

Abstract
The segregation of prokaryotic plasmids typically requires a centromere-like site and two proteins, a centromere-binding protein (CBP) and an NTPase. By contrast, a single 245 residue Par protein mediates partition of the prototypical staphylococcal multiresistance plasmid pSK1 in the absence of an identifiable NTPase component. To gain insight into centromere binding by pSK1 Par and its segregation function we performed structural, biochemical and in vivo studies. Here we show that pSK1 Par binds a centromere consisting of seven repeat elements. We demonstrate this Par-centromere interaction also mediates Par autoregulation. To elucidate the Par centromere binding mechanism, we obtained a structure of the Par N-terminal DNA-binding domain bound to centromere DNA to 2.25 Å. The pSK1 Par structure, which harbors a winged-helix-turn-helix (wHTH), is distinct from other plasmid CBP structures but shows homology to the B. subtilis chromosome segregation protein, RacA. Biochemical studies suggest the region C-terminal to the Par wHTH forms coiled coils and mediates oligomerization. Fluorescence microscopy analyses show that pSK1 Par enhances the separation of plasmids from clusters, driving effective segregation upon cell division. Combined the data provide insight into the molecular properties of a single protein partition system.
Keywords: pSK1 plasmid, DNA segregation, Par protein, X-ray crystallography, S. aureus
Introduction
Most naturally-occurring low-copy number plasmids possess an active partitioning system that facilitates the efficient segregation of plasmid copies during cell division. Generally, these systems encode a cis-acting centromere-like site, a centromere-binding protein (CBP), and an NTPase1–5. To date three main types of plasmid partitioning systems have been recognized in prokaryotes (types I to III), classified according to the NTPase protein they encode1. Type I systems encode Walker box NTPase proteins1–5, 6–8 and are further divided into type Ia and Ib systems. Type Ia systems contain CBPs with helix-turn-helix (HTH) motifs1–5,9–10 while type Ib encode CBPs with ribbon-helix-helix (RHH) motifs1–5,11. Unlike type Ib NTPases, the type Ia NTPases contain an extra N-terminal helix-turn-helix (HTH) domain that binds to the par promoter region and functions to regulate transcription of the par operon12–14. In the type Ib system this transcription autoregulatory function is mediated by the CBP protein11. Type II partition systems encode an actin-like15–16 NTPase motor protein (ParM) and a RHH DNA-binding protein (ParR)17–18. In addition to its role in plasmid partitioning, ParR contributes to autoregulation of the par operon by binding to the centromere-like site, which overlaps the promoter region16,19. Type III systems encode an NTPase, called TubZ, with a tubulin like fold and a CBP, TubR, with a HTH motif20–22.
The ParB/ParR/TubR CBPs bind to the centromere-like site and form a nucleoprotein complex called the partition complex. More recent work has shown that type I ParB proteins are cytosine triphosphate (CTP) dependent molecular switches wherein CTP binding and hydrolysis effects DNA spreading and partition complex formation23–28. The CBPs also act as adaptors to link the centromere (and hence the plasmid itself) to the partner NTPase via protein-protein interactions to form a so-called segrosome1–5. NTPases typically drive the physical separation of plasmid substrates. In type I and II systems, the multimerization of the NTPase proteins in the presence of a nucleotide cofactor leads to the formation of filaments, which physically segregate plasmids to opposite cell poles using either an insertional polymerization mechanism (type II)16,29 or a tramming like process (type III)21–22. Although initially all type I NTPase proteins were thought to form functional polymers30–33, studies have shown that these systems employ diffusion ratchet like mechanisms whereby the NTPase binds the nucleoid DNA, using it in a piggy-back type mechanism34–40. Super resolution data indicated that ParA NTPases equilibrate to high density regions of the nucleoid formed near the two poles of dividing cells, leading to equipartitioning of plasmids39.
More recently, partitioning systems distinct from type I, II and III, have been identified on the Escherichia coli conjugative plasmid R388 and the Staphylococcus aureus pSK1 multiresistance plasmid41–44. Notably, these systems appear to require only one protein to mediate partition. In the R388 system the StbA protein binds to a set of upstream direct repeats, stbDRs and seems sufficient for plasmid retention42. Indeed, although stbB, encoded on the same operon, encodes a putative NTPase, it has no apparent role in plasmid segregation. Interestingly, StbA also contributes to conjugative transfer of the plasmid, and hence in this system, plasmid partitioning and conjugative transfer are interdependent processes42. The S. aureus pSK1 stability determinant contains a single gene encoding a 245-amino acid protein, Par, which is essential for pSK1 retention43. Genes homologous to pSK1 par are widely conserved on staphylococcal plasmids, particularly multiresistance plasmids, commonly carried by clinical S. aureus isolates44. In each case, par is found upstream of, and transcribed divergently from, the plasmid’s replication initiation gene, rep45. Homologs of par have also been identified in other Gram-positive genera including Lactococcus, Lactobacillus, Streptococcus, Tetragenococcus and Clostridium, highlighting the significance of this unique system in plasmid maintenance45.
The pSK1 Par protein shows no homology to other plasmid partitioning proteins identified thus far, including the StbA protein. Thus, to gain insight into the function of pSK1 Par, we carried out biochemical, structural and in vivo studies. We demonstrate that pSK1 Par binds to repeat elements located in the par-rep intergenic region and that DNA-binding also mediates autoregulation at the par promoter, Ppar. We obtained the structure of the DNA-binding domain of pSK1 Par bound to a centromere repeat, revealing that it binds DNA using a winged helix-turn-helix (wHTH) motif that is distinct from any previous structure of a plasmid CBP protein. Instead, the pSK1 Par DNA-binding domain structure shows similarity to members of the MerR family of regulators. But it displays the strongest structural homology to the B. subtilis DNA segregation protein, RacA46, that functions in chromosomal DNA partition during sporulation47–49. We also show that a region C-terminal to the DNA-binding domain, which contains a predicted coiled coil (CC) mediates Par multimerization. Cellular studies suggest that Par contributes to plasmid segregation stability by dividing plasmid clusters, apparently without the involvement of a NTPase activity, to mediate efficient plasmid partitioning to future daughter cells.
Results
Identification of pSK1 Par promoter.
pSK1 Par was found to be essential for pSK1 segregational stability43. However, the promoter regulating its expression, the DNA sites it binds and the molecular mechanism by which it binds DNA are unknown. Thus, we initiated this study by identifying and characterizing the pSK1 par promoter. Simpson et al.43 previously identified four putative pSK1 par promoters (Ppar-1, Ppar-2, Ppar-3 and Ppar-4) via transcript mapping. Ppar-4 has subsequently been shown to be the promoter for the antisense regulator of the rep gene and has been renamed PrnaI (Figure 1(A))50. To assess the functionality of these promoters with respect to par transcription, we examined the effect of promoter mutations using chloramphenicol acetyltransferase (CAT) reporter gene assays. The reporter gene constructs were derived from pSK5336, which contains the 0.42 kb par-rep intergenic region fragment encompassing the putative par promoters, cloned upstream of the promoterless cat reporter gene of the E. coli-S. aureus shuttle vector pRB394. We conducted site-directed mutagenesis on pSK5336 to individually alter the −10 region of each of the four putative par promoters (Figure 1(A)). The CAT assays showed that plasmids containing mutations in Ppar-2 (pSK6149), Ppar-3 (pSK6150) or PrnaI (pSK6134) produced CAT activities of 1328±159, 1515±213 and 1506±87 units, respectively, which are not substantially different to the 1499±184 units of CAT activity produced from the wild-type (WT) par-rep region (pSK5336). In contrast, the CAT activity from the Ppar-1 mutant (pSK6138) was 103±12 units, which represents a 93% decrease in activity compared to WT, thereby implicating it as the source of par transcription.
Figure 1.
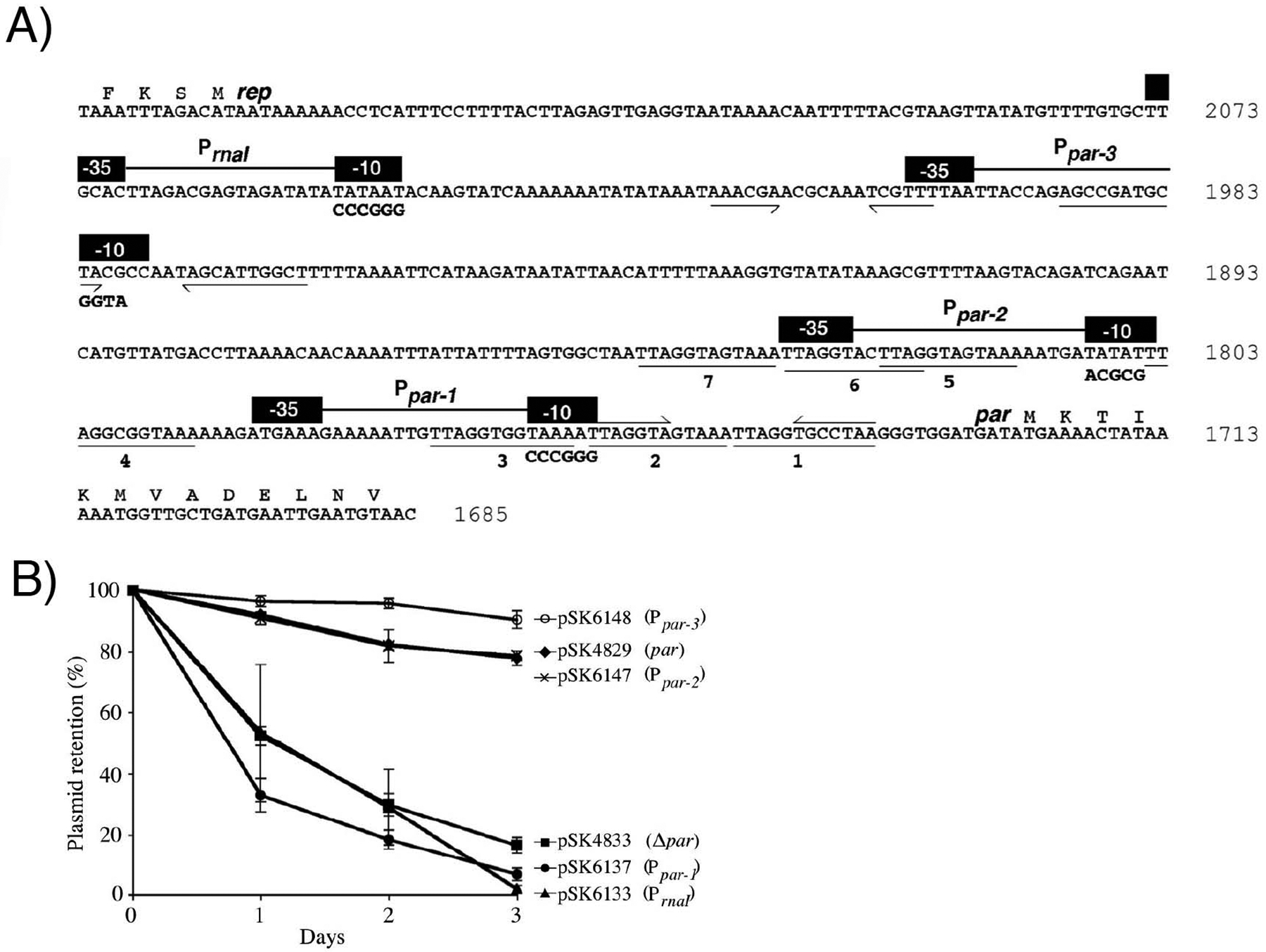
Organization of the pSK1 par-rep intergenic region and the effect of promoter mutations on plasmid segregational stability. (A) Sequence of the par-rep intergenic region, including the sequence of the 5′ ends of par and rep. Numbers at the end of each line indicate the pSK1 sequence position (GenBank entry GU565967). Black boxes represent −10 and −35 sequences of PrnaI and the putative par promoters Ppar-1–Ppar-3; nucleotides altered by site-directed mutagenesis are shown in bold text below the WT sequence. Directly repeated sequences (numbered 1–7) are underlined. Half arrows denote inverted repeats. (B) Segregational stability of plasmids containing mutations to the putative par promoters. Each data point is the mean of three independent assays, each normalized to 100% plasmid retention on Day 0. Error bars indicate standard error of the mean.
We next examined the contribution of the putative promoters to par function via plasmid segregational stability assays. Promoter mutations identical to those on pSK5336 described above were generated45 and the promoter mutant derivatives electroporated into S. aureus RN4220 cells for segregational stability assays. After three days of subculture (~40 generations), pSK4829 (par) was retained by 78±2% of the cell population (Figure 1(B)). In contrast, only 17±3% of the cell population retained the pSK4833 (Δpar) plasmid. Of the pSK4829 promoter mutant derivatives, only those harboring a mutation in Ppar-1 (pSK6137) or PrnaI (pSK6133) exhibited a significant reduction in segregational stability, with 7±2% and 2±1% of the respective cell populations retaining the plasmid. Combined with the CAT assay results, these data indicate that Ppar-1 is the only promoter directing transcription of par, and as such, Ppar-1 will henceforth be referred to as Ppar (Figure S1). The reduced segregational stability of the PrnaI mutant is likely due to increased plasmid copy number in the absence of RNAI50, which reduced the growth rate of plasmid-containing S. aureus cells so that plasmid-free cells in the population outgrow plasmid-containing counterparts.
Par binds to seven 12-bp repeats located in the par-rep intergenic region.
A region of pSK1 upstream of par, which contains seven 12-bp repeats, has previously been shown to mediate incompatibility43, similar to that observed for centromere-like sites of characterized partitioning systems. Thus, this region represented a putative binding site for Par. To examine the DNA-binding activity of Par, we performed electrophoretic mobility shift assays (EMSAs) by incubating purified 6xHis-tagged WT Par with a 212-bp end-labeled par-rep DNA probe that encompasses these repeats (nt 1689–1900, GenBank entry GU565967; Figure 1(A)). EMSAs showed that the migration of labeled probe (par-rep intergenic DNA) was increasingly retarded with increasing amounts of WT Par (Figure 2(A)). Furthermore, competition experiments showed that WT Par-probe DNA binding could be titrated with an unlabeled par-rep intergenic DNA fragment (specific competitor) but not with a non-specific competitor DNA of similar length (Figure 2(B)).
Figure 2.
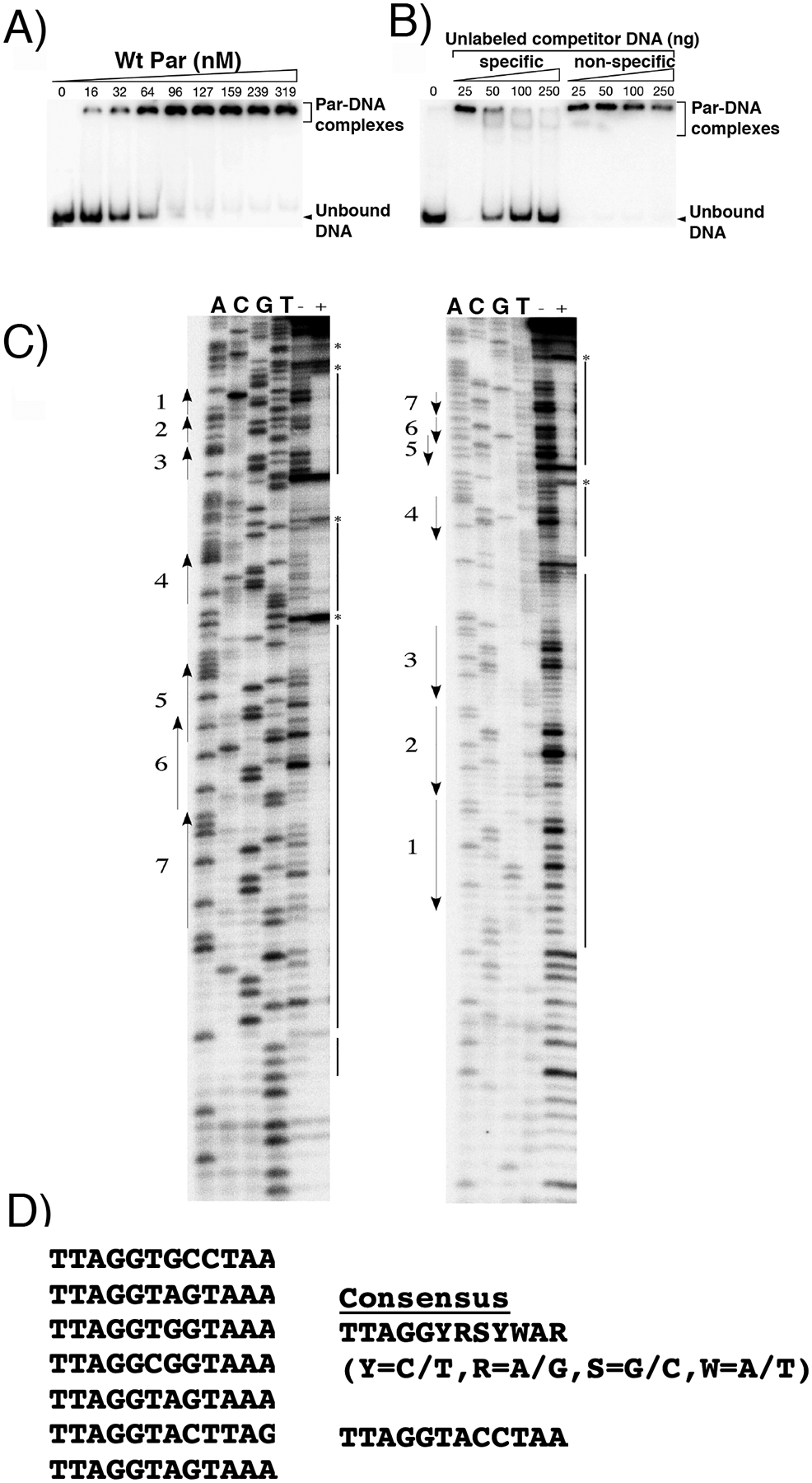
Identification of pSK1 Par DNA centromere site. (A) EMSAs of increasing amounts of Par binding to a DNA probe containing the par-rep intergenic region. (B) Competition EMSA performed as in (A) using a fixed amount of WT Par (556 nM) and increasing amounts of unlabeled competitor DNA, either specific (same as labeled probe) or non-specific (S. epidermidis DNA fragment of similar length). (C) DNase I footprinting analysis of the par-rep intergenic region incubated with (+) and without (−) Par. par sense and antisense strands are shown at left and right, respectively. Numbered arrows indicate repeats. Solid lines indicate regions of DNA protected by WT Par, and asterisks denote nucleotides hypersensitive to DNase I digestion. DNA sequencing reactions (ACGT) were performed with the same primer as end-labeled for the PCR. (D) Par binding motifs in the pSK1 centromere. Shown aligned are the motifs protected by pSK1 Par addition and, to the right, a consensus motif derived from these sequences.
To delimit the sequences to which Par binds, we performed DNase I footprinting experiments. The 212-bp par-rep fragment described above was PCR-amplified using primers that were alternately end-labeled to enable analysis of both DNA strands. Comparison of the DNase I footprints with co-electrophoresed sequencing ladders generated using the same primers revealed that Par bound across the segment containing all seven 12-bp repeats (Figure 1(A); Figure 2(C), (D); Figure S1), which have the consensus sequence TTAGGYRSYWAR (where Y=C/T, R=A/G, S=G/C, W=A/T) (Figure 2(D)) and contains a putative palindrome (TTAG(X)4CTAA) (Figure 2(D)). The appearance of hypersensitive sites (denoted by asterisks in Figure 2(C)) suggests a topological change in the DNA upon Par binding. The fact that the seven repeats in the centromere are separated by different numbers of nucleotides suggests that each is likely bound by a Par DNA-binding entity.
Crystal structure of pSK1 Par DNA-binding domain bound to centromere DNA.
The pSK1 Par protein contains an N-terminal domain with a putative HTH motif, a centrally-located domain (residues ~78–170) predicted to contain a coiled coil (CC) and a C-terminal region from residues 171–245 that is predicted to be disordered (Figure S2). For structural studies, we utilized a construct encoding Par residues 1–170, and co-crystallized the protein with centromere DNA (Experimental Procedures). We obtained crystals by mixing Par(1–170) with an 18-mer DNA site, ATGTTAGGTACCTAACTA, that contains a repeat element (with two potential half sites, ATGTTAGGT•ACCTAACTA). The structure was solved by platinum single wavelength anomalous diffraction (SAD) (Experimental Procedures; Table S2) and contains two Par subunits and one DNA duplex. In the structure each Par protomer is bound to a half site and there are no contacts between the subunits (Figure 3(A)). Only Par residues 1–56 were visible for each subunit and subsequent analyses revealed that the protein had broken down during the 6 months that it took for the crystals to be produced. This is consistent with secondary structure analyses, which indicated that the region encompassing Par residues 57–77 are predicted to be disordered (Figure S2). The final structure has Rwork/Rfree values of 21.4%/24.6% to 2.25 Å resolution (Table S2).
Figure 3.
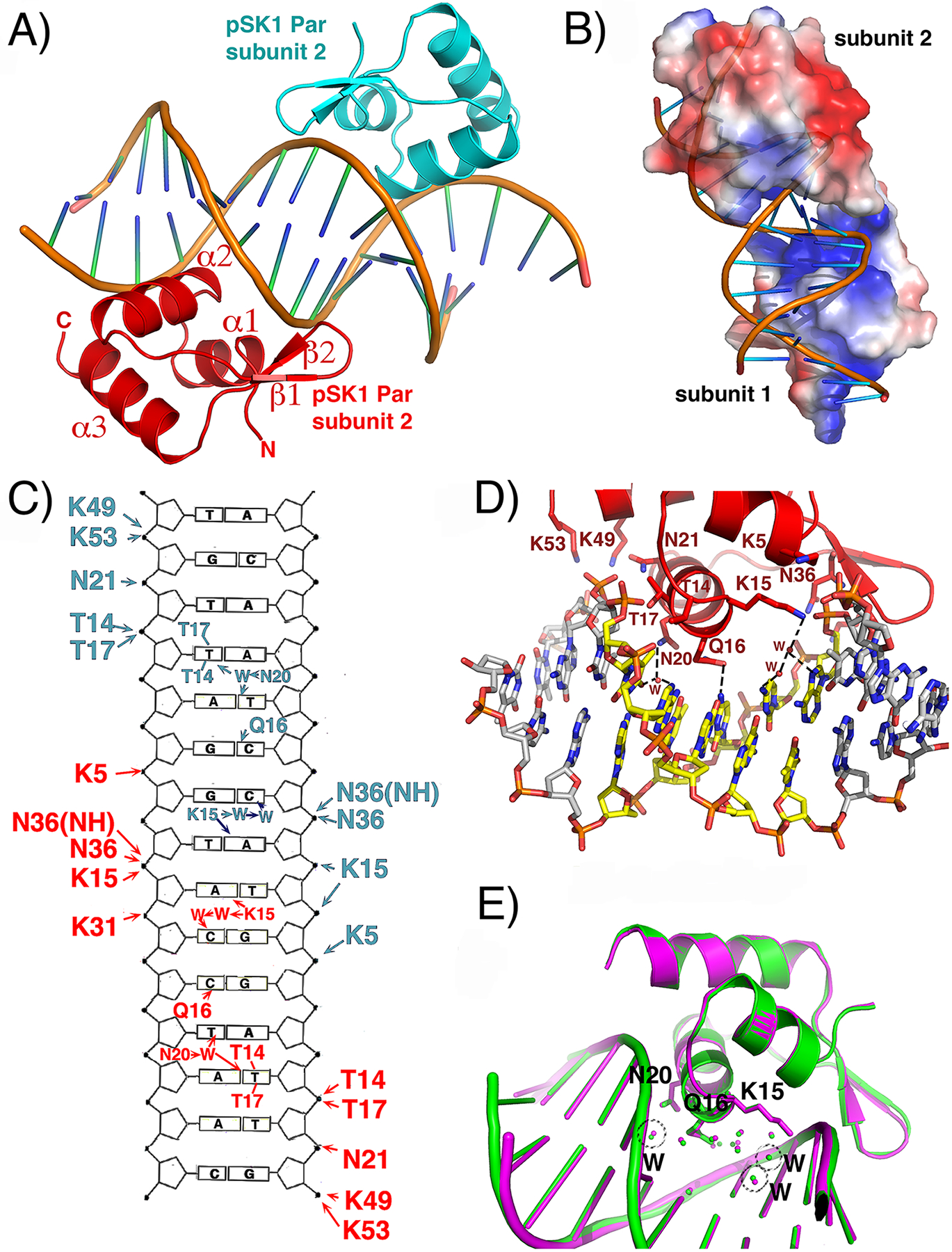
Crystal structure of pSK1 Par(1–170)-centromere complex. (A) Ribbon diagram of the structure showing the two Par subunits in the crystallographic asymmetric unit, each bound to a half site of the centromere repeat. Secondary structural elements are labeled for one subunit. (B) Electrostatic surface representation of pSK1 Par where electropositive and electronegative regions are colored blue and red, respectively. Each DNA binding face of the subunits in the crystallographic ASU are the same (electropositive) and bind DNA. Only the electropositive face of subunit is shown. (C) Schematic DNA ladder showing the contacts from each Par subunit to the DNA. Hydrogen bond and hydrophobic interactions are indicated by arrows and lines, respectively. (D) Close up of the Par-DNA interactions showing the contacts from one subunit (the contacts from the other subunit are the same). (E) Superimposition of the two Par-DNA half site complexes including the ordered solvent in the protein-DNA interface, showing that the water molecule positions are conserved.
The Par structure contains the topology: α1(residues 3–11)-α2(14–25)-β1(28–32)-β2(34–38)-α3(40–53) (Figure 3(A)). Helices 1 and 2 form an HTH motif with helix 2 inserting into the major groove. Examination of the electrostatic surface potential of Par reveals that the DNA binding face is electropositive while the opposite side of the molecule is electronegative (Figure 3(B)). DALI (structural homology) searches reveal that the pSK1 Par DNA-binding domain shows the strongest structural similarity to the winged HTH (wHTH) of the Bacillus subtilis “remodeling and anchoring of the chromosome” (RacA) protein46–50; 51 Cα atoms of the two structures can be superimposed with a root mean square deviation (rmsd) of 1.8 Å (and Z-score of 6.0), despite the fact that RacA and pSK1 Par share only ~16% sequence identity overall (Figure S3(A), (B)). The structures with the next strongest homologies revealed in DALI searches are the B. subtilis GlnR and TnrA structures51, with which 51 Cα atoms of the Par wHTH can be superimposed with rmsds of 2.2 Å and 2.3 Å, respectively. The sequence of the DNA-binding domains of TnrA and GlnR share less than 15% identity with that of pSK1 Par.
Both RacA and TnrA/GlnR are distant members of the MerR family of DNA binding regulators52. However, instead of a CC directly attached to their DNA-binding domains as in canonical MerR proteins, TnrA/GlnR members contain a disordered region that is attached to a short C-terminal tail, which folds into a helix upon binding to their effector protein, glutamine synthetase51. Similarly, RacA has a flexible region following its wHTH but is predicted to contain a CC C-terminal to the flexible domain46. Interestingly, RacA is an essential DNA segregation protein utilized during sporulation to ensure accurate segregation of the B. subtilis chromosome46. RacA binds to centromere-like “RacA binding motifs” (ram) DNA sites near the B. subtilis origin of replication to anchor the chromosome to the pole regions during sporulation49. The C-terminal region of RacA was shown to bind the pole protein DivIVA in B. subtilis, which indicates how RacA may anchor its bound chromosomal DNA to the poles48. Notably, pSK1 Par appears to harbor similarity in its overall domain architecture to RacA as it is also predicted to contain a CC C-terminal to the flexible region that follows its DNA-binding domain. However, our yeast two-hybrid studies did not reveal an interaction between Par and the S. aureus DivIVA protein. Thus, while pSK1 Par shows some structural homology to RacA, it clearly harbors differences as it is a plasmid encoded partition protein and mediates DNA segregation by a mechanism that does not involve DivIVA. RacA also lacks a C-terminal disordered region (Figure S3(B)).
pSK1 Par contacts to the centromere site.
The pSK1 Par-DNA structure shows that all Par contacts to DNA bases are to the 12-bp element comprising the centromere repeat. However, Par makes numerous contacts to the phosphate backbone of the entire 18-bp DNA site, which serves to dock the Par wHTH motifs onto the DNA. These contacts likely explain why crystals were only obtained with DNA sites 18-bp or longer. Phosphate contacts from pSK1 Par include contacts from the wHTH as well as α3, which harbors several basic residues. In particular, α3 residues Lys49 and Lys53 interact with DNA phosphates located at the ends of the DNA site (Figure 3(C)). Lys5 from α1 and Thr14, Thr17, Lys15 and Asn21 from the recognition helix, α2, and Asn36 and Lys31 from the wing region provide DNA phosphate contacts to both DNA strands of each site (Figure 3(D)). The structure shows that the base contacts from each Par subunit are primarily to the TAGG bps in each half of the repeat element (Figure 3(C), (D)). Interestingly, Par-centromere base contacts are largely hydrophobic and water mediated, with only one direct hydrogen bond to a base from a Par side chain. This hydrogen bond is from Gln16, which contacts the N4 atom of the cytosine paired with the first G in the motif (TAGG). However, this interaction is not likely highly specific as the glutamine side chain harbors both Oε and Nε atoms on its side chain, and the side chain can rotate. By contrast, the hydrophobic contacts by Thr14 and Thr17 from the recognition helix made to the second T in the motif (TTAGG) specify the thymine base (Figure 3(C), (D)). These contacts likely explain why this thymine is one of the most conserved bp among the Par repeats (Figure 2(D)). The side chain of Asn20 contacts a solvent molecule that interacts with this thymine as well as the thymine on the opposite strand (TTAGG) (Figure 3(C), (D)). Finally, Lys15, which makes anchoring phosphate contacts, is also bridged via a water molecule to the adenine base that pairs with the thymine just 3’ of the last conserved guanine in the motif (TTAGGT). In addition, the water molecule contacted by the Lys15 side chain is further bridged to another water molecule that interacts with the cytosine paired with the last guanine of the DNA repeat (TTAGGT). Unlike the base contacting residues, the Par residues that provide phosphate contacts are highly conserved among pSK1 homologs (Figure S2). In particular, Lys15 and Lys31 are completely conserved, while Lys5 and Asn21 are strongly conserved. The base contacting residues are likely less conserved because they are involved in providing specificity to their specific centromere sequence.
To gain insight into possible contributions from indirect readout or the influence of DNA structure on binding by pSK1 Par, we used w3DNA and Curves programs to analyze the properties of the Par bound centromere DNA53–54. Curves analyses indicate that the DNA in the structure is significantly bent at the center of the site (~40°). This structural change in the DNA suggests that binding of each subunit generates a distortion in the DNA, as observed in our footprinting experiments. The two Par DNA-binding domains don’t interact and hence the DNA bend must be generated through the DNA. The analyses show that the individual repeats bound by each Par subunit display primarily B-DNA characteristics, with rise and twist values of 3.2 Å and 33.6°, respectively, compared to 3.3 Å and 34.3° for B-DNA. The AT-rich regions of Par binding sites, however, show significant propeller twist (−15°). These base conformations permit contacts to the key threonine side chains as well as permitting water mediated interactions with the protein. AT-rich DNA sites are known to exhibit high degrees of propeller twist and hence may contribute to the recognition by pSK1 Par to its AT-rich DNA sites. Notably, when each subunit of Par-DNA is overlaid, the water molecules that interface between the protein and the DNA are conserved in each half site interaction (Figure 3(E)). Thus, the water mediated contacts along with the thymine contacts provide some level of base specific binding to the DNA. However, the lack of highly specific hydrogen bond-base interactions suggests that pSK1 Par might be able to contact DNA sites that deviate from the centromere consensus, perhaps at reduced affinity, that could have implications for its function.
Fluorescence polarization (FP) binding studies of pSK1 Par binding to centromere DNA and centromere DNA mutants.
Our pSK1 Par-DNA structure was obtained with a palindromic DNA repeat site. To assess the binding affinity of the protein for this site compared to the in vivo repeat elements we performed fluorescence polarization (FP) binding assays using fluoresceinated 18-mer DNA sites corresponding to the site used for crystallization, repeat 1, repeat 2 (which is identical to repeats 5 and 7 and hence referred to as repeat 2/5/7), repeat 3, repeat 4 and repeat 6 (Figure 2(D)). These data showed that the palindromic site displayed a modest enhancement in binding compared to the in vivo sites, but all sites were bound with high affinity by Par(1–170). The Kds were 43.4 ± 1.5 nM, 67.5 ± 3.2 nM, 66.5 ± 4.9 nM, 62.7 ± 1.3 nM, 140 ± 10 nM and 90.2 ± 5.6 nM for the site used for crystallization, repeat 1, repeat 2/5/7, repeat 3, repeat 4 and repeat 6, respectively (Figure 4(A)). The presence of 5 mismatches between the palindrome, as in repeat 4, showed a moderate, 3-fold, reduction in binding while the remaining repeats bound pSK1 Par(1–170) within 1–2 fold of the palindrome.
Figure 4.
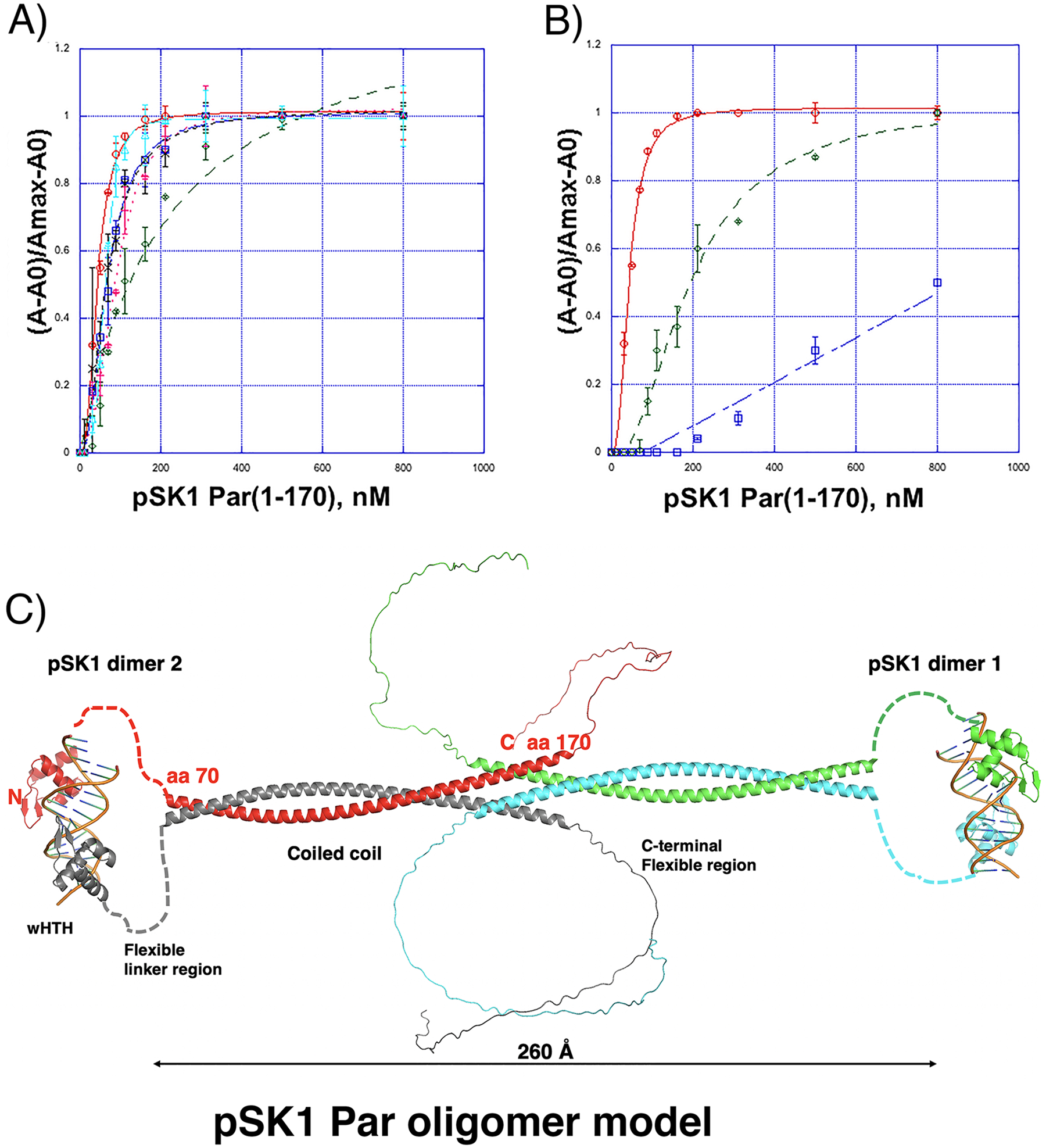
pSK1 Par oligomerization and DNA binding. (A) FP binding isotherms showing interaction of pSK1 Par(1–170) to fluoresceinated 18-mer DNA containing the site used for crystallization, ATGTTAGGTACCTAACTA (top strand) (red open circles), centromere repeat 1, ATGTTAGGTGCCTAACTA (blue open squares), centromere repeat 2/5/7, ATGTTAGGTAGTAAACTA (black crosses), centromere repeat 3 ATGTTAGGTGGTAAACTA (cyan open triangles), centromere repeat 4, ATGTTAGGCGGTAAACTA (green open diamonds), centromere repeat 6 ATGTTAGGTACTTAGCTA (magenta crosses). The Kds were 43.4 ± 1.5 nM, 67.5 ± 3.2 nM, 66.5 ± 4.9 nM, 62.7 ± 1.3 nM, 140 ± 10 nM and 90.2 ± 5.6 nM for the site used for crystallization, repeat 1, repeat 2/5/7, repeat 3, repeat 4 and repeat 6, respectively. The x axis is pSK1 Par(1–170) concentration in nM and the y axis is normalized mP. The curves are representative curves from three technical repeats. The error bars represent SD. Data are presented as mean values +/− SD. (B) FP binding isotherms showing interaction of pSK1 Par(1–170) to fluoresceinated 18-mer DNA containing the crystallization site, TTAGGTACCTAA (red open circles), half site mutant, TTAGGTACCCGA (green open diamonds) and both half site mutant, TCGGGGTACCGA (blue open squares). The resulting Kds were 43.4 ± 1.5 nM and 195 ± 10.8 nM and no measurable binding (Kd not detectable in the measurement range), respectively. The x axis is pSK1 Par(1–170) concentration in nM and the y axis is normalized mP. The curves are representative curves from three technical repeats. The error bars represent SD. Data are presented as mean values +/− SD. (C) Model of full length pSK1 Par bound to DNA. The structures of the DNA-binding domains bound to DNA were from our crystal structure. Residues 78–245 were modeled in alphafold 2 (as a dimer). A dimer-of-dimers model was generated by docking the ends of the CC from each dimer.
The structure revealed that key base contacts mediated by Par are to the thymine and the 3’ thymine base paired with the adenine in the TTAGGT motif (underlined in the half site). Indeed, the thymine (TA) is conserved in all par repeats and is specifically contacted by Par (Figure 3(C), (D)). The adenine (TA) is highly conserved and its base paired 3’ thymine is contacted by several water mediated contacts (Figure 3(C), (D)). These data suggest these nucleotides as important in pSK1 Par binding. Thus, using FP we tested the effect of substituting the TA motifs in one or both half sites. The data showed that substitution of these bps in both half sites, while still permitting weak binding, abrogated high affinity interactions, while substitution of one motif on the right side of the palindrome weakened high affinity binding, resulting in a Kd of 195 ± 10.8 nM (Figure 4(B)). Collectively, these FP experiments indicate that pSK1 Par, while displaying some DNA binding promiscuity, requires at least one centromere consensus half site for high affinity binding.
Analysis of pSK1 Par oligomerization.
As noted, the region of Par C-terminal to the wHTH is predicted to contain a short, disordered region followed by a coiled coil (CC). There are no interactions between the wHTH domains in the structure, however CC regions are known to be involved in mediating oligomerization. To assess if pSK1 Par(1–170), which harbors the CC domain, forms oligomers we performed size exclusion chromatography (SEC) experiments. These studies revealed a molecular weight of ~90 kDa for Par(1–170), which is most consistent with a Par(1–170) tetramer or dimer-of-dimers (Figure S4(A)). However, to further assess if the CC region is responsible for oligomer formation we purified His6-tagged Par constructs encompassing the wHTH, (Par(1–65)), and the CC, Par(78–170), and examined these proteins by SEC. The SEC analyses showed that the wHTH eluted at a volume consistent with a monomer (~12 kDa), while the CC region eluted at a predicted MW consistent with a dimer-of-dimer or tetramer (~65 kDa) (Figure S4(B), (C)), in line with the notion that the CC domain, indeed, mediates oligomer formation. We next used Alphafold 2 to generate a model55–57 of FL pSK1 Par (Figure 4(D)). Alphafold 2 indicated that pSK1 Par residues 1–56 adopt a wHTH fold similar to our structure (rmsd = 0.40 Å for superimposition of 51 Cα atoms of the wHTH crystal structure and the Alphafold wHTH prediction) with high confidence [predicted local Distance Difference Test (pLDDT) > 90] (Figure S4(D)). The structure of residues 57–78, was modeled by Alphafold 2 as a flexible loop-like region, consistent with our data showing this region is flexible and proteolytically sensitive, but the exact loop structure was predicted with low confidence (<30%). Notably, the Par region from residues 78–178 was predicted to form an extended CC with an even higher degree of confidence than the wHTH (pLDDT of > 95%) (Figure S4(D)). The CC region was also predicted to favorably dimerize by Alphafold 2 (Figure 4(C)). Finally, Alphafold 2 modeled the Par C-terminal region as an extended loop-like structure, but several loop structures were predicted and with low reliability. Docking experiments further revealed that two Par dimers could interact to form a dimer-of-dimer through the CC dimer domains, as indicated by our SEC analyses (Figure 4(B), (C)).
Circular dichroism (CD) can be used to detect the presence of CC as structures containing CC give a characteristic spectra with a 220 nm/209 nm ratio of 1 or greater than 1 for proteins or domains composed entirely of CC. By contrast, folded, non-CC helical proteins58–59 produce spectra with 220 nm/209 nm of 0.6–0.8. The prediction that ~100 residues of Par(1–170) contain a CC indicated that this should be observable by CD analyses. Indeed, CD analyses on Par(1–170) resulted in a 220 nm/209 nm ratio of 0.97, strongly supporting the presence of the CC in the region C-terminal to the wHTH (Figure S5). However, to further test this hypothesis, we analysed (Par(1–65)) and Par(78–170) by CD. Consistent with the prediction that Par residues 78–170 contain a CC, the 220 nm/209 nm from the CD spectra for Par(78–170) was 1.01, while the ratio for Par(1–65), which encompasses only the wHTH, was 0.75 (Figure S5).
Probing the role of Par domains in DNA binding.
To next interrogate the role of specific Par residues and domains in binding to the FL centromere-like site we performed EMSAs studies with a series of Par constructs encoding domains and mutant proteins (Figure 5(A)–(E)). The structure revealed that conserved residue Lys15 makes key contacts to the centromere phosphate backbone as well as DNA bases. Thus, we substituted this residue to alanine. The ParK15A mutant protein showed no binding to the probe over the concentration range tested compared to WT (Figure 2(A); Figure 5(B)), consistent with the key role of this residue in DNA binding revealed in the structure. The Par C-terminal disordered region is not predicted to be involved in DNA binding (Figure 4(C)). Indeed, a Par deletion mutant (missing residues 171–245 (ParΔCTD)) bound the centromere site (Figure 5(C)). We next generated a truncated form of Par, ParΔCC, in which the predicted CC region, residues 83–155, was removed. This protein bound the DNA probe weakly (Figure 5(D)). Our pSK1 Par model predicts that residue L132 would be key for dimer formation as the residue lies in the center of the CC dimer interface. Hence, a L132A mutation was generated. The ParL132A mutant protein also showed reduced binding to the centromere (Figure 5(E)). The finding that residues and regions involved in pSK1 Par oligomer formation impact DNA binding are in line with previous studies showing that oligomerization by a domain distant from the DNA-binding domain of a DNA binding protein can significantly increase DNA binding affinity through mass action3,9.
Figure 5.
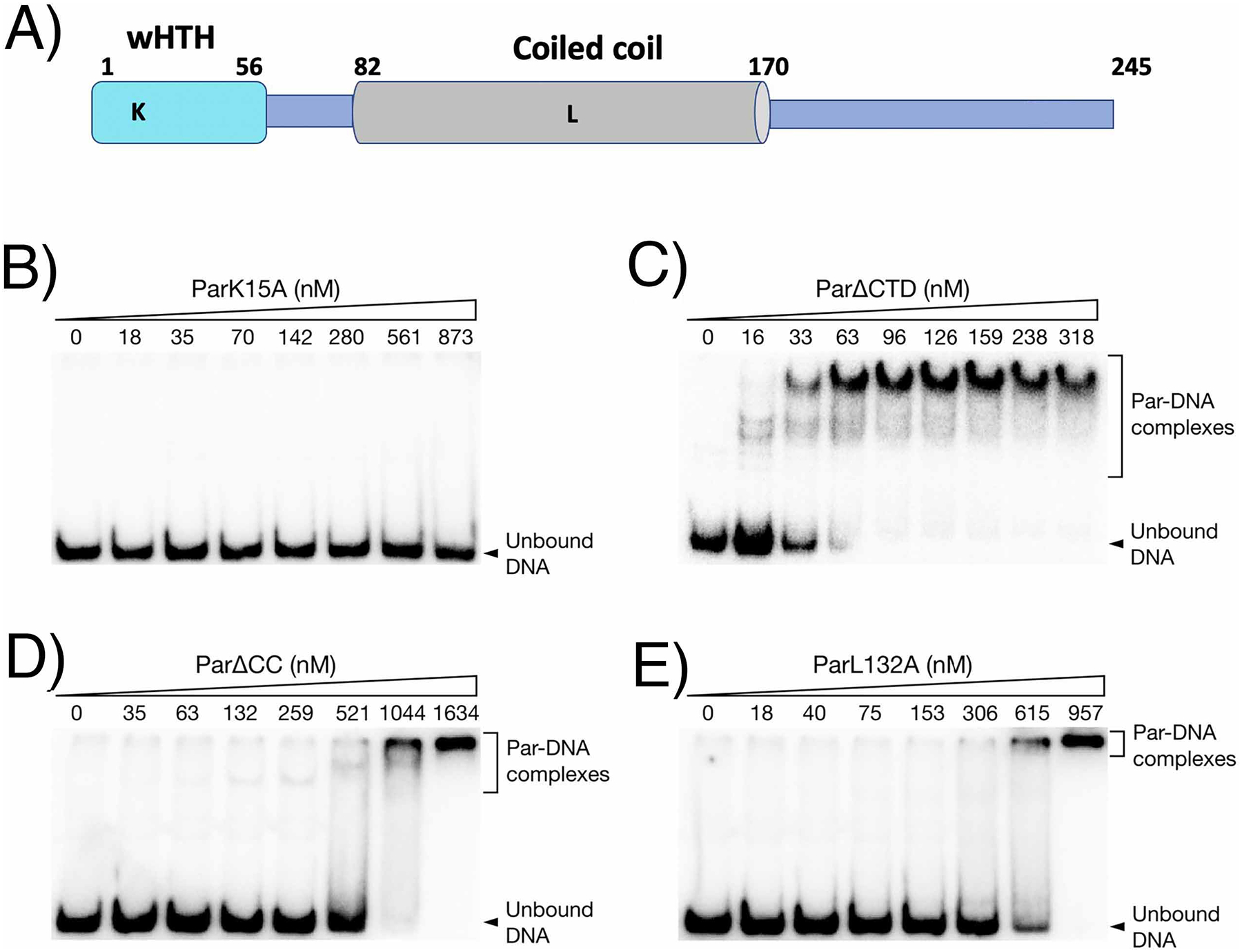
EMSAs of mutant Par binding to the par-rep intergenic region. (A) schematic showing pSK1 Par domains and locations of residues mutated for biochemical and in vivo studies. EMSA analyses for: (B) ParK15A, (C) ParΔCTD, (D) ParΔCC and (E) ParL132A.
Par autoregulates its own expression.
Partition protein levels are known to be tightly regulated as altered expression levels of these proteins results in segregation defects4,6,13–14. Par-DNA footprinting experiments showed that a segment of the protected region overlaps Ppar (Figure 2(C), Figure S1), suggesting that Par might regulate its own expression. To investigate this possibility, a 1.7 kb fragment (nt 985–2727, GenBank entry GU565967), which encompasses par and the upstream repeats, was cloned into the promoter probe vector pRB394. Two stop codons were introduced into the resultant plasmid (pSK6144), producing the non-functional derivative pSK6152. These plasmids were individually electroporated into S. aureus RN4220 cells and CAT reporter assays were performed. The CAT activities from pSK6144 and pSK6152 were 142.7±17.1 and 923.3±63.3 units, respectively. Therefore, the presence of an intact par gene in pSK6144, resulted in an 84.5% decrease in transcriptional activity, indicating that par is autoregulated.
Par and pSK1 plasmid localization in S. aureus.
To determine the localization of Par in live S. aureus cells we next constructed a Par-GFP fusion (pSK9088, Ppar::par-gfp), which expresses GFP at the C-terminus of Par. Because Par-GFP was only partially functional (Figure S6), we provided WT Par from a compatible IPTG-inducible expression plasmid. When provided in trans, plasmid segregational stability increased in a dose-dependent manner to levels approaching pSK4829 (Figure 6(A)). Importantly, since Par alone was unable to fully complement pSK4833 (Δpar) plasmid instability in trans (Figure S7), these data suggest that cis-encoded Par-GFP was contributing to the stability observed when Par was supplied in trans (Figure 6(A)). Par-GFP was visualized as foci, with typically one to four foci observed per cell (Figure 6(B), upper). ParΔCC-GFP, which lacks the CC oligomerization domain, did not produce fluorescent foci (Figure 6(B), middle), suggesting that foci were not artefacts of GFP aggregation. The DNA-binding mutant, ParK15A-GFP, formed fluorescent foci similar to Par-GFP although the foci were more diffuse than in the WT (Figure 6(B), lower), perhaps reflecting Par multimerization in the absence of DNA binding.
Figure 6.
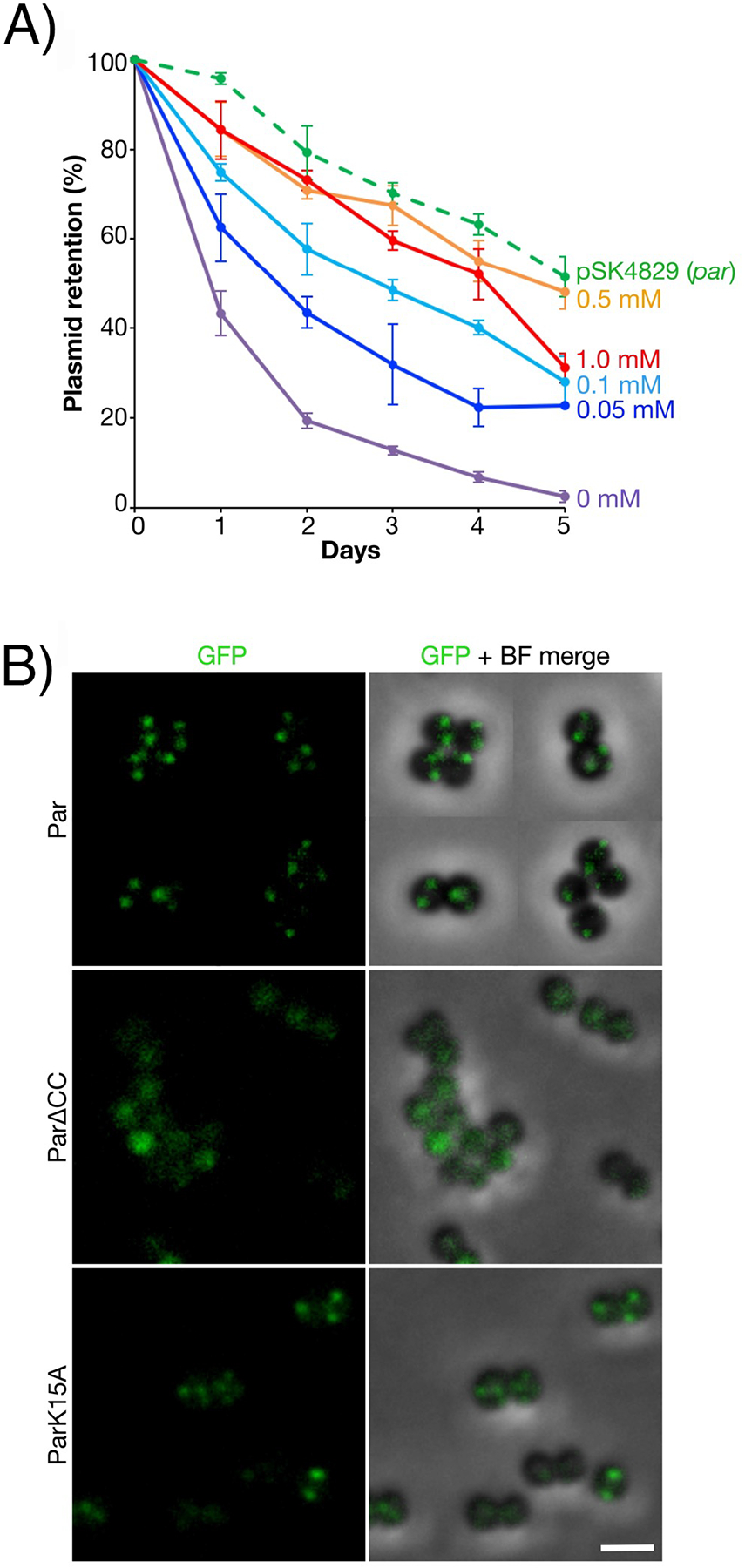
Fluorescence localization of Par-GFP in S. aureus. (A) Segregational stability of pSK1 minireplicons expressing Par-GFP (pSK9088, Ppar::par-gfp) in the presence of untagged Par, provided in trans from pSK9104 (Pspac::par), induced with 0 mM (purple), 0.05 mM (dark blue), 0.1 mM (light blue), 0.5 mM (orange) or 1.0 mM (red) IPTG. The segregational stability of pSK4829 is shown for comparison. Each data point is the mean of three independent assays, each normalized to 100% plasmid retention on Day 0. Error bars indicate standard error of the mean. (B) Fluorescence localization of Par-GFP in S. aureus cells. Par-GFP (pSK9088, Ppar::par-gfp) was expressed in the presence of untagged Par, supplied in trans from a compatible, co-resident expression plasmid (pSK9104, Pspac::par), induced with 1.0 mM IPTG (upper). Fluorescence localization of ParΔCC-GFP (pSK9103, Pspac::parΔCC, middle) and ParK15A-GFP (pSK9102, Pspac::parK15A, lower) in S. aureus, induced with 0.1 mM IPTG in trans to ParΔCC (pSK7721, Ppar::parΔCC) and ParK15A (pSK7764, Ppar::parK15A), respectively. Shown are GFP (left) and merged images of GFP and bright-field (BF) channels. Scale bar = 2 μm.
We next localized pSK1 minireplicons in live S. aureus cells. Since it could not be assumed that all Par-GFP foci were bound to centromere-DNA, we used a TetR-GFP/tetO fluorescent repressor-operator system (FROS) to fluorescently label plasmid DNA. tetO arrays were inserted in pSK1 minireplicons with and without par, while TetR-GFP was expressed from an IPTG-inducible co-resident plasmid. The presence of par enhanced the segregational stability of pSK1 minireplicons carrying the tetO array (Figure 7(A)). Importantly, we observed no dramatic effect of TetR-GFP production on the segregational stability of tetO-carrying plasmids (Figure 7(A)).
Figure 7.
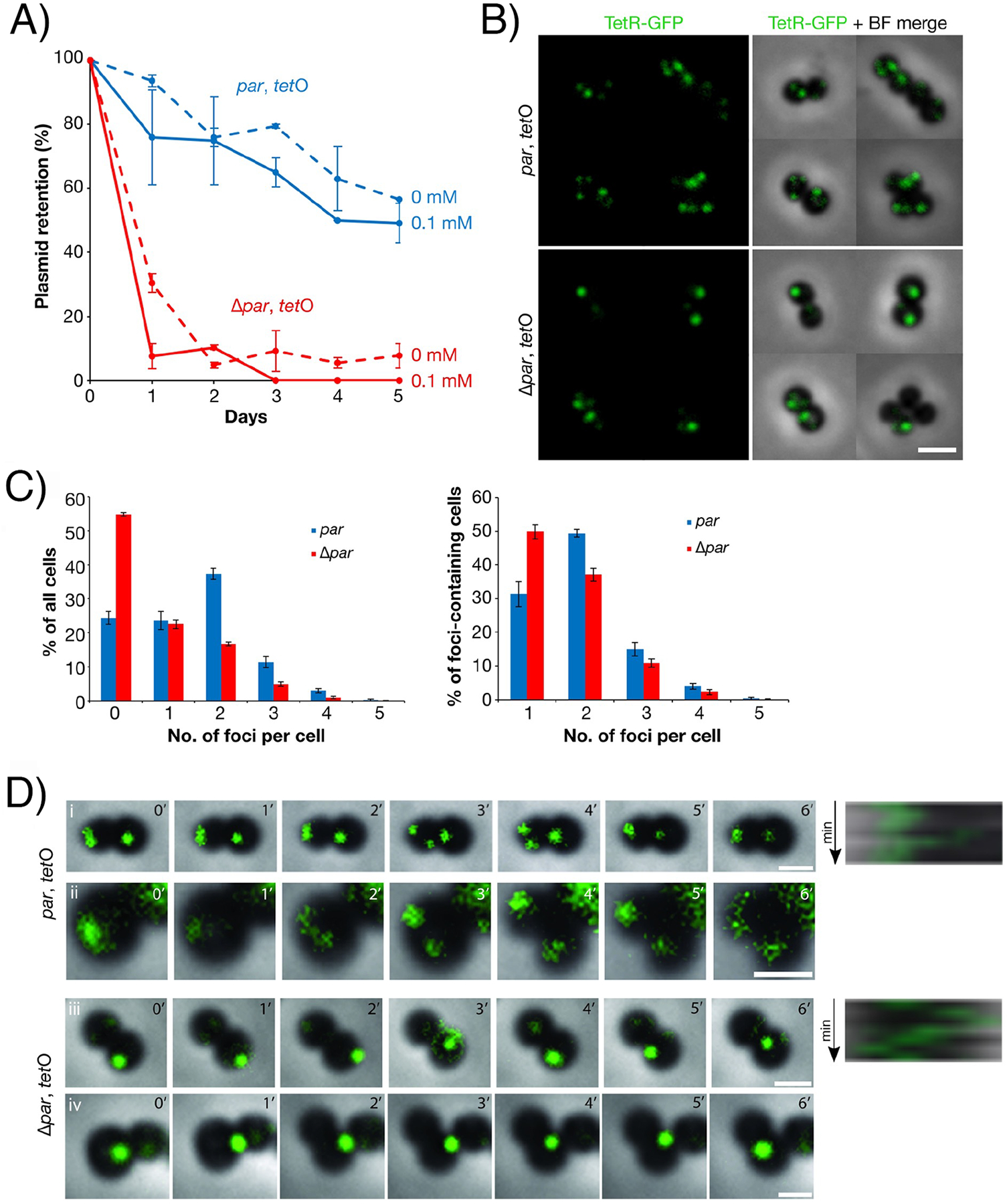
Fluorescence localization of pSK1 minireplicons in S. aureus using a TetRGFP/tetO fluorescent repressor-operator system. (A) Plasmid segregational stability assays of pSK1 minireplicons carrying tetO arrays in the presence (pSK9144, par, blue) and absence (pSK9145, Δpar, red) of par. Expression of TetR-GFP was induced from a co-resident plasmid, pSK9142 (Pspac::tetR-gfp), with 0 mM IPTG (dashed lines) or 0.1 mM IPTG (solid lines). Each data point is the mean of three independent assays, each normalized to 100% plasmid retention on Day 0. Error bars indicate standard error of the mean. (B) Fluorescence localization of pSK1 minireplicons pSK9144 (par, tetO, upper) and pSK9145 (Δpar, tetO, lower) in S. aureus cells. Plasmids were visualized using TetRGFP induced from pSK9142 (Pspac::tetR-gfp) with 0.1 mM IPTG. Scale bar = 2 μm. (C) Number of fluorescent plasmid foci per cell, as detected by epifluorescence microscopy, in S. aureus cells harboring pSK1 minireplicons pSK9144 (par, tetO, blue bars; ntotal = 1,362) or pSK9145 (Δpar, tetO, red bars; ntotal = 1,616). Histograms show the percentage of all cells (left) and foci-containing cells (right) against the number of plasmid foci per cell. Values shown are the mean of four independent experiments. Error bars represent standard error of the mean. (D) Time-lapse epifluorescence microscopy of pSK1 minireplicons pSK9144 (par, tetO) and pSK9145 (Δpar, tetO) in S. aureus cells, labeled using TetR-GFP as in (B). Images were captured at 1 min intervals for six minutes (0′–6′). Cells in (i and ii) and (iii and iv) are biological replicates. Images shown are overlays of GFP and bright-field channels. Kymographs for the right-most cell in (i) and (iii) are shown on the right and were measured anti-clockwise along the cell circumference from the point indicated by the red arrowheads. Scale bars = 1 μm. Movies of time-lapse micrographs are shown in Movies S1–S4.
Fluorescence microscopy revealed that S. aureus cells harboring tetO array plasmids commonly contained one to four fluorescent foci (Figure 7(B), upper panel). Only diffuse TetR-GFP fluorescence was observed in the absence of tetO arrays (Figure S8), indicating that TetR-GFP foci were formed only by binding specifically to tetO arrays. The distribution of plasmid focus numbers in cells with and without par was significantly different (p<2.2×10−16), with a substantially higher proportion of cells containing no plasmid foci in cells lacking par compared to cells containing par (55±1% and 24±2%, respectively, Figure 7(B), lower and Figure 7(C), left), reflecting the segregational instability of Δpar plasmids (Figure 7(A)). When considering only foci-containing cells, the distribution of plasmid focus numbers in cells with and without par was also significantly different (p<2.6×10−9). Specifically, in the absence of par, approximately half of the foci-containing cells contained only one plasmid focus (50±2%), which was significantly higher than the proportion for cells that contained par (31±4%, p<0.05, Figure 7(C), right). Also, the single plasmid focus in Δpar cells was often of high fluorescence intensity (Figure 7(B), lower). Compared to cells lacking par, a higher proportion of cells carrying par-encoding plasmids contained two plasmid foci (49±1% compared to 37±2% without par; p<0.05), which was also the most frequent number of observed foci for par-containing plasmids (Figure 7(C), right).
pSK1 plasmid segregation.
We next performed time-lapse epifluorescence microscopy on S. aureus cells carrying pairs of FROS plasmids, with and without par, to track the localization of pSK1 minireplicons during cell division. In the presence of par, TetR-GFP foci, and hence plasmid DNA, were either static or appeared to have a restricted trajectory confined to a small region (Figure 7(D)i kymograph and Movie S1). Plasmid foci were also observed to converge and separate during the time-lapse experiments, and thus the number of plasmid foci varied during the cell cycle (Figure 7(D)i and Movie S1). In dividing cells, TetR-GFP appeared as a single larger, more intense fluorescent focus, possibly representing a pair or cluster of replicated plasmids (Figure 7(D)ii, panel 0′ and Movie S2). In par+ cells, the large focus then separated into two or more foci, which were segregated into individual daughter cells upon cell division (Figure 7(D)ii, panels 2′–4′ and Movie S2).
In contrast to the movement of par+ pSK1 minireplicons, the movement of Δpar plasmids was less restricted, and foci often appeared to traverse the cell diameter (Figure 7(D)iii kymograph and Movie S3). Because many cells carrying Δpar plasmids contained no, or only one, TetR-GFP focus (Figure 7(C)), we expected that plasmid segregation would be inefficient. Indeed, when dividing cells contained a single focus, the Δpar plasmid focus did not separate into more foci, unlike what was observed for par+ plasmids (Figure 7(D)iv and Movie S4). Instead, the single focus remained within one cell and was not inherited by the other daughter cell (Figure 7(D)iv and Movie S4). Even when two plasmid foci were observed, the foci were not equally segregated (Movie S5), highlighting the reduced plasmid segregational stability of par-deficient pSK1 minireplicons.
Discussion
The mechanism of pSK1 par function in plasmid segregational stability is of significant genetic difference from characterized active plasmid partitioning systems. Namely, that pSK1 par encodes only a single protein and hence lacks an identifiable NTPase component that could drive plasmid segregation. pSK1 Par also shows no sequence homology to other plasmid CBP proteins. That includes the E. coli R388 StbA protein, which also appears to mediate segregation of its plasmid without the assistance of an NTPase. Hence, to characterize the pSK1 Par protein we performed biochemical, structural and in vivo studies. Our pSK1 Par-centromere DNA crystal structure combined with mutagenesis and DNA binding assays reveal the mechanism of DNA binding by Par. In particular, these studies demonstrate that DNA binding is mediated by the pSK1 Par N-terminal wHTH domain. This domain is attached to a flexible linker, followed by predicted CC and C-terminal disordered regions. Consistent with the lack of sequence homology, the pSK1 Par DNA-binding domain structure revealed no homology to other plasmid CBP proteins. But strikingly, it showed structural similarity to RacA, which is a B. subtilis protein required for chromosome segregation during sporulation46–49. RacA forms dimers and dimer-of-dimer oligomers via interactions mediated by a CC C-terminal to its DNA-binding domain46–49. Similarly, our biochemical experiments revealed that the Par CC forms oligomers. Combined DNA binding and in vivo results suggest that this oligomerization is important in Par segregation function. Indeed, oligomerization by a distant, physically separated domain from the DNA-binding domain of a regulator has been shown to be one way to increase DNA binding affinity through enhanced proximity or mass action,9. In particular, DNA binding by bacterial segregation ParB proteins containing HTH DNA-binding motifs have been shown to be significantly enhanced by the oligomerization of domains connected to the HTH domains by flexible linkers9.
From a mechanistic perspective, the sequence of events that occurs subsequent to Par centromere-binding, in particular, the physical segregation of plasmids to daughter cells, remains to be revealed. However, our study showed a significant difference in the distribution of plasmid focus numbers between cell populations carrying plasmids with and without par. Specifically, time-lapse fluorescence imaging of plasmid localization revealed that in par+ cells, pSK1 minireplicons separate into two or more foci, at least one of which is inherited by each daughter cell (Figure 7(D)ii). Plasmids lacking par failed to separate into new foci and did not segregate evenly into daughter cells (Figure 7(D)iv); Figure 7(C)).
In most plasmid partitioning systems described to date, NTPase activity plays a key role in plasmid segregation dynamics either through the formation of filaments or the ability of the NTP bound protein, in the case of type I systems, to bind nucleoid DNA and attract plasmid bound ParB27–33. However, pSK1 Par harbors no known NTPase motifs and shares no sequence or structural similarity with known NTPases and we did not observe any filament formation by Par-GFP. We do not exclude the possibility that interactions between Par and host-encoded proteins, such as nucleoid associated proteins, might contribute to plasmid segregation (Figure 8(A)). However, our yeast two-hybrid screening of an S. aureus genomic DNA prey library using FL Par as bait did not reveal any biologically meaningful interaction partners. RacA, which shows structural homology to pSK1 Par and which functions in B. subtilis chromosome segregation during sporulation binds the architectural protein DivIVA to enable partition. Such a mechanism involving an architectural protein(s) may be at play for pSK1 Par (Figure 8(B)). But, as noted, we observed no interactions between pSK1 Par and DivIVA. And though these studies lacked centromeric DNA binding sites and S. aureus chromosomal DNA, we note that interactions identified between RacA and DivIVA did not require DNA substrates to be present46.
Figure 8.
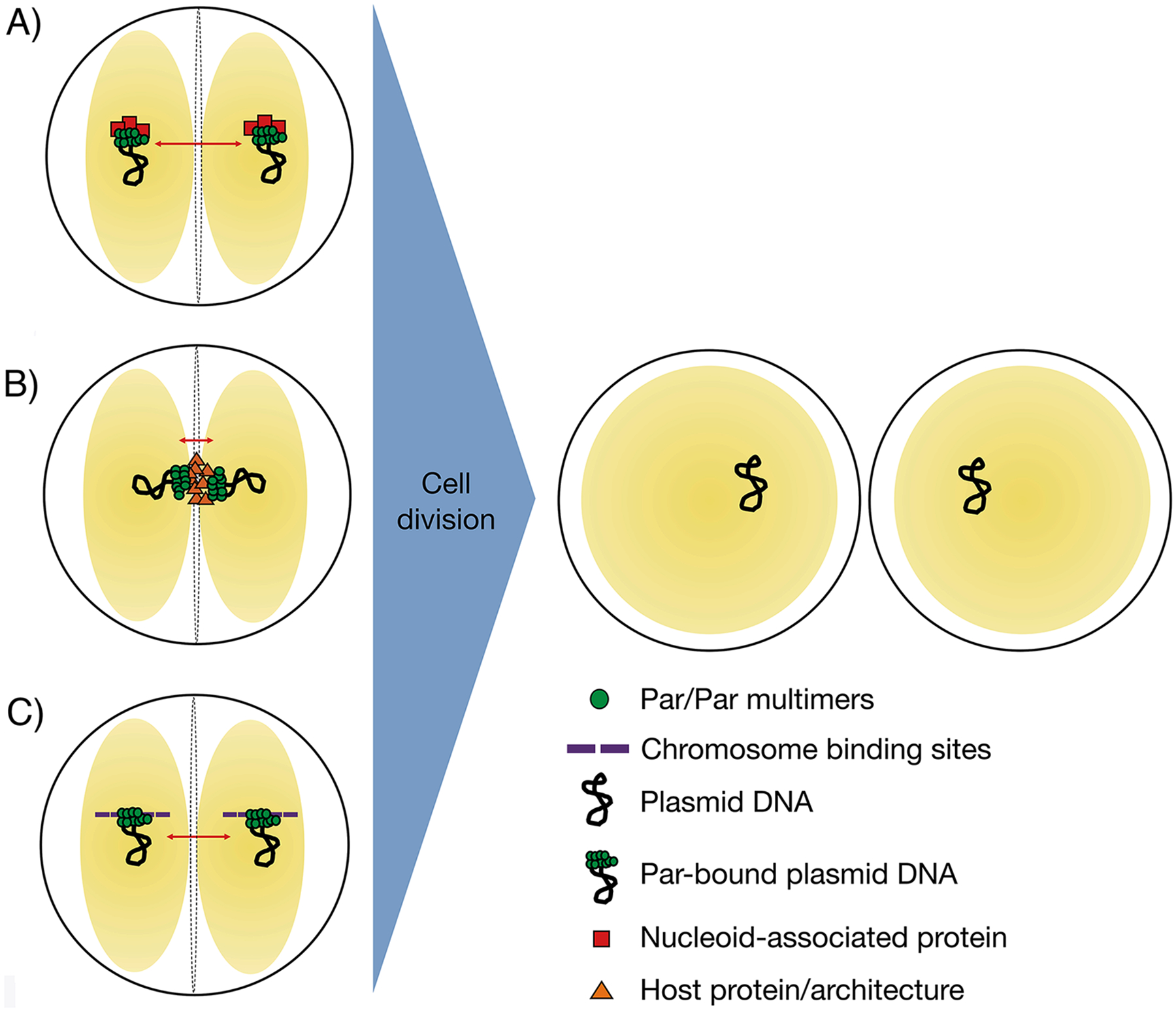
Proposed possible models for plasmid segregation by pSK1 par. (A) Par acts as an adapter between plasmid DNA and nucleoid-associated proteins. Par-bound plasmid DNA is therefore tethered to the nucleoid (yellow spheres), such that plasmids are segregated (red arrows) together with chromosomes. (B) Par-bound plasmid DNA is anchored to specifically-localized host-encoded proteins or components of the host cell architecture, such as the division septum or cell poles. The nucleoid DNA is represented as yellow spheres. (C) Par binds to the plasmid centromere-like site and interacts directly with the nucleoid (yellow spheres), thus piggy backing with its plasmid along with the nucleoid DNA during segregation of chromosomal DNA. Correct subcellular localization of plasmid DNA at predetermined positions ensures accurate plasmid segregation upon cell division. Right, S. aureus cells are represented by black circles, the nucleoid DNA as yellow spheres.
Perhaps the most plausible pSK1 plasmid segregation mechanism is the utilization of the host nucleoid DNA. In such a hitch-hiking mechanism (Figure 8(C)), one pSK1 Par dimer could bind the pSK1 Par plasmid via interactions with centromere DNA, while the other pSK1 Par dimer could interact with the S. aureus host nucleoid (Figure 4(C); Figure 8(C)). The high concentration of DNA in the S. aureus nucleoid, which is AT-rich, could possibly enable such binding even if low affinity. In fact, a search of the S. aureus genome revealed multiple (nineteen) matches to 10 bp of the centromere repeat, TTAGGTGCCT, and more than 20 sequences with 9 of the 12 bp conserved. Our FP data showed Par can bind DNA sites that contain one half site. Thus, pSK1 Par may be capable of binding these sites within the host genome if the sites are accessible. However, it is not clear how this mechanism would explain efficient segregation to new daughter cells; how it would be assured that pSK1 Par would bind to both segregating chromosomes. Hence, further analyses will be needed to elucidate the in vivo means of Par-mediated plasmid distribution and any proposed mechanism needs to account for better than random plasmid segregation at a pSK1 copy number of only around five per cell60. Thus, a hitch-hiking mechanism might only be feasible if the number of potential segregating “hitching” sites are limiting. In summary, our studies have shown that pSK1 Par is necessary for pSK1 plasmid segregation, described the specific properties of the system and revealed the structure of the Par DNA-binding domain and DNA binding mechanism for this unique one-protein segregation system. Future studies will unveil the in vivo steps and means of physical separation of pSK1 plasmids.
Materials and Methods
Bacterial strains, growth conditions, plasmids and oligonucleotides.
Bacterial strains, plasmids and oligonucleotides used are listed in Table S1. Electrocompetent S. aureus cells were prepared using B2 medium as described previously61.
DNA manipulations.
Plasmid DNA was isolated from E. coli using the alkaline lysis method62 or the Quantum Prep plasmid miniprep kit (Bio-Rad, Hercules, California). Cloning in E. coli was performed using standard methods63 and restriction enzymes, calf alkaline phosphatase and T4 DNA ligase were purchased from New England Biolabs (Ipswich, Massachusetts). DNA fragments were PCR-amplified using Taq (New England Biolabs) or Pfu Turbo DNA polymerase (Stratagene, La Jolla, California) and site-directed mutagenesis was performed using Stratagene’s QuickChange® method. Primers for mutagenesis are described in Table S1. Automated DNA sequencing was performed by the Australian Genome Research Facility.
Construction of plasmids.
Plasmids were constructed as described below.
pSK9088 (Ppar::par-gfp).
Primers 3130BamHI and 994gfp were used to PCR-amplify a 2.1 kb fragment encompassing pSK1 rep and par from pSK4829 template DNA. A separate reaction was used to amplify gfpmut-1 from pLOW-GFP template DNA using primers gfp_linker and gfpHindIII. Primers 994gfp and gfp_linker contain complementary sequences encoding an SCGAS linker64 to be introduced between Par and GFP such that overlap extension PCR using primers 3130BamHI and gfpHindIII fused the GFP coding region downstream of the rep-par fragment, producing a 2.9 kb fusion product containing pSK1 rep and par-gfp in the same genetic organisation as rep and par on pSK1. The 2.9 kb overlap extension PCR product encoding Par-GFP was then ligated to the restricted and dephosphorylated BamHI and HindIII sites of pWE180.
pSK9102 (Pspac::parK15A-gfp) and pSK9103 (Pspac::parΔCC-gfp).
A DNA fragment containing the par ribosome binding site (RBS) and ParK15A or ParΔCC coding region, excluding the stop codon, was amplified by PCR from pSK7764 and pSK7721 template DNA, respectively, using primers 1743SalI and 990BamHI. Subsequent ligation of the amplicon to the restricted SalI and BamHI sites of pJEG015 resulted in an N-terminal fusion in-frame with the GFP coding sequence present on the vector.
pSK9104 (Pspac::par).
A 0.8 kb DNA fragment containing the par ribosome binding site (RBS) and Par coding region, including the stop codon, was amplified by PCR from pSK4829 template DNA using primers 1743SalI and 988HindIII, and cloned into the SalI and SmaI sites of pJEG015 such that the stop codon was located between the par and gfp sequences.
pSK9142 (Pspac::tetR-gfp).
The ermC erythromycin resistance gene on the S. aureus expression plasmid pSK9067 was first replaced with the aadD neomycin resistance gene. The aadD gene was obtained by restriction digestion of pSK9065 with NcoI and SphI. This fragment was subsequently ligated with the blunted KpnI and ClaI sites of pSK9067 after removal of overhangs by the Klenow fragment of DNA polymerase I, generating pSK9140. Tn10 tetR was amplified from pSK9065 using primers tetRSalI and tetRBamHI, which contain the strong S. aureus superoxide dismutase (SOD) RBS and a five-codon linker (SCGAS). The tetR amplicon was ligated with pSK9140 at the SalI and BamHI sites to generate pSK9142.
pSK9144 (par, tetO) and pSK9145 (Δpar, tetO).
A tetO array was amplified from pLAU44 using primers tetOKasIF and tetOKasIR. The amplicon was then restricted with KasI, resulting in a 2.2 kb fragment consisting of approximately 60 copies of tetO, which was cloned into the KasI site of pSK4829 (par) and pSK4833 (Δpar), generating pSK9144 and pSK9145, respectively.
Plasmid segregational stability assays.
Strains to be assayed were grown overnight in LB medium supplemented with appropriate selection for the plasmid, subcultured (10−4 dilution) into selective LB medium and grown for 4 h. A viable count was then performed using non-selective LB plates and 100 μl of the culture was used to inoculate 10 ml of LB medium. After overnight growth without selection, a subculture (10−4 dilution) and viable count was again performed and the process repeated as necessary. Each day 100 colonies from the viable count plates were patched onto selective and non-selective LB plates to determine the proportion of the population that retained the plasmid. Plasmid DNA was isolated from selected colonies to confirm the presence or absence of the plasmid.
Chloramphenicol acetyltransferase (CAT) reporter gene assays.
CAT reporter gene assays were performed using soluble protein extracts from S. aureus RN4220 cells as described previously65. Lysostaphin, acetyl Coenzyme A and 5–5’-dithio-bis[2-nitrobenzoic acid] were purchased from Sigma-Aldrich and bovine serum albumin (BSA) from New England Biolabs. CAT units are expressed as nanomoles of chloramphenicol acetylated mg−1 of protein min−1 at 37 °C and are the average of at least three independent assays.
Protein production and purification.
The pSK1 par coding region was PCR-amplified using the primer pair 991HindIII and 1722BamHI, digested with HindIII and BamHI, and cloned into the respective sites of the expression vector pQE-30 (Qiagen, Hilden, Germany) such that a 6x histidine tag would be located at the N-terminal end of FL pSK1 Par to facilitate protein purification. The resulting construct, pSK5344, was checked by DNA sequencing and used to transform E. coli M15 containing pREP4. Plasmid DNA extracted from this strain was subsequently used to generate, via site-directed mutagenesis, Par overexpression constructs that contain the domain mutations described in Figure 5. All these Par constructs used in analyses shown in Figure 2(A)–(C) and Figure 5 were purified as described below.
An overnight culture of E. coli M15 containing pREP-4 and pSK5344 (or the mutant derivatives thereof) was diluted 1:50 in LB medium and grown to an OD600 of 0.6. Protein expression was induced by the addition of isopropyl β-D-thiogalactopyranoside (IPTG) to a final concentration of 0.1 mM. Growth was continued at 30 °C for an additional 3.5 h. FL pSK1 Par was found in inclusion bodies and hence had to be purified via a denaturation/renaturation process. Similarly the proteins utilized in experiments shown in Figure 5 were purified via this method. For purification, cells were pelleted and resuspended in denaturing lysis buffer (8 M urea, 100 mM NaH2PO4, 10 mM Tris-HCl, pH 8.0) and lysed using a French press at 20,000 psi. The soluble fraction was collected and incubated with Ni2+-NTA metal chelate affinity agarose at 4°C, washed twice with denaturing wash buffer I (8 M urea, 100 mM NaH2PO4, 10 mM Tris-HCl, pH 6.3) at 4 °C and washed twice with denaturing wash buffer II (8 M urea, 100 mM NaH2PO4, 10 mM Tris-HCl, pH 5.5) at room temperature (rt). Recombinant Par was eluted with denaturing elution buffer (8 M urea, 100 mM NaH2PO4, 10 mM Tris-HCl, pH 4.5) at rt and aliquots of each purification step were analysed by SDS-PAGE. Purified Par was then renatured via exchange into DNA binding buffer (10 mM Tris-HCl pH 7.5, 10 mM MgCl2, 100 mM NaCl, 0.2 mM DTT, 10% glycerol) or phosphate buffered saline (PBS, pH 7.4) using PD-10 desalting columns. Since FL pSK1 Par migrates larger than its predicted size, as previously noted43, the identity of the purified protein was confirmed using peptide mass fingerprinting.
EMSA DNA-binding experiments.
The par-rep intergenic region was PCR-amplified using primers 1689 and 1900, and end-labeled using [γ−32P]ATP (Perkin-Elmer, Waltham, Massachusetts) and T4 polynucleotide kinase (New England Biolabs). The end-labeled DNA probe fragment was purified using the illustra™ DNA and Gel Purification Kit (GE Healthcare), eluted in water and stored at −20°C. EMSAs were performed by incubating the end-labeled probe (6000 cpm) with 2 μg of poly[dI-dC] (Sigma-Aldrich) and increasing amounts of purified Par in DNA binding buffer. Binding reactions (20 μl total volume) were incubated for 30 min at rt and analysed by high ionic strength PAGE, as described by Ausubel et al.66.
DNase I footprinting experiments.
DNase I footprinting was performed using the end-labeled par-rep intergenic fragment (30,000 cpm; both strands were separately labeled and tested), which was incubated with and without purified Par (0.8 μM) using the EMSA conditions described above. The volume of each reaction was brought to 200 μl with DNase I buffer (10 mM Tris-HCl pH 8.0, 5 mM MgCl2, 1 mM CaCl2, 100 mM KCl, 2 mM DTT, 50 μg ml−1 BSA, 2 μg ml−1 salmon sperm DNA). DNase I (Sigma-Aldrich) was added at a concentration predetermined to nick approximately 50% of the DNA once, and digestion was allowed to proceed for 2 min at rt. Digestion was terminated with the addition of 700 μl of DNase I stop solution (92% ethanol, 3 M sodium acetate, 10 μg ml−1 salmon sperm DNA). DNA samples were ethanol precipitated and analysed using denaturing 8% (w/v) polyacrylamide sequencing gels. Sequencing ladders were prepared using the SequiTherm EXCEL II DNA sequencing kit (Epicentre Technologies, Madison, Wisconsin) and gels were imaged using a Molecular Imager FX (Bio-Rad).
Microscopy.
S. aureus cells harboring the relevant plasmids were grown overnight in 10 ml of selective LB-broth at 30°C with aeration. Overnight cultures were used to inoculate 25 ml of fresh selective LB-broth (1:50), containing the appropriate concentrations of IPTG when necessary, and grown until mid-exponential phase (OD600nm approximately 0.6). A 750 μl aliquot of cells was harvested by centrifugation (16,060 ×g / 1 min / RT) and resuspended in 250 μl of PBS. 3 μl of the cell suspension were mounted on 2% (w/v) agarose pads prepared either directly on the slide, or within a Gene Frame (Thermo Fisher Scientific) assembled on the slide.
Epifluorescence microscopy was performed at 100× magnification using a Zeiss AxioImager Z.1 microscope equipped with an AxioCam MRm cooled charge-coupled device (CCD) camera controlled using Zen software (Blue edition, 2012, Carl Zeiss, Oberkochen, Germany). GFP fluorescence was visualized using filter set 44 (Carl Zeiss), and cells were visualized using bright-field microscopy. Image analysis and processing were performed using Zen software (Blue edition, 2012, Carl Zeiss) and Adobe Photoshop CS3 Extended. Cell counts were performed using the Cell Tracker plugin in ImageJ. Kymographs were generated using the KymoResliceWide plugin in ImageJ, using a line width of 50.
Statistical analyses.
A Pearson’s Chi-squared test for homogeneity was performed to determine whether the distribution of par and Δpar cells was equal for the number of foci per cell. Welch’s two-sample t-tests were performed to determine whether the proportion of par+ and Δpar cells was equal for each number of foci per cell. p-values less than 0.05 were interpreted as statistically significant.
Purification of pSK1 Par(1–170), ParCC and Par wHTH for biochemical and structural studies.
Attempts to crystallize refolded FL Par in the absence or presence of centromere DNA failed. Hence, we obtained constructs encoding pSK1 Par(1–170), Par(1–65) and Par(78–170) from Genscript that were codon optimized for protein expression in E. coli. These optimized constructs could be expressed in soluble form. The constructs were transformed into E. coli C41(DE3) cells. For protein expression, par expressing cells were grown to an OD600 of 0.6–0.8 and induced with 1 mM IPTG for 4 hr at 15 °C. The proteins were purified via the same protocol. Specifically, cells were lysed in buffer A (25 mM Tris pH 7.5, 300 mM NaCl, 5% glycerol, 1 mM β-mercaptoethanol) using a microfluidizer and cell debris removed by centrifugation at 17,000 rpm. Lysates were loaded onto a Cobalt NTA column and the column washed with increasing concentrations of imidazole in buffer A. The proteins were eluted in batch mode with 30, 40, 50, 75, 100, 200, 300 500 mM imidazole fractions and samples containing protein were concentrated prior to loading onto an S75 size exclusion chromatography (SEC) column for final purification. Pure fractions were combined and proteins were buffer exchanged into 50 mM Tris pH 7.5, 200 mM NaCl, 5% glycerol and 1 mM DL-dithiothreitol (DTT).
Crystallization and structure determination of pSK1 Par(1–170)-centromere complex.
To form the protein-DNA complexes for crystallization, Par(1–170) was mixed with DNA sites containing centromere repeats at a 1:1.5 ratio and multiple hanging drop vapor diffusion screens were performed at rt. Several deoxyoligonucleotides were used in crystallization with Par(1–170) ranging from the minimal site (12-bp) to 21-bp. Several crystals were formed with DNA sites that were 18-bp or longer. However, the only crystals that showed diffraction beyond 10 Å were obtained in a drop in which the complex had been mixed with a 18-mer DNA site containing a palindrome centromere repeat, (with the top strand having the sequence, 5´-ATGTTAGGTACCTAACTA-3´), and this mixture combined 1:1 with a crystallization reagent consisting of 24% PEG 5000, 0.2 M ammonium sulphate and 0.1 M Tris pH 7.5. The crystals took 6 months to grow. Subsequent to solving the structure some of the solution in the crystallization drop was run on a gel and revealed that the protein had been proteolyzed. The crystals, which take the space group C2, were cryo-preserved from the drop. Data were collected at the Advanced Light Source (ALS) beamline 8.3.1 and processed with MOSFLM and scaled with SCALA67–68. A native data set was collected first to 2.25 Å. After the native data set was collected, potassium tetrachloroplatinate was added to the drop at saturating concentrations to obtain a heavy atom derivative. A fluorescence scan revealed a peak at the platinum (Pt) edge and a single wavelength anomalous diffraction (SAD) data set was collected to 2.79 Å (Table S2). Four clear Pt sites were identified in Autosol confirming that the crystals had been derivatized. Phase determination and density modification resulted in an experimental SAD map that was adequate to construct 80% of the model in O69. For final refinement and model construction, the high-resolution 2.25 Å native data set was utilized. After multiple rounds of model rebuilding in O and refinement in Phenix the model converged to final Rwork/Rfree values of 21.4%/24.6% to 2.25 Å resolution70. The final model contains residues 1–56 of the two pSK1 Par subunits and one DNA duplex. Two 3´ nucleotides on one strand and one on the other strand had weak density and were not included in the final model. X-ray intensity data collection and model refinement statistics are presented in Table S2.
Fluorescence polarization (FP) binding experiments.
To quantify DNA repeat element binding by pSK1 Par(1–170), 18-mer DNA sites were used. Specifically, fluoresceinated versions of the 18-mer DNA site used for crystallization, repeat elements from the centromere site and mutant forms of the DNA were analyzed for pSK1 Par binding. For the experiment, increasing concentrations of the pSK1 Par(1–170) protein was titrated into the sample cell containing 1 nM of the DNA in a buffer of 25 mM Tris pH 7.5, 100 mM NaCl, 5 mM MgCl2. The resultant data were plotted using KaleidaGraph Version 4.5 for Mac; serial # 8011073 (Synergy Software) and the curves fit to deduce binding affinities. Three technical repeats were performed for each curve.
Circular dichroism.
Far-UV CD spectra of pSK1 Par(1–170), Par(1–65) and Par(78–170) were recorded on an AVIV 435 Circular Dichroism Spectrophotometer in a 1 mm sample cell. Measurements were taken from 200–260 nm with a wavelength step of 1.00 nm and a 1.00 s averaging time. The spectra presented are averages of 5 scans. The protein concentrations were ~0.2 mg/ml and the buffer composition was 20 mM NaH2PO4 (pH 7.5), 300 mM NaF, 5% glycerol, and 1 mM βME.
Size exclusion chromatography (SEC) analyses.
SEC studies were carried out on pSK1 Par(1–170) at 0.5 mg/ml. For this analysis, the protein was injected onto a Superdex S75 column (Fisher) with a mobile phase of 50 mM Tris pH 7.5, 300 mM NaCl, 5%(v/v) glycerol. The elution volumes of each sample were compared to a series of protein standards to determine the molecular weights. The standards used for calculation of the standard curve are cytochrome c oxidase (12.4 kDa), carbonic anhydrase (29.0 kDa), albumin (66.0 kDa), alcohol dehydrogenase (150.0 kDa) and β-amylase (200 kDa).
Supplementary Material
Acknowledgements
We thank Ian Grainge and David Sherratt for providing plasmid pLAU44AII. X-ray intensity data were collected at beamline 8.3.1 at the advanced light source (ALS). Beamline 8.3.1 is operated by the University of California Office of the President, Multicampus Research Programs and Initiatives grant MR-328599 and the National Institute of Health (R01 GM124149 and P30 GM124169).
Funding
This work was supported by Project Grants 307620 (to R.A.S. and N.F.) and APP1030003 (to N.F. and S.O.J.) from the National Health and Medical Research Council (Australia) and a National Institutes of Health grant R35GM130290 and Nanaline H Duke Endowed chair (to M.A.S.).
Footnotes
Accession numbers
Coordinates and structure factor amplitudes for the pSK1 Par-DNA complex have been deposited in the Protein Data Bank under the accession code 8CSH.
Conflict of Interest
The authors declare no competing interests.
References
- 1.Gerdes K, Møller-Jensen J, Bugge Jensen R, (2000). Plasmid and chromosome partitioning: surprises from phylogeny. Mol Microbiol. 37, 455–466. [DOI] [PubMed] [Google Scholar]
- 2.Oliva MA (2016). Segrosome complex formation during DNA trafficking in bacterial cell division. Front. Mol. Biosci 3, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Million-Weaver S, Camps M, (2014). Mechanisms of plasmid segregation: have multicopy plasmids been overlooked? Plasmid 75, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baxter JC, Funnel BE, (2014). Plasmid partition mechanisms. Microbiol. Spectr 2, PLAS-0023–2014. [DOI] [PubMed] [Google Scholar]
- 5.Møller-Jensen J, Gerdes K, (2007). Plasmid segregation: spatial awareness at the molecular level. J. Cell. Biol 179, 813–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davey MJ, Funnell BE, (1997). Modulation of the P1 plasmid partition protein ParA by ATP, ADP, and P1 ParB. J. Biol. Chem 272,15286–15292. [DOI] [PubMed] [Google Scholar]
- 7.Lim GE, Derman AI, Pogliano J, (2005). Bacterial DNA segregation by dynamic SopA polymers. Proc. Natl. Acad. Sci. USA 102,17658–17663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayes F, (2000). The partition system of multidrug resistance plasmid TP228 includes a novel protein that epitomizes an evolutionarily distinct subgroup of ParA superfamily. Mol. Microbiol 37, 528–541. [DOI] [PubMed] [Google Scholar]
- 9.Pratto F, Cicek A, Weihofen WA, Lurz R, Saenger W, Alonso JC, (2008). Streptococcus pyogenes SM19035 requires dynamic assembly of ATP-bound ParA and ParB on parS DNA during plasmid segregation. Nucleic Acids Res. 36, 3676–3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ravin NV, Rech J, Lane D, (2003). Mapping of functional domains in F plasmid partition proteins reveals a bipartite SopB-recognition domain in SopA. J. Mol. Biol 329, 875–889. [DOI] [PubMed] [Google Scholar]
- 11.Golovanov AP, Barillà D, Golovanova M, Hayes F, Lian L-Y, (2003). ParG, a protein required for active partition of bacterial plasmids, has a dimeric ribbon–helix–helix structure. Mol. Microbiol 50,1141–1153. [DOI] [PubMed] [Google Scholar]
- 12.Davis MA, Martin KA, Austin SJ, (1992). Biochemical activities of the ParA partition protein of the P1 plasmid. Mol. Microbiol 6,1141–1147. [DOI] [PubMed] [Google Scholar]
- 13.Hirano M et al. , (1998). Autoregulation of the partition genes of the mini-F plasmid and the intracellular localization of their products in Escherichia coli. Mol. Gen. Genet 257, 392–403. [DOI] [PubMed] [Google Scholar]
- 14.Friedman SA, Austin SJ, (1988). The P1 plasmid-partition system synthesizes two essential proteins from an autoregulated operon. Plasmid 19,103–112. [DOI] [PubMed] [Google Scholar]
- 15.Bork P, Sander C, Valencia A, (1992). An ATPase domain common to prokaryotic cell cycle proteins, sugar kinases, actin, and hsp70 heat shock proteins. Proc. Natl. Acad. Sci. USA 89, 7290–7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Møller-Jensen J, Borch J, Dam M, Jensen RB, Roepstorff P, Gerdes K, (2003). Bacterial mitosis: ParM of plasmid R1 moves plasmid DNA by an actin-like insertional polymerization mechanism. Mol. Cell 12, 1477–1487. [DOI] [PubMed] [Google Scholar]
- 17.Schumacher MA, et al. , (2007). Segrosome structure revealed by a complex of ParR with centromere DNA. Nature 450, 1268–1271. [DOI] [PubMed] [Google Scholar]
- 18.Jensen RB, Lurz R, Gerdes K, (1998). Mechanism of DNA segregation in prokaryotes: replicon pairing by parC of plasmid R1. Proc. Natl. Acad. Sci. USA 95, 8550–8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jensen RB, Dam M, Gerdes K, (1994). Partitioning of plasmid R1. The parA operon is autoregulated by ParR and its transcription is highly stimulated by a downstream activating element. J. Mol. Biol 236,1299–1309. [DOI] [PubMed] [Google Scholar]
- 20.Tang M, Bidesh DK, Park H-W, Federici BA, (2006). Minireplicon from pBtoxis of Bacillus thuringiensis subsp. israelensis. Appl. Environ. Microbiol 72, 6948–6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aylett CHS, Löwe J, (2012). Superstructure of the centromeric complex of TubZRC plasmid partitioning systems. Proc. Natl. Acad. Sci. USA 109,16522–16527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ni L, Xu W, Kumaraswami M, Schumacher MA, (2010). Plasmid protein TubR uses a distinct mode of HTH-DNA binding and recruits the prokaryotic tubulin homolog TubZ to effect DNA partition. Proc. Natl. Acad. Sci. USA 107, 11763–11768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soh YM, et al. , (2019). Self-organization of parS centromeres by the ParB CTP hydrolase. Science 366, 1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Osorio-Valeriano M, Altegoer F, Steinchen W, Urban S, Liu Y, Bange G, Thanbichler M, (2019). ParB-type DNA segregation proteins are CTP-dependent molecular switches. Cell 179, 1512–1524. [DOI] [PubMed] [Google Scholar]
- 25.Jalal AS, et al. , (2021). A CTP-dependent gating mechanism enables ParB spreading on DNA. Elife 10, e69676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osorio-Valeriano M, et al. , (2021). The CTPase activity of ParB determines the size and dynamics of prokaryotic DNA partition complexes. Mol. Cell 81, 3992–4007. [DOI] [PubMed] [Google Scholar]
- 27.Antar H, et al. , (2021). Relief of ParB autoinhibition by parS DNA catalysis and recycling of ParB by CTP hydrolysis promote bacterial centromere assembly. Sci. Adv 7, eabj2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taylor JA, Seol Y, Budhathoki J, Neuman KC, Mizuuchi K, (2021). CTP and parS coordinate ParB partition complex dynamics and ParA-ATPase activation for ParABS-mediated DNA partitioning. Elife 10, e65651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garner EC, Campbell CS, Weibel DB, Mullins RD, (2007). Reconstitution of DNA segregation driven by assembly of a prokaryotic actin homolog. Science 315,1270–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ringgaard S, van Zon J, Howard M, Gerdes K, (2009). Movement and equipositioning of plasmids by ParA filament disassembly. Proc. Natl. Acad. Sci. USA 106, 19369–19374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ebersbach G, et al. , (2006). Regular cellular distribution of plasmids by oscillating and filament-forming ParA ATPase of plasmid pB171. Mol. Microbiol 61,1428–1442. [DOI] [PubMed] [Google Scholar]
- 32.Barillà D, Rosenberg MF, Nobbmann U, Hayes F, (2005). Bacterial DNA segregation dynamics mediated by the polymerizing protein ParF. EMBO J. 24, 1453–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bouet JY, Ah-Seng Y, Benmeradi N, Lane D, (2007). Polymerization of SopA partition ATPase: regulation by DNA binding and SopB. Mol. Microbiol 63, 468–481. [DOI] [PubMed] [Google Scholar]
- 34.Vecchiarelli AG, Mizuuchi K, Funnell BE, (2012). Surfing biological surfaces: exploiting the nucleoid for partition and transport in bacteria. Mol. Microbiol 86, 513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vecchiarelli AG, Havey JC, Ing LL, Wong EOY, Waples WG, Funnell BE, (2013). Dissection of the ATPase active site of P1 ParA reveals multiple active forms essential for plasmid partition. J. Biol. Chem 288, 17823–17831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chu CH, et al. , (2019). Crystal structures of HpSoj-DNA complexes and the nucleoid-adaptor complex formation in chromosome segregation. Nucleic Acids Res. 47, 2113–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vecchiarelli AG, Neuman KC, Mizuuchi K, (2014). A propagating ATPase gradient drives transport of surface-confined cellular cargo. Proc. Natl. Acad. Sci. USA 111, 4880–4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vecchiarelli AG, Taylor JA, Mizuuchi K, (2015). Reconstituting ParA/ParB-mediated transport of cargo DNA. Methods Cell Biol. 128, 243–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Le Gall A, et al. , (2016). Bacterial partition complexes segregate within the volume of the nucleoid. Nat. Commun 7, 12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang H, Schumacher MA, (2017). Structures of partition protein ParA with nonspecific DNA and ParB effector reveal molecular insights into principles governing Walker-box segregation. Genes Dev. 31, 481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guynet C, de la Cruz F, (2011). Plasmid segregation without partition. Mob. Genet. Elements 1, 236–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guynet C, Cuevas A, Moncalián G, de la Cruz F, (2011). The stb operon balances the requirements for vegetative stability and conjugative transfer of plasmid R388. PLoS Genet. 7,e1002073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simpson AE, Skurray RA, Firth N, (2003). A single gene on the staphylococcal multiresistance plasmid pSK1 encodes a novel partitioning system. J. Bacteriol 185, 2143–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shearer JES, et al. , (2011). Major families of multiresistant plasmids from geographically and epidemiologically diverse staphylococci. G3 (Bethesda) 1, 581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Firth N, et al. , (2000). Replication of staphylococcal multiresistance plasmids. J. Bacteriol 182, 2170–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schumacher MA, Lee J, Zeng W, (2016). Molecular insights into DNA binding and anchoring by the Bacillus subtilis sporulation kinetochore-like RacA protein. Nucleic Acids Res. 44, 5438–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ben-Yehuda S, Rudner DZ, Losick R, (2003). RacA, a bacterial protein that anchors chromosomes to the cell poles. Science 299, 532–536. [DOI] [PubMed] [Google Scholar]
- 48.van Baarle S, Celik IN, Kaval KG, Bramkamp M, Hamoen LW, Halbedel S, (2013). Protein-protein interaction domains of Bacillus subtilis DivIVA. J. Bacteriol 195, 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ben-Yehuda S, et al. , (2005). Defining a centromere-like element in Bacillus subtilis by identifyin the binding sites for the chromosome anchoring protein RacA. Mol. Cell 17, 773–782. [DOI] [PubMed] [Google Scholar]
- 50.Kwong SM, Lim R, LeBard RJ, Skurray RA & Firth N (2008). Analysis of the pSK1 replicon, a prototype from the staphylococcal multiresistance plasmid family. Microbiology 154, 3084–3094. [DOI] [PubMed] [Google Scholar]
- 51.Schumacher MA, Chinnam NB, Cuthbert B, Tonthat NK, Whitfill T, (2015). Structures of regulatory machinery reveal novel molecular mechanisms controlling B. subtilis nitrogen homeostasis. Genes Dev. 29, 451–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brown NL, Stoyanov JV, Kidd SP, Hobman JL, (2003). The MerR family of transcriptional regulators. FEMS Microbiol. Rev 27, 145–163. [DOI] [PubMed] [Google Scholar]
- 53.Lavery R, Moakher M, Maddocks JH, Petkeviciute D, Zakrzewska K, (2009). Conformational analysis of nucleic acids revisited: Curves+. Nucleic Acids Res. 37, 5917–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li S, Olson WK, Lu X-J, (2019). Web 3DNA 2.0 for the analysis, visualization, and modeling of 3D nucleic acid structures. Nucleic Acids Res. 47, W26–W34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jumper J, et al. , (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Varadi M, et al. , (2022). AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M, (2022). ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou NE, Kay CM, Hodges RS, (1994). The net energetic contribution of interhelical electrostatic attractions to coiled-coil stability. Protein Eng. 7, 1365–1372. [DOI] [PubMed] [Google Scholar]
- 59.Sutherland TA, Church JS, Hu X, Huson MG, Kaplan DL, Weisman S, (2011). Single honeybee silk protein mimics properties of multi-protein silk. PLoS One 6, e16489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grkovic S, Brown MH, Hardie MH, Firth N, Skurray RA (2003). Stable low-copy-number Staphylococcus aureus shuttle vectors. Microbiology 149, 785–794. [DOI] [PubMed] [Google Scholar]
- 61.Schenk S, Laddaga RA, (1992). Improved method for electroporation of Staphylococcus aureus. FEMS Microbiol. Lett 73,133–138. [DOI] [PubMed] [Google Scholar]
- 62.Bimboim HC, Doly J, (1979). A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 7,1513–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sambrook J, Russell DW, (2001). Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbour Laboratory Press, Cold Spring Harbour, N. Y. [Google Scholar]
- 64.Veiga H, Jorge AM, Pinho MG, (2011). Absence of nucleoid occlusion effector Noc impairs formation of orthogonal FtsZ rings during Staphylococcus aureus cell division. Mol. Microbiol 80, 1366–1380. [DOI] [PubMed] [Google Scholar]
- 65.Kwong SM, Skurray RA, Firth N, (2004). Staphylococcus aureus multiresistance plasmid pSK41: analysis of the replication region, initiator protein binding and antisense RNA regulation. Mol. Microbiol 51, 497–509. [DOI] [PubMed] [Google Scholar]
- 66.Ausubel FM, (2002). Short protocols in molecular biology: a compedium of methods from Current protocols in molecular biology, 5th ed. John Wiley & Sons, Inc., New York. [Google Scholar]
- 67.Leslie AG, (2006). The integration of macromolecular diffraction data. Acta Crystallogr. D. Biol. Crystallogr 26(Pt 1), 48–57. [DOI] [PubMed] [Google Scholar]
- 68.Potterton E, Briggs P, Turkenburg M, Dodson E, (2003). A graphical user interface to the CCP4 program suite. Acta Crystallogr. D. Biol. Crystallogr 59 (Pt 7), 1131–1137. [DOI] [PubMed] [Google Scholar]
- 69.Jones TA, Zou J-Y, Cowan W, Kjeldgaard M, (1991). Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. A47, 110–119. [DOI] [PubMed] [Google Scholar]
- 70.Adams PD, et al. , (2010). PHENIX: a comprehensive Phyton-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.