

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest of all cancer types with a constant rise in global incidence. Therefore, better understanding of PDAC biology, in order to design more efficient diagnostic and treatment modalities, is a priority.
Here we found that the expression levels of cystathionine β-synthase (CBS), a transsulfuration enzyme, is markedly elevated in metastatic PDAC cells compared to cell lines isolated from non-metastatic primary tumors. On human immunohistochemical samples from PDAC patients we also found higher CBS staining in cancerous ductal cells compared to in non-tumor tissue, which was further elevated in the lymph node metastasis of the same patients. In mice, orthotopically injected CBS-silenced T3M4 cells induced fewer liver metastases compared to control cells indicating important roles for CBS in PDAC cancer cell invasion and malignant transformation. Wound healing and colony formation assays in cell culture confirmed that CBS-deficient metastatic T3M4 and non-metastatic BxPC3 primary tumor cells migrate slower and have impaired anchorage-independent growth capacities compared to control T3M4 cells. CBS silencing in T3M4 cells lowered WNT5a and SNAI1 gene expression down to levels that were observed in BxPC3 cells as well as resulted in an increase in E-cadherin and a decrease in Vimentin signals in mouse tumors when injected orthotopically. These observations suggested a primary role for the epithelial to mesenchymal transformation of cancer cells in CBS-mediated tumor aggressiveness. Under normal conditions, STAT3, an upstream regulator of Wnt signaling pathways, was less phosphorylated and more oxidized in shCBS T3M4 and BxPC3 compared to control T3M4 cells, which is consistent with decreased transcriptional activity at lower CBS levels due to less protection against oxidation. Sulfur metabolome analyses suggested that this CBS-mediated protection against oxidative modifications is likely to be related to persulfide/sulfide producing activities of the enzyme rather than its canonical function to produce cystathionine for cysteine synthesis. Taken together, CBS overexpression through regulation of the EMT plays a significant role in PDAC cancer cell invasion and metastasis.
Keywords: Hydrogen sulfide, Persulfide, Pancreatic ductal adenocarcinoma, Cystathionine β-synthase, Epithelial-to-mesenchymal transition
Graphical abstract
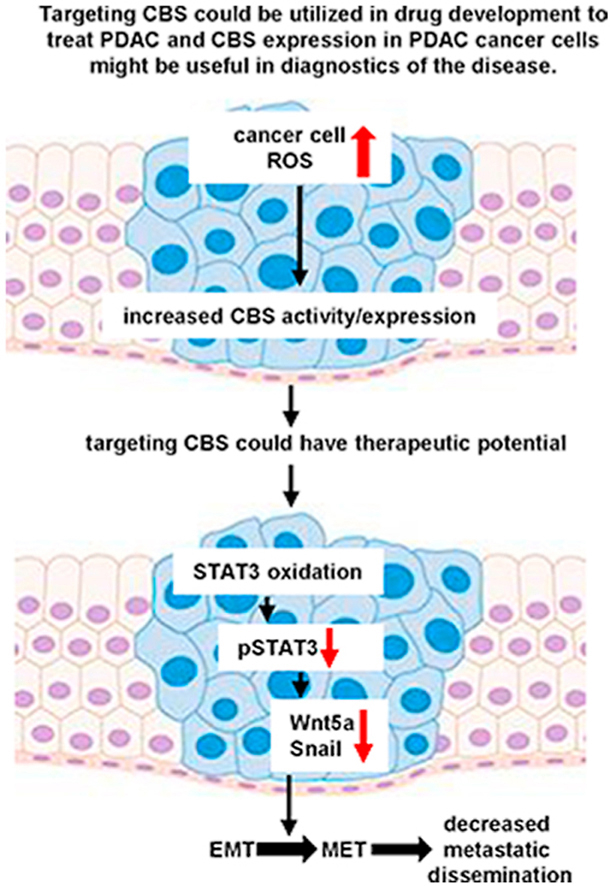
Highlights
-
•
CBS is involved in metastatic dissemination of PDAC.
-
•
CBS is important in PDAC cancer cell invasion and malignant transformation.
-
•
CBS overexpression is a key player in PDAC cancer cell invasion through regulation of epithelial-to-mesenchymal transition.
-
•
CBS protects STAT3 from oxidation induced loss of transcriptional activity.
-
•
CBS mediated cysteine persulfidation may contribute to protection against oxidative inactivation of STAT3.
List of abbreviations
- PC
pancreatic cancer
- EMT
epithelial-to-mesenchymal transition
- Fzd2
Frizzled-2 protein
- STAT3
signal transducer and activator of transcription 3
- CBS
cystathionine β-synthetase
- CSE
cystathionine γ-Lyase
- PDAC
pancreatic ductal adenocarcinoma
- BLBC
Basal like Breast Cancer
- HPE-IAM
β-(4-hydroxyphenyl) ethyl iodoacetamide
- SNAI1
Snail Family Transcriptional Repressor 1
- WNT5a
Recombinant protein of human wingless-type MMTV integration site family, member 5A
- FBS
fetal bovine serum
- DMSO
Dimethyl Sulfoxide
- shRNA
Short hairpin RNA
- BSA
Bovine Serum Albumine
- TBST
Tris Buffered Saline with Tween 20
- GAPDH
Glyceraldehyde-3-phosphate dehyrogenase
- IgG
Immunoglobulin G
- RNA
Ribonucleic acid
- DNase I
Deoxyribonuclease 1
- EDTA
Ethylenediaminetetraacetic Acid Tetrasodium Salt
- cDNA
Complementary DNA
- DNA
Deoxyribonucleic acid
- PCR
polymerase chain reaction
- mRNA
messenger RNA
- DTT
Dithiothreitol
- LC-MS/MS
Liquid Chromatography with Mass Spectrometry
- PBS
Phosphate Buffered Saline
- RT
Room Temperature
- RIPA
Radioimmunoprecipitation Assay Buffer
- TCA
Trichloroacetic acid
- SDS
Sodium Dodecyl Sulfate
- MS/MS
Tandem mass spectrometry
- MET
Mesenchymal-to-epithelial transition
- SDS-PAGE
Sodium dodecyl-sulfate polyacrylamide gel electrophoresis
- H&E
hematoxylin&eosin
- RT-qPCR
real-time PCR
1. Introduction
The overall survival of pancreatic cancer (PC) patients is among the lowest of all cancer types. This poor prognosis is associated with outstanding aggressiveness, lack of typical symptoms and a consequent high degree of late diagnosis, which together with the unfavorable anatomical location result in a low resection rate and a high recurrence rate. In addition, there are no effective targeted or immunotherapy drugs or radiotherapy interventions available to treat PC. On average, 80% of patients at diagnosis already have local or distant metastases [1,2]. The early event of metastatic dissemination is mainly attributed to an evolutionary biological process called epithelial-to-mesenchymal transition (EMT). EMT is associated with improvement in cell migratory and invasive properties, which involves the disruption of cell–cell adhesion and cellular polarity, remodeling of the cytoskeleton and changes in cell–matrix adhesion [3].
Wnt signaling pathways are generally viewed as major drivers of EMT. Wnt proteins constitute a large family of secreted lipid-modified glycoproteins. These proteins are implicated in a variety of cellular processes, such as proliferation, apoptosis, differentiation, and migration [4]. Each family member exhibits unique expression patterns and distinct biological functions. Wnt signaling can be broadly divided into two categories: the canonical, β-catenin-dependent pathway and the non-canonical β-catenin-independent pathway [5]. Wnt-5a has been identified as a non-canonical Wnt protein and as the cognate ligand for the Fzd2 receptor. A crucial role for Wnt-5a in cancer progression and metastasis via contributing to cancer cell migration and invasion is well documented [6,7].
Wnt-induced activation of Fzd2 was associated with SNAIL gene expression to produce the Snail protein, a prominent EMT activator, which strongly represses E-cadherin (epithelial marker) and provoke vimentin (mesenchymal marker) expression. This E-cadherin/vimentin expression pattern was associated with poor clinical outcome; therefore, E-cadherin repressors and vimentin inducers are regarded as markers of malignancy [[8], [9], [10]].
Fzd2-dependent EMT and cell migration involves STAT3 activation via phosphorylation on Tyr705. Moreover, Fzd2 and STAT3 physically associate, most likely in a complex involving additional proteins [11].
More importantly, STAT3 is an upstream transcriptional regulator of Wnt signaling. It is considered as an oncoprotein and its constitutive activation is described in a variety of human cancers. STAT3 is redox-regulated, its transcriptional activity is inhibited through reversible oxidation of conserved cysteine residues at the STAT3 DNA binding domain [12,13]. Both S-glutathionylation and S-nitrosylation were reported to impair STAT3 phosphorylation and down-modulate STAT3 signaling pathways [13,14]. Redox regulation also occurs via reversible induction of inter- and intra-molecular disulfide linkages among a number of STAT3 Cys residues through a redox relay system involving peroxiredoxin-2 and thioredoxin-1. The resultant disulfide-linked STAT3 oligomers and monomers having extra intramolecular disulfide bridges also attenuate transcriptional activity [15,16].
Extensive rewiring of cellular metabolism has emerged as a hallmark of cancer [17]. Adaptation to the microenvironment and oncogenes are key drivers of metabolic mechanisms that drive tumor progression via providing nutrient support and mitigating cellular stress. Pancreatic cancer cells have an outstanding adaptation capacity to strive under stress conditions, however the underlying molecular mechanisms of these processes are scarcely understood. Here we provide evidence that realignment of the transsulfuration pathway contributes to the malignancy of PC cells via promoting the EMT process. The transsulfuration pathway can contribute to de novo cysteine synthesis in order to promote cell survival upon extracellular cysteine deprivation [18]. In recent years it was demonstrated that one of the key enzymes in the transsulfuration pathway, cystathionine β-synthetase (CBS), plays an important role in the protecting functions of cancer cells from oxidative insults. Increased CBS expression in cancer was first demonstrated in patients undergoing colorectal tumor resection. Higher expression of CBS was observed in the tumors compared to the surrounding normal tissue [19]. In our previous study comprehensive proteomic analyses revealed a rewired Cys metabolic profile with a significant increase in CBS expression in the most aggressive Basal like Breast Cancer (BLBC) subtype compared to other breast cancer subtypes, which we proved to play an integral role in the progression of BLBC [20]. In their recent review, Kelly Ascencao and Csaba Szabó summarize preclinical and clinical data underlying the pathogenetic roles of CBS in several cancer types [21].
CBS has a number of different enzymatic activities. The canonical activity of the enzyme is to convert serine and homocysteine into cystathionine, which is utilized by Cystathionine γ-Lyase (CSE) to produce cysteine [22] (Scheme 1, reaction 1). Furthermore, CBS was shown to be a key enzyme in the production of hydrogen sulfide, a small signaling molecule, via reverse transsulfuration pathways (Scheme 1, reaction 2 and 3) [23] as well as it can utilize cystine (the disulfide of cysteine) to produce cysteine persulfide and induce persulfidation on protein cysteine residues via transpersulfidation pathways (Scheme 1, reaction 4) [24]. Overexpression of CBS was proposed to contribute to protection of cancer cells from oxidative stress and promote tumor progression via all of its above-mentioned activities: i.) in neuroblastoma via an elevated production of cysteine and glutathione [18], ii.) in colon cancer via increased synthesis of H2S or ii.) in BLBC via persulfide generation on protein cysteine residues [20].
Scheme 1.

CBS mediated enzymatic pathways. 1. The canonical activity of CBS is to produce cystathionine using serine and homocysteine as a substrates, cystathionine than can be converted to cysteine but this latter reaction is catalyzed by CSE. 2–3. CBS can also catalyze the generation of H2S by using homocysteine and cysteine as a substrates and/or using two cysteines to produce lanthionine. 4. Direct generation of Cys persulfide is catalyzed by CBS when cystine is used as a substrate, in this case Cys persulfide is generated without any involvement of H2S.
The roles of CBS in pancreatic cancer have not been studied to date. Here we show a significant increase in CBS expression in metastatic pancreatic ductal adenocarcinoma cells compared to cell lines isolated from non-metastatic primary tumors. We present evidence that CBS overexpression has an important functional role in PDAC aggressiveness and metastasis formation. We observed a decrease in Wnt-5a, Snail expression and STAT3 phosphorylation in CBS silenced cells under normal conditions. Our studies have shown that formation of intra- and intermolecular disulfide bridges due to STAT3 oxidation in CBS silenced cells lead to a weakening of STAT3 transcriptional activity. Sulfur metabolome analyses suggest that the observed CBS-mediated cellular protection against oxidative protein modifications is related to protein persulfidation rather than an elevated production of cysteine and/or glutathione. Taken together, CBS overexpression through regulation of epithelial-mesenchymal transition plays a significant role in PDAC cancer cell invasion and metastasis via persulfidation-mediated protection against the formation of inhibitory oxidative posttranslational modifications on STAT3.
2. Experimental procedures
2.1. Matertials
HPE-IAM (β-(4-hydroxyphenyl) ethyl iodoacetamide) was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Dimethyl sulfoxide (DMSO), Sodium selenite, 2-Mercaptoethanol, Protease Inhibitor Cocktail, CHAPS, Giemsa, May-Grünwald Stain were purchased from Sigma (St. Louis, MO, USA). Fetal bovine serum (FBS) was purchased from EuroClone (Pero (MI), Italy), RPMI 1640 medium, l-glutamine, penicillin/streptomycin from Lonza (Basel, Swiss). Phosphatase Inhibitor Cocktail was purchased from Merck (Darmstadt, Germany).
Stable isotope labeled standards were produced by Cambridge Isotope Laboratories, Inc (Tewksbury, MA, USA).
2.2. Cell culture
T3M4 and BxPC3 [25,26] cell lines were maintained in a 5% CO2 incubator at 37 °C using RPMI1640 containing 10% fetal bovine serum (FBS), 100 U/mL penicillin/streptomycin, 2 mM l-glutamine and 100 nM Na-selenite.
2.3. Lentiviral transduction
For shRNA mediated stable knockdown we used the Mission Lentiviral-mediated gene specific shRNA system (Sigma, St. Louis, MO, USA). For CBS knockdown we used MISSION shRNA Lentiviral Transduction Particles (SHCLNV-NM_000071, ID: TRCN0000291407). Its corresponding control was MISSION pLKO.1-puro empty vector control transduction particle (SHC001V). Lentiviral particles were used at 5 multiplicity of infection (MOI) value. After 48 h of transduction cells were selected with 5 μg/mL puromycin for 48 h, two times. For single cell colonies, cells were seeded into 96 well plates by diluting one cell per well. After colony formation they were transferred into 24, 12, 6 well plates, respectively.
2.4. Orthotopic injection
Animals used in our study were taken care of according to the “Guiding Principles for the Care and Use of Animals” based on the Helsinki declaration and they were approved by the Institutional Ethics Committee at the National Institute of Oncology (Budapest, Hungary). Animal housing density was according to the regulations and recommendations from directive 2010/63/EU of the European Parliament and of the Council of the European Union on the protection of animals used for scientific purposes (permission number: PE/EA/1461–7/2020). Adult male NODSCID (NOD.Cg-Prkdcscid) mice were bred in a specified pathogen free (SPF) environment in the National Institute of Oncology. Mice were kept in a sterile environment in Makrolon® cages at 22–24 °C (40–50% humidity), with light regulation of 12/12 h light/dark. The animals had free access to tap water and were fed with sterilized standard diet (VRF1, autoclavable, Akronom Kft.) ad libitum. 10 mice from our colony (two groups: 5 control and 5 shCBS injected) were used for the experiments.
The orthotopic tumor cell injection into the pancreas was performed as published previously [28,29]. After intraperitoneal anesthesia (mixture of Zoletil, Xilazin and Butorfanol), a left paramedian laparotomy was made in 10–12 mm length, the spleen with the attached pancreatic tail was exteriorized with cotton swabs, and 5 × 105 tumor cells in 40 μl FBS free RPMI medium were gently injected into the pancreatic parenchyma with an Insulin syringe (26G). After the organs were returned into the abdominal cavity, peritoneum and skin were closed by 4–0 surgical suture (Ethicon, Johnson & Johnson Medical GmbH, Norderstedt, Germany). After the surgical procedure wound healing, body weight and physical condition of the mice were monitored daily. Mice were euthanized when they reached the predetermined endpoint (14th postoperative day), tumors were harvested, weighed and samples were collected for further analyses (immunohistochemistry to see liver metastases). This experiment was repeated 3 times.
2.5. Western blot
Cells were harvested in RIPA (10 mM Tris/HCl, pH = 7.4, 1% NP-40, 0.1% Sodium deoxycholate, 0.1% SDS, 150 mM NaCl) buffer in the presence of protease and phosphatase inhibitors. After 10 s of sonication, cellular debris were removed by centrifugation at 14000g for 10 min at 4 °C. Protein concentrations were determined from supernatants with the bicinchoninic acid (BCA) method using bovine serum albumin (BSA) as a standard. 15 μg protein samples were reduced with 5% beta mercapthoethanol or not, and denatured at 95 °C for 10 min than loaded on a polyacrylamide gel. After size separation by electrophoresis, proteins were transferred onto nitrocellulose membranes (Trans-Blot Turbo Blotting System, Bio-Rad, Hercules, CA, USA). Transfer efficiencies were verified with Ponceau staining. Membranes were blocked with 3% nonfat-milk in TBST (0.05% Tween 20) for 1 h at room temperature. Primary antibodies against CBS (ab140600), GAPDH (ab181602), beta-Actin (ab8227), Snail (ab216347), Wnt-5a (ab229200) were purchased from Abcam (Cambridge, UK); STAT3 (79D7), phospho-STAT3 (Y705) (D3A7) from Cell Signaling (Danvers, MA, USA). Antibodies were diluted in 3% nonfat-milk in TBST and incubated with the membranes overnight at 4 °C. After washing in TBST (3 × 10 min) membranes were incubated with a secondary antibody (horseradish peroxidase conjugated anti mouse/rabbit IgG (DAKO, Santa Clara, CA, USA) at a 1:4000 dilution for 1 h at room temperature. After washing, membranes were incubated with the ECL reagent (Bio-Rad, Hercules, CA, USA) for 1–5 min and the signal was detected using a gel documentation system (Syngene, Cambridge, UK). Immunostaining for GAPDH and beta-Actin was used to normalize protein expression levels. Densitometry was performed using ImageJ software.
2.6. RNA isolation and RT-qPCR
Cells were cultured in T25 flasks. Total RNA was isolated using TRIzol reagent (Applied Biosystem, Waltham, MA, USA) according to the manufacturer's instructions. Concentrations and purities were measured spectrophotometrically using a Nanodrop machine. 3 μg RNA samples were treated with 2U of DNase I (ThermoScientific, Waltham, MA, USA) at 37 °C for 30 min. Reactions were stopped by adding 20 mM EDTA followed by heat inactivation at 70 °C for 10 min and placing the samples on ice immediately. RNA samples were reverse transcribed using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Waltham, MA, USA) according to the manufacturer's instructions. DNA-RNA hybrid products were diluted 10 fold in RNase free water and used as a template for real time quantitative PCR. Analyses were performed by using a 2x qPCRBIO SyGreen Mix reagent (PCR Biosystems, London, UK) according to the manufacturer's instructions on a Roche Light Cycler 480 machine. Relative abundances of mRNA were calculated by the ΔCT method using GAPDH and actin as invariant controls. The primer sequences are listed in Table 1. RT2 Profiler™ PCR Arrays for Human Epithelial to Mesenchymal Transition (EMT) (PAHS-090Z) was used according to the manufacturer's instructions. For the array we used 2 μg total RNA.
Table 1.
The primer sequences used in RT-qPCR measurements.
| Primer | Sequence |
|---|---|
| WNT5A forward | ACATCGACTATGGCTACCGC |
| WNT5A reverse | GATGCGAGCACTCTCGTAGG |
| SNAI1 forward | GACCCCAATCGGAAGCCTAA |
| SNAI1 reverse | AGGGCTGCTGGAAGGTAAAC |
| CDH2 forward | GCATGGTGTATGCCGTGAGA |
| CDH2 reverse | CTGCCACTTGCCACTTTTCC |
2.7. CBS activity assay
Cells were washed with HBSS and collected from T25 flasks or 6 well plates followed by lysis in CHAPS buffer (150 mM KCl, 50 mM HEPES pH 7.4, 0.1% CHAPS, protease inhibitors). After 10 s sonication cell debris were removed by centrifugation (14000 g 10 min, 4 °C). Protein concentrations were estimated from the supernatant using the BCA method. For CBS activity 20 μl of the lysates diluted to 1 mg/ml protein content were prepared as published previously [30]. The procedure briefly: 280 mM homocysteine in 100 mM Tris-HCl (pH 8.6) was prepared by the addition of 10 mM DTT to homocysteine thiolactone and increasing the pH with 6 M NaOH solution to cleave the thiolactone ring. After incubating the alkaline mixture for 5 min at 37 °C the pH was set to 8.6 with 1:1 HCl. Initiation of the assay was carried out by adding 5 μl of this solution to the sample mixture that was made by mixing 20 μl of the diluted lysate with 25 μl of solution containing 200 mM Tris-HCl pH 8.6, 1 mM pyridoxal 5′-phosphate, 0.5 mM S-adenosyl-methionine and 40 mM 2,3,3-D-labeled serine. After 4 h of incubation at 37 °C reactions were stopped by acidification with Reagent 1 of EZ:Faast kit (Phenomenex, Torrance, CA, USA) containing 3.3 μM of 3,3,4,4-D labeled cystathionine as an internal standard. All consequent sample preparation steps of the EZ:Faast kit were carried out exactly as instructed in the manual. LC-MS/MS measurements were performed on a Thermo Vanquish UHPLC coupled to a Thermo Q Exactive Focus mass spectrometer.
2.8. Cys and CSSC measurement
Cells were washed with HBSS and collected from 6 well plate followed by lysis in CHAPS buffer (150 mM KCl, 50 mM HEPES pH 7.4, 0.1% CHAPS, protease inhibitors). After 10 s sonication cell debris was removed by centrifugation (14000 g 10 min, 4 °C). Protein concentration was estimated from the supernatant using the BCA method. 50 μl of the lysate at 1 mg/ml protein concentration was derivatized with the EZ:Faast kit (Phenomenex, Torrance, CA, USA) following the manufacturer's instructions and measured with a Thermo Vanquish UHPLC system coupled to a Thermo Q Exactive Focus mass spectrometer.
2.9. Immunohistochemistry
After routine formalin fixation and paraffin embedding, 3 and 5 μm thick tissue sections were prepared. Sections on microscopic slides were deparaffinized and rehydrated followed by hematoxylin&eosin (H&E) staining. Sizes of necrotic areas were evaluated by Olympus cellSense dimension software. For immunohistochemistry, deparaffinized sections underwent antigen epitope retrieval in citrate buffer at pH: 6.0 (VectorLaboratories, Newark, CA, USA) in a microwave oven for 20 min. After cooling to room temperature endogenous peroxidase inactivation occurred in 0.3% H2O2 for 10 min. Immunohistochemistry was performed by using an ImmPRESS Peroxidase Polymer Detection Kit (Vector Laboratories, Newark, CA, USA) according to the manufacturer's instructions. Primary antibodies from Abcam anti-CBS (ab140600) and anti-E cadherin (ab40772) were diluted 100-; 500- and 500-fold in a 0.1% BSA solution, respectively and incubated with the sections overnight at 4 °C in a humidified chamber. Immune complexes were detected by diaminobenzidine (ImmPact DAB Peroxidase Substrate). Hematoxylin was used as a nuclear counterstain. Sections were dehydrated in ethanol, cleared in xylene and mounted. Tumor tissue sections using Vimentin IHC kit (Proteintech, Manchester, UK) were stained for vimentin according to the manufacturer's instructions. Images were acquired using an Olympus BX43 microscope equipped with a DP74 camera. Formalin-fixed paraffin-embedded tissue sections of human PDAC patient tumor samples were collected from the biobank of the National Institute of Oncology (the procedure was approved by the Hungarian National Ethics Committee under file number IV/10441–1/2020/EKU).
2.10. Soft agar colony assay
Colony formation was performed as described in Ref. [31]. Briefly, the bottom of 6 well plates were layered with sterile 1% noble agar. Cells were harvested by trypsinization and counted. 5000 cells were diluted in 2x RPMI1640 cell culture medium (prepared from solid powder, Auro-Science, Budapest, Hungary) and mixed in 1:1 ratio in 0.6% sterile noble agar and immediately layered on the bottom layer of the 6 well plates. To prevent desiccation the upper layer of the noble agar was covered with RPMI1640 cell culture medium and exchanged two times per week. Upon colonies became visible under the microscope, cells were stained with 1 mg/ml nitroblue tetrazolium chloride solution overnight. After staining photographs were taken.
2.11. Wound healing assay
T3M4 and BxPC3 cells were seeded in 12-well plates on glass coverslip in media containing 1% FBS at a final concentration of 4 × 105 cell per well. After 24 h, the cell layers were scraped with 200 μl pipette tip to form straight wounds. After removing suspended cells with PBS, cells were grown for 48 h. Cells on the coverslip were stained with Giemsa-May Grünwald staining according to the manufacturer's instructions. Pictures of the wound areas were taken using an Olympus BX43 microscope equipped with a DP74 camera at 0 and 48 h. The difference between the wound areas (measured the distance) at 48 h and 0 h were used to calculate the wound closure. Each experiment were performed in triplicate.
2.12. Transwell migration assay
The migration assay was performed as published previously [32]. Briefly, T3M4 (control and shCBS) cells suspended in FBS free RPMI were seeded (2 × 104 cells/well) into the upper chamber of polyvinylpyrrolidone-free, 8-μm pore size transwells (Osmonics, Minnetonka, MN, USA). Fibronectin (1:10 dilution in cell culture medium) was used for cells to attach. The whole chamber was incubated at 37 °C and 5% CO2 for 7 h.
2.13. Sequential non-reducing/reducing two-dimensional SDS-PAGE
T3M4 and BxPC3 cells after 48 h culture were harvested. Free thiols were blocked with 5 mM HPE-IAM for 1 h. Cells were lysed and separated in the first dimension under non-reducing conditions on 4–12% gradient polyacrylamide gel. To reduce disulfide bonds, lanes were incubated in 20 mg/mL DTT containing equilibration buffer (6 M urea, 0.375 M Tris/HCl pH 8.8, 2% SDS and 20% glycerol) for 20 min, followed by incubation with 25 mg/mL IAM containing equilibration buffer for 20 min at room temperature (RT). After the second-dimension electrophoresis, gels were subjected to immunoblotting using a STAT3 (79D7) antibody (Abcam, Cambridge, UK).
2.14. LC-MS/MS measurement of high molecular weight (protein) per/polysulfides
Measurements were based on the method published by Akaike et al. [33] and performed as described here: Cells seeded in 12-well plates were washed once with HBSS before the addition of 5 mM HPE-IAM in RIPA buffer. Cells were scraped, collected and sonicated. Following centrifugation at 14000g for 10 min at RT, 100 μl of the supernatants were desalted using Zeba spin-columns (7K MWCO, 0.5 mL). Protein content of the collected flow-through was measured using BCA, followed by the addition of 4 μl of 100 mM HPE-IAM in DMSO to the flow-through. Protein levels of the desalted samples were brought to equal using RIPA buffer and digested with pronase (3 mg/ml) in 35 mM Na-acetate buffer (pH 5.0) at 37 °C for 1 h. Digestion was quenched by the addition of 10% TCA, samples were centrifuged for 10 min at 14000 g and the supernatants were injected on the LC-MS/MS.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) measurements were done on Thermo Q Exactive Focus mass spectrometer coupled to a Thermo Vanquish UHPLC (ultra-high performance liquid chromatography). The derivatives were separated with Phenomenex Kinetex C18 (50 × 2.1 mm, 2.6 μm) column using a linear gradient elution. Eluent (A) contained 0.1% FA/H2O and eluent (B) 0.1% FA/MeOH. Flow rate was 0.3 ml/min at 30 °C. The initial 5% eluent B content was linearly increased to 95% in 15 min then lowered back to 5% B in 2 min and held there for 3 min before the next injection. Ionization was carried out using a heated ESI (electrospray ionization) head at 1.5 kV and 250 °C in positive ionization mode. Parent ions were fragmented using higher-energy collisional dissociation (HCD) and the derivatives were detected by selected reaction monitoring (SRM). Parent and fragment m/z values that were used can be found in our previous work [20].
2.15. LC-MS/MS measurement of GSH
Measurement is based on the method published by Akaike et al. [33] as described here: Cells seeded in 12 well plates were washed twice with PBS before the addition of ice-cold 5 mM β-(4-hydroxyphenyl)ethyl iodoacetamide (HPE-IAM) in 75% methanol. Cells were scraped, collected on ice and sonicated. Lysates were incubated at 37 °C for 20 min followed by centrifugation at 14000g for 10 min at 4 °C. 100 μl of the supernatant was acidified with 5 μl of 10% formic acid and diluted two-fold with 0.1% FA/H2O before injection. Cell pellets were dissolved in 150 μl 1% SDS/PBS and protein content was measured using the BCA assay. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) measurements were carried out on a Thermo Orbitrap Focus mass spectrometer coupled to a Thermo Vanquish UHPLC (ultra-high-performance liquid chromatograph). The derivatives were separated with Phenomenex Kinetex C18 (50 × 2.1 mm, 2.6 μm) column using a linear gradient elution. Eluent (A) contained 0.1% FA/H2O and eluent (B) 0.1% FA/MeOH. Flow rate was 0.3 ml/min at 30 °C. The initial 5% eluent B content was linearly increased to 95% in 15 min then lowered back to 5% B in 2 min and held there for 3 min before the next injection. MS/MS detection in positive ionization mode and higher-energy collisional dissociation (HCD) was used to detect the 356 m/z fragment from the 485 m/z precursor ion.
2.16. Statistical analyses
GraphPad Prism (Version 9.0, San Diego, CA, USA) was used for graphical representation and statistical analyses of data. All statistical analyses were performed on the basis of at least three independent experiments. We used two-tailed Student's t-test (paired and unpaired), one sample t-test and one-way ANOVA to analyze differences in the normalized values between cell lines. Statistical significance is denoted in figures as *P < 0.05, **P < 0.01 and ***P < 0.001.
3. Results
3.1. Increased CBS expression in metastatic ductal adenocarcinoma of the pancreas
We observed an increase in CBS expression on mRNA as well as on protein levels in metastatic pancreatic ductal adenocarcinoma cells compared to cell lines isolated from non-metastatic primary tumors (Fig. 1A–C). Increased cellular expression levels correlated with increased CBS activities as measured by an LC-MS/MS based method (Fig. 1D). Importantly, immunohistochemical staining of formalin-fixed paraffin-embedded tissue sections of human PDAC patient tumor samples showed higher CBS expression in ductal cells compared to those in non-tumor tissues (Fig. 2A, CBS staining, brown color, solid arrows pointing to ductal cells). We have also investigated lymph node metastases from the same patients and confirmed stronger CBS expression in these samples compared to primary tumor samples (Fig. 2B, CBS staining, brown color, solid arrows pointing to ductal cells).
Fig. 1.

Increased CBS expression in metastatic ductal adenocarcinoma of the pancreas. (A). Molecular characteristics of pancreatic cancer cell lines isolated from PDAC metastases and primary tumors [[25], [26],52]. (B). CBS mRNA levels in PDAC and non-metastatic primary tumor cell lines as measured by RT-qPCR. (C). Western blot analyses of CBS protein expression levels in PDAC and non-metastatic primary tumor cell lines. (D). CBS activities in PDAC cell lysates as measured by an LC-MS/MS based method using the EZ:faast kit (for more details see methods). The data are shown as mean ± SEM of 2 independent experiments.
Fig. 2.

Elevated expression of CBS in ductal cells of metastatic pancreatic cancer. (A). Immunohistochemical staining for CBS protein using tissue sections from 3 representative human PDAC tumors and from the matching non-tumor tissues (patient 1-2-3). (B). Immunohistochemistry of the lymph node metastases from the same patients (1-2-3). CBS protein in ductal cells is visualized by brown staining and indicated by the solid line arrows. Tumor sections of the indicated tissues were also stained with H&E to reveal tissue architecture. (Scale bars: 50 μm). Images are representative of 10 independent tissue samples. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.2. CBS is involved in metastatic dissemination of PDAC
In order to study whether the above observed differences in CBS expression levels are pure markers of tumor progression or have a biological function in PDAC we used shRNA technology to silence CBS expression in the aggressive metastatic PDAC cell line T3M4. CBS silencing was confirmed by Western blot analyses as well as by CBS activity measurements in lysates using a mass spectrometry-based method (see materials and methods, Fig. 3A). To assess how CBS silencing affects tumor growth in vivo, we injected the control and CBS silenced T3M4 cells into the pancreas of immune deficient mice orthotopically. Mice were monitored and after termination we measured tumor weights. We did not observe significant differences in tumor size or in necrotic areas in shCBS tumors compared to controls (Fig. 3B). However, CBS silenced T3M4 tumors gave significantly fewer distant metastases compared to tumors induced by the injection of control T3M4 cells (Fig. 3C).
Fig. 3.

CBS is involved in metastatic dissemination of PDAC. (A). Confirmation of CBS silencing in a T3M4 cell line by Western blot analyses and CBS activity measurements. (B). Tumor weights and histological assessment of intratumoral necrosis of tumors induced by orthotopical injection of control and shCBS T3M4 cells into the pancreas. Necrotic areas were evaluated by the Olympus cellSense Dimension software. Representative images are shown, experiments were repeated tree times. (C). Assessment of liver metastases 14 days after orthotopic injection of control and shCBS T3M4 cells into the pancreas. Liver sections were stained with H&E and the number of metastases per animal liver were evaluated with Olympus BX43 microscope equipped with a DP74 camera. The data are shown as mean ± SEM of 3 independent experiments with n = 5 animals in each group, **P < 0.001 compared to control.
3.3. CBS is important in PDAC cancer cell invasion and malignant transformation
To evaluate the role of CBS in PDAC aggressiveness we performed the soft agar colony formation assay. This method provides information on the anchorage-independent growth of cancer cells in vitro, which is the ability of transformed cells to grow independently of a solid surface and is a hallmark of carcinogenesis. We seeded the control T3M4, its CBS silenced version and the primary PDAC tumor BxPC3 cells (which were harvested before metastatic dissemination of a patient tumor) on soft agar plates. We followed the colony formation capability of these cells for 21 days. No colonies were found in BxPC3 cells, and significantly less colonies were detected in T3M4 shCBS cells compared to controls (Fig. 4A). In addition, both the scratch and trans-well migration assays suggested that CBS knock down indeed inhibited the migration capacity of pancreatic cancer cells. Importantly the sulfide donor GYY4137 rescued this capability in shCBS cells (Fig. 4B–C), which led us to hypothesize that the sulfide/persulfide inducing functions of CBS are potentially more important in promoting PDAC cell migration than its canonical function to produce cystathionine for cysteine synthesis.
Fig. 4.

CBS overexpression is an important factor in PDAC cancer cell invasion and malignant transformation. (A). Anchorage-independent growth of BxPC3, T3M4 control and shCBS cells were measured by a soft agar colony assay. Colonies were visualized after 21 day with nitroblue tetrazolium sodium salt solution staining. The data are shown as mean ± SEM of 4 independent experiments, *P < 0.05 compared to control. (B). Representative tracking of migration of T3M4 control, shCBS and BxPC3 cells with or without the sulfide donor GYY4137 for 48 h in the wound healing assay. Bar graph shows quantitative analyses of wound healing assays with or without GYY4137 after 48 h (relative wound closure compared to 0 h). The data are shown as mean ± SEM of 3 independent experiments, *P < 0.05, **P < 0.001 compared to control (C). Transwell migration assay showed a significant decrease in the migration capacity of shCBS cells compared to control T3M4 cells. The data are shown as mean ± SEM of 3 independent experiments, *P < 0.05 compared to control. Representative images are shown.
3.4. CBS overexpression plays a significant role in PDAC cancer cell invasion through regulation of epithelial-to-mesenchymal transition
Next, in order to better understand the underlying molecular mechanisms behind the observed role of CBS in PDAC aggressiveness we investigated whether CBS silencing could have induced changes in epithelial-to-mesenchymal transition (EMT) of PC cells. In CBS silenced T3M4 cells significant decreases of WNT5A, SNAIL and CDH2 (N-cadherin) gene expressions were observed compared to control and to BxPC3 primary tumor cells (Fig. 5A). These observations were confirmed at the protein level for Wnt-5a and Snail using Western blot analyses (Fig. 5B). Importantly, we observed an increase in E-cadherin and a decrease in Vimentin signals in mouse tumors upon orthotopic injection of shCBS compared to control T3M4 cells, respectively. This observation corroborated the involvement of CBS in EMT-induced metastatic dissemination of PDAC in vivo (Fig. 5C). Taken together our results indicate a shift from the transformed (mesenchymal) towards the normal (epithelial) state (termed as mesenchymal to epithelial transition, MET) in response to silencing CBS and could explain the concomitant reduced motility of PDAC cells (see Fig. 4B–C). Therefore, targeting CBS in PDAC to induce MET could hamper its aggressive metastatic phenotype.
Fig. 5.

CBS overexpression plays a significant role in PDAC cancer cell invasion through regulation of epithelial-to-mesenchymal transition. (A). RT-qPCR measurement of endogenous mRNA levels of WNT5A, SNAI1 and CDH2 (N-cadherin) using RT2 Profiler™ PCR Arrays for Human Epithelial to Mesenchymal Transition. The data are shown as mean ± SEM of 3 independent experiments, *P < 0.05 compared to control. (B). Western blot analysis of Wnt-5a and Snail protein expression in T3M4 and BxPC3 cells. The data are shown as mean ± SEM (n = 7), *P < 0.05 compared to control. Representative images of the 7 independent experiments are shown. (C). Immunohistochemistry of T3M4 control and shCBS mouse pancreatic tumor tissues. CBS protein presence and silencing were confirmed by brown CBS staining. EMT was examined with E-cadherin an epithelial and Vimentin a mesenchymal marker. (Scale bars: 50 μm; brown color). Images are representative of 5 independent tumor tissues. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.5. CBS protects STAT3 from oxidation induced loss of transcriptional activity
Next, we investigated the effects of CBS silencing on STAT3 activation, which is a transcriptional upstream mediator of Wnt signaling. Under normal conditions, STAT3 phosphorylation, which is a necessary posttranslational modification for transcriptional activation, was decreased in CBS-deficient T3M4 cells compared to controls (Fig. 6A). In addition, we observed an increase in steady-state STAT3 oligomerization in shCBS T3M4 cells compared to controls. Furthermore, lower levels of phosphorylation and elevated oligomerization of STAT3 was also apparent in primary BxPC3 cells, which have less intracellular CBS activities, compared to the metastatic T3M4 cells (Fig. 6A–B). These observations led us to hypothesize that high expression of CBS could protect STAT3 from intracellular oxidation induced disulfide-linked oligomer formation. Two-dimensional sequential nonreducing-reducing diagonal SDS-PAGE followed by immunoblotting with a STAT3 specific antibody corroborated that the observed elevated oligomerization in shCBS T3M4 cells compared to control T3M4 cells indeed correspond to oxidation-induced formation of disulfide bridges among STAT3 monomers (Fig. 6C-D). Loss of protection against STAT3 oxidation upon CBS silencing may also explain the diminished phosphorylation of STAT3 in shCBS cells (Fig. 6A). Taken together, our results suggest that elevated CBS levels protect STAT3 from oxidation in PDAC cells, which allows elevated STAT3 phosphorylation leading to an increase in transcriptional activity. In turn, this could trigger Wnt signaling and EMT to aid the metastatic dissemination of PDAC.
Fig. 6.

CBS protects STAT3 from oxidation induced loss of transcriptional activity. (A). Quantification of STAT3 phosphorylation at Tyr 705 by Western blot analyses. In our quantification we determined the ratios between STAT3/GAPDH and p-STAT3/GAPDH and then determined the ratio between (p-STAT3/GAPDH)/(STAT3/GAPDH) and finally calculate the % of control. The data are presented as mean ± SEM of 7 independent experiments, ***P < 0.001, ****P < 0.0001 compared to control. (B). Western blot analyses of STAT3 oligomerization in the presence and absence of H2O2 (100 μM for 2 min) after separation by SDS-PAGE gel electrophoresis in control and shCBS T3M4 as well as in BxPC3 cells. Image is representative of 3 independent experiments. (C). Visualization of STAT3 disulfide-linked dimers after separation by two dimensional sequential nonreducing-reducing SDS-PAGE and immunoblotting of the diagonal gel with a STAT3 specific antibody. Arrows indicate intermolecular disulfide linked STAT3 oligomers. Image is representative of 3 independent experiments. (D). Quantification of oxidized STAT3 oligomers. The data are presented as mean ± SEM of 4 independent experiments, *P < 0.05 compared to control.
3.6. CBS mediated cysteine persulfidation may contribute to protection against STAT3 oligomerization
Mass spectrometry-based metabolome analyses revealed no differences in GSH or steady-state cysteine levels among control and shCBS T3M4 cells (Fig. 7A–B.). However, we found that shCBS T3M4 and BxPC3 cells exhibited significantly lower levels of steady-state protein persulfidation compared to T3M4 control cells (Fig. 7C). In addition, a significant increase in the oxidized cystine levels in shCBS compared to control T3M4 cells was observed (Fig. 7D). These results together suggest that protein persulfidation is likely to contribute to the observed protective effects of CBS against oxidative processes.
Fig. 7.

CBS mediated cysteine persulfidation may contribute to protection against oxidative inactivation of STAT3. (A-B and D.) Metabolome analyses of steady-state GSH, cysteine and cystine levels, respectively. (C). Protein cysteine persulfidation levels in T3M4 control, shCBS and primary tumor (BxPC3) cells. Metabolites were measured by LC-MS/MS. *P < 0.05, **P < 0.01 show significant differences in shCBS and BxPC3 compared to control T3M4 cells. The data are means ± SEM of 3 independent experiments.
4. Discussion
Pancreatic ductal adenocarcinoma (PDAC) is the most prevalent neoplastic disease of the pancreas accounting for more than 90% of all pancreatic malignancies [34]. To date, PDAC is the fourth most frequent cause of cancer-related deaths worldwide with a 5-year overall survival of less than 8% [35]. Over the past decade, advances in diagnostic approaches, perioperative management, radiotherapy techniques, and systemic therapies for advanced disease have made relevant but only modest incremental progress in patient outcomes. New strategies for screening high-risk patients to detect pancreatic tumors at earlier stages and novel targeted therapeutic interventions that can stop or slow down the dissemination of the disease are desperately needed to make a clinically significant impact. To reach these goals, a better understanding of PDAC tumor biology is vital.
In the present work we discovered an oncogenic role for cystathionine β-synthase (CBS) in metastatic PDAC. In normal healthy pancreas tissues, CBS was detected in acinar cells [36], with negligible expression in ductal cells. However, a significant increase in CBS expression was observed in metastatic pancreatic ductal adenocarcinoma cells compared to cell lines isolated from non-metastatic primary tumors. Analyses of primary tumor samples from PDAC patients supported increased CBS production in cancerous ductal cells compared to in the non-tumor tissues with further elevation of CBS expression in the lymph node metastasis of the same patients. Using an orthotopic injection mouse model, we demonstrated that CBS overexpression in PDAC cells is not a consequence, but a promoting factor in the formation of liver metastases in vivo.
Cancer cells must undergo changes to be able to detach from the tumor mass and colonize in distant organs. Changes include a decrease in cellular junctions and adhesion, and an increase in cell motility. With CBS silencing we observed diminished WNT5A and SNAIL gene expression in the aggressive metastatic T3M4 pancreatic cancer cells. Both Wnt-5a and Snail play important roles in the regulation of epithelial to mesenchymal transformation of PDAC cells. Overexpression of Snail is crucial for cancer invasiveness and metastasis and elevated Wnt-5a activation induces cell migration via the non-canonical Wnt signaling pathway [10,11]. Parallel with the diminished Wnt-5a and Snail expressions we indeed observed impaired migratory and colony formation capabilities of CBS silenced T3M4 cells, which was close to the minimal invasive capacities of BxPC3 primary tumor cells. The potential involvement of CBS and other H2S producing enzymes in promoting cancer cell migration and invasion were previously mentioned in other cancer types (e.g. [[37], [38], [39]]).
Then the obvious question was raised: What is the link between CBS and Wnt-5a/Snail-induced EMT in PDAC?
Aberrant Snail expression can be initiated by phosphorylated STAT3 binding to the SNAI1 promoter [40] and Wnt signaling is also mediated via STAT3 [11]. In our experiments we observed lower STAT3 phosphorylation in CBS silenced metastatic and primary tumor cells compared to control T3M4 cells. Oxidation of STAT3 cysteines were shown to reversibly inhibit STAT3-induced gene expression and cell proliferation activities [12] and CBS was demonstrated to play a role in the protection of cancer cells against oxidative modifications on protein Cys residues [20]. STAT3 oxidation, among others, results in disulfide bridge formation and oligomerization of the protein [16]. The extent by which the steady-state levels of disulfide linked STAT3 oligomers in control T3M4 compared to shCBS cells was diminished was similar to when peroxiredoxin 2 (the protein that reacts directly with the added H2O2 and transmits the redox equivalents to STAT3) was knocked down in HEK293T cells [15]. This observation together with an increase in STAT3 phosphorylation compared to in shCBS T3M4 and to BxPC3 primary tumor cells is consistent with an increased transcriptional activity in more aggressive cells due to CBS-mediated protection of this transcription factor against oxidation. STAT3 activation has been implicated in the context of CBS/H2S in some studies in other cancer types [41,42].
The role of CBS in cancer progression is gaining increasing attention [21]. It was shown to be involved in angiogenesis, cancer cell proliferation, response to hypoxic conditions, protection against ferroptotic cell death etc. [20,21]. However, the role of CBS in regulating EMT in cancer has only been mentioned in colon cancer [43] but to our knowledge no molecular mechanistic studies or any mention on the roles of transsulfuration enzymes have been published to date in relation to pancreatic cancer.
Another largely unresolved question in the field is: Which of the many enzymatic activities of the enzyme is most important in the progression of the disease? The canonical function of CBS is to supplement the cellular cysteine demand of the cell in linked enzymatic activities with CSE (see Scheme 1, reaction 1). This can obviously be associated with cellular protection against oxidative stress via providing cysteine to thiol protein and glutathione synthesis, which play pivotal roles in cellular antioxidant capacities. This function was demonstrated -by the elegant study from Craig Thompson's group-to be important in melanoma, glioblastoma, neuroblastoma and prostate cancer [18]. In addition, the role of CBS in reactive sulfur species production -including hydrogen sulfide and cysteine persulfide (Cys–SSH)– is now gaining increasing attention in tumor biology. In cancer types where CBS was found to be up-regulated, increases in H2S as well as persulfide levels were also observed (e.g. [19,20,44]). Although CBS can directly catalyze the formation of both species (using Cys or cystine as substrates, respectively; see Scheme 1, reactions 2–3 or 4, respectively), they can also be generated from each other so their formation is intimately interlinked. This together with the appreciated limitations of current detection protocols [45,46] render it currently impossible to elucidate the specific roles of sulfur metabolic pathways in producing persulfides under different pathophysiological conditions. Importantly, both H2S and Cys persulfidation can protect cells from oxidative stress via interactions with metalloproteins or via protecting oxidation sensitive protein thiols through transpersulfidation [[47], [48], [49], [50]]. Our sulfur metabolome analyses suggest that CBS-mediated antioxidant protection in PDAC cells is likely to be related to protein cysteine persulfidation rather than CBS-mediated more intense cysteine and glutathione supply. Although the intracellular prevalence and enzyme protecting/regulatory functions of protein Cys persulfidation is now well documented [[49], [50], [51]], the mechanism of CBS-related STAT3 protection against oxidative inactivation or its effect on STAT3 transcriptional activity remains to be elucidated. We are currently developing novel technologies in our laboratory to uncover this missing link, but these investigations are outside the scope of the present study.
We conclude that CBS overexpression through regulation of the EMT plays a significant role in PDAC cancer cell invasion and metastasis. Therefore, targeting CBS could increase STAT3 oxidation in cancer cells and induce mesenchymal-to epithelial transition (MET), which could be utilized in the medical oncology stratagem to treat patients with pancreatic cancer.
Author contributions
Á.C. was responsible for most of the biology experiments, analyzed data and wrote the manuscript; K.E. did biology experiments on pancreatic cancer cell lines, made the CBS silenced T3M4 cell line and analyzed data; T.D. conducted most metabolome analyses and analyzed data; N. Sz. was involved in the biological experimental work; E.P.J did the Cys persulfide measurements; Sz. Sz. performed the orthotopic tumor cell injection into the pancreas of mice; J.T, supervised the animal experiments and provided essential intellectual input; T.S. provided the PDAC human tissue samples and intellectual input; P.N. conceived and oversaw the study, analyzed data and wrote the manuscript. All authors edited and/or approved the manuscript.
Declaration of competing interest
The authors whose names are listed immediately below certify that they have NO affiliations with or involvement in any organization or entity with any financial interest (such as honoraria, educational grants, participation in speakers’ buruaus, membership, employment, consultancies, stock ownership or other equity interest, and expert testimony or patent-licensing arrangement) or non-financial interest (such as personal or professional relationship, affiliation, knowledge or beliefs) in the subject matter or materials discussed in this manuscript.
Acknowledgements
This work was supported by the National Laboratories Program (under the National Tumor Biology Laboratory (2022-2.1.1-NL-2022-00010)) and the Hungarian Thematic Excellence Program (under project TKP2020-NKA-26 and TKP2021-EGA-44) from the Hungarian National Research, Development and Innovation Office. Human pancreatic carcinoma cell lines (AsPc-1, BxPc-3, Capan-1, Colo-357, MiaPaca-2, Panc-1, Su.86.86 and T3M4) were a kind gift from PD Dr. Klaus Felix (University of Heidelberg, European Pancreas Center, Germany). The excellent technical assistance for Anna Mária Tóth and Anita Hidvégi are greatly appreciated.
Data availability
Data will be made available on request.
References
- 1.Balaban E.P., Mangu P.B., Khorana A.A., Shah M.A., Mukherjee S., Crane C.H., Javle M.M., Eads J.R., Allen P., Ko A.H., Engebretson A., Herman J.M., Strickler J.H., A.B.B., Urba S., Yee N.S. Locally advanced, unresectable pancreatic cancer: American society of clinical oncology clinical practice guideline. J. Clin. Oncol. 2016;34(22):2654–2668. doi: 10.1200/JCO.2016.67.5561. [DOI] [PubMed] [Google Scholar]
- 2.Vaccaro V., Sperduti I., Vari S., Bria E., Melisi D., Garufi C., Nuzzo C., Scarpa A., Tortora G., Cognetti F., Reni M., Milella M. Metastatic pancreatic cancer: is there a light at the end of the tunnel? World J. Gastroenterol. 2015;21(16):4788–4801. doi: 10.3748/wjg.v21.i16.4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roche J. The epithelial-to-mesenchymal transition in cancer. Cancers. 2018;10(2):52. doi: 10.3390/cancers10020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ng L.F., Kaur P., Bunnag N., Suresh J., Sung I.C.H., Tan Q.H., Gruber J., Tolwinski N.S. WNT signaling in disease. Cells. 2019;8(8):826. doi: 10.3390/cells8080826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serra R., Easter S.L., Jiang W., Baxley S.E. Wnt5a as an effector of TGFβ in mammary development and cancer. J. Mammary Gland Biol. Neoplasia. 2011;16(2):157–167. doi: 10.1007/s10911-011-9205-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ren D., Minami Y., Nishita M. Critical role of Wnt5a–Ror2 signaling in motility and invasiveness of carcinoma cells following Snail-mediated epithelial–mesenchymal transition. Gene Cell. 2011;16(3):304–315. doi: 10.1111/j.1365-2443.2011.01487.x. [DOI] [PubMed] [Google Scholar]
- 7.Jin Z., Zhao C., Han X., Han Y. Wnt5a promotes ewing sarcoma cell migration through upregulating CXCR4 expression. BMC Cancer. 2012;12(1):480. doi: 10.1186/1471-2407-12-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barrallo-Gimeno A., Nieto M.A. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132(14):3151–3161. doi: 10.1242/dev.01907. [DOI] [PubMed] [Google Scholar]
- 9.Thiery J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer. 2002;2(6):442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y., Shi J., Chai K., Ying X., Zhou B.P. The role of Snail in EMT and tumorigenesis. Curr. Cancer Drug Targets. 2013;13(9):963–972. doi: 10.2174/15680096113136660102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gujral Taranjit S., Chan M., Peshkin L., Sorger Peter K., Kirschner Marc W., MacBeath G. A noncanonical Frizzled 2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell. 2014;159(4):844–856. doi: 10.1016/j.cell.2014.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li L., Cheung S.-h., Evans E.L., Shaw P.E. Modulation of gene expression and tumor cell growth by redox modification of STAT3. Cancer Res. 2010;70(20):8222–8232. doi: 10.1158/0008-5472.CAN-10-0894. [DOI] [PubMed] [Google Scholar]
- 13.Kim J., Won J.-S., Singh A.K., Sharma A.K., Singh I. STAT3 regulation by S-nitrosylation: implication for inflammatory disease. Antioxidants Redox Signal. 2014;20(16):2514–2527. doi: 10.1089/ars.2013.5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie Y., Kole S., Precht P., Pazin M.J., Bernier M. S-glutathionylation impairs signal transducer and activator of transcription 3 activation and signaling. Endocrinology. 2009;150(3):1122–1131. doi: 10.1210/en.2008-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sobotta M.C., Liou W., Stöcker S., Talwar D., Oehler M., Ruppert T., Scharf A.N.D., Dick T.P. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2015;11(1):64–70. doi: 10.1038/nchembio.1695. [DOI] [PubMed] [Google Scholar]
- 16.Li L., Shaw P.E. A STAT3 dimer formed by inter-chain disulphide bridging during oxidative stress. Biochem. Biophys. Res. Commun. 2004;322(3):1005–1011. doi: 10.1016/j.bbrc.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 17.Hanahan D., Weinberg Robert A. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 18.Zhu J., Berisa M., Schwörer S., Qin W., Cross J.R., Thompson C.B. Transsulfuration activity can support cell growth upon extracellular cysteine limitation. Cell Metabol. 2019;30(5):865–876. doi: 10.1016/j.cmet.2019.09.009. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szabo C., Coletta C., Chao C., Módis K., Szczesny B., Papapetropoulos A., Hellmich M.R. Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc. Natl. Acad. Sci. USA. 2013;110(30):12474–12479. doi: 10.1073/pnas.1306241110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Erdélyi K., Ditrói T., Johansson H.J., Czikora Á., Balog N., Silwal-Pandit L., Ida T., Olasz J., Hajdú D., Mátrai Z., Csuka O., Uchida K., Tóvári J., Engebraten O., Akaike T., Dale A.-L.B., Kásler M., Lehtiö J., Nagy P. Reprogrammed transsulfuration promotes basal-like breast tumor progression via realigning cellular cysteine persulfidation. Proc. Natl. Acad. Sci. USA. 2021;118(45) doi: 10.1073/pnas.2100050118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ascenção K., Szabo C. Emerging roles of cystathionine β-synthase in various forms of cancer. Redox Biol. 2022;53 doi: 10.1016/j.redox.2022.102331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kraus J.P. Academic Press; 1987. Cystathionine β-synthase (Human), Methods in Enzymology; pp. 388–394. [DOI] [PubMed] [Google Scholar]
- 23.Kabil O., Banerjee R. Enzymology of H2S biogenesis, decay and signaling. Antioxidants Redox Signal. 2014;20(5):770–782. doi: 10.1089/ars.2013.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ida T., Sawa T., Ihara H., Tsuchiya Y., Watanabe Y., Kumagai Y., Suematsu M., Motohashi H., Fujii S., Matsunaga T., Yamamoto M., Ono K., Devarie-Baez N.O., Xian M., Fukuto J.M., Akaike T. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. U. S. A. 2014;111(21):7606–7611. doi: 10.1073/pnas.1321232111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okabe T., Yamaguchi N., Ohsawa N. Establishment and characterization of a carcinoembryonic antigen (CEA)-producing cell line from a human carcinoma of the exocrine pancreas. Cancer. 1983;51(4):662–668. doi: 10.1002/1097-0142(19830215)51:4<662::aid-cncr2820510419>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 26.Tan M.H., Nowak N.J., Loor R., Ochi H., Sandberg A.A., Lopez C., Pickren J.W., Berjian R., Douglass H.O., Jr., Chu T.M. Characterization of a new primary human pancreatic tumor line. Cancer Invest. 1986;4(1):15–23. doi: 10.3109/07357908609039823. [DOI] [PubMed] [Google Scholar]
- 28.Dobiasch S., Szanyi S., Kjaev A., Werner J., Strauss A., Weis C., Grenacher L., Kapilov-Buchman K., Israel L.L., Lellouche J.P., Locatelli E., Franchini M.C., Vandooren J., Opdenakker G., Felix K. Synthesis and functionalization of protease-activated nanoparticles with tissue plasminogen activator peptides as targeting moiety and diagnostic tool for pancreatic cancer. J. Nanobiotechnol. 2016;14(1):81. doi: 10.1186/s12951-016-0236-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosenberger I., Strauss A., Dobiasch S., Weis C., Szanyi S., Gil-Iceta L., Alonso E., González Esparza M., Gómez-Vallejo V., Szczupak B., Plaza-García S., Mirzaei S., Israel L.L., Bianchessi S., Scanziani E., Lellouche J.P., Knoll P., Werner J., Felix K., Grenacher L., Reese T., Kreuter J., Jiménez-González M. Targeted diagnostic magnetic nanoparticles for medical imaging of pancreatic cancer. J. Contr. Release. 2015;214:76–84. doi: 10.1016/j.jconrel.2015.07.017. [DOI] [PubMed] [Google Scholar]
- 30.Krijt J., Kopecká J., Hnízda A., Moat S., Kluijtmans L.A.J., Mayne P., Kožich V. Determination of cystathionine beta-synthase activity in human plasma by LC-MS/MS: potential use in diagnosis of CBS deficiency. J. Inherit. Metab. Dis. 2011;34(1):49–55. doi: 10.1007/s10545-010-9178-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borowicz S., Van Scoyk M., Avasarala S., Karuppusamy Rathinam M.K., Tauler J., Bikkavilli R.K., Winn R.A. The soft agar colony formation assay. JoVE. 2014;92 doi: 10.3791/51998. e51998-e51998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen H.-C. In: Cell Migration: Developmental Methods and Protocols. Guan J.-L., editor. Humana Press; Totowa, NJ: 2005. Boyden chamber assay; pp. 15–22. [Google Scholar]
- 33.Akaike T., Ida T., Wei F.Y., Nishida M., Kumagai Y., Alam M.M., Ihara H., Sawa T., Matsunaga T., Kasamatsu S., Nishimura A., Morita M., Tomizawa K., Nishimura A., Watanabe S., Inaba K., Shima H., Tanuma N., Jung M., Fujii S., Watanabe Y., Ohmuraya M., Nagy P., Feelisch M., Fukuto J.M., Motohashi H. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 2017;8(1):1177. doi: 10.1038/s41467-017-01311-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kleeff J., Korc M., Apte M., La Vecchia C., Johnson C.D., Biankin A.V., Neale R.E., Tempero M., Tuveson D.A., Hruban R.H., Neoptolemos J.P. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016;2(1) doi: 10.1038/nrdp.2016.22. [DOI] [PubMed] [Google Scholar]
- 35.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2018. CA A Cancer J. Clin. 2018;68(1):7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 36.Tamizhselvi R., Moore P.K., Bhatia M. Hydrogen sulfide acts as a mediator of inflammation in acute pancreatitis: in vitro studies using isolated mouse pancreatic acinar cells. J. Cell Mol. Med. 2007;11(2):315–326. doi: 10.1111/j.1582-4934.2007.00024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phillips C.M., Zatarain J.R., Nicholls M.E., Porter C., Widen S.G., Thanki K., Johnson P., Jawad M.U., Moyer M.P., Randall J.W., Hellmich J.L., Maskey M., Qiu S., Wood T.G., Druzhyna N., Szczesny B., Módis K., Szabo C., Chao C., Hellmich M.R. Upregulation of cystathionine-β-synthase in colonic epithelia reprograms metabolism and promotes carcinogenesis. Cancer Res. 2017;77(21):5741–5754. doi: 10.1158/0008-5472.CAN-16-3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo S., Li J., Huang Z., Yue T., Zhu J., Wang X., Liu Y., Wang P., Chen S. The CBS-H(2)S axis promotes liver metastasis of colon cancer by upregulating VEGF through AP-1 activation. Br. J. Cancer. 2022;126(7):1055–1066. doi: 10.1038/s41416-021-01681-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Augsburger F., Randi E.B., Jendly M., Ascencao K., Dilek N., Szabo C. Role of 3-mercaptopyruvate sulfurtransferase in the regulation of proliferation, migration, and bioenergetics in murine colon cancer cells. Biomolecules. 2020;10(3) doi: 10.3390/biom10030447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou J.J., Meng Z., He X.Y., Cheng D., Ye H.L., Deng X.G., Chen R.F. Hepatitis C virus core protein increases Snail expression and induces epithelial-mesenchymal transition through the signal transducer and activator of transcription 3 pathway in hepatoma cells. Hepatol. Res. 2017;47(6):574–583. doi: 10.1111/hepr.12771. [DOI] [PubMed] [Google Scholar]
- 41.Youness R.A., Gad A.Z., Sanber K., Ahn Y.J., Lee G.J., Khallaf E., Hafez H.M., Motaal A.A., Ahmed N., Gad M.Z. Targeting hydrogen sulphide signaling in breast cancer. J. Adv. Res. 2021;27:177–190. doi: 10.1016/j.jare.2020.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L., Han H., Liu Y., Zhang X., Shi X., Wang T. Cystathionine β-synthase induces multidrug resistance and metastasis in hepatocellular carcinoma. Curr. Mol. Med. 2018;18(7):496–506. doi: 10.2174/1566524019666181211162754. [DOI] [PubMed] [Google Scholar]
- 43.Ascenção K., Dilek N., Augsburger F., Panagaki T., Zuhra K., Szabo C. Pharmacological induction of mesenchymal-epithelial transition via inhibition of H2S biosynthesis and consequent suppression of ACLY activity in colon cancer cells. Pharmacol. Res. 2021;165 doi: 10.1016/j.phrs.2020.105393. [DOI] [PubMed] [Google Scholar]
- 44.Bhattacharyya S., Saha S., Giri K., Lanza I.R., Nair K.S., Jennings N.B., Rodriguez-Aguayo C., Lopez-Berestein G., Basal E., Weaver A.L., Visscher D.W., Cliby W., Sood A.K., Bhattacharya R., Mukherjee P. Cystathionine beta-synthase (CBS) contributes to advanced ovarian cancer progression and drug resistance. PLoS One. 2013;8(11) doi: 10.1371/journal.pone.0079167. e79167-e79167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bianco C.L., Akaike T., Ida T., Nagy P., Bogdandi V., Toscano J.P., Kumagai Y., Henderson C.F., Goddu R.N., Lin J., Fukuto J.M. The reaction of hydrogen sulfide with disulfides: formation of a stable trisulfide and implications for biological systems. Br. J. Pharmacol. 2019;176(4):671–683. doi: 10.1111/bph.14372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bogdándi V., Ditrói T., Bátai I.Z., Sándor Z., Minnion M., Vasas A., Galambos K., Buglyó P., Pintér E., Feelisch M., Nagy P. Nitrosopersulfide (SSNO(-)) is a unique cysteine polysulfidating agent with reduction-resistant bioactivity. Antioxidants Redox Signal. 2020;33(18):1277–1294. doi: 10.1089/ars.2020.8049. [DOI] [PubMed] [Google Scholar]
- 47.Garai D., Ríos-González B.B., Furtmüller P.G., Fukuto J.M., Xian M., López-Garriga J., Obinger C., Nagy P. Mechanisms of myeloperoxidase catalyzed oxidation of H(2)S by H(2)O(2) or O(2) to produce potent protein Cys-polysulfide-inducing species. Free Radic. Biol. Med. 2017;113:551–563. doi: 10.1016/j.freeradbiomed.2017.10.384. [DOI] [PubMed] [Google Scholar]
- 48.Nagy P. Mechanistic chemical perspective of hydrogen sulfide signaling. Methods Enzymol. 2015;554:3–29. doi: 10.1016/bs.mie.2014.11.036. [DOI] [PubMed] [Google Scholar]
- 49.Dóka É., Ida T., Dagnell M., Abiko Y., Luong N.C., Balog N., Takata T., Espinosa B., Nishimura A., Cheng Q., Funato Y., Miki H., Fukuto J.M., Prigge J.R., Schmidt E.E., Arnér E.S.J., Kumagai Y., Akaike T., Nagy P. Control of protein function through oxidation and reduction of persulfidated states. Sci. Adv. 2020;6(1) doi: 10.1126/sciadv.aax8358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zivanovic J., Kouroussis E., Kohl J.B., Adhikari B., Bursac B., Schott-Roux S., Petrovic D., Miljkovic J.L., Thomas-Lopez D., Jung Y., Miler M., Mitchell S., Milosevic V., Gomes J.E., Benhar M., Gonzalez-Zorn B., Ivanovic-Burmazovic I., Torregrossa R., Mitchell J.R., Whiteman M., Schwarz G., Snyder S.H., Paul B.D., Carroll K.S., Filipovic M.R. Selective persulfide detection reveals evolutionarily conserved antiaging effects of S-sulfhydration. Cell Metabol. 2020;31(1):207. doi: 10.1016/j.cmet.2019.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paul B.D., Snyder S.H. H2S signalling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell Biol. 2012;13(8):499–507. doi: 10.1038/nrm3391. [DOI] [PubMed] [Google Scholar]
Further reading
- 52.Moore P.S., Sipos B., Orlandini S., Sorio C., Real F.X., Lemoine N.R., Gress T., Bassi C., Klöppel G., Kalthoff H., Ungefroren H., Löhr M., Scarpa A. Genetic profile of 22 pancreatic carcinoma cell lines. Analysis of K-ras, p53, p16 and DPC4/Smad4. Virchows Arch. 2001;439(6):798–802. doi: 10.1007/s004280100474. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available on request.