS/MS spectra acquisition in the linear ion trap using quadrupole isolation. A 3 s cycle time was selected between master full scans in Fourier transform (FT) and the ions selected for fragmentation in the HCD cell by higher-energy collisional dissociation with a normalized collision energy of 27%. Selected ions were dynamically excluded for 45 s and exclusion mass by mass width was set to +/− 10 ppm.
Data analysis was performed using Proteome Discoverer 2.2 (Thermo Scientific, San Jose, CA). The data were searched against Tbrucei427 version 4.2, Tbrucei427 version 9.0 and cRAP (http://www.thegpm.org/crap/) Fasta files. Trypsin was set as the enzyme with maximum missed cleavages set to 2. The precursor ion tolerance was set to 10 ppm and the fragment ion tolerance was set to 0.6 Da. Variable modifications included SILAC (+6.020 Da) on lysine and arginine, oxidation on methionine (+15.995 Da), carbamidomethyl on cysteine (+57.021 Da), and acetylation at protein N-terminus (+42.011 Da). Data were searched using Sequest HT and results were run through Percolator for scoring. Quantification was performed using the canned SILAC method in Proteome Discoverer (Orsburn, 2021; Palomba et al., 2021). Data are available via ProteomeXchange with identifier PXD036393.
Proteins with ≥ 2-fold change in abundance ratio of heavy (SCYX-7158 treated): light (DMSO-treated) or medium (NEU-4438 treated): light (DMSO-treated) in at least two out of three biological replicates are presented in Tables S1–S6. The data were filtered at the peptide level for a false discovery rate of 1% or less. The sum posterior error probability (PEP) of protein score was calculated as the sum of the negative logarithms of the PEP values of connected peptide spectrum match (PSM) (Kall et al., 2008, 2009).
Epitope tagging and western blotting of AMPK-γ protein
The C-terminus of AMPK-γ subunit (Tb427.10.3700) was tagged by homologous recombination (Oberholzer et al., 2006). Primers containing overhangs for the C-term and 3′ UTR of the gene were used to amplify the C-terminus along with a myc-tag and puromycin resistance coding sequence on pMOTag plasmid (Oberholzer et al., 2006) using ExpandTM High Fidelity PCR (Dean et al., 2015). Primers used were:
AMPKγ_myc_F: 5′- CAG CTG AAC ATA TCG GAA GTG GTC TTT TTC TTA GTG TTC GGC ACA ACA AAT ACC AAC AAC CCA AGT AAG AGT CAG TGC GGC TGT GGT ACC GGT ACC GGG CCC CCC CTC GAG -3′
and AMPKγ_myc_R: 5′- TAA ATC AAC AAA CTT TAG ATG TCG ATG TTA CGC TCC TCC ATT TAT CTA TCG TAT TTT CAT ATT CCT TTA TCT TCA TTA TCA TTT CGC ACG TGG CGG CCG CTC TAG AAC TAG TGG AT -3’. PCR conditions were initial denaturation, 94°C, 5 min; denaturation, 94°C- 15 s; annealing, 63°C- 30 s; extension, 72°C- 2 min (32 cycles); and final extension, 72°C, 7 min. PCR products were ethanol precipitated and resuspended in 10 μL nuclease-free water. For transfection of T. brucei single marker line (Poon et al., 2012) with the linear DNA obtained, 4×107 cells were nucleofected using a Lonza Nucleofector V with program X-001 (Burkard et al., 2007). Recombinant clonal populations were selected after serial dilution and maintained under puromycin (0.1 μg/mL) pressure (McLatchie et al., 2013; Oberholzer et al., 2006).
Trypanosomes harboring the AMPK-γ C-term Myc-tagged protein (5×105/mL) were treated with DMSO, or DCC25 NEU-4438 (150 nM) or DCC90 NEU-4438 (1.5 μM) (Table 1). Clathrin heavy chain C-term Myc-tagged trypanosomes (from Dr. Gaurav Kumar, Mensa-Wilmot Lab, KSU) were used as a control and were treated similarly. After 6 h incubation, 4×106 or 1×106 cells were harvested for respective cell lines, pelleted, washed with PBS (PBS), and lysed in SDS-PAGE sample buffer (25 μL total volume) after which proteins were resolved by SDS-PAGE (12%).
For visualization of total proteins, polypeptides were transferred to Bio-Rad Trans-Blot® TurboTM PVDF membrane using a Bio-Rad Trans-Blot® TurboTM system (Bio-Rad Laboratories, Hercules, CA). The membrane was stained with LI-COR RevertTM 700 total protein stain (LI-COR Biosciences Lincoln, NE) for 5 min and washed twice with wash solution (6.7% (v/v) glacial acetic acid, 30% (v/v) methanol, in water), rinsed with ultra-pure water, and images captured on LI-COR® Odyssey CLx Near-Infrared Fluorescence Imaging System (LI-COR Biosciences Lincoln, NE).
To detect Myc-tagged proteins, the membrane was blocked with LI-COR® InterceptTM blocking buffer-PBS (LI-COR Biosciences Lincoln, NE) for 2 h and incubated with rabbit anti-c-Myc primary antibody (Sigma) (1: 2000 dilution in LI-COR® InterceptTM blocking buffer containing 0.1% Tween 20). The membrane was then washed with PBS containing 0.2% Tween 20 (5 min, three times) and incubated in goat anti-rabbit secondary antibody IR Dye® 800 CW (1:10,000 dilution in blocking buffer containing 0.1% Tween 20 and 0.01% SDS) for 1 h. After washing (5 min, three times), the membrane was rinsed with ultra-pure water and images were obtained with an LI-COR® Odyssey CLx Near-Infrared Fluorescence Imaging System and analyzed on Empiria Studio® Software (LI-COR Biosciences Lincoln, NE).
Basal body detection
Trypanosomes treated with DMSO, or drug for 6 h were washed once with PBS (PBS) (1 mL) and resuspended in 4% paraformaldehyde (PFA) (60 μL) in PBS. The resuspension was applied to poly-L-lysine–coated coverslips and immunofluorescence detection of basal body with antibody YL1/2 was performed as previously documented (Andre et al., 2014; Sullenberger et al., 2017). Fluorescence signals from DAPI and the secondary antibody (Alexa Fluor 594) (Andre et al., 2014) were visualized on a microscope (Keyence BZ-X800). A minimum of 120 cells were imaged and sorted based on the number of basal bodies (B), kinetoplasts (K) and nuclei (N) (as 1B1K1N, 2B1K1N, 2B2K1N or 2B2K2N). Statistical significance of differences in distributions of cell populations of drug-treated and DMSO-treated samples was calculated with a Chi-square test (in Prism 9, GraphPad Software; La Jolla, CA).
DNA synthesis
Trypanosomes pre-treated with DMSO or drugs for 6 h were pelleted, resuspended in drug-free HMI-9 medium and 5-ethynyl-2′-deoxyuridine (EdU) (300 μM final concentration) (abcam) added for 1 h (Sullenberger et al., 2017). Cells were washed once with PBS (1 mL) and fixed in 4% paraformaldehyde (in PBS) (60 μL). The resuspension was applied onto poly-L-lysine–coated coverslips and incorporated EdU was detected microscopically (Sullenberger et al., 2017). Fluorescence signal of DAPI and Alexa Fluor™ 488 was visualized on a fluorescence microscope (Keyence BZ-X800). Images from a minimum of 120 cells were captured and analyzed with BZ-X800 analyzer (Keyence). Statistical significance of differences in distribution of intensity of EdU signal per nucleus between control and experimental samples was analyzed with a Kolmogorov-Smirnov test in Prism 9 (GraphPad).
Tracking incorporation of L-homopropargylglycine
T.b. brucei (5×105/mL) was incubated with L-homopropargylglycine (HPG) (4 μM) in methionine-free RPMI medium (ThermoFisher #A1451701) supplemented with 10% fetal bovine serum and 25 mM HEPES (pH 7.4) (Doering et al., 1990b) for 1 h. After labeling, 2×106 cells were pelleted, washed in PBS (PBS) and fixed in 4% paraformaldehyde (200 μL) for 5 min at room temperature. Incorporated HPG was reacted with Azide-PEG3-biotin in a Click-iT reaction [1× Tris-buffered saline (20 mM Tris base (Genesee Scientific, San Diego, CA) and 0.14 M NaCl (Sigma-Aldrich) containing 4 mM copper sulfate (Sigma-Aldrich), 10 μM Azide-PEG3-biotin conjugate (Sigma- Aldrich), and 300 mM ascorbic acid (Avantor Performance Materials, Center Valley, PA)] for 30 min. Cell pellets were washed with PBS twice, lysed in 30 μL SDS loading buffer, and electrophoresed on SDS-PAG (12%).
For visualization proteins were transferred to a PVDF membrane (Bio-Rad) using Bio-Rad Trans-Blot® TurboTM system. The membrane was stained with LI-COR RevertTM 700 total protein stain (LI-COR Biosciences Lincoln, NE) from 5 min and rinsed twice with wash solution (6.7% (v/v) glacial acetic acid, 30% (v/v) methanol in water). The membrane was rinsed with ultra-pure water and imaged on LI-COR® Odyssey CLx Near-Infrared Fluorescence Imaging System.
For visualization of HPG-incorporation, the membrane was blocked with LI-COR® InterceptTM blocking buffer-PBS (LI-COR Biosciences Lincoln, NE) for 1 h. Azide-PEG3-biotin (covalently linked to HPG) was detected with Streptavidin IRDye® 800CW (1: 2500 dilution in blocking buffer containing 0.1% Tween 20 and 0.01% SDS). After washing (5 min, three times), the membrane was rinsed with ultra-pure water, images were captured with an LI-COR® Odyssey CLx Near-Infrared Fluorescence Imaging System and analyzed on Empiria Studio® Software (LI-COR Biosciences Lincoln, NE).
Protein synthesis
Trypanosomes (5×105/mL) in HMI-9 medium were treated with DMSO, test drug or cycloheximide (DCC25 = 800 nM, Figure S3) for 6 h. Trypanosomes were rinsed, and resuspended (at 5×105 cells/mL) in methionine-free RMPI medium (ThermoFisher #A1451701) supplemented 10% fetal bovine serum and 25 mM HEPES (pH 7.4) (Doering et al., 1990a) for 1 h. The culture was supplemented with L-homopropargylglycine (HPG, Invitrogen) (4 μM) along with respective drugs. At the end of 1 h labeling period, cells (2×106) from each sample were washed with PBS and fixed in paraformaldehyde (4% in PBS) (200 μL) for 5 min at room temperature. Fixed trypanosomes were pelleted (3000g for 10 min) and incubated in the dark for 30 min in a click-iT reaction cocktail [1× Tris-buffered saline (20 mM Tris base (Genesee Scientific, San Diego, CA) and 0.14 M NaCl (Sigma-Aldrich) containing 4 mM copper sulfate (Sigma-Aldrich), 10 μM Alexa Fluor™ 647-Azide (Life Technologies), and 300 mM ascorbic acid (Avantor Performance Materials, Center Valley, PA)]. Cells were washed twice with 1 mL PBS, pelleted at 3000 g for 10 min, resuspended in 500 μL PBS (PBS), and analyzed with a flow cytometer (Beckman Coulter CyAn). Data obtained from cells gated by forward and side scatter was analyzed on FlowJo (Tree Star, Ashland, OR). Three independent biological experiments were performed. Student’s T-test was used to determine significance of differences between mean fluorescence intensities of drug and solvent treated trypanosome populations (10,000 events).
Endocytosis of proteins
Drug or DMSO treated trypanosomes (5×105/mL) were resuspended in 100 μL serum-free HMI-9 medium. Trypanosomes were incubated with fluorescent protein cargo for 15 min at 37°C, 5% CO2. Proteins used were: (i) BSA-AlexaFluor647 conjugate (50 μg Invitrogen, Eugene, OR)), (ii) transferrin-AlexaFluor647 conjugate (50 μg, Invitrogen, Eugene, OR)) and (iii) Haptoglobin phenotype 1-1-AlexaFluor647 complexed with hemoglobin (Sigma) 1:1 by weight (5 μg) (Guyett et al., 2016). Haptoglobin was labeled using AlexaFluor protein labeling kit (Life Technologies, Invitrogen, Eugene, OR). Cells were transferred to an ice-water bath, washed with cold PBS/G at 4°C (3000g for 5 min), resuspended in PBS/G (500 mL) containing propidium iodide (3 μM), and analyzed on a flow cytometer (Beckman Coulter Cyan): FlowJo software (FlowJo, LLC) was used to gate trypanosomes based on size and shape (forward and side scatter features). Fluorescence intensity of endocytosed cargo was measured only in viable cells (negative for propidium iodide uptake). FlowJo software was used to determine the median fluorescence intensity of each cargo in cells (at least 15,000 events). Three independent biological experiments were performed. Statistical significance of differences in fluorescence intensities was analyzed by two-tailed Student’s T-test with unequal variance.
NEU-4438 efficacy in a murine model of HAT
Mice were infected with 5 × 104 T brucei AnTat1.1 AmLuc (in 100 μL of culture medium) and were randomly distributed in two groups. One uninfected mouse was used as a control for background signal from substrate. Mice in the control group received vehicle alone. Treated mice received a single daily dose of NEU-4438 (150 mg/kg body weight (bw) from 1 to 4 days post infection (DPI) and 120 mg/kg bw 5 to 7 DPI. For whole body bioluminescence imaging, mice were given an intraperitoneal injection of D-Luciferin (GoldBio) (150 mg/kg) (McLatchie et al., 2013), anesthetized under 2.5% isoflurane mixed with medical grade oxygen and imaged on an IVIS Lumina II system after 12 min. Images were obtained using the same exposure conditions, grouped, and analyzed with Living Image® software (PerkinElmer) for progression of infection. Total flux (p/s) from the whole body of each mouse was expressed as mean ± SD and the significance of differences between treated and untreated mice was determined with a Student’s T-test. Control mice were removed from the study on day 16, and remaining mice were monitored until day 30. Mice without bioluminescence signal for 30 days are considered cured.
Quantification and statistical analysis
All assays were performed thrice with technical duplicates in each independent experiment. Representation of data and statistical tests for each experiment have described in respective sections. Data were analyzed using Graph Pad Prism version 9.0 (GraphPad, San Diego, CA). A p value < 0.05 was considered statistically significant.
Acknowledgment
We thank Dr. Phil Gafken (Fred Hutchinson Cancer Research Center, Seattle, WA) for mass spectrometry, and Ms. Efytchia M. Mavrogiannaki and Dr. Michael Pollastri (Northeastern University) for providing SCYX-7158. This work was conducted under NIH funded projects R01AI126311 and R01AI124046. Justin Wiedeman provided an image of a trypanosome used in the graphical abstract.
Author contributions
Conceptualization, A.S. and K.M-W.; methodology, A.S., S.L.H., M.C., K.M-W.; investigation, A.S; writing – original draft, A.S. and K.M-W.; writing – review & editing, A.S., L.F., and K.M-W.; funding acquisition, K.M-W; resources, L.F., M.C., and S.L.H.; supervision, S.L.H. and K.M-W.
Declaration of interests
The authors declare no competing interests.
Published: November 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105302.
Supplemental information
References
- Abo-Rady M., Bellmann J., Glatza M., Marrone L., Reinhardt L., Tena S., Sterneckert J. Phenotypic screening using mouse and human stem cell-based models of neuroinflammation and gene expression analysis to study drug responses. Methods Mol. Biol. 2019;1888:21–43. doi: 10.1007/978-1-4939-8891-4_2. [DOI] [PubMed] [Google Scholar]
- Andre J., Kerry L., Qi X., Hawkins E., Drizyte K., Ginger M.L., McKean P.G. An alternative model for the role of RP2 protein in flagellum assembly in the African trypanosome. J. Biol. Chem. 2014;289:464–475. doi: 10.1074/jbc.M113.509521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang M.L.T., Murima P., Pethe K. Next-generation antimicrobials: from chemical biology to first-in-class drugs. Arch Pharm. Res. (Seoul) 2015;38:1702–1717. doi: 10.1007/s12272-015-0645-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anighoro A., Bajorath J., Rastelli G. Polypharmacology: challenges and opportunities in drug discovery. J. Med. Chem. 2014;57:7874–7887. doi: 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]
- Aslett M., Aurrecoechea C., Berriman M., Brestelli J., Brunk B.P., Carrington M., Depledge D.P., Fischer S., Gajria B., Gao X., et al. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 2010;38:D457–D462. doi: 10.1093/nar/gkp851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachovchin K.A., Sharma A., Bag S., Klug D.M., Schneider K.M., Singh B., Jalani H.B., Buskes M.J., Mehta N., Tanghe S., et al. Improvement of aqueous solubility of lapatinib-derived analogues: identification of a quinolinimine lead for human african trypanosomiasis drug development. J. Med. Chem. 2019;62:665–687. doi: 10.1021/acs.jmedchem.8b01365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangs J.D. Evolution of antigenic variation in african trypanosomes: variant surface glycoprotein expression, structure, and function. Bioessays. 2018;40:e1800181. doi: 10.1002/bies.201800181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begolo D., Vincent I.M., Giordani F., Pohner I., Witty M.J., Rowan T.G., Bengaly Z., Gillingwater K., Freund Y., Wade R.C., et al. The trypanocidal benzoxaborole AN7973 inhibits trypanosome mRNA processing. PLoS Pathog. 2018;14:e1007315. doi: 10.1371/journal.ppat.1007315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behera R., Thomas S.M., Mensa-Wilmot K. New chemical scaffolds for human african trypanosomiasis lead discovery from a screen of tyrosine kinase inhibitor drugs. Antimicrob. Agents Chemother. 2014;58:2202–2210. doi: 10.1128/AAC.01691-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellini V., Swale C., Brenier-Pinchart M.P., Pezier T., Georgeault S., Laurent F., Hakimi M.A., Bougdour A. Target identification of an antimalarial oxaborole identifies AN13762 as an alternative chemotype for targeting CPSF3 in apicomplexan parasites. iScience. 2020;23:101871. doi: 10.1016/j.isci.2020.101871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz C., Dondelinger F., McKean P.G., Urbaniak M.D. Cell cycle synchronisation of Trypanosoma brucei by centrifugal counter-flow elutriation reveals the timing of nuclear and kinetoplast DNA replication. Sci. Rep. 2017;7:17599. doi: 10.1038/s41598-017-17779-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernacchia L., Paris A., Gupta A.R., Moores A.A., Kad N.M. Identification of the target and mode of action for the prokaryotic nucleotide excision repair inhibitor ATBC. Biosci. Rep. 2022;42 doi: 10.1042/BSR20220403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berriman M., Ghedin E., Hertz-Fowler C., Blandin G., Renauld H., Bartholomeu D.C., Lennard N.J., Caler E., Hamlin N.E., Haas B., et al. The genome of the african trypanosome trypanosoma brucei. Science. 2005;309:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- L Bolognesi M. Polypharmacology in a single drug: multitarget drugs. Curr. Med. Chem. 2013;20:1639–1645. doi: 10.2174/0929867311320130004. [DOI] [PubMed] [Google Scholar]
- Brehmer D., Greff Z., Godl K., Blencke S., Kurtenbach A., Weber M., Muller S., Klebl B., Cotten M., Keri G., et al. Cellular targets of gefitinib. Cancer Res. 2005;65:379–382. doi: 10.1158/0008-5472.379.65.2. [DOI] [PubMed] [Google Scholar]
- Buckner F.S., Buchynskyy A., Nagendar P., Patrick D.A., Gillespie J.R., Herbst Z., Tidwell R.R., Gelb M.H. Phenotypic drug discovery for human african trypanosomiasis: a powerful approach. Trop. Med. Infect. Dis. 2020;5:23. doi: 10.3390/tropicalmed5010023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkard G., Fragoso C.M., Roditi I. Highly efficient stable transformation of bloodstream forms of Trypanosoma brucei. Mol. Biochem. Parasitol. 2007;153:220–223. doi: 10.1016/j.molbiopara.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Burton N.R., Kim P., Backus K.M. Photoaffinity labelling strategies for mapping the small molecule-protein interactome. Org. Biomol. Chem. 2021;19:7792–7809. doi: 10.1039/d1ob01353j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capewell P., Cren-Travaille C., Marchesi F., Johnston P., Clucas C., Benson R.A., Gorman T.A., Calvo-Alvarez E., Crouzols A., Jouvion G., et al. The skin is a significant but overlooked anatomical reservoir for vector-borne African trypanosomes. Elife. 2016;5 doi: 10.7554/eLife.17716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carolino K., Winzeler E.A. The antimalarial resistome - finding new drug targets and their modes of action. Curr. Opin. Microbiol. 2020;57:49–55. doi: 10.1016/j.mib.2020.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldi M., Gaudino A., Lariccia V., Russo M., Amoroso S., di Renzo G., Annunziato L. Imatinib-mesylate blocks recombinant T-type calcium channels expressed in human embryonic kidney-293 cells by a protein tyrosine kinase-independent mechanism. J. Pharmacol. Exp. Ther. 2004;309:208–215. doi: 10.1124/jpet.103.061184. [DOI] [PubMed] [Google Scholar]
- Cerisier N., Petitjean M., Regad L., Bayard Q., Reau M., Badel A., Camproux A.C. High impact: the role of promiscuous binding sites in polypharmacology. Molecules. 2019;24:2529. doi: 10.3390/molecules24142529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatelain E., Ioset J.R. Phenotypic screening approaches for Chagas disease drug discovery. Expet Opin. Drug Discov. 2018;13:141–153. doi: 10.1080/17460441.2018.1417380. [DOI] [PubMed] [Google Scholar]
- Chen L., Shu Y., Liang X., Chen E.C., Yee S.W., Zur A.A., Li S., Xu L., Keshari K.R., Lin M.J., et al. OCT1 is a high-capacity thiamine transporter that regulates hepatic steatosis and is a target of metformin. Proc. Natl. Acad. Sci. USA. 2014;111:9983–9988. doi: 10.1073/pnas.1314939111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou D.H.C., Vetere A., Choudhary A., Scully S.S., Schenone M., Tang A., Gomez R., Burns S.M., Lundh M., Vital T., et al. Kinase-independent small-molecule inhibition of JAK-STAT signaling. J. Am. Chem. Soc. 2015;137:7929–7934. doi: 10.1021/jacs.5b04284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C.L., Tiwari A.K., Wu C.P., Su X.D., Wang S.R., Liu D.G., Ashby C.R., Jr., Huang Y., Robey R.W., Liang Y.J., et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008;68:7905–7914. doi: 10.1158/0008-5472.CAN-08-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M.I., Hunt J.P., Herrgard S., Ciceri P., Wodicka L.M., Pallares G., Hocker M., Treiber D.K., Zarrinkar P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011;29:1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Dean S., Sunter J., Wheeler R.J., Hodkinson I., Gluenz E., Gull K. A toolkit enabling efficient, scalable and reproducible gene tagging in trypanosomatids. Open Biol. 2015;5:140197. doi: 10.1098/rsob.140197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering T.L., Masterson W.J., Hart G.W., Englund P.T. Biosynthesis of glycosyl phosphatidylinositol membrane anchors. J. Biol. Chem. 1990;265:611–614. doi: 10.1016/s0021-9258(19)40092-6. [DOI] [PubMed] [Google Scholar]
- Doering T.L., Masterson W.J., Hart G.W., Englund P.T. Biosynthesis of glycosyl phosphatidylinositol membrane anchors. J. Biol. Chem. 1990;265:611–614. doi: 10.1016/s0021-9258(19)40092-6. [DOI] [PubMed] [Google Scholar]
- Dolloff N.G., Mayes P.A., Hart L.S., Dicker D.T., Humphreys R., El-Deiry W.S. Off-target lapatinib activity sensitizes colon cancer cells through TRAIL death receptor up-regulation. Sci. Transl. Med. 2011;3:86ra50. doi: 10.1126/scitranslmed.3001384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling R.J., Zakikhani M., Fantus I.G., Pollak M., Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67:10804–10812. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- Dunne G., Breen L., Collins D.M., Roche S., Clynes M., O'Connor R. Modulation of P-gp expression by lapatinib. Invest. N. Drugs. 2011;29:1284–1293. doi: 10.1007/s10637-010-9482-7. [DOI] [PubMed] [Google Scholar]
- Eder J., Sedrani R., Wiesmann C. The discovery of first-in-class drugs: origins and evolution. Nat. Rev. Drug Discov. 2014;13:577–587. doi: 10.1038/nrd4336. [DOI] [PubMed] [Google Scholar]
- Ege N., Bouguenina H., Tatari M., Chopra R. Phenotypic screening with target identification and validation in the discovery and development of E3 ligase modulators. Cell Chem. Biol. 2021;28:283–299. doi: 10.1016/j.chembiol.2021.02.011. [DOI] [PubMed] [Google Scholar]
- El-Sayed N.M., Myler P.J., Bartholomeu D.C., Nilsson D., Aggarwal G., Tran A.N., Ghedin E., Worthey E.A., Delcher A.L., Blandin G., et al. The genome sequence of trypanosoma cruzi, etiologic agent of chagas disease. Science. 2005;309:409–415. doi: 10.1126/science.1112631. [DOI] [PubMed] [Google Scholar]
- Eperon G., Balasegaram M., Potet J., Mowbray C., Valverde O., Chappuis F. Treatment options for second-stage gambiense human African trypanosomiasis. Expert Rev. Anti Infect. Ther. 2014;12:1407–1417. doi: 10.1586/14787210.2014.959496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairlamb A.H., Gow N.A.R., Matthews K.R., Waters A.P. Drug resistance in eukaryotic microorganisms. Nat. Microbiol. 2016;1:16092. doi: 10.1038/nmicrobiol.2016.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria J., Moraes C.B., Song R., Pascoalino B.S., Lee N., Siqueira-Neto J.L., Cruz D.J.M., Parkinson T., Ioset J.R., Cordeiro-da-Silva A., Freitas-Junior L.H. Drug discovery for human african trypanosomiasis: identification of novel scaffolds by the newly developed HTS SYBR green assay for trypanosoma brucei. J. Biomol. Screen. 2015;20:70–81. doi: 10.1177/1087057114556236. [DOI] [PubMed] [Google Scholar]
- Futaki S. Functional peptides that target biomembranes: design and modes of action. Chem. Pharm. Bull. (Tokyo) 2021;69:601–607. doi: 10.1248/cpb.c21-00140. [DOI] [PubMed] [Google Scholar]
- Givan S.A., Sprague G.F., Jr. The ankyrin repeat-containing protein Akr1p is required for the endocytosis of yeast pheromone receptors. Mol. Biol. Cell. 1997;8:1317–1327. doi: 10.1091/mbc.8.7.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldshmidt H., Matas D., Kabi A., Carmi S., Hope R., Michaeli S. Persistent ER stress induces the spliced leader RNA silencing pathway (SLS), leading to programmed cell death in Trypanosoma brucei. PLoS Pathog. 2010;6:e1000731. doi: 10.1371/journal.ppat.1000731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A.K., Foley K.A., Versteeg S.G. New antifungal agents and new formulations against dermatophytes. Mycopathologia. 2017;182:127–141. doi: 10.1007/s11046-016-0045-0. [DOI] [PubMed] [Google Scholar]
- Guyett P.J., Behera R., Ogata Y., Pollastri M., Mensa-Wilmot K. Novel effects of lapatinib revealed in the african trypanosome by using hypothesis-generating proteomics and chemical biology strategies. Antimicrob. Agents Chemother. 2017;61 doi: 10.1128/AAC.01865-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyett P.J., Xia S., Swinney D.C., Pollastri M.P., Mensa-Wilmot K. Glycogen synthase kinase 3β promotes the endocytosis of transferrin in the african trypanosome. ACS Infect. Dis. 2016;2:518–528. doi: 10.1021/acsinfecdis.6b00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman E.A. Regulated cell death signaling pathways and marine natural products that target them. Mar. Drugs. 2019;17:76. doi: 10.3390/md17020076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M., Mills C.E., Subramanian K., Chen C., Chung M., Boswell S.A., Everley R.A., Liu C., Walmsley C.S., Juric D., Sorger P.K. Multiomics profiling establishes the polypharmacology of FDA-approved CDK4/6 inhibitors and the potential for differential clinical activity. Cell Chem. Biol. 2019;26:1067–1080.e8. doi: 10.1016/j.chembiol.2019.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao G., Li H., Yang F., Dong D., Li Z., Ding Y., Pan W., Wang E., Liu R., Zhou H. Discovery of benzhydrol-oxaborole derivatives as Streptococcus pneumoniae leucyl-tRNA synthetase inhibitors. Bioorg. Med. Chem. 2021;29:115871. doi: 10.1016/j.bmc.2020.115871. [DOI] [PubMed] [Google Scholar]
- Hao H., Zhang D., Shi J., Wang Y., Chen L., Guo Y., Ma J., Jiang X., Jiang H. Sorafenib induces autophagic cell death and apoptosis in hepatic stellate cell through the JNK and Akt signaling pathways. Anticancer Drugs. 2016;27:192–203. doi: 10.1097/CAD.0000000000000316. [DOI] [PubMed] [Google Scholar]
- Hartmann S., Nusbaum D.J., Kim K., Alameh S., Ho C.L.C., Cruz R.L., Levitin A., Bradley K.A., Martchenko M. Role of a small molecule in the modulation of cell death signal transduction pathways. ACS Infect. Dis. 2018;4:1746–1754. doi: 10.1021/acsinfecdis.8b00231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez V., Crepin T., Palencia A., Cusack S., Akama T., Baker S.J., Bu W., Feng L., Freund Y.R., Liu L., et al. Discovery of a novel class of boron-based antibacterials with activity against gram-negative bacteria. Antimicrob. Agents Chemother. 2013;57:1394–1403. doi: 10.1128/AAC.02058-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano Y., Murata S., Tanaka K., Shimizu M., Sato R. Sterol regulatory element-binding proteins are negatively regulated through SUMO-1 modification independent of the ubiquitin/26 S proteasome pathway. J. Biol. Chem. 2003;278:16809–16819. doi: 10.1074/jbc.M212448200. [DOI] [PubMed] [Google Scholar]
- Hirumi H., Hirumi K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 1989;75:985–989. doi: 10.2307/3282883. [DOI] [PubMed] [Google Scholar]
- Horn D. Genome-scale RNAi screens in African trypanosomes. Trends Parasitol. 2022;38:160–173. doi: 10.1016/j.pt.2021.09.002. [DOI] [PubMed] [Google Scholar]
- Hunter S., Apweiler R., Attwood T.K., Bairoch A., Bateman A., Binns D., Bork P., Das U., Daugherty L., Duquenne L., et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37:D211–D215. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs R.T., Nare B., Wring S.A., Orr M.D., Chen D., Sligar J.M., Jenks M.X., Noe R.A., Bowling T.S., Mercer L.T., et al. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human african trypanosomiasis. PLoS Negl. Trop. Dis. 2011;5:e1151. doi: 10.1371/journal.pntd.0001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs R.T., Plattner J.J., Nare B., Wring S.A., Chen D., Freund Y., Gaukel E.G., Orr M.D., Perales J.B., Jenks M., et al. Benzoxaboroles: a new class of potential drugs for human African trypanosomiasis. Future Med. Chem. 2011;3:1259–1278. doi: 10.4155/fmc.11.80. [DOI] [PubMed] [Google Scholar]
- Jia Z., Jia Y., Liu B., Zhao Z., Jia Q., Liang H., Zhang H. Genistein inhibits voltage-gated sodium currents in SCG neurons through protein tyrosine kinase-dependent and kinase-independent mechanisms. Pflugers Arch. 2008;456:857–866. doi: 10.1007/s00424-008-0444-2. [DOI] [PubMed] [Google Scholar]
- Jones D.C., Foth B.J., Urbaniak M.D., Patterson S., Ong H.B., Berriman M., Fairlamb A.H. Genomic and proteomic studies on the mode of action of oxaboroles against the african trypanosome. PLoS Negl. Trop. Dis. 2015;9:e0004299. doi: 10.1371/journal.pntd.0004299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kall L., Storey J.D., Noble W.S. Non-parametric estimation of posterior error probabilities associated with peptides identified by tandem mass spectrometry. Bioinformatics. 2008;24:i42–i48. doi: 10.1093/bioinformatics/btn294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kall L., Storey J.D., Noble W.S. QVALITY: non-parametric estimation of q-values and posterior error probabilities. Bioinformatics. 2009;25:964–966. doi: 10.1093/bioinformatics/btp021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaman M.W., Herrgard S., Treiber D.K., Gallant P., Atteridge C.E., Campbell B.T., Chan K.W., Ciceri P., Davis M.I., Edeen P.T., et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotech. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Katiyar S., Kufareva I., Behera R., Thomas S.M., Ogata Y., Pollastri M., Abagyan R., Mensa-Wilmot K. Lapatinib-binding protein kinases in the african trypanosome: identification of cellular targets for kinase-directed chemical scaffolds. PLoS One. 2013;8:e56150. doi: 10.1371/journal.pone.0056150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur J., Bhardwaj A., Huang Z., Knaus E.E. Aspirin analogues as dual cyclooxygenase-2/5-lipoxygenase inhibitors: synthesis, nitric oxide release, molecular modeling, and biological evaluation as anti-inflammatory agents. ChemMedChem. 2012;7:144–150. doi: 10.1002/cmdc.201100460. [DOI] [PubMed] [Google Scholar]
- Kennedy C.R., Goya Grocin A., Kovacic T., Singh R., Ward J.A., Shenoy A.R., Tate E.W. A probe for NLRP3 inflammasome inhibitor MCC950 identifies carbonic anhydrase 2 as a novel target. ACS Chem. Biol. 2021;16:982–990. doi: 10.1021/acschembio.1c00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkut A., Wang W., Demir E., Aksoy B.A., Jing X., Molinelli E.J., Babur O., Bemis D.L., Onur Sumer S., Solit D.B., et al. Perturbation biology nominates upstream-downstream drug combinations in RAF inhibitor resistant melanoma cells. Elife. 2015;4 doi: 10.7554/eLife.04640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourbeli V., Chontzopoulou E., Moschovou K., Pavlos D., Mavromoustakos T., Papanastasiou I.P. An overview on target-based drug design against kinetoplastid Protozoan infections: human african trypanosomiasis, chagas disease and leishmaniases. Molecules. 2021;26:4629. doi: 10.3390/molecules26154629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota K., Funabashi M., Ogura Y. Target deconvolution from phenotype-based drug discovery by using chemical proteomics approaches. Biochim. Biophys. Acta Proteins Proteom. 2019;1867:22–27. doi: 10.1016/j.bbapap.2018.08.002. [DOI] [PubMed] [Google Scholar]
- Lackey K.E. Lessons from the drug discovery of lapatinib, a dual ErbB1/2 tyrosine kinase inhibitor. Curr. Top. Med. Chem. 2006;6:435–460. doi: 10.2174/156802606776743156. [DOI] [PubMed] [Google Scholar]
- Larsson O., Morita M., Topisirovic I., Alain T., Blouin M.J., Pollak M., Sonenberg N. Distinct perturbation of the translatome by the antidiabetic drug metformin. Proc. Natl. Acad. Sci. USA. 2012;109:8977–8982. doi: 10.1073/pnas.1201689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A., Giuliano C.J., Palladino A., John K.M., Abramowicz C., Yuan M.L., Sausville E.L., Lukow D.A., Liu L., Chait A.R., et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aaw8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W.C., Hsu F.S., Kuo K.L., Liu S.H., Shun C.T., Shi C.S., Chang H.C., Tsai Y.C., Lin M.C., Wu J.T., et al. Trichostatin A, a histone deacetylase inhibitor, induces synergistic cytotoxicity with chemotherapy via suppression of Raf/MEK/ERK pathway in urothelial carcinoma. J. Mol. Med. (Berl.) 2018;96:1307–1318. doi: 10.1007/s00109-018-1697-7. [DOI] [PubMed] [Google Scholar]
- Love M.S., McNamara C.W. Phenotypic screening techniques for Cryptosporidium drug discovery. Expet Opin. Drug Discov. 2021;16:59–74. doi: 10.1080/17460441.2020.1812577. [DOI] [PubMed] [Google Scholar]
- Lytton S.D., Mester B., Dayan I., Glickstein H., Libman J., Shanzer A., Cabantchik Z.I. Mode of action of iron (III) chelators as antimalarials: I. Membrane permeation properties and cytotoxic activity. Blood. 1993;81:214–221. doi: 10.1182/blood.v81.1.214.bloodjournal811214. [DOI] [PubMed] [Google Scholar]
- Ma K., Chen W., Yan S.Q., Liu Z.Z., Lin X.Q., Zhang J.B., Gao Y., Wang T., Zhang J.G., Yang Y.J. Purification, characterization, mode of action, and application of jileicin, a novel antimicrobial from paenibacillus jilinensis YPG26. J. Agric. Food Chem. 2022;70:5570–5578. doi: 10.1021/acs.jafc.2c01458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madiraju A.K., Erion D.M., Rahimi Y., Zhang X.M., Braddock D.T., Albright R.A., Prigaro B.J., Wood J.L., Bhanot S., MacDonald M.J., et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510:542–546. doi: 10.1038/nature13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrid A.S., Mancuso J., Cande W.Z., Weis K. The role of the integral membrane nucleoporins Ndc1p and Pom152p in nuclear pore complex assembly and function. J. Cell Biol. 2006;173:361–371. doi: 10.1083/jcb.200506199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maric M., Mukherjee P., Tatham M.H., Hay R., Labib K. Ufd1-Npl4 recruit Cdc48 for disassembly of ubiquitylated CMG helicase at the end of chromosome replication. Cell Rep. 2017;18:3033–3042. doi: 10.1016/j.celrep.2017.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLatchie A.P., Burrell-Saward H., Myburgh E., Lewis M.D., Ward T.H., Mottram J.C., Croft S.L., Kelly J.M., Taylor M.C. Highly sensitive in vivo imaging of Trypanosoma brucei expressing "red-shifted" luciferase. PLoS Negl. Trop. Dis. 2013;7:e2571. doi: 10.1371/journal.pntd.0002571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mensa-Wilmot K. How physiological targets can Be distinguished from drug-binding proteins. Mol. Pharmacol. 2021;100:1–6. doi: 10.1124/molpharm.120.000186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesu V.K.B.K., Kalonji W.M., Bardonneau C., Mordt O.V., Blesson S., Simon F., Delhomme S., Bernhard S., Kuziena W., Lubaki J.P.F., et al. Oral fexinidazole for late-stage African Trypanosoma brucei gambiense trypanosomiasis: a pivotal multicentre, randomised, non-inferiority trial. Lancet. 2018;391:144–154. doi: 10.1016/S0140-6736(17)32758-7. [DOI] [PubMed] [Google Scholar]
- Meyer K.J., Shapiro T.A. Cytosolic and mitochondrial Hsp90 in cytokinesis, mitochondrial DNA replication, and drug action in trypanosoma brucei. Antimicrob. Agents Chemother. 2021;65:e0063221. doi: 10.1128/AAC.00632-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugge F.L.B., Silva A.M. Aspirin metabolite sodium salicylate selectively inhibits transcriptional activity of ATF6α and downstream target genes. Sci. Rep. 2017;7:9190. doi: 10.1038/s41598-017-09500-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam H.J., Kim Y.E., Moon B.S., Kim H.Y., Jung D., Choi S., Jang J.W., Nam D.H., Cho H. Azathioprine antagonizes aberrantly elevated lipid metabolism and induces apoptosis in glioblastoma. iScience. 2021;24:102238. doi: 10.1016/j.isci.2021.102238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nare B., Wring S., Bacchi C., Beaudet B., Bowling T., Brun R., Chen D., Ding C., Freund Y., Gaukel E., et al. Discovery of novel orally bioavailable oxaborole 6-carboxamides that demonstrate cure in a murine model of 8late stage central nervous system African trypanosomiasis. Antimicrob. Agents Chemother. 2010 doi: 10.1128/AAC.00498-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes D.C.d.O.S., Costa M.S., Bispo-da-Silva L.B., Ferro E.A.V., Zoia M.A.P., Goulart L.R., Rodrigues R.S., Rodrigues V.d.M., Yoneyama K.A.G. Mitochondrial dysfunction on Leishmania (Leishmania) amazonensis induced by ketoconazole: insights into drug mode of action. Mem. Inst. Oswaldo Cruz. 2022;117:e210157. doi: 10.1590/0074-02760210157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyman E., Stein R.R., Jing X., Wang W., Marks B., Zervantonakis I.K., Korkut A., Gauthier N.P., Sander C. Perturbation biology links temporal protein changes to drug responses in a melanoma cell line. PLoS Comput. Biol. 2020;16:e1007909. doi: 10.1371/journal.pcbi.1007909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hare T., Eide C.A., Agarwal A., Adrian L.T., Zabriskie M.S., Mackenzie R.J., Latocha D.H., Johnson K.J., You H., Luo J., et al. Threshold levels of ABL tyrosine kinase inhibitors retained in chronic myeloid leukemia cells define commitment to apoptosis. Cancer Res. 2013 doi: 10.1158/0008-5472.CAN-12-3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberholzer M., Morand S., Kunz S., Seebeck T. A vector series for rapid PCR-mediated C-terminal in situ tagging of Trypanosoma brucei genes. Mol. Biochem. Parasitol. 2006;145:117–120. doi: 10.1016/j.molbiopara.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Ooi C.P., Rudenko G. How to create coats for all seasons: elucidating antigenic variation in African trypanosomes. Emerg. Top. Life Sci. 2017;1:593–600. doi: 10.1042/ETLS20170105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsburn B.C. Proteome discoverer-A community enhanced data processing suite for protein informatics. Proteomes. 2021;9:15. doi: 10.3390/proteomes9010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palencia A., Li X., Bu W., Choi W., Ding C.Z., Easom E.E., Feng L., Hernandez V., Houston P., Liu L., et al. Discovery of novel oral protein synthesis inhibitors of Mycobacterium tuberculosis that target leucyl-tRNA synthetase. Antimicrob. Agents Chemother. 2016;60:6271–6280. doi: 10.1128/AAC.01339-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomba A., Abbondio M., Fiorito G., Uzzau S., Pagnozzi D., Tanca A. Comparative evaluation of MaxQuant and proteome discoverer MS1-based protein quantification tools. J. Proteome Res. 2021;20:3497–3507. doi: 10.1021/acs.jproteome.1c00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit S.G., Mekala K.P.R., Puttananjaiah M.H., Peddha M.S., Dhale M.A. Dual mode of action of talaromyces purpureogenus CFRM02 pigment to ameliorate alcohol induced liver toxicity in rats. Appl. Biochem. Biotechnol. 2022;194:4258–4265. doi: 10.1007/s12010-022-03973-x. [DOI] [PubMed] [Google Scholar]
- Panka D.J., Wang W., Atkins M.B., Mier J.W. The Raf inhibitor BAY 43-9006 (Sorafenib) induces caspase-independent apoptosis in melanoma cells. Cancer Res. 2006;66:1611–1619. doi: 10.1158/0008-5472.can-05-0808. [DOI] [PubMed] [Google Scholar]
- Patel G., Karver C.E., Behera R., Guyett P.J., Sullenberger C., Edwards P., Roncal N.E., Mensa-Wilmot K., Pollastri M.P. Kinase scaffold repurposing for neglected disease drug discovery: discovery of an efficacious, lapatanib-derived lead compound for trypanosomiasis. J. Med. Chem. 2013;56:3820–3832. doi: 10.1021/jm400349k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelfrene E., Harvey Allchurch M., Ntamabyaliro N., Nambasa V., Ventura F.V., Nagercoil N., Cavaleri M. The European Medicines Agency's scientific opinion on oral fexinidazole for human African trypanosomiasis. PLoS Negl. Trop. Dis. 2019;13:e0007381. doi: 10.1371/journal.pntd.0007381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y., Bai J., Bandla C., Garcia-Seisdedos D., Hewapathirana S., Kamatchinathan S., Kundu D.J., Prakash A., Frericks-Zipper A., Eisenacher M., et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022;50:D543–D552. doi: 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon S.K., Peacock L., Gibson W., Gull K., Kelly S. A modular and optimized single marker system for generating Trypanosoma brucei cell lines expressing T7 RNA polymerase and the tetracycline repressor. Open Biol. 2012;2:110037. doi: 10.1098/rsob.110037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proschak E., Stark H., Merk D. Polypharmacology by design: a medicinal chemist's perspective on multitargeting compounds. J. Med. Chem. 2019;62:420–444. doi: 10.1021/acs.jmedchem.8b00760. [DOI] [PubMed] [Google Scholar]
- Ridgley E.L., Xiong Z.H., Ruben L. Reactive oxygen species activate a Ca2+-dependent cell death pathway in the unicellular organism Trypanosoma brucei brucei. Biochem. J. 1999;340:33–40. doi: 10.1042/bj3400033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson J.G. Enzymes as a special class of therapeutic target: clinical drugs and modes of action. Curr. Opin. Struct. Biol. 2007;17:674–679. doi: 10.1016/j.sbi.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Sanz-Rodriguez C.E., Hoffman B., Guyett P.J., Purmal A., Singh B., Pollastri M.P., Mensa-Wilmot K. Physiologic targets and modes of action for CBL0137, a lead for human african trypanosomiasis drug development. Mol. Pharmacol. 2022;102:1–16. doi: 10.1124/molpharm.121.000430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M. Phenotypic screening using large‑scale genomic libraries to identify drug targets for the treatment of cancer (Review) Oncol. Lett. 2020;19:3617–3626. doi: 10.3892/ol.2020.11512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer A.B., Wenzel M. A how-to guide for mode of action analysis of antimicrobial peptides. Front. Cell. Infect. Microbiol. 2020;10:540898. doi: 10.3389/fcimb.2020.540898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singaraja R.R., Hadano S., Metzler M., Givan S., Wellington C.L., Warby S., Yanai A., Gutekunst C.A., Leavitt B.R., Yi H., et al. HIP14, a novel ankyrin domain-containing protein, links huntingtin to intracellular trafficking and endocytosis. Hum. Mol. Genet. 2002;11:2815–2828. doi: 10.1093/hmg/11.23.2815. [DOI] [PubMed] [Google Scholar]
- Srivastava A., Badjatia N., Lee J.H., Hao B., Gunzl A. An RNA polymerase II-associated TFIIF-like complex is indispensable for SL RNA gene transcription in Trypanosoma brucei. Nucleic Acids Res. 2018;46:1695–1709. doi: 10.1093/nar/gkx1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steketee P.C., Vincent I.M., Achcar F., Giordani F., Kim D.H., Creek D.J., Freund Y., Jacobs R., Rattigan K., Horn D., et al. Benzoxaborole treatment perturbs S-adenosyl-L-methionine metabolism in Trypanosoma brucei. PLoS Negl. Trop. Dis. 2018;12:e0006450. doi: 10.1371/journal.pntd.0006450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suenaga H., Kagaya N., Kawada M., Tatsuda D., Sato T., Shin-Ya K. Phenotypic screening system using three-dimensional (3D) culture models for natural product screening. J. Antibiot. (Tokyo) 2021;74:660–666. doi: 10.1038/s41429-021-00457-8. [DOI] [PubMed] [Google Scholar]
- Sullenberger C., Pique D., Ogata Y., Mensa-Wilmot K. AEE788 inhibits basal body assembly and blocks DNA replication in the african trypanosome. Mol. Pharmacol. 2017;91:482–498. doi: 10.1124/mol.116.106906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swalley S.E. Expanding therapeutic opportunities for neurodegenerative diseases: a perspective on the important role of phenotypic screening. Bioorg. Med. Chem. 2020;28:115239. doi: 10.1016/j.bmc.2019.115239. [DOI] [PubMed] [Google Scholar]
- Swinney D.C. Phenotypic vs. target-based drug discovery for first-in-class medicines. Clin. Pharmacol. Ther. 2013;93:299–301. doi: 10.1038/clpt.2012.236. [DOI] [PubMed] [Google Scholar]
- Swinney D.C., Lee J.A. Recent advances in phenotypic drug discovery. F1000Res. 2020;9 doi: 10.12688/f1000research.25813.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C., Luo H., Luo D., Yang H., Zhou X. Src homology phosphotyrosyl phosphatase 2 mediates cisplatin-related drug resistance by inhibiting apoptosis and activating the Ras/PI3K/Akt1/survivin pathway in lung cancer cells. Oncol. Rep. 2018;39:611–618. doi: 10.3892/or.2017.6109. [DOI] [PubMed] [Google Scholar]
- Tarral A., Blesson S., Mordt O.V., Torreele E., Sassella D., Bray M.A., Hovsepian L., Evene E., Gualano V., Felices M., Strub-Wourgaft N. Determination of an optimal dosing regimen for fexinidazole, a novel oral drug for the treatment of human african trypanosomiasis: first-in-human studies. Clin. Pharmacokinet. 2014;53:565–580. doi: 10.1007/s40262-014-0136-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S.M., Purmal A., Pollastri M., Mensa-Wilmot K. Discovery of a carbazole-derived lead drug for human African trypanosomiasis. Sci. Rep. 2016;6:32083. doi: 10.1038/srep32083. https://www.nature.com/articles/srep32083#supplementary-information [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trindade S., Rijo-Ferreira F., Carvalho T., Pinto-Neves D., Guegan F., Aresta-Branco F., Bento F., Young S.A., Pinto A., Van Den Abbeele J., et al. Trypanosoma brucei parasites occupy and functionally adapt to the adipose tissue in mice. Cell Host Microbe. 2016;19:837–848. doi: 10.1016/j.chom.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulloch L.B., Menzies S.K., Coron R.P., Roberts M.D., Florence G.J., Smith T.K. Direct and indirect approaches to identify drug modes of action. IUBMB Life. 2018;70:9–22. doi: 10.1002/iub.1697. [DOI] [PubMed] [Google Scholar]
- Vanhollebeke B., De Muylder G., Nielsen M.J., Pays A., Tebabi P., Dieu M., Raes M., Moestrup S.K., Pays E. A haptoglobin-hemoglobin receptor conveys innate immunity to Trypanosoma brucei in humans. Science. 2008;320:677–681. doi: 10.1126/science.1156296. [DOI] [PubMed] [Google Scholar]
- Venkatesan P., Bhutia S.K., Singh A.K., Das S.K., Dash R., Chaudhury K., Sarkar D., Fisher P.B., Mandal M. AEE788 potentiates celecoxib-induced growth inhibition and apoptosis in human colon cancer cells. Life Sci. 2012;91:789–799. doi: 10.1016/j.lfs.2012.08.024. [DOI] [PubMed] [Google Scholar]
- Vennemann A., Hofmann T.G. SUMO regulates proteasome-dependent degradation of FLASH/Casp8AP2. Cell Cycle. 2013;12:1914–1921. doi: 10.4161/cc.24943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter J.C., Smith H.O., Adams M.D. The sequence of the human genome. Clin. Chem. 2015;61:1207–1208. doi: 10.1373/clinchem.2014.237016. [DOI] [PubMed] [Google Scholar]
- Verma N.K., Singh G., Dey C.S. Miltefosine induces apoptosis in arsenite-resistant Leishmania donovani promastigotes through mitochondrial dysfunction. Exp. Parasitol. 2007;116:1–13. doi: 10.1016/j.exppara.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Wall R.J., Rico E., Lukac I., Zuccotto F., Elg S., Gilbert I.H., Freund Y., Alley M.R.K., Field M.C., Wyllie S., Horn D. Clinical and veterinary trypanocidal benzoxaboroles target CPSF3. Proc. Natl. Acad. Sci. USA. 2018;115:9616–9621. doi: 10.1073/pnas.1807915115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Zhang C.J., Zhang J., He Y., Lee Y.M., Chen S., Lim T.K., Ng S., Shen H.M., Lin Q. Mapping sites of aspirin-induced acetylations in live cells by quantitative acid-cleavable activity-based protein profiling (QA-ABPP) Sci. Rep. 2015;5:7896. doi: 10.1038/srep07896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.E., Hunt C.R., Chen J., Taylor J.S. Biotinylated quercetin as an intrinsic photoaffinity proteomics probe for the identification of quercetin target proteins. Bioorg. Med. Chem. 2011;19:4710–4720. doi: 10.1016/j.bmc.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Yao B., Wang Y., Zhang M., Fu S., Gao H., Peng R., Zhang L., Tang J. Increased FoxM1 expression is a target for metformin in the suppression of EMT in prostate cancer. Int. J. Mol. Med. 2014;33:1514–1522. doi: 10.3892/ijmm.2014.1707. [DOI] [PubMed] [Google Scholar]
- Win S., Than T.A., Kaplowitz N. The regulation of JNK signaling pathways in cell death through the interplay with mitochondrial SAB and upstream post-translational effects. Int. J. Mol. Sci. 2018;19:3657. doi: 10.3390/ijms19113657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wring S., Gaukel E., Nare B., Jacobs R., Beaudet B., Bowling T., Mercer L., Bacchi C., Yarlett N., Randolph R., et al. Pharmacokinetics and pharmacodynamics utilizing unbound target tissue exposure as part of a disposition-based rationale for lead optimization of benzoxaboroles in the treatment of Stage 2 Human African Trypanosomiasis. Parasitology. 2014;141:104–118. doi: 10.1017/S003118201300098X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin M.J., Yamamoto Y., Gaynor R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of IκB kinase-β. Nature. 1998;396:77–80. doi: 10.1038/23948. [DOI] [PubMed] [Google Scholar]
- Zuhl A.M., Nolan C.E., Brodney M.A., Niessen S., Atchison K., Houle C., Karanian D.A., Ambroise C., Brulet J.W., Beck E.M., et al. Chemoproteomic profiling reveals that cathepsin D off-target activity drives ocular toxicity of beta-secretase inhibitors. Nat. Commun. 2016;7:13042. doi: 10.1038/ncomms13042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request. Mass spectrometry proteomics data have been deposited at the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2022) partner repository with the dataset identifier PXD036393 and 10.6019/PXD036393. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.