

Abstract

The preparation of metastable zeolites is often restricted to a limited range of synthesis conditions, which is exemplified in commercial syntheses lacking organics to stabilize the crystal structure. In the absence of an organic structure-directing agent, interzeolite transformation is a common phenomenon that can lead to undesirable products or impurities. Many studies have investigated the substitution of Si and Al in zeolite frameworks with alternative elements (heteroatoms) as a means of tailoring the properties of zeolites; however, relatively few studies have systematically explored the impact of heteroatoms on interzeolite transformations and their concomitant effects on zeolite crystallization. In this study, we examine methods to prepare isostructures of faujasite (FAU), which is one of the most commercially relevant zeolites and also a thermodynamically metastable structure. A survey of multivalent elements revealed that zinc is capable of stabilizing FAU at high temperatures and inhibiting its frequent transformation to zeolite gismondine (GIS). Using combined experimental and computational studies, we show that zinc alters the chemical nature of growth mixtures by sequestering silicates. Zinc heteroatoms incorporate in the FAU framework with a loading-dependent coordination. Our collective findings provide an improved understanding of driving forces for the FAU-to-GIS interzeolite transformation where we observe that heteroatoms (e.g., zinc) can stabilize zeolite FAU over a broad range of synthesis conditions. Given the growing interest in heteroatom-substituted zeolites, this approach to preparing zinc-containing FAU may prove applicable to a broader range of zeolite structures.
Keywords: zeolite nucleation, interzeolite transformation, zinc, heteroatoms, faujasite, DFT
Introduction
The versatility of zeolites as stable solid-acid catalysts has led to their widespread implementation across multiple industries. These nanoporous aluminosilicates can be synthesized with over 250 distinct framework topologies possessing diverse pore geometries, compositions, and crystal morphologies. Tetrahedrally coordinated aluminum sites are balanced by extra-framework cations, often hydrogen (i.e., Brønsted acid sites), where the concentration and siting1 of Al atoms can strongly influence catalyst performance and stability.2,3 Zeolites can also be synthesized with a wide range of framework compositions, referred to as zeotypes (or zeolite isostructures) when tetrahedral sites (T-sites) are occupied by “heteroatoms”, which are elements other than Si and Al. Heteroatom substitution with diverse species has garnered significant interest in recent years4 as a method to enhance zeolite properties—most notably the nature and strength of Brønsted and/or Lewis acid sites.5 For example, tin,6 hafnium,7 and zirconium8 have been incorporated into the *BEA structure for applications in biomass conversion. Previous studies have also shown that heteroatoms, such as titanium,9 zinc,10 and gallium,11 can enhance the catalytic activity of zeolite MFI. Furthermore, incorporation of some elements (e.g., Fe,12 Sn,13 and Zn14,15) can lead to mesoporous structures by promoting defects. The ability to synthesize zeolites with isomorphically substituted heteroatoms is specific to both the element and framework of interest,16 where many combinations can only be synthesized after prohibitively long synthesis times.4 It has been suggested that the addition of heteroatom precursors to zeolite syntheses alters crystallization kinetics by modifying physicochemical properties of the growth mixture17 or facilitating the formation of crystal structures that are thermodynamically less stable owing to heteroatom incorporation.18 Investigating these hypotheses, however, is nontrivial since complex synthesis media and nonclassical crystallization mechanisms obfuscate attempts to identify causal relationships between synthesis parameters and growth kinetics or properties of zeolites/zeotypes.19,20
Owing to the relatively high geologic abundance of zinc and its ability to coordinate to zeolites as framework sites or extra-framework species, zinc is commonly investigated for its potential to enhance zeolite properties. The nature of zinc in zeolite crystals is difficult to establish since it can be present in many forms: zinc oxide21 or zinc hydroxide22 occlusion, framework sites,22,23 or extra-framework cations (Zn2+, ZnOH+, or [Zn–O–Zn]2+).24 Zinc incorporation in zeolites can alter Si/Al ratios,17 promote intergrowths (i.e., hierarchical architectures),14 or introduce Lewis acidity.10,22 Zinc can be incorporated top-down by either impregnation or ion exchange, or it can be introduced bottom-up by adding a precursor (e.g., Zn(NO3)2) to the synthesis mixture,17 which leads to better dispersion and stability.15 Understanding of the effects of zinc on crystallization pathways can lead to more effective strategies for preparing Zn-zeolites without deleterious effects.
One topic that has received little attention is the impact of heteroatoms on zeolite growth mechanisms. This is particularly relevant for organic-free syntheses where polymorphism and interzeolite transformations (IZTs) are common phenomena.4,19,25 IZT is frequently encountered when using inorganic structure-directing agents (ISDAs), which are less selective for syntheses of pure framework types than their organic counterparts. Transformations purportedly abide by the Ostwald rule of stages where the sequence of events in the direction of increased thermodynamic stability20,25 involves the initial formation of a metastable (parent) zeolite followed by the subsequent nucleation and growth of a second (daughter) phase with concomitant disappearance of the parent. The sequence of IZT stages can be difficult to predict. Numerous parent–daughter pairings involve zeolites with at least one shared composite building unit (CBU),19 while other cases comprise zeolites with no apparent structural similarity.25 One example of the latter is the transformation from faujasite (FAU) to gismondine (GIS) for synthesis temperatures approaching 100 °C (Figure 1a). Both of these structures form naturally and are also industrially relevant.26 FAU is the most widely used fluidized catalytic cracking (FCC) catalyst.27 Syntheses of FAU often lead to impurity phases, such as GIS, which crystallizes as polymorphs P1 or P2 (Figure 1b).28 Kinetics of FAU, GIS, and other zeolites in complex systems are also of interest in nuclear waste glass disposal as the acceleration or inhibition of growth of certain zeolites can impact the alteration behavior of a glass.29
Figure 1.

(a) FAU-to-GIS interzeolite transformation (IZT) involves a reduction in free energy for two zeolites that do not share a common CBU. (b) Side-by-side comparison of P1 and P2 polymorphs of GIS. P1 exhibits identical pore mouths (yellow), whereas alternating 4-membered rings in P2 lead to slight variations in pore mouth geometry (green and orange).
In zeolite syntheses, the partitioning of solute between solid (amorphous precursor) and solution (supernatant) leads to changes in the growth environment over the course of zeolite crystallization. The impact of these changes on IZT events and the ability to manipulate these processes via the introduction of heteroatoms is elusive. In this study, we report methods to prepare Zn-FAU in organic-free media and investigate the impact of zinc and other multivalent metals on the FAU-to-GIS transformation. Our findings reveal that zinc is an efficient inhibitor of IZT, which enables Zn-FAU to be prepared over a broad range of synthesis conditions.
Results and Discussion
Controlling FAU-to-GIS Transformation
We investigated the effects of diverse multivalent species, M, on the FAU-to-GIS IZT using synthesis gels with a molar composition (9SiO2/0.5Al2O3/zMn+/11NaOH/190H2O) that is typical for the preparation of GIS-P2.30 The nominal condition for synthesis (z = 0, control) was used to benchmark crystallization kinetics for comparison with growth mixtures containing multivalent additives. The FAU-to-GIS transformation is monitored quantitatively by powder X-ray diffraction (XRD) and qualitatively by electron microscopy owing to the distinct size and shape of FAU (Figure 2a) and GIS (Figure 2b) crystals. In the control synthesis, FAU forms within 4 h of hydrothermal treatment at 100 °C (Figure S1). Within 24 h, we observe the emergence of Bragg peaks in powder XRD patterns that are indicative of GIS-type zeolite nucleation. Additional hydrothermal treatment results in the reduction of FAU peaks in XRD patterns with a concomitant shift to pure GIS-P2 within 4 days (Figures S1 and S2). The same synthesis was repeated multiple times with the addition of various multivalent species to the synthesis gel (z = 0.2) using a fixed hydrothermal treatment of 4 days at 100 °C to investigate which multivalent species alter crystallization time and/or polymorph selection. Multivalent species selected for this study included four divalent alkaline earth metals (Ca, Mg, Sr, Ba), a divalent transition metal (Zn), and four higher valence species (B, Ga, Ge, In) to assess a diverse range of chemical properties. As shown in Figure 2c, screening multivalent additives revealed differences in the final product being FAU (green shading), GIS-P2 (purple shading), or mixtures of both.
Figure 2.

(a, b) Representative scanning electron micrographs of zeolites (a) FAU and (b) GIS-P1. The morphology of GIS-P2 is nearly identical to the latter (see Figure 8d). (c) Screening of group II, III, and IV heteroatoms used in zeolite synthesis mixtures compared to Zn (the sole transition metal investigated in this study). The effects of various multivalent metals were assessed using a fixed molar composition of 9SiO2/0.5Al2O3/0.2M/11NaOH/190H2O, where M represents the highlighted multivalent species. Growth mixtures were hydrothermally treated at 100 °C for 4 days. Color coding is purple (GIS), green (FAU), and gray (amorphous) with mixed colors indicating binary combinations (with shaded areas representing approximate percentages of each framework). The principal components of each zeolite (Si and Al) are shown for reference.
An interesting observation is the dichotomy between alkaline earth metals (Figure 2c, left) and multivalent species with higher valence states (Figure 2c, right). Group II elements do not suppress the FAU-to-GIS IZT, whereas other assayed elements either partially or completely suppress GIS-P2 formation (Figure S3). Among group III (B, Ga, In) and fourth row (Zn, Ga, Ge) elements, IZT is generally suppressed with increasing atomic size of multivalent species. When Zn is used, transformation to GIS is suppressed for longer than 28 days, regardless of whether Zn(NO3)2 or ZnO is used as the zinc source (Figure S4). This is consistent with studies by Schwieger and co-workers14 who showed that zinc stabilized FAU at a synthesis temperature of 75 °C, which is higher than many reported syntheses. Prior studies indicate that metallosilicate condensation varies depending on metal selection31 wherein the angle and flexibility of Si–O–M bonds varies for distinct multivalent species. Indeed, some heteroatoms tend to condense silicates more readily than others,31,32 which may be related to the ability of each species to suppress FAU-to-GIS IZT. We surmise these effects could be attributed to several factors: (i) different bond angles of metallosilicates relative to aluminosilicates may render the former less amenable to GIS nucleation and/or growth, potentially due to the fact that GIS is a denser structure; (ii) metallosilicate condensation may unfavorably alter the chemical potential of the growth solution; or (iii) heteroatoms may alter parent (FAU) properties such as crystal stability (vide infra).
Trends in Figure 2c can be rationalized by differences in the ionic structures these elements adopt in the highly alkaline solutions used in zeolite synthesis. Under these conditions, alkaline earth metals dissociate (either completely or partially) into their constituent ions. The latter can function as ISDAs in addition to sodium, which is present at a much higher concentration than the additives (i.e., molar ratio Na+/M2+ = 55). When the ratio of Na+/M2+ is small, alkaline earth metals can inhibit FAU crystallization by reducing the pH;33 however, in this study, the effect of sodium on zeolite crystallization seemingly far outweighs that of any alkaline earth metal. Conversely, higher valence species (hereafter referred to as heteroatoms) are not present as cations, but rather as anionic metal hydroxide complexes in high-pH media31,32 akin to those formed by silicon (silicates) and aluminum (aluminates). We posit the primary role of these species is less that of an ISDA and more related to a pseudo sequestering agent based on their ability to promote metallo-(alumino)silicate oligomerization. Moreover, isomorphic substitution of metal heteroatoms in zeolite frameworks can alter crystallization. Herein we analyze heteroatom impact on IZT and polymorphism, focusing on zinc as the sole multivalent additive.
Stabilization of Zeolite FAU
We compiled kinetic phase diagrams of sodium-containing syntheses (7 days, 100 °C) in the absence (Figure 3a) and presence (Figure 3b) of zinc. In a previous study,30 we showed that zeolite GIS is the dominant phase in Si-rich media with polymorphs P1 and P2 forming in more and less alkaline conditions, respectively. The addition of zinc at a constant concentration (molar ratio Zn/H2O = 0.001) completely suppresses the GIS phase (Figure 3b) and transforms the kinetic phase diagram into one resembling low-temperature (i.e., 65 °C) Na-zeolite syntheses25 in the Si-rich region. The Al-rich region remains characteristic of 100 °C Na-zeolite syntheses, irrespective of zinc addition, with sodalite (SOD) formed in more alkaline media. Interestingly, both FAU and LTA are metastable zeolites known for their rapid conversion to more stable (i.e., higher framework density) phases when the temperature is raised from 65 °C to 100 °C; however, in the presence of zinc, both phases are stabilized and the LTA region shifts toward higher Si/Al ratios (Figure S5). Overall, these results reveal that zinc inhibits GIS formation regardless of synthesis gel compositions tested.
Figure 3.
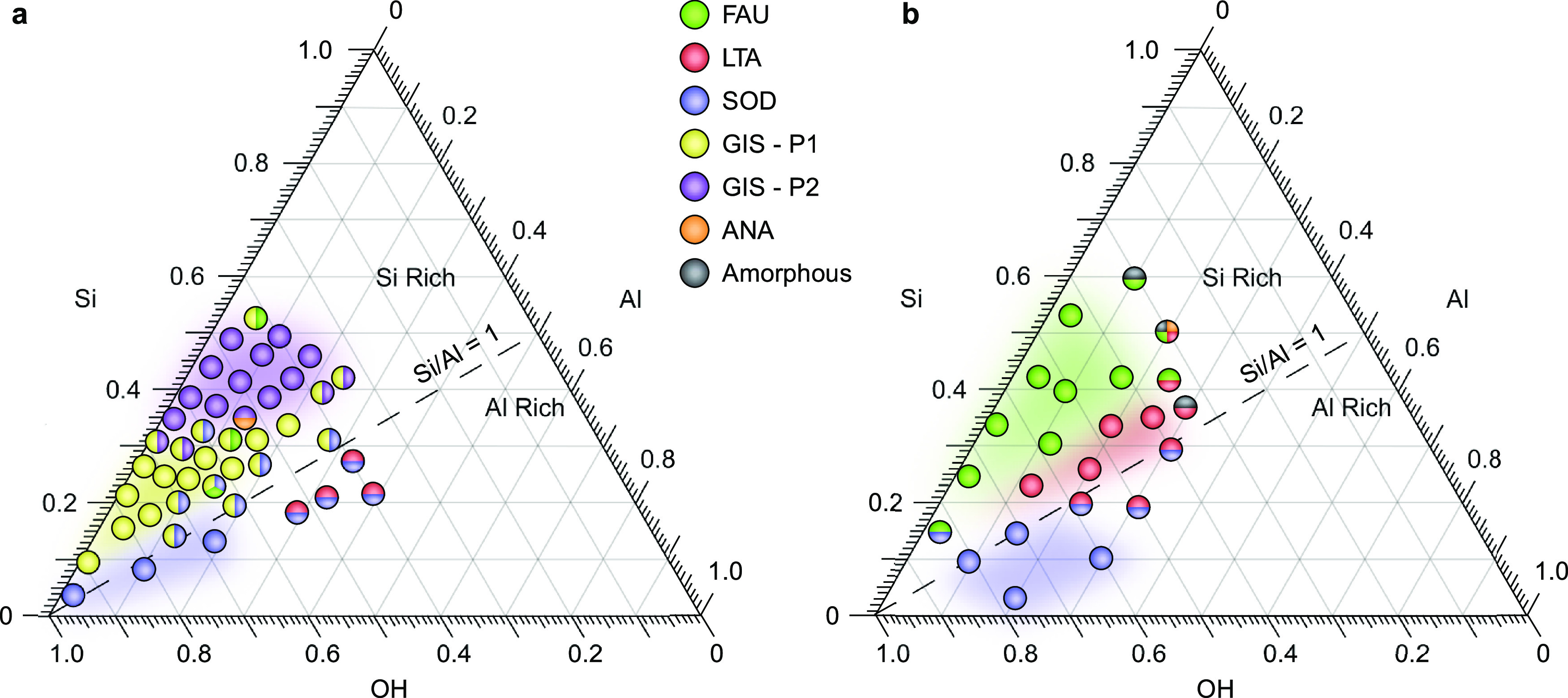
Kinetic ternary phase diagrams with molar compositions xSi/yAl/zZn/11NaOH/190H2O comparing conditions with (a) no zinc (z = 0) and (b) a fixed zinc concentration (z = 0.2). Data in (a) were previously reported by Oleksiak et al.30 and Maldonado et al.25 Symbols are color-coded based on the final zeolite structure after 7 days of synthesis at 100 °C. Symbols of a single color are pure zeolite phases, whereas mixed symbols indicate binary or ternary mixtures based on analysis of powder XRD patterns.
We investigated the impact of zinc concentration on the final zeolite product after hydrothermal treatment at 100 °C for 4 days, which is the time required to achieve fully crystalline GIS in the absence of zinc (Figure S1). Comparison of powder XRD patterns at increasing Zn/Al molar ratio (Figure S6) reveal a threshold zinc concentration (Zn/Al ∼ 0.02) below which GIS-P2 formation is unhindered and above which we observe residual FAU, consistent with our observation that zinc suppresses the FAU-to-GIS IZT. The first evidence of GIS inhibition shows that the presence of zinc also shifts the GIS polymorph to P1 (rather than the nominal P2). We posit that this effect is related to the ability of zinc to sequester silicates, thus leading to a growth mixture with a lower effective Si/Al ratio, which favors P1 (Figure 3a).30 Further increase in zinc concentration (Zn/Al ∼ 0.1) results in pure FAU, whereas higher zinc content stunts zeolite nucleation and leads to a purely amorphous product at Zn/Al > 1. This indicates there is an optimum amount of zinc to stabilize FAU, which depends on synthesis time and temperature. The full sequence of heteroatom effects on fractions of zeolite/amorphous products is highlighted in Figure 4a.
Figure 4.

(a) Phase purity of solids extracted from syntheses after 4 days at 100 °C using a molar composition of 9SiO2/0.5Al2O3/11NaOH/zZn/190H2O. The approximate percentage of each zeolite is based on relative peak area for each phase in powder XRD patterns (see Figure S6). (b) Fraction of FAU converted to GIS during a synthesis at 100 °C, where Zn(NO3)2 is added to the gel (to yield a molar ratio of 0.2Zn/190H2O) after 0.5 (i), 1 (ii), 2 (iii), and 3 (iv) days (indicated by arrows). Fractions of GIS are based on powder XRD analysis of extracted solids (see Figure S7). Dashed lines are a visual guide to the eye.
We performed a series of experiments where zinc was introduced into the synthesis gel at periodic times of heating to assess whether its presence prior to FAU and/or GIS nucleation impacted the ability of zinc to suppress the FAU-to-GIS transformation. We selected synthesis times corresponding to complete FAU crystallization (prior to IZT) and periodic stages during GIS growth. In all cases, mid-synthesis addition of zinc (arrows in Figure 4b) caused immediate cessation of GIS formation. This seems to indicate that the presence of zinc impacts the chemical potential of the synthesis medium. For instance, it would be expected that GIS being a more thermodynamically stable zeolite than FAU would continue to grow if the chemical potential of the medium remained favorable for IZT; however, our findings reveal that zinc’s mode of action is independent of the properties of the growth mixture prior to its addition.
Analysis of Zn-Mediated GIS Suppression
Here we explore whether the presence of zinc in solution or in the solid state (i.e., amorphous precursor or FAU parent crystals) has a more critical influence on the chemical evolution of the growth mixture. Inductively coupled plasma mass spectrometry (ICP-MS) was used to track the elemental composition of supernatant solutions extracted at periodic times during FAU synthesis. For these studies we used a lower synthesis temperature of 80 °C to reduce the kinetics of zeolite crystallization, thereby enabling more samples to be collected as a function of reaction progress. Prior to heating, zinc and aluminum are present at similar concentrations in the supernatant (Figure S8). During the induction period, both of these elements are gradually incorporated into the solid phase prior to the onset of Bragg peaks detected in powder XRD patterns of extracted solids. This resulting decrease in supernatant Zn and Al concentrations reaches an apparent plateau around the time of nucleation (ca. 1.7 days, Figure S8). Beyond this time, we observe a stark difference in temporal variations of these two species in the supernatant with increasing time during FAU crystallization. Notably, zinc gradually relocates from solid to solution as aluminum incorporates into FAU crystals (i.e., the Zn/Al ratio of the supernatant increases with time).
A prior study by Anseau and co-workers32 revealed the importance of the Zn/Al molar ratio on oligomer speciation in highly alkaline solutions containing zinc, aluminum, and silicon at similar ratios as those used in our study. They found that although zinc and aluminum do not oligomerize with one another, they compete for available silicon to generate zincosilicate and aluminosilicate oligomers, respectively. This was demonstrated using 29Si NMR spectroscopy to show that silicates have a slight preference to form oligomers with zinc. Previous research has shown that there are clear differences in the oligomerization behavior and crystallization of zincosilicates relative to aluminosilicates.32 For example, it has been demonstrated that zincosilicate zeotypes possess 3-membered rings (3R), which are not typically observed in aluminosilicate zeolites.22,23 Furthermore, zinc-containing zeotypes typically require high synthesis temperatures, suggesting larger energetic barriers to nucleation compared to those for aluminosilicates. For the synthesis conditions used in this study, increased temperature (from 100 to 150 °C) and replacement of aluminum with zinc results in the formation of VPI-7 (Figure S10), which is a zincosilicate zeotype (VSV type).23
We used density functional theory (DFT) calculations to compare the condensation and oligomerization energetics of silicate, zincosilicate, and aluminosilicate species. Formation energies of experimentally identified silicate oligomers34,35 were calculated for n = 1–4 (Figure 5) where n is the number of tetrahedral species (T = Si, Al, or Zn). Growth of linear complexes (monomer (M) → dimer (D) → trimer (Tr) → tetramer (T)) in the pure-silicate system shows a downhill energetic trend (exothermic). The formation of cyclic structures (3-membered ring (3R), 3-membered ring with one acyclic silicon ((3-1)R), and 4-membered ring (4R)) from their corresponding linear structures is also calculated and appears to be generally favorable (i.e., cyclic complexes are more stable than linear). Formation thermodynamics agree well with oligomerization energies reported by Mora-Fonz et al.36 assuming an alkaline condition (i.e., considering the presence of negatively charged species) with an implicit solvation model. The 4R silicate species is the most stable among cyclic structures investigated, which is consistent with observations of its dominant population in aqueous solutions34 and in many zeolite frameworks.
Figure 5.

Formation Gibbs free energies of oligomers (up to n = 4, where n is the number of tetrahedral species, i.e., Si, Al, or Zn) calculated using DFT (M = monomer, D = dimer, Tr = trimer, 3R = 3-membered ring, T = tetramer, (3-1)R = 3-membered ring with one acyclic silicon, and 4R = 4-membered ring). Oligomer growth via condensation is represented by solid lines, and cyclization is represented by dotted lines. Aluminosilicate and zincosilicate isomers with the most exothermic formation energy values are reported. Optimized molecular structures of pure-silicate oligomers are provided with color-coded balls representing silicon (gray), oxygen (red), and hydrogen (white) atoms. Additional details are given in Figures S11 and S12.
The presence of Al or Zn significantly affects oligomerization thermodynamics. Overall, zincosilicate species (Figure 5, blue) show more exothermic formation energy than corresponding aluminosilicates (Figure 5, red), followed by the least exothermic silicate species (Figure 5, black). Freeman et al.37 similarly reported a more exothermic formation of aluminosilicate oligomers relative to their pure-silicate counterparts. Energetic differences in reaction pathways containing Al or Zn are primarily owing to the monomer addition step rather than condensation or cyclization (Figure S12). As expected from the thermodynamic calculations, kinetics of oligomerization is found to be fastest for zincosilicates, followed by that for aluminosilicates (Figure S13). Detailed analysis of oligomerization energetics is found in the Methods section. The observed oligomerization thermodynamics are consistent with solution 29Si NMR spectra reported by Anseau et al.32 showing a higher percentage of silicate dimerization with zincate species than those of aluminate. While the presence of Al or Zn significantly affects the oligomerization thermodynamics, it does not alter the preference among different species; thus the energetic preferences of pure-silicate species (4R > (3-1)R > T > 3R > Tr > D > M) is true for all systems with one exception: the inversion between aluminosilicate Tr and 3R (although with a small energy difference). These findings indicate that aluminate and zincate species play an important role in prenucleation chemistry and oligomer composition, but less so for oligomer size and structure.
Zinc Coordination in FAU
Here we use a combination of experimental and computational techniques to examine how Zn heteroatoms incorporate in zeolite crystals. Elemental analysis of Zn,Na-FAU crystals reveals a nonuniform distribution of Zn and Al atoms throughout the crystal. As shown in Table 1, two methods were used to evaluate the Si/Al and Si/Zn ratios. X-ray photoelectron spectroscopy (XPS) was used to extract the molar ratios at the outermost surface (i.e., depth of ca. 8 nm), whereas energy dispersive spectroscopy (EDS) was used to evaluate the overall composition (i.e., depth of ca. 1 μm) of each sample. Comparison of EDS and XPS data for Na-FAU reveals a similar Si/Al ratio, indicating a homogeneous distribution of Al without evidence of either Al- or Si-zoning.3 For Zn,Na-FAU we observe a higher Si/Al ratio from XPS relative to EDS, which indicates minor Si-zoning in the outer rim of these crystals. The loss of Al sites at the exterior surface is compensated by the increased occlusion of zinc, which is confirmed by the lower Si/Zn ratio from XPS measurements (Table 1). This anticorrelated relationship between zinc and aluminum was observed previously in Zn-FAU samples.17 Potential explanations for Zn-zoning in FAU and its putative role in suppressing GIS nucleation are provided in the following sections.
Table 1. Composition of FAU Crystals.
| molar ratio | method | Na-FAUa | Zn,Na-FAU (Zn/Al = 0.2) | Zn,Na-FAU (Zn/Al = 0.4) |
|---|---|---|---|---|
| Si/Al | EDS | 2.2 ± 0.3 | 2.0 ± 0.1 | 2.4 ± 0.3 |
| Si/Al | XPS | 2.1 ± 0.2 | 3.5 ± 0.3 | 4.6 ± 0.3 |
| Si/Zn | EDS | n.a. | 40 ± 20 | 11 ± 3 |
| Si/Zn | XPS | n.a. | 4.3 ± 0.8 | 5.5 ± 0.6 |
Solids were extracted from growth mixtures by decanting the supernatant solutions prior to washing with DI water (Methods section).
There are multiple possibilities for zinc coordination in zeolites. We performed tests to eliminate several options for zinc coordination in Zn,Na-FAU samples. X-ray pair distribution function (PDF) analysis was performed on as-synthesized samples (Figure 6a), revealing the absence of O–Zn2+ bonds, which are signatures of extra-framework Zn2+ ions associated with framework oxygen. We attempted to remove any potential extra-framework zinc complexes by ion exchange with monovalent (Na+ and K+) and divalent (Co2+) cations; however, these treatments had a minimal effect on Si/Zn ratios, indicating the absence of extra-framework zinc species.
Figure 6.
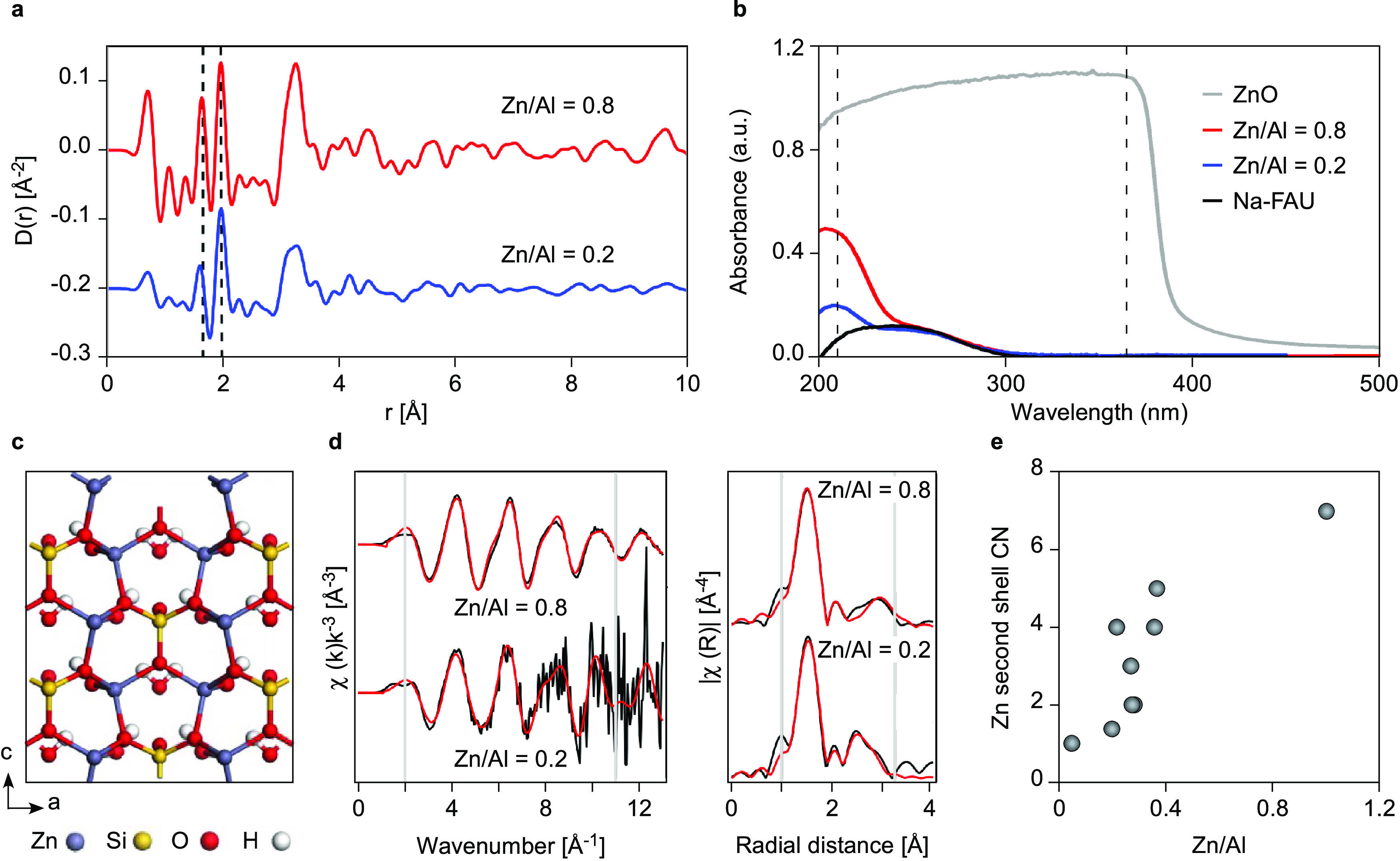
(a) Differential X-ray PDF (dPDF) of Zn,Na-FAU samples relative to Na-FAU. Dashed lines indicate the Al/Si–O distance in faujasite (1.64 Å) and the Zn–O distance in hemimorphite (1.96 Å). The loss of intensity beyond 4 Å indicates the Zn-associated disorder is primarily short to medium order in scale, extending to approximately 5 Å at low Zn and to approximately 10 Å at high Zn. (b) UV–vis spectra of Na-FAU (black), Zn,Na-FAU synthesized with Zn/Al ratios of 0.8 (red) and 0.2 (blue), and commercial ZnO (gray) as a reference. Dashed lines indicate peaks at 210 and 365 nm. (c) Crystal structure of hemimorphite (nonporous zincosilicate). Reproduced with permission from ref (38) copyright 2010 Elsevier. (d) Extended X-ray absorption spectroscopy (EXAFS) measurements of Zn,Na-FAU samples washed with deionized water prior to centrifugation. (Left) Zn K-edge EXAFS spectra (black) for Zn,Na-FAU samples. Shell-by-shell fits (red) were produced using FAU framework and hemimorphite reference structures for low and high Zn/Al samples, respectively. (Right) Fourier transforms of the Zn EXAFS data. Gray lines define the fit window of 2–11 Å–1 (k space) and 1–3.25 Å (R space). (e) Dependence of zinc coordination number (CN) on the amount of zinc present in the Zn,Na-FAU synthesis mixture. Raw and analyzed data used to generate (e) are shown in Figure S14 and Table S1, respectively.
The potential incorporation of ZnO particles was also investigated by UV–Vis spectroscopy, which can detect relatively small amounts of metals.39 Spectra were assessed for Zn,Na-FAU samples prepared with two Zn/Al ratios (Figure 6b). Low zinc content (Zn/Al = 0.2) results in only one peak at 210 nm, which is commonly associated with framework zinc, although this assignment is ambiguous.22,24 For samples with high zinc content (Zn/Al = 0.8), the peak associated with Zn framework species increases while extra-framework species are absent with the proper washing of solids during product isolation (see Figure S15 in the Supporting Information for more details).
The nature of zinc incorporation in FAU samples was further assessed using extended X-ray absorption spectroscopy (EXAFS) in combination with PDF analysis. The Zn EXAFS spectra of Zn,Na-FAU samples were dependent on zinc loading (Figure 6d). For samples with Zn/Al < 0.2, the EXAFS data can be fit with the FAU structure; however, samples with Zn/Al > 0.2 are best fit with a hemimorphite-like structure. For all samples, hemimorphite is not evident in XRD patterns (Figure S6), consistent with PDF profiles (Figure S16) indicating these species do not possess long-range order. Hemimorphite (Zn4(Si2O7)(OH)2·H2O) is a naturally occurring zincosilicate crystal with a nonporous structure (Figure 6c). The appropriateness of using hemimorphite to fit the EXAFS Zn spectra is due to the increased Zn–O–Zn bond length within the zeolite structure. As shown in Figure 6e, the Zn coordination number increases with increasing zinc loading, consistent with the presence of a more ZnO-like phase, and the Zn–O–Zn bond length increases from 2.9 Å (FAU) to 3.3 Å (hemimorphite, Table S1). However, the lack of shell-by-shell fit sensitivity (p(F) value > 0.05) to the inclusion of Zn–O–Si bonds in the coordination environment of zinc supports that an ordered hemimorphite phase is not present and that this model fit structure is likely best at capturing the increase in the Zn–O–Zn bond length (Table 2).
Table 2. Shell-by-Shell Fits to Zn EXAFS Spectraa.
| sample | neighbor | CN | R + ΔR (Å) | σ2 | p(F) | ΔE0 | R factor | fit profile |
|---|---|---|---|---|---|---|---|---|
| Zn,Na-FAU (Zn/Al = 0.2) | O | 4b | 1.945(6) | 0.0029(4) | <0.001 | 4(1) | 0.012 | FAU |
| Zn | 1b | 2.920(9) | 0.003(1) | <0.001 | ||||
| Zn,Na-FAU (Zn/Al = 0.8)c | O | 5.30b | 1.947(5) | 0.0060(4) | <0.001 | 5.6(7) | 0.007 | hemimorphite40 |
| Zn | 7(3) | 3.32(1) | 0.017(4) | <0.001 | ||||
| Zn,Na-FAU (Zn/Al = 0.8)c | O | 5.30b | 1.947(4) | 0.0058(5) | <0.001 | 5.6(6) | 0.004 | hemimorphite40 |
| Si | 1.33b | 3.14(3) | 0.009(5) | 0.176 | ||||
| Zn | 7.70b | 3.32(2) | 0.021(3) | 0.003 |
So2 = 0.8; coordination number (CN); k space fit window 2–11 Å–1, Fourier transform fit window 1–3.25 Å; interatomic distance (R + ΔR); disorder parameter (σ2); F-test [p(F)]; energy shift (ΔE0); goodness-of-fit parameter (R factor).
Fixed value.
Na,Zn-FAU (Zn/Al = 0.8) was fit using two methods where Si next-nearest neighbors were ignored (top) or included (bottom). FAU framework standard generated in VASP.41
Shell-by-shell fitting of EXAFS spectra to determine the precise Zn coordination environment was particularly challenging due to heterogeneous or disordered environments. The proposed increase in bond length of the Zn/Al = 0.8 sample is corroborated using PDF, which shows that when zinc is present in low quantities, its effect on crystal order is limited to distances less than 10 Å (Figure S16). These short-range shifts in Si–O and Zn–O bond lengths are more prominent with higher Zn/Al ratios (Figures 6a and S16) and indicate the presence of a hemimorphite-like structure with medium-range disorder. Collectively, EXAFS and PDF results suggest that a small quantity of zinc can be incorporated into the FAU crystal, but the zinc coordination environment changes as the Zn/Al molar ratio increases (Figure 6e), approaching a partially disordered hemimorphite-like structure at the highest loadings tested.
The apparent imprecision suggests that the zinc coordination is challenging to identify in these structures owing to nonhomogeneous or disordered environments. Further evidence of this possibility involves the lack of shell-by-shell fit sensitivity to the inclusion of Zn–O–Si bonds in the coordination environment of zinc (Table 2). As zinc loading increases, the Zn coordination increases (Figure 6e), consistent with the presence of a ZnO-like phase with a Zn–O–Zn bond of 3.3 Å (Table 2), in good agreement with hemimorphite.
To investigate whether zinc incorporation is thermodynamically feasible in FAU and GIS, we calculated energetic changes of replacing a tetrahedral Si site by Al and Zn. Here, DFT calculations are combined with a semiempirical method (see Methods sections) to compare Al and Zn substitution energies at different T-sites. Both frameworks were evaluated using two DFT regimes in the crystal: one at the surface and one in the bulk where all other atoms were included in the semiempirical regime (Figure S17). As shown in Figure 7a, regardless of framework type (FAU or GIS) and substitution location (surface or bulk), the substitution energy of aluminum is around −55 kJ/mol, which is consistent with experimental observations of homogeneously distributed Al in the absence of Zn (Table 1). On the other hand, Zn substitution energetics depend on the location of the T-site; in both frameworks, bulk T-sites do not strongly favor zinc incorporation, whereas the surface shows increased exothermicity. Hence, aluminum is preferred over zinc in the bulk, but zinc is preferentially located near the exterior surface (Figure 7a), which is consistent with elemental analysis (Table 1) showing Zn,Na-FAU crystals with Zn-rich and Al-poor surfaces relative to their overall (or bulk) composition.
Figure 7.
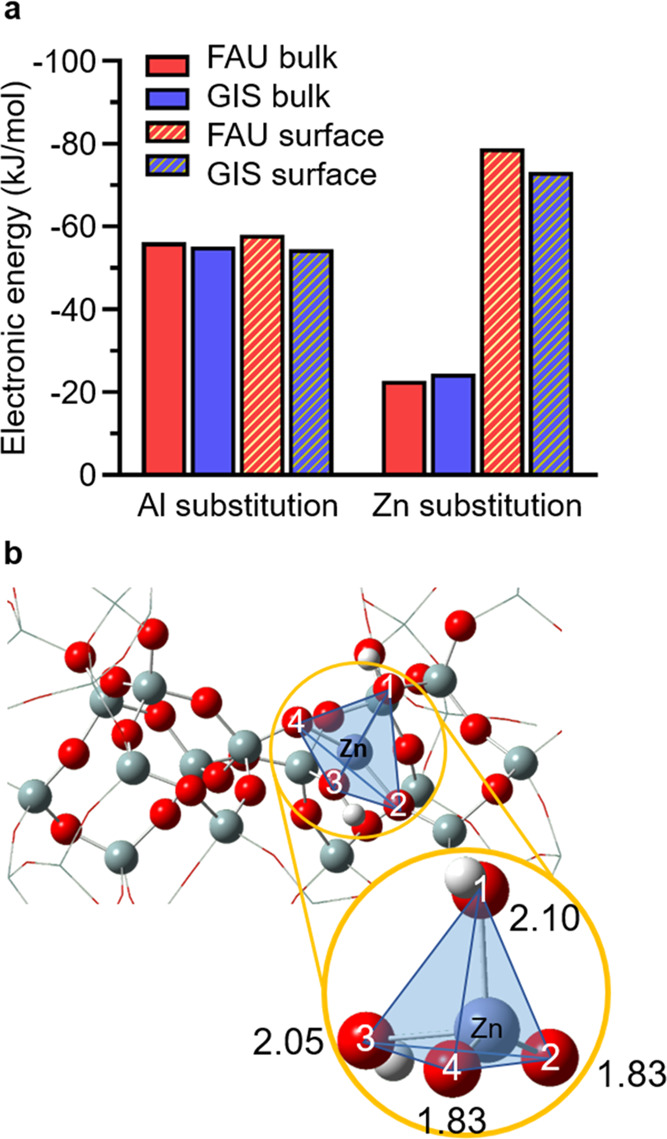
(a) Calculated energies of Al and Zn substitution on FAU and GIS frameworks in bulk (solid bars) and surface (striped bars) locations. Additional details are provided in Figure S18. (b) Optimized local geometry around Zn in the bulk position of the FAU framework with Zn–O bond lengths labeled (in Å). A similar illustration for GIS is provided in Figure S20.
The strong preference of zinc to populate surface sites is attributed to changes in the local coordination environment. We observed increased bond lengths wherein oxygen atoms move further away from a T-site after substitution with Zn compared to Al (Table S3). This bond elongation is greater for oxygens with the Brønsted acid. For example, an Al site has three Al–O bonds at around 1.72 Å and one Al–O(H) bond at 1.89 Å, which is consistent with values reported in the literature.42 Conversely, Zn sites have two Zn–O bonds, which are around 1.83 Å and two Zn–O(H) bonds at 2.05 and 2.10 Å (Figure 7b), the average of which (1.95 Å) corresponds well with results from EXAFS (Table 2). This configuration breaks the symmetry of the regular tetrahedron (SiO4) and distorts the local geometry (Figure S19). The average distortion of the tetrahedral (TO4) bond upon Al or Zn substitution (Table S3) is quantified based on the deviation of positions of oxygen atoms from the four vertices of a corresponding regular tetrahedron43 (see the Methods section). The original site (SiO4) is very close to a regular tetrahedron with a consistent Si–O length of 1.6 Å. Aluminum substitution results in a longer average T–O bond length with minimal distortion (<5%), which does not depend on location. On the other hand, zincate tetrahedra are substantially enlarged and distorted from the silicate tetrahedron, especially at T-sites on external surfaces. Indeed, a T-site located within the bulk is more constrained than a surface site since the former is embedded in the rigid crystalline structure, which promotes selective substitution of Zn on surface sites.
Considering the computational evidence regarding favorable zinc incorporation on zeolite surfaces, Zn-zoning is predicted, consistent with results in Table 1 showing a lower Si/Zn ratio at the exterior surface of FAU. The presence of framework zinc in FAU is not fully resolved; however, collective results from ONIOM calculations (DFT combined with a semiempirical method) and EXAFS measurements suggest zinc can incorporate in the framework with a coordination less than that of aluminum. These sites introduce defects in the FAU crystal structure that may be responsible for changes in bulk crystal morphology. For instance, synthesis of FAU in the presence of zinc leads to apparent intergrowths (Figure S21).
Bypassing Interzeolite Transformation
ONIOM calculations revealing energetic equivalence of zinc incorporation in FAU and GIS (Figure 7a) supports the hypothesis that zinc in the solution phase is responsible for disrupting the FAU-to-GIS transformation. Here we tested this idea by deconvoluting the effects of zinc in the solid (FAU) and solution (supernatant) phases. We synthesized FAU crystals in the presence (Zn,Na-FAU) and absence (Na-FAU) of zinc and in each case isolated their respective supernatants (Table S4). A series of four seeded growth experiments were performed where all combinations of FAU crystals and supernatant solutions were combined and heated for 3 days at 100 °C to determine under which conditions we observed a FAU-to-GIS transformation. All combinations where zinc was present in the supernatant did not result in GIS formation. For the case where Zn,Na-FAU seeds were suspended in a zinc-free supernatant, we observed partial transformation of FAU to GIS, indicating zinc within the solid crystal was insufficient to suppress the onset of IZT; however, release of zinc from the FAU crystal into solution (Table S4) may be the reason for FAU-to-GIS IZT to cease within the timeframe of the experiment. In another set of experiments, we heated only the supernatant solutions (without seed addition) and observed no crystals after 3 days for the zinc-containing solution; however, the supernatant without zinc resulted in direct GIS crystallization, thus corroborating previous suggestions that GIS does not nucleate epitaxially on FAU surfaces.44,45
In a recent perspective article,19 we postulated that IZTs can be bypassed if the chemical potential Δμ of the synthesis medium could be altered in a way that favors nucleation of the more thermodynamically stable zeolite. Zeolite synthesis mixtures are heterogeneous with the majority of silica retained within undissolved sources; thus, the inaccessibility of silica at early stages of synthesis creates a pseudo environment in both solution and solid states that differs from the overall molar composition of the synthesis mixture.46 Although chemical potential is a nebulous parameter that is difficult to quantify, we proposed a general trend in Δμ as a function of synthesis progress (Figure 8a) where the driving force leads to the initial metastable zeolite FAU but changes later in the synthesis to favor growth of GIS. We hypothesized that it could be possible to directly nucleate GIS if Δμ of the initial solution was altered. Here we show that the FAU supernatant solution is still sufficiently supersaturated to crystallize GIS. We prepared growth mixtures with identical concentrations as the supernatant solution after Na-FAU synthesis: growth mixture β (Figure 8a) has a molar composition of 164SiO2/0.5Al2O3/209NaOH/5115H2O. Preparation and hydrothermal treatment of growth mixture β revealed direct GIS crystallization in less than 3 days (Figure 8b), whereas the addition of a small amount of zinc (i.e., Zn/Al = 0.2) inhibited zeolite nucleation. Most notably, FAU intermediates are absent, unlike the nominal growth conditions in this study (growth mixture α, Figure 8a,c). We also observe that growth mixtures α and β lead to crystals with subtly distinct morphological features (Figure 8d,e, respectively).
Figure 8.

(a) Hypothetical changes in chemical potential, Δμ, with synthesis progress, adapted from the IZT concept proposed8 for different growth solutions (α and β) and zeolite products (FAU and GIS). Growth mixture α refers to the nominal synthesis condition resulting in a FAU-to-GIS transformation. Growth mixture β is the supernatant of the previous mixture extracted at a time corresponding to full FAU crystallinity prior to the onset of GIS nucleation, where continued heating results in direct crystallization of GIS. (b, c) Time-resolved powder XRD patterns of solids extracted from growth mixtures β (b) and α (c). Select peaks of FAU, GIS-P2, and impurity phases are indicated by green, purple, and orange circles, respectively. (d, e) Scanning electron micrographs of GIS-P2 products obtained after 7 days of hydrothermal treatment of growth mixtures (d) α and (e) β. Scale bars represent 1 μm.
Conditions in this study that lead to the direct formation of GIS are similar to the few studies in the literature reporting the direct nucleation of GIS without first observing crystalline FAU as a metastable intermediate.47,48 In such cases, the growth medium is dilute with low solute concentration44,47,49 and often has a high Si/Al ratio.44,50 When we compare GIS nucleation during IZT to that in the extracted supernatant, the induction times are approximately identical (Figure S22). This indicates that FAU crystals do not appreciably accelerate GIS nucleation. During synthesis progress, FAU crystallization leads to substantive changes in the growth mixture composition and distribution of Si and Al species (i.e., the growth medium progressively becomes more Si-rich). As postulated in Figure 8a, these changes alter the chemical potential in ways that favor GIS nucleation, and several experiments (Figures S24 and S25) confirmed that increased Si availability promotes GIS crystallization. Interestingly, Okubo and co-workers51 modeled Al incorporation in the GIS framework by molecular mechanics and reported it is most thermodynamically stable at Si/Al molar ratios ≥ 3, compared to FAU that reaches its maximum stability at Si/Al < 3.52 This is consistent with findings in this study that Zn acts as an inhibitor of GIS crystallization. GIS nucleates in media with a higher Si/Al ratio and given that Zn is an efficient scavenger of silicates (i.e., reduces supernatant Si/Al ratio), this inhibits GIS crystallization.
Conclusions
Multivalent metals have diverse effects on zeolite crystallization. In this study, we specifically addressed their impact on FAU-to-GIS IZT by assessing how the addition of various metals to synthesis media impact nucleation of both parent and daughter zeolites. Elements selected from group III and IV as well as zinc (the sole transition metal selected for this study) can serve as heteroatoms in zeolite frameworks and have a more pronounced impact on crystallization pathways than group II elements. Here we focused on zinc as an effective modifier of IZT pathways.
Working in the absence of zinc we showed that the nucleation of GIS (daughter) is independent of the presence of FAU (parent). The direct nucleation of GIS from the supernatant solution of a FAU synthesis tested the hypothesis that changes in the initial composition (or chemical potential) can alter the trajectory of IZT. This also suggests that metastable FAU has a less active role in GIS nucleation, and likely serves only as a source of aluminosilicate solute to allow continued growth of GIS crystals. Introduction of zinc into the growth medium can have several effects based on its concentration. At high concentration, zinc suppresses zeolite nucleation and leads to an amorphous product. At low concentration, zinc inhibits the FAU-to-GIS IZT and also shifts the final GIS product from polymorph P2 to P1. At an intermediate concentration, zinc completely suppresses GIS nucleation leading to a stable FAU product. Surprisingly, zinc can stabilize FAU crystals at temperatures around 100 °C, which is much higher than typical synthesis conditions for this metastable zeolite. The ability of zinc to suppress GIS nucleation is independent of when the heteroatom is added to the synthesis medium.
In the presence of zinc, we posit that aluminate and zincate species compete to condense alumino-/zincosilicate oligomers, where the extent of zincosilicate formation depends on the Zn/Al ratio. At intermediate zinc compositions (0.1 < Zn/Al < 0.2), zinc prevents the level of chemical evolution necessary for GIS nucleation by sequestering much of the remaining silicate species. This draws greater attention to the role of the solution phase where soluble zincosilicate species seemingly have a more pronounced effect on zeolite phase transition. This was confirmed by DFT calculations, which show that zincosilicate oligomerization is more favorable than that of (alumino)silicate species. Using EXAFS measurements, we showed that zinc heteroatoms are not fully coordinated when incorporated into the FAU framework. Our findings have not conclusively resolved the local structure of zinc species, but have revealed that framework zinc has a local coordination that more closely resembles a zincosilicate mineral, hemimorphite, and is preferentially located near the surface of zeolite crystals. The selective incorporation of Zn on the crystal surface is rationalized by ONIOM calculations, which showed that Zn substitution on the surface is promoted by the more flexible coordination environment on the surface relative to the bulk crystal.
In summary, we have used a combination of experimental and computational approaches to develop a deeper understanding of how heteroatoms can influence zeolite crystallization. There is increased interest in designing zeolites with a variety of heteroatoms as potential sites for catalytic reactions. Here, we focus on zinc and highlight the challenges of conclusively identifying heteroatom coordination in zeolites. One previously unrecognized benefit of heteroatoms in zeolite synthesis that we observe in this study is their ability to stabilize FAU over a broad temperature range, thus avoiding unwanted impurities and expanding the synthesis space of this commercially relevant zeolite. Two key observations in this study related to IZTs are that daughter nucleation does not necessarily occur on the surface of the parent crystal; and that the pathway of zeolite nucleation can be altered by changing the solution state, which is counter to many nonclassical pathways of zeolite crystallization that focus on the role of precursors (i.e., gel or solid state). Future studies of other heteroatoms and zeolite frameworks may prove how generally these observations apply to syntheses of other materials.
Methods
Materials
The following reagents were used for zeolite synthesis as purchased from Sigma-Aldrich without further purification: LUDOX AS-40 colloidal silica (40 wt % suspension in water), sodium aluminate (anhydrous), sodium hydroxide (98% pellets), zinc nitrate hydrate (98%), gallium nitrate hydrate (99.9%), indium nitrate hydrate (99.9%), germanium oxide (99.99%), boric acid (99.5%), magnesium hydroxide (95%), calcium hydroxide (95%), strontium hydroxide octahydrate (95%), barium hydroxide octahydrate (95%), and zinc oxide (99.9%). Deionized (DI) water was produced with an Aqua Solutions purification system.
Zeolite Synthesis
Growth mixtures were prepared with molar compositions 9SiO2/0.5Al2O3/xMaOb/5.5Na2O/190H2O where x ranges from 0 to 1, representing the M/Al ratio (M = Zn, Ga, Ge, In, Sn, B, Mg, Ca, Sr, Ba). Growth solutions were prepared in a polypropylene bottle. First, sodium hydroxide and sodium aluminate were dissolved in DI water under magnetic stirring. Next, metal reagents were added (when M ≠ 0 except those where zinc is added mid-synthesis—see below) and stirred either for 10 min or until dissolved. Then, LUDOX AS-40 was added using a plastic pipette, whereupon the synthesis mixture coagulates. The gel was immediately shaken, then stirred for 24 h at room temperature with a magnetic stir bar. After this, the synthesis mixture was transferred to a Teflon-lined autoclave and placed in a 100 °C oven for a period of hydrothermal treatment. In some cases, the synthesis mixture was briefly removed from the oven for mid-synthesis addition of zinc nitrate in a fume hood. For the latter, polypropylene bottles were used to allow for mid-synthesis addition of zinc without the need to cool mixtures to room temperature.
After hydrothermal treatment, solid crystals were recovered as follows. Synthesis mixtures were washed with 1–30 mL of DI water in a centrifuge tube. After centrifugation (5 min, 13,000 rpm), the supernatant solution was decanted. This procedure was repeated twice more with ∼40 mL DI water. The gel product was dried overnight in an incubator (∼60 °C). In some instances, the supernatant solution was recovered after the first round of centrifugation for further analysis. One observation made during solids extraction in Zn-zeolite syntheses is that diluting the sample with DI water prior to first extracting the supernatant leads to the incorporation of additional zincosilicate solid within the sample, thereby decreasing product purity. This phenomenon likely occurs because zincosilicate precipitation is sensitive to pH32. Indeed, we confirmed that this outcome can be minimized by centrifuging the mixture and decanting the supernatant prior to washing the solid with DI water. We characterized samples that were washed before or after separating the solids from the supernatant. When prewashing leads to hemimorphite formation there is a significant increase in solid yield (by as much as a factor of two for samples with high Zn/Al ratios) compared to samples washed after separation. The inclusion of hemimorphite in the product also results in a marked reduction in Brunauer–Emmett–Teller (BET) surface area (see Figure S15). Collectively, these findings highlight the sensitivity of zeolite growth media to the presence of zinc species, which can easily precipitate soluble silicates and produce impurities in syntheses of zinc-containing zeolites.
Ion Exchange
Samples were ion-exchanged using a 1 M solution of NaNO3, KNO3, Co(NO3)2, or NH4NO3 with 5 wt % zeolite. Slurries were continuously stirred (magnetic stir bar) for 2 h in a glass vial under constant temperature maintained at 80 °C using an oil bath. The slurry was then washed and centrifuged, and the supernatant was decanted. The entire process was repeated a total of three times. Subsequently, the remaining ion-exchanged solid was left in an incubator at 60 °C overnight to dry.
Materials Characterization
Powder X-ray diffraction (XRD) patterns were collected on a Rigaku diffractometer using Cu Kα radiation (40 kV, 40 mA). Data were collected with a step size of 0.02 (2θ) at a rate of 8.33 steps per second. Fractions of crystalline phases were calculated by measuring their peak area using Rigaku PDXL Software. The fraction of amorphous phase was calculated by comparing the combined peak area for a fully crystalline sample to the combined peak area of partially amorphous samples. Relative quantities of crystalline and amorphous phases were calculated based on peak areas. Scanning electron microscopy (SEM) was conducted on a FEI-235 Dual-Beam Focused Ion Beam instrument operated at 15 kV and a 5 mm working distance. Samples for SEM analysis were prepared by affixing dried crystals to a SEM sample holder with carbon tape. Electron dispersive spectroscopy (EDS) was performed with a FEI 235 dual-beam (focused ion beam) system operated at 12 kV and a 15 mm working distance. X-ray photoelectron spectroscopy (XPS) was performed on a PHI 5800 ESCA (Physical Electronics) system, which is equipped with a standard achromatic Al Kα X-ray source (1486.6 eV) operating at 300 W (15 kV and 20 mA) and a concentric hemispherical analyzer. Since zeolites are insulating materials, the equipment neutralizer component was utilized to prevent charging effects. Diffuse reflectance ultraviolet visible spectra (UV-Vis DRS) were obtained using a PerkinElmer Lambda 35 UV–Vis scanning spectrophotometer. BET surface area was determined using a Micromeritics 3Flex instrument (N2 adsorption); microporous volume was determined from the t-plot method. Supernatant composition was determined using a Thermo Scientific iCAP RQ ICP-MS with an approximate dilution factor of 10,000. Extended X-ray adsorption fine structure (EXAFS) spectra and X-ray pair distribution functions (PDF) were collected in the transmission mode in the bending magnet beamline 20-BM-B and 11-ID-B, respectively, at the advanced photon source (APS), Argonne National Laboratory. Additional details regarding EXAFS and PDF analysis can be found in the Supporting Information.
First-Principles Calculations
Density functional theory (DFT) calculations were performed using the Gaussian 09 software package53 to investigate the initial oligomerization/cyclization thermodynamics of small zeolite precursor species. The global hybrid Minnesota functional with a high percentage of Hartree–Fock exchange (M062X) was used as it accurately describes main-group chemistry54 with Pople’s split-valance triple-ζ basis set with polarizable functions (6-311G(d,p)). Frequency calculations were performed after full optimizations to verify that the obtained structures were minima of the potential energy surface. To account for the hydrothermal environment in the prenucleation stage, the conductor-like polarizable continuum model (CPCM)55 was applied using water as a solvent. In addition, the Gibbs free energy of reactions was calculated by applying thermochemistry including enthalpic and entropic corrections to the electronic energy at elevated temperature (373 K, similar to experiments). The growth process of oligomers is assumed to occur via (1) monomer addition, (2) condensation, and (3) water removal. Cyclic species are assumed to form by intramolecular condensation (i.e., cyclization) of linear species,56 followed by the release of water to the liquid phase. A 3-membered ring is produced by cyclization of a linear trimer and tetramer cyclization results in a (3-1)-ring or a 4-membered ring. Full pathways including monomer addition, condensation, cyclization reaction, and water removal steps are found in Figure S12. For aluminosilicate and zincosilicate systems, no more than one aluminate (Al(OH)3(H2O)) or zincate monomer (Zn(OH)2(H2O)2) is considered, which is added to a silicate monomer (Si(OH)4) at the first addition step. All possible positions of aluminum and zinc were examined on the complexes larger than dimer, and the formation energies of the most stable species are reported. A pH-neutral environment is assumed to maintain a charge-neutral growth system which does not require charge balancing counter-ions and explicit solvent molecules to provide charge stabilization. Although this may not represent a typical experimental setting for zeolite synthesis, it is proven to be an effective approach to obtain general trends of aluminosilicate species growth while maintaining reasonable computational cost.3 Additional details regarding DFT calculations can be found in the Supporting Information.
Acknowledgments
The authors acknowledge Mahalingam Balasubramanian from APS Sector 20 (now at Oak Ridge National Laboratory) for collecting the EXAFS dataset. They also acknowledge computational support from the Center for Research Computing at the University of Pittsburgh and Dr. Boris Makarenko and Muhammad Fiji for assistance with data collection.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.2c00325.
Zeolite characterization techniques and detailed description of computational studies (DFT and ONIOM) (PDF)
Author Contributions
The project was conceived by J.D.R., G.M., J.J.N., and R.K.M. Experimental studies were performed by A.J.M. Computational chemistry calculations were performed by S.H. and E.E.F. Characterization and analysis of samples by EXAFS and PDF were performed by S.A.S. and S.M. The original draft of the manuscript was prepared by A.J.M. and J.D.R., in cooperation with S.H. and G.M. All authors contributed to the compilation and editing of the manuscript.
This work was primarily supported by the U.S. Department of Energy (DOE), Nuclear Energy University Program (DOE-NEUP) under Award 18-15496. J.D.R. received additional support from The Welch Foundation under Award E-1794. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
The authors declare no competing financial interest.
Supplementary Material
References
- Knott B. C.; Nimlos C. T.; Robichaud D. J.; Nimlos M. R.; Kim S.; Gounder R. Consideration of the aluminum distribution in zeolites in theoretical and experimental catalysis research. ACS Catal. 2018, 8, 770–784. 10.1021/acscatal.7b03676. [DOI] [Google Scholar]
- Dedecek J.; Lucero M. J.; Li C.; Gao F.; Klein P.; Urbanova M.; Tvaruzkova Z.; Sazama P.; Sklenak S. Complex analysis of the aluminum siting in the framework of silicon-rich zeolites. A case study on ferrierites. J. Phys. Chem. C 2011, 115, 11056–11064. 10.1021/jp200310b. [DOI] [Google Scholar]
- Le T. T.; Chawla A.; Rimer J. D. Impact of acid site speciation and spatial gradients on zeolite catalysis. J. Catal. 2020, 391, 56–68. 10.1016/j.jcat.2020.08.008. [DOI] [Google Scholar]
- Devos J.; Shah M. A.; Dusselier M. On the key role of aluminium and other heteroatoms during interzeolite conversion synthesis. RSC Adv. 2021, 11, 26188–26210. 10.1039/D1RA02887A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Corma A.; Yu J. Synthesis of new zeolite structures. Chem. Soc. Rev. 2015, 44, 7112–7127. 10.1039/C5CS00023H. [DOI] [PubMed] [Google Scholar]
- Moliner M.; Román-Leshkov Y.; Davis M. E. Tin-containing zeolites are highly active catalysts for the isomerization of glucose in water. Proc. Natl. Acad. Sci. 2010, 107, 6164–6168. 10.1073/pnas.1002358107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T.; Kamiya Y.; Otomo R. A rapid synthesis of Hf-Beta zeolite as highly active catalyst for Meerwein-Ponndorf-Verley reduction by controlling water content of precursor gel. Microporous Mesoporous Mater. 2022, 333, 111743 10.1016/j.micromeso.2022.111743. [DOI] [Google Scholar]
- Wang J.; Jaenicke S.; Chuah G.-K. Zirconium–Beta zeolite as a robust catalyst for the transformation of levulinic acid to γ-valerolactone via Meerwein–Ponndorf–Verley reduction. RSC Adv. 2014, 4, 13481–13489. 10.1039/C4RA01120A. [DOI] [Google Scholar]
- Xu W.; Zhang T.; Bai R.; Zhang P.; Yu J. A one-step rapid synthesis of TS-1 zeolites with highly catalytically active mononuclear TiO6 species. J. Mater. Chem. A 2020, 8, 9677–9683. 10.1039/C9TA13851J. [DOI] [Google Scholar]
- Mehdad A.; Lobo R. F. Ethane and ethylene aromatization on zinc-containing zeolites. Catal. Sci. Technol. 2017, 7, 3562–3572. 10.1039/C7CY00890B. [DOI] [Google Scholar]
- Zhou Y.; Thirumalai H.; Smith S. K.; Whitmire K. H.; Liu J.; Frenkel A. I.; Grabow L. C.; Rimer J. D. Ethylene dehydroaromatization over Ga-ZSM-5 catalysts: nature and role of gallium speciation. Angew. Chem., Int. Ed. 2020, 59, 19592–19601. 10.1002/anie.202007147. [DOI] [PubMed] [Google Scholar]
- Li M.; Zhai Y.; Zhang X.; Wang F.; Lv G.; Li M.; Zhang Q.; Liu Y. Ferric nitrate nonahydrate induced synthesis of hollow zeolite with high framework iron content. Microporous Mesoporous Mater. 2021, 327, 111410 10.1016/j.micromeso.2021.111410. [DOI] [Google Scholar]
- Zhang Y.; Zhou Y.; Huang L.; Xue M.; Zhang S. Sn-modified ZSM-5 as support for platinum catalyst in propane dehydrogenation. Ind. Eng. Chem. Res. 2011, 50, 7896–7902. 10.1021/ie1024694. [DOI] [Google Scholar]
- Inayat A.; Schneider C.; Schwieger W. Organic-free synthesis of layer-like FAU-type zeolites. Chem. Commun. 2015, 51, 279–281. 10.1039/C4CC07947G. [DOI] [PubMed] [Google Scholar]
- Wang L.; Sang S.; Meng S.; Zhang Y.; Qi Y.; Liu Z. Direct synthesis of Zn-ZSM-5 with novel morphology. Mater. Lett. 2007, 61, 1675–1678. 10.1016/j.matlet.2006.07.097. [DOI] [Google Scholar]
- Gabelica Z.; Valange S. Synthesis of MFI metallosilicate zeolites using metallic amino complexes as mineralizing agents: an overview. Microporous Mesoporous Mater. 1999, 30, 57–66. 10.1016/S1387-1811(99)00021-9. [DOI] [Google Scholar]
- Guo Y.; Sun T.; Gu Y.; Liu X.; Ke Q.; Wang S. Organic-Free, ZnO-Assisted Synthesis of Zeolite FAU with Tunable SiO2/Al2O3 Molar Ratio. Chem. - Asian J. 2018, 13, 1114–1118. 10.1002/asia.201800178. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Yu J.; Xu R. Needs and trends in rational synthesis of zeolitic materials. Chem. Soc. Rev. 2012, 41, 1729–1741. 10.1039/C1CS15150A. [DOI] [PubMed] [Google Scholar]
- Jain R.; Mallette A. J.; Rimer J. D. Controlling Nucleation Pathways in Zeolite Crystallization: Seeding Conceptual Methodologies for Advanced Materials Design. J. Am. Chem. Soc. 2021, 143, 21446–21460. 10.1021/jacs.1c11014. [DOI] [PubMed] [Google Scholar]
- De Yoreo J. J.; Gilbert P. U. P. A.; Sommerdijk N. A. J. M.; Penn R. L.; Whitelam S.; Joester D.; Zhang H.; Rimer J. D.; Navrotsky A.; Banfield J. F.; et al. Crystallization by particle attachment in synthetic, biogenic, and geologic environments. Science 2015, 349, aaa6760 10.1126/science.aaa6760. [DOI] [PubMed] [Google Scholar]
- Bouvy C.; Marine W.; Sporken R.; Su B.-L. Nanosized ZnO confined inside a Faujasite X zeolite matrix: Characterization and optical properties. Colloids Surf., A 2007, 300, 145–149. 10.1016/j.colsurfa.2006.12.043. [DOI] [Google Scholar]
- Koike N.; Chaikittisilp W.; Iyoki K.; Yanaba Y.; Yoshikawa T.; Elangovan S. P.; Itabashi K.; Okubo T. Organic-free synthesis of zincoaluminosilicate zeolites from homogeneous gels prepared by a co-precipitation method. Dalton Trans. 2017, 46, 10837–10846. 10.1039/C7DT02001E. [DOI] [PubMed] [Google Scholar]
- Suzuki Y.; Wakihara T.; Kohara S.; Itabashi K.; Ogura M.; Okubo T. Mechanistic study on the synthesis of a porous zincosilicate VPI-7 containing three-membered rings. J. Phys. Chem. C 2011, 115, 443–446. 10.1021/jp109373z. [DOI] [Google Scholar]
- Gao D.; Zhi Y.; Cao L.; Zhao L.; Gao J.; Xu C.; Ma M.; Hao P. Influence of zinc state on the catalyst properties of Zn/HZSM-5 zeolite in 1-hexene aromatization and cyclohexane dehydrogenation. Chin. J. Chem. Eng. 2022, 43, 124–134. 10.1016/j.cjche.2022.01.005. [DOI] [Google Scholar]
- Maldonado M.; Oleksiak M. D.; Chinta S.; Rimer J. D. Controlling Crystal Polymorphism in Organic-Free Synthesis of Na-Zeolites. J. Am. Chem. Soc. 2013, 135, 2641–2652. 10.1021/ja3105939. [DOI] [PubMed] [Google Scholar]
- Dusselier M.; Davis M. E. Small-pore zeolites: synthesis and catalysis. Chem. Rev. 2018, 118, 5265–5329. 10.1021/acs.chemrev.7b00738. [DOI] [PubMed] [Google Scholar]
- Degnan T. F. Jr The implications of the fundamentals of shape selectivity for the development of catalysts for the petroleum and petrochemical industries. J. Catal. 2003, 216, 32–46. 10.1016/S0021-9517(02)00105-7. [DOI] [Google Scholar]
- Van Tendeloo L.; Gobechiya E.; Breynaert E.; Martens J. A.; Kirschhock C. E. A. Alkaline cations directing the transformation of FAU zeolites into five different framework types. Chem. Commun. 2013, 49, 11737–11739. 10.1039/c3cc47292b. [DOI] [PubMed] [Google Scholar]
- Strachan D. M.; Neeway J. J. Effects of alteration product precipitation on glass dissolution. Appl. Geochem. 2014, 45, 144–157. 10.1016/j.apgeochem.2014.03.013. [DOI] [Google Scholar]
- Oleksiak M. D.; Ghorbanpour A.; Conato M. T.; McGrail B. P.; Grabow L. C.; Motkuri R. K.; Rimer J. D. Synthesis Strategies for Ultrastable Zeolite GIS Polymorphs as Sorbents for Selective Separations. Chem. - Eur. J. 2016, 22, 16078–16088. 10.1002/chem.201602653. [DOI] [PubMed] [Google Scholar]
- Fricke R.; Kosslick H.; Lischke G.; Richter M. Incorporation of gallium into zeolites: syntheses, properties and catalytic application. Chem. Rev. 2000, 100, 2303–2406. 10.1021/cr9411637. [DOI] [PubMed] [Google Scholar]
- Anseau M. R.; Leung J. P.; Sahai N.; Swaddle T. W. Interactions of Silicate Ions with Zinc(II) and Aluminum(III) in Alkaline Aqueous Solution. Inorg. Chem. 2005, 44, 8023–8032. 10.1021/ic050594c. [DOI] [PubMed] [Google Scholar]
- Singh R.; Dutta P. K. Stabilization of natural Faujasite zeolite: possible role of alkaline earth metal ions. Microporous Mesoporous Mater. 1998, 21, 103–109. 10.1016/S1387-1811(97)00055-3. [DOI] [Google Scholar]
- Swaddle T. W. Silicate complexes of aluminum (III) in aqueous systems. Coord. Chem. Rev. 2001, 219–221, 665–686. 10.1016/S0010-8545(01)00362-9. [DOI] [Google Scholar]
- Knight C. T. G.; Balec R. J.; Kinrade S. D. The Structure of Silicate Anions in Aqueous Alkaline Solutions. Angew. Chem. 2007, 119, 8296–8300. 10.1002/ange.200702986. [DOI] [PubMed] [Google Scholar]
- Mora-Fonz M. J.; Hamad S.; Catlow C. R. A. Modelling nucleation and nano-particle structures. Mol. Phys. 2007, 105, 177–187. 10.1080/00268970701211626. [DOI] [Google Scholar]
- Freeman E. E.; Neeway J. J.; Motkuri R. K.; Rimer J. D.; Mpourmpakis G. Understanding initial zeolite oligomerization steps with first principles calculations. AIChE J. 2020, 66, e17107 10.1002/aic.17107. [DOI] [Google Scholar]
- Ding Z.; Yin Z.; Hu H.; Chen Q. Comparison of thermal property and dissolution behavior of synthetic compound and natural hemimorphite. Thermochim. Acta 2010, 511, 168–173. 10.1016/j.tca.2010.08.010. [DOI] [Google Scholar]
- Göltl F.; Conrad S.; Wolf P.; Müller P.; Love A. M.; Burt S. P.; Wheeler J. N.; Hamers R. J.; Hummer K.; Kresse G.; et al. UV–Vis and photoluminescence spectroscopy to understand the coordination of Cu cations in the zeolite SSZ-13. Chem. Mater. 2019, 31, 9582–9592. 10.1021/acs.chemmater.9b01439. [DOI] [Google Scholar]
- Libowitzky E.; Kohler T.; Armbruster T.; Rossman G. R. Proton disorder in dehydrated hemimorphite-IR spectroscopy and X-ray structure refinement at low and ambient temperatures. Eur. J. Mineral. 1997, 9, 803–810. 10.1127/ejm/9/4/0803. [DOI] [Google Scholar]
- Downward L.; Booth C. H.; Lukens W. W.; Bridges F. In A variation of the F-test for determining statistical relevance of particular parameters in EXAFS Fits, American Institute of Physics Conference; American Institute of Physics, 2007; Vol. 882, pp 129–131.
- Joyner R. W.; Smith A. D.; Stockenhuber M.; van den Berg M. W. E. The local structure of aluminium sites in zeolites. Phys. Chem. Chem. Phys. 2004, 6, 5435–5439. 10.1039/b411001c. [DOI] [Google Scholar]
- Stoiber D.; Niewa R. PolyDis: simple quantification tool for distortion of polyhedra in crystalline solids. Z. Kristallogr. - Cryst. Mater. 2019, 234, 201–209. 10.1515/zkri-2018-2115. [DOI] [Google Scholar]
- Sharma P.; Yeo J.-g.; Han M. H.; Cho C. H. Knobby surfaced, mesoporous, single-phase GIS-NaP1 zeolite microsphere synthesis and characterization for H 2 gas adsorption. J. Mater. Chem. A 2013, 1, 2602–2612. 10.1039/c2ta01311h. [DOI] [Google Scholar]
- Subotić B.; Šmit I.; Madžija O.; Sekovanić L. Kinetic study of the transformation of zeolite A into zeolite P. Zeolites 1982, 2, 135–142. 10.1016/S0144-2449(82)80015-8. [DOI] [Google Scholar]
- Zaarour M.; Dong B.; Naydenova I.; Retoux R.; Mintova S. Progress in zeolite synthesis promotes advanced applications. Microporous Mesoporous Mater. 2014, 189, 11–21. 10.1016/j.micromeso.2013.08.014. [DOI] [Google Scholar]
- Sathupunya M.; Gulari E.; Wongkasemjit S. ANA and GIS zeolite synthesis directly from alumatrane and silatrane by sol-gel process and microwave technique. J. Eur. Ceram. Soc. 2002, 22, 2305–2314. 10.1016/S0955-2219(02)00042-0. [DOI] [Google Scholar]
- Ali I. O.; El-Sheikh S. M.; Salama T. M.; Bakr M. F.; Fodial M. H. Controllable synthesis of NaP zeolite and its application in calcium adsorption. Sci. China Mater. 2015, 58, 621–633. 10.1007/s40843-015-0075-9. [DOI] [Google Scholar]
- Pal P.; Das J. K.; Das N.; Bandyopadhyay S. Synthesis of NaP zeolite at room temperature and short crystallization time by sonochemical method. Ultrason. Sonochem. 2013, 20, 314–321. 10.1016/j.ultsonch.2012.07.012. [DOI] [PubMed] [Google Scholar]
- Garcia G.; Cardenas E.; Cabrera S.; Hedlund J.; Mouzon J. Synthesis of zeolite Y from diatomite as silica source. Microporous Mesoporous Mater. 2016, 219, 29–37. 10.1016/j.micromeso.2015.07.015. [DOI] [Google Scholar]
- Muraoka K.; Chaikittisilp W.; Okubo T. Energy analysis of aluminosilicate zeolites with comprehensive ranges of framework topologies, chemical compositions, and aluminum distributions. J. Am. Chem. Soc. 2016, 138, 6184–6193. 10.1021/jacs.6b01341. [DOI] [PubMed] [Google Scholar]
- Oleksiak M. D.; Muraoka K.; Hsieh M.-F.; Conato M. T.; Shimojima A.; Okubo T.; Chaikittisilp W.; Rimer J. D. Organic-Free Synthesis of a Highly Siliceous Faujasite Zeolite with Spatially Biased Q4 (nAl) Si Speciation. Angew. Chem. 2017, 129, 13551–13556. 10.1002/ange.201702672. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A. Gaussian 09, Revision D. 01, Gaussian, Inc., Wallingford, CT, 2009.
- Wang Y.; Verma P.; Jin X.; Truhlar D. G.; He X. Revised M06 density functional for main-group and transition-metal chemistry. Proc. Natl. Acad. Sci. 2018, 115, 10257–10262. 10.1073/pnas.1810421115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone V.; Cossi M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. 10.1021/jp9716997. [DOI] [Google Scholar]
- Trinh T. T.; Jansen A. P. J.; van Santen R. A. Mechanism of oligomerization reactions of silica. J. Phys. Chem. B 2006, 110, 23099–23106. 10.1021/jp063670l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.