

Abstract
Most primary thyroid tumours are of epithelial origin. Primary thyroid mesenchymal tumours are rare but are being increasingly detected. A vast majority of thyroid mesenchymal tumours occur between the fourth and seventh decades of life, presenting as progressively enlarging thyroid nodules that often yield non-diagnostic results or spindle cells on fine needle aspiration biopsy. Surgery is the preferred mode of treatment, with adjuvant chemoradiotherapy used for malignant thyroid mesenchymal tumours. Benign thyroid mesenchymal tumours have excellent prognosis, whereas the outcome of malignant thyroid mesenchymal tumours is variable. Each thyroid mesenchymal tumour is characterised by its unique histopathology and immunohistochemistry. Because of the rarity and aggressive nature of malignant thyroid mesenchymal tumours, a multidisciplinary team-based approach should ideally be used in the management of these tumours. Comprehensive guidelines on the management of thyroid mesenchymal tumours are currently lacking. In this Review, we provide a detailed description of thyroid mesenchymal tumours, their clinical characteristics and tumour behaviour, and provide recommendations for the optimal management of these tumours.
Introduction
Palpable thyroid nodules are prevalent in up to 5% of women and 1% of men in iodine-sufficient regions,1 and up to 25% of individuals in endemic-goitre regions.2 Incidental thyroid nodules are detected by ultrasonography in about 20–76% of the general population.3 Fine needle aspiration biopsy is considered in nodules that meet sonographic characteristics and size criteria according to the guidelines of the American Thyroid Association or American College of Radiology.1,4,5 The Bethesda system for reporting thyroid cytopathology is widely used to determine the likelihood of malignancy in a fine needle aspiration biopsy sample.1,6 Based on the cytopathological category, management options include clinical follow-up, repeat fine needle aspiration biopsy, molecular diagnostics, lobectomy, or total thyroidectomy.1,6
The most common form of benign thyroid nodule encountered on histopathological examination is a hyperplastic nodule (also known as adenomatous, adenomatoid, or follicular nodules), whereas the most common benign thyroid neoplasm is follicular adenoma.6 The most common forms of thyroid cancer are the differentiated thyroid cancers, which include papillary thyroid carcinoma and follicular thyroid carcinoma, which comprise more than 90% of cases of thyroid cancer. Medullary thyroid carcinoma comprises 1–5% and anaplastic thyroid carcinoma comprises 1∙3–9∙8% of thyroid cancers, and both are rarer than differentiated thyroid cancers.7,8 Although robust guidelines exist for the diagnosis, risk stratification, treatment, and follow-up of typical thyroid tumours that are of epithelial origin,1,8 insufficient data for thyroid tumours of mesenchymal origin remain. The goal of this Review is to summarise the current knowledge on thyroid mesenchymal tumours and analyse patterns of clinical presentation, diagnostic procedures, and therapeutic approaches and their associated outcomes in order to propose recommendations for management of these tumours.
Thyroid tumours of mesenchymal origin
Mesenchymal tissues are formed by mesenchymal cells (ie, cells derived predominantly from embryonic mesoderm and, to a small extent, from embryonic ectoderm), which interact with epithelial tissue to form nearly every organ in the body.9 Mesenchymal cells eventually give rise to connective tissues, bones, cartilage, striated and smooth muscles, and the circulatory and lymphatic systems.6,9 A thyroid mesenchymal tumour is defined as a tumour that arises from the thyroid mesenchymal tissue, that is, the stromal and blood vessel elements of the thyroid gland.6 In the 2017 WHO guidelines on the classification of thyroid tumours, thyroid mesenchymal tumours are grouped under “paraganglioma and stromal/mesenchymal tumors”.10 Although robust epidemiological data for the incidence and prevalence of thyroid mesenchymal tumours are currently lacking, these tumours comprise approximately 0∙3% of all malignant thyroid tumours.11 However, over the past few decades, thyroid mesenchymal tumours have been increasingly reported.12,13 More than 30 different types of thyroid mesenchymal tumours have been described in the literature (panel). A systematic review of data from 142 patients with primary malignant thyroid mesenchymal tumours between 1990 and 2014 revealed angiosarcomas in 20·4% of patients, epithelioid haemangioendothelioma in 16·3%, undifferentiated pleomorphic sarcoma (previously known as malignant fibrous histiocytoma) in 14·1%, leiomyosarcoma in 11·3%, and fibrosarcoma in 9·2%.14 However, metastatic mesenchymal tumours into the thyroid (from gastrointestinal stromal tumours, uterine leiomyosarcomas, and malignant breast phyllodes tumour) are more common than primary thyroid mesenchymal tumours.6 The focus of this Review is solely on primary thyroid mesenchymal tumours.
Data collection
Search strategy and selection criteria
We searched PubMed, Embase, and MEDLINE. Data were reviewed from full-length articles, including case reports, case series, observational retrospective studies, systematic reviews, and meta-analyses to identify thyroid mesenchymal tumours reported on any date before Jan 1, 2020. We used the search terms “thyroid mesenchymal tumor”, “thyroid stromal tumor”, “thyroid sarcoma”, and “mesenchymal tumors of the thyroid gland”. For the individual thyroid mesenchymal tumours, we coupled the word “thyroid” and the name of the specific thyroid mesenchymal tumour; for example “thyroid paraganglioma” or “thyroid chondrosarcoma”. We also used an alternative search phrase for specific thyroid mesenchymal tumours, which was “TMT name” followed by the phrase “of the thyroid”; for example, “paraganglioma of the thyroid” or “chondrosarcoma of the thyroid”. Older literature used terminologies that are no longer valid, such as haemangiopericytoma and malignant fibrous histiocytoma. In these situations, we grouped these reports with those that used updated terminologies in order to count the total number of cases. We excluded haematological malignancies and lymphomas, which are separately categorised as haematolymphoid tumours under the WHO 2017 classification.10 We excluded reports on thyroid lymphangioma, a condition previously considered as a thyroid mesenchymal tumour, now reclassified as a vascular malformation. Spindle epithelial tumours with thymus-like differentiation, which predominantly contains epithelial components, was also excluded. The final reference list was created based on the originality of the articles as well as their relevance to the broad scope of this Review. We did not apply any language restriction for our search.
Search results
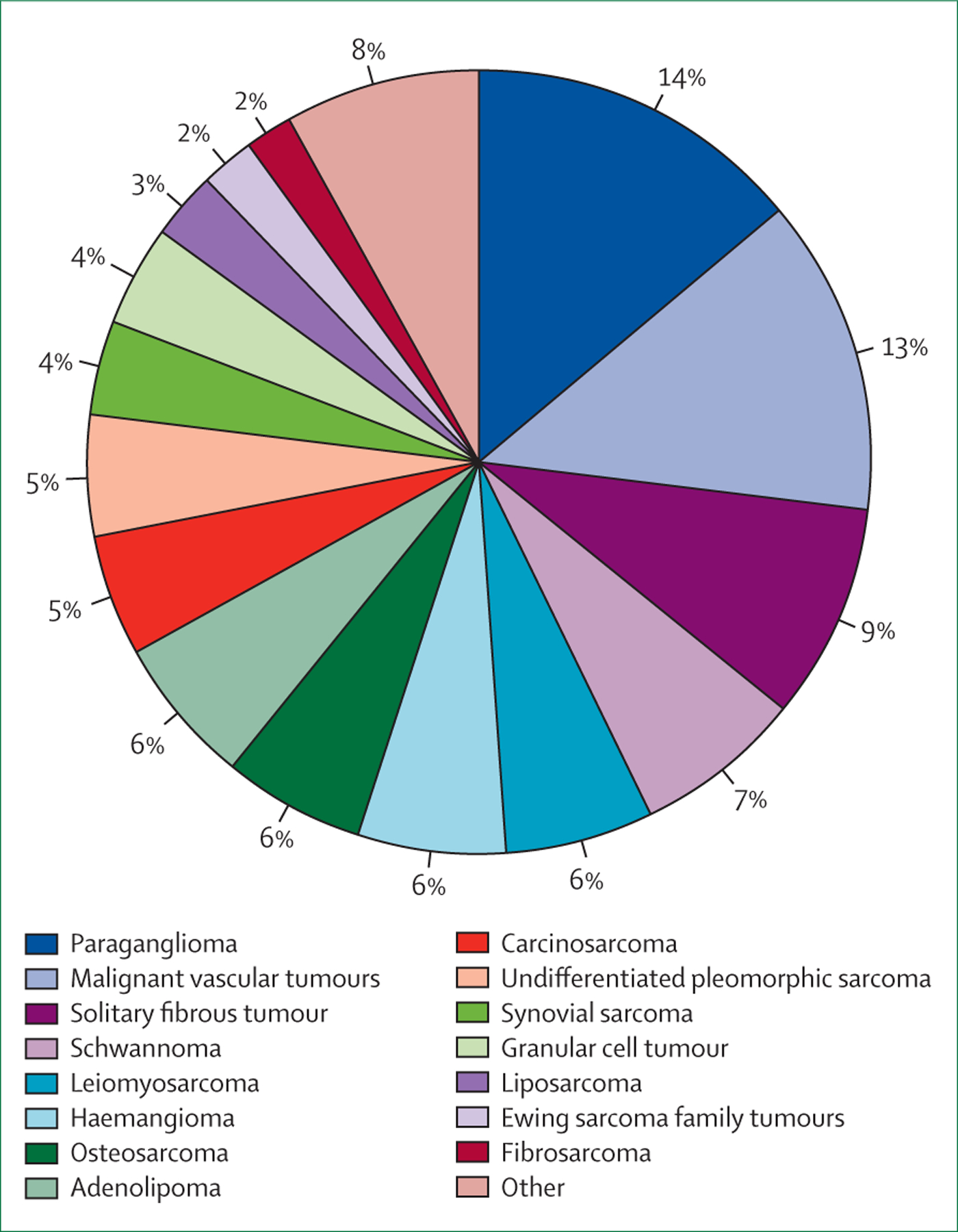
In total, we identified at least 530 cases of thyroid mesenchymal tumours described in the literature (appendix pp 1–10), out of which about 290 (55%) thyroid mesenchymal tumours were malignant, either manifesting as locally aggressive disease or as distant metastases. The vast majority of thyroid mesenchymal tumours were represented by paragangliomas (76 [14%] of 530 cases), followed by malignant vascular tumours, which include angiosarcomas and epithelioid haemangioendotheliomas (68 [13%]), solitary fibrous tumours or haemangiopericytomas (47 [9%]), benign schwannomas (37 [7%]), leiomyosarcomas (32 [6%]), haemangiomas (31 [6%]), osteosarcomas (31 [6%]), and adenolipomas (30 [6%]; figure 1). A comprehensive tabulated summary of each thyroid mesenchymal tumour is provided in the appendix (pp 1–4).
Figure 1: Frequency of occurrence of primary thyroid mesenchymal tumours identified in the literature search.
Clinical features
A vast majority of thyroid mesenchymal tumours occur between the fourth and seventh decades of life; however, these tumours have also been described at the extremes of age, ranging from 7 months to 97 years.12,14–16 Several thyroid mesenchymal tumours, including paragangliomas, adenolipomas, leiomyomas, schwannomas, malignant peripheral nerve sheath tumours, granular cell tumours, angiosarcomas, leiomyosarcomas, undifferentiated pleomorphic sarcomas, and carcinosarcomas are predominantly observed in females, whereas rhabdomyosarcomas, synovial sarcomas, and the Ewing sarcoma family of tumours are more frequently observed in males, and the rest of the thyroid mesenchymal tumours do not have a clear gender predilection (appendix pp 1–4).14,15,17,18 We did not observe any significant association between family history and incidence of thyroid mesenchymal tumours. There are reports on thyroid mesenchymal tumours such as liposarcoma, angiosarcoma, and fibrosarcoma developing after exposure to radiation.19–21 Angiosarcoma has also been associated with a history of exposure to vinyl chloride and thorium dioxide.21 Kaposi’s sarcoma is likely to be found in patients with HIV infection, although its occurrence in an HIV-negative patient has also been reported.22 In addition, malignant vascular tumours show a unique geographical distribution, with a majority of cases occurring in the iodine-deficient Alpine regions of Europe, and comprise up to 15–20% of all thyroid malignancies in the mountainous regions of Switzerland, Austria, and northern Italy.21
The most common clinical presentation of thyroid mesenchymal tumours is in the form of a progressively enlarging neck mass, often associated with compressive symptoms such as dysphagia, globus sensation, or hoarseness of voice.14,23,24 A more rapid growth along with systemic symptoms such as weight loss can be observed with malignant thyroid mesenchymal tumours.25–27 Although the size tends to vary, many thyroid mesenchymal tumours reach 5–6 cm in diameter.14 Certain benign thyroid mesenchymal tumours such as adenolipoma and haemangioma have been reported to grow over 20 cm.24,28 Most individuals with thyroid mesenchymal tumours are clinically euthyroid. Only few patients have shown histological evidence of concomitant autoimmune thyroiditis.29 Among patients with thyroid paraganglioma, either sporadic or associated with familial syndromes, plasma or 24-h urine metanephrines are usually reported as normal.30
Imaging characteristics
The features of thyroid sarcomas on routine imaging studies, including ultrasonography, CT, MRI, and functional imaging studies are similar to that of the more common forms of thyroid nodules.1 In a cohort of 8849 patients with various thyroid disorders treated between 1997 and 2013 at a medical centre in Germany, thyroid sarcomas were identified in 0∙1% of the patients, and ultrasonography features of thyroid sarcomas in 90% of these patients were described as inhomogeneous hypoechoic to hyperechoic lesions, of which 80% had indistinct margins.31 On CT scan, the tumours were hypodense, and on MRI the tumours showed inhomogeneous increase in signal on T2-weighted images and a decrease in signal with inhomogeneous enhancement on T1-weighted images.31 In another study consisting of 142 patients with primary malignant thyroid mesenchymal tumours, sonographic data from 36 patients revealed that these tumours had a mixed hypoechoic to hyperechoic appearance in 58%, and hypoechoic in 36%, of patients.14 Among the 31 patients who received a CT scan, the thyroid mesenchymal tumours appeared as heterogeneous hypodense to hyperdense masses in 59%, isodense in 17%, and hypodense in 24% of patients.14 Data for the use of functional imaging such as 18F-fluorodeoxyglucose PET-CT in the evaluation of malignant thyroid mesenchymal tumours is exceedingly rare.32–34 Moreover, data for the use of other functional imaging modalities used to diagnose paragangliomas, such as gallium-68 dodecanetetraacetic acid-DPhe1, Tyr3-octreotate (68Ga-DOTATATE) PET–CT, or 18-fluorodeoxyphenylalanine (18F-DOPA) PET–CT, in the diagnosis of thyroid paraganglioma are also sparse.35,36 One study reported the performance of various functioning imaging studies, including 68Ga-DOTATATE PET–CT, 18F-DOPA PET–CT, iodine-131 meta-iodobenzylguanidine (¹³¹I-MIBG) scan, and indium-111 octreotate scintigraphy postoperatively in four patients who had undergone either total or hemithyroidectomy for non-metastatic thyroid paraganglioma, and the results were normal in all of these scans.30 However, the utility of functional imaging has been documented in patients with extrathyroidal paragangliomas.
Histopathology, immunohistochemistry, and molecular landscape
On fine needle aspiration biopsy, cytological results have been reported across all classes of the Bethesda reporting system, ranging from non-diagnostic (category I) to malignant (category VI); however, most reports of fine needle aspiration biopsy have been either non-diagnostic or have described spindle cells on cytology.26,37–39 The differential diagnoses of thyroid mesenchymal tumours include medullary thyroid carcinoma, anaplastic thyroid carcinoma, and its atypical histological variants, Hurtle cell neoplasms, lymphoma, squamous cell carcinoma, and spindle cell lesions in the thyroid gland, such as the spindle cell variants of papillary thyroid carcinoma, anaplastic thyroid carcinoma, hyalinising trabecular tumour, spindle epithelial tumour with thymus-like differentiation, Riedel thyroiditis, and reactive lesions such as post-fine needle aspiration biopsy spindle cell nodules.40 It is important to do a flow cytometry analysis in the lymphocyte-rich cytology specimens to assess for lymphoma, as the management strategy of lymphoma is substantially different to the therapy of other solid thyroid tumours, with chemotherapy being the first-line treatment modality. By contrast, a diagnostic surgery might have a key role in differential diagnosis of thyroid mesenchymal tumours, as a clear distinction between thyroid mesenchymal tumours and other more common thyroid nodules can be successfully made only through histopathology and immunohistochemistry in most cases.
A summary of histopathological and immunohistochemistry findings of thyroid mesenchymal tumours along with the differential diagnoses is presented in the appendix (pp 1–4). Malignant thyroid mesenchymal tumours might show extensive fibrosis, evidence of extracapsular invasion, or firm adhesion to thyroid parenchyma or adjacent structures (eg, the trachea or oesophagus).14 Histological variants of anaplastic thyroid carcinoma (eg, the sarcomatoid or paucicellular, and rhabdoid variants) can mimic malignant thyroid mesenchymal tumours and present a diagnostic challenge. Malignant thyroid mesenchymal tumours can be differentiated from these atypical anaplastic thyroid carcinoma variants by the lack of a well differentiated thyroid carcinoma component, absence of neoplastic cell infiltration of large-sized vessels, absence of palisading necrosis, absence of epithelial markers on immunohistochemistry, and identification of an extrathyroidal primary mesenchymal tumour in cases of metastatic disease.41 Occasionally, keratins can be expressed by synovial sarcomas or malignant vascular tumours, but the presence of PAX8 and p63 on immunohistochemistry favours a diagnosis of anaplastic thyroid carcinoma.42 Calcitonin-negative and carcinoembryonic antigen-negative medullary thyroid carcinomas can histologically mimic paragangliomas. However, the morphological features of paraganglioma do not often overlap with those of medullary thyroid carcinomas, and a panel of immunohistochemistry markers that include TTF-1 for medullary thyroid carcinoma in addition to calcitonin and carcinoembryonic antigen, and GATA3 and SOX10 for paraganglioma would be helpful in distinguishing calcitonin-negative and carcinoembryonic antigen-negative medullary thyroid carcinomas from paragangliomas.7,43,44 Molecular analysis of the specimen might help distinguish between paraganglioma and calcitonin-negative and carcinoembryonic antigen-negative de-differentiated medullary thyroid carcinoma, as the former is characterised by somatic mutations in the SDHx genes, whereas medullary thyroid carcinoma does not harbour SDHx variants but contains RET and RAS pathogenic variants instead.7,30
Although the molecular landscape of thyroid epithelial tumours is well studied, this information about thyroid mesenchymal tumours remains largely unknown, mainly due to the paucity of molecular analysis data among reported cases. Molecular analysis of primary thyroid tumours of epithelial origin has categorised them into BRAF-like and RAS-like tumours.45 The BRAF-like tumours harbour BRAFV600E (ie, Val600Glu) as the primary driving mutation (60% of all pathogenic variants in papillary thyroid carcinoma), and are characterised by classic papillary thyroid carcinoma morphology and high MAPK-pathway signalling, whereas the RAS-like tumours harbouring RAS and other non-BRAF mutations are characterised by a follicular morphology (follicular variant of papillary thyroid carcinoma, follicular thyroid carcinoma), and low MAPK-pathway signalling.45 However, the pathways affected in mesenchymal tumours can be distinct from the ones affected in thyroid epithelial tumours (appendix pp 11–12).
Several mesenchymal tumours have pathogenic variants in genes involved in the Akt and mTOR pathway and in genes involved in crosstalk with the hippo pathway, controlling cell proliferation and apoptosis.46,47 Paragangliomas are characterised by the presence of somatic mutations in the SDHx genes (appendix pp 11–12). For instance, molecular analysis of a thyroid paraganglioma tumour in a 39-year-old woman identified several single nucleotide polymorphisms in SDHB, SDHC, and SDHD genes.48 Gene fusion is another commonly observed molecular defect in mesenchymal tumours, such as NAB2-STAT6 fusion in solitary fibrous tumours, EWSR1-ETS family gene-related fusions in the Ewing sarcoma family of tumours, SYT-SSX fusion in synovial sarcomas, WWTR1-CAMTA1 and YAP1-TFE3 fusions in epithelioid haemangioendotheliomas, FUS-DDIT3 and EWSR1-DDIT3 fusions in myxoid liposarcomas, ASPSCR1-TFE3 fusions in alveolar soft part sarcomas, gene fusions between NR4A3 and EWSR1, TAF15, TCF12, or TFG in myxoid chondrosarcomas, and HEY1-NCOA2 fusion in mesenchymal chondrosarcomas (appendix pp 1–4, 11–12).12,49–56 In addition, a DICER1 (p.Glu1705Lys) pathogenic variant was identified in the lung metastases in a 45-year-old female with an aggressive form of primary thyroid carcinosarcoma.57 A comprehensive genomic analysis of a tumour and its surrounding normal tissues, as well as patient-matched blood in a 44-year-old woman with a thyroid follicular dendritic cell sarcoma identified 81 somatic variants exclusively in the tumour, including a truncating variant in the CLTCL1 gene, which is involved in mitotic spindle stabilisation, missense variants in ATM and TP53, and a splice-site variant in VEGFR1.58 Additionally, a local chromothripsis event between chromosomes 15 and 17 was identified in the same tumour, comprising more than 24 structural variants eventually resulting in expression of six fusion genes. Certain benign thyroid mesenchymal tumours such as adenolipomas can be associated with pathogenic variants in the PTEN gene.59 Germline analysis in a 28-year-old woman with multiple thyroid adenolipomas revealed a heterozygous deletion of adenine (827delA) in exon 8 of the PTEN gene.59
About 40% of paragangliomas might present as an inherited disorder associated with one of 14 susceptibility genes (MEN1, NF1, RET, VHL, SDHA, SDHB, SDHC, SDHD, SDHAF2, TMEM127, EGLN1, HIF2A, KIF1Bb, and MAX).13 Among thyroid paragangliomas, the pathogenic variants in SDHB and SDHA are the most commonly observed. Molecular analysis in five patients with thyroid paragangliomas identified from a population-based registry identified germline variants in SDHx in four (80%) of the five patients, with two patients harbouring variants in SDHB (c.530G→A, p.Arg177His; and c.201–1339_239delinsAluYb8), and two other patients harbouring variants in SDHA (c.394T→C, p.Trp132Arg; and c.1799G→A, p.Arg600Gln), and insilico analyses showed that these variants were likely to be pathogenic.30
Treatment and prognosis
Benign thyroid mesenchymal tumours (adenolipomas, leiomyomas, benign peripheral nerve sheath tumours, and haemangiomas) have a favourable prognosis and are cured with either hemithyroidectomy or total thyroidectomy, and there have been no reported recurrences after surgical excision.60,61 Thyroid paragangliomas can manifest as locally invasive disease (either thyroid capsule adherence or extension into adjacent structures) in about 30% of cases, or rarely (one case report) as a metastatic disease with hepatic and peritoneal metastases.13,62,63 All patients with locally invasive disease were successfully treated with aggressive surgery without documented recurrence, even in the familial forms, except for one patient with SDHA c.1799G→A (p.Arg600Gln) germline variant who had local recurrence 12 years after thyroidectomy.30,62 One patient underwent adjuvant peptide receptor radionuclide therapy after surgical removal of a locally aggressive thyroid paraganglioma without distant metastases, and no evidence of recurrent disease upon 3 years of follow-up was observed.64 Most thyroid solitary fibrous tumours are benign and are surgically cured, with recurrent and metastatic disease being reported only in two patients.12 Similarly, almost all reported thyroid granular cell tumours were benign, with locally aggressive disease reported in one patient who was eventually cured with surgery.18
When it comes to frankly malignant thyroid mesenchymal tumours, treatment and prognostication can become more complex. On the basis of a systematic review by Surov and colleagues,14 evaluating primary malignant thyroid mesenchymal tumours, up to 70·4% of patients with malignant thyroid mesenchymal tumours can harbour disease within the thyroid without evidence of local or distant metastases. Based on the same report, evidence of infiltration into the trachea or oesophagus was frequently observed in patients with undifferentiated pleomorphic sarcoma (up to 70%) and with liposarcoma (up to 57%). Lymph node metastases are frequently observed with patients in fibrosarcoma (12%) and epithelioid haemangioendothelioma (10·5%), while distant metastases were more common in patients with leiomyosarcoma (20%), osteosarcoma (16·7%), and angiosarcoma (15·4%).14 Among the reports with available data for treatment and follow-up, surgery was used as the sole treatment option in 75 (52·8%) patients, and a combination of surgery with or without chemoradiotherapy was used in 53 (37·3%) patients.14 Observation time in these patients ranged from 0∙5 months to 120 months, during which 45 (31·7%) patients were alive and 73 (51·7%) died.14 Adjuvant radiotherapy has shown excellent efficacy as indicated by several case reports of thyroid malignant vascular tumours, with one study showing local control in 92% of patients who were treated with radiotherapy, often in combination with razoxane.21,65 Doxorubicin, ifosfamide, epirubicin, and taxanes are chemotherapeutic agents commonly used in the treatment of thyroid malignant vascular tumours, either in a neoadjuvant or an adjuvant setting.21 In addition, other regimens have been used in the treatment of malignant thyroid mesenchymal tumours, including doxorubicin and ifosfamide along with radiotherapy for liposarcoma and leiomyosarcoma,66,67 cisplatin and doxorubicin for osteosarcoma,32 and adjuvant or neoadjuvant cyclophosphamide, ifosfamide, doxorubicin, vincristine, and etoposide with or without radiotherapy for the Ewing sarcoma family of tumours.17 On the one hand, liposarcomas, synovial sarcomas, and the Ewing sarcoma family of tumours are associated with relatively favourable long-term outcomes after surgery and chemoradiotherapy in most patients,51,66,68 whereas on the other hand, malignant peripheral nerve sheath tumours, malignant vascular tumours, osteosarcomas, leiomyosarcomas, undifferentiated pleomorphic sarcomas, and carcinosarcomas are associated with poor outcomes and high mortality due to local disease burden or distant metastasis, or both (appendix pp 1–4).21,27,67,69–71
Guidance on diagnosis and management
Figure 2 summarises the key points that can be used in the management of thyroid mesenchymal tumours. These recommendations are based on the data described in this Review and on expert opinion of the authors. The initial evaluation and management of a thyroid mesenchymal tumour nodule should be done based on findings from ultrasonography and fine needle aspiration biopsy as per the American Thyroid Association guidelines.1 Apart from thyroid function tests and autoimmune antibody (anti-thyroid peroxidase and anti-thyroglobulin) evaluation as routine pre-surgical laboratory investigations, all authors of this Review believe that serum calcitonin should be measured in those patients whose thyroid nodules show spindle cells on a fine needle aspiration biopsy sample in order to evaluate for medullary thyroid carcinoma. On the basis of association between calcitonin concentration and loco-regional and distant metastases, the disease burden could be predicted guiding additional imaging with CT and MRI.7 The cytology specimen should also be stained for calcitonin and carcinoembryonic antigen if the morphology is not entirely typical for medullary thyroid carcinoma. In non-diagnostic aspirates, a repeat fine needle aspiration biopsy, a core biopsy, a diagnostic surgery or surgical biopsy, or serial monitoring through repeat ultrasonography can be considered based on the previous ultrasound features of the lesion, duration and type of symptoms, as well as patient’s age and comorbidities. If the cytology specimen shows an abundance of lymphocytes, flow cytometry needs to be done to rule in or rule out thyroid lymphoma, particularly in patients with a history of Hashimoto’s thyroiditis. If the fine needle aspiration biopsy shows cytologic features suggesting a paraganglioma, further biochemical testing in the form of plasma or 24-h urine metanephrines, or both, should be undertaken, and pre-operative adrenergic blockade should be implemented if there is over-production of catecholamines. Furthermore, CT, MRI, or functional imaging studies such as a 68Ga-DOTATATE PET–CT, 18F-DOPA PET–CT, or an 123I-MIBG scan could be considered for the evaluation of extrathyroidal paragangliomas and phaeochromocytomas.35,36 Although 18F-DOPA PET–CT has shown promising results for the diagnosis of head and neck paragangliomas,36 68Ga-DOTATATE PET–CT might be superior for diagnosing metastatic paragangliomas.35 Therefore, clinical judgement should be applied in choosing the optimal functional imaging while also taking into consideration the availability of these studies in a given geographical region.
Figure 2: Key points on the diagnosis and management of primary thyroid mesenchymal tumours.
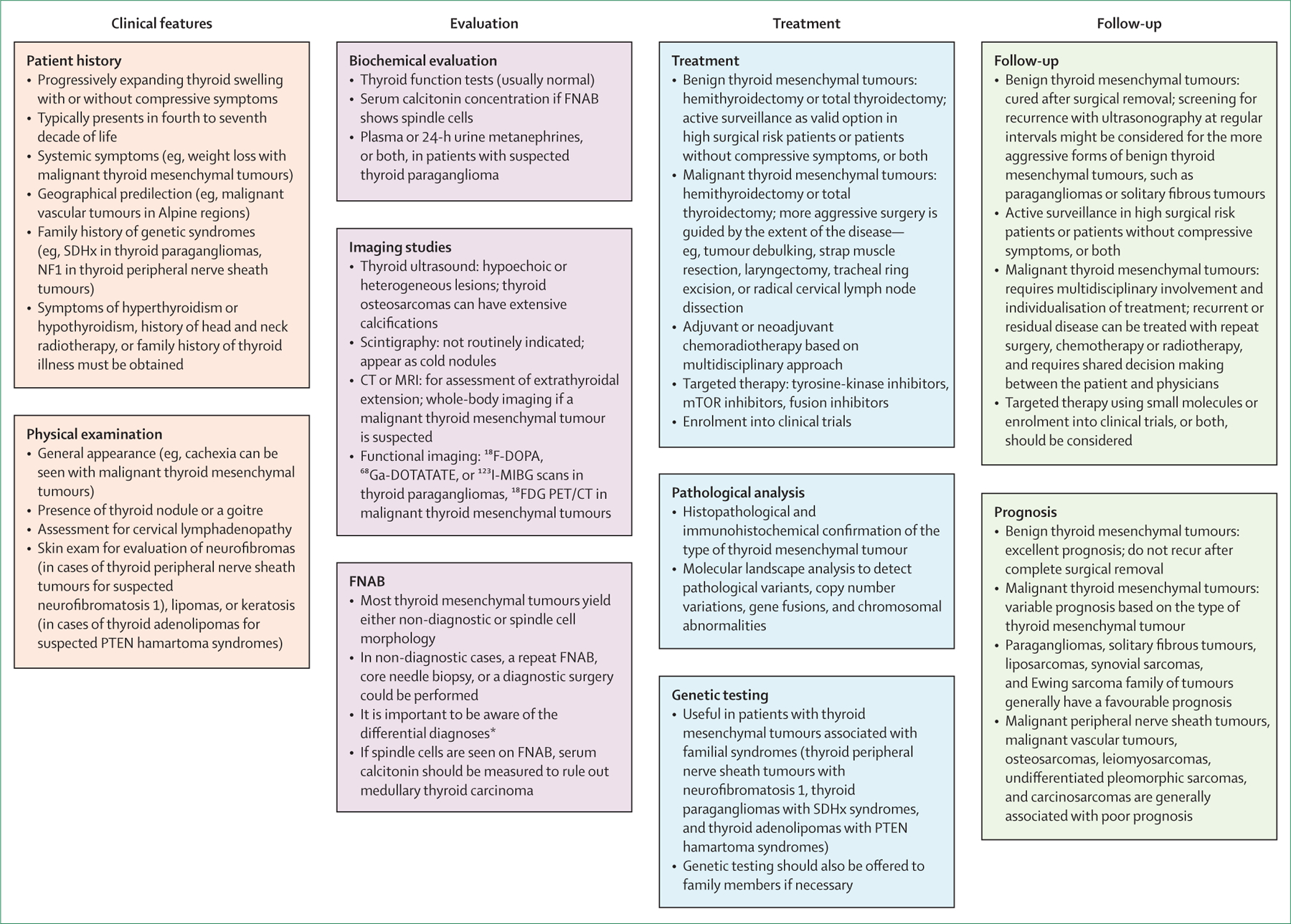
18FDG=18F-fluorodeoxyglucose. 18F-DOPA=18-fluorodeoxyphenylalanine. 68Ga-DOTATATE=gallium-68 dodecanetetraacetic acid-DPhe1, Tyr3-octreotate. 123I-MIBG=iodine-123 metaiodobenzylguanidine. FNAB=fine needle aspiration biopsy. *Differential diagnoses: spindle cell variants of thyroid carcinomas, medullary thyroid carcinoma, squamous cell carcinoma, hyalinising trabecular tumour, spindle epithelial tumour with thymus-like differentiation, Riedel thyroiditis, lymphoma, and reactive lesions such as post-FNAB spindle cell nodules.
Genetic testing for germline pathogenic variants should be offered to all patients with thyroid paragangliomas.72 The most efficient way to achieve this outcome is with a multi-gene panel that includes sequencing and deletion or duplication analysis of SDHB, SDHC, SDHD, SDHAF2, SDHA, VHL, and TMEM127.30 Patients with malignant peripheral nerve sheath tumours should be evaluated for neurofibromatosis 1 (von Recklinghausen’s disease) and neurofibromatosis 2 syndromes, and germline genetic testing of NF1 and NF2 might be indicated. Similarly, patients with adenolipomas of the thyroid should be evaluated for PTEN hamartoma syndromes, and potentially be offered germline sequencing and deletion or duplication analysis of the PTEN gene, if the syndrome is suspected. The Cleveland Clinic’s PTEN Risk Calculator is a helpful tool to evaluate the likelihood of this syndrome in a patient.73 Germline genetic testing might be also offered, if the tumour tissue reveals a potentially causative somatic variant in SDHB, SDHC, SDHD, SDHAF2, SDHA, VHL, TMEM127, NF1, NF2, or PTEN genes. At-risk relatives should only be offered targeted testing for a known pathogenic or a likely pathogenic variant documented in the proband. All families with pathogenic or likely pathogenic gene variants should undergo appropriate genetic counselling, including a discussion of the syndrome being tested for, the likelihood of a positive result, and the surveillance and management that would be recommended if the gene test result is positive.
Because of the favourable prognosis of benign thyroid mesenchymal tumours and a low chance of recurrence, a hemithyroidectomy or a total thyroidectomy should suffice in most cases. Surgery could especially be considered if the tumours are big enough to cause compressive symptoms. Among older patients or patients with comorbidities with benign thyroid mesenchymal tumours without compressive symptoms, active surveillance, rather than surgical treatment, might be a reasonable option. For tumours with likelihood of recurrence or more aggressive behaviour, such as paragangliomas or solitary fibrous tumours, a follow-up neck ultrasound could be done. The timing and frequency of ultrasonography should be individualised depending on tumour size and location determining the likelihood of extrathyroidal extension—initially every 3–6 months, and if stable, with lower frequency (eg, yearly) thereafter.
If there is sufficient concern for a malignant thyroid mesenchymal tumour, further imaging studies, including whole-body CT scan or MRI and functional imaging such as 18F-fluorodeoxyglucose PET-CT scan should be considered, not only to evaluate for potential metastatic spread of the thyroid mesenchymal tumour, but also to identify a potential extrathyroidal primary malignant mesenchymal tumour that might have metastasised to the thyroid. Physicians should be aware that metastatic lesions from malignant mesenchymal tumours of extrathyroidal origin into the thyroid are more common than primary thyroid mesenchymal tumours.6 Mesenchymal tumours that commonly metastasise to the thyroid include uterine leiomyosarcoma, malignant phyllodes tumour of the breast, and gastrointestinal stromal tumour.6 Management of malignant thyroid mesenchymal tumours can be more complex and a multidisciplinary approach with input from oncologists, surgeons, endocrinologists, pathologists, and radiologists is crucial. The pathologist’s role remains central in recognising the type of thyroid mesenchymal tumour and differentiating it from the more common forms of malignant thyroid tumours, and from other rare tumours such as atypical anaplastic thyroid carcinoma variants, and calcitonin-negative and carcinoembryonic antigen-negative medullary thyroid carcinomas. The differential diagnosis for each major type of thyroid mesenchymal tumour is listed in the appendix (pp 1–4). Furthermore, standard treatment protocols for treating malignant thyroid mesenchymal tumours have been difficult to develop because of the rarity of these tumours. Thyroidectomy with or without lymph node dissection and removal of adjacent structures, when appropriate, should be done. For thyroid malignant vascular tumours, thyroidectomy is suggested; however, the tumour’s high vascularity and evidence of extrathyroidal extensions could lead to life-threatening haemorrhages during and after surgery. The potential risks of morbidity from extensive surgery must be weighed against potential benefits of improvements in quality of life and life-expectancy in patients with large tumours or extensive regional tumour burden. In general, the decisions on chemotherapy or radiotherapy, or both, should be considered based on standards of care for extrathyroidal malignant mesenchymal tumours, and the medical oncologist has a crucial role in driving these decisions. Adjuvant radiotherapy might improve local disease control in malignant vascular tumours, and neoadjuvant or adjuvant chemotherapy with or without radiotherapy could also be considered based on local invasion or distant metastasis.65 For the Ewing sarcoma family of tumours, systemic therapy after surgical resection is the standard of care, as it is for metastatic disease. The current recommended regimen is vincristine, doxorubicin, and cyclophosphamide alternating with ifosfamide and etoposide.74 For most malignant thyroid mesenchymal tumours, adjuvant radiotherapy is indicated for aggressive disease (high mitotic count, abundant nuclear pleomorphism, extracapsular spread, or surgically unresectable disease). In patients with high-risk localised soft tissue sarcomas, adjuvant or neoadjuvant chemotherapy is also considered with anthracyclines combined with ifosfamide.75 Adjuvant chemotherapy is also recommended for aggressive subtypes of otherwise indolent malignant thyroid mesenchymal tumours such as follicular dendritic cell sarcoma. The second-line approach should involve consideration of recruitment into a clinical trial. As of March, 2020, there are 214 phase 2 and phase 3 clinical trials listed on ClinicalTrials.gov that are currently recruiting patients for management of sarcomas, including thyroid sarcomas.
Molecular analysis on the excised tumour can facilitate the identification of drug targets for small molecules such as tyrosine-kinase inhibitors (TKIs), mTOR inhibitors, or fusion inhibitors. For instance, the fusion gene HDGRFP3-SHC4 identified in a thyroid follicular dendritic cell sarcoma led to a 200-fold increase in the expression of SHC4, an oncogene that is a potential target of the TKI dasatinib.58 For malignant vascular tumours, TKIs such as sunitinib and pazopanib with action on VEGF receptors, or VEGF inhibitors such as bevacizumab, or phosphatidylinositol 3-kinase (PI3K) inhibitors could have a potential benefit.21 The use of targeted therapies utilising small molecules in sarcoma are being investigated in clinical trials, including mTOR inhibitors such as sirolimus (NCT02821507, NCT02584647), temsirolimus (NCT02567435), everolimus (NCT03114527), and nanoparticle albumin-bound rapamycin (NCT03190174); TKIs such as sunitinib (NCT01391962), anlotinib (NCT03815474, NCT03890068), famitinib (NCT04044378), cediranib (NCT01391962), apatinib (NCT04012827), PLX3397 (NCT02584647), and sitravatinib (NCT02978859); fusion protein inhibitors, including ALK or TRK inhibitors such as entrectinib (NCT02568267), larotrectinib (NCT03213704, NCT02576431), the EWS-FLI1 inhibitor TK216 (NCT02657005); and the selective EZH2 methyltransferase inhibitor tazemetostat (NCT03213665).
Conclusion
This Review constitutes the most extensive and up-to-date literature review on primary thyroid mesenchymal tumours. Because of the aggressive nature of malignant thyroid mesenchymal tumours and the paucity of their occurrence in clinical practice, a multidisciplinary team-based approach consisting of endocrinologists, surgeons, oncologists, pathologists, and radiologists should ideally be used in the management of these tumours. Currently, the molecular landscape of most thyroid mesenchymal tumours is vastly unknown. However, data for the molecular landscape of extrathyroidal forms of these tumours are growing. The genetic and molecular characterisation of thyroid mesenchymal tumours constitute a pivotal field for future research that might help with developing targeted therapies against the more aggressive forms of thyroid mesenchymal tumours. Moreover, molecular profiling could potentially provide valuable insights into the histogenesis and biology of these rare thyroid tumours.
Supplementary Material
Panel: Classification of known primary thyroid mesenchymal tumours.
Benign
Paraganglioma
- Benign peripheral nerve sheath tumours
- Schwannoma
- Neurofibroma
- Granular cell tumour
- Vascular tumours
- Haemangioma
- Striated and smooth muscle tumours
- Rhabdomyoma
- Leiomyoma
- Adipose tissue tumours
- Adenolipoma
Solitary fibrous tumour
Malignant
Malignant paraganglioma
Malignant peripheral nerve sheath tumours
- Vascular tumours
- Angiosarcoma
- Epithelioid haemangioendothelioma
- Kaposi’s sarcoma
- Striated and smooth muscle tumours
- Rhabdomyosarcoma
- Leiomyosarcoma
- Adipose tissue tumours
- Liposarcoma
- Bone tumours
- Osteosarcoma
- Chondrosarcoma
- Ewing sarcoma
- Adamantinoma-like Ewing sarcoma
Malignant solitary fibrous tumour or haemangiopericytoma
Malignant granular cell tumour
Synovial sarcoma
Fibrosarcoma
Ameloblastic fibrosarcoma
Undifferentiated pleomorphic sarcoma (malignant fibrous histiocytoma) or myxofibrosarcoma
Alveolar soft part sarcoma
Malignant triton tumour
Follicular dendritic cell sarcoma
Malignant glomus tumour
Malignant mesenchymoma
Carcinosarcoma*
The data shown in this panel is a modified version of the classification of thyroid mesenchymal tumours provided by the 2017 WHO guidelines.10 *Carcinosarcoma is classified as a variant of anaplastic carcinoma as per the 2017 WHO guidelines.
Acknowledgments
This Review was funded by the Intramural Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (DK047053–12).
Footnotes
Declaration of interests
SG, ST, ACF, MR, JW, JDR, and JK-G are employees of the National Institutes of Health. All other authors declare no competing interests.
Contributor Information
Sriram Gubbi, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD, USA.
Shilpa Thakur, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD, USA.
Shirisha Avadhanula, Department of Endocrinology, Diabetes, and Metabolism, Cleveland Clinic, Cleveland, OH, USA.
Katherine A Araque, Department of Endocrinology, Pacific Neuroscience Institute, John Wayne Cancer Institute, Santa Monica, CA, USA.
Armando C Filie, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA.
Mark Raffeld, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA.
James Welch, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD, USA.
Jaydira Del Rivero, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA.
Electron Kebebew, Department of General Surgery, Stanford University School of Medicine, Stanford, CA, USA.
Kenneth D Burman, Department of Endocrinology, MedStar Washington Hospital Center, Washington, DC, USA.
Leonard Wartofsky, Department of Endocrinology, MedStar Health Research Institute, Washington, DC, USA.
Joanna Klubo-Gwiezdzinska, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD, USA.
References
- 1.Haugen BR, Alexander EK, Bible KC, et al. 2015 American Thyroid Association management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: the American Thyroid Association Guidelines Task Force on thyroid nodules and differentiated thyroid cancer. Thyroid 2016; 26: 1–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parsa AA, Gharib H. Thyroid nodule: current evaluation and management. In: Luster M, Duntas LH, Wartofsky L, eds. The thyroid and its diseases: a comprehensive guide for the clinician Cham: Springer, 2019: 493–516. [Google Scholar]
- 3.Gharib H, Papini E, Paschke R, Duick DS, Valcavi R, Hegedus L. Medical guidelines for clinical practice for the diagnosis and management of thyroid nodules. Endocr Pract 2006; 12: 63–102. [DOI] [PubMed] [Google Scholar]
- 4.Grant EG, Tessler FN, Hoang JK, et al. Thyroid ultrasound reporting lexicon: white paper of the ACR Thyroid Imaging, Reporting and Data System (TIRADS) committee. J Am Coll Radiol 2015; 12: 1272–79. [DOI] [PubMed] [Google Scholar]
- 5.Burman KD, Wartofsky L. Thyroid nodules. N Engl J Med 2015; 373: 2347–56. [DOI] [PubMed] [Google Scholar]
- 6.LiVolsi VA, Montone KT, Baloch ZW. Pathology of the thyroid: a review. In: Luster M, Duntas LH, Wartofsky L, eds. The thyroid and its diseases: a comprehensive guide for the clinician Cham: Springer, 2019: 455–92. [Google Scholar]
- 7.Wells SA Jr, Asa SL, Dralle H, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma: the American Thyroid Association Guidelines Task Force on medullary thyroid carcinoma. Thyroid 2015; 25: 567–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smallridge RC, Ain KB, Asa SL, et al. American Thyroid Association guidelines for management of patients with anaplastic thyroid cancer. Thyroid 2012; 22: 1104–39. [DOI] [PubMed] [Google Scholar]
- 9.Mesenchyme MacCord K. The Embryo project encyclopedia 2012. https://embryo.asu.edu/handle/10776/3941 (accessed Aug 29, 2020).
- 10.Lloyd RV, Osamura RY, Klöppel G, et al. WHO classification of tumours of endocrine organs Lyon: International Agency for Research on Cancer, 2017. [Google Scholar]
- 11.Bula G, Waler J, Niemiec A, Trompeta J, Steplewska K, Gawrychowski J. Unusual malignant thyroid tumours—a clinical study of 20 cases. Acta Chirurgica Belgica 2008; 108: 702–07. [DOI] [PubMed] [Google Scholar]
- 12.Thompson LD, Wei C, Rooper LM, Lau SK. Thyroid gland solitary fibrous tumor: report of 3 cases and a comprehensive review of the literature. Head Neck Pathol 2019; 13: 597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mishra P, Padhi S, Behera G. Thyroid paraganglioma: a case-based systematic review of literature. J Cancer Res Ther 2019; published online March 1. 10.4103/jcrt.JCRT_713_18 (preprint). [DOI] [PubMed]
- 14.Surov A, Gottschling S, Wienke A, Meyer HJ, Spielmann RP, Dralle H. Primary thyroid sarcoma: a systematic review. Anticancer Res 2015; 35: 5185–91. [PubMed] [Google Scholar]
- 15.Furze AD, Lehman DA, Roy S. Rhabdomyosarcoma presenting as an anterior neck mass and possible thyroid malignancy in a seven-month-old. Int J Pediatr Otorhinolaryngol 2005; 69: 267–70. [DOI] [PubMed] [Google Scholar]
- 16.Tong GX, Hamele-Bena D, Liu JC, Horst B, Remotti F. Fine-needle aspiration biopsy of primary osteosarcoma of the thyroid: report of a case and review of the literature. Diagn Cytopathol 2008; 36: 589–94. [DOI] [PubMed] [Google Scholar]
- 17.Kabata P, Kaniuka-Jakubowska S, Kabata W, et al. Primary Ewing sarcoma of the thyroid—eight cases in a decade: a case report and literature review. Front Endocrinol 2017; 8: 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Almaghrabi M, Almaghrabi H, Al-Maghrabi H. Granular cell tumor of thyroid: challenging pitfalls and mimickers in diagnosis. J Microsc Ultrastruct 2020; 8: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Griem KL, Robb PK, Caldarelli DD, Templeton AC. Radiation-induced sarcoma of the thyroid. Arch Otolaryngol Head Neck Surg 1989; 115: 991–93. [DOI] [PubMed] [Google Scholar]
- 20.Tamada A, Makimoto K, Tasaka Y, Nakamoto Y, Iwasaki H, Yamabe H. Radiation-induced fibrosarcoma of the thyroid. J Laryngol Otol 1984; 98: 1063–66. [DOI] [PubMed] [Google Scholar]
- 21.De Felice F, Moscatelli E, Orelli S, Bulzonetti N, Musio D, Tombolini V. Primary thyroid angiosarcoma: a systematic review. Oral Oncol 2018; 82: 48–52. [DOI] [PubMed] [Google Scholar]
- 22.Poniecka A, Ghorab Z, Arnold D, Khaled A, Ganjei-Azar P. Kaposi’s sarcoma of the thyroid gland in an HIV-negative woman: a case report. Acta cytologica 2007; 51: 421–23. [DOI] [PubMed] [Google Scholar]
- 23.Pennisi M, Conti A, Farina R, et al. Thyroid adenolipoma: a case report. J Ultrasound 2018; 21: 165–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maciel LMZ, Gomes PM, Magalhães PKR, Filho FVM, Conti-Freitas LC. A giant primary hemangioma of the thyroid gland. J Clin Endocrinol Metab 2011; 96: 1623–24. [DOI] [PubMed] [Google Scholar]
- 25.Guarda V, Pickhard A, Boxberg M, Specht K, Buchberger AMS. Liposarcoma of the thyroid: a case report with a review of the literature. Eur Thyroid J 2018; 7: 102–08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kandil E, Khalek MA, Abdullah O, et al. Primary peripheral nerve sheath tumors of the thyroid gland. Thyroid 2010; 20: 583–86. [DOI] [PubMed] [Google Scholar]
- 27.Nechifor-Boilă A, Decaussin-Petrucci M, Varga-Ilyés A, Chinezu L, Caraşca C, Borda A. Angioinvasion as a factor for predicting aggressive outcome in primary thyroid angiosarcoma: three case reports and literature review. Pol J Pathol 2018; 69: 53–61. [DOI] [PubMed] [Google Scholar]
- 28.Trites A. Thyrolipoma, thymolipoma and pharyngeal lipoma: a syndrome. Can Med Assoc J 1966; 95: 1254–59. [PMC free article] [PubMed] [Google Scholar]
- 29.Hansen T, Gaumann A, Ghalibafian M, Höferlin A, Heintz A, Kirkpatrick C. Haemangiopericytoma of the thyroid gland in combination with Hashimoto’s disease. Virchows Archiv 2004; 445: 315–19. [DOI] [PubMed] [Google Scholar]
- 30.Von Dobschuetz E, Leijon H, Schalin-Jäntti C, et al. A registry-based study of thyroid paraganglioma: histological and genetic characteristics. Endocr Relat Cancer 2015; 22: 191–204. [DOI] [PubMed] [Google Scholar]
- 31.Surov A, Holzhausen HJ, Machens A, Dralle H. Imaging findings of thyroidal sarcoma. Clin Imaging 2014; 38: 826–30. [DOI] [PubMed] [Google Scholar]
- 32.Makis W, Novales-Diaz J-A, Hickeson M. Primary thyroid osteosarcoma: staging and evaluation of response to therapy with F-18 FDG PET-CT. Clin Nucl Med 2010; 35: 517–20. [DOI] [PubMed] [Google Scholar]
- 33.Treglia G, Bongiovanni M, Paone G, Ceriani L, Giovanella L. Metastatic undifferentiated spindle cell sarcoma of the thyroid gland evaluated by 18F-FDG PET/CT. Clin Nucl Med 2015; 40: e208–10. [DOI] [PubMed] [Google Scholar]
- 34.Kishimoto Y, Ohno Kishimoto A, Yamada Y, et al. Dedifferentiated liposarcoma of the thyroid gland: a case report. Mol Clin Oncol 2019; 11: 219–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Janssen I, Blanchet EM, Adams K, et al. Superiority of [68Ga]-DOTATATE PET/CT to other functional imaging modalities in the localization of SDHB-associated metastatic pheochromocytoma and paraganglioma. Clin Cancer Res 2015; 21: 3888–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.King KS, Chen CC, Alexopoulos DK, et al. Functional imaging of SDHx-related head and neck paragangliomas: comparison of 18F-fluorodihydroxyphenylalanine, 18F-fluorodopamine, 18F-fluoro-2-deoxy-D-glucose PET, 123I-metaiodobenzylguanidine scintigraphy, and 111In-pentetreotide scintigraphy. J Clin Endocrinol Metab 2011; 96: 2779–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dutta M, Chatterjee I, Roy S, Gure PK. Primary embryonal rhabdomyosarcoma of the anterior neck and thyroid: report of a new case with review of the literature. Laryngoscope 2013; 123: 2072–76. [DOI] [PubMed] [Google Scholar]
- 38.Vujosevic S, Krnjevic D, Bogojevic M, et al. Primary leiomyosarcoma of the thyroid gland with prior malignancy and radiotherapy: a case report and review of literature. World J Clin Cases 2019; 7: 473–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Al-Ghamdi S, Fageeh N, Dewan M. Malignant schwannoma of the thyroid gland. Otolaryngol Head Neck Surg 2000; 122: 143–44. [DOI] [PubMed] [Google Scholar]
- 40.Papi G, Corrado S, LiVolsi VA. Primary spindle cell lesions of the thyroid gland: an overview. Am J Clin Pathol 2006; 125 (suppl 1): 95–123. [DOI] [PubMed] [Google Scholar]
- 41.Ragazzi M, Ciarrocchi A, Sancisi V, Gandolfi G, Bisagni A, Piana S. Update on anaplastic thyroid carcinoma: morphological, molecular, and genetic features of the most aggressive thyroid cancer. Int J Endocrinol 2014; 2014: 790834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feng G, Laskin WB, Chou PM, Lin X. Anaplastic thyroid carcinoma with rhabdoid features. Diagn Cytopathol 2015; 43: 416–20. [DOI] [PubMed] [Google Scholar]
- 43.Miettinen M, Cue PAM, Sarlomo-Rikala M, et al. GATA 3: a multispecific but potentially useful marker in surgical pathology: a systematic analysis of 2500 epithelial and non-epithelial tumors. Am J Surg Pathol 2014; 38: 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lau N, Hari D, French S. SOX10 expression in a gangliocytic paraganglioma—a case report. Exp Mol Pathol 2015; 98: 99–101. [DOI] [PubMed] [Google Scholar]
- 45.Agrawal N, Akbani R, Aksoy BA, et al. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014; 159: 676–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abeshouse A, Adebamowo C, Adebamowo SN, et al. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell 2017; 171: 950–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mohamed AD, Tremblay AM, Murray GI, Wackerhage H. The Hippo signal transduction pathway in soft tissue sarcomas. Biochim Biophys Act 2015; 1856: 121–29. [DOI] [PubMed] [Google Scholar]
- 48.Costinean S, Balatti V, Bottoni A, Old M, Croce C, Wakely PE Jr. Primary intrathyroidal paraganglioma: histopathology and novel molecular alterations. Hum Pathol 2012; 43: 2371–75. [DOI] [PubMed] [Google Scholar]
- 49.Bishop JA, Alaggio R, Zhang L, Seethala RR, Antonescu CR. Adamantinoma-like Ewing family tumors of the head and neck: a pitfall in the differential diagnosis of basaloid and myoepithelial carcinomas. Am J Surg Pathol 2015; 39: 1267–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tu J, Huo Z, Gingold J, Zhao R, Shen J, Lee D-F. The histogenesis of Ewing sarcoma. Cancer Rep Rev 2017; 1: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Owen C, Constantinidou A, Miah AB, et al. Synovial sarcoma of the thyroid gland, diagnostic pitfalls and clinical management. Anticancer Res 2018; 38: 5275–82. [DOI] [PubMed] [Google Scholar]
- 52.Antonescu C. Malignant vascular tumors—an update. Mod Pathol 2014; 27 (suppl 1): 30–38. [DOI] [PubMed] [Google Scholar]
- 53.Demicco EG. Molecular updates in adipocytic neoplasms. Semin Diagn Pathol 2019; 36: 85–94. [DOI] [PubMed] [Google Scholar]
- 54.Agaram NP, Zhang L, Sung Y-S, Singer S, Antonescu CR. Extraskeletal myxoid chondrosarcoma with non-EWSR1-NR4A3 variant fusions correlate with rhabdoid phenotype and high-grade morphology. Hum Pathol 2014; 45: 1084–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang L, Motoi T, Khanin R, et al. Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes Chromosomes Cancer 2012; 51: 127–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Whaley RD, Thompson LD. Primary thyroid gland alveolar soft part sarcoma. Head Neck Pathol 2019; published online Nov 28. 10.1007/s12105-019-01099-x. [DOI] [PMC free article] [PubMed]
- 57.Yang J, Sarita-Reyes C, Kindelberger D, Zhao Q. A rare malignant thyroid carcinosarcoma with aggressive behavior and DICER1 gene mutation: a case report with literature review. Thyroid Res 2018; 11: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davila JI, Starr JS, Attia S, et al. Comprehensive genomic profiling of a rare thyroid follicular dendritic cell sarcoma. Rare Tumors 2017; 9: 6834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cameselle-Teijeiro J, Fachal C, Cabezas-Agrícola JM, et al. Thyroid pathology findings in Cowden syndrome: a clue for the diagnosis of the PTEN hamartoma tumor syndrome. Am J Clin Pathol 2015; 144: 322–28. [DOI] [PubMed] [Google Scholar]
- 60.Thompson LD, Wenig BM, Adair CF, Heffess CS. Peripheral nerve sheath tumors of the thyroid gland: a series of four cases and a review of the literature. Endocr Pathol 1996; 7: 309–18. [DOI] [PubMed] [Google Scholar]
- 61.Miao J, Chen S, Li Y, Fu L, Li H. A primary cavernous hemangioma of the thyroid gland: a case report and literature review. Medicine 2017; 96: e8651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Armstrong MJ, Chiosea SI, Carty SE, Hodak SP, Yip L. Thyroid paragangliomas are locally aggressive. Thyroid 2012; 22: 88–93. [DOI] [PubMed] [Google Scholar]
- 63.Reznick D, Scharpf J, Doshi K, et al. Malignant primary paraganglioma of the thyroid gland: the first reported case. AACE Clin Case Rep 2016; 2: e70–75. [Google Scholar]
- 64.Filipović A, Vučković L, Pejakov L. Paraganglioma of the thyroid gland: a case report. Vojnosanit Pregl 2014; 71: 875–78. [PubMed] [Google Scholar]
- 65.Rhomberg W, Boehler F, Eiter H, Fritzsche H, Breitfellner G. Treatment options for malignant hemangioendotheliomas of the thyroid. Int J Radiat Oncol Biol Phys 2004; 60: 401–05. [DOI] [PubMed] [Google Scholar]
- 66.Palla AR, Bollig CA, Jorgensen JB. Well-differentiated liposarcoma localized to the thyroid gland. Eur Thyroid J 2018; 7: 179–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Canu G, Bulla J, Lai M, et al. Primary thyroid leiomyosarcoma: a case report and review of the literature. G Chir 2018; 39: 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ongkeko M, Zeck J, de Brito P. Molecular testing uncovers an adamantinoma-like Ewing family of tumors in the thyroid: case report and review of literature. AJSP Rev Rep 2018; 23: 8–12. [Google Scholar]
- 69.Tang T, Li C, Chen J, Yang G, Liao Q, Wang Z. Primary malignant peripheral nerve sheath tumor of the thyroid—an extremely rare case report of thyroid carcinoma. Cancer Cell Res 2017; 13: 316–19. [Google Scholar]
- 70.Al-Sobhi SS, Novosolov F, Sabançi Ü, Epstein HD, Greenspan FS, Clark OH. Management of thyroid carcinosarcoma. Surgery 1997; 122: 548–52. [DOI] [PubMed] [Google Scholar]
- 71.Zeng Q, Tang P, Xu Z, Qi Y, Wu X, Liu W. Primary malignant fibrous histiocytoma of the thyroid. Eur J Surg Onc 2009; 35: 649–53. [DOI] [PubMed] [Google Scholar]
- 72.Kantorovich V, King KS, Pacak K. SDH-related pheochromocytoma and paraganglioma. Best Pract Res Clin Endocrinol Metab 2010; 24: 415–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eng C PTEN hamartoma tumor syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al. , eds. GeneReviews Seattle, WA: University of Washington, Seattle, 2016. [Google Scholar]
- 74.Grier HE, Krailo MD, Tarbell NJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 2003; 348: 694–701. [DOI] [PubMed] [Google Scholar]
- 75.O’Connor J, Chacón M, Petracci F, Chacón R. Adjuvant chemotherapy in soft tissue sarcoma (STS): a meta-analysis of published data. J Clin Oncol 2008; 26 (suppl 15): 10526. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.