

Abstract
T-cell immunoglobulin and mucin domain 3 (Tim-3) is an immune checkpoint receptor that dampens effector functions and causes terminal exhaustion of cytotoxic T-cells. Tim-3 inhibitors are under investigation in immuno-oncology (IO) trials, because blockade of T-cell-Tim-3 enhances antitumor immunity. Here, we identify an additional role for Tim-3 as a growth-suppressive receptor intrinsic to melanoma cells. Inhibition of melanoma cell-Tim-3 promoted tumor growth in both immunocompetent and immunocompromised mice, while melanoma-specific Tim-3 overexpression attenuated tumorigenesis. Antibody (Ab)-mediated Tim-3 blockade inhibited growth of immunogenic murine melanomas in T-cell-competent hosts, consistent with established antitumor effects of T-cell Tim-3 inhibition. In contrast, Tim-3 Ab administration stimulated tumorigenesis of both highly and lesser immunogenic murine and human melanomas in T-cell-deficient mice, confirming growth-promoting effects of melanoma-Tim-3 antagonism. Melanoma-Tim-3 activation suppressed, while its blockade enhanced, phosphorylation of pro-proliferative downstream mitogen-activated protein kinase (MAPK) signaling mediators. Finally, pharmacologic MAPK inhibition reversed unwanted Tim-3 Ab-mediated tumorigenesis in T-cell-deficient mice and enhanced desired antitumor activity of Tim-3 interference in T-cell-competent hosts. These results identify melanoma-Tim-3 blockade as a mechanism that antagonizes T-cell-Tim-3-directed IO therapeutic efficacy. They further reveal MAPK targeting as a combination strategy for circumventing adverse consequences of unintended melanoma-Tim-3 inhibition.
Keywords: Melanoma, Tim-3, immune checkpoint, MAPK, therapy, resistance
INTRODUCTION
Immune checkpoint inhibitors (ICIs) have shown unprecedented clinical activity in patients with advanced stage cancers of diverse etiology (1). Nevertheless, clinical benefit is highly variable among patients, highlighting the need for more effective treatment modalities. A promising approach in this regard is to overcome mechanisms of resistance to ICI therapy (2). For example, induction of other immune checkpoints, such as Tim-3, has been associated with resistance to ICI regimens targeting programmed cell death-1 (PD-1) (3,4). Moreover, Tim-3 inhibition synergizes with PD-1:PD-L1 axis antagonists in preclinical IO models (5–7) and can improve tumor-specific T-cell immunity in some cancer patients compared to PD-1 blockade alone (8). Together, these findings have generated excitement regarding Tim-3 as a promising IO target (9). Thus, several Tim-3 inhibitors have entered cancer clinical trials, either alone or in combination with PD-1 therapy, for the treatment of hematologic malignancies and solid tumors, including melanoma (8).
Tim-3 is a member of the TIM family of immunoregulatory proteins primarily studied in T-cells (9). In cancer, engagement of the Tim-3 receptor by its predominant ligand, Galectin-9, dampens effector functions and causes terminal exhaustion of cytotoxic T-lymphocytes (10). Consequently, Tim-3 blocking antibodies (Abs) reverse T-cell exhaustion to reinvigorate antitumor immunity (6,7). However, preclinical efficacy of Tim-3 monotherapy is modest compared to PD-1 interference (8), raising potential concerns regarding the clinical benefit of Tim-3-based ICI regimens, and especially single agent Tim-3 blockade, in patients with solid tumors.
Several aspects of Tim-3 immunobiology have been recognized as potential confounders impacting clinical success of Tim-3 checkpoint inhibition (8). For instance, Tim-3 interacts at distinct binding pockets with multiple ligands, including Galectin-9, high-mobility group box 1 (HMGB1), phosphatidylserine (PtdSer), and carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM-1), with diverse roles in antitumor immunity (9). Accordingly, reported differences in ligand-neutralizing activity of Tim-3 inhibitors (8,11) could result in divergent clinical outcomes. Tim-3 therapeutic efficacy could also depend on the relative frequency of Tim-3-expressing cell types in the tumor microenvironment (TME). Indeed, Tim-3 is not only expressed by T-cells, but also by multiple additional immune and non-immune cell types with varying ligand positivity, resultant Tim-3 receptor signaling and functions (8). For example, in leukemic stem cells (LSCs), Tim-3:Galectin-9 interactions promote self-renewal (12). Thus, direct targeting of LSC-Tim-3 is thought to contribute to the observed preliminary efficacy of Tim-3 therapy in patients with leukemia (8). Tim-3 expression by cancer cells has also been reported in solid malignancies (13–15), although little is known about the significance of cancer cell-intrinsic Tim-3 in solid tumor progression and therapy. Because melanoma cells are known to express immune checkpoints (16–19) as well as Tim-3 ligands, including Galectin-9 (20–22), we sought to characterize Tim-3 functional expression by melanoma cells and investigate possible effects of melanoma cell-directed Tim-3 targeting on tumorigenesis.
Here, we report expression of Tim-3 directly on melanoma cells in established murine and human cell lines and patient tumor biospecimens. RNAi-mediated inhibition of melanoma cell-Tim-3 in both immunocompetent and immunocompromised, but not Galectin-9 null mice, significantly increases tumor growth. Conversely, enforced expression of melanoma cell-Tim-3 suppresses tumorigenesis in wildtype, but not Galectin-9-deficient hosts. Tim-3 Ab blockade inhibits growth of immunogenic murine melanomas in T-cell-competent, but promotes tumorigenesis of both highly and lesser immunogenic murine and human melanomas in T-cell-deficient, mice. Melanoma cell-Tim-3 activation reduces, while Tim-3 blockade enhances phosphorylation of pro-proliferative Tim-3 signaling mediators (12,23), including the MAPK effectors, MEK1/2 and ERK1/2. Consistently, pharmacologic MAPK inhibition, using trametinib, reverses unwanted Tim-3 Ab-mediated tumorigenesis in immunocompromised, and enhances desired antitumor effects of Tim-3 inhibition in immunocompetent, hosts. Our results identify Tim-3 as a growth-inhibitory receptor intrinsic to melanoma cells. By stimulating tumorigenesis, inadvertent melanoma-Tim-3 blockade could thus counteract clinical benefit from T-cell-directed Tim-3 therapy. We uncover MAPK inhibition as a combination strategy for circumventing such adverse effects of melanoma cell-Tim-3 interference.
MATERIALS AND METHODS
Melanoma Cell Lines, Culture Methods, and Clinical Melanoma Specimens.
Authenticated, Mycoplasma-free human and murine melanoma cell lines were obtained from ATCC or Millipore Sigma between 2016–2018 and cultured no longer than 3 weeks after thawing, as described in the Supplementary Methods. Human PBMCs were obtained from healthy donors, and clinical tumor biospecimens from melanoma patients in accordance with protocols approved by the Mass General Brigham IRB, under protocol numbers 2019P001246, 2018P001983, and 2013P001014. Informed consent was obtained from all subjects, and all studies were conducted in accordance with the Declaration of Helsinki. Single-cell suspensions were generated from human melanoma grafts using collagenase digestion, as described (17).
RT-PCR, RT-qPCR, and Flow Cytometry.
The full-length coding sequences of human and murine Tim-3 mRNAs (HAVCR2 and Havcr2, respectively) were amplified and sequenced following reverse transcription of total mRNA using Tim-3 gene-specific primer pairs. The full coding sequences (CDS) of HAVCR2 (NM_032782.5) expressed by the human A2058 and G361 melanoma cell lines and of Havcr2 (NM_134250.2) expressed by the murine B16-F10 melanoma cell line were submitted to the GenBank database under the following accession numbers: MW055427 (A2058), MW055428 (G361), and MW055429 (B16-F10). Relative HAVCR2 and Havcr2, or LGALS9 and Lgals9 were determined by RT-qPCR and calculated using the 2(−ΔΔCt) method (primers described in the Supplementary Methods). Tim-3 surface and Galectin-9 protein expression and binding of recombinant soluble murine Galectin-9 (R&D Systems, 3535-GA-050) by established melanoma lines and/or immune cell positive controls were analyzed by flow cytometry, as described (17). Tim-3+ and Tim-3− melanoma cell subsets were isolated by FACS, following Tim-3 surface staining as above.
Western Blot and Phosphoprotein Array Analyses.
Cells were lysed, total protein separated by SDS-PAGE and transferred to a PVDF membrane by electroblotting, as described in detail in the Supplementary Methods. Expression levels of human and murine Tim-3 and of phosphorylated versus total ERK1/2 or MEK1/2 (Cell Signaling Technology, clones D13.14.4E and 137F5, 41G9 and 61B12, respectively) were determined using enhanced chemiluminescence or the Odyssey CLx imaging system (LI-COR Biosciences). Expression levels of phosphorylated versus respective total protein controls in Tim-3 variant and/or Ab-treated melanoma lines were quantified by the Phospho Explorer Antibody Array (Full Moon Biosystems, PEX100) and/or via densitometric quantification of protein bands using ImageJ (National Institutes of Health).
Immunofluorescence Staining.
Immunofluorescence double labeling for Tim-3 and SOX-10, Galectin-9 and SOX-10, or Galectin-9 and CD45 in formalin-fixed, paraffin-embedded tumor biospecimens obtained from patients with melanoma was performed, as described (17).
Generation of Stable Tim-3 Knockdown and Tim-3-Overexpressing, or nuclear GFP-labeled Melanoma Cell Line Variants.
Stable Tim-3 knockdown (KD) melanoma lines were generated, as described in detail in the Supplementary Methods, using lentiviral transduction particles containing short hairpin RNAs (shRNA) against human HAVCR2 or murine Havcr2, and Tim-3–overexpressing (OE) melanoma lines by infection with viral particles containing the full-length CDS of HAVCR2 or Havcr2. Melanoma lines stably expressing nuclear GFP were generated by fusing a nuclear localization signal with GFP in the MSCV-N-Flag-HA-GFP vector (Addgene, 41034), followed by retroviral infection, puromycin selection, and single-cell sorting of melanoma cells for high GFP expression.
Three-Dimensional Melanoma Culture.
Melanoma tumor sphere cultures of native or melanoma-Tim-3 variant lines were maintained, as described (17), in standard culture medium, as above, containing 0.5% (w/v) methyl cellulose, in the presence or absence of anti-human (F38–2E2, BioLegend, 345010) or anti-mouse (RMT3–23, BioLegend, 119734) Tim-3 blocking or respective isotype control mAbs.
In Vivo Tumorigenicity Studies.
C57BL/6 and NSG mice (17) mice were purchased from The Jackson Laboratory (Bar Harbor, ME), and Lgals9(−/−) knockout (KO) C57BL/6 (24) obtained from Dr. Michael Croft (La Jolla Institute for Allergy and Immunology, La Jolla, CA). All mice were female, at least 6 weeks of age, age-matched between experimental groups, maintained and housed at the animal facility of Brigham and Women’s Hospital, and used in accordance with the National Institutes of Animal Healthcare Guidelines. All animal experiments were conducted under protocols approved by the Institutional Animal Care and Use Committee of Brigham and Women’s Hospital. For tumorigenicity studies, wildtype or HAVCR2/Havcr2 variant melanoma cells were injected subcutaneously into flanks of recipient mice, as described (17,25). For Tim-3 Ab targeting experiments, melanoma cells were grafted, mice intraperitoneally (i.p.) injected with anti-human (F38–2E2) or anti-mouse (RMT3–23) Tim-3 blocking or isotype control mAbs (200 μg, respectively) every three days starting the day of and/or 15 days after melanoma cell inoculation, and tumor formation/growth assessed, as described (17). For MEK inhibitor studies, mice were fed a submaximal dose (0.15 mg/kg/d, p.o.) of trametinib (Selleckchem, S2673) or vehicle control (19) incorporated into rodent chow (Research Diets, Inc.).
Statistical Analysis.
Gene and protein expression levels, tumor spheroid and in vivo melanoma growth were compared statistically using the unpaired Student t test, the nonparametric Mann-Whitney test (comparison of two experimental groups), one-way ANOVA with Dunnett post-test, repeated-measures two-way ANOVA, or Mixed Model followed by the Bonferroni correction (comparison of three or more experimental groups). The Spearman rank correlation test was used to measure the degree of association between two variables. Data was tested for normal distribution using the D’Agostino and Pearson omnibus normality test. A two-sided value of p < 0.05 was considered statistically significant.
Study Approval.
All studies involving human specimens were approved by the Mass General Brigham IRB, under protocol numbers 2019P001246, 2018P001983, and 2013P001014. Written informed consent was obtained from all subjects and all studies were conducted in accordance with the Declaration of Helsinki. All animal experiments were conducted under protocols approved by the Institutional Animal Care and Use Committee of Brigham and Women’s Hospital.
Data Availability.
The data generated in this study are publicly available in Genbank under accession numbers MW055427–29 or from the corresponding authors upon reasonable request.
See also the Supplementary Methods.
RESULTS
Melanomas contain Tim-3-expressing cancer cells
Analysis of a single-cell RNA-sequencing dataset (26) revealed Tim-3 (HAVCR2, Fig. 1A) and Galectin-9 (LGALS9) mRNA expression by melanoma cells in all patient lesions (n = 14) examined, at expression ranges overlapping HAVCR2 or LGALS9 levels observed in tumor-infiltrating T-cells, B-cells, NK cells, macrophages, endothelial cells, and cancer-associated fibroblasts (Fig. S1A–B). Co-expression of both genes was found in 42.6% of melanoma and 79.4% of T-cells (Fig. S1C). In the TCGA PanCancer Atlas (27,28), HAVCR2 expression in melanomas was similar to that in positive control acute myeloid leukemia (AML), while LGALS9 levels were significantly higher in AML versus melanoma (Fig. S2A). HAVCR2 and LGALS9 expression levels correlated significantly with each other (Fig. S2B) and also tended to correlate inversely with melanoma stage (Fig. S2C), but not metastasis (Fig. S2D) or overall survival (Fig. S2E). Immunofluorescence double labeling of clinical melanoma biospecimens for Tim-3 with the melanocytic lineage marker, SOX-10, confirmed Tim-3 protein expression by up to 10% of SOX-10+ melanoma cells in 7 of 15 (Fig. 1B). Galectin-9 was expressed by 12% ± 3% of SOX-10+ cells (mean ± SEM) and 11 ± 3% of CD45+ lymphocyte positive controls in 11 of 12 patient samples examined (Fig. S3A–B). Galectin-9 expression tended to be greater in tumors containing Tim-3+ cancer cells compared to those without detectable melanoma cell-Tim-3 (Fig. S3C). Flow cytometric (FACS) analyses revealed Tim-3 surface and Galectin-9 protein expression by 5/5 human melanoma cell lines, with greatest surface-Tim-3 positivity (mean ± SEM) found in A2058 (3.9 ± 0.5%) and G361 cells (5.0 ± 0.8%, Fig. 1C), and >85% Galectin-9 positivity across all human lines tested (Fig. S3D). Tim-3 surface and Galectin-9 protein expression was also detected in 7/7 murine melanoma lines, with highest Tim-3+ cancer cell frequencies observed for B16-F10 (13.5 ± 0.8%) and YUMMER1.7D4 (15.4 ± 2.7%) melanoma cells (mean ± SEM, Fig. 1D) and 42.4–90.6% Galectin-9 positivity across all murine melanoma lines examined (Fig. S3E). RT-PCR amplification and sequencing of the full CDS of the human Tim-3 (HAVCR2) and mouse Tim-3 (Havcr2) genes revealed Tim-3 mRNA expression by human A2058 and G361 and murine B16-F10 melanoma cells, respectively (Fig. 1E). FACS-based mean fluorescence intensity (MFI, Fig. S4A) and immunoblot analyses further validated Tim-3 protein expression by human A2058 and G361 and/or murine B16-F10 melanoma cells, at molecular weights corresponding to respective positive controls (Fig. 1F and Fig. S4B). RT-qPCR independently confirmed Tim-3 gene expression at varying levels by 13/13 human and 8/8 murine melanoma lines analyzed (Fig. S5A). FACS-purified Tim-3+ melanoma cells showed >2–21-fold increased HAVCR2 or Havcr2 levels compared to Tim-3− A2058, G361, B16-F10 or YUMMER1.7D4 tumor cell subsets, respectively (Fig. S5B). Similarly, all human and murine melanoma lines examined showed significant expression (Fig. S6A) and enrichment of LGALS9 and Lgals9 among Tim-3+ versus Tim-3− melanoma subpopulations (Fig. S6B). Finally, Tim-3 and Galectin-9 gene co-expression significantly correlated across tumor cell lines (Fig. S6C).
Figure 1. Tim-3 expression by melanoma cells.
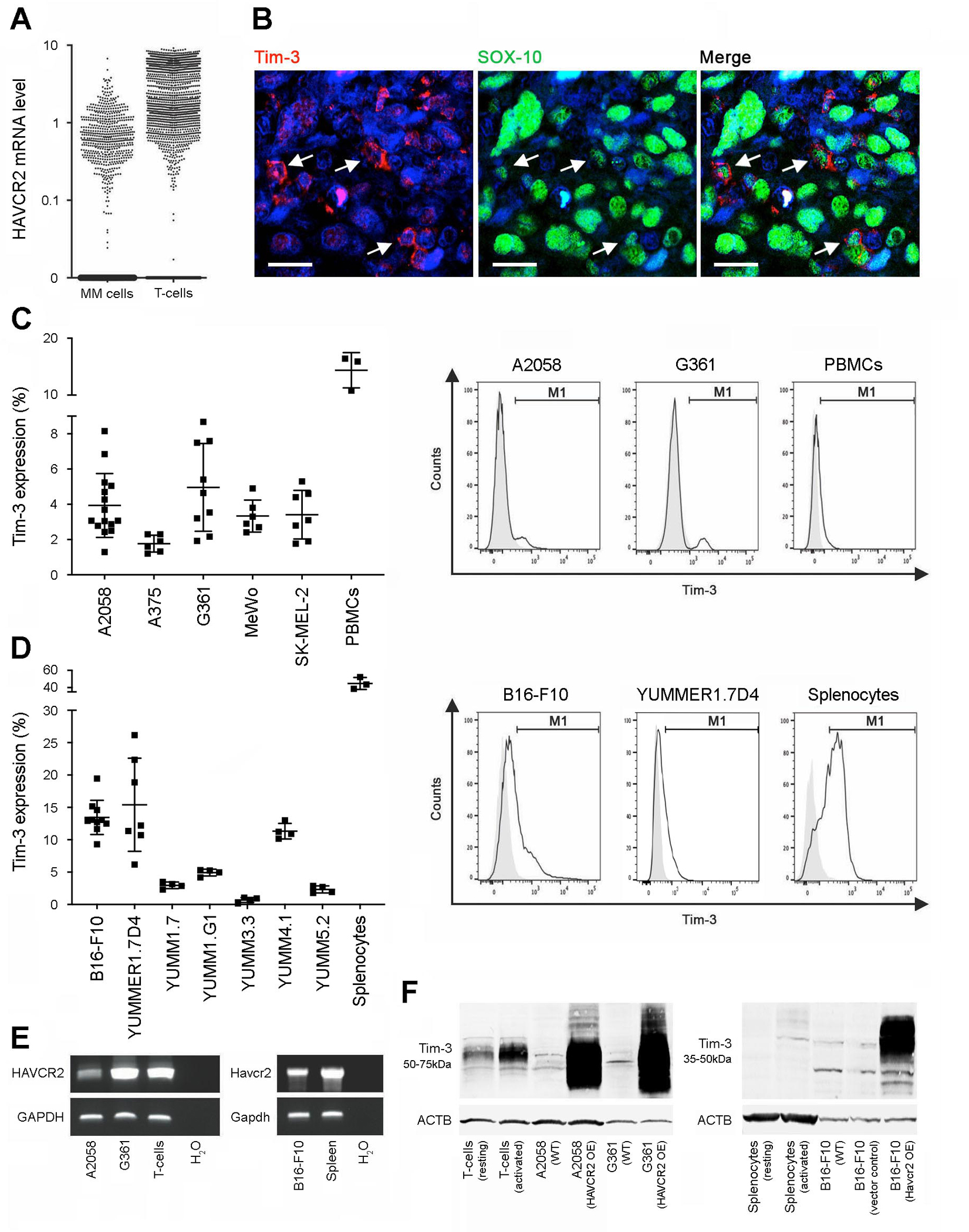
A, Single-cell RNA-seq analysis of human Tim-3 gene (HAVCR2) expression in patient melanoma (MM) cells versus tumor-infiltrating T-cells. B, Representative dual immunofluorescence staining of a clinical melanoma biopsy for co-expression (arrows) of Tim-3 (red) and the melanocytic marker, SOX-10 (green). Nuclei were counterstained with DAPI (blue). Size bars, 20 μm. C-D, Percentages (mean ± SD, left) and representative flow cytometric histograms (right) of Tim-3 surface protein expression by C, human melanoma lines and PBMCs and by D, murine melanoma lines and C57BL/6-derived splenocytes (n = 3–10 independent experiments). E, RT-PCR expression analysis of the full-length HAVCR2 coding sequence by human A2058 and G361 (left) or murine B16-F10 (Havcr2, right) melanoma lines and respective T-cell or splenocyte positive controls. F, Immunoblot of Tim-3 protein expression by human (left) or murine (right) wild-type (WT), vector control and/or Tim-3 (HAVCR2/Havcr2)-overexpressing (OE) melanoma cells, resting versus activated T-cell or splenocyte positive controls. See also Figures S1–S6.
Melanoma cell-Tim-3 inhibits murine tumor growth in a Galectin-9-dependent manner
To dissect roles of melanoma cell-Tim-3 in tumorigenesis, we generated stable Havcr2 knockdown (KD) B16-F10 lines, using two independent short hairpin (sh) RNAs (Fig. S7A), and Havcr2-overexpressing (OE) B16-F10 melanoma variants (Fig. S7B). Compared to respective controls, melanoma-specific Havcr2-KD resulted in increased and Havcr2-OE in decreased B16-F10 tumor growth in both immunocompetent C57BL/6 (Fig. 2A) and NSG mice lacking adaptive immunity (Fig. 2B). Consistent with our in vivo findings, Havcr2-KD promoted (Fig. S7C), while Havcr2-OE impaired in vitro three-dimensional B16-F10 culture growth versus controls (Fig. S7D). We next examined whether ligation of melanoma-Tim-3 to its predominant ligand, Galectin-9, is required for Tim-3-mediated inhibition of tumorigenesis. Compared to vector control cells, Havcr2-OE B16-F10 melanoma variants bound significantly higher levels of recombinant Galectin-9 (Fig. S7E), confirming Galectin-9 binding by melanoma-expressed Tim-3. The enhancement or suppression of B16-F10 melanoma growth in C57BL/6 (Fig. 2A) or NSG mice (Fig. 2B) resulting from tumor cell-intrinsic Havcr2-KD or -OE, respectively, was abrogated in Galectin-9 null (Lgals9−/−) C57BL/6 recipients (Fig. 2C), underscoring Galectin-9 involvement in melanoma-Tim-3-dependent regulation of tumorigenesis. These findings uncover melanoma cell-Tim-3-Galectin-9 as a growth-suppressive axis.
Figure 2. Melanoma cell-Tim-3 inhibits murine tumor growth in a Galectin-9-dependent fashion.
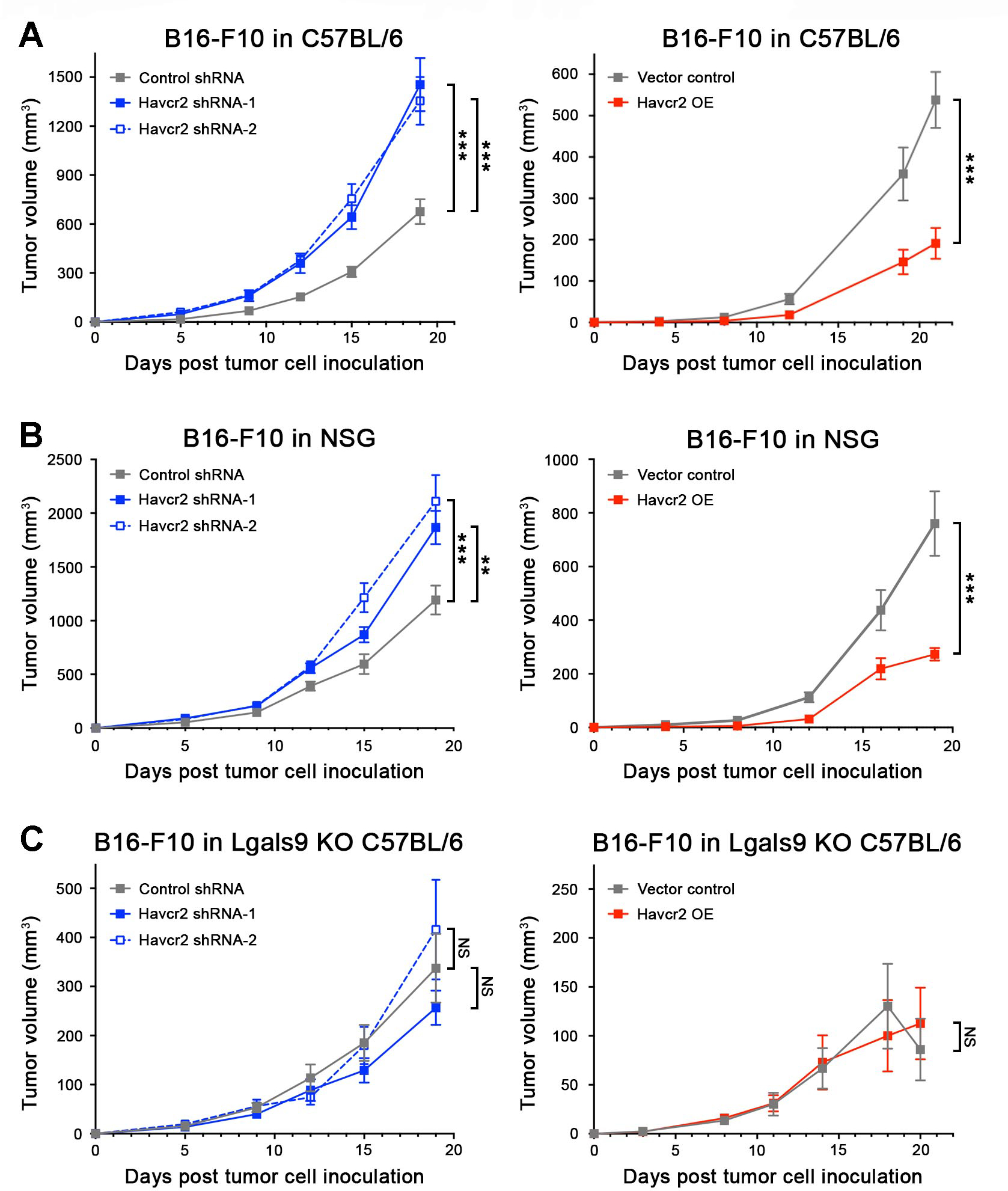
Tumor growth kinetics (mean ± SEM) of Tim-3 knockdown (Havcr2 shRNA-1/-2, left) and Tim-3-overexpressing (Havcr2 OE, right) versus shRNA or vector control B16-F10 cells, respectively, in A, immunocompetent C57BL/6, B, immunocompromised NSG, and C, Galectin-9 knockout (Lgals9−/− KO) C57BL/6 mice. All experiments were performed in n ≥ 10 mice per group on n = 2–3 independent occasions. **, p < 0.01; ***, p < 0.001; NS, not significant. See also Figure S7.
Tim-3 antibody blockade inhibits growth of immunogenic murine melanomas in T-cell-competent but promotes tumorigenesis in T-cell-deficient mice
Administration of a Tim-3 blocking Ab known to inhibit Galectin-9 binding (11) did not significantly alter growth of B16-F10 melanomas of poor immunogenicity (25) in T-cell-competent C57BL/6 mice (Fig. 3A), consistent with previous reports (6). In contrast, Tim-3 Ab treatment significantly inhibited growth of highly immunogenic (25) YUMMER1.7D4 tumors in C57BL/6 recipients, compared to controls (Fig. 3A). Conversely, the same Tim-3 blocking Ab promoted growth of three-dimensional in vitro tumor cultures (Fig. S7F) and tumorigenesis of both B16-F10 and YUMMER1.7D4 melanomas in T-cell-deficient NSG mice (Fig. 3B) expressing high tumoral levels of Havcr2 and Lgals9 (Fig. S8A–B). These results support the possibility that growth stimulation resulting from Ab-based melanoma cell-Tim-3 blockade might antagonize desired antitumor efficacy of T-cell-directed Tim-3 therapy.
Figure 3. Antibody-mediated Tim-3 blockade inhibits growth of immunogenic melanomas in immunocompetent mice but promotes tumorigenesis in T-cell-deficient hosts.
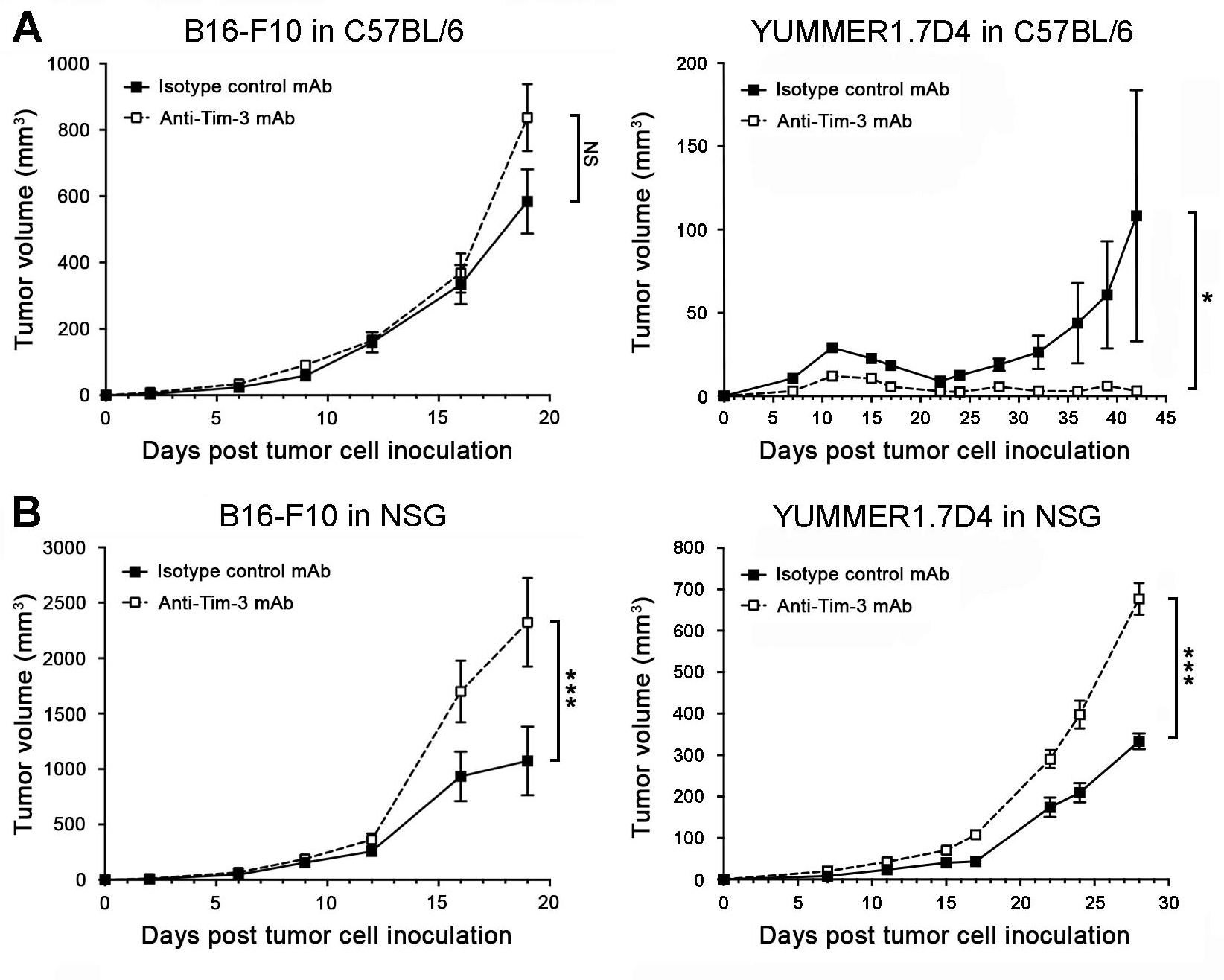
Tumor growth kinetics (mean ± SEM) of B16-F10 (left) or YUMMER1.7D4 (right) wild-type cells in A, C57BL/6 and B, NSG mice treated with Tim-3 blocking versus isotype-matched control monoclonal antibodies (mAbs). All experiments were performed in n ≥ 10 mice per group on n = 2 independent occasions. *, p < 0.05; ***, p < 0.001; NS, not significant. See also Figures S7–S8.
Melanoma-Tim-3 inhibits and its blockade promotes human tumor xenograft growth
We next examined the effects of melanoma-specific Tim-3-KD versus Tim-3-OE on human melanoma xenograft growth. Transduction of human A2058 and G361 melanoma cells with two distinct HAVCR2 shRNAs significantly inhibited (Fig. S9A), while infection with HAVCR2-OE constructs resulted in marked upregulation of both Tim-3 mRNA and protein expression (Fig. S9B). Similar to our findings in murine melanoma models (Fig. 2), HAVCR2-KD significantly increased (Fig. 4A), while HAVCR2-OE decreased human melanoma xenograft growth in NSG mice (Fig. 4B) and three-dimensional culture growth (Fig. S9C–D) compared to respective A2058 and G361 controls. Administration of an anti-human Tim-3 blocking Ab known to inhibit Galectin-9 binding (12) significantly increased human tumor xenograft growth in NSG mice (Fig. 5A) and also promoted A2058 and G361 in vitro three-dimensional growth (Fig. S9E). Consistent with the more pronounced growth-promoting effect of this human-specific (Fig. S9F) Tim-3 blocking Ab on G361 versus A2058 melanoma xenografts, particularly at early time points (<3 weeks, Fig. 5A), binding of the Ab to tumor target cells was significantly higher in G361 (23.9 ± 6.1%) compared to A2058 tumors (6.9 ± 2.5%) 3 weeks post tumor cell inoculation (mean ± SEM, Fig. 5B). Both HAVCR2 and LGALS9 were detected at significant levels in A2058 and G361 tumor xenografts (Fig. S10A–B). These results further support Tim-3 as a growth suppressor intrinsic to cancer cells, the inhibition of which stimulates tumorigenesis in both murine and human melanomas.
Figure 4. Tim-3 expression by human melanoma cells inhibits tumorigenesis.
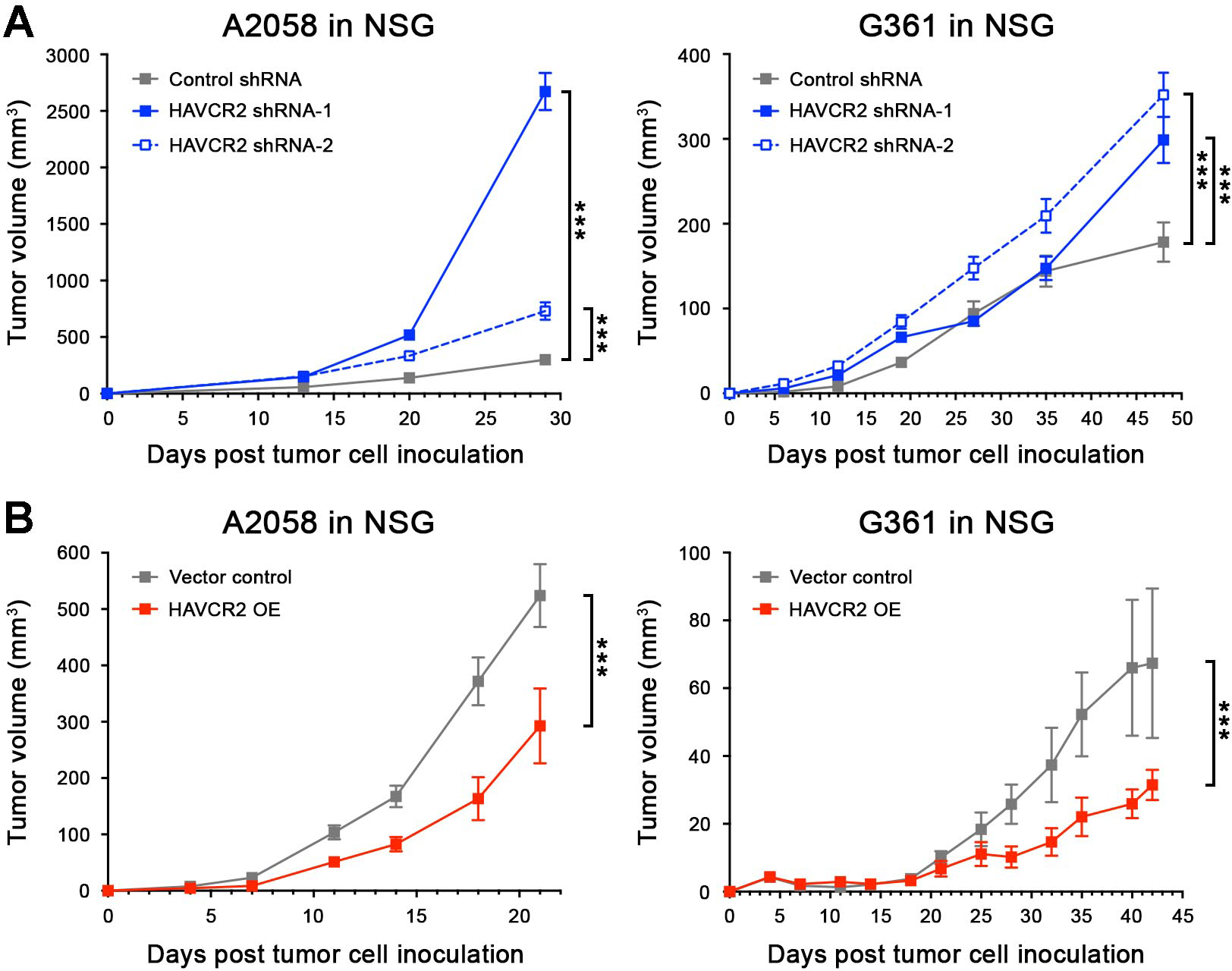
Tumor growth kinetics (mean ± SEM) in NSG mice of A, Tim-3 knockdown (HAVCR2 shRNA-1/-2) and B, Tim-3-overexpressing (HAVCR2 OE) versus respective control human A2058 (left) and G361 (right) melanoma cell inoculates (n = 10 each, representative of n = 3 independent experiments, respectively). ***, p < 0.001. See also Figure S9.
Figure 5. Antibody-mediated human melanoma-Tim-3 blockade promotes tumorigenesis.
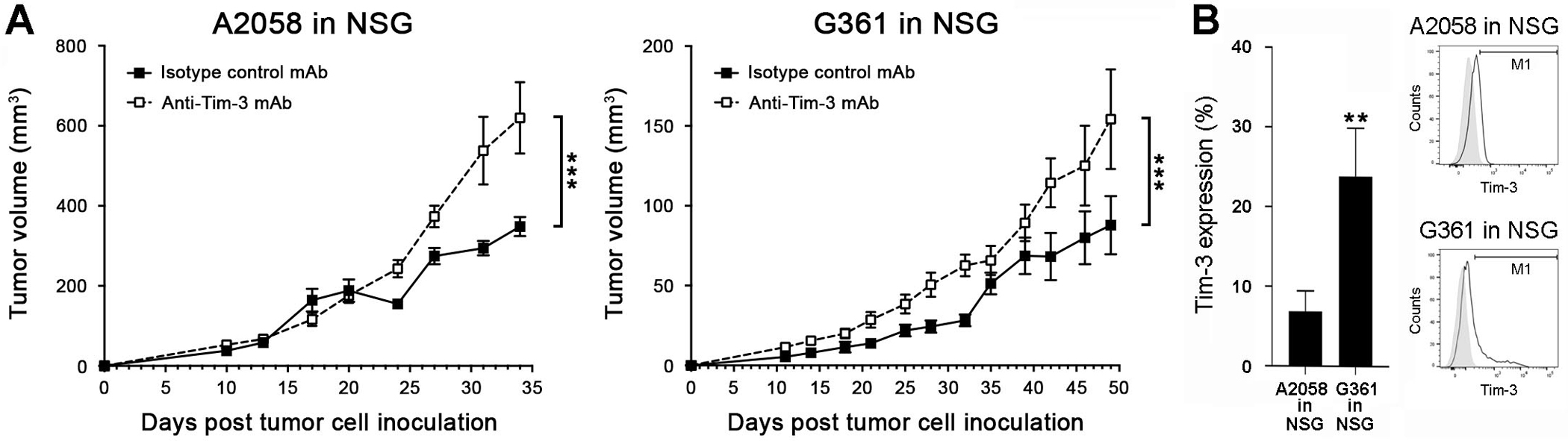
Tumor growth kinetics (mean ± SEM) of wild-type A, A2058 (left) and G361 cells (right) in NSG mice treated with human-specific anti-Tim-3 blocking versus isotype-matched control monoclonal antibodies (mAbs, n = 4–10 each, representative of n = 3 independent experiments). B, In vivo reactivity (% positive cells, mean ± SEM, left) of the anti-human Tim-3 blocking mAb used in A-B with nuclear GFP-labeled A2058 and G361 melanoma cells isolated from tumor xenografts 3 weeks post tumor cell inoculation into NSG mice (n = 4–6 each). Representative histogram plots are shown on the right. **, p < 0.01; ***, p < 0.001. See also Figure S10.
Inhibition of the melanoma-Tim-3 downstream signaling pathway, MAPK, reverses unwanted growth stimulation of melanoma cell-Tim-3 blockade
We next systematically analyzed melanoma cell-intrinsic Tim-3 effector pathways to identify candidate targets for reversal of unwanted protumorigenic effects of melanoma-Tim-3 inhibition. Specifically, we subjected human HAVCR2-OE A2058 and G361 and murine Havcr2-OE B16-F10 versus respective vector control cells to an unbiased phosphoarray encompassing 1,318 phospho-specific Abs covering more than 30 signaling pathways (29), followed by STRING protein association network analysis. We found three major clusters associated with melanoma-Tim-3 downstream signaling pathway activity, MAPK, NF-κB, and cell cycle regulators (Fig. 6A). The predominant phospho-protein cluster enriched among HAVCR2-OE melanoma cells consisted of pro-proliferative MAPK members with decreased phosphorylation relative to controls, including MEK1 (Fig. 6A), consistent with the observed growth-inhibitory activity of melanoma cell-Tim-3. Immunoblotting confirmed MAPK pathway suppression in HAVCR2-OE versus vector control A2058 (top) and G361 (bottom) cells, as evidenced by reduced phosphorylation of the MAPK effector molecules, MEK1/2 and ERK1/2 (Fig. 6B and Fig. S11A–B). Consistently, shRNA- or Tim-3 blocking Ab-mediated inhibition of melanoma cell-Tim-3 increased MEK1/2 and ERK1/2 phosphorylation compared to respective controls (Fig. 6B and Fig. S11A–B). We next examined whether MAPK pathway inhibition, using the FDA-approved MEK1/2 inhibitor, trametinib (30), can reverse unwanted growth stimulation of Ab-mediated melanoma-Tim-3 blockade. Accordingly, we treated NSG mice with a submaximal trametinib dose that significantly inhibits melanoma growth, but does not fully eliminate tumors (19), to enable assessment of Tim-3-dependent, pro-proliferative MAPK effects. This regimen significantly reversed Tim-3 Ab-induced growth of human A2058 and G361 melanoma xenografts (Fig. 6C). We next assessed the effect of combined Tim-3 Ab and MEK inhibition on melanoma growth in immunocompetent hosts. Similar to our findings in human melanoma cells (Fig. 6B), Ab-mediated Tim-3 blockade of YUMMER1.7D4 cells increased MEK1/2 phosphorylation compared to isotype control (Fig. 6D and Fig. S11C). Consistently, combined trametinib and Tim-3 Ab administration inhibited YUMMER1.7D4 tumor growth more effectively than either treatment alone in immunocompetent C57BL/6 mice when therapy was initiated the day of or fifteen days post tumor cell engraftment (Fig. 6E–F). Combined trametinib and Tim-3 Ab therapy completely eradicated tumors at the experimental endpoint in 7 of 10 and 2 of 10 mice, respectively, but only in 1 of 20 mice receiving Tim-3 monotherapy and none in any other control group (Fig. 6E–F). Together, these results identify MAPK as a melanoma-intrinsic Tim-3 receptor target, which is induced via melanoma cell-Tim-3 blockade to promote tumorigenesis. Consequently, MAPK inhibition counteracts the unwanted growth stimulation from melanoma-Tim-3 antagonism in T-cell null hosts and enhances desired antitumor activity of Tim-3 interference in T-cell-competent mice.
Figure 6. MEK inhibition reverses melanoma-Tim-3 blockade-mediated growth stimulation.
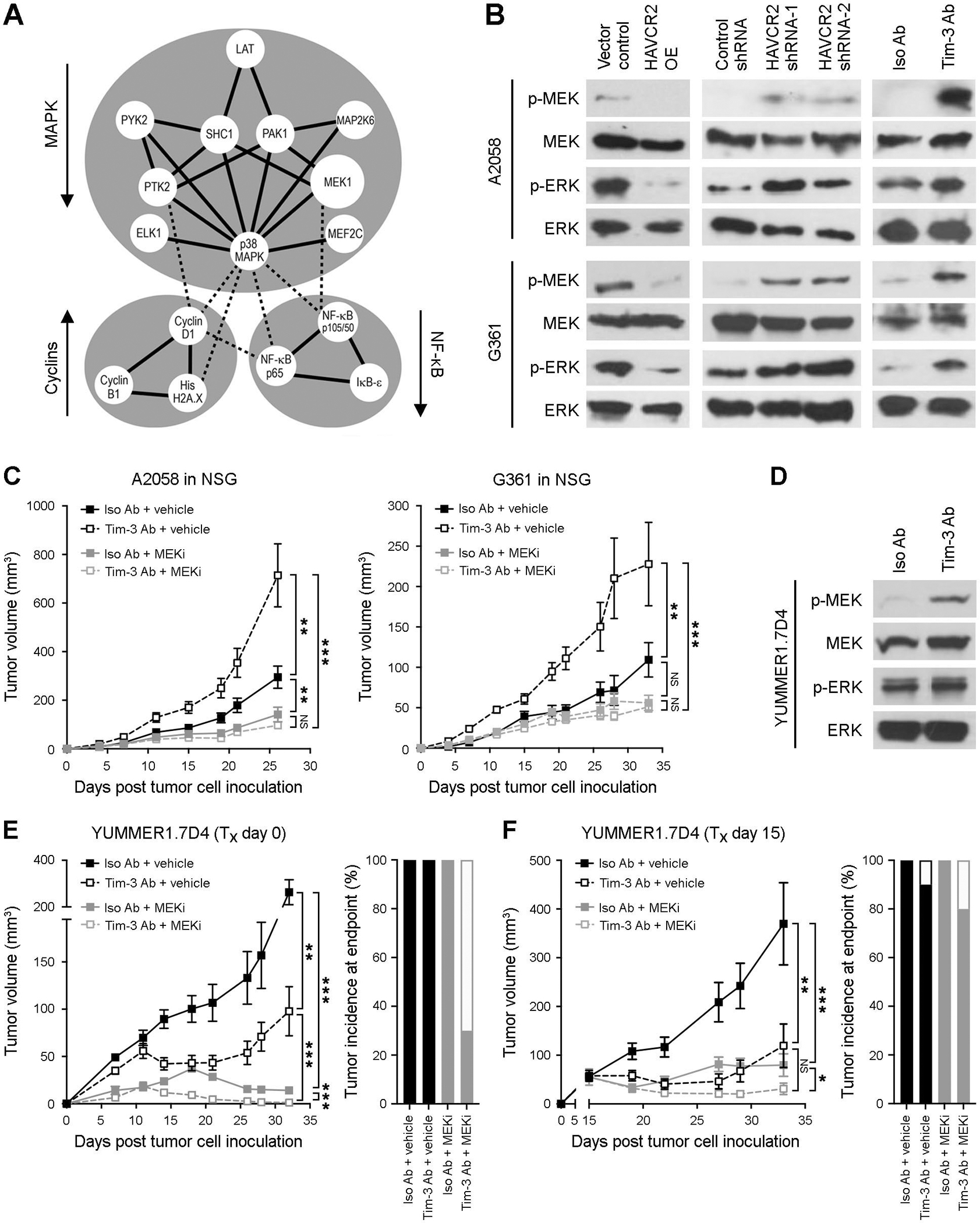
A, Protein-protein interaction map (STRING) of differentially phosphorylated proteins (Phospho Explorer Antibody Array) in Tim-3 (HAVCR2/Havcr2)-overexpressing (OE) versus vector control human A2058 and G361 or murine B16-F10 melanoma cells. Arrows indicate pathway activation (up) or inhibition (down); solid lines, strong protein-protein interactions; dashed lines, weaker protein–protein interactions. B, Immunoblots of phosphorylated (p) and total MEK1/2 and ERK1/2 in vector control versus HAVCR2 OE (left), control shRNA versus HAVCR2 knockdown (shRNA-1/-2, middle), and Tim-3 blocking versus isotype control Ab-treated (right) human A2058 (top) and G361 (bottom) cells. C, Tumor growth kinetics (mean ± SEM) of human A2058 (left) and G361 (right) cells in NSG mice treated with human-specific Tim-3 blocking versus isotype control mAbs, with or without submaximal dosage (0.15 mg/kg/d, p.o.) of the MEK inhibitor, trametinib. D, Immunoblots of p- and total MEK1/2 and ERK1/2 in anti-Tim-3 versus isotype control Ab-treated murine YUMMER1.7D4 melanoma cells. E-F, Tumor growth kinetics (left, mean ± SEM) and tumor incidence at experimental endpoint (right) of murine YUMMER1.7D4 cells in C57BL/6 mice treated (Tx) with anti-murine Tim-3 blocking versus isotype control mAbs, with or without trametinib (as in C) starting on E, day 0 or F, day 15 (n = 10 mice per treatment group, respectively). * p < 0.05; **, p < 0.01; ***, p < 0.001; NS, not significant. See also Figure S11.
DISCUSSION
Tim-3 is an immune checkpoint receptor that triggers T-effector cell exhaustion to promote tumor immune escape (9). Accordingly, Tim-3 has mainly been studied on tumor-infiltrating T-cells (31). Using several independent techniques, our study reveals functional Tim-3 expression directly by cancer cells, and co-expression with Galectin-9, in established murine and human lines and patient melanomas. These findings add another layer of complexity to Tim-3 immunobiology, by recognizing tumor cell-intrinsic, in addition to T-cell immunoregulatory Tim-3:Galectin-9 pathway functions, in the TME of solid malignancies. Similarly, Tim-3 is expressed by both LSCs and normal T-cells in hematologic disorders (32). Tim-3:Galectin-9 interactions maintain LSC self-renewal through an autocrine stimulatory loop in preclinical models of myeloproliferative disease (12). As in leukemias, we found that Tim-3 expression is restricted to subpopulations of melanoma cells, which are nevertheless involved in tumor maintenance. In contrast to LSC-Tim-3, however, the melanoma cell-Tim-3:Galectin-9 axis suppresses tumor growth in multiple models, thus more closely resembling its anti-proliferative effects in T-cells (9). Moreover, the Tim-3 receptor stimulates downstream NF-κB and MAPK signaling in LSCs (12), while inhibiting said pathways in T-cells (8) and melanoma cells, as shown in our study (Fig. 7). Consistently, whereas Tim-3 and Galectin-9 are associated with tumor virulence in leukemias (32), Tim-3 and Galectin-9 expression in melanomas correlate with improved patient survival in some datasets (21), and decreases with tumor stage, as found herein. We are cognizant that these analyses do not discriminate between transcript expression among cell types. Nevertheless, this correlative data is supportive of growth-suppressive Tim-3:Galectin-9 functions in melanoma.
Figure 7. Therapeutic implications of the melanoma cell intrinsic-Tim-3 signaling axis.
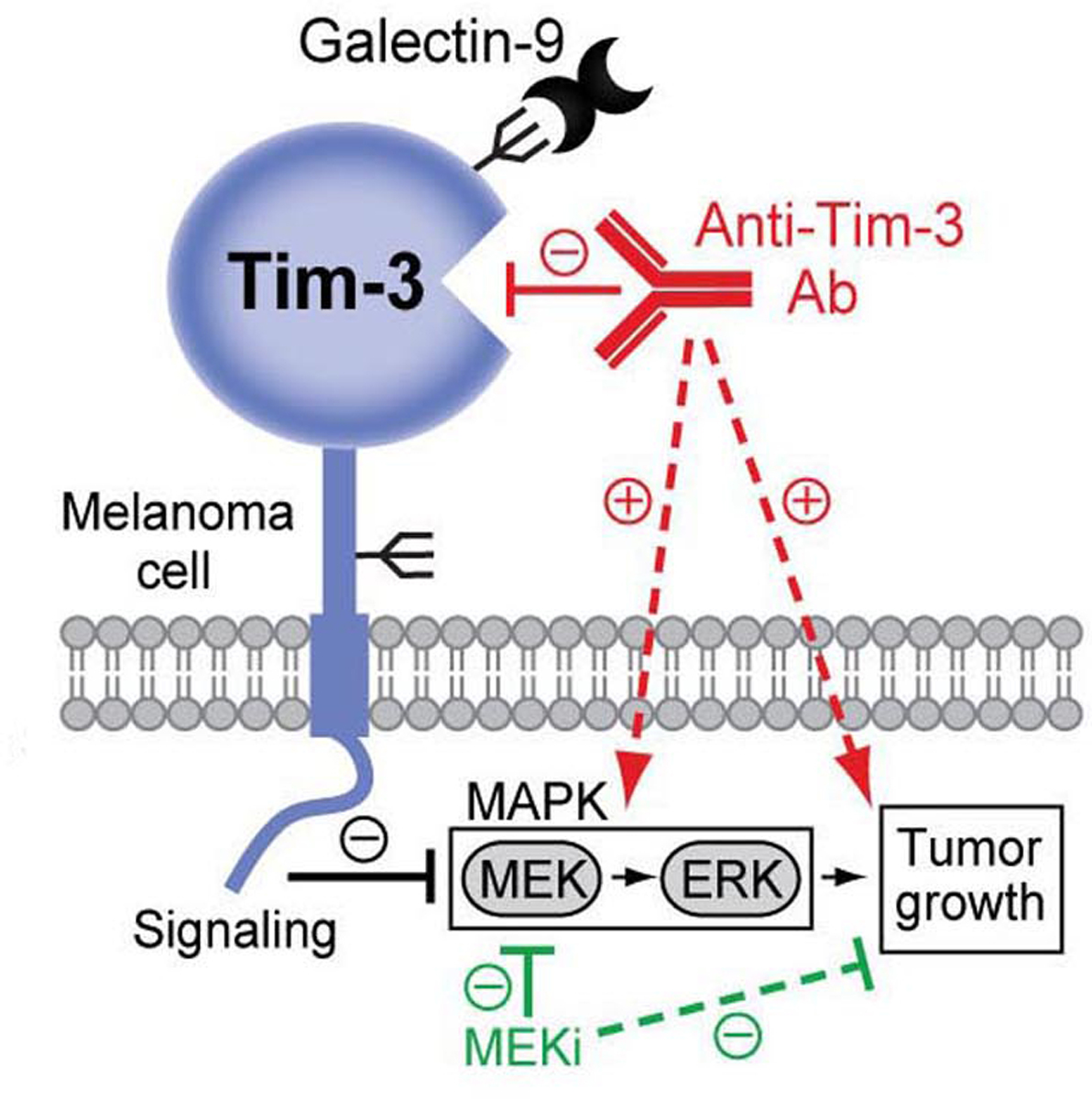
The melanoma cell-Tim-3 receptor binds Galectin-9 and suppresses MAPK signaling to inhibit tumor growth (black, solid line). Ab-based melanoma-Tim-3 blockade (red, solid line) activates MAPK effectors, MEK and ERK, thereby unintentionally promoting tumorigenesis (red, dashed lines). Combination therapy with MEK inhibitors (MEKi) reverses Tim-3 Ab-mediated MEK activation (green, solid line), leading to desirable melanoma growth suppression (green, dashed line).
Disease-associated and cell context-dependent differences in Tim-3 receptor signaling and function could result from tissue-associated variations in Tim-3 ligand expression and possibly also immunoregulatory activity. Indeed, Tim-3 interacts with multiple ligands with divergent tissue distribution and roles in cell proliferation and immune homeostasis, including Galectin-9, CEACAM-1, HMGB1, and PtdSer (9). Whether differences in Tim-3 ligand expression, binding, or immunomodulation contribute to opposing Tim-3 signaling between distinct cell types or tissues will require careful dissection in a future dedicated study. Nevertheless, our work involving Galectin-9-deficient mice clearly identifies Galectin-9 as a critical mediator of Tim-3-dependent growth suppression in melanoma cells, like in T-cells (9), and in contrast to protumorigenic Galectin-9 effects in leukemic cells (12). Intriguingly, melanoma growth was markedly suppressed in Galectin-9 KO versus wildtype mice, but stimulated via melanoma-Tim-3 inhibition, thus implying additional Galectin-9 roles in tumorigenesis independent of melanoma-Tim-3. Indeed, Galectin-9 interacts with multiple alternative receptors, some of which have known protumorigenic functions, including PD-1 (22).
Several ICIs targeting Tim-3 have entered clinical IO trials for melanoma and other cancers (8). Therefore, our findings that melanoma-Tim-3 interference stimulates tumorigenesis are concerning, because inadvertent blockade of the melanoma cell-Tim-3:Galectin-9 axis in patients could antagonize desired efficacy of T-cell-directed Tim-3 therapy. One might thus predict that clinical-grade Tim-3 inhibitors with low affinity for melanoma cell-Tim-3 would elicit greater clinical benefit than those with high melanoma-Tim-3 binding. Because unwanted blockade of melanoma-Tim-3 by clinical inhibitors might pose an obstacle to successful T-cell-Tim-3 therapy, we believe that consideration in trial design and interpretation is warranted. It should be noted that therapeutic outcomes might not merely depend on relative Tim-3 Ab affinity for T-cells versus melanoma cells, but also on melanoma immunogenicity or reactivity with other Tim-3-expressing TME cell types, including dendritic cells (33), myeloid (14), and natural killer cells (34). Moreover, while inhibition of cancer cell-Tim-3 promotes tumor growth in some malignancies, as reported herein, it suppresses tumorigenesis in other cancers, such as leukemias (12), to synergize with T-cell-Tim-3 blockade. Tim-3 therapeutic efficacy could thus vary between tumor entities based on disease-specific variations in cancer cell-intrinsic or TME cell-Tim-3 pathway functions.
In scenarios where inadvertent antagonism of cancer cell-intrinsic Tim-3 stimulates growth, concurrent targeting of protumorigenic Tim-3 downstream pathways could improve outcomes. Indeed, we found that pharmacologic inhibition of the melanoma-Tim-3 signaling effector, MEK1/2, reverses unwanted tumorigenesis caused by melanoma-Tim-3 blockade. Because MEK inhibitors, including trametinib, have been approved by the FDA for the treatment of melanomas harboring BRAF-activating mutations (30), they should be considered for Tim-3-based combination therapy in patients. In support, combined Tim-3 Ab and trametinib administration inhibited both tumor initiation and growth of established immunogenic YUMMER1.7D4 melanomas in immunocompetent hosts to a greater extent than either therapy alone. In addition to thwarting tumor-intrinsic oncogenic signaling, MEK inhibitors synergistically promote T-cell immunity when combined with other ICI regimens (35), further underscoring the promise of dual Tim-3-MEK inhibitor modalities. In melanomas lacking BRAF mutations, targeting of other Tim-3 signaling mediators (12,23), such as NF-κB, might provide an alternative strategy for reversing unwanted consequences of tumor cell-Tim-3 blockade.
In summary, we identify an additional role for Tim-3 as a growth-inhibitory receptor intrinsic to melanoma cells. Because melanoma-Tim-3 blockade stimulates MAPK-dependent tumorigenesis, it could antagonize benefit from T-cell-directed Tim-3 therapy. This provides a therapeutic rationale for implementing Tim-3 Abs with greater T-cell selectivity or combining Tim-3 with MAPK pathway inhibitors (Fig. 7). Such cell type-directed Tim-3 targeting strategies could help overcome unintended protumorigenic effects of melanoma-Tim-3 interference and maximize desired anticancer T-cell activity.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Tim-3 is a growth-suppressive receptor intrinsic to melanoma cells, the blockade of which promotes MAPK-dependent tumorigenesis and thus counteracts antitumor activity of T-cell-directed Tim-3 inhibition.
ACKNOWLEDGMENTS
We thank Dr. Michael Croft (La Jolla Institute for Allergy and Immunology, La Jolla, CA) for providing Galectin-9-deficient mice for our study.
FINANCIAL SUPPORT:
This work was supported by a Merck-Melanoma Research Alliance Young Investigator Award, a V Foundation for Cancer Research Scholar Grant, an Outrun the Sun Melanoma Research Scholar Award, a Fund to Sustain Research Excellence from the Brigham Research Institute, Brigham and Women’s Hospital (to S.R.B.), a Developmental Project Grant from the Harvard Stem Cell Institute, and NIH/NCI grants R01CA247957, R01CA258637 (to S.R.B. and T.S.), and R01CA190838 (to T.S.). Partial support was provided by a Mizutani Foundation for Glycoscience Research Grant, NIH/NCI grant U01CA225644, and NIH/NIAID grant R21AI146368 (to C.J.D.), a Walter Benjamin Fellowship from the German Research Foundation (DFG, to E.R.), a Martin Mihm Fellowship from the German Society of Dermatopathology (ADH, to M.V.H.), and a DFG Scholarship (to A.B.) as part of Germany’s ImmunoSensation2 Excellence Initiative (EXC 2151).
Footnotes
CONFLICT OF INTEREST: The authors declare no potential conflict of interest.
REFERENCES
- 1.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 2015;161:205–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalbasi A, Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol 2020;20:25–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun 2016;7:10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shayan G, Srivastava R, Li J, Schmitt N, Kane LP, Ferris RL. Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunol 2017;6:e1261779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med 2010;207:2175–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res 2011;71:3540–51 [DOI] [PubMed] [Google Scholar]
- 7.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med 2010;207:2187–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Acharya N, Sabatos-Peyton C, Anderson AC. Tim-3 finds its place in the cancer immunotherapy landscape. J Immunother Cancer 2020;8:e000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wolf Y, Anderson AC, Kuchroo VK. TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol 2020;20:173–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev 2017;276:97–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sabatos-Peyton CA, Nevin J, Brock A, Venable JD, Tan DJ, Kassam N, et al. Blockade of Tim-3 binding to phosphatidylserine and CEACAM1 is a shared feature of anti-Tim-3 antibodies that have functional efficacy. Oncoimmunol 2018;7:e1385690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kikushige Y, Miyamoto T, Yuda J, Jabbarzadeh-Tabrizi S, Shima T, Takayanagi S, et al. A TIM-3/Gal-9 Autocrine Stimulatory Loop Drives Self-Renewal of Human Myeloid Leukemia Stem Cells and Leukemic Progression. Cell Stem Cell 2015;17:341–52 [DOI] [PubMed] [Google Scholar]
- 13.Cao Y, Zhou X, Huang X, Li Q, Gao L, Jiang L, et al. Tim-3 expression in cervical cancer promotes tumor metastasis. PLoS One 2013;8:e53834. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Komohara Y, Morita T, Annan DA, Horlad H, Ohnishi K, Yamada S, et al. The Coordinated Actions of TIM-3 on Cancer and Myeloid Cells in the Regulation of Tumorigenicity and Clinical Prognosis in Clear Cell Renal Cell Carcinomas. Cancer Immunol Res 2015;3:999–1007 [DOI] [PubMed] [Google Scholar]
- 15.Zhuang X, Zhang X, Xia X, Zhang C, Liang X, Gao L, et al. Ectopic expression of TIM-3 in lung cancers: a potential independent prognostic factor for patients with NSCLC. Am J Clin Pathol 2012;137:978–85 [DOI] [PubMed] [Google Scholar]
- 16.Clark CA, Gupta H, Sareddy GR, Pandeswara S, Lao S, Yuan B, et al. Tumor-intrinsic PD-L1 signals regulate cell growth, pathogenesis and autophagy in ovarian cancer and melanoma. Cancer Res 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, et al. Melanoma Cell-Intrinsic PD-1 Receptor Functions Promote Tumor Growth. Cell 2015;162:1242–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mo X, Zhang H, Preston S, Martin K, Zhou B, Vadalia N, et al. Interferon-gamma Signaling in Melanocytes and Melanoma Cells Regulates Expression of CTLA-4. Cancer Res 2018;78:436–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanlorenzo M, Vujic I, Floris A, Novelli M, Gammaitoni L, Giraudo L, et al. BRAF and MEK Inhibitors Increase PD-1-Positive Melanoma Cells Leading to a Potential Lymphocyte-Independent Synergism with Anti-PD-1 Antibody. Clin Cancer Res 2018;24:3377–85 [DOI] [PubMed] [Google Scholar]
- 20.Wiersma VR, de Bruyn M, van Ginkel RJ, Sigar E, Hirashima M, Niki T, et al. The glycan-binding protein galectin-9 has direct apoptotic activity toward melanoma cells. J Invest Dermatol 2012;132:2302–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holderried TAW, de Vos L, Bawden EG, Vogt TJ, Dietrich J, Zarbl R, et al. Molecular and immune correlates of TIM-3 (HAVCR2) and galectin 9 (LGALS9) mRNA expression and DNA methylation in melanoma. Clin Epigenetics 2019;11:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang R, Sun L, Li CF, Wang YH, Yao J, Li H, et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat Commun 2021;12:832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee J, Su EW, Zhu C, Hainline S, Phuah J, Moroco JA, et al. Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol Cell Biol 2011;31:3963–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seki M, Oomizu S, Sakata KM, Sakata A, Arikawa T, Watanabe K, et al. Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T cells, and regulates experimental autoimmune arthritis. Clin Immunol 2008;127:78–88 [DOI] [PubMed] [Google Scholar]
- 25.Wang J, Perry CJ, Meeth K, Thakral D, Damsky W, Micevic G, et al. UV-induced somatic mutations elicit a functional T cell response in the YUMMER1.7 mouse melanoma model. Pigment Cell Melanoma Res 2017;30:428–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016;352:189–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018;173:400–16 e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nold-Petry CA, Lo CY, Rudloff I, Elgass KD, Li S, Gantier MP, et al. IL-37 requires the receptors IL-18Ralpha and IL-1R8 (SIGIRR) to carry out its multifaceted anti-inflammatory program upon innate signal transduction. Nat Immunol 2015;16:354–65 [DOI] [PubMed] [Google Scholar]
- 30.Flaherty KT, Hodi FS, Fisher DE. From genes to drugs: targeted strategies for melanoma. Nat Rev Cancer 2012;12:349–61 [DOI] [PubMed] [Google Scholar]
- 31.Liu Y, Cai P, Wang N, Zhang Q, Chen F, Shi L, et al. Combined blockade of Tim-3 and MEK inhibitor enhances the efficacy against melanoma. Biochem Biophys Res Commun 2017;484:378–84. [DOI] [PubMed] [Google Scholar]
- 32.Jan M, Chao MP, Cha AC, Alizadeh AA, Gentles AJ, Weissman IL, et al. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc Natl Acad Sci 2011;108:5009–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Mingo Pulido A, Gardner A, Hiebler S, Soliman H, Rugo HS, Krummel MF, et al. TIM-3 Regulates CD103(+) Dendritic Cell Function and Response to Chemotherapy in Breast Cancer. Cancer Cell 2018;33:60–74 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.da Silva IP, Gallois A, Jimenez-Baranda S, Khan S, Anderson AC, Kuchroo VK, et al. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol Res 2014;2:410–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016;44:609–21 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are publicly available in Genbank under accession numbers MW055427–29 or from the corresponding authors upon reasonable request.
See also the Supplementary Methods.