

Abstract
Pre-eclampsia (PE) is a hypertensive condition that occurs during pregnancy and complicates up to 4% of pregnancies. PE exhibits several circadian-related characteristics, and the placenta possesses a functioning molecular clock. We examined the associations of 17 core circadian gene transcripts in placenta with PE vs. non-PE (a mixture of pregnant women with term, preterm, small-for-gestational-age, or chorioamnionitis) using two independent gene expression datasets: GSE75010-157 (80 PE vs. 77 non-PE) and GSE75010-173 (77 PE and 96 non-PE). We found a robust difference in circadian gene expression between PE and non-PE across the two datasets, where CRY1 mRNA increases and NR1D2 and PER3 transcripts decrease in PE placenta. Gene set variation analysis revealed an interplay between co-alterations of circadian clock genes and PE with altered hypoxia, cell migration/invasion, autophagy, and membrane trafficking pathways. Using human placental trophoblast HTR-8 cells, we show that CRY1/2 and NR1D1/2 regulate trophoblast migration. A subgroup study including only term samples demonstrated that CLOCK, NR1D2, and PER3 transcripts were simultaneously decreased in PE placenta, a finding supported by CLOCK protein downregulation in an independent cohort of human term PE placenta samples. These findings provide novel insights into the roles of the molecular clock in the pathogenesis of PE.
Subject terms: Computational biology and bioinformatics, Data mining, Gene regulatory networks, Microarrays, Cell migration
Introduction
Pre-eclampsia (PE) is a hypertensive condition of pregnancy and complicates up to 4% of pregnancies in the US1,2. PE is a leading cause of maternal, fetal, and neonatal morbidity and mortality3,4. Women with a history of PE have an increased risk of cardiovascular diseases, type II diabetes, and chronic kidney disease later in life3,5. The involved risk factors for pregnant women to develop PE include: a history of preeclampsia, chronic hypertension, pregestational diabetes mellitus, multiple pregnancy, antiphospholipid syndrome, obesity, advanced maternal age, nulliparity, history of chronic kidney disease, use of assisted reproductive technologies, and genetic susceptibility3,6,7. PE is typically defined as the presence of hypertension and either proteinuria or signs of severe multiorgan dysfunction after 20 weeks' gestation in a previously normotensive woman3,8. Based on the timing of disease onset, PE can be grouped into two subtypes: early-onset (occurring at < 34 weeks' gestation) and late-onset (occurring at ≥ 34 weeks' gestation)9. The two subtypes of PE may have different clinical characteristics and pathophysiology10, as well as different associated risk factors and outcomes9,11. However, the stratification of PE by the timing of disease onset still cannot fully explain the heterogeneity of PE’s clinical presentation, which might at least partially account for the lack of robust predictive biomarkers and effective treatments. To date, the only effective treatment for PE still remains the delivery of the placenta and fetus3,4, although low-dose aspirin has been recommended for reducing the risk of PE in high-risk pregnant women8,12 and novel potential therapies are emerging8,13,14.
Light-induced circadian dysregulation has been associated with multiple pregnancy complications including miscarriage15–17, preterm birth17,18, and gestational diabetes19. There is compelling evidence that pathophysiological functions regulated by the circadian system contribute to PE, where pregnant women with PE frequently are diagnosed with an abnormal circadian blood pressure rhythm20. The abnormal blood pressure associated with PE is often referred to as ‘non-dipping nocturnal high blood pressure’, in which blood pressure drops by less than 10% at night21. Pregnant women with early-onset PE present with a significantly higher prevalence of non-dipping nocturnal hypertension compared to late-onset PE22. Meanwhile, in women who had PE with severe features, non-dipping nocturnal blood pressure was associated with adverse maternal/perinatal outcomes such as preterm birth, retinopathy, HELLP (Hemolysis, Elevated Liver enzymes and Low Platelets) syndrome, low birth weight, and fetal growth restriction23. In addition, in a randomized chronotherapy trial, where the time-of-day of drug intake was recorded and controlled for, administration of low-dose aspirin at bedtime significantly reduced the ambulatory blood pressure and the incidence of preeclampsia, as compared to the ingestion of aspirin in the morning24. Together, these data suggest that at least some subtypes and/or specific clinical presentations of PE are related to circadian rhythms. Whether these circadian-related aspects of PE are controlled by either maternal central or placental peripheral clock systems or both remain unknown.
Preeclampsia is caused by placental dysfunction3,8,25. Studies on global placental gene expression profiling have significantly improved our understanding of the heterogeneity of PE’s clinical presentations, etiology, and pathophysiology26–30. However, our current understanding of placental PE-related gene expression lacks studies on the potential role of clock genes. This is surprising, since PE has numerous circadian-related characteristics and the placenta possesses a functioning molecular clock, which drives endogenous cellular circadian rhythms31–36. At the cellular level, circadian rhythms are generated by a complex cell-autonomous transcription-translation feedback mechanism, generating 24 h gene expression rhythms. Among the core circadian transcriptional regulators are: Aryl hydrocarbon receptor nuclear translocator-like protein 1 (ARNTL1, also known as BMAL1), Circadian Locomotor Output Cycles Kaput (CLOCK), Period 1/2/3 (PER1/2/3), Cryptochrome 1/2 (CRY1/2) and the nuclear receptor subfamily 1 group D members 1/2 (NR1D1/2, also known as REV-ERBα/β). Circadian clock genes are expressed in female reproductive tissues, such as the ovary, oviducts, uterus, and placenta33,37–42. While the role of circadian genes in the ovary is well-understood43, their role in the placenta remains elusive.
The goal of this study is to identify specific clock genes associated with PE by examining the mRNA expression patterns of 17 core circadian transcripts in PE and non-PE placentas. Using existing gene expression datasets together with functional and molecular approaches, we identified differentially expressed circadian genes in PE placentas whose expression affects trophoblast migration in vitro. These results will allow future studies to address the role of these clock genes in placental development and function for the etiology/pathogenesis and therapeutics of PE.
Materials and methods
Human research
All methods were performed in accordance with the relevant guidelines and regulations. According to the IRB guidelines and the HIPPA Privacy Rule, the analysis of de-identified, publicly available data does not constitute human subjects research as defined at 45 CFR 46.102 and thus, the present study does not require IRB review. Placentas for molecular analysis were collected in accordance with protocols approved by the Institutional Review Boards of Michigan State University, Mount Sinai Hospital, and the University of Kansas Medical Center. Placentas from gestational age- and delivery-matched healthy and PE pregnancies were collected from the Research Center for Women’s and Infant’s Health BioBank at the Samuel Lunenfeld Research Institute (Mount Sinai Hospital, Toronto, Ontario, Canada).
Study design, selection of patients, and demographics
Publicly available human placenta microarray gene expression data (GSE75010, released in 2016) from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) were analyzed. The GSE75010 data were composed of two independent datasets. One dataset, GSE75010-157, was a PE microarray dataset generated from a total of 157 placenta samples (80 PE vs. 77 non-PE) that were obtained from the Research Centre for Women’s and Infants’ Health BioBank (Mount Sinai Hospital, Toronto, Canada)44. The second dataset, GSE75010-173, was a combination of 173 placental samples (77 PE vs. 96 non-PE) from seven independent studies conducted in five different countries (Canada, China, Japan, Norway, US)44.
For the dataset ‘GSE75010-157’, PE was defined as the onset of systolic pressure ≥ 140 mmHg and/or diastolic pressure ≥ 90 mmHg after the 20th week of gestation, with proteinuria (> 0.3 g protein/day, or ≥ 2 + on dipstick)44. Fetal sex, preterm (< 34 weeks gestation), and small-for-gestational-age infants (SGA, neonatal birth weight < 10th percentile for gestational age and sex) were approximately balanced between PE and non-PE groups. Two groups (PE vs. non-PE) were designated as ‘Overall PE’ vs. ‘Overall non-PE’ due to such heterogeneity of the patients’ composition. Only samples from singleton pregnancies were included, and patients with pre-existing or gestational diabetes, sickle cell anemia, or severe obesity (BMI ≥ 40) were excluded. The PE group was composed of 44.9% of Caucasian, 75.0% with maternal age ≥ 30 years old, 18.2% having obesity before pregnancy (30 < BMI < 40), and 21.3% having previous miscarriage (Table S1). There were no significant differences in these maternal characteristics between PE and non-PE groups (Table S1).
For the dataset ‘GSE75010-173’, among seven different studies45–51, four of them used the definition of PE similar to that in the dataset ‘GSE75010-157’; two studies used severe PE only, which was defined as blood pressure of at least 160 mmHg (systolic) and/or 110 mmHg (diastolic) after the 20th week of gestation, with proteinuria (> 2 g protein/day, or > 2 + on dipstick); one study used both44. Patients in the PE group consisted of 10.4% from Canada, 14.3% from China, 23.4% from Japan, 22.1% from Norway, and 29.9% from the US (Table S2).
Placental tissue microarray gene expression data
The dataset ‘GSE75010-157’ was generated with the Human Gene 1.0 ST Array chips (Affymetrix). The dataset ‘GSE75010-173’ was a combination of the microarray data from seven different studies, in which five different commercial microarray kits from Applied Biosystems, Agilent Technologies, Affymetrix, Roche NimbleGen Arrays, and Illumina were applied44. According to Leavey et al., both datasets were harmonized by normalization and batch effect correction with the virtual Array R package52 to improve the reproducibility of the downstream statistical analyses. Finally, all data were converted into log2 values using R Affy library53. A total of 14,651 genes with expression values were detectable in both datasets44. The harmonized datasets ‘GSE75010-157’ and ‘GSE75010-173’ were used as discovery set and replication set, respectively.
Bioinformatics and statistical analyses
A bioinformatics pipeline that we developed to identify PE-associated circadian genes and their correlated pathways with two independent datasets is presented in Fig. 1. Briefly, we retrieved 17 core circadian transcripts (ARNTL, ARNTL2, CLOCK, CRY1, CRY2, NPAS1, NPAS2, NPAS3, NR1D1, NR1D2, PER1, PER2, PER3, RORA, RORB, RORC, and TIMELESS) from two independent datasets (GSE75010-157 and GSE75010-173). We selected these core clock genes based on their critical roles in circadian rhythms54 and the detectability of their mRNAs in the available microarray data. We summarized the mRNA levels (log2-transformed) of all 17 core circadian genes in placenta as means with standard deviations (SDs). The mean differences in gene mRNA levels between the non-PE and PE groups were examined with a two-sample, two-tailed t-test and a false discovery rate (FDR) to adjust for multiple comparisons. The cut-off values for statistical significance were set as p < 0.05 and FDR q < 0.1055. Then, we overlapped the two pools of significant candidate genes identified from two independent datasets and obtained common significant PE-associated circadian genes across two datasets (Fig. 1). By referring to our previous method56, we linearly combined these common significant PE-associated circadian genes to generate a risk score of PE (Fig. 1). A Firth binary logistic regression model57 was applied to assess the association strength between this risk score (categorized into a binary variable using the median of the risk score as a cut-off value) and PE.
Figure 1.

Bioinformatics pipeline to analyze circadian genes-correlated, PE-associated pathways.
Next, with our previous methods56, we conducted the Gene Set Variation Analysis (GSVA) using two independent datasets to explore the biological pathways that are involved in PE-clock gene relationship, respectively (Fig. 1). The pathway sub-collection—c2.cp.v7.4.symbols.gmt in the Molecular Signatures Database (MSigDB) was used. We applied limma R package58 to identify differential pathways between non-PE and PE with the cut-offs of p < 0.05 and Benjamini and Hochberg adjusted p < 0.10 (Fig. 1). Then, we further examined the correlations of the identified clock genes-based risk score with the PE-associated pathways with Pearson Correlation statistic (p < 0.05 and Benjamini and Hochberg adjusted p < 0.10) (Fig. 1). Finally, two pools of PE-associated pathways correlated with circadian genes-based risk score were overlapped to generate common PE-associated pathways that were correlated with the circadian genes-based risk score (Fig. 1).
PE can be divided into preterm PE and term PE59,60. To disentangle the possible confounding effect of preterm birth (PTB, defined as birth before 37 weeks of gestation) on the relationship between circadian clock genes and PE, a subgroup analysis was performed by removing the individuals who were diagnosed as PTB from overall PE and non-PE groups in the dataset GSE75010-157. There was no available information about PTB diagnosis in the dataset GSE75010-173. The analytic methods for identifying PE-associated circadian genes and their correlated pathways after removing patients diagnosed with PTB were the same as those used in the analyses of the GSE75010-157 dataset.
We conducted all data management and statistical analyses described above using R (R Development Core Team) and SAS v9.4 (SAS Institute, Cary, North Carolina).
HTR-8 cell culture and wound healing assay
Trophoblast cells derived from a 6–12 weeks-old human placenta (HTR-8 SVNeo, referred to as HTR-8 cells, ATCC #CRL-3271) were cultured in RPMI 1640 Medium (Gibco, #11875093) with 10% FBS (Sigma-Aldrich, #F4135) and 1% Penicillin–Streptomycin (Sigma-Aldrich Cat# P4333). HTR-8 cells were plated at 0.5 million cells/ml per well in a 24-well wound healing assay (Abcam, #ab242285). Twenty-four h after plating, the HTR-8 cells formed a monolayer and the inserts were removed, where after the cells were treated with vehicle (DMSO 1/500 or 1/5000 dilution, no difference in HTR-8 cell migration in response to DMSO concentration was found and data were pooled), KL001 (Tocris Bioscience, # 46-851-0), PF670462 (Tocris Bioscience™ Cat# 33-161-0), SR9009 (Tocris Bioscience, # 5855), SR8278 (Tocris Bioscience, # 4463), or KL101 (Sigma Aldrich, #SML3014) at 1 μM and 10 μM. Bright field image acquisition of the wound healing area was done using a light microscope (Leica DMi1) 0, 24 and 48 h after treatment. Data were analyzed using ImageJ/Fiji® version 1.53 (NIH).
Westernblot
Human placenta samples were matched for gestation age and mode of delivery (C-section, C-section + labor, Vaginal delivery + labor) between PE and normal placentas. After collection, samples were rapidly frozen in liquid nitrogen before being stored at − 80 °C until use. Proteins were extracted by homogenization for 15–30 s in RIPA buffer containing protease and phosphatase inhibitors using a Bead Beater (MP Biomedicals, Irvine, CA, USA). Lysates were centrifuged for 10–20 min at 4 °C, 15,000g. Supernatant was recovered and protein concentration determined using the Pierce BCA Assay (Thermo Scientific, Waltham, MA, USA). Proteins were then denatured by boiling in Laemmli buffer at 95 °C for 5 min, and 20 µg were electrophoresed on a 10% SDS–polyacrylamide gel at 120 V for 45 min. Protein was then transferred to a nitrocellulose membrane (Cytiva Amersham™ Protran™; Fisher Scientific, Pittsburgh, PA, USA) by electroblotting at 200 V for 45 min. The membrane was blocked in 3% milk for 1 h at RT and incubated with rabbit anti-human CLOCK primary antibody (1:1000, ab3517, Abcam, Cambridge, UK, RRID: AB_303886) for 16 h at 4 °C. After washing, the blot was incubated in goat anti-rabbit IgG-HRP secondary antibody (1:2000, BioRad, 1706515, RRID:AB_11125142) in 3% milk for 1 h at RT. HRP activity of bound secondary antibody was visualized in an iBright (ThermoFisher Scientific) using Enhanced Chemiluminescent reagent (Amersham) according to the manufacturer’s instructions. To stain for beta ACTIN, the membrane was stripped with stripping buffer (Restore Western Blot Stripping Buffer, 21059, Thermo Scientific), washed 3 × 5 min in TBS and 1 × 5 min in TBS-Tween 0.05%, blocked with buffer containing 3% milk for 1 h at RT, and incubated with a rabbit anti-beta ACTIN antibody (1:2000 Invitrogen, MA5-32479, RRID: AB_2809756). Secondary antibody and visualization with ECL were similar to those for CLOCK antibody. We verified the proteins by their molecular weight using 10 µl of a protein marker (Precision Plus Protein Standards, Bio-Rad, 161-0374). We confirmed the used CLOCK antibody had non-specific bands below 50 kDa. These non-specific bands did not interfere with CLOCK analysis at 100 kDa. Using Image J (NIH), the integral densities of CLOCK protein bands were normalized to the integral density of beta-ACTIN.
Results
The clock genes CRY1, NR1D2, and PER3 are differentially expressed in the placentas of preeclamptic patients in two independent datasets
To determine if expression of the 17 candidate circadian genes is dysregulated in PE placentas, we analyzed their expression using two independent publicly available datasets, comparing placentas from PE versus non-PE patients. To find the differentially expressed circadian genes that commonly exist in two datasets, we overlapped the results from the separate analyses with two datasets. We found that the expression of CRY1, NR1D2, and PER3 gene transcripts is robustly different between overall PE and non-PE across the two datasets (Table 1). The average expression of NR1D2 and PER3 transcripts in placenta is lower in PE than in non-PE across the two datasets, respectively (the decrease percentages are 2.4%-4.0% for NR1D2 and 3.0%-3.6% for PER3 genes in the two datasets, all p ≤ 0.0030 and FDR ≤ 0.0225, Table 1). In contrast, the mean of CRY1 gene transcript in placenta is significantly higher in PE than in non-PE (the increase percentages are 1.5–1.7%, p ≤ 0.0250 and FDR ≤ 0.0711, Table 1). The remaining circadian gene transcripts are not significantly different between PE vs. non-PE in either dataset (e.g., ARNTL, CRY2, NPAS2, NPAS3, NR1D1, PER1, and RORC, p > 0.05 and FDR > 0.10), or are different in one, but not the other dataset (e.g., ARNTL2, NPAS1, PER2, RORB, and TIMELESS are different in GSE75010-157, but not in GSE75010-173; CLOCK and RORA are the opposite, Table 1).
Table 1.
Descriptive statistics of 17 candidate circadian clock genes’ expression levels in placenta from PE vs. non-PE, independent of gestation length (overall PE vs overall non-PE, respectively), in two independent datasets.
| Gene | GSE75010-157 samples | GSE75010-173 samples | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Non-PE | PE | pb | FDRc | Non-PE | PE | p | FDR | |||||
| n | Meana (SD) | n | Mean (SD) | n | Meana (SD) | n | Mean (SD) | |||||
| ARNTL | 77 | 7.36 (0.38) | 80 | 7.38 (0.42) | 0.7006 | 0.8507 | 96 | 7.38 (0.39) | 77 | 7.35 (0.32) | 0.5509 | 0.7128 |
| ARNTL2 | 77 | 7.95 (0.40) | 80 | 8.09 (0.42) | 0.0346 | 0.0735 | 96 | 8.01 (0.41) | 77 | 8.01 (0.43) | 0.9995 | 0.9995 |
| CLOCK | 77 | 8.88 (0.50) | 80 | 8.73 (0.53) | 0.0766 | 0.1302 | 96 | 8.88 (0.53) | 77 | 8.71 (0.40) | 0.0181 | 0.0711 |
| CRY1 | 77 | 8.39 (0.41) | 80 | 8.53 (0.39) | 0.0250 | 0.0607 | 96 | 8.40 (0.40) | 77 | 8.53 (0.36) | 0.0209 | 0.0711 |
| CRY2 | 77 | 8.39 (0.27) | 80 | 8.38 (0.28) | 0.9098 | 0.9098 | 96 | 8.40 (0.29) | 77 | 8.38 (0.26) | 0.6289 | 0.7128 |
| NPAS1 | 77 | 6.56 (0.36) | 80 | 6.76 (0.39) | 0.0008 | 0.0045 | 96 | 6.68 (0.44) | 77 | 6.61 (0.45) | 0.2943 | 0.5440 |
| NPAS2 | 77 | 9.06 (0.37) | 80 | 8.96 (0.32) | 0.0759 | 0.1302 | 96 | 8.99 (0.38) | 77 | 9.03 (0.32) | 0.4382 | 0.6607 |
| NPAS3 | 77 | 6.55 (0.42) | 80 | 6.45 (0.37) | 0.1226 | 0.1788 | 96 | 6.51 (0.40) | 77 | 6.48 (0.39) | 0.6276 | 0.7128 |
| NR1D1 | 77 | 7.23 (0.40) | 80 | 7.32 (0.34) | 0.1262 | 0.1778 | 96 | 7.28 (0.36) | 77 | 7.22 (0.41) | 0.2638 | 0.5440 |
| NR1D2 | 77 | 8.80 (0.48) | 80 | 8.45 (0.51) | < 0.0001 | 0.0008 | 96 | 8.70 (0.51) | 77 | 8.49 (0.41) | 0.0030 | 0.0255 |
| PER1 | 77 | 7.40 (0.37) | 80 | 7.41 (0.47) | 0.8812 | 0.9098 | 96 | 7.37 (0.44) | 77 | 7.43 (0.39) | 0.3200 | 0.5440 |
| PER2 | 77 | 7.93 (0.38) | 80 | 8.08 (0.32) | 0.0096 | 0.0272 | 96 | 8.05 (0.33) | 77 | 7.95 (0.36) | 0.0737 | 0.2088 |
| PER3 | 77 | 7.30 (0.32) | 80 | 7.04 (0.30) | < 0.0001 | 0.0008 | 96 | 7.25 (0.30) | 77 | 7.03 (0.27) | < 0.0001 | 0.0017 |
| RORA | 77 | 8.54 (0.43) | 80 | 8.52 (0.40) | 0.7598 | 0.8611 | 96 | 8.59 (0.42) | 77 | 8.45 (0.36) | 0.0204 | 0.0711 |
| RORB | 77 | 6.64 (0.67) | 80 | 7.00 (0.93) | 0.0063 | 0.0214 | 96 | 6.79 (0.67) | 77 | 6.87 (0.82) | 0.4664 | 0.6607 |
| RORC | 77 | 6.56 (0.35) | 80 | 6.64 (0.35) | 0.1711 | 0.2237 | 96 | 6.55 (0.39) | 77 | 6.62 (0.38) | 0.2341 | 0.5440 |
| TIMELESS | 77 | 7.92 (0.40) | 80 | 7.73 (0.32) | 0.0015 | 0.0064 | 96 | 7.82 (0.37) | 77 | 7.83 (0.36) | 0.9138 | 0.9709 |
aMean of gene expression values, normalized and log2-transformed. The mean change percentages of 3 circadian genes’ expression levels are: for CRY1 gene, (8.53–8.39)*100/8.39 = 1.7% in GSE75010-157 Samples and (8.53–8.40)*100/8.40 = 1.5% in GSE75010-173 Samples; for NR1D2 gene, (8.80–8.45)*100/8.80 = 4.0% in GSE75010-157 Samples and (8.70–8.49)*100/8.70 = 2.4% in GSE75010-173 Samples; and for PER3 gene, (7.30–7.04)*100/7.30 = 3.6% in GSE75010-157 Samples and (7.25–7.03)*100/7.25 = 3.0% in GSE75010-173 Samples.
bTwo-sample, two-tailed t-tests were used to generate p-values.
Bold values denote statistical significance (p < 0.05 and FDR < 0.10) in both datasets.
Association strength of the differentially expressed circadian genes-based risk score with overall PE in two independent datasets
To determine if co-alteration of the differentially expressed genes CRY1, NR1D2, and PER3 occurs in PE placentas, we first generated a risk score of PE by using a linear combination of these 3 gene transcripts: Risk score = CRY1 + (11-NR1D2) + (9-PER3), where the maximum values of NR1D2 and PER3 were < 11 and < 9, respectively. Then, we categorized the risk score into a binary variable using the median of the risk score as a cut-off value and calculated the odds-ratio (OR) of PE with the categorized risk score using a Firth binary logistic regression model. Maternal characteristic variables were not included in the model because they are similar between non-PE vs PE in GSE75010-157, and not available for GSE75010-173. As shown in Table 2, the OR (95% confidence interval) of PE for the risk score > median (< median as the reference) is 5.35 (2.70, 10.57) in GSE75010-157 and 5.46 (2.83, 10.54) in GSE75010-173 (both p values < 0.0001), respectively (Table 2). Collectively, these results show that co-alteration of CRY1, NR1D2, and PER3 in the placenta is strongly associated with the risk of PE.
Table 2.
Combined effect of the DE clock genes CRY1, NR1D2 and PER3 in placenta on the risk of overall PE with firth logistic regression model for two independent datasets.
| Risk score* | N (%) | Non-PE, n (%) | PE, n (%) | ORPE vs. Non-PE (95% CI) | p |
|---|---|---|---|---|---|
| GSE75010-157 | |||||
| < Median | 78 (100.0) | 54 (69.2) | 24 (30.8) | Ref | |
| > Median | 79 (100.0) | 23 (29.1) | 56 (70.9) | 5.35 (2.70, 10.57) | < 0.0001 |
| GSE75010-173 | |||||
| < Median | 86 (100.0) | 65 (75.6) | 21 (24.4) | Ref | |
| > Median | 87 (100.0) | 31 (35.6) | 56 (64.4) | 5.46 (2.83, 10.54) | < 0.0001 |
*Risk score = CRY1 + (11-NR1D2) + (9-PER3).
Bold values denote statistical significance (p < 0.05).
PE-associated biological pathways correlate with CRY1, NR1D2, and PER3 in two independent datasets
We next used GSVA and Pearson correlation analyses to identify PE-associated pathways that correlate with co-alteration of CRY1, NR1D2, and PER3. We found 299 common PE-associated pathways that are correlated with the three circadian clock genes-based risk score, among which 212 are down-regulated and 87 are up-regulated across the two independent datasets (the range of absolute correlation coefficients is 0.15–0.57, all p < 0.05 and FDR < 0.10, Table S3).
To better interpret the most represented pathways that are up- and down-regulated in PE, we referred to the literature (regardless of cell origin and mammalian species). We manually grouped the top-most representative pathways into hypoxia-related and cell migration/invasion-related pathways (Table 3), where the top hypoxia-related pathways7,61,62 include galactose metabolism, hypoxia inducible factor (HIF) 1 transcription factor (TF) pathway, HIF2 pathway and Cori Cycle. The top identified cell migration/invasion-related pathways8,63–68 include transcriptional regulation by Runt-related transcription factor 1 (Runx1), deubiquitinating enzymes (DUBs), negative regulation of mesenchymal-epithelial transition factor (Met) activity, signaling by Notch4, β-catenin independent WNT signaling and PCP/CE polar (planar cell polarity/convergent extension) pathway, and signaling by Hedgehog and Hedgehog on State.
Table 3.
Top 10 pathways correlated with CRY1, NR1D2 and PER3-based risk score in placenta (overall PE vs. overall non-PE) in two independent datasets (cut-off: p < 0.05 and FDR < 0.10).
| Risk score*-correlated pathway | GSE75010-157 | GSE75010-173 | ||||
|---|---|---|---|---|---|---|
| r (95% CI) | p | FDR | r (95% CI) | p | FDR | |
| Top 10 decreased pathways in overall PE: | ||||||
| REACTOME_TRANSCRIPTIONAL_REGULATION_BY_RUNX1 | −0.57 (−0.67, −0.45) | < 0.0001 | 0.0002 | −0.36 (−0.49, −0.23) | < 0.0001 | 0.0009 |
| REACTOME_SIGNALING_BY_HEDGEHOG | −0.54 (−0.64, −0.42) | < 0.0001 | 0.0002 | −0.42 (−0.53, −0.29) | < 0.0001 | 0.0009 |
| REACTOME_NEGATIVE_REGULATION_OF_MET_ACTIVITY | −0.53 (−0.63, −0.40) | < 0.0001 | 0.0002 | −0.29 (−0.42, −0.14) | 0.0001 | 0.0009 |
| REACTOME_BETA_CATENIN_INDEPENDENT_WNT_SIGNALING | −0.51 (−0.62, −0.39) | < 0.0001 | 0.0002 | −0.32 (−0.45, −0.18) | < 0.0001 | 0.0009 |
| REACTOME_HEDGEHOG_ON_STATE | −0.50 (−0.61, −0.37) | < 0.0001 | 0.0002 | −0.38 (−0.50, −0.24) | < 0.0001 | 0.0009 |
| KEGG_BETA_ALANINE_METABOLISM | −0.50 (−0.61, −0.37) | < 0.0001 | 0.0002 | −0.33 (−0.46, −0.19) | < 0.0001 | 0.0009 |
| REACTOME_ANTIGEN_PROCESSING_CROSS_PRESENTATION | −0.50 (−0.60, −0.37) | < 0.0001 | 0.0002 | −0.27 (−0.40, −0.13) | 0.0003 | 0.0019 |
| REACTOME_PCP_CE_PATHWAY | −0.49 (−0.60, −0.37) | < 0.0001 | 0.0002 | −0.29 (−0.42, −0.15) | 0.0001 | 0.0009 |
| REACTOME_SIGNALING_BY_NOTCH4 | −0.49 (−0.60, −0.36) | < 0.0001 | 0.0002 | −0.30 (−0.43, −0.15) | < 0.0001 | 0.0009 |
| REACTOME_METALLOPROTEASE_DUBS | −0.49 (−0.60, −0.36) | < 0.0001 | 0.0002 | −0.25 (−0.38, −0.10) | 0.001 | 0.0047 |
| Top 10 increased pathways in overall PE: | ||||||
| KEGG_GALACTOSE_METABOLISM | 0.54 (0.42, 0.65) | < 0.0001 | 0.0002 | 0.39 (0.26, 0.51) | < 0.0001 | 0.0009 |
| PID_HIF1_TFPATHWAY | 0.52 (0.39, 0.62) | < 0.0001 | 0.0002 | 0.36 (0.22, 0.48) | < 0.0001 | 0.0009 |
| WP_TRANSCRIPTIONAL_CASCADE_REGULATING_ADIPOGENESIS | 0.50 (0.37, 0.61) | < 0.0001 | 0.0002 | 0.32 (0.18, 0.45) | < 0.0001 | 0.0009 |
| KEGG_RIG_I_LIKE_RECEPTOR_SIGNALING_PATHWAY | 0.49 (0.36, 0.60) | < 0.0001 | 0.0002 | 0.35 (0.22, 0.48) | < 0.0001 | 0.0009 |
| REACTOME_TRANSCRIPTIONAL_REGULATION_OF_WHITE_ADIPOCYTE_DIFFERENTIATION | 0.48 (0.35, 0.59) | < 0.0001 | 0.0002 | 0.23 (0.09, 0.37) | 0.0019 | 0.0078 |
| PID_P38_MKK3_6PATHWAY | 0.46 (0.32, 0.57) | < 0.0001 | 0.0002 | 0.24 (0.10, 0.38) | 0.0012 | 0.0054 |
| WP_CORI_CYCLE | 0.46 (0.33, 0.57) | < 0.0001 | 0.0002 | 0.38 (0.25, 0.50) | < 0.0001 | 0.0009 |
| PID_HIF2PATHWAY | 0.45 (0.32, 0.57) | < 0.0001 | 0.0002 | 0.30 (0.16, 0.43) | < 0.0001 | 0.0009 |
| KEGG_ADIPOCYTOKINE_SIGNALING_PATHWAY | 0.44 (0.31, 0.56) | < 0.0001 | 0.0002 | 0.30 (0.16, 0.43) | < 0.0001 | 0.0009 |
| REACTOME_ACTIVATION_OF_AMPK_DOWNSTREAM_OF_NMDARS | 0.44 (0.31, 0.56) | < 0.0001 | 0.0002 | 0.17 (0.02, 0.31) | 0.0234 | 0.0515 |
*Risk score = CRY1 + (11-NR1D2) + (9-PER3).
Pharmacological stabilization of CRY1/2 and antagonism of NR1D1/2 slows down migration of the human trophoblast cell line HTR-8
Based on the above PE-associated pathways, co-alteration of CRY1, NR1D2, and PER3 in PE are predicted to impact cell migration. To assess whether CRY1, PER3 and NR1D2 regulate cell migration, we used the human-derived migratory trophoblast cell line HTR-8 to test drugs targeting CRY1, CRY1/2, PER1/2/3 and NR1D1/2. No drugs specifically targeting PER3 or NR1D2 are currently commercially available, thus only drugs targeting PER1/2/3 and NR1D1/2 were used for the study. Stabilizing/upregulating CRY1/2, using KL001 slowed HTR-8 migration at 24 h (Fig. 2A,B), whereas stabilizing/upregulating CRY1 with KL101, did not impact HTR-8 migration (Fig. 2A); these results suggest that CRY2 or CRY1/2 together impact trophoblast cell migration. Upregulating PER1/2/3 by preventing their degradation using PF670462 did not impact HTR-8 migration (Fig. 2C,D). In contrast, the NR1D1/2 agonist, SR9009, did not significantly change migration (Fig. 2E,F), whereas the NR1D1/2 antagonist/inverse agonist using SR8278, slowed migration of HTR-8 cells in a dose-dependent manner (Fig. 2E,G).
Figure 2.

HTR-8 cell migration is regulated by ligands targeting CRY1/2 and NR1D1/2. Wound healing assays in HTR-8 cells, show that (A, B) KL001, a drug stabilizing CRY1/2, but not KL101, a drug stabilizing CRY1, reduced HTR-8 cell migration. (C, D) PF670462, a drug preventing PER1/2/3 degradation, did not impact HTR-8 cell migration. (E–G) The NR1D1/2 agonist SR9009, had no significant effect on migration, whereas the NR1D1/2 antagonist/inverse agonist, SR8278, decreased HTR-8 cell migration. All drugs were tested at 1 and 10 µM. The data were pooled if the two doses were not significantly different. (B, D, F, G) Illustrative wound healing assay images. Dotted line on histograms represents approximate wound area in control at 24 h. Data is expressed as % change in average wound size from 0 h ± SEM. N = 3–6, in duplicate. One-way ANOVA repeated measures, *, p < 0.05; ***, p < 0.001.
CLOCK, NR1D2, and PER3 are downregulated in the term PE placenta—a subgroup analysis after removing patients diagnosed as PTB from GSE75010-157
PE frequently leads to PTB, particularly in severe cases of PE. To exclude the potential confounding effect of gestation length from PE-specific effects, we reanalyzed the data presented in Table 1 after removing patients diagnosed with PTB. In this reanalyzed data set, the expression of CRY1 becomes non-significant between PE and non-PE, while CLOCK transcript is significantly lower in PE without PTB (Table 4, p = 0.0168 and FDR = 0.0952). Interestingly, both NR1D2 (p = 0.0120, FDR = 0.0952) and PER3 (p ≤ 0.0137 and FDR = 0.0952) transcripts in placenta are consistently lower in term PE (Table 4). This identifies NR1D2 and PER3 transcripts to be downregulated in PE, independent of gestation length. All other circadian gene transcripts in placental tissue were not significantly associated with term PE (p > 0.05 and FDR > 0.10, Table 4).
Table 4.
Descriptive statistics of 17 candidate circadian genes’ expression levels in placenta from PE without PTB (Term-PE) vs. non-PE without PTB (Term) in GSE75010-157 dataset.
| Gene | Term | Term-PE | pb | FDRc | ||
|---|---|---|---|---|---|---|
| n | Meana (SD) | n | Mean (SD) | |||
| ARNTL | 42 | 7.22 (0.33) | 31 | 7.39 (0.41) | 0.0619 | 0.1754 |
| ARNTL2 | 42 | 8.03 (0.37) | 31 | 7.96 (0.39) | 0.3919 | 0.5626 |
| CLOCK | 42 | 9.06 (0.33) | 31 | 8.73 (0.69) | 0.0168 | 0.0952 |
| CRY1 | 42 | 8.55 (0.34) | 31 | 8.52 (0.51) | 0.7561 | 0.8034 |
| CRY2 | 42 | 8.44 (0.24) | 31 | 8.34 (0.30) | 0.0943 | 0.2004 |
| NPAS1 | 42 | 6.57 (0.37) | 31 | 6.77 (0.46) | 0.0449 | 0.1527 |
| NPAS2 | 42 | 9.07 (0.29) | 31 | 8.93 (0.34) | 0.0724 | 0.1758 |
| NPAS3 | 42 | 6.58 (0.44) | 31 | 6.49 (0.41) | 0.3971 | 0.5626 |
| NR1D1 | 42 | 7.33 (0.34) | 31 | 7.36 (0.33) | 0.6501 | 0.7368 |
| NR1D2 | 42 | 8.92 (0.40) | 31 | 8.61 (0.56) | 0.0120 | 0.0952 |
| PER1 | 42 | 7.35 (0.26) | 31 | 7.31 (0.38) | 0.6164 | 0.7368 |
| PER2 | 42 | 8.05 (0.32) | 31 | 7.99 (0.29) | 0.3812 | 0.5626 |
| PER3 | 42 | 7.34 (0.30) | 31 | 7.16 (0.30) | 0.0137 | 0.0952 |
| RORA | 42 | 8.77 (0.32) | 31 | 8.58 (0.41) | 0.0308 | 0.1309 |
| RORB | 42 | 6.55 (0.58) | 31 | 6.84 (0.97) | 0.1535 | 0.2899 |
| RORC | 42 | 6.61 (0.35) | 31 | 6.59 (0.36) | 0.8158 | 0.8158 |
| TIMELESS | 42 | 7.78 (0.35) | 31 | 7.72 (0.40) | 0.5378 | 0.7033 |
aMean of gene expression values, normalized and log2-transformed.
Bold values denote statistical significance (p < 0.05 and FDR < 0.10).
To identify the PE-associated pathways that correlate with the co-alteration of the CLOCK, NR1D2, and PER3 genes as a whole in placenta, we first generated a linear combination of these three clock genes as a risk score, which was calculated as (10-CLOCK) + (11-NR1D2) + (9-PER3) based on the maximum values of the 3 genes, followed by the GSVA. The results demonstrate that a total of 41 term PE (PE without PTB)-associated pathways are significantly correlated with the CLOCK, NR1D2, and PER3-based risk score, among which 14 and 27 pathways are down- and up-regulated in the case group, respectively (the range of the absolute correlation coefficients is 0.25–0.53, all p ≤ 0.0344 and FDR ≤ 0.0520, Table S4).
Table 5 presents the top 10 down- and up-regulated pathways that are significantly correlated with CLOCK, NR1D2, and PER3-based risk score in placenta for term PE in GSE75010-157. Similarly, to better interpret the most represented pathways that are up- and down-regulated in PE in the sensitivity analysis, we manually collapsed the top-most regulated pathways into three broad categories: hypoxia-related pathways, membrane trafficking pathways, and autophagy-related pathways (Table 5). The increased hypoxia-related pathways include Cori Cycle, glycolysis and gluconeogenesis, computational model of aerobic glycolysis, HIF1 TF pathway, and reversible hydration of carbon dioxide. The decreased membrane trafficking pathways include intra Golgi traffic, RAB (Ras-associated binding) GEFs (GDP-GTP exchange factors), SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) interactions in vesicular transport, RAB Regulation of Trafficking, and VxPx (a cilium localization motif) cargo targeting to cilium. The decreased autophagy-related pathways include pexophagy, nanoparticle triggered autophagic cell death, and autophagy in PE patients (Table 5).
Table 5.
Top 10 pathways correlated with CRY1, NR1D2 and PER3-based risk score in placenta (PE without PTB vs. non-PE without PTB) in GSE75010-157 dataset. Cut-off: p < 0.05 and FDR < 0.10.
| Risk score*-correlated pathway | r | p | FDR |
|---|---|---|---|
| Top 10 decreased pathways in PE without PTB | |||
| REACTOME_INTRA_GOLGI_TRAFFIC | − 0.53 | < 0.0001 | 0.0007 |
| REACTOME_RAB_GEFS_EXCHANGE_GTP_FOR_GDP_ON_RABS | − 0.47 | < 0.0001 | 0.0007 |
| KEGG_SNARE_INTERACTIONS_IN_VESICULAR_TRANSPORT | − 0.44 | < 0.0001 | 0.0007 |
| BIOCARTA_IGF1MTOR_PATHWAY | − 0.40 | 0.0004 | 0.0019 |
| REACTOME_PEXOPHAGY | − 0.39 | 0.0005 | 0.0022 |
| WP_NANOPARTICLE_TRIGGERED_AUTOPHAGIC_CELL_DEATH | − 0.39 | 0.0006 | 0.0023 |
| REACTOME_RAB_REGULATION_OF_TRAFFICKING | − 0.35 | 0.0023 | 0.0068 |
| REACTOME_VXPX_CARGO_TARGETING_TO_CILIUM | − 0.34 | 0.003 | 0.0078 |
| REACTOME_TRANSLATION_OF_REPLICASE_AND_ASSEMBLY_OF_THE_REPLICATION_TRANSCRIPTION_COMPLEX | − 0.34 | 0.003 | 0.0078 |
| WP_AUTOPHAGY | − 0.32 | 0.0055 | 0.0126 |
| Top 10 increased pathways in PE without PTB | |||
| WP_CORI_CYCLE | 0.52 | < 0.0001 | 0.0007 |
| WP_GLYCOLYSIS_AND_GLUCONEOGENESIS | 0.50 | < 0.0001 | 0.0007 |
| BIOCARTA_ACH_PATHWAY | 0.46 | < 0.0001 | 0.0007 |
| WP_COMPUTATIONAL_MODEL_OF_AEROBIC_GLYCOLYSIS | 0.45 | < 0.0001 | 0.0007 |
| PID_HIF1_TFPATHWAY | 0.44 | < 0.0001 | 0.0007 |
| KEGG_ARACHIDONIC_ACID_METABOLISM | 0.44 | < 0.0001 | 0.0007 |
| WP_ASPIRIN_AND_MIRNAS | 0.42 | 0.0002 | 0.0012 |
| WP_PHOTODYNAMIC_THERAPYINDUCED_HIF1_SURVIVAL_SIGNALING | 0.40 | 0.0003 | 0.0016 |
| REACTOME_ACTIVATION_OF_AMPK_DOWNSTREAM_OF_NMDARS | 0.40 | 0.0003 | 0.0016 |
| REACTOME_REVERSIBLE_HYDRATION_OF_CARBON_DIOXIDE | 0.39 | 0.0006 | 0.0023 |
*Risk score = (10-CLOCK) + (11-NR1D2) + (9-PER3).
Reduced CLOCK protein in term PE placenta
To determine if the small decrease in placenta CLOCK transcript in term PE samples (Table 4) translated into a change in CLOCK protein, we analyzed an independent cohort of 10 human placenta samples by westernblot (Fig. 3A). To obtain homogeneous group sizes, our “term” group was composed of late preterm + term placenta samples (35–40 weeks of gestation), whereas the PTB group was composed of placenta samples of < 35 weeks of gestation. Despite the small sample size (n = 3–6/group, total of 10 samples), we found that when excluding one term PE outlier (labeled in orange, Fig. 3B, sample not included in statistical analysis), term PE was associated with a significant reduction in CLOCK in the human placenta, as compared to term (Fig. 3B). Interestingly, the Two-way ANOVA showed that gestation length trended towards a reduction in CLOCK [F(1,5) = 6.020, p = 0.0577], supporting our previous work that identified that PTB correlated with reduced maternal blood CLOCK mRNA56.
Figure 3.
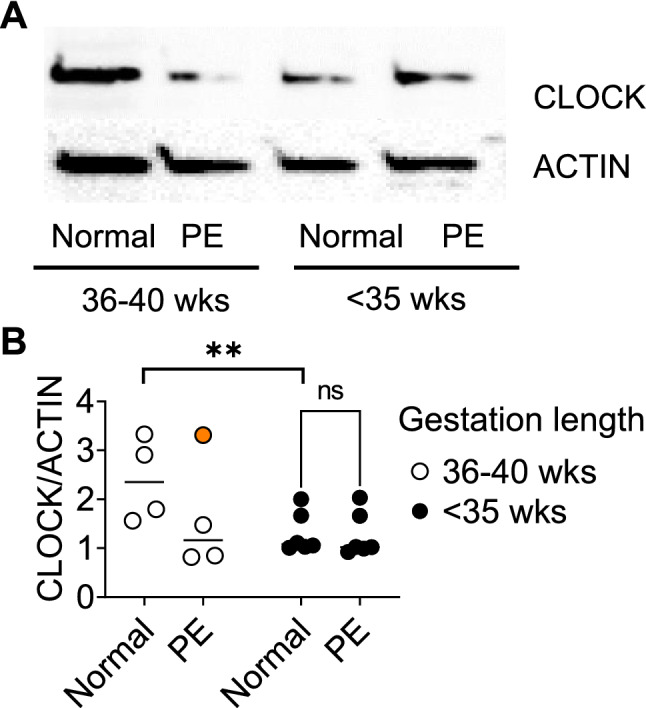
CLOCK protein is reduced in term PE placenta. (A) Example westernblot image for CLOCK and beta-ACTIN (ACTIN), of indicated human placenta samples. (B) Histogram of CLOCK/ACTIN in human placenta. Each dot represents a sample. Two-way ANOVA, **, p < 0.001. n = 3–6/group. Sample labeled in orange was an outlier and excluded from statistical analysis.
Discussion
In the present study, we found that the circadian gene transcripts for CRY1, NR1D2, and PER3 in human placenta were robustly different between PE and non-PE across two independent datasets. Importantly, NR1D2 and PER3 were downregulated in placentas of PE patients independent of gestation length, CLOCK was downregulated in PE patients who delivered at term, and CRY1 was upregulated in overall PE. Further analysis demonstrated that co-alteration of CRY1, NR1D2, and PER3 transcripts were strongly associated with the risk of PE (the OR of PE was increased more than fivefold with the risk score > median). In vitro migration assays further identified a role of CRY1/2 and NR1D1/2 in trophoblast migration. Together, this identifies CRY1, NR1D2, and PER3 as genes of particular interest in overall PE, as well as CLOCK, NR1D2, and PER3 in term PE.
CRY1, NR1D2, PER3 and CLOCK dysregulation in PE placenta
Clock genes are important in homeostatic regulation of cells and provide a molecular mechanism allowing tissues to anticipate daily changes in physiological functions. Despite the well-established role of circadian rhythms and clock genes in female reproductive function and pregnancy maintenance37,39,42,43,69, our understanding of the contribution of clock genes in placental development and function in relation to PE is in its infancy. Here, we find that among the 17 clock genes analyzed, only CRY1, NR1D2 and PER3 are consistently up- (CRY1) and down- (NR1D2 and PER3) regulated in the PE placenta, independent of PE subtypes. It should be noted that the differential expression of 7 out of 17 studied clock genes (ARNTL2, CLOCK, NPAS1, PER2, RORA, RORB, and TIMELESS) were inconsistent between the two independent datasets. Specifically, CLOCK was significantly lower in PE patients than in controls from the dataset GSE75010-173, but not in the dataset GSE75010-157. Interestingly, our subgroup analysis demonstrated that after removing patients with PTB from the GSE750-157, the decreased expression of the CLOCK transcript in placenta of the term PE group became statistically significant, which is similar to the result from the dataset GSE75010-173. In contrast, removing patients with PTB from GSE75010-157 drives the CRY1 level to be non-significant between term PE and term non-PE. These results suggest that alteration of the CLOCK transcript might be more specific to term PE, CRY1 might be more specific to heterogeneous PE, whereas the expression patterns of NR1D2 and PER3 are more robustly associated with PE, independent of gestation length and PE subtype.
CRY1, NR1D2, and PER3 associated pathways and their potential role in the pathophysiology and etiology of PE through regulation of trophoblast migration
To further understand how dysregulation of CRY1, NR1D2, and PER3 might impact placental function in relation to PE, we performed pathway analyses. The results revealed that among the top pathways that correlated with the co-alteration of the CRY1/NR1D2/PER3 in overall PE (including preterm PE), the most enriched pathways include the hypoxia-related pathways, which were increased, and the cell migration and invasion pathways, which were decreased. Because abnormal trophoblast migration is a hallmark feature of PE8,63–68, we asked if pharmacological targeting of these clock proteins in the human migratory trophoblast cell line, HTR-8, would impact their migration. Due to the limited availability of drugs specifically targeting the studied proteins, we were unable to directly test the role of NR1D2 and PER3. Pharmacologically mimicking CRY1 upregulation, using KL101, did not significantly impact HTR-8 migration, whereas pharmacologically upregulating CRY1/2, using KL001, slowed HTR-8 migration. Although KL101 did not slow HTR-8 migration, studies in cancer cell lines have shown that down-regulation of CRY1 increases cell migration, indicating CRY1, at least under specific conditions, does regulate cell migration70. Future studies using primary migratory extravillous trophoblasts will determine if KL101 might slow migration in primary trophoblast cells, which more precisely reflect extravillous trophoblast migratory properties.
Previous work in mice and cell lines has shown a role of PER3 in cell migration, where Per3 knock-down reduced migration71–73. Such reduction in cell migration when PER3 is low agrees with the predictions of our bioinformatics analysis. As no PER3 antagonist or drug downregulating PER3 are available, we asked if upregulation of PER1/2/3, using PF670462, would increase HTR-8 cell migration. In our assay, PF670462 did not impact HTR-8 migration. The lack of effect of PF670462 does not refute that low PER3 in HTR-8 cells would slow migration. Indeed, the fact that PF670462 upregulates PER1/2/3 might mask a specific effect of PER3 on HTR-8 cell migration; however, due to experimental constraints, we were unable to test whether PER3 downregulation would decrease HTR-8 migration. Another possibility is that PER3 might signal gestation length, as supported by our maternal blood study56, and preliminary GWAS study74, both of which suggest that deregulated or low PER3 levels might be linked to gestation length and predispose women to PTB. Mechanisms that PER3 may engage to signal gestation length remain unknown.
The last target protein for our migration studies was NR1D2, and again our study was limited by the availability of drugs, as both drugs used targeted NR1D1/2. In agreement with our bioinformatics prediction, an NR1D1/2 agonist did not significantly impact migration, whereas NR1D1/2 antagonism/inverse agonism slowed migration of HTR-8 cells, indicating that the level of NR1D1/2 defines increased vs decreased trophoblast migration. Together, our migration assays, combined with published work indicate that reduced PER3, NR1D(1)/2, and potentially increased CRY1, might impair trophoblast migration, a well-established feature of PE. Future work establishing how these clock proteins regulate trophoblast migration and function will be key to understand if their deregulation is the cause or a symptom of PE.
While more rigorous studies are needed to further clarify the relationships among placental clock genes, PE, and these biological pathways, based on our current understanding of the pathogenesis of PE, the clock genes identified in this study are plausible candidates for the pathogenesis and etiology of PE. For example, we identified NR1D2 and PER3 to be downregulated in the PE placenta independent of gestation length, and we show functional migration data, supporting the idea that reduced PER3 and NR1D1/2 reduces trophoblast migration, a feature known to cause PE.
CLOCK, NR1D2, and PER3 associated pathways and their potential role in the pathophysiology and etiology of term PE
In term PE patients, the most enriched pathways that were correlated with CLOCK/NR1D2/PER3-based risk score were hypoxia-related pathways and the membrane trafficking and autophagy-related pathways, which increased or decreased, respectively. Placental oxidative stress has been suggested to be central to the pathogenesis of preeclampsia7,61,62, whereas impaired trophoblast migration/invasion and spiral artery remodeling are also documented as hallmarks of PE8,63–68. While autophagy is regulated by membrane trafficking pathways in many species from yeast to mammals (reviewed by 75,76), the failure of placental autophagy has been reviewed as a risk factor of PE77. In contrast to known functional roles of NR1D2 and PER3 proteins outside their capacity to generate circadian rhythms, less is known about CRY1. The CRY1 gene encodes a flavin adenine dinucleotide-binding protein that is best known for its role in altered sleep patterns. A dominant coding variation in the CRY1 gene has been linked to familial delayed sleep phase disorder78, while PE has been associated with sleep-disordered breathing79. However, whether a similar mutation of the CRY1 gene can be found in PE patients with sleep-disordered breathing and subsequently cause the change of CRY1 transcript in placenta remains unclear.
More rigorous studies are needed to further clarify the relationships among placental clock genes, PE, and these biological pathways. Based on our current understanding of the pathogenesis of PE, the clock genes identified in this study are plausible candidates for the pathogenesis and etiology of PE. For example, we identified NR1D2 and PER3 to be downregulated in the PE placenta independent of gestation length. The nuclear receptor NR1D2 promotes expression of the inflammatory mediator, interleukin 6, a pathway that has been described to be involved in trophoblast hypoxia, arterial hypertension, placental inflammation, and autophagy in gestational diabetes mellitus80–82.
In support of our finding that term PE was associated with reduced CLOCK mRNA and protein expression in the term placenta, another study showed reduced CLOCK protein in human PE placenta83. Interestingly, this study also identified an increase in CLOCK mRNA, suggesting that mRNA processing or stability of CLOCK might be dysregulated in some cases of PE83. Like our study, these authors also used HTR-8 cells, which possess a functional molecular clock35 and respond to hypoxia by reducing CLOCK expression83.
Strengths and limitations of the present study
Our study presents several strengths and limitations. We used 17 core clock gene candidates and conducted integrated pathway analysis in two independent datasets with > 75 individual samples per group in each. These analyses improved the biological plausibility of the gene-disease relationship as well as our understanding and interpretation of the final model. Our findings are robust thanks to the use of two independent datasets that have different definitions of PE and different race/ethnicity as well as the application of both ‘in silico and molecular approaches. The datasets analyzed are publicly available (NCBI GEO GSE75010-157 and GSE75010-173). On the other hand, these datasets do not report information of time-of-onset of PE, time-of-day, or time-of-year of sample collection. Future studies controlling for time-of-day and time-of-year in both clinical/epidemiological and laboratory settings will be important to further validate the presented findings. In addition, we also lack the information on the regions of the placentas from which the tissues were sampled for RNA extraction, as central vs. peripheral cotyledons, and basal plate vs. interior may be subject to different stressors and/or contain different cellular composition. More rigorous studies with the single-cell RNA sequencing technique are needed for further clarification.
Conclusion
We here identify co-alterations of placental CRY1/NR1D2/PER3 in overall PE (a mixture of PE subtypes including term and preterm PE) and CLOCK/NR1D2/PER3 in term PE (excluding the samples from the patients with PTB) in human using bioinformatics analysis. We identify PE-associated pathways correlated with these clock genes, including increased hypoxia-related pathways as well as the decreased cell migration/invasion, autophagy, and membrane trafficking pathways in the PE placenta. We demonstrate that pharmacological targeting of CRY1/2 and NR1D1/2 in the human HTR-8 trophoblast cell line impacts migration and support the reduction of CLOCK protein in term PE through westernblot. Together these studies highlight CRY1, CLOCK, NR1D2, and PER3 as novel genes of interest to study in placental development and function in relation to PE.
Supplementary Information
Acknowledgements
We would like to thank Soo Ahn and Thu V. Duong for assistance with data generation and analysis. This research was funded by Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under Award Number R00 HD084759 (H.M.H.), the USDA National Institute of Food and Agriculture Hatch project MICL1018024 (H.M.H.) and the National Institutes of Health 5T35OD016477-19 (E.W).
Author contributions
G.Z., E.W., M.P., D.N., and H.H. conceived and designed the study. G.Z., and E.W., carried out gene expression data analysis experiments. E.W. and D.N. completed westernblot and cell migration assays. G.Z., E.W., M.P., D.N., E.P.K. and H.H. drafted the manuscript. All authors had final approval of the submitted and published versions.
Data availability
The datasets analyzed during the current study are available from the corresponding author on reasonable request. The public data (GSE75010) underlying this article are available from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE75010.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Guoli Zhou, Email: zhoug@msu.edu.
Hanne M. Hoffmann, Email: hanne@msu.edu
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-022-22507-3.
References
- 1.Bibbins-Domingo K, et al. Screening for preeclampsia. JAMA. 2017;317:1661. doi: 10.1001/jama.2017.3439. [DOI] [PubMed] [Google Scholar]
- 2.Ananth CV, Keyes KM, Wapner RJ. Pre-eclampsia rates in the United States, 1980–2010: Age-period-cohort analysis. BMJ. 2013;347:f6564. doi: 10.1136/bmj.f6564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pauli JM, Repke JT. Preeclampsia. Obstet. Gynecol. Clin. N. Am. 2015;42:299–313. doi: 10.1016/j.ogc.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Henderson JT, Thompson JH, Burda BU, Cantor A. Preeclampsia screening. JAMA. 2017;317:1668. doi: 10.1001/jama.2016.18315. [DOI] [PubMed] [Google Scholar]
- 5.Heidrich M-B, Wenzel D, von Kaisenberg CS, Schippert C, von Versen-Höynck FM. Preeclampsia and long-term risk of cardiovascular disease: What do obstetrician-gynecologists know? BMC Pregnancy Childbirth. 2013;13:61. doi: 10.1186/1471-2393-13-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeyabalan A. Epidemiology of preeclampsia: Impact of obesity. Nutr. Rev. 2013;71:S18–S25. doi: 10.1111/nure.12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rana S, Lemoine E, Granger JP, Karumanchi SA. Preeclampsia. Circ. Res. 2019;124:1094–1112. doi: 10.1161/CIRCRESAHA.118.313276. [DOI] [PubMed] [Google Scholar]
- 8.Phipps EA, Thadhani R, Benzing T, Karumanchi SA. Author Correction: Pre-eclampsia: Pathogenesis, novel diagnostics and therapies. Nat. Rev. Nephrol. 2019;15:386. doi: 10.1038/s41581-019-0156-1. [DOI] [PubMed] [Google Scholar]
- 9.Lisonkova S, Joseph KS. Incidence of preeclampsia: Risk factors and outcomes associated with early-versus late-onset disease. Am. J. Obstet. Gynecol. 2013;209(544):e1–544.e12. doi: 10.1016/j.ajog.2013.08.019. [DOI] [PubMed] [Google Scholar]
- 10.Iacobelli S, Bonsante F, Robillard P-Y. Comparison of risk factors and perinatal outcomes in early onset and late onset preeclampsia: A cohort based study in Reunion Island. J. Reprod. Immunol. 2017;123:12–16. doi: 10.1016/j.jri.2017.08.005. [DOI] [PubMed] [Google Scholar]
- 11.Lisonkova S, et al. Maternal morbidity associated with early-onset and late-onset preeclampsia. Obstet. Gynecol. 2014;124:771–781. doi: 10.1097/AOG.0000000000000472. [DOI] [PubMed] [Google Scholar]
- 12.LeFevre ML. Low-dose aspirin use for the prevention of morbidity and mortality from preeclampsia: US Preventive Services Task Force Recommendation Statement. Ann. Intern. Med. 2014;161:819. doi: 10.7326/M14-1884. [DOI] [PubMed] [Google Scholar]
- 13.Karumanchi SA. Angiogenic factors in preeclampsia. Hypertension. 2016;67:1072–1079. doi: 10.1161/HYPERTENSIONAHA.116.06421. [DOI] [PubMed] [Google Scholar]
- 14.Armaly Z, Jadaon JE, Jabbour A, Abassi ZA. Preeclampsia: Novel mechanisms and potential therapeutic approaches. Front. Physiol. 2018;9:1–15. doi: 10.3389/fphys.2018.00973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stocker LJ, Macklon NS, Cheong YC, Bewley SJ. Influence of shift work on early reproductive outcomes. Obstet. Gynecol. 2014;124:99–110. doi: 10.1097/AOG.0000000000000321. [DOI] [PubMed] [Google Scholar]
- 16.Yaw AM, McLane-Svoboda AK, Hoffmann HM. Shiftwork and light at night negatively impact molecular and endocrine timekeeping in the female reproductive axis in humans and rodents. Int. J. Mol. Sci. 2021;22:1–28. doi: 10.3390/ijms22010324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cai C, et al. The impact of occupational shift work and working hours during pregnancy on health outcomes: A systematic review and meta-analysis. Am. J. Obstet. Gynecol. 2019 doi: 10.1016/j.ajog.2019.06.051. [DOI] [PubMed] [Google Scholar]
- 18.Matteo, B., Palmer, K. T., David, C., Michele, C. & Antonella, C. Europe PMC Funders Group Shift work and pregnancy outcomes: A systematic review with meta-analysis of currently available epidemiological studies. 118, 1429–1437 (2012). [DOI] [PMC free article] [PubMed]
- 19.Weschenfelder F, Lohse K, Lehmann T, Schleußner E, Groten T. Circadian rhythm and gestational diabetes: Working conditions, sleeping habits and lifestyle influence insulin dependency during pregnancy. Acta Diabetol. 2021;58:1177–1186. doi: 10.1007/s00592-021-01708-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bouchlariotou S, et al. Melatonin secretion is impaired in women with preeclampsia and an abnormal circadian blood pressure rhythm. Ren. Fail. 2014;36:1001–1007. doi: 10.3109/0886022X.2014.926216. [DOI] [PubMed] [Google Scholar]
- 21.Ingabire PM, et al. High prevalence of non-dipping patterns among Black Africans with uncontrolled hypertension: A secondary analysis of the CREOLE trial. BMC Cardiovasc. Disord. 2021;21:254. doi: 10.1186/s12872-021-02074-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salazar MR, et al. Nocturnal hypertension and risk of developing early-onset preeclampsia in high-risk pregnancies. Hypertens. Res. 2021;44:1633–1640. doi: 10.1038/s41440-021-00740-z. [DOI] [PubMed] [Google Scholar]
- 23.Zhong L, et al. The relationship between circadian blood pressure variability and maternal/perinatal outcomes in women with preeclampsia with severe features. Hypertens. Pregnancy. 2020;39:405–410. doi: 10.1080/10641955.2020.1797777. [DOI] [PubMed] [Google Scholar]
- 24.Ayala DE, Ucieda R, Hermida RC. Chronotherapy with low-dose aspirin for prevention of complications in pregnancy. Chronobiol. Int. 2013;30:260–279. doi: 10.3109/07420528.2012.717455. [DOI] [PubMed] [Google Scholar]
- 25.Roberts JM, Escudero C. The placenta in preeclampsia. Pregnancy Hypertension. 2012;2:72–83. doi: 10.1016/j.preghy.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Founds SA, et al. Altered global gene expression in first trimester placentas of women destined to develop preeclampsia. Placenta. 2009;30:15–24. doi: 10.1016/j.placenta.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Louwen F, Muschol-Steinmetz C, Reinhard J, Reitter A, Yuan J. A lesson for cancer research: Placental microarray gene analysis in preeclampsia. Oncotarget. 2012;3:759–773. doi: 10.18632/oncotarget.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaiman D, Calicchio R, Miralles F. Landscape of transcriptional deregulations in the preeclamptic placenta. PLoS ONE. 2013;8:e65498. doi: 10.1371/journal.pone.0065498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kleinrouweler CE, et al. differentially expressed genes in the pre-eclamptic placenta: A systematic review and meta-analysis. PLoS ONE. 2013;8:e68991. doi: 10.1371/journal.pone.0068991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaartokallio T, et al. Erratum: Corrigendum: Gene expression profiling of pre-eclamptic placentae by RNA sequencing. Sci. Rep. 2016;6:17245. doi: 10.1038/srep17245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Waddell BJ, Wharfe MD, Crew RC, Mark PJ. A rhythmic placenta? Circadian variation, clock genes and placental function. Placenta. 2012;33:533–539. doi: 10.1016/j.placenta.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 32.Ditisheim AJ, Dibner C, Philippe J, Pechére-Bertschi A. Biological rhythms and preeclampsia. Front. Endocrinol. (Lausanne) 2013;4:1–8. doi: 10.3389/fendo.2013.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pérez S, et al. Evidence for clock genes circadian rhythms in human full-term placenta. Syst. Biol. Reprod. Med. 2015;61:360–366. doi: 10.3109/19396368.2015.1069420. [DOI] [PubMed] [Google Scholar]
- 34.Mark PJ, Crew RC, Wharfe MD, Waddell BJ. Rhythmic three-part harmony: The complex interaction of maternal, placental and fetal circadian systems. J. Biol. Rhythms. 2017;32:534–549. doi: 10.1177/0748730417728671. [DOI] [PubMed] [Google Scholar]
- 35.Frigato E, Lunghi L, Ferretti ME, Biondi C, Bertolucci C. Evidence for circadian rhythms in human trophoblast cell line that persist in hypoxia. Biochem. Biophys. Res. Commun. 2009;378:108–111. doi: 10.1016/j.bbrc.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 36.Akiyama S, et al. The uterus sustains stable biological clock during pregnancy. Tohoku J. Exp. Med. 2010;221:287–298. doi: 10.1620/tjem.221.287. [DOI] [PubMed] [Google Scholar]
- 37.Yaw AM, McLane-Svoboda AK, Hoffmann HM. Shiftwork and light at night negatively impact molecular and endocrine timekeeping in the female reproductive axis in humans and rodents. Int. J. Mol. Sci. 2020;22:1–28. doi: 10.3390/ijms22010324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mereness AL, et al. Conditional deletion of Bmal1 in ovarian theca cells disrupts ovulation in female mice. Endocrinology. 2016;157:913–927. doi: 10.1210/en.2015-1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoffmann HM, et al. The transcription factors SIX3 and VAX1 are required for suprachiasmatic nucleus circadian output and fertility in female mice. J. Neurosci. Res. 2021 doi: 10.1002/jnr.24864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown TM, Colwell CS, Waschek JA, Piggins HD. Disrupted neuronal activity rhythms in the suprachiasmatic nuclei of vasoactive intestinal polypeptide-deficient mice. J. Neurophysiol. 2007;97:2553–2558. doi: 10.1152/jn.01206.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakamura TJ, Sellix MT, Menaker M, Block GD. Estrogen directly modulates circadian rhythms of PER2 expression in the uterus. Am. J. Physiol. Endocrinol. Metab. 2008;295:1025–1031. doi: 10.1152/ajpendo.90392.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kennaway DJ, Boden MJ, Varcoe TJ. Circadian rhythms and fertility. Mol. Cell. Endocrinol. 2012 doi: 10.1016/j.mce.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 43.Sen, A. & Hoffmann, H. M. H. M. Role of core circadian clock genes in hormone release and target tissue sensitivity in the reproductive axis. Mol. Cell. Endocrinol.501, (2020). [DOI] [PMC free article] [PubMed]
- 44.Leavey K, et al. Unsupervised placental gene expression profiling identifies clinically relevant subclasses of human preeclampsia. Hypertension. 2016;68:137–147. doi: 10.1161/HYPERTENSIONAHA.116.07293. [DOI] [PubMed] [Google Scholar]
- 45.Nishizawa H, et al. Microarray analysis of differentially expressed fetal genes in placental tissue derived from early and late onset severe pre-eclampsia. Placenta. 2007;28:487–497. doi: 10.1016/j.placenta.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 46.Sitras V, et al. Differential placental gene expression in severe preeclampsia. Placenta. 2009;30:424–433. doi: 10.1016/j.placenta.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 47.Nishizawa H, et al. Comparative gene expression profiling of placentas from patients with severe pre-eclampsia and unexplained fetal growth restriction. Reprod. Biol. Endocrinol. 2011;9:107. doi: 10.1186/1477-7827-9-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsai S, et al. Transcriptional profiling of human placentas from pregnancies complicated by preeclampsia reveals disregulation of sialic acid acetylesterase and immune signalling pathways. Placenta. 2011;32:175–182. doi: 10.1016/j.placenta.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meng T, et al. Identification of differential gene expression profiles in placentas from preeclamptic pregnancies versus normal pregnancies by DNA microarrays. Omi. A J. Integr. Biol. 2012;16:301–311. doi: 10.1089/omi.2011.0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiang Y, et al. Up-regulated expression and aberrant DNA methylation of LEP and SH3PXD2A in pre-eclampsia. PLoS ONE. 2013;8:e59753. doi: 10.1371/journal.pone.0059753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blair JD, et al. Widespread DNA hypomethylation at gene enhancer regions in placentas associated with early-onset pre-eclampsia. Mol. Hum. Reprod. 2013;19:697–708. doi: 10.1093/molehr/gat044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heider A, Alt R. virtualArray: A R/bioconductor package to merge raw data from different microarray platforms. BMC Bioinform. 2013;14:75. doi: 10.1186/1471-2105-14-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gautier L, Cope L, Bolstad BM, Irizarry RA. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 54.Albrecht U. Timing to perfection: The biology of central and peripheral circadian clocks. Neuron. 2012;74:246–260. doi: 10.1016/j.neuron.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 55.Park CS, et al. Hepatocyte gene expression and DNA methylation as ancestry-dependent mechanisms in African Americans. NPJ Genomic Med. 2019;4:1–12. doi: 10.1038/s41525-019-0102-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou G, Duong TV, Kasten EP, Hoffmann HM. Low CLOCK and CRY2 in 2nd trimester human maternal blood and risk of preterm birth: A nested case-control study. Biol. Reprod. 2021 doi: 10.1093/biolre/ioab119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walker, D. A. & Smith, T. J. Logistic regression under sparse data conditions. J. Mod. Appl. Stat. Methods18, (2019).
- 58.Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47–e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chandra I, Sun L. Preterm and term preeclampsia: Differences in biochemical parameter and pregnancy outcomes. Postgrad. Med. 2018;130:703–707. doi: 10.1080/00325481.2018.1527169. [DOI] [PubMed] [Google Scholar]
- 60.Poon LC, Sahota D, Li YL, Pan Y. Screening and prevention of preeclampsia. Matern. Med. 2019;1:25–30. [Google Scholar]
- 61.Albers RE, et al. Trophoblast-specific expression of Hif-1α results in preeclampsia-like symptoms and fetal growth restriction. Sci. Rep. 2019;9:2742. doi: 10.1038/s41598-019-39426-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Iriyama T, et al. Hipoxia independent up-regulation of placenta HIF-1A contributes to the pathogenesis of preeclampsia. Hypertension. 2016;65:1307–131565. doi: 10.1161/HYPERTENSIONAHA.115.05314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Steegers EAP, von Dadelszen P, Duvekot JJ, Pijnenborg R. Pre-eclampsia. Lancet. 2010;376:631–644. doi: 10.1016/S0140-6736(10)60279-6. [DOI] [PubMed] [Google Scholar]
- 64.Lyall F, Robson SC, Bulmer JN. Spiral artery remodeling and trophoblast invasion in preeclampsia and fetal growth restriction. Hypertension. 2013;62:1046–1054. doi: 10.1161/HYPERTENSIONAHA.113.01892. [DOI] [PubMed] [Google Scholar]
- 65.Pollheimer J, Vondra S, Baltayeva J, Beristain AG, Knöfler M. Regulation of placental extravillous trophoblasts by the maternal uterine environment. Front. Immunol. 2018;9:1–18. doi: 10.3389/fimmu.2018.02597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Canfield J, et al. Decreased LIN28B in preeclampsia impairs human trophoblast differentiation and migration. FASEB J. 2019;33:2759–2769. doi: 10.1096/fj.201801163R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Staff AC, et al. Failure of physiological transformation and spiral artery atherosis: Their roles in preeclampsia. Am. J. Obstet. Gynecol. 2020 doi: 10.1016/j.ajog.2020.09.026. [DOI] [PubMed] [Google Scholar]
- 68.Chen Q, et al. Association of lncRNA SH3PXD2A-AS1 with preeclampsia and its function in invasion and migration of placental trophoblast cells. Cell Death Dis. 2020;11:583. doi: 10.1038/s41419-020-02796-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Miller BH, et al. Circadian Clock mutation disrupts estrous cyclicity and maintenance of pregnancy. Curr. Biol. 2004;14:1367–1373. doi: 10.1016/j.cub.2004.07.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhou L, Yu Y, Sun S, Zhang T, Wang M. Cry 1 regulates the clock gene network and promotes proliferation and migration via the Akt/P53/P21 pathway in human osteosarcoma cells. J. Cancer. 2018;9:2480–2491. doi: 10.7150/jca.25213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tang W, et al. Period 3, a tumor suppressor in non-small cell lung cancer, is silenced by hypermethylation. Int. J. Clin. Exp. Pathol. 2018;11:120–128. [PMC free article] [PubMed] [Google Scholar]
- 72.Hong Z, Feng Z, Sai Z, Tao S. PER3, a novel target of miR-103, plays a suppressive role in colorectal cancer in vitro. BMB Rep. 2014;47:500–505. doi: 10.5483/BMBRep.2014.47.9.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Noda M, et al. Role of Per3, a circadian clock gene, in embryonic development of mouse cerebral cortex. Sci. Rep. 2019;9:1–11. doi: 10.1038/s41598-019-42390-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guerrero, R. F. et al. Genetic polymorphisms associated with adverse pregnancy outcomes in nulliparas. 0–16 (2022). [DOI] [PMC free article] [PubMed]
- 75.Ao X, Zou L, Wu Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 2014;21:348–358. doi: 10.1038/cdd.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou C, et al. Regulation of mATG9 trafficking by Src- and ULK1-mediated phosphorylation in basal and starvation-induced autophagy. Cell Res. 2017;27:184–201. doi: 10.1038/cr.2016.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nakashima A, et al. Placental autophagy failure: A risk factor for preeclampsia. J. Obstet. Gynaecol. Res. 2020;46:2497–2504. doi: 10.1111/jog.14489. [DOI] [PubMed] [Google Scholar]
- 78.Patke, A. et al. Mutation of the human circadian clock gene CRY1 in familial delayed sleep phase disorder. Cell169, (2017). [DOI] [PMC free article] [PubMed]
- 79.Yinon D. Pre-eclampsia is associated with sleep-disordered breathing and endothelial dysfunction. Eur. Respir. J. 2006;27:328–333. doi: 10.1183/09031936.06.00010905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li Y, et al. Gestational diabetes mellitus in women increased the risk of neonatal infection via inflammation and autophagy in the placenta. Medicine (Baltimore) 2020;99:e22152. doi: 10.1097/MD.0000000000022152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shirasuna K, et al. Moderate hypoxia down-regulates interleukin-6 secretion and TLR4 expression in human Sw.71 placental cells. Cell. Physiol. Biochem. 2015;36:2149–2160. doi: 10.1159/000430181. [DOI] [PubMed] [Google Scholar]
- 82.Hashimoto-Kataoka T, et al. Interleukin-6/interleukin-21 signaling axis is critical in the pathogenesis of pulmonary arterial hypertension. Proc. Natl. Acad. Sci. USA. 2015;112:E2677–E2686. doi: 10.1073/pnas.1424774112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li Y, et al. Circadian clock gene Clock is involved in the pathogenesis of preeclampsia through hypoxia. Life Sci. 2020;247:117441. doi: 10.1016/j.lfs.2020.117441. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets analyzed during the current study are available from the corresponding author on reasonable request. The public data (GSE75010) underlying this article are available from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE75010.