

Abstract
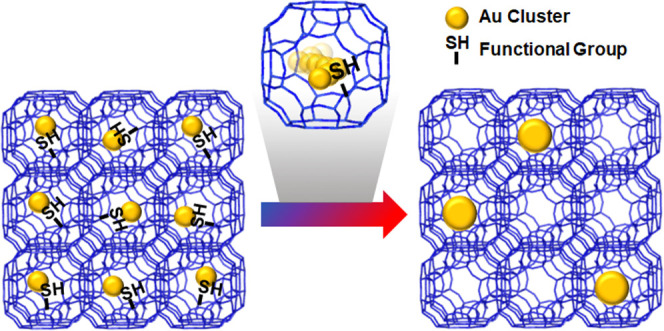
Confining Au nanoparticles (NPs) in a restricted space (e.g., zeolite micropores) is a promising way of overcoming their inherent thermal instability and susceptibility to aggregation, which limit catalytic applications. However, such approaches involve complex, multistep encapsulation processes. Here, we describe a successful strategy and its guiding principles for confining small (<2 nm) and monodisperse Au NPs within commercially available beta and MFI zeolites, which can oxidize CO at 40 °C and show size-selective catalysis. This protocol involves post-synthetic modification of the zeolite internal surface with thiol groups, which confines AuClx species inside microporous frameworks during the activation process whereby Au precursors are converted into Au nanoparticles. The resulting beta and MFI zeolites contain uniformly dispersed Au NPs throughout the void space, indicating that the intrinsic stability of the framework promotes resistance to sintering. By contrast, in situ scanning transmission electron microscopy (STEM) studies evidenced that Au precursors in bare zeolites migrate from the matrix to the external surface during activation, thereby forming large and poorly dispersed agglomerates. Furthermore, the resistance of confined Au NPs against sintering is likely relevant to the intrinsic stability of the framework, supported by extended X-ray absorption fine structure (EXAFS), H2 chemisorption, and CO Fourier transform infrared (FT-IR) studies. The Au NPs supported on commercial MFI maintain their uniform dispersity to a large extent after treatment at 700 °C that sinters Au clusters on mesoporous silicas or beta zeolites. Low-temperature CO oxidation and size-selective reactions highlight that most gold NPs are present inside the zeolite matrix with a diameter smaller than 2 nm. These findings illustrate how confinement favors small, uniquely stable, and monodisperse NPs, even for metals such as Au susceptible to cluster growth under conditions often required for catalytic use. Moreover, this strategy may be readily adapted to other zeolite frameworks that can be functionalized by thiol groups.
Keywords: supported catalyst, gold, zeolite, confinement, nanoparticles
Introduction
Metallic gold has been considered chemically inert historically, but some studies have shown that gold nanoparticles (typically smaller than 10 nm) are exceptionally active in a wide range of chemical reactions.1−3 Since then, considerable research efforts have been devoted to developing methods for the synthesis of gold nanoparticles. As a result, various applications, such as drug delivery, sensing, and catalysis, have emerged from the synthesis of gold nanoparticles.4−6
Synthesizing gold nanoparticles requires stabilizing gold atoms at the surface. Using soluble ligands that strongly interact with gold atoms during preparation is a straightforward way of synthesizing stable gold nanoparticles.7 For this purpose, thiol-containing organic molecules are the most widely used stabilizing agents.8 However, in practical catalytic applications, where the phase of the catalyst (mostly solid) differs from that of the reactants or products (mostly gaseous), the gold catalysts are primarily supported on high-surface-area solid materials. Such heterogeneous gas-phase reactions remain mostly overlooked owing to difficulties in preparing supported gold nanoparticles.9 Furthermore, metallic gold has a low Hüttig temperature, which facilitates the agglomeration of small nanoparticles and consequently rapid deactivation during catalytic cycles. For this reason, thermally stable ultrasmall Au nanoparticles (i.e., ∼1 nm in size) are much more difficult to prepare than their counterparts of platinum group metals.
To overcome this lack of thermal stability, many researchers have sought to develop methods for confining Au nanoparticles within a restricted space by embedding them into porous materials, such as MCM-41, porous Al2O3, and zeolites.10−12 Among these materials, zeolites stand out as crystalline microporous aluminosilicates with uniform micropores smaller than 1 nm and regularly arranged throughout the framework.13,14 Aluminosilicate zeolites have negatively charged surfaces that can accommodate positively charged cations (e.g., Pt(NH3)42+ and Pd2+) inside the microporous network. As such, metal-supported zeolites can be easily prepared by loading cationic metal species into the micropores via cation-exchange and subsequent thermal activation processes.15 However, the lack of affinity between the negatively charged zeolite surface and the similarly negative AuCl4–-derived gold complexes hinders the adsorption of gold species onto zeolites.16 Consequently, conventional ion-exchange/reduction or impregnation/reduction methods have persistently failed to promote high Au dispersion on zeolites.17 Furthermore, when using the standard deposition–precipitation method, the gold hydroxide species (i.e., Au(OH)4–) that are precipitated at an elevated pH are not deposited onto the zeolite matrix due to charge repulsion, as described above.18 In other attempts to deposit gold species onto supports at a pH of ∼2, which is close to the isoelectric point of zeolite supports,10,19 the residual Cl– on the catalysts resulted in fast deactivation in CO oxidation.20 Yet, despite these limitations, Wan et al. were able to reverse the negative charge of commercial HY zeolites by partial ion exchange with Na+, thereby favoring gold deposition onto micropore walls, with Y zeolite-supported Au exhibiting high activity toward CO oxidation.21 For now, this phenomenon remains exclusive to Y zeolite, but if a versatile method for supporting gold nanoparticles on a wide range of preformed or commercially available zeolites were to be developed, such a system would support a wide range of key catalytic reactions.
Several alternative methods for improving gold dispersion have been proposed recently, including direct synthesis of zeolites in the presence of Au nanoparticles or gold precursors and the use of mesoporous zeolites as supports. For example, Christensen et al. first prepared an Au nanoparticle/amorphous silica composite matrix and then hydrothermally crystallized the amorphous silica into the zeolite phase while retaining the initial structure.22,23 The resulting MFI crystals had 1–2 nm gold nanoparticles within the zeolite matrix although some gold nanoparticles were also present on the external surface. Iglesia et al. were able to directly synthesize gold nanoparticles encapsulated in LTA and MFI from chloroauric acid-containing aluminosilicate gels using mercaptosilanes as the simultaneous encapsulating agents and framework building units.24−26 In turn, Kegnæs et al. reported that generating constricted mesopores in bulk MFI crystals markedly enhanced the initial dispersion of Au and its resistance against sintering due to the confinement effect of constricted mesopores.27 In addition to this, both strategies, however, involve complex, multistep in situ synthesis processes, which may limit their practical applications.
In the present study, we describe a versatile method for confining gold nanoparticles within preformed zeolites by post-synthetic modification of their zeolite internal surface with thiol groups, which have a high affinity to gold species. For this purpose, commercially available beta zeolites (denoted by “u-beta”) with Si/Al = 12.5 (CP814E*) were mixed with 4-thiolbenzene diazonium salts in the acetonitrile solvent under reflux. The resulting functionalized zeolites, hereafter denoted by “f-beta”, contained thiol moieties at the zeolite micropores for AuClx confinement in the zeolite framework. During the activation process, gold precursors were reduced to gold nanoparticles. Thus, small Au nanoparticles can be supported on commercially available beta zeolites with high thermal stability, as shown in Scheme 1 (top). In contrast, Au precursors on bare beta zeolites easily migrate from the internal to the external surface of the zeolite crystal. Because this commercial zeolite has a very low external surface area, Au species formed large and poorly dispersed agglomerates with low thermal stability (Scheme 1, bottom). Moreover, this strategy may be extended to other commercial zeolites, such as MFI zeolite (CBV2314).
Scheme 1. Comparative Thermal Stabilities of Au Nanoparticles Supported on Surface-Modified (Top) and Unmodified (Bottom) Commercially Available Beta Zeolites.

Methods
Materials
Beta and MFI (CBV2314) zeolites were purchased from Zeolyst International. Hydrogen tetrachloroaurate(III) hydrate (99.9%) and 4-aminothiophenol (97.0%) were purchased from Alfa-Aesar. Tetrafluoroboric acid solution (48.0 wt % in H2O), tert-butyl nitrite (90.0%), silicon dioxide (0.85–4.75 mm, 99.9%), and 3,5-di-tert-butyl-4-hydroxybenzylalcohol were purchased from Sigma-Aldrich. Benzyl alcohol (99.0%) and dodecane (99.0%) were purchased from Tokyo Chemical Industry. Acetonitrile (99.5%), diethyl ether (99.5%), acetone (99.5%), and chloroform (99.8%) were purchased from Samchun. All reagents were used as received.
Synthesis of 4-Thiolbenzene Diazonium Tetrafluoroborate
The diazonium salt was prepared according to the method reported by Moloney et al.28 In a typical procedure, 4-aminothiophenol (1 equiv), tetrafluoroboric acid solution (2 equiv), and ethanol (2 equiv) were added to a 100 mL round-bottomed flask (RBF). The RBF was sealed with a rubber stopper and then placed in an ice bath under continuous stirring for 30 min, followed by dropwise addition of tert-butyl nitrite (2 equiv). The final reaction mixture was then aged for 2 h. After this period, an appropriate amount of sample was taken, filtered, and washed with diethyl ether. The resulting material was dried in a vacuum oven at room temperature.
Functionalization of Preformed Aluminosilicate Zeolites
Commercial zeolite (1 g, beta or MFI) was placed into a convection oven for 3 h, which was heated at 150 °C under atmosphere. The dried zeolites were then added to a glass bottle containing acetonitrile (10 mL) and 4-thiobenzene diazonium tetrafluoroborate (200 mg). Subsequently, the glass bottle was placed to a Personal Organic Synthesizer (company: EYELA, model number: ChemiStation PPS-CTRL). Then, the solution was aged for 2 h at room temperature (RT) and then for 8 h at 70 °C, while stirring at 300 rpm. After cooling to RT, the solid products were filtered with filter paper (pore size: 5 μm), washed twice with acetonitrile, and dried at RT for 1 h. After dispersing the solid precipitates in acetonitrile (15 mL), the resulting mixture was stirred for 4 h at RT. The solid powders were filtered and dried at RT in a vacuum oven.
Preparation of Au Particles Supported on Zeolites
Gold nanoparticles were supported on zeolites using the wet-impregnation method or the incipient-wetness method, as detailed in the following paragraphs.
For the wet-impregnation method, zeolites (1 g) were dispersed in distilled water (50 mL), followed by dropwise addition of deionized water (12 mL) containing an appropriate amount of HAuCl4 under vigorous stirring at RT. After agitation for 4 h at RT, the solid products were filtered, washed with distilled water, and dried overnight in a vacuum oven at RT. The dried samples were then subjected to calcination at 0.88 °C min–1 for 4 h under a flow of O2 (100 mL min–1). Note that Au/f-beta was calcined at 550 °C for complete decomposition of the thiolbenzene (TB) moiety in the zeolite frameworks. After cooling to RT, the O2-activated zeolites were reduced at 350 °C for 2 h under a flow of H2 (20 mL min–1). Prior to sample collection, the Au-supported catalysts were passivated in air at RT for 1 h.
For the incipient-wetness method, the required amount of metal source was dissolved in an amount of distilled water equivalent to the pore volume of the support. The solution was then impregnated into zeolite using a mortar and pestle, and the sample was added into a polypropylene bottle. The bottle was then closed tightly and left standing for 1 h at RT. Subsequently, the sample was dried in a vacuum oven at RT and activated using the method followed for the wet impregnated sample.
Characterization
X-ray diffraction (XRD) patterns were analyzed at 30 kV and 15 mA under Cu Kα radiation using a Rigaku Miniflex diffractometer. Solid-state 13C{1H} cross-polarization (CP) nuclear magnetic resonance (NMR) spectra were recorded at a magic angle spinning (MAS) rate of 12 kHz on a Bruker Avance 400 WB spectrometer. The samples were degassed at 250 °C for 6 h before the measurements using the following 13C{1H} CP MAS NMR measurement conditions: 13C{1H} resonance frequency = 100.61 MHz, pulse width = 2.5 μs (CP pulse sequence), relaxation delay = 5 s, and number of scans = 16,000. The C and H contents of zeolite were determined on a FlashSmart Elemental Analyzer (Thermo Fisher Scientific) equipped with a thermal conductivity detector. Thermogravimetric analysis (TGA) was performed using a Brucker TG-DTA2000 analyzer under a 20 mL min–1 flow of oxygen at a heating rate of 5 °C min–1 up to 700 °C. For beta zeolite, the argon adsorption–desorption isotherm was measured using an ASAP 2020 instrument (Micromeritics) at −186 °C. Before the analysis, the samples were degassed at 250 °C for 4 h. The volume of the micropores was calculated from adsorption isotherm data following Horvath–Kawazoe (HK) analysis. In MFI zeolite, nitrogen adsorption–desorption isotherms were obtained on a Micromeritics Tristar II 3020 analyzer at −196 °C. Before the measurements, the samples were degassed at 250 °C for 4 h. The micropore volume was derived from the t-plot method. The amount of gold in zeolites was determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES) on an iCAP 6300 Duo spectrometer (Thermo Scientific Co., U.K.). Scanning electron microscopy (SEM) imaging was performed under a JEOL IT-800 HR-SEM using a backscattered electron (BSE) detector. STEM images were acquired under a JEOL NEOARM 200F atomic resolution analytical electron microscope operating at 200 kV, using an ADF detector. In situ heating and imaging in a transmission electron microscope were performed using a Gatan 652 double-tilt heating holder.
Hydrogen chemisorption was measured using a volumetric method and a laboratory-built volumetric glass rig. Before the measurements, the Au-loaded samples were reduced under H2 flow (99.999%, passed through a MnO/SiO2 trap) at 200 °C for 2 h. Subsequently, the reduced samples were evacuated for 2 h at the same temperature for hydrogen desorption. After cooling down to 60 °C, hydrogen chemisorption was measured in the pressure range from 20 to 200 torr. The ratio of hydrogen atoms to Au metal atoms (H/Au) was calculated by H2 adsorption isotherm extrapolation to zero pressure.
Extended X-ray absorption fine structure (EXAFS) spectra were recorded at Pohang Accelerator Laboratory (10C) using synchrotron X-rays. For EXAFS measurements, zeolite powders were pressed into round-shaped pellets at ∼1 ton for 1 min. After loading the pellet on a holder, the measurements were conducted around the Au L3 edge at 11,919 eV. The EXAFS function χ(E) was derived from absorption spectroscopy data by preedge adsorption subtraction, normalization to the adsorption edge, and post-edge background subtraction. The resulting χ(E) was converted into k-space, with k being the wave vector. The χ(k) was weighted by k3, and k3χ(k) was Fourier transformed into R-space with a region of wave vector 3 < k < 14 Å–1 by applying the window function of Hanning. Subsequently, the resulting k3χ(k) and |χ(R)| were fitted using the program Artemis. All fits were performed in k- and R-space to determine the set of coordination numbers (N), bond distance (R), Debye–Waller factor (σ2), and phase-shift adjustment parameters (ΔE0). The fitting ranges for k- and R-space are 3 < k < 14 Å–1 and 1.5 < R < 3.0 nm, respectively. All samples were fitted only with the nearest-neighbor Au coordination shell.
Prior to Fourier transform infrared (FT-IR) studies, all zeolites were pressed into self-supporting wafers (∼8 to 12 mg cm–2) and activated at 450 °C for 2 h (ramping rate: 5 °C min–1) in a quartz in situ cell. To obtain a detailed insight into the nature of gold sites, low-temperature CO adsorption was performed. That is, subsequent doses of CO were first adsorbed on activated zeolites at −150 °C for 5 min. After full saturation of sites present in the zeolites under study, the physisorbed probe molecules were removed by evacuation. All IR spectra were recorded with a Nicolet iS50 spectrometer equipped with an MCT detector at −150 °C and normalized to the same density of sample (10 mg cm–2). The spectral resolution was 2 cm–1.
Catalytic Reactions
CO oxidation was performed in a continuous fixed-bed down-flow reaction system equipped with a laboratory-made stainless steel vertical reactor (inner diameter = 0.74 cm, length = 36 cm). Typically, 0.1 g of the sieved catalyst (0.36–0.60 mm) diluted in quartz (1.05 g) was loaded at the center of the reactor. The reaction temperature was measured using a thermocouple placed just above the catalyst bed. The catalysts were activated at 350 °C with a ramping rate of 0.9 °C min–1 for 2 h under a flow of H2 (20 mL min–1) and then cooled to 40 °C, while the H2 flow was changed to an Ar flow. Subsequently, a gas mixture consisting of 0.5 vol % CO, 5 vol % H2O, 10 vol % O2, and balance Ar was introduced into the reactor at a total flow rate of 16.5 mL min–1. CO conversion was lower than 10% under these reaction conditions. The feed gas was passed through a water bubbler to add water content by controlling the temperature of the bubbler. The outlet gas passing through drierite was regularly analyzed every 10 min on a gas chromatograph (Agilent 7890A) equipped with a GS GASPRO column to obtain the CO conversion. CO and CO2 concentrations of the outlet gas were analyzed using a flame ionization detector after passing the product gas stream through a methanizer. CO conversion and reaction rate were calculated as follows
| 1 |
| 2 |
Benzyl alcohol and 3,5-di-tert-butyl-4-hydroxybenzylalcohol oxidation reactions were performed in an organic synthesizer using a tube glass reactor sealed with a rubber stopper. In total, 10 mg of the catalyst was suspended in 31.6 g of the reaction mixture (1 mol % benzyl alcohol/1 mol % 3,5-di-tert-butyl-4-hydroxybenzylalcohol/1 mol % dodecane (internal standard)/20 mol % chloroform/77% p-xylene) under continuous stirring at 25 °C. O2 gas was bubbled into the reaction mixture. For analysis, 0.5 mL of the reaction mixture was collected each hour. The reaction mixture was filtered through a 0.45 μm syringe filter (Whatman, PVDF). This aliquot was analyzed on a GC system (Agilent 7890A) equipped with a flame ionization detector and a HP-5 column. The conversion of aromatic alcohol was calculated from the ratio of the decreased peak area of aromatic alcohols with that of the peak area of dodecane. The reaction rate was calculated using eq 3
| 3 |
To calculate the reaction rate of benzyl alcohol and 3,5-di-tert-butyl-4-hydroxybenzylalcohol, data were collected only until conversion reached 10%.
Acetylene hydrogenation reactions were performed in a purge flow reactor system using a 45 cm long Pyrex tube reactor with an inner diameter of 0.91 cm. The sieved catalysts (0.1 g, 0.36–0.60 mm) were placed onto a fritted disc and positioned at the middle of a Pyrex tube. The gold-supported catalysts were activated under a H2 flow (20 mL min–1) at a heating rate of 0.9 °C min–1 at 350 °C. After treating the sample at 350 °C for 4 h, H2 gas was replaced by inert N2 gas, and the temperature was decreased to 250 °C. When the temperature was stabilized, the catalyst samples were subjected to a flow of gases (1 C2H2/10 H2/19 N2) at 20 mL min–1, and the temperature was maintained at 250 °C for 4 h. The outlet gas stream was regularly analyzed using a gas chromatograph (Agilent 7890A) equipped with a flame ionization detector and a GS GASPRO column.
Results and Discussion
Strong Covalent Attachment of Thiol Groups on the Internal Surface of Zeolites Using Thiolbenzene Diazonium
Figure 1a shows XRD patterns of f-beta and u-beta samples, demonstrating that zeolite crystallinity is retained after functionalization using organics. Elemental analysis (EA) indicated that f-beta contains 4.9 wt % C atoms and 1.2 wt % S atoms, that is, a perceptible amount of TB moieties. These TB moieties are covalently attached to the zeolite frameworks, as evidenced by the NMR spectra and TGA profiles shown in Figure 1b,c, respectively. The 13C solution NMR spectrum of TB-N2+ showed four resonances at 114, 128, 134, and 152 ppm (Figure S1). However, after the reaction of TB-N2+ with u-beta, a new signal appeared at ∼161 ppm in the solid-state 13C{1H} CP MAS NMR spectrum of the f-beta sample (red line). This result suggests that the C–N2+ bond is replaced by the C–O bond through a substitution reaction. The strong covalent attachment of −TB to the zeolite beta framework was further demonstrated by the TGA profiles in Figure 1c. Here, the u-beta sample exhibited mass loss between 50 and 150 °C, resulting from water desorption. Although the f-beta sample also exhibited mass loss due to water desorption, an additional weight loss of ∼6.2 wt % was observed in the high-temperature range of 250–500 °C, resulting from the decomposition of organic species by air. In this regard, TB is most likely strongly attached to the zeolite framework by chemical bonding. This grafting of TB onto the interior pore walls was further evidenced by the Ar isotherms in Figure 1d. Both samples exhibited a well-resolved upward step in the region of 0.0 < P/P0 < 0.01 due to micropore filling. The f-beta sample had a cumulative micropore volume of 0.165 cm3 g–1, which is 0.25 times lower than that of u-beta. Such a decrease in micropore volume indicated that micropore availability decreased due to micropore-wall functionalization.29 Overall, these findings confirm that TB moieties are attached to the interior micropore walls.
Figure 1.
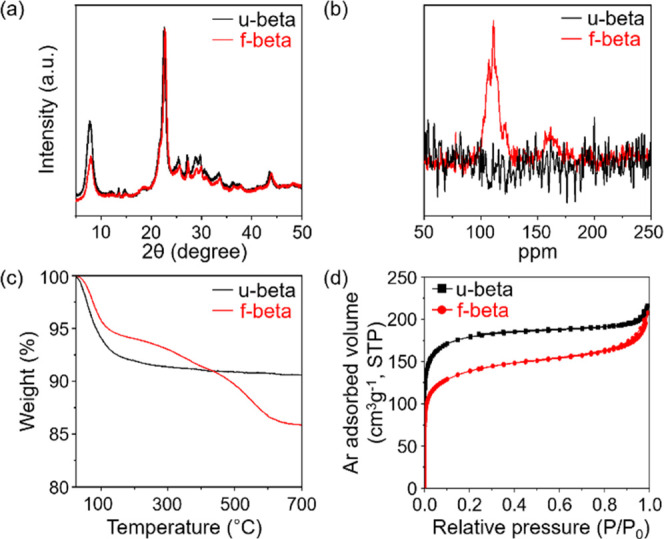
XRD patterns (a), solid-state 13C{1H} CP MAS NMR spectra (b), TGA profiles (c), and Ar isotherms (d) of f-beta and u-beta samples.
Post-Synthetic Modification Enables the Confinement of Gold Nanoparticles within Zeolite Micropores
As shown in Figure S2, no change was observed in the UV spectrum of the AuCl4– solution after continuous mixing with u-beta for 8 h at 25 °C for wet impregnation. In addition, as detected by visual examination of the precursor solution before and after mixing with u-beta, the yellow color was retained (Figure S3, top row). Furthermore, based on our ICP-OES analysis, only negligible amounts of gold species were absorbed into u-beta due to weak interactions between AuCl4– ions and zeolite pore walls in the neutral environment (pH ∼ 6). The low isoelectric point of the aluminosilicate zeolite is known to hinder AuCl4– deposition onto the support.21 By contrast, when the AuCl4– solution was continuously stirred with f-beta, the solution changed from yellowish to transparent (bottom row, Figure S3). After filtration, the characteristic UV absorption peak of the AuCl4– ions in pure solution (black line, Figure S4) was significantly reduced in the filtrate (red line, Figure S4). The resulting f-beta sample is hereafter denoted by Au/f-beta. Our ICP-OES analysis also indicated that Au/f-beta contains 1.1 wt % gold. These results demonstrate that AuCl4– ions in an aqueous solution are effectively adsorbed onto f-beta during the aging process.
For comparison, u-beta zeolites were impregnated with the AuCl4– precursors using the incipient-wetness impregnation (IWI) method and are denoted by Au/u-beta hereafter. The IWI process uses a minimum amount of metal precursor solution to fill the entire pore volume of the support. After filling the pores with the precursor solution, the solvent was evaporated by heating to support gold precursors as AuCl4– species within the microporous network.
Figure 2 shows XRD patterns of Au/u-beta and Au/f-beta samples after each step in the Au loading process (i.e., O2 calcination (at 350 °C for Au/u-beta, at 550 °C for Au/f-beta), and H2 reduction at 350 °C). Note that Au/f-beta was calcined at 550 °C for complete decomposition of TB moieties in zeolite frameworks. As shown in Figure 2a, after O2 calcination, the XRD pattern of Au/u-beta exhibited two narrow peaks at 38.2 and 44.4°, which can be assigned to the (111) and (200) crystal planes of metallic gold (red line, Figure 2a). The crystal size, calculated using the Scherrer equation, was 29 nm. After H2 treatment, the Au species remained in large agglomerates in this bulk crystal sample (blue line, Figure 2a). By contrast, the Au species supported on f-beta were found in small gold nanoparticles after O2 calcination at 550 °C, as confirmed by the broad XRD peaks in Figure 2b (red line). The calculated crystal size was ∼2.5 nm. Furthermore, the Au nanoparticles maintained their size after H2 treatment (blue line, Figure 2b).
Figure 2.
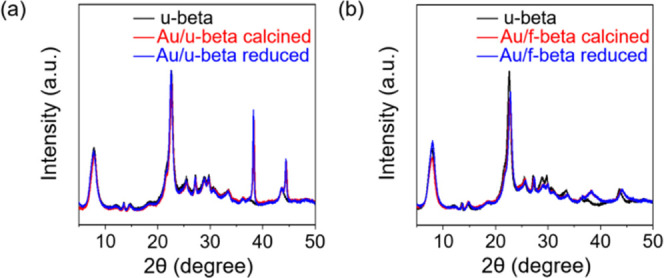
XRD patterns of Au/u-beta (a) and Au/f-beta (b) samples taken after each successive step in the Au loading process: O2 calcination (at 350 °C for Au/u-beta, at 550 °C for Au/f-beta) and H2 reduction at 350 °C. In each case, the XRD pattern of u-beta is displayed in black for comparison.
These XRD results indicate a marked difference in Au dispersity after the O2 activation step, even though Au/u-beta was calcined at a much lower temperature than Au/f-beta. Hence, this high dispersion of Au nanoparticles was facilitated by the −SH group attachment on the internal surface of the zeolite. The effect of the thiol moiety on the size and location of the Au species was assessed by SEM and STEM analyses of supported gold nanoparticles at various treatment stages, as shown in Figures S5 and 3. In addition, O2-calcined Au/u-beta and Au/f-beta samples were tested in probe reactions (see Methods) to determine the extent to which the host zeolites prevent molecules larger than their apertures from accessing active Au surfaces.
Figure 3.

Representative STEM images of Au/u-lab-beta (top row) and Au/f-lab-beta (bottom row) samples after AuCl4– impregnation (a, d), O2 calcination at 350 °C (b, e), O2 calcination at 550 °C (f), and H2 reduction at 350 °C (c, g).
EM was performed using 500–700 nm beta zeolites prepared in the laboratory, as shown in Figure S6, rather than the commercial zeolite, for a clearer cross-sectional image. These samples are hereafter denoted by Au/u-lab-beta and Au/f-lab-beta. The XRD pattern presented in Figure S7 indicates that using the large-sized beta crystallites instead of the commercial beta leads to similar gold dispersion results.
After the initial impregnation step, the Au/u-lab-beta sample exhibited a homogeneous distribution of Au species throughout the microporous network (Figure 3a). However, after the O2-calcination step, gold nanoparticles with diameters ranging from tens to hundreds of nanometers were observed on external crystal surfaces (Figures 3b and S5b), but not in the interior of the Au/u-beta sample, as evidenced by the cross-sectional STEM image in Figure S8. Accordingly, during the O2 calcination process, gold species migrate from the matrix to the external surface, where they agglomerate into large Au crystallites. A previous study has revealed that HAuCl4 is first decomposed to AuCl3 at 190 °C, with the simultaneous release of H2O and HCl and then transformed to AuCl at 240 °C, with Cl2 release, and ultimately converted into Au at 320 °C.30 The intermediate Au species (i.e., AuCl3, AuCl) have low melting and boiling points, and AuCl, in particular, has a very low boiling point of 298 °C.31 These intermediate Au species may be highly mobile during the activation process, thereby explaining their potential to migrate away from intracrystalline void space and toward the external surface.
We further performed in situ heating STEM studies to track the evolution, location, and stability of metal nanoparticles in Au/u-lab-beta. Images of unfunctionalized, as-synthesized Au/u-lab-beta showed no large metal nanoparticles (Figure S9a). After, in situ heating in an electron microscope for 2 h at 550 °C, we observed the evolution of relatively large gold nanoparticles located on the surface of zeolite crystals (Figure S9b). This evidence supports the discussion above, proving that calcining unfunctionalized zeolites produces mobile nanoparticles, located mostly at the external crystal surface. The samples were further heated (at 700 °C for 2 h) in the electron microscope to track the thermal stability and sintering effects of Au at the crystal surface. Figure S9c shows that the nanoparticles previously observed at the surface of the crystals grew significantly and that larger Au species simultaneously evolved on the surface. These findings also demonstrate that the unfunctionalized support did not stabilize the nanoparticles, in addition to significant sintering effects at high temperatures.
The high-resolution STEM image of the Au/f-lab-beta sample in Figure 3d also reveals high dispersion of gold precursors throughout the zeolite matrix. As shown by X-ray fluorescence, the Cl/Au ratio is lower in gold species supported on Au/f-lab-beta (Cl/Au = 0.6) than in AuCl4– supported on u-beta (Cl/Au = 4.9), most likely because the Cl– ligand was partly replaced by the thiol and OH– group during wet impregnation.32 After O2 calcination at 350 °C (Figure 3e), Au/f-lab-beta contained highly monodisperse Au clusters with diameters smaller than ∼1 nm throughout the zeolite crystals, in sharp contrast to the O2-calcined Au/u-lab-beta. When further increasing the activation temperature to 500 °C, the size of the gold nanoparticles slightly increased from 0.5 to 1.8 nm, as determined by comparing Au nanoparticles in several STEM images (Figures 3f and S10). Nevertheless, these nanoparticles were nearly monodispersed, thus indicating that the cluster growth proceeded without any detectable loss of uniformity under these conditions. The cross-sectional STEM image in Figure S11 also shows the uniform distribution of 1.5–2.5 nm gold nanoparticles inside zeolite particles. This uniform distribution demonstrates that the gold species remain embedded in the zeolite matrix after calcination. These results can be explained based on the EA and 13C{1H} CP MAS NMR spectrum in Figure S12, which indicate the continued presence of thiolbenzene moieties on the internal surface of Au/f-beta even after calcination at 350 °C. These −SH groups effectively prevent the migration of intermediate gold species to the external surface, thus resulting in a large and uniform spacing between precursors and, hence, limited cluster growth during conversion to metallic Au. When increasing the O2-calcination temperature to 550 °C, however, thiolbenzene completely decomposed. Under these conditions, cluster growth, which was mainly caused by the tendency of small particles to minimize their surface energy, was sterically hindered by the surrounding framework, including both kinetic hurdles and thermodynamic barriers. The kinetic hurdles were imposed by the large numbers and small sizes of the intervening apertures, and the thermodynamic barriers were imposed by the energy required to locally disrupt the zeolite framework to accommodate larger clusters.24 In other words, the clusters may continue to grow until the driving force for their expansion is offset by these barriers to further disruption of the framework.
Based on XRD and STEM results described above, the uniform dispersion of Au+ cations throughout the zeolite framework is not sufficient to preserve the encapsulation and monodispersity of small (∼1 to 2 nm) Au clusters.33,34 Supporting highly monodisperse Au nanoparticles in a zeolite matrix requires promoting strong interactions between micropore walls and intermediate gold species during the activation process, thereby confining AuClx species in the zeolite framework before and during their conversion to Au clusters.
The present strategy can be extended to other commercial zeolites with an MFI structure, as confirmed by thiol-functionalization of commercial MFI zeolite shown in the solid-state 13C{1H} CP MAS NMR results in Figure S13. In turn, Figure S14 shows that the thiol-functionalized MFI zeolite, denoted by f-MFI, exhibits a pore volume of 0.077 cm3 g–1, which is smaller than that of u-MFI (0.13 cm3 g–1). After impregnating the gold precursor solution into f-MFI using the wet-impregnation method and into u-MFI using the IWI process, we converted the Au precursors into Au nanoparticles by calcination in air at 350 °C for u-MFI and at 550 °C for f-MFI. In both samples of Au/f-MFI and Au/u-MFI, ICP indicated an Au loading of 0.48 wt %, and the XRD patterns compared in Figure 4 exhibited XRD peaks matching the structure of the MFI zeolite. However, Au/u-MFI exhibits two additional peaks centered at 38.2 and 44.4°, which can be assigned to the (111) and (200) peaks of metallic Au, with an estimated average particle size of ∼27 nm, according to the Scherrer equation, but these Au peaks were not observed in Au/f-MFI. These results indicate that Au is much better dispersed as tiny metal nanoparticles into the thiol-functionalized MFI support than into u-MFI.
Figure 4.
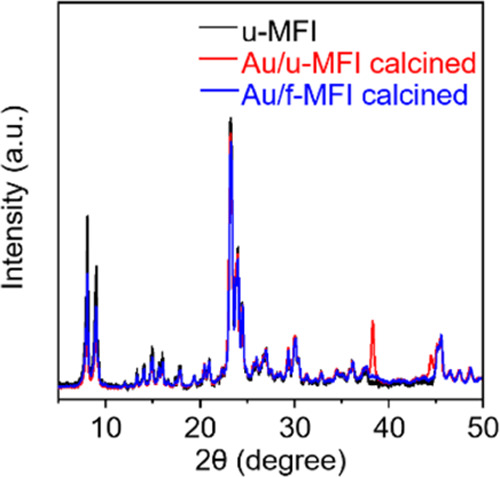
XRD patterns of Au/f-MFI (blue) and Au/u-MFI (red); the XRD pattern of the commercial MFI is shown in black for comparison.
Enhanced Thermal Stability of Au Nanoparticles Supported on Functionalized Zeolites
The high sintering resistance of calcined Au/f-beta and Au/f-MFI at 500, 600, and 700 °C is further demonstrated by the XRD patterns in Figure 5. Both Au/f-beta and Au/f-MFI contained the same amounts of Au (∼0.48 wt %). In the following discussion, the thermally treated samples are denoted by Au/f-beta-t and Au/f-MFI-t, where t indicates the treatment temperature. In the Au/f-beta-t series, the XRD peaks corresponding to the (111) and (200) facets of Au nanoparticles become gradually narrower with the increase in sintering temperature. This correlation indicates a slight increase in the diameters of the supported gold particles with the temperature. According to the Scherrer equation, the calculated average diameters are approximately 4.8, 5.4, and 7.3 nm for Au/f-beta-500, Au/f-beta-600, and Au/f-beta-700, respectively. Nevertheless, Au/f-beta-700 exhibits a better dispersion of Au nanoparticles than Au/u-beta.
Figure 5.
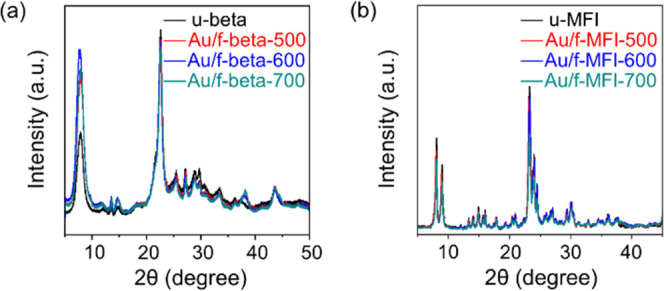
XRD patterns of calcined Au/f-beta (a) and Au/f-MFI (b) after heat treatment at 500, 600, and 700 °C. Both samples contained 0.48 wt % Au.
In the Au/f-MFI-t series, no XRD peaks corresponding to metallic Au are observed even after heating at 700 °C (Figure 5b). These results suggest that the gold nanoparticles embedded in MFI zeolites are not significantly sintered by heating at 700 °C under a N2 atmosphere. This hypothesis is further supported when examining the extent to which surface gold atoms can catalyze the hydrogenation reaction of acetylene to ethylene and ethane. Here, the loss of activity due to thermal sintering was less extreme in Au/f-MFI than in Au/f-beta (the catalytic activities of Au/f-beta-500, Au/f-beta-600, and Au/f-beta-700 and Au/f-MFI-500, Au/f-MFI-600, and Au/f-MFI-700 are described in Figure S15). More specifically, Au/f-beta-500 exhibited a 33% acetylene conversion, whereas Au/f-beta-600 and Au/f-beta-700 reached 32 and 20% conversion, respectively. Yet, both Au/f-MFI-500 and Au/f-MFI-600 maintained similar acetylene conversions of ∼50%, which is 1.5 times higher than that of Au-f-beta. Further increasing the temperature to 700 °C decreased the catalytic activity only to a small extent (i.e., down to ∼40%), thereby confirming that gold nanoparticles supported on MFI zeolites did not sinter considerably. Note that no catalytic activity was observed in Au/u-beta and Au/u-MFI samples. Based on these results, Au nanoparticles supported on MFI are more resistant to sintering than those supported on zeolite beta.
As outlined earlier, the driving force for metal particle growth is likely sterically hindered by the surrounding framework. If so, Au nanoparticles may continue to grow only until the driving force for their expansion is offset by this barrier to further framework disruption. In line with this assumption, framework stability should be a key factor in the sintering resistance of supported Au nanoparticles.
Framework stability varies with topology. For example, anomalously large numbers of defect sites (silanol groups) are present in zeolite beta samples.35,36 These defects have been shown to account for the structural instability of such materials. Accordingly, the higher structural stability of MFI zeolites increases the resistance of supported Au nanoparticles to sintering.
Catalytic Activity and Selectivity of Zeolite-Supported Au Nanoparticles
As demonstrated above, we achieved high monodispersion of Au nanoparticles on commercially available beta and MFI zeolites by attaching thiol groups to the zeolite internal surface. Following the aforementioned procedures, different amounts of Au nanoparticles (i.e., 0.12–1.3 wt %) were supported on f-beta and f-MFI. The resulting supported Au catalysts were characterized by EXAFS and H2 chemisorption to determine the dispersion of Au nanoparticles (Table 1). Figure S16 shows the Au L3-edge EXAFS spectra and their Fourier transforms of x Au/f-beta and x Au/f-MFI samples, where x indicates the contents of Au (wt %) in zeolites measured by the ICP method. The experimental EXAFS spectra were fitted including only single-scattering paths (i.e., Au–Au bond) in the fitting model to calculate the average coordination number and Au–Au distance (Table S1). In six samples, fitting 0.18 Au/f-MFI yielded chemically implausible fitting parameters owing to their very low values of Δμ at absorption edge energies, so their results were excluded. The coordination numbers were used to determine the average metal particle sizes, assuming spherical particles (Table 1).37 In the case of x Au/f-beta samples, 1.3 Au/f-beta had the largest average particle diameter of 4.1 nm, in line with the TEM measurements. When decreasing the Au content in f-beta from 1.3 to 0.28 wt %, the average particle diameters progressively decreased from 4.1 to 2.7 nm. In turn, 0.48 Au/f-MFI had an average particle diameter of 3.3 nm, which is slightly smaller than that of 0.48 Au/f-beta. In summarizing the EXAFS results, we concluded that the average particle diameters of supported Au decreased in the following order: 1.3 Au/f-beta > 0.48 Au/f-beta > 0.48 Au/f-MFI > 0.28 Au/f-beta > 0.28 Au/f-MFI. The number of surface Au atoms measured by H2 chemisorption should follow the reverse particle diameter trend. Of the Au supported on f-beta and f-MFI samples, 1.3 Au/f-beta had the lowest H/Au values (6.8%), with values increasing in the following order: 1.3 Au/f-beta (6.8%) < 0.48 Au/f-beta (7.4%) < 0.48 Au/f-MFI (7.7%) < 0.28 Au/f-beta (7.8%) < 0.28 Au/f-MFI (8.3%) < 0.12 Au/f-MFI (10%). The trends observed in EXAFS and H2 chemisorption matched, but the H/Au values were excessively low considering the Au particle diameters derived from EXAFS. This is because hydrogen adsorption and dissociation is limited to gold atoms on corner and edge positions and the dissociated hydrogen atoms could not spill over to face sites, which was proposed by Bokhoven et al.38 In this context, we replaced the number of total Au atoms by the number of Au atoms located at corner and edge sites when calculating H/Au values (denoted by H/[AuEd + AuCo] in Table 1). The number of edge and corner sites of Au nanoparticles depending on the particle size was calculated according to the previous papers, assuming that Au NPs have truncated octahedron geometry39,40 (see the Supporting Information for calculation details). As the result shows, 1.3 Au/f-beta exhibited a H/[AuEd + AuCo] value of 100%, while 0.48 Au/f-beta and 0.28 Au/f-beta showed 80 and 49%, respectively. Overall, as the dEXAFS value is decreased, the H/[AuEd + AuCo] value is decreased. The smaller nanoparticles have more opportunities to close contact with the micropore walls than their large counterparts, which might lead to more loss of accessible surface atoms. This is one possibility to interpret those results. Another possibility of lower values is attributed to that Au NPs within zeolite matrix have different morphologies from truncated octahedron geometry.
Table 1. Characterization Results and Catalytic Properties of Au/f-beta, Au/f-MFI, Au/u-beta, and Au/u-MFIa.
| reaction rate (molreactant min–1 molAu–1) |
||||||||
|---|---|---|---|---|---|---|---|---|
| sample | metal content (wt %)a | CNb | Au cluster diameter (dEXAFS) (nm)c | H/Au (%)d | H/[AuEd + AuCo] (%)e | RCOf | RBzOHg | R3,5-DTBBzOHh |
| 1.3 Au/f-beta | 1.3 | 10.7 | 4.1 | 6.8 | ∼100 | 0.016 | 2.7 | 0 |
| 0.48 Au/f-beta | 0.48 | 10.3 | 3.5 | 7.4 | 80 | 0.042 | 3.9 | |
| 0.28 Au/f-beta | 0.28 | 9.58 | 2.7 | 7.8 | 49 | 0.056 | 7.8 | |
| 1.1 Au/u-beta | 1.1 | 11.4 | 5.8 | 0.76 | 0 | 0 | 0 | |
| 0.48 Au/f-MFI | 0.48 | 10.1 | 3.3 | 7.7 | 79 | 0.053 | 0 | |
| 0.28 Au/f-MFI | 0.28 | 8.42 | 1.6 | 8.3 | 22 | 0.13 | 0 | |
| 0.18 Au/f-MFI | 0.18 | n.d. | n.d. | 10.3 | 0.29 | 0 | ||
| 0.48 Au/u-MFI | 0.48 | 11.8 | 8.8 | 0.73 | 0 | |||
Au contents obtained by ICP.
The average number of neighbors for the first coordination shell calculated using amplitude reduction factor of S02 = 0.832.
Au nanoparticle diameter determined from EXAFS fitting results.
The ratio of the amount of chemisorbed H atom to total Au atoms determined from H2 chemisorption at 60 °C.
The ratio of the amount of chemisorbed H atom to the number of Au atoms located at edge and corner sites. The calculation method for the number of edge and corner Au atoms are described in the Supporting Information.
CO oxidation: catalyst amount = 0.1 g, temperature = 40 °C, CO/H2O/O2/Ar = 0.5:5:10:84.5.
Benzyl alcohol oxidation: catalyst amount = 0.01 g, temperature = 25 °C, benzyl alcohol/3,5-di-tert-butyl-4-hydroxybenzylalcohol/dodecane/p-xylene/chloroform = 1:1:1:77:20.
3,5-Di-tert-butyl-4-hydroxybenzylalcohol oxidation: catalyst amount = 0.01 g, temperature = 25 °C, benzyl alcohol/3,5-di-tert-butyl-4-hydroxybenzylalcohol/dodecane/p-xylene/chloroform = 1:1:1:77:20.
In contrast to Au/f-zeolite samples, both Au/u-beta and Au/u-MFI had coordination numbers of 11.4 and 11.8, respectively, which are close to those of bulk Au materials and had very low H/Au values of less than 1%. These results indicate the poor dispersion of Au particles on u-beta and u-MFI zeolites.
Given this high dispersion, x Au/f-zeolites should have a higher mass activity (i.e., the reaction rate normalized to the mass of Au employed) than Au/u-zeolites. Inert gold is known to become catalytically active only when dispersed in the form of nanoparticles. For this reason, CO oxidation using a gold catalyst has been intensively studied as a model reaction for understanding this phenomenon.41,42 According to the previous papers, improvement of Au dispersion increases the Au mass activity.21,43 Nevertheless, it is still controversial for the nature of active sites involved in CO oxidation.44,45 This is because gold species at high dispersion state can exist in the forms of monocation species, sub-nanometer-sized Au clusters, and/or Au nanoparticles.
Au nanoparticles encapsulated into irreducible oxide supports (e.g., SiO2, Al2O3, zeolites) can catalyze CO oxidation despite the typically low ability to adsorb or store oxygen of these supports.46 Consequently, oxygen adsorption and dissociation should be possible on gold nanoparticles, but DFT calculations suggest only weak interactions on all but a few facets of these small nanoparticles.47 Furthermore, in these irreducible supports, CO oxidation activity critically depends on the size of the gold nanoparticles.43 Due to the enhanced dissociative adsorption of oxygen on small gold nanoparticles, only particles smaller than 2 nm and/or cationic Au species can act as catalytically highly active species.46,48,49
Figure 6a shows the Au mass-specific activities (molCO molAu–1 min–1) of the series of x Au/f-beta in the CO oxidation reaction under humid conditions (5 vol % H2O) at 40 °C in comparison with that of 1.1 Au/u-beta, which was measured at 10 min. As the results show, the CO conversion rate at 10 min decreased in the following order: 0.28 Au/f-beta (0.056) > 0.48 Au/f-beta (0.042) > 1.3 Au/f-beta (0.016) ≫ 1.1 Au/u-beta (0). The negligible catalytic activity of Au/u-beta is consistent with the characterization results discussed above, which revealed large Au agglomerates on the zeolite external surface. In contrast to the unfunctionalized zeolite, the CO conversion rate of x Au/f-beta was noticeable thanks to the presence of very small Au nanoparticles. Thus, our CO oxidation analysis strongly suggests that the thiol-functionalization of commercially available beta zeolites markedly improves the dispersity of Au nanoparticles. Among the Au/f-beta samples, 0.28 Au/f-beta had a higher CO conversion rate normalized by Au content (0.056 molCO molAu–1 min–1) than 0.48 Au/f-beta and 1.3 Au/f-beta. As evidenced by EXAFS and H2 chemisorption, described in the paragraph above, this high catalytic activity can be explained by the high dispersion of Au nanoparticles.
Figure 6.
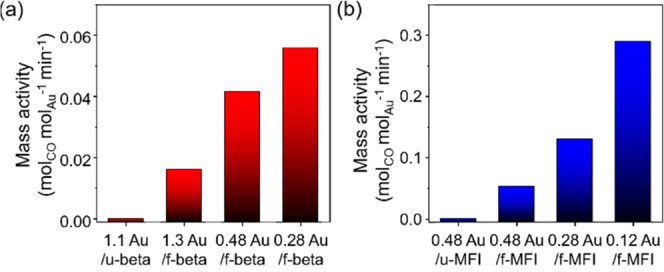
Au mass-specific activities (molCO molAu–1 min–1) calculated from experiments at 10 min in CO oxidation reaction under humid conditions: (a) 1.3 Au/f-beta, 0.48 Au/f-beta, 0.28 Au/f-beta, and 1.1 Au/u-beta and (b) 0.48 Au/f-MFI, 0.28 Au/f-MFI, 0.12 Au/f-MFI, and 0.48 Au/u-MFI. Reaction conditions: weight hour space velocity (WHSV) = 0.0619 h–1, temperature = 40 °C, CO/H2O/O2/Ar = 0.5:5:10:84.5.
Similar Au content and Au mass-specific activity trends were also observed in x Au/f-MFI catalysts (Figure 6b). More specifically, 0.12 Au/f-MFI showed the best catalytic activity of 0.29 molCO molAu–1 min–1, and their activity decreased with the increase in Au content [0.28 Au/f-MFI (0.13 molCO molAu–1 min–1), 0.48 Au/f-MFI (0.053 molCO molAu–1 min–1)]. At similar Au contents, the catalytic activity of 0.28 Au/f-MFI was two times higher than that of 0.28 Au/f-beta, which can be explained by higher dispersion. In the previous section, Au nanoparticles supported on MFI were more resistant to sintering. Therefore, supported Au nanoparticles were more highly dispersed on f-MFI than on f-beta, as evidenced by EXAFS and H2 chemisorption. A comparative study of our catalysts with the results reported in the literature is listed in Table S2 by calculating the CO conversion rate of the catalyst with respect to gold content (Table S2).
Figures S17 shows the FT-IR spectra of adsorbed CO on 1.3, 0.48, and 0.28 Au/f-beta and, 0.48, 0.28, and 0.12 Au/f-MFI with comparison to that of the calcined f-MFI and f-beta samples. To avoid CO-induced reduction of gold species, the CO dosing and subsequent FT-IR spectral measurement were performed at −150 °C. The calcined f-beta showed three distinguished IR bands at 2186, 2175, and 2166 cm–1. The most intensive band at 2175 cm–1 represents an interaction between CO and strongly acidic bridging hydroxyl groups Si(OH)Al.50 The band at 2186 cm–1 can be assigned to the vibration of CO molecule bonded to extra-framework aluminum species.50 The band at 2166 cm–1 can represent carbonyls, which are the result of the interaction between CO molecules and silanol nests.51,52 As compared to the calcined f-beta, two bands have been distinguished in the calcined f-MFI at 2186 and 2166 cm–1. The CO band related to extra-framework Al species is not observed.
The collected spectra for the six x Au/zeolite samples after adsorption of CO at −150 °C show complex adsorption bands, characteristic for interactions between CO molecules and different types of both gold and acid sites. As shown in Figure S17, the dotted lines represent bands assigned to carbonyls associated with acid sites/silanols, the colored curves represent the gold sites found upon the deconvolution process. The band at 2156 cm–1 represents (i) the isolated Au+ ions.51 Another three bands have been distinguished in the range of 2150–2130 cm–1, which can also be assigned to carbonyls associated to gold species. The gold species was mostly present in the form of metallic Au, according to the X-ray absorption near-edge structure spectra (Figure S18). Boronat et al. reported that the calculated CO stretching could be related to the net atomic charge on the gold atom to which CO binds.53 That is, CO vibration is progressively increased with the net atomic charge on gold. Referring to previous literatures,51−53 the band at 2148 cm–1 can be assigned to the vibration of CO molecule bonded to Au+ ions located on gold particles, while the other two bands (2140, 2132 cm–1) can represent carbonyls associated with partially positively charged Au+δ located on gold nanoparticles. When comparing MFI and beta zeolite with the same metal loading, 0.48 Au/f-beta showed a more intense CO band at 2156 cm–1 corresponding to the isolated Au+ ion, while 0.48 Au/f-MFI displayed a more intense CO band at 2140 cm–1 assigned to Au+δ located on gold nanoparticles. Considering the 0.48 Au/f-MFI exhibited higher CO oxidation activity than 0.48 Au/f-beta, it is highly probable that Au+δ species seemed to be responsible for the high activity of CO oxidation reaction.
In terms of catalytic performance, both Au/f-beta and Au/f-MFI exhibited CO conversion rates, which are about 100 times less than one observed in acetylene hydrogenation reaction (see Table 1 and Figure S15). One possibility is that the active sites might not be the same and therefore not comparable between those two reactions. Another possibility is that we performed the CO oxidation in the presence of 5 vol % H2O vapor. In the previous literature, the presence of water can lower the activation energy, leading to an increase in the reaction rate.54,55 However, the catalytic activity is dramatically decreased in the presence of the excess amount of water (e.g., 6000 ppm), which might be due to the blocking of the active sites.54 In the present study, (1) aluminosilicate zeolites typically prefer to adsorb the steam, and (2) CO oxidation is performed in the presence of 50,000 ppm H2O. Under this situation, CO accessibility to Au nanoparticles might be blocked by the H2O molecules adsorbed by the aluminosilicate zeolites, resulting in the loss of catalytic activity in the CO oxidation reaction.
Lastly, gold-catalyzed size-selective reactions were performed to assess whether as-synthesized supported Au nanoparticles are definitely located inside the zeolite matrix or not. To this end, two organic molecules with different molecular sizes were used simultaneously as the reactants. The organic molecules were selected based on the diameter of the zeolite micropores so that one organic molecule was smaller and the other larger than the zeolite micropore diameter. More specifically, benzyl alcohol and 3,5-di-tert-butyl-4-hydroxybenzylalcohol were used as reactants for shape-selective catalysis over Au/f-beta.
The results outlined in Table 1 indicate that Au/f-beta selectively oxidizes benzyl alcohol without showing any activity to 3,5-di-tert-butyl-4-hydroxybenzylalcohol. As such, the diffusion rate of benzyl alcohol in beta zeolite should be much faster than that of 3,5-di-tert-butyl-4-hydroxybenzylalcohol. Accordingly, confining Au NPs in beta selectively converts benzyl alcohol into oxygenated products in the presence of 3,5-di-tert-butyl-4-hydroxybenzylalcohol due to molecular sieving effects. These results clearly demonstrate that zeolites can prevent molecules larger than their apertures from accessing active Au surfaces and that the active Au surface resides in zeolite voids.
In the case of liquid-phase benzyl alcohol oxidation, the series of x Au/f-beta and x Au/f-MFI was further investigated (Table 1). Interestingly, all of the Au/f-MFI samples exhibited no catalytic activity in benzyl alcohol oxidation. The effective aperture size of MFI zeolite is larger than the benzyl alcohol and their oxidized products. In this regard, it is highly probable that Au NPs located at the microporous channel and/or channel intersection space might restrict the accessibility of benzyl alcohol to Au species, resulting in no catalytic activity in benzyl alcohol oxidation. As compared to MFI zeolite, beta zeolites have an intersecting three-dimensional pore structure and 12-MR windows of ∼0.7 nm. The three-dimensional structure and large-pore diameters may enable the diffusion of benzyl alcohol through the microporous channel despite the presence of Au NPs in the zeolite matrix. In the case of the series of Au/f-beta, 0.28 Au/f-beta exhibited the highest mass activity of 7.8 molbenzyl alcohol molAu–1 min–1 and decreased in the following order: 0.28 Au/f-beta (7.8) > 0.48 Au/f-beta (3.9) > 1.3 Au/f-beta (2.7). These results are well matched with the Au dispersity, which are derived from EXAFS and H2 chemisorption.
Conclusions
Small Au NPs can be confined within a preformed zeolite matrix via post-synthetic modification, as exemplified by confining Au NPs within commercially available beta and MFI zeolites. Our protocol involves the post-synthetic modification of the zeolite internal surface with thiol groups, followed by loading and activation of gold precursors. During activation, in air, at high temperatures, up to ∼350 °C (the thermal decomposition temperature (i.e., ∼350 °C) of the thiol groups), these groups effectively encapsulate AuClx species inside the microporous frameworks, thereby enabling the conversion of the precursor into metallic Au clusters inside the zeolite framework. Above 350 °C, further crystal growth is inhibited, most likely due to steric hindrance by the surrounding framework. As a result, Au NPs (1–2 nm) are uniformly dispersed throughout the void space. Furthermore, the intrinsic framework stability promotes resistance to sintering. These confined Au nanoparticles show high catalytic activity toward CO oxidation at a low temperature (40 °C) and size-based selectivity in catalytic reactions such as alcohol oxidation and triple-bond hydrogenation due to constraints of the zeolite matrix. Our strategy may have a wide applicability for Au confinement in preformed zeolites with various pore topologies and compositions and may also extend currently available Au encapsulation techniques to other host materials, including aluminosilicate zeolites, for which impregnation or cation-exchange protocols are not feasible.
Acknowledgments
This work was supported by “Carbon Upcycling Project for Platform Chemicals” (Project No. 2022M3J3A1085579) through the National Research Foundation (NRF) funded by the Ministry of Science and ICT, Republic of Korea. This work was supported by the Technology Innovation Program (Industrial Strategic Technology Development Program) funded by the Ministry of Trade, Industry & Energy of the Republic of Korea (Project No. 20017393). The authors thank Dr. N.S. Kim for his help with STEM measurements. D.N.R. and M.M. acknowledge OP VVV “Excellent Research Teams” Project No. CZ.02.1.01/0.0/0.0/15_003/0000417—CUCAM. K.G. acknowledges the Czech Science Foundation for the ExPro Project (19-27551X). The authors also thank the Laboratory of Electron Microscopy, Imaging Methods Core Facility of Biology Section, Faculty of Science (IMCF Viničná) for SEM images and Dr. Carlos V. Melo for editing the manuscript.
Glossary
Abbreviations
- XRD
X-ray diffraction
- STEM
scanning transmission electron microscopy
- SEM
scanning electron microscopy
- ICP-OES
inductively coupled plasma-optical emission spectroscopy
- WHSV
weight hour space velocity
- BET
Brunauer–Emmett–Teller
- HK
Horvath–Kawazoe
- EA
elemental analysis
- TGA
thermogravimetric analysis
- NMR
nuclear magnetic resonance
- EXAFS
extended X-ray absorption fine structure
- FT-IR
Fourier transform infrared
- UV
ultraviolet
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.2c00380.
UV, SEM, XRD, and STEM of cross-sectioned samples; size distributions of metal particles; solid-state 13C NMR; EA results; catalytic results; EXAFS; and FT-IR (PDF)
Author Contributions
All authors have approved the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Haruta M. When gold is not noble: catalysis by nanoparticles. Chem. Rec. 2003, 3, 75–87. 10.1002/tcr.10053. [DOI] [PubMed] [Google Scholar]
- Ishida T.; Murayama T.; Taketoshi A.; Haruta M. Importance of Size and Contact Structure of Gold Nanoparticles for the Genesis of Unique Catalytic Processes. Chem. Rev. 2020, 120, 464–525. 10.1021/acs.chemrev.9b00551. [DOI] [PubMed] [Google Scholar]
- Taketoshi A.; Haruta M. Size- and Structure-specificity in Catalysis by Gold Clusters. Chem. Lett. 2014, 43, 380–387. 10.1246/cl.131232. [DOI] [Google Scholar]
- Higaki T.; Li Y.; Zhao S.; Li Q.; Li S.; Du X. S.; Yang S.; Chai J.; Jin R. Atomically Tailored Gold Nanoclusters for Catalytic Application. Angew. Chem. 2019, 131, 8377–8388. 10.1002/ange.201814156. [DOI] [PubMed] [Google Scholar]
- Saha K.; Agasti S. S.; Kim C.; Li X.; Rotello V. M. Gold nanoparticles in chemical and biological sensing. Chem. Rev. 2012, 112, 2739–2779. 10.1021/cr2001178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztandera K.; Gorzkiewicz M.; Klajnert-Maculewicz B. Gold Nanoparticles in Cancer Treatment. Mol. Pharmaceutics 2019, 16, 1–23. 10.1021/acs.molpharmaceut.8b00810. [DOI] [PubMed] [Google Scholar]
- Maity P.; Xie S.; Yamauchi M.; Tsukuda T. Stabilized gold clusters: from isolation toward controlled synthesis. Nanoscale 2012, 4, 4027–4037. 10.1039/c2nr30900a. [DOI] [PubMed] [Google Scholar]
- Jadzinsky P. D.; Calero G.; Ackerson C. J.; Bushnell D. A.; Kornberg R. D. Structure of a thiol monolayer-protected gold nanoparticle at 1.1 Å resolution. Science 2007, 318, 430–433. 10.1126/science.1148624. [DOI] [PubMed] [Google Scholar]
- Haruta M. Gold as a novel catalyst in the 21st century: Preparation, working mechanism and applications. Gold Bull. 2004, 37, 27–36. 10.1007/BF03215514. [DOI] [Google Scholar]
- Cai J.; Ma H.; Zhang J.; Song Q.; Du Z.; Huang Y.; Xu J. Gold nanoclusters confined in a supercage of Y zeolite for aerobic oxidation of HMF under mild conditions. Chem. - Eur. J. 2013, 19, 14215–14223. 10.1002/chem.201301735. [DOI] [PubMed] [Google Scholar]
- Chi Y.-S.; Lin H.-P.; Mou C.-Y. CO oxidation over gold nanocatalyst confined in mesoporous silica. Appl. Catal., A 2005, 284, 199–206. 10.1016/j.apcata.2005.01.034. [DOI] [Google Scholar]
- Wang J.; Lu A.-H.; Li M.; Zhang W.; Chen Y.-S.; Tian D.-X.; Li W.-C. Thin porous alumina sheets as supports for stabilizing gold nanoparticles. ACS Nano 2013, 7, 4902–4910. 10.1021/nn401446p. [DOI] [PubMed] [Google Scholar]
- Weitkamp J. Zeolites and catalysis. Solid State Ionics 2000, 131, 175–188. 10.1016/S0167-2738(00)00632-9. [DOI] [Google Scholar]
- Dusselier M.; Davis M. E. Small-Pore Zeolites: Synthesis and Catalysis. Chem. Rev. 2018, 118, 5265–5329. 10.1021/acs.chemrev.7b00738. [DOI] [PubMed] [Google Scholar]
- Gallezot P.Preparation of Metal Clusters in Zeolites. In Post-Synthesis Modification I; Molecular Sieves (Science and Technology); Springer: Berlin, Heidelberg, 2002; Vol. 3, pp 275–305. [Google Scholar]
- Corma A.; Garcia H. Supported gold nanoparticles as catalysts for organic reactions. Chem. Soc. Rev. 2008, 37, 2096–2126. 10.1039/b707314n. [DOI] [PubMed] [Google Scholar]
- Walkowiak A.; Wolski L.; Ziolek M. Lights and Shadows of Gold Introduction into Beta Zeolite. Molecules 2020, 25, 5781 10.3390/molecules25245781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F.; Peng L.; Yao X.; Zhang Y.; Zhang C.; Gu X. Hollow fiber supported Au-Zr/FAU catalytic membranes for H2 purification by CO preferential oxidation. Energy Technol. 2017, 5, 2283–2293. 10.1002/ente.201700331. [DOI] [Google Scholar]
- Kang Y.-M.; Wan B.-Z. Preparation of gold in Y-type zeolite for carbon monoxide oxidation. Appl. Catal., A 1995, 128, 53–60. 10.1016/0926-860X(95)00076-3. [DOI] [Google Scholar]
- Oh H.-S.; Yang J. H.; Costello C. K.; Wang Y. M.; Bare S. R.; Kung H. H.; Kung M. C. Selective catalytic oxidation of CO: effect of chloride on supported Au catalysts. J. Catal. 2002, 210, 375–386. 10.1006/jcat.2002.3710. [DOI] [Google Scholar]
- Chen Y.-H.; Mou C.-Y.; Wan B.-Z. Ultrasmall gold nanoparticles confined in zeolite Y: Preparation and activity in CO oxidation. Appl. Catal., B 2017, 218, 506–514. 10.1016/j.apcatb.2017.06.083. [DOI] [Google Scholar]
- Laursen A. B.; Højholt K. T.; Lundegaard L. F.; Simonsen S. B.; Helveg S.; Schüth F.; Paul M.; Grunwaldt J.-D.; Kegnæs S.; Christensen C. H.; Egeblad K. Substrate size-selective catalysis with zeolite-encapsulated gold nanoparticles. Angew. Chem. 2010, 122, 3582–3585. 10.1002/ange.200906977. [DOI] [PubMed] [Google Scholar]
- Højholt K. T.; Laursen A. B.; Kegnæs S.; Christensen C. H. Size-Selective Oxidation of Aldehydes with Zeolite Encapsulated Gold Nanoparticles. Top. Catal. 2011, 54, 1026 10.1007/s11244-011-9722-x. [DOI] [Google Scholar]
- Otto T.; Zones S. I.; Iglesia E. Challenges and strategies in the encapsulation and stabilization of monodisperse Au clusters within zeolites. J. Catal. 2016, 339, 195–208. 10.1016/j.jcat.2016.04.015. [DOI] [Google Scholar]
- Choi M.; Wu Z.; Iglesia E. Mercaptosilane-assisted synthesis of metal clusters within zeolites and catalytic consequences of encapsulation. J. Am. Chem. Soc. 2010, 132, 9129–9137. 10.1021/ja102778e. [DOI] [PubMed] [Google Scholar]
- Otto T.; Ramallo-López J. M.; Giovanetti L. J.; Requejo F. G.; Zones S. I.; Iglesia E. Synthesis of stable monodisperse AuPd, AuPt, and PdPt bimetallic clusters encapsulated within LTA-zeolites. J. Catal. 2016, 342, 125–137. 10.1016/j.jcat.2016.07.017. [DOI] [Google Scholar]
- Mielby J.; Abildstrøm J. O.; Wang F.; Kasama T.; Weidenthaler C.; Kegnæs S. Oxidation of bioethanol using zeolite-encapsulated gold nanoparticles. Angew. Chem. 2014, 126, 12721–12724. 10.1002/ange.201406354. [DOI] [PubMed] [Google Scholar]
- Choong C.; Foord J.; Griffiths J.-P.; Parker E. M.; Baiwen L.; Bora M.; Moloney M. G. Post-polymerisation modification of surface chemical functionality and its effect on protein binding. New. J. Chem. 2012, 36, 1187–1200. 10.1039/c2nj00002d. [DOI] [Google Scholar]
- Lee H. S.; Kim N. S.; Kwon Di.; Lee S. K.; Numan M.; Jung T.; Cho K.; Mazur M.; Cho H. S.; Jo C. Post-Synthesis Functionalization Enables Fine-Tuning the Molecular-Sieving Properties of Zeolites for Light Olefin/Paraffin Separations. Adv. Mater. 2021, 33, 2105398 10.1002/adma.202105398. [DOI] [PubMed] [Google Scholar]
- Otto K.; Acik I. O.; Krunks M.; Tõnsuaadu K.; Mere A. Thermal decomposition study of HAuCl4·3H2O and AgNO3 as precursors for plasmonic metal nanoparticles. J. Therm. Anal. Calorim. 2014, 118, 1065–1072. 10.1007/s10973-014-3814-3. [DOI] [Google Scholar]
- Rose T. K. XC.–Some physical properties of the chlorides of gold. J. Chem. Soc., Trans. 1895, 67, 905–906. 10.1039/CT8956700905. [DOI] [Google Scholar]
- Barngrover B. M.; Aikens C. M. The golden pathway to thiolate-stabilized nanoparticles: following the formation of gold(I) thiolate from gold (III) chloride. J. Am. Chem. Soc. 2012, 134, 12590–12595. 10.1021/ja303050s. [DOI] [PubMed] [Google Scholar]
- Guillemot D.; Polisset-Thfoin M.; Fraissard J. Preparation of nanometeric gold particles on NaHY. Catal. Lett. 1996, 41, 143–148. 10.1007/BF00811481. [DOI] [Google Scholar]
- Zeng S.; Ding S.; Li S.; Wang R.; Zhang Z. Controlled growth of gold nanoparticles in zeolite L via ion-exchange reactions and thermal reduction processes. Inorg. Chem. Commun. 2014, 47, 63–66. 10.1016/j.inoche.2014.07.013. [DOI] [Google Scholar]
- Newsam J.; Treacy M. M.; Koetsier W.; Gruyter C. D. Structural characterization of zeolite beta. Proc. R. Soc. Lond. A 1988, 420, 375–405. 10.1098/rspa.1988.0131. [DOI] [Google Scholar]
- Wright P. A.; Zhou W.; Pérez-Pariente J.; Arranz M. Direct Observation of Growth Defects in Zeolite Beta. J. Am. Chem. Soc. 2005, 127, 494–495. 10.1021/ja043948s. [DOI] [PubMed] [Google Scholar]
- Jentys A. Estimation of mean size and shape of small metal particles by EXAFS. Phys. Chem. Chem. Phys. 1999, 1, 4059–4063. 10.1039/a904654b. [DOI] [Google Scholar]
- Bus E.; Miller J. T.; van Bokhoven J. A. Hydrogen chemisorption on Al2O3-supported gold catalysts. J. Phys. Chem. B 2005, 109, 14581–14587. 10.1021/jp051660z. [DOI] [PubMed] [Google Scholar]
- Murzin D. Y. Kinetics of cluster shape sensitive heterogeneous catalytic reactions. Chem. Eng. J. 2021, 425, 130642 10.1016/j.cej.2021.130642. [DOI] [Google Scholar]
- Mori T.; Hegmann T. Determining the composition of gold nanoparticles: a compilation of shapes, sizes, and calculations using geometric considerations. J. Nanopart. Res. 2016, 18, 295 10.1007/s11051-016-3587-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruta M.; Tsubota S.; Kobayashi T.; Kageyama H.; Genet M. J.; Delmon B. Low-temperature oxidation of CO over gold supported on TiO2, α-Fe2O3, and Co3O4. J. Catal. 1993, 144, 175–192. 10.1006/jcat.1993.1322. [DOI] [Google Scholar]
- Haruta M.; Yamada N.; Kobayashi T.; Iijima S. Gold catalysts prepared by coprecipitation for low-temperature oxidation of hydrogen and of carbon monoxide. J. Catal. 1989, 115, 301–309. 10.1016/0021-9517(89)90034-1. [DOI] [Google Scholar]
- Guczi L.; Petö G.; Beck A.; Frey K.; Geszti O.; Molnár G.; Daróczi C. Gold nanoparticles deposited on SiO2/Si (100): Correlation between size, electron structure, and activity in CO oxidation. J. Am. Chem. Soc. 2003, 125, 4332–4337. 10.1021/ja0213928. [DOI] [PubMed] [Google Scholar]
- Guzman J.; Gates B. C. Catalysis by supported gold: correlation between catalytic activity for CO oxidation and oxidation states of gold. J. Am. Chem. Soc. 2004, 126, 2672–2673. 10.1021/ja039426e. [DOI] [PubMed] [Google Scholar]
- Costello C. K.; Guzman J.; Yang J.; Wang Y.; Kung M.; Gates B.; Kung H. Activation of Au/γ-Al2O3 catalysts for CO oxidation: Characterization by X-ray absorption near edge structure and temperature programmed reduction. J. Phys. Chem. B 2004, 108, 12529–12536. 10.1021/jp048643e. [DOI] [Google Scholar]
- Okumura M.; Nakamura S.; Tsubota S.; Nakamura T.; Azuma M.; Haruta M. Chemical vapor deposition of gold on Al2O3, SiO2, and TiO2 for the oxidation of CO and of H2. Catal. Lett. 1998, 51, 53–58. 10.1023/A:1019020614336. [DOI] [Google Scholar]
- Sun K. Theoretical investigations on CO oxidation reaction catalyzed by gold nanoparticles. Chin. J. Catal. 2016, 37, 1608–1618. 10.1016/S1872-2067(16)62476-2. [DOI] [Google Scholar]
- Schubert M. M.; Hackenberg S.; Van Veen A. C.; Muhler M.; Plzak V.; Behm R. J. CO oxidation over supported gold catalysts– “Inert” and “active” support materials and their role for the oxygen supply during reaction. J. Catal. 2001, 197, 113–122. 10.1006/jcat.2000.3069. [DOI] [Google Scholar]
- Mavrikakis M.; Stoltze P.; Nørskov J. K. Making gold less noble. Catal. Lett. 2000, 64, 101–106. 10.1023/A:1019028229377. [DOI] [Google Scholar]
- Tarach K.; Góra-Marek K.; Tekla J.; Brylewska K.; Datka J.; Mlekodaj K.; Makowski W.; López M. I.; Triguero J. M.; Rey F. Catalytic cracking performance of alkaline-treated zeolite Beta in the terms of acid sites properties and their accessibility. J. Catal. 2014, 312, 46–57. 10.1016/j.jcat.2014.01.009. [DOI] [Google Scholar]
- Mihaylov M.; Knoezinger H.; Hadjiivanov K.; Gates B. C. Characterization of the oxidation states of supported gold species by IR spectroscopy of adsorbed CO. Chem. Ing. Tech. 2007, 79, 795–806. 10.1002/cite.200700029. [DOI] [Google Scholar]
- Hadjiivanov K. I.; Vayssilov G. N. Characterization of oxide surfaces and zeolites by carbon monoxide as an IR probe molecule. Adv. Catal. 2002, 47, 307–511. 10.1016/S0360-0564(02)47008-3. [DOI] [Google Scholar]
- Boronat M.; Concepción P.; Corma A. Unravelling the nature of gold surface sites by combining IR spectroscopy and DFT calculations. Implications in catalysis. J. Phys. Chem. C 2009, 113, 16772–16784. 10.1021/jp905157r. [DOI] [Google Scholar]
- Daté M.; Haruta M. Moisture effect on CO oxidation over Au/TiO2 catalyst. J. Catal. 2001, 201, 221–224. 10.1006/jcat.2001.3254. [DOI] [Google Scholar]
- Saavedra J.; Doan H. A.; Pursell C. J.; Grabow L. C.; Chandler B. D. The critical role of water at the gold-titania interface in catalytic CO oxidation. Science 2014, 345, 1599–1602. 10.1126/science.1256018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.