

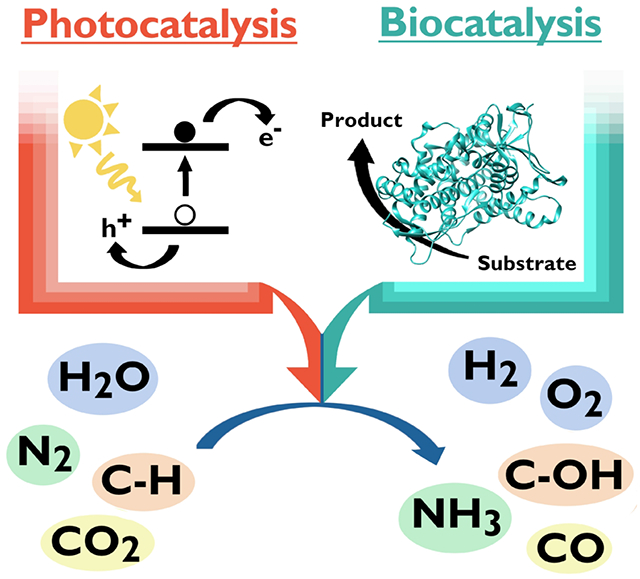
Graphical Abstract
Keywords: Biocatalysis, photocatalysis, C-H activation, small-molecule activation, artificial photosynthesis
1. Introduction
Biocatalysis is a booming field racing to meet challenges in the research lab and in industry. Generally, biocatalysis is defined as the use of biological systems to catalyze chemical reactions. The biological systems are most often purified enzymes, but also can be components of cell lysate or even living organisms (1). Interest in developing biocatalysis is longstanding, and these processes already enhance our lives in a number of ways (2–4). Enzyme chemistry has been tied to industry for centuries or more, namely in the processing of food, drinks, and paper products. An increased understanding of enzyme chemistry and the development of technologies including recombinant DNA technology, heterologous protein expression, and, most recently, directed evolution has led to significant growth in the use of biocatalysis in industry (1,5–10). Today, the industrial enzyme market is worth billions of dollars, with projected growth of billions more over the coming decade (8) Expanding this industry to new reactions, including those not known in nature, with engineered enzymes is increasingly possible (7,11,12).
Compared to traditional industrial methods, one appeal of biocatalysis is the mild conditions under which the chemistry is carried out (4). While many industrial processes use harsh conditions, equivalent reactions in nature are accomplished at mild temperatures and one atmosphere of pressure using earth-abundant metals. For example, the reduction of N2 to NH3 (nitrogen fixation) is a critically important reaction for sustaining life. The reaction is carried out at ambient temperatures and pressures by nitrogenase enzymes in select microorganisms, providing a useable source of nitrogen for life on earth (13). In contrast, the Haber-Bosch process, the industrial equivalent of biological nitrogen fixation, produces NH3 from N2 and H2 on a scale capable of sustaining modern society, but requires high temperatures (650-750 K), high pressures (100 atm), and non-renewable natural gas to generate hydrogen (14,15).
Another appeal of biocatalysis is the ability to carry out reactions with high selectivity while maintaining the aforementioned benefits (10). An example that has recently penetrated industry is the synthesis of sitagliptin, the active ingredient in a drug used to treat diabetes. The best performing large-scale synthesis of sitagliptin relied on a rhodium catalyst that functioned under high pressures (18 atm) (10,16,17). Recently, the synthesis was reinvented to be catalyzed by a transaminase created through directed evolution. This transaminase route circumvents the drawbacks of the rhodium-based synthesis while maintaining high selectivity (>99.95% enantiomeric excess of the desired product) and improving overall yields (16).
While the ability of enzymes to accomplish selective reactions under benign conditions attracts the admiration of chemists, complex mechanisms in biological systems underlie this success. For example, in natural biocatalytic processes, “helper” proteins and energy-carrying molecules are needed to drive processes forward. The aforementioned fixation of N2 by nitrogenase enzymes requires eight sequential electron transfers that are dependent on cofactors, reducing agents, and electron-transfer proteins to generate two moles of NH3. Furthermore, 16 moles of ATP are hydrolyzed in this process. The need to use, recycle, or replace expensive components like NAD(P)H and ATP that allow enzymes to function is a major challenge to the broader application of enzyme chemistry (10,18). A promising approach to overcome this limitation is to couple enzymatic processes with light absorption to create photocatalytic systems that draw on the favorable properties of enzymes while minimizing consumption of expensive cofactors and the use of “helper” proteins such as those that donate electrons.
In photocatalysis, the absorption of light is used to drive a catalytic reaction. Photocatalysis is a large and varied field that, alongside biocatalysis, is showing significant growth (19–21). Driving thermodynamically uphill chemical reactions with light has enabled diverse catalysis, including a range of synthetic organic transformations and small-molecule activations (22–28). Light-driven reactions often have auspicious properties not present in dark reactions. Light absorption can create excited states that are highly oxidizing or reducing (29,30), renew oxidants and reductants in a catalytic cycle to eliminate stoichiometric additives (23,31), and create access to radical species (32) that drive reactions (23,28). An inspiration for the continuing growth of photocatalysis research is at the convergence of photochemistry and biocatalysis: photosynthesis. It has long been desired that the sun, a renewable resource, would replace the non-renewable feedstocks that drive human life and industry (20,25,26,33–35).
While there are many ways in which light may provide the energy to drive a reaction, the process as inspired by photosynthesis contains several fundamental steps: light absorption (1), charge separation (2), charge accumulation (3), catalysis (4), and replenishment of electrons (5) (numbers indicate steps as illustrated in Fig. 1) (20,21,34,36–38). Reactions are initiated through light absorption (1). In natural photosynthetic reactions, absorption of visible light is facilitated by light-harvesting pigment-protein complexes that efficiently capture and funnel low-energy solar irradiation (39). These complexes inspire the properties of light-absorbing components, commonly metal complexes or nanoparticles, used in light-driven catalysis [Fig. 2]. Strong and size-tunable light absorption is desired, and the light-absorbing molecule should possess a long-lived excited state to facilitate charge separation (2). Formation and efficient separation of an electron-hole pair are carried out using complex molecular architectures in natural photosynthesis (39). A variety of strategies have been implemented in artificial systems, such as directly tethering molecules, to mitigate energy loss and charge recombination (34,40). As light is absorbed, charge is accumulated to fuel catalysis (3). Catalysis of the desired reaction is carried out using reducing or oxidizing equivalents produced (4). Often, a sacrificial electron donor is used (5), but when possible, it is desired to replace sacrificial components by coupling reductive and oxidative processes (for example, coupling water oxidation and proton reduction) (37).
Fig. 1.
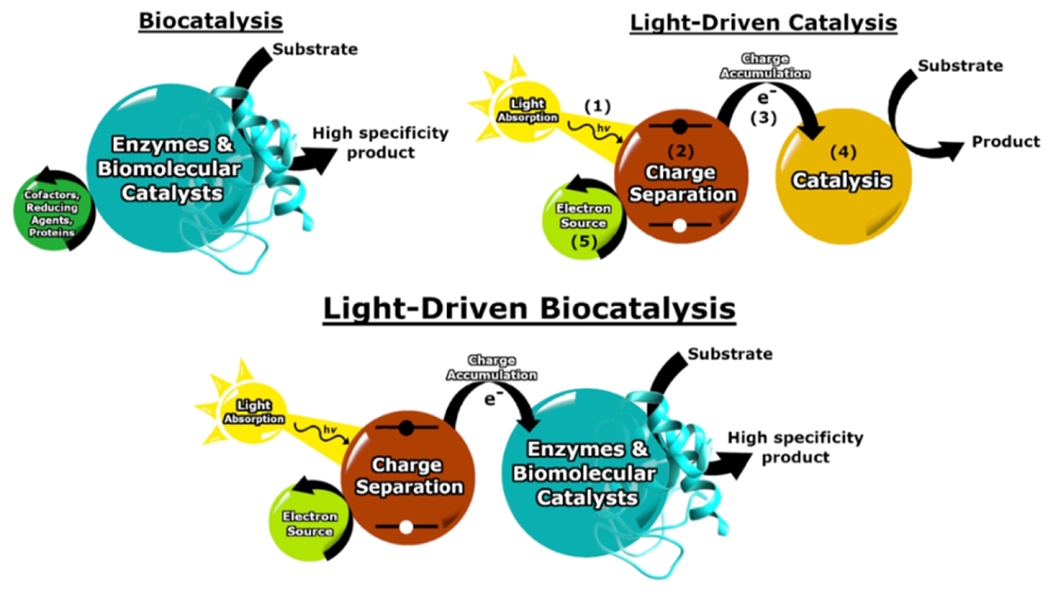
A schematic summary of biocatalysis, light-driven catalysis, and the merging of both processes. While these processes are represented by common steps for simplicity, in general the steps may be coupled in a variety of ways, such as charge separation directly fueling catalysis.
Fig. 2.
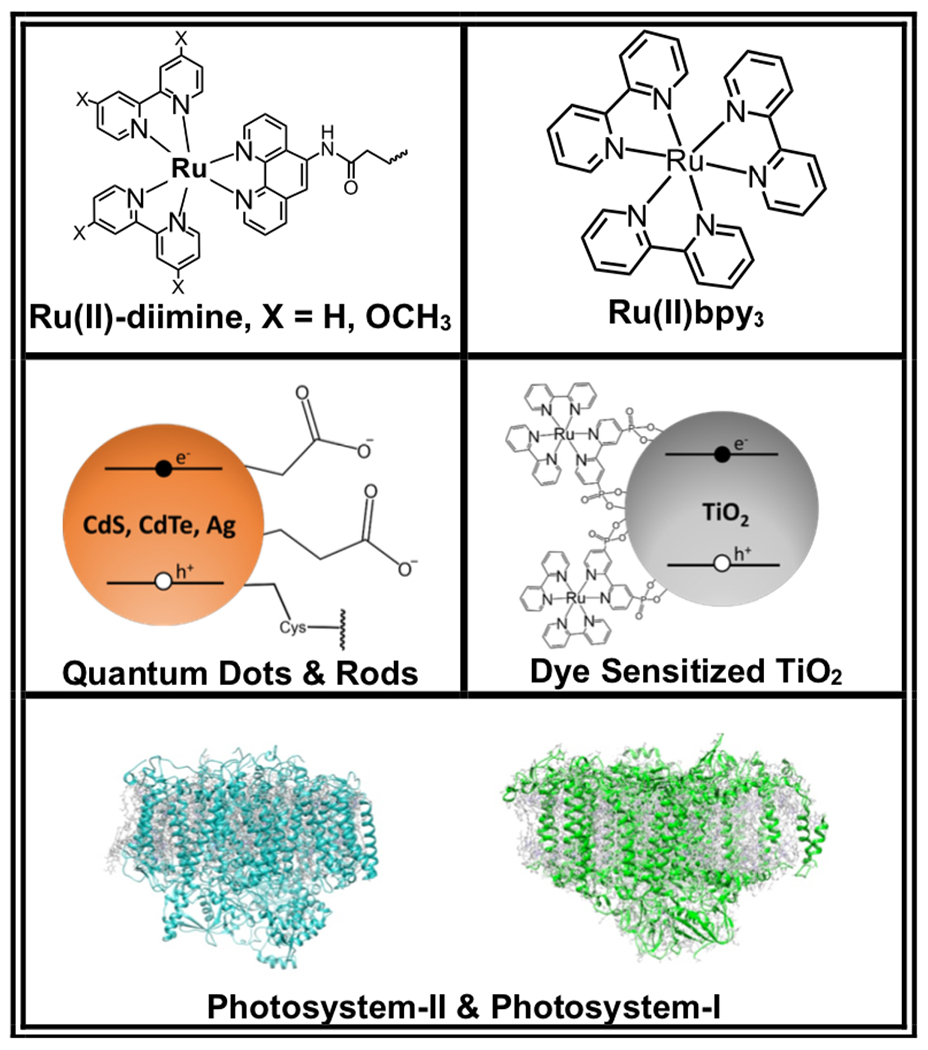
Examples of photosensitizers used in work discussed herein.
The convergence of photocatalysis and biocatalysis capitalizes on the advantages of both fields [Fig. 1]. Fueling biocatalytic reactions with light can overcome the need for costly cofactors while providing a potentially environmentally friendly route to important high specificity chemical conversions. Recently, several aspects of this topic have been reviewed. These reviews include strategies toward pairing natural enzymes with a variety of photosensitizers, using light to fuel cofactor regeneration, and building in vivo mechanisms for photobiocatalysis (41–44). In addition, new directions to combine photocatalysis and biocatalysis are constantly emergent, such as using photoexcitation to promote promiscuous activities in naturally occurring enzymes (45,46). In this mini-review, we place an emphasis on chemistry facilitated by engineered enzymes and biomolecular catalysts that are activated by electron transfer from a photosensitizer. The selected examples included herein are sorted into reactions of importance to research and human industry, including C-H oxidation, proton reduction, water oxidation, CO2 reduction, and N2 reduction. These examples illustrate the ability of light-driven biocatalysis to rise to the occasion of catalyzing challenging transformations.
2. C-H Oxidation
The activation and functionalization of C-H bonds is one of the most important reactions in chemistry. Over several decades, there have been many leaps forward in efforts to selectively and efficiently perform these reactions (47,48). The strength and abundance of C-H bonds in a wide range of target molecules makes selective functionalization a major challenge; this is particularly the case for C(sp3)-H bonds (49). While challenging, these reactions have critical importance for installation of C-O, C-N, C-C, or C-X bonds with regio- and stereo-selectivity. Research in synthetic organic chemistry has uncovered elegant techniques for selective C-H functionalization in complex molecules, often focusing on modifications within precursor molecules that lead to correct final products (49–52). In addition, catalysts have been created to promote selective functionalization, with many 4d, 5d, (53–55) and some 3d transition metal catalysts (56,57) developed to carry out various reactions. Recently, attention has cycled back to perhaps the most traditional method, drawing on nature’s incredible ability to selectively catalyze C-H activation (12,49). Pioneering work in the field of directed evolution has given a massive boost to push biocatalytic C-H activation beyond nature’s substrates (12,58,59). The use of enzymes tailored to functionalize complex scaffolds has already begun permeating industrial pharmaceutical syntheses (10,16,17).
Many classes of enzymes can perform C-H oxidation reactions. Among the best studied are the cytochrome P450s. The enzymes in this large family have a variety of roles in nature (48,60). The defining characteristic of P450 enzymes is a heme (iron protoporphyrin IX) prosthetic group coordinated to the enzyme through a proximal Cys thiol(ate). Nearly all P450 enzymes are monooxygenases, using molecular oxygen to insert an oxygen atom into a substrate [Equation 1] (60). One of their most commonly performed reactions is hydrocarbon hydroxylation [Fig. 3] (61,62). Catalysis relies on reducing equivalents from NAD(P)H, with electrons delivered to the heme active site by a variety of redox partners depending on the particular P450. The need for stoichiometric addition of costly nucleotide reductants to drive catalysis is a limitation to realizing the full potential of cytochrome P450 enzymes.
| (Equation 1) |
Fig. 3.
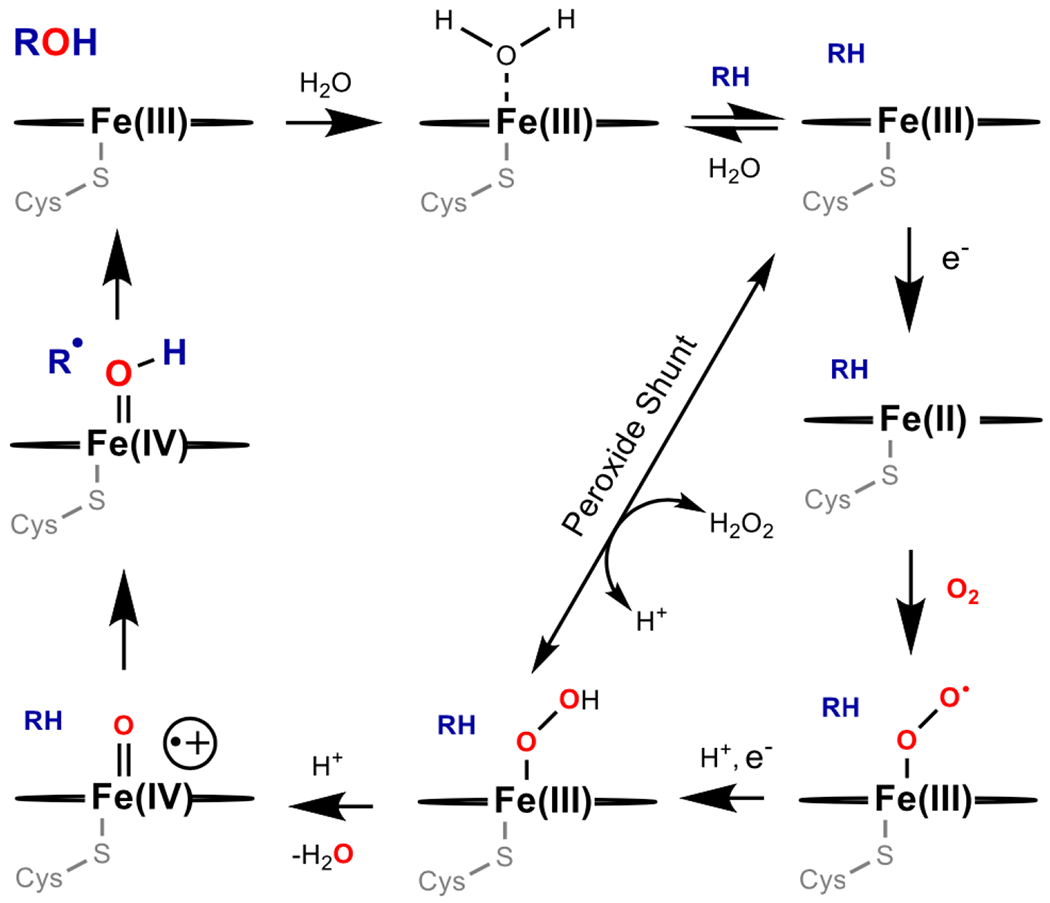
A general mechanism for C-H hydroxylation catalyzed by cytochrome P450.
Pioneering work demonstrated that direct coordination of a ruthenium(II) diimine photosensitizer to a P450 enzyme promoted photooxidation (63). Since this promising discovery, progress has been made toward using light to drive the oxidation of a range of substrates using Ru(II)-modified P450s (18,63–68). Cytochrome P450 BM3 (CYP102A1) from Bacillus megaterium has been an attractive target in this work because it demonstrates a particularly high rate for hydroxylation reactions among reported P450 enzymes (65,69). The thermostability of P450 BM3 makes it an attractive candidate for translation to industrial use (61). In order to drive P450 BM3 chemistry using light rather than NAD(P)H, photoactive Ru(II)-diimine complexes [Fig. 2] were designed to be linked covalently to the enzyme through reaction of a surface cysteine with iodoacetamide derivates of a phenanthroline ligand (PhenA) bound to Ru(II).
Two hybrid P450 BM3 enzymes, Ru-K97C-BM3 and Ru-Q397C-BM3, were created to carry out the light-driven hydroxylation of lauric acid (64,70). The triple-mutant hybrids contain non-native cysteine residues, Cys97 and Cys397, each of which is positioned near the heme for binding of an Ru(II)-diimine sensitizer. Two more mutations of native cysteine residues (C62A and C156S) ensure site-selective installation of the sensitizer (63). When the hybrids were combined with sodium diethyldithiocarbamate as a sacrificial reductant and dioxygen as an oxygen atom source, turnover numbers (TONs) up to 80 (mol product/mol enzyme) were observed in the light-driven regio-selective hydroxylation reaction (70). Results suggest that the generation of reactive oxygen species (ROS) by the hybrids drives catalysis, rather than direct reduction of heme by the reductively-quenched Ru(I) sensitizer. The ancillary ligands on the Ru(II)-diimine sensitizer were modified to produce an Ru(I) species capable of directly reducing the ferric heme, and binding location of the sensitizer was varied.
The result of optimization was a more active and thermostable enzyme, singly mutated Ru-sL407C-BM3, in which the sensitizer selectively binds to Cys407 [Fig. 4] (65,64). With visible light irradiation and in the presence of diethyldithiocarbamate, Ru-sL407C-BM3 shows TONs up to 935 (mol product/mol enzyme) and rates up to 125 mol product/(mol enzyme/min) for the light-driven hydroxylation of lauric acid, yielding three monohydroxylated products in the same ratio as the wild type enzyme [Fig. 4]. The TON and rate rival many alternative electron delivery systems for driving catalysis with P450 BM3, including chemical reduction with NAD(P)H (71), light-driven reduction with deazaflavins (72), and electrochemical reduction with a Pt electrode (65,73). Mutants on the L407C background were also studied for the hydroxylation of other long-chain fatty acids (68) and trifluoromethylated arenes (67), targets of interest in the production of pharmaceuticals [Fig. 4] (74,75). The ability of both enzyme and sensitizer to be tailored to accommodate regio- and stereo-selective reactions with multiple substrates is a significant advantage.
Fig. 4.
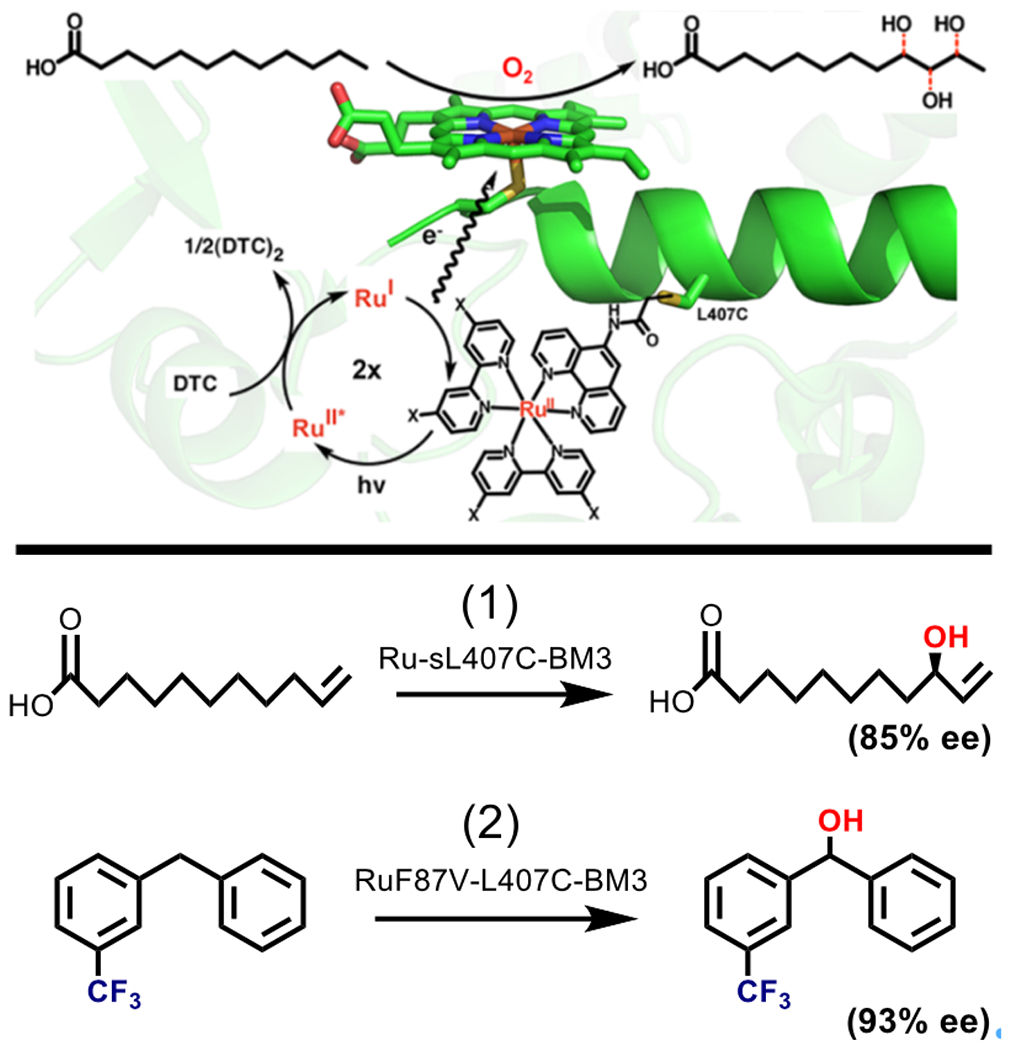
Top: Diagram showing the Ru(II) diimine sensitizer (X = H, OMe) attached via 407C in close proximity to the P450 BM3 active site. Excitation of the Ru (II) sensitizer generates an Ru (I) species that donates an electron to the heme, fueling the production of three monohydroxylated products of lauric acid. Adapted with permission from Tran N, Nguyen D, Dwaraknath S, Mahadevan S, Chavez G, Nguyen A, Dau T, Mullen S, Nguyen T-A, Cheruzel L (2013) J. Am. Chem. Soc. 135, 14484–14487. Copyright (2013) American Chemical Society. Bottom: Scheme showing other reactions catalyzed by mutants on the Ru-L407C-BM3 background. (1) The light driven hydroxylation of 10-undecenoic acid is catalyzed by Ru-sL407C-BM3 with 85% enantiomeric excess. (2) L407C mutants have also been used to hydroxylate trifluoromethylated substrates, for example meta-trifluoromethylated diphenylmethane was hydroxylated with 93% ee by RuF87V-L407C-BM3.
A related strategy has been used to drive hydroxylation reactions with P450 BSβ (CYP152A1) using more robust photosensitizers, CdS quantum dots (CdS QDs) [Fig. 2] (76,77). Nanocrystalline sensitizers like CdS QDs have tunable electronic and surface properties, both of which can be advantageous in preparing QD-biocatalyst hybrids (78,79). P450 BSβ catalyzes the hydroxylation of fatty acids at the α and β positions using hydrogen peroxide as an oxidant (80). In order to create a hybrid of P450 BSβ-CdS QDs, a P450 BSβ containing a positively charged 6x-His-tagged C-terminus was prepared. CdS nanoparticles capped with mercaptoacetic acid, providing a negative surface charge, promote adsorption of P450 BSβ (76). When irradiated, the P450 BSβ-CdS QD hybrid carry out the hydroxylation of myristic acid at the α and β positions in the same ratio as the peroxide shunt [Fig. 3] reaction of the native enzyme. Since QDs produce ROS upon irradiation (81), no additional oxidants or cofactors are required to fuel the reaction. The activity is switchable by UV-light, providing control over the reaction (77). However, due to enzyme degradation from light and ROS, total TONs (turnovers of product/minute) are lower in the hybrid enzyme compared to the native enzyme (76). Further work demonstrated that coating CdS with tobacco etch virus protease (TEV) minimizes short-lived radical ROS and promotes production of H2O2 (82). Combining CdS-TEV with P450 BSβ leads to higher product yields than the P450 BSβ-CdS QD hybrid system (82).
The previous examples pair biological catalysts with synthetic molecular or nanocrystalline photosensitizers. However, engineered systems that draw on nature’s apparatus for converting light into chemical energy have also been reported. Assembling enzymes, like cytochrome P450, into chloroplasts could be a valuable method for self-sustaining and scalable syntheses (83). The inspiration for these systems stems from oxygenic photosynthesis that occurs in chloroplasts and yields light-dependent water oxidation, NAD(P)H formation, and ATP formation. These light-driven reactions are mediated by thylakoid membrane-bound photosystems I and II (PSI, PSII) [Fig. 2] (84). While reducing equivalents provided by the water-splitting reaction typically create chemical fuels like NAD(P)H, the photochemistry of PSI could also provide electrons directly to expressed enzymes. The synthesis of dhurrin, a cyanogenic glycoside, was used as a model to demonstrate the possibility of transferring light-driven P450 reactivity to a chloroplast [Fig. 5] (83). Plants synthesize dhurrin as a cyanide-containing defense against pests, and its production pathway has been the focus of research aiming to increase insect-resistance in plants that do not naturally produce dhurrin (85,86). The biosynthesis of dhurrin in Sorghum bicolor is therefore well understood and the genes for encoding the pathway are cloned, making it a viable model system (87).
Fig. 5.
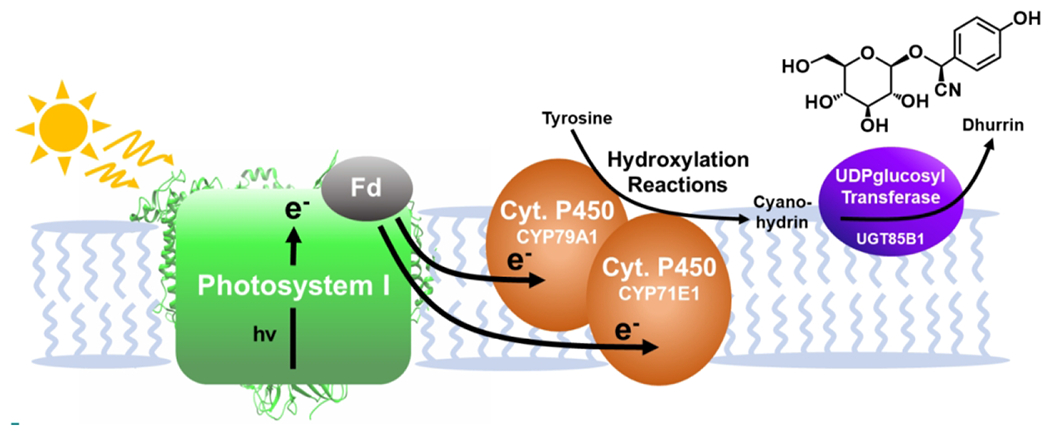
A schematic representation of the process used to drive Cytochrome P450 enzymes using light. Reducing equivalents produced by excitation of PSI fuel the hydroxylation reactions of Cyt. P450 and ultimately the full synthesis of dhurrin.
The full synthesis of dhurrin requires a precursor cyanohydrin molecule prepared through hydroxylation of tyrosine by two P450 enzymes (CYP79A14 and CYP71E15), which were expressed in the chloroplast. Upon excitation with light, PSI fuels their hydroxylation reactions (83). While typically a P450 oxidoreductase would be used to provide reducing equivalents from NAD(P)H, expression in a chloroplast allowed the needed reducing equivalents to be provided directly by excitation of PSI, successfully synthesizing the cyanohydrin product (p-hydroxymandelonitrile). Expression of a final enzyme in the pathway, a UDP-glucosyl transferase (UGT85B1), facilitated the full light-driven synthesis of dhurrin both in vitro and in vivo [Fig. 5]. The relative amount of dhurrin produced upon irradiation is enhanced by approximately 12-fold over the minimal amount of dhurrin synthesized in the dark (83). Overcoming the need for stoichiometric reductants and using water as the source of electrons, the dhurrin synthesis demonstrates the potential of using the machinery of photosynthesis to catalyze non-native reactions. One significant challenge to overcome in vivo is competitive endogenous electron sinks. To combat this possible source of inefficiency, the P450 enzyme (CYP79A1) expressed in the chloroplast was covalently linked to ferredoxin (Fd) (88,89). The covalent attachment of ferredoxin better directs electron transfer from PSI to cytochrome P450, illustrated by higher hydroxylated product yields in the P450-ferredoxin hybrid.
Significant progress has been made on driving cytochrome P450 enzymes with light using molecular, nanocrystalline, and biological photosensitizers. Using these strategies, the need for costly chemical reductants is eliminated. A number of strategies for light-driven generation of ROS species to fuel the catalysis of P450 enzymes, as opposed to direct electron transfer from a photosensitizer, have also been detailed elsewhere (18). This strategy has been used to fuel hydroxylation reactions of both P450 and peroxygenase enzymes (90,91). Using a water oxidation photocatalyst to generate H2O2 can renewably fuel peroxygenase reactivity, opening another route for light energy and enzymatic catalysis to be combined and scaled up (90).
The potential to engineer the enzymes for product selection demonstrates promise to expand the chemistry in many useful directions. While light may replace cofactors, a remaining challenge is the difficulty of high-yielding expression of cytochrome P450 enzymes (88,89,92). A number of advances have been made in optimizing gene sequence, secondary structure, and co-expression to overcome this issue (89). It is worth noting that P450 enzymes have also inspired an arsenal of catalysts capable of performing oxidation reactions that may be more easily synthesized. A full discussion is outside the scope of biomolecular catalysts highlighted in this review, but metalloporphyrin catalysts have a long history of performing a variety of C-H oxidation reactions, taking advantage of high-valent metal species inspired by P450 (93,94). In addition, a number of non-heme systems with high-valent iron centers have been created (95–99). While these fields have mostly existed in isolation, merging the reactivity of these synthetic iron catalysts with the lessons taken from light-driven P450 catalysis could be a step into the future.
3. Proton Reduction
The production of hydrogen (H2), a clean-burning and energy-dense fuel, has been an important goal of chemists for decades (36,100–102). The light-driven proton reduction reaction has been of particular interest because driving this reaction using sunlight would mimic photosynthesis, generating hydrogen as a solar fuel (25,35,103,104). Currently, hydrogen is produced on an industrial scale from non-renewable fossil fuels (101). Hydrogen is not just a fuel, but also an important feedstock in industrial reactions like the Haber-Bosch process (14,15,105). Many heterogeneous, molecular, and nanocrystalline catalysts have been developed for hydrogen generation (36,106–111). Platinum surfaces and nanostructures are a benchmark for hydrogen evolution catalysts, operating with high efficiencies (109,112). However, the high cost and low abundance of precious metals have stimulated the development of other heterogeneous catalysts, often comprised of first-row metals (109,113). An alternative approach is to turn to nature’s catalysts, the hydrogenases, that function with high rates and high efficiencies but without precious metals. Here, we focus on the use of these enzymes and other biomolecular systems for solar hydrogen production.
3.1. Hydrogenase Enzymes
Hydrogenase enzymes reversibly catalyze the reduction of protons to form dihydrogen [Equation 2]. Hydrogenases, using earth-abundant iron and nickel active sites, rival platinum metal for the efficiency and rate at which they catalyze the proton reduction reaction (114–116). [FeFe] and [FeNi] hydrogenase enzymes contain specialized reaction centers with an open coordination site where proton reduction can occur [Fig. 6] (115,117). The enzymes have scaffolds that facilitate delivery of protons and H2, and also transfer of electrons through a series of [Fe-S] clusters [Fig. 6] (117). [FeFe] hydrogenases tend to show the highest activity toward the H2 production reaction, while [FeNi] hydrogenases have higher oxygen tolerance (117). Generally, the enzymes are robust to poisoning agents like CO and H2S, maintaining specificity for H2 production (116). Given the efficient and specific activity, there is significant interest in exploring light-activated hydrogenase enzymes and their many functional mimics (115).
| (Equation 2) |
Fig. 6.
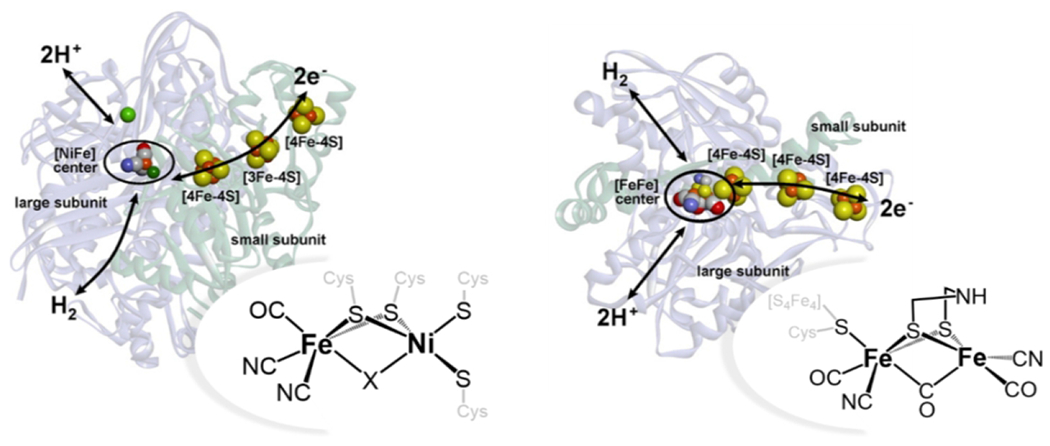
A diagram displaying the active sites of [FeNi] (DvMF (233)) and [FeFe] (Db (234)) active sites, along with electron transport chains. Adapted with permission from Lubitz W, Ogata H, Rüdiger O, Reijerse E. (2014) Chem. Rev. 114, 4081-4148. Copyright (2014) American Chemical Society
Hydrogenase enzymes in many phototropic microorganisms evolve hydrogen metabolically (118,119). However, hydrogen generation is sensitive to oxygen and competes with other endogenous electron transfer pathways (120), spurring the development of alternative approaches. As described previously for cytochrome P450, PSI is capable of providing electrons to enzymes other than its natural partners (83), and this approach also has been successful to activate hydrogenase enzymes (121). Taking advantage of an oxygen-tolerant [NiFe] hydrogenase from Ralstonia eutropha, a fusion of hydrogenase to a subunit of PSI (PsaE from Thermosynechococcus elongatus) was created to fuel light-driven hydrogen generation (122). PSI is comprised of multiple subunits, and PsaE helps form a binding site for ferredoxin (84). The PsaE unit spontaneously associates with PSI, integrating hydrogenase into the PSI architecture. The full assembly is competent for light-driven hydrogen evolution when paired with reducing agents. The hydrogenase-PsaE-PSI hybrid produces hydrogen at a rate 5.3 times higher than a mixture of hydrogenase and PSI. In further work, the assembly was attached to a gold electrode to produce hydrogen as a photocurrent was passed (123). Notably, the nanodevice operates under mild conditions, pH 7.5 and 20 °C, achieving high specific activities. While in vivo application is desired (119), in the presence of excess natural electron acceptors like ferredoxin, the activity of hydrogenase-PsaE-PSI is inhibited (122).
An approach to activating hydrogenase with light in a way that circumvents unproductive electron transfer is to connect PSI to hydrogenase via a molecular wire (124). In this work, researchers wired an [FeFe] hydrogenase (Clostridium acetobutylicum) via its distal [4Fe-4S] cluster, the site of electron injection into the enzyme [Fig. 6], to the PsaC subunit of PSI (Synechococcus spongiarum), which harbors the terminal cluster for electron transport to acceptors (Fb). A single surface-located cysteine on the hydrogenase distal [4Fe-4S] cluster was replaced with glycine (C97G) to open up a site for ligand binding. Glycine was also installed (C13G) at the Fb cluster pf PsaC subunit of PSI. These modifications allowed 1,6-hexanedithiol to be attached to electron transport chains on both proteins, thus directly wiring the electron donor (PsaC Fb cluster) to the acceptor (hydrogenase distal cluster). The PSI-wire-hydrogenase enzyme complex self assembles, and when paired with a combination of electron donors – cytochrome c6, ascorbate, and phenazine methosulfate – it achieves faster rates of hydrogen production than hydrogenase-PsaE-PSI without direct wiring (30.3 μmol H2 hr−1 mg chlorophyll−1 versus 0.58 μmol H2 hr−1 mg chlorophyll−1). Flexibility in both enzyme and tethering choices could allow further improvement upon these pioneering PSI-hydrogenase studies.
Using semiconducting nanoparticles as photosensitizers provides a robust photodonor that is amenable to direct attachment of photosensitizers and enzymes, as aforementioned. A [FeNiSe]-hydrogenase from Desulfomicrobium baculatum (Db-[FeNiSe]) was chosen to probe hydrogen production activity while adsorbed on TiO2 particles [Fig. 7] (116,125). This hydrogenase is notable for having relatively high oxygen tolerance and good activity toward of H2 production compared to other [NiFe] variants. TiO2 particles were modified with a stable ruthenium dye to facilitate visible light sensitization, as well as the hydrogenase, which readily adsorbs onto TiO2. The hydrogenase-TiO2 hybrid shows activity for hydrogen production when irradiated with visible light under ambient conditions (25 °C, pH 7). Db-[FeNiSe] was the most successful of several hydrogenase enzymes vetted for assembly with TiO2, possessing the fastest rates and highest TONs for H2 evolution. Importantly, Db-[FeNiSe] outperformed other O2-tolerant and active hydrogenases such as Escherichia coli [NiFe] hydrogenase (Ec-[NiFe]-Hyd-2). Stable adsorption of a hydrogenase enzyme to TiO2 in the most electroactive configuration, where the distal [4Fe-4S] cluster is positioned near the surface, is critical for H2 production [Fig. 7] (116). Directed electron transfer via attachment to the nanoparticle surface, as opposed to diffusion or non-specific interaction, produced the best results. In further work, probing similar interactions between hydrogenase enzymes and TiO2 electrodes is providing insight into how hybrids may be used to build photoelectrochemical devices (126,127).
Fig. 7.
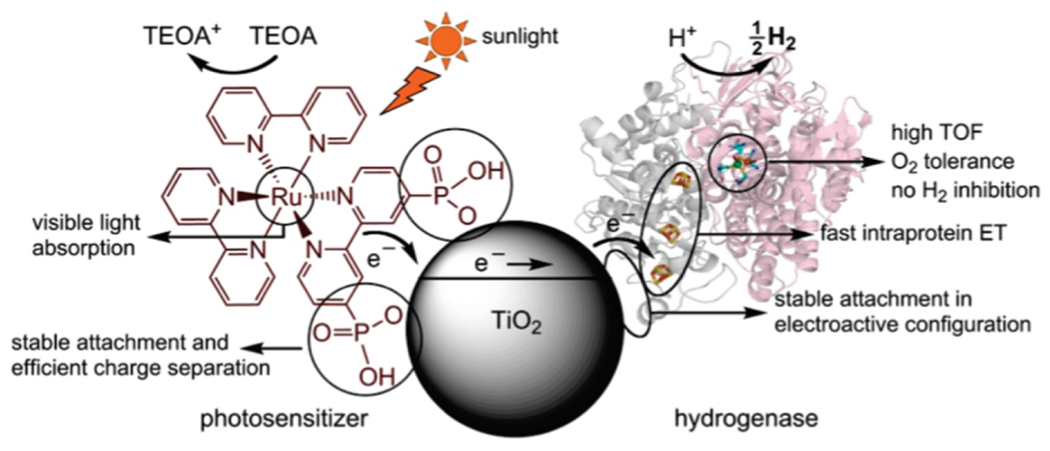
A schematic of the hydrogenase TiO2 system for hydrogen evolution. Visible light sensitization fuels electron transfer from TiO2 to hydrogenase enzymes. This occurs most efficiently when the distal [4Fe-4S] cluster of hydrogenase is near the surface of TiO2, the electroactive configuration. Reprinted with permission from Reisner E, Powell DJ, Cavazza C, Fontecilla-Camps JC, Armstrong FA. (2009) J. Am. Chem. Soc. 131, 18457-18466. Copyright (2009) American Chemical Society
The importance of directed electron transfer and controlled orientation of enzymes was explored further with an [FeFe] hydrogenase (Clostridium acetobutylicum) on the surface of CdTe nanocrystals (128,129). Mercaptopropionic acid (MPA) ligands were chosen to cap the nanocrystals, giving the nanoparticle surfaces an overall negative charge [Fig. 2]. A negative surface charge mimics ferredoxin, which interacts with a positively charged patch on the enzyme surface to facilitate electron transfer in vivo [Fig. 8]. In the presence of ascorbic acid under visible light irradiation, the CdTe nanocrystal-hydrogenase hybrids produce hydrogen. The efficiency of electron transfer was altered by changing the ratio of nanoparticles to enzymes and improved at higher nanoparticle-to-enzyme ratios. An optimal ratio of 4:1 CdTe nanocrystals:[FeFe] hydrogenase was discovered for activity (128). Additionally, a decrease in activity was observed at high salt concentrations, which screens electrostatic interactions between the enzyme and nanocrystal. Results observed in this study emphasize the importance of surface interactions for efficient electron transfer. Further work has contributed to understanding the architecture and optimization of the hydrogenase-nanoparticle assemblies (130–133). A [NiFe] hydrogenase from Thiocapsa roseopersicina and MPA-capped CdTe quantum dots assemble into a similar complex (130). A comparison between the electron transfer from CdTe QDs and molecular sensitizer [Ru(bpy)3]2+ corroborate ideas about surface interaction with nanocrystals. [Ru(bpy)3]2+ is a single-electron reductant and interacts non-specifically with hydrogenase. Lower solar-to-hydrogen conversion efficiency for H2, 0.02% versus 4% with nanocrystals, and different intermediates generated by [Ru(bpy)3]2+, suggest that the ability for fast multielectron reductions and specific binding to nanocrystals bolster the function of QD-hydrogenase assemblies (130).
Fig. 8.
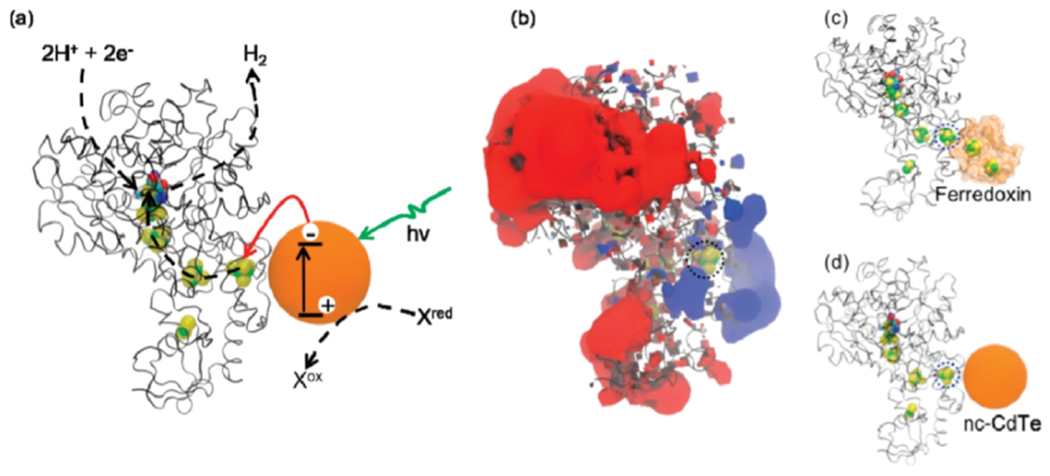
Upon irradiation, electron transfer from a CdTe nanocrystal fuels hydrogen evolution with hydrogenase (a). The positively charged region on hydrogenase (b) interacts with ferredoxin (c) in vivo. In these experiments, the enzyme interacts with a negatively charged nanocrystal instead of ferredoxin (d). Reprinted with permission from Brown KA, Dayal S, Ai X, Rumbles G, King PW. (2010) J. Am. Chem. Soc. 132, 9672-9680. Copyright (2010) American Chemical Society.
The role of the capping ligand in facilitating electron transfer was also probed by varying the length of carboxylate capping ligands on CdS quantum rods (134). With increasing ligand length, and therefore a longer tether between enzyme and nanorod, the rate of electron transfer decreased. This study reveals that not only do ligands impact the photophysics of the sensitizer, but also the degree of electronic coupling that can be achieved between the nanocrystal and enzyme. Using longer capping ligands resulted in less efficient hydrogenase adsorption and slower electron transfer, resulting in an overall decrease in hydrogen production. A recurring theme in enzyme-nanocrystal hybrids is the critical impact of efficient and directed electron transfer on hydrogen production, where controlling ratios between enzymes and nanocrystals and selecting capping ligands that facilitate optimal nanocrystal-enzyme association play key roles (135). This high degree of tunability could be powerful in shaping the future of light-driven enzyme catalysis, especially because enzymes can also be tuned to facilitate interactions with nanoparticles by altering surface residues around key electron transfer sites.
Enzymes that are amenable to site-directed mutagenesis can be engineered to alter or enhance activity. For example, variants of oxygen-tolerant Ec-[NiFe]-Hyd-2 were engineered to enhance their interaction with silver nanoclusters [Fig. 9] (136). Assisting this work was the structural characterization of the enzyme and the development of a system for high-yield expression (137). Three primary mutants of Ec-[NiFe]-Hyd-2 were prepared to catalyze hydrogen evolution [Fig. 9]. In 3S-Hyd-2, the enzyme’s three naturally occurring surface cysteines were replaced with serine. In mutant Y’-222C-Hyd-2, a tyrosine in close proximity to the distal [4Fe-4S] cluster is replaced with cysteine. In the final variant, 3S1C-Hyd-2, the three surface cysteines are replaced with serines, and the distal tyrosine with cysteine. Light-driven hydrogen production experiments revealed a significant difference between the surface cysteine-containing variants and 3S-Hyd-2. In the presence of visible light irradiation and triethanolamine as an electron donor, the cysteine-containing variants 3S1C-Hyd-2 and Y’-222C-Hyd-2 have turnover numbers and rates significantly higher than 3S-Hyd-2. 3S1C-Hyd-2 has the greatest interaction with the nanoclusters as indicated via luminescence quenching, and the engineered cysteine residue ensures this interaction occurs near the distal [4Fe-4S] cluster. Adding metal oxide nanoparticles like TiO2 that could bind to the silver nanoclusters greatly enhances the rate and total hydrogen production (136). Notably, the variant with no cysteines (3S-Hyd-2) still underperforms compared to the other variants. “Hard wiring” the hydrogenase variants via a cysteine residue is a versatile method for promoting enzyme-nanocrystal interactions.
Fig. 9.
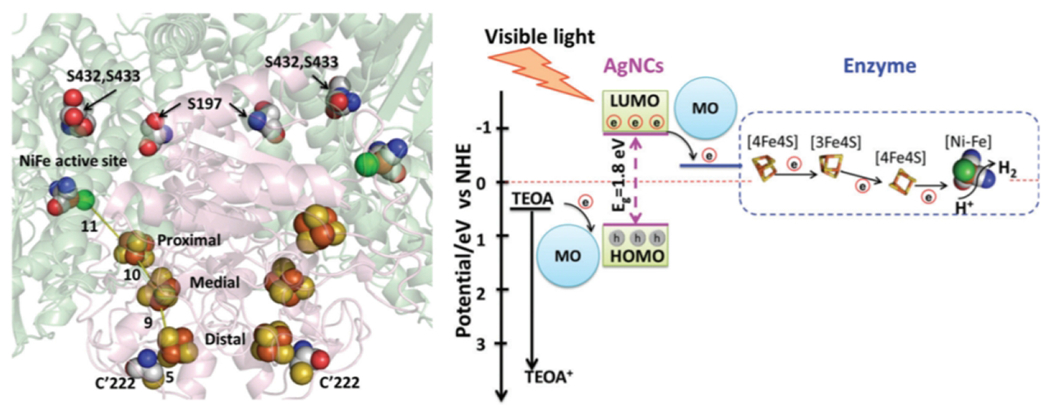
In [NiFe]-Hyd-2, there are electron transport chains leading to active sites (left). Locations of mutations discussed in the text are shown. The overall schematic for electron transport from silver nanocrystals to the active site of hydrogenase is also shown (right). Reprinted with permission from Zhang L, Beaton SE, Carr SB, Armstrong FA. (2018) Energy Environ. Sci. 11, 3342-3348 – Published by the Royal Society of Chemistry
3.2. Structural Mimics of Hydrogenase
Many molecules have been engineered to mimic hydrogenase, and reviews on these systems are available (114,138,139). Here, we discuss several noteworthy examples of light-driven hydrogen production using engineered biomolecular catalysts. These systems take advantage of biomolecular motifs, which confer water solubility and can facilitate the introduction of second-sphere interactions at the active site. While notable progress has been made with using light to activate hydrogenase enzymes, limitations like low density of active sites and difficulty of achieving high-yielding enzyme expression remain (117,140). Innovative work has been carried out over several decades to mimic the structure of [FeFe] and [NiFe] hydrogenase active sites with synthetic molecular catalysts (114,141,142). These mimics have been instrumental in providing insight into catalytic mechanisms and intermediates (117,143). Conveying high catalytic activity to these molecular mimics is of interest, but presents unique challenges such as achieving water solubility and stability (114,117). The combination of structural mimics with peptide and protein scaffolds has been one route to overcoming some of these challenges (114).
The first incorporation of an [FeFe] hydrogenase active-site mimic into a peptide scaffold was reported in 2007 (144). The complex was coordinated to a simple α-helical peptide via two cysteine residues in a CXXC motif. This method for ligation is versatile and powerful, and allowed researchers to capitalize on the native CXXC motif in cytochrome c in subsequent work to prepare a similar system using a cytochrome c polypeptide (145). The reaction of apo-cytochrome c with an Fe2(CO)6 cluster, a simple mimic of the [FeFe] hydrogenase active site, incorporates the active-site mimic into the protein scaffold via the two cysteine residues. This hydrogenase mimic, H-apocyt c, facilitates photocatalytic hydrogen production in water when paired with [Ru(bpy)3]2+ as a photosensitizer and ascorbate as an electron donor at both acidic and neutral pH. Prior to this work, similar iron clusters participated in photocatalytic hydrogen evolution only in organic solvents or mixtures (146–149). H-apocyt c is active for two hours under visible light irradiation, reaching a TON (mol H2/mol H-apocyt c) of 82 at pH 4.7. H-apominicyt c, a catalyst containing Fe2(CO)6 ligated to a shorter oligopeptide fragment of cytochrome c, is less active, achieving a TON (mol H2/mol H-apocyt c) of 13 under similar conditions. A larger protein scaffold was proposed to convey higher activity to H-apocyt c (145). These studies opened the door for experimentation with different protein scaffolds.
The use of a more robust protein scaffold, nitrobindin, to host the active-site mimic proved more effective than use of apo-cytochrome c. Nitrobindin typically possesses a heme within a beta barrel, and the barrel maintains its structure in the apo form (150). The apo form was reacted with a (μ–S2)Fe2(CO)6 derivative containing a dithiolate ligand with a maleimide group. This group reacts to form a covalent bond with cysteine, facilitating ligation of the iron complex to nitrobindin. Using [Ru(bpy)3]2+ as a sensitizer and ascorbate as an electron donor, the nitrobindin active site mimic facilitated a TON (mol H2/mol catalyst) of 130 in completely aqueous solution. While the basis for slightly higher activity of the complex with nitrobindin is not clear, it is possible that the more defined structure of the beta-barrel compared to the apocytochrome c peptide enhances reactivity by providing a more defined active site or stabilizing the interaction between the protein-donated ligand and the iron complex.
The conjugation of synthetic catalysts to proteins is not limited to the use of naturally-occurring metal cofactor-binding motifs. Introduction of an unnatural amino acid containing a 1,3-dithiol moiety (Dt) demonstrates how this approach may be extended to many protein scaffolds (151). While aforementioned work had relied on an existing CXXC motif to bind an active site hydrogenase mimic, the unnatural Dt amino acid could potentially be introduced anywhere to facilitate chelation of a cofactor or active-site mimic. When Dt was incorporated into an alanine-rich helical peptide, it reacted to form a complex with the Fe2(CO)6 cluster. Under visible light in the presence of [Ru(bpy)3]2+ and ascorbate, the Fe2CO6-peptide achieves TONs (mol H2/mol catalyst) nearing 100. Importantly, this work opens an avenue for the incorporation of the active site mimic into a variety of engineered protein scaffolds. In addition, versatility of ligation to protein scaffolds allows for attachment of sensitizers (152). An octadecapeptide from cytochrome c, containing the CXXCH motif, was used to coordinate both a ruthenium sensitizer, through histidine, and an Fe2(CO)6 active-site mimic. Facilitating directed electron transfer to the active site through close coordination of the sensitizer was important for activity; the separated components did not facilitate hydrogen production.
While most hydrogenase mimicry has focused on the diiron hydrogenase, one of the earliest systems was based on the [NiFe] hydrogenase (153–159). Substitution of Ni in rubredoxin yields a catalyst, Ni-Rd, with a tetrathiolate active site mimicking the primary coordination sphere of the Ni site in [NiFe] hydrogenase (153,159). When combined with [Ru(bpy)3]2+ and ascorbate and irradiated with visible light, Ni-Rd catalyzes hydrogen evolution (153). A TON greater than 100 nmol/mg catalyst was observed, and the biomimetic catalyst is stable through catalysis in aqueous solution, allowing greater than 90% catalyst recovery. The protein structure is amenable to many different mutations, opening the possibility for optimization (158). In further work, a ruthenium sensitizer was covalently attached to the protein scaffold, creating an integrated hydrogen evolution system (157).
3.3. Functional Mimics of Hydrogenase
While many artificial hydrogenases have been based on structural mimics of hydrogenase active sites, successes have been found in mimicking function using complexes that are not related to any hydrogenase active-site structure. In particular, a variety of molecular cobalt catalysts have been developed for hydrogen production even though cobalt is not used in nature for this reaction. Furthermore, Ni-based catalysts have been successful, even though many of these systems do not resemble the enzyme active site (107,160–162). One of the most well-studied families of molecular cobalt catalysts are “cobaloximes,” mimics of vitamin B12 that also catalyze proton reduction [Fig. 10] (107,163,164). Challenges with the cobaloxime catalysts include poor water solubility and stability, with loss of the dimethylglyoxime noted during catalysis (163).
Fig. 10.
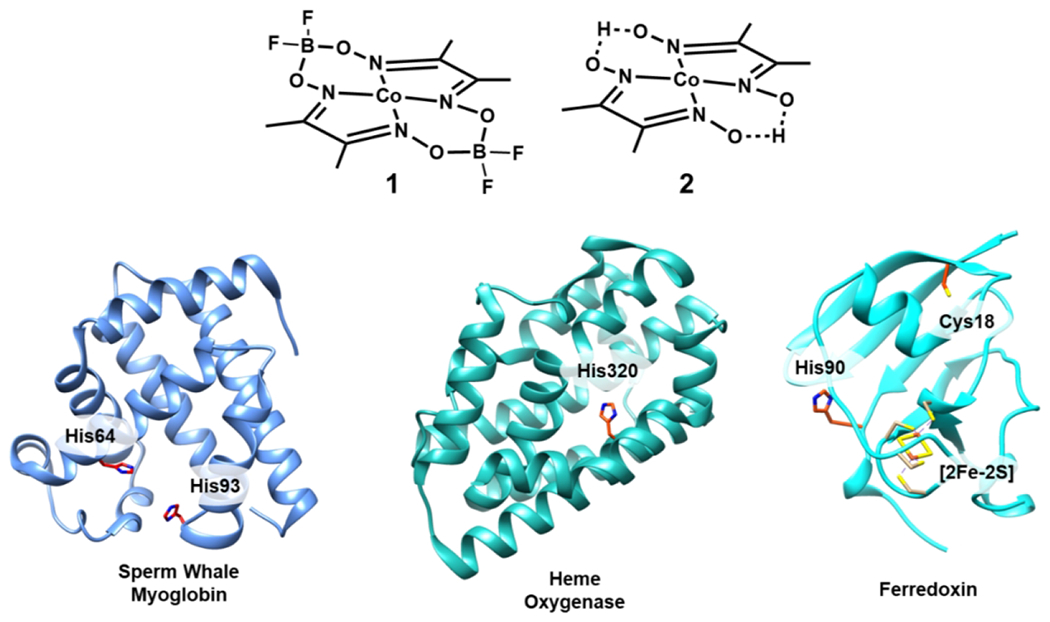
Common cobaloxime catalysts (1, difluoroboronyl bridged and 2, proton bridged) have been paired with protein scaffolds like sperm whale myoglobin (PDB 1UFJ), heme oxygenase (PDB 1IW0), and ferredoxin (PDB 1A70). Highlighted in each protein are key residues including the histidine binding sites for the cobaloxime catalysts.
To address these issues, cobaloxime catalysts have been incorporated into protein scaffolds; among the first were myoglobin and heme oxygenase (165,166). Two cobaloxime catalysts were reacted with sperm whale apo-myoglobin [Fig. 10]. Sperm whale myoglobin typically binds heme through a histidine residue, making it a good candidate to accommodate square planar metal catalysts with similar dimensions to heme. Spectroscopic and computational evidence supports the binding of the cobaloximes to His93 through axial coordination at the cobalt center. The cobaloxime catalysts support hydrogen production in the presence of [Ru(bpy)3]2+ and ascorbate, notably doing so in water near neutral pH. However, the catalysts perform similarly in terms of longevity and total activity both within and without the protein scaffold, achieving modest TONs (mol H2/mol Co) between 3 – 8 over the course of minutes. Incorporating the same cobaloximes into Corynebacterium diphteriae heme oxygenase provides improvements over myoglobin, increasing TONs up to threefold. Unlike in sperm whale myoglobin, the cobaloxime catalysts can bind heme oxygenase in different conformations that provide a mixture of hydrophobic and hydrophilic exposure. These results suggest that modest modifications to catalyst binding environments are sufficient to promote different active-site activity and stability (166).
Cobaloxime 1 [Fig. 10] has also been prepared in a complex with electron-transfer protein ferredoxin (Spinacia oleracea) (167). This ferredoxin contains a [2Fe-2S] cluster used to mediate electron transfer. The cobaloxime catalyst was ligated axially to His90 of ferredoxin, similar to the installation in myoglobin. In addition, a ruthenium sensitizer was covalently linked through Cys18, creating a Ru-Fd-Co hybrid. A second complex, Ru-ApoFd-Co, was prepared in the same style, but lacking the [2Fe-2S] cluster of ferredoxin. In the presence of ascorbate in aqueous solution, Ru-Fd-Co facilitates a TON up to 320 (mol H2/mol sensitizer) under irradiation, remaining active over six hours. Spectroscopic evidence indicates that the ruthenium sensitizer could pass electrons to the [2Fe-2S] cluster of ferredoxin. Ru-ApoFd-Co does not produce hydrogen under the same optimum conditions, demonstrating the importance of the [2Fe-2S] cluster for electron transfer from sensitizer to catalyst (167). The role of the cluster in electron transfer was further confirmed in subsequent work that replaced the [2Fe-2S] cluster iron ions with redox-inactive gallium (168). Hydrogen evolution is inhibited in Ru-GaFd-Co, demonstrating that the activity is linked to electron transfer rather than changes in protein structure. A second biohybrid, Ru-ApoFld-Co was prepared in the same style as Ru-Fd-Co, using flavodoxin from S. lividus as a protein scaffold. In Ru-ApoFld-Co relative to Ru-ApoFd-Co, the distance between Ru and Co is shortened from 16.3 Å to 10.2 Å, and the complex is competent for hydrogen evolution. Ru-ApoFld-Co produces less hydrogen than Ru-Fd-Co, achieving TONs (mol H2/mol sensitizer) on average of 85. These works collectively demonstrate the tunability of activity with site-specific installation of sensitizer, catalyst, and redox-active moieties to optimize electron transfer. Ru-Fd also facilitates hydrogen evolution when combined with a cobaloxime-bound variant of its partner protein, ferredoxin-NADP+-reductase (169). The ability to instill and expand tunability to multi-protein systems is a valuable step toward replicating natural photosynthetic behaviors.
Other cobalt-containing proteins and protein derivatives have been developed for catalyzing light-driven hydrogen production (170). Cobalt MP11-Ac, a porphyrin-peptide complex derived from horse cytochrome c, was reported as an electrochemical proton reduction catalyst (171), and the closely related CoPPIX was chosen to incorporate into a protein scaffold [Fig. 11] (170). When reacted with apomyoglobin, CoPPIX binds axially through His-93, the heme-binding residue [Fig 10]. In the presence of [Ru(bpy)3]2+ and ascorbic acid, hydrogen production is observed after irradiation with visible light. Compared to CoPPIX, lacking the myoglobin scaffold, CoMyo shows a several-fold enhancement in TON (mol H2/mol catalyst), from approximately 120 up to 518 at pH 7. Complexes of three mutants were also prepared, H64A, H97A, and H64/97A, where histidine residues in close proximity to the active site were exchanged for alanine. His97 plays an important role in reducing the lability of the cofactor, forming a hydrogen bond with a propionic acid residue of CoPPIX. The H97A mutant’s activity is similar to free CoPPIX, which may indicate dissociation from the protein scaffold. The H64A and H64/97A mutants, on the other hand, have increased activity at pH 6.5. His64 is a competitor for proton binding, and removing His64 also potentially creates a change in the charge near the active site. This series of experiments demonstrates nicely how small modulations in the protein scaffold can provide control over engineered protein catalyst stability and activity.
Fig. 11.
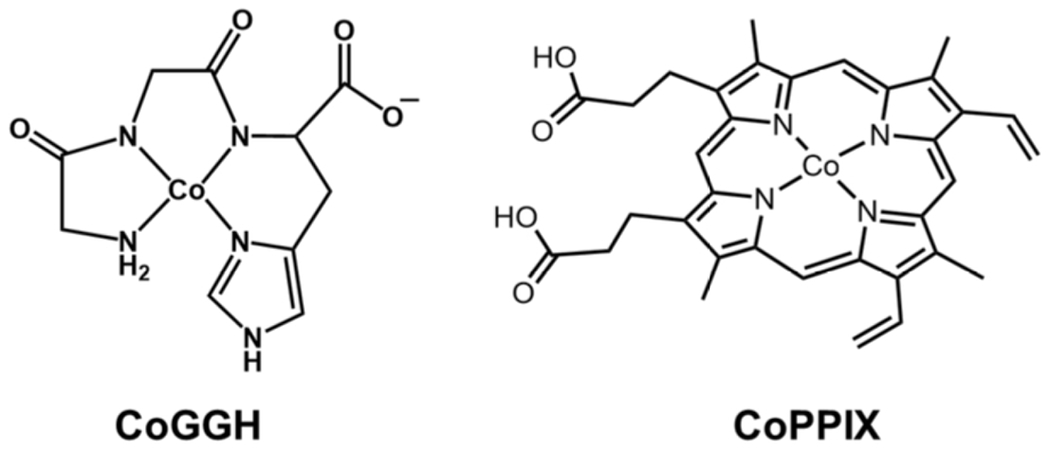
Cobalt catalysts CoGGH and CoPPIX.
A recent study demonstrates that a simple cobalt peptide complex mimicking a biological metal-binding site can serve in light-driven hydrogen evolution. Cobalt Gly-Gly-His (CoGGH), uses a tripeptide model of the metal-binding “ATCUN” (amino-terminal copper and nickel-binding) motif found in some proteins [Fig. 11]. CoGGH was previously reported to be active for electrochemical proton reduction (172), and subsequently was incorporated into a photochemical system by pairing with [Ru(bpy)3]2+ and ascorbic acid in water near neutral pH (173). Although it is not a full protein scaffold, the metal derivatives of the GGH peptide are water soluble. This motif also has the potential for second-sphere interactions with the cobalt active site via the N-terminus or the carboxylate moiety. Upon irradiation with visible light, the catalyst achieves TONs (mol H2/mol catalyst) up to 2200 at pH 7.1. The amenability of the peptide to modification leaves room to expand in many directions, both to promote activity of the catalyst and interaction with more robust sensitizers.
The same general strategies used for cobalt catalysts have been used to incorporate nickel catalysts into protein scaffolds. Some nickel catalysts, denoted DuBois catalysts after their inventor, have activities that rival hydrogenase, and are relatively stable and amenable to modification (174,175). A nickel DuBois-type catalyst was incorporated into PSI via self-assembly (176). Hybrids with 1-2 Ni per PSI monomer produce the most hydrogen in a light-driven assembly. Although the self-assembled system functions well, achieving TONs (mol H2/mol PSI) up to 1870 near neutral pH, more powerful is the directed insertion of the Ni catalyst using flavodoxin (Fld). By replacing the flavin mononucleotide (FMN) cofactor of Fld with the Ni catalyst, generating Ni-ApoFld, the nickel catalyst could be delivered to PSI by Fld. Docking the catalyst via Fld provides enhanced TON (mol H2/mol PSI) values of up to 2825, and also extends longevity slightly. This method opens the possibility of introducing a number of non-native cofactors and catalysts into nature’s PSI machinery for light energy conversion and storage.
The chemistry of proton reduction is a rich field that will continue expanding. Other bioinspired catalysts in the field of proton reduction have been reported that may be developed for light-driven activity. For example, a short seven-mer peptide has been used to create a scaffold for nickel and iron (177), and peptide fragments have been interfaced with nickel DuBois catalysts to facilitate high electrochemical activity in water (178–180). Although not biomolecular in nature, many proton reduction catalysts take other lessons from nature. In particular, scaffolds like hangman porphyrins or the DuBois-type catalysts promote proton transfer as an enzyme scaffold might (139,162,174,181). Going forward, continued merging of inorganic and heterogeneous catalysts with biomolecular catalysis may convey many of the advantages discussed here – namely, functioning in fully aqueous solution under mild conditions and tailoring the scaffold to promote different activities – with enhanced robustness, which is a limitation for may existing systems.
4. Water Oxidation
For proton reduction to become a viable route to producing hydrogen as a solar fuel for widespread use, an integral part of the overall process is water oxidation, which allows water to serve as a source of the needed protons and electrons, similar to natural photosynthesis (34,35,84,182). Transforming this abundant feedstock as plants do is challenging, and en route to the selective four-electron, four-proton oxidation to O2 are reactive partially reduced oxygen species (25,34,38). The development of catalysts of the water oxidation reaction has utilized inorganic bulk or nanocrystalline materials for heterogeneous catalysis (183,35) and inorganic molecular systems for homogeneous catalysis (21,36). In nature, the reaction is catalyzed by the oxygen-evolving complex (OEC) in photosystem II (PSII) [Equation 3, Fig. 12] (38). The OEC contains an Mn4Ca cluster that carries out the water-splitting reaction. This structure has been the inspiration for the design of synthetic structural and functional mimics, especially clusters containing cobalt (184) and manganese (185), that have provided insight into mechanism (183). However, the use of biocatalytic water oxidation reactions driven by light has focused on utilizing the apparatus of PSII for water oxidation, and coupling this system to another for proton reduction.
| (Equation 3) |
Fig. 12.
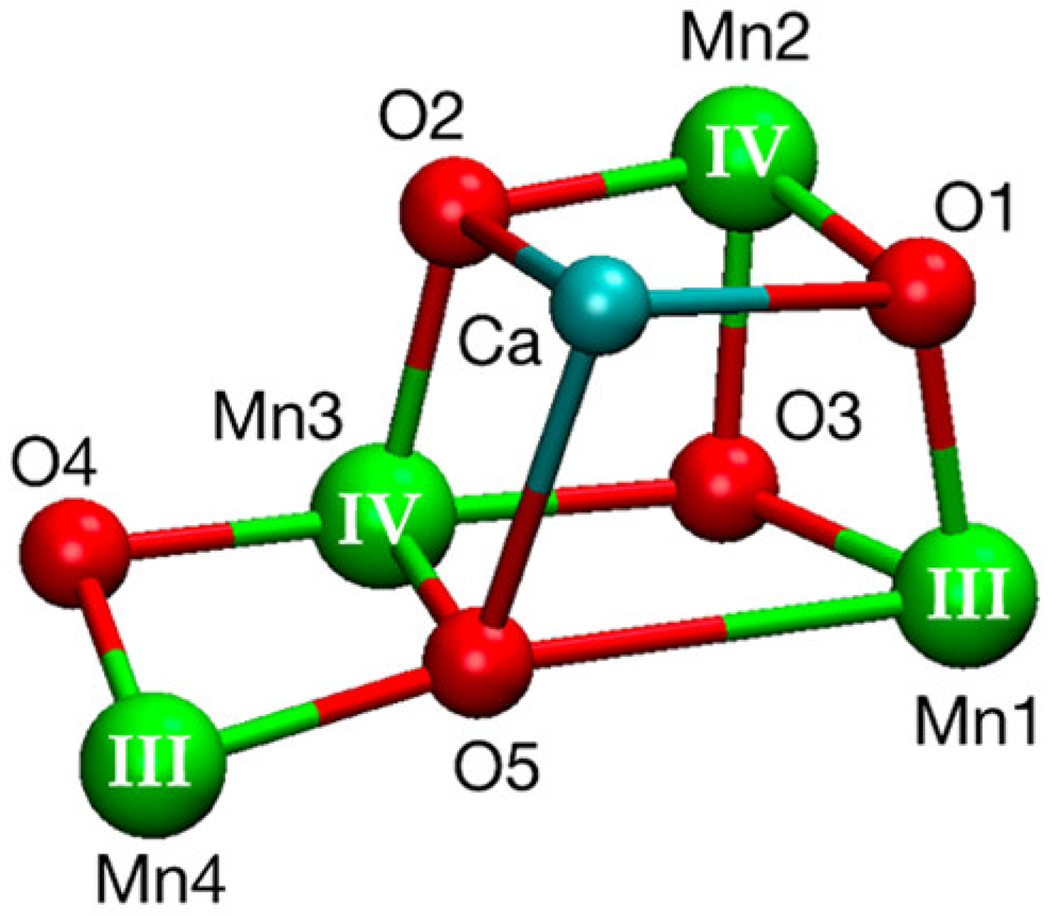
The oxygen-evolving complex (OEC) of photosystem II is a Mn4Ca cluster. Reprinted with permission from Takahashi H, Suzuoka D, Sakuraba S, Morita A (2019) J. Phys. Chem. B 123, 7081-7091. Copyright (2019) American Chemical Society
In order to perform light-driven water splitting to O2 and H2, a PSII-photocatalyst hybrid was prepared (104). PSII was assembled on the surface of an inorganic proton reduction catalyst (Ru2S3/CdS or Ru/SrTiO3:Rh). This assembly catalyzes the overall water-splitting reaction in the presence of an electron mediator and visible light irradiation. The half-reaction in the presence of only PSII and an electron acceptor, [Fe(CN)6]3−, produces the desired product O2. Once the inorganic photocatalyst is present, the acceptor delivers electrons to catalyze the proton reduction reaction, and both hydrogen and oxygen evolution are monitored simultaneously. When fueled by visible light, the PSII-Ru/SrTiO3:Rh hybrid produces 2,489 mol H2/mol PSII and 1,334 mol O2/mol PSII, with overall activity lasting approximately three hours. The system was primarily limited by degradation of PSII, likely due to oxidative damage and photodamage. PSII has previously been attached covalently to a photoelectrode (186). The methods proposed for directed attachment to mesoITO provide a good stepping stone to future studies of PSII coupled with inorganic catalysts.
As previously discussed, catalysts, nanoparticles, and proteins have been integrated with PSI for proton reduction (121,124,176). If these systems could be coupled to the a catalyst for the water oxidation reaction, the need for a sacrificial donor would be removed. Using abiotic catalysts – platinum nanoparticles and metal complexes – assembled with PSI, the prospect of coupling proton reduction to PSII-driven water oxidation in a membrane was explored [Fig. 13] (187). Platinum nanoparticles stabilized with mercaptosuccinic acid were self-assembled with thylakoid membranes from spinach and cyanobacteria. The best activity was observed when interfacing the nanoparticles with thylakoid membranes from cyanobacterium S. leopoliensis. Spectroscopic evidence indicated the docking of platinum nanoparticles to PSI, and upon irradiation the membrane system carried out overall water splitting, producing a 1:1 ratio of O2:H2 over eight hours. Electron flow through the plastoquinone (PQ) pool was important for the overall activity observed, and the addition of cytochrome c6 also facilitated electron transfer. After the promising demonstration of activity with platinum nanoparticles, a cobaloxime and nickel DuBois-type catalyst were also interfaced with PSI [Fig. 13]. When the molecular catalysts were used, overall water splitting was observed, albeit at less efficient rates than the platinum nanoparticles. While nonspecific catalyst binding likely hinders the activity of molecular catalysts, the ability to use a carrier protein to dock a related catalyst has been explored (176) and could be used to improve specificity.
Fig. 13.
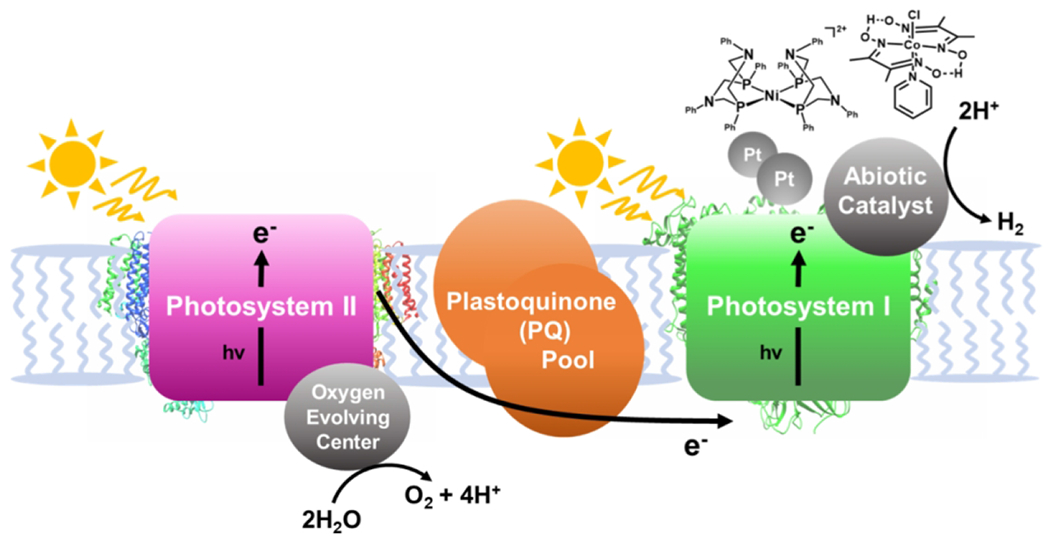
A simplified schematic showing the coupling of water oxidation at PSII to proton reduction with abiotic catalysts at PSI.
The use of PSII for water oxidation will prove difficult in large-scale applications. However, lessons can still be taken from the work done here. For example, a Mn12 cluster, using a bulky hydrophilic ligand that mimics the peptides in the Mn4Ca cluster, was recently used for electrochemical water oxidation (185). While it has not been reported to achieve light-driven water oxidation yet, the cluster is a promising start to translating active-site mimics to eventual light-driven water splitting.
5. CO2 Reduction
Among potential abundant feedstocks for chemical conversion, CO2 is one of the most attractive. Accumulation of CO2, a greenhouse gas, is a growing issue actively contributing to climate change (188,189). Recycling CO2 as a feedstock for large-scale reactions or to generate a fuel could mitigate net CO2 emissions by using an alternative carbon source to fossil fuels and by providing renewable fuel resources (190–194). The reduction of CO2 in multi-electron, multi-proton reactions can produce a variety of products. Furthermore, protons are always required, and thus proton reduction to H2 is a competing reaction. Thus, achieving substrate and product selectivity in CO2 reduction are major challenges (195). Of particular interest are reduction products like carbon monoxide, formic acid, formaldehyde, and alcohols [Equations 4 – 7]. TiO2 nanoparticles and other heterogeneous catalysts have been used to directly reduce CO2, along with a variety of metal-based catalysts and metal-organic frameworks (195–200). However, selectivity for desired products is difficult to achieve.
| (Equation 4) |
| (Equation 5) |
| (Equation 6) |
| (Equation 7) |
Enzymes that fix CO2 have high product selectivity that has been difficult to replicate with synthetic catalysts. There are several biological routes to CO2 fixation, including the Calvin cycle which accounts for roughly 90% of biological CO2 fixation (201,202). Some anaerobic organisms have other CO2 fixation pathways to support metabolism (203,204). Nickel carbon monoxide dehydrogenase (Ni-CODH) enzymes in anaerobic organisms reversibly catalyze the reduction of CO2 to CO. Most Ni-CODH enzymes contain an [NiFe3S4] cluster at the active site, along with two [Fe4S4 clusters] (205). Ni-CODH enzymes function in junction with acetyl-CoA synthase in microbial organisms to use CO as a building block. CO is also a critical building block in industry, and is used in the Monsanto process, Fischer-Tropsch process, and the water-gas shift reaction, among others. Driving enzymes or biomimetics like CODH with light could provide a clean pathway to converting a greenhouse gas into valuable feedstocks.
5.1. CODH Enzyme
Headway has been made on driving the reactions of CODH enzymes photochemically to generate CO from CO2 (206–208). CODH I from Carboxydothermus hydrogenoformans (Ch) has a [NiFe4-S4] cluster that is active for both the oxidation and reduction reaction, along with [Fe4S4] clusters for electron transport. To activate the enzyme with light, CODH I was adsorbed to TiO2 nanoparticles sensitized with a ruthenium dye [Fig. 14]. The resulting Ru-TiO2-CODH construct carries out the reduction of CO2 to CO upon irradiation in the presence of a sacrificial electron donor (MES) under mild conditions (pH 6, 20 °C). CO is produced selectively, and activity is lost over time, ending after approximately four hours (207). Enzymes were oriented on the surface of TiO2 in random fashion. Consequently, the distal iron-sulfur cluster near the surface of the enzyme may not always be oriented toward the nanocrystal in order to receive electrons, which may decrease overall efficiency of the assembly (206,207).
Fig. 14.
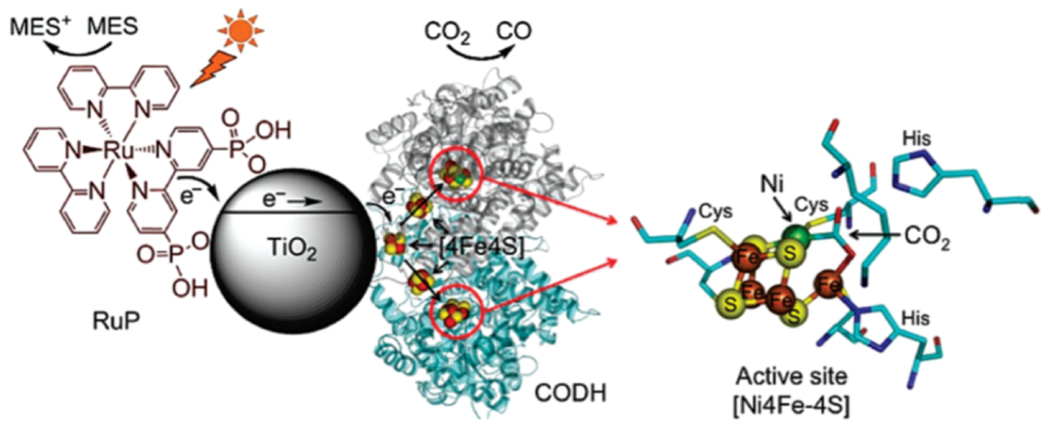
Schematic demonstrating the TiO2 CODH I hybrid system. Reprinted with permission from Woolerton TW, Sheard S, Reisner E, Pierce E, Ragsdale SW, Armstrong FA. (2010) J. Am. Chem. Soc. 132, 2132-2133. Copyright (2010) American Chemical Society
Modifications to the enzyme and ruthenium sensitizer were made in an attempt to better control their assembly and orientation on TiO2, but these were not successful (206). To simplify the process of light sensitization, CODH I was attached to CdS nanocrystals, eliminating the need for dye sensitization (208). Interestingly, the size and surface of nanocrystals played a large role in the overall activity observed, with nanorods facilitating higher TONs (mol CO/mol attached enzyme) than quantum dots. With both dye-sensitized TiO2 and CdS, rapid multi-electron transfer events can facilitate catalysis in a way that single sequential electron transfers would not (209). An additional improvement to the CODH-nanoparticle systems was made by adding poly(methacrylic acid) (PMAA)-stabilized silver nanocrystals to the CODH-TiO2 assembly (209). While the exact interactions of the ternary complex between PMAA-Ag nanocrystals, CODH I, and TiO2 have not been fully extracted, the rate for CO evolution when all components are present is double that reported for dye-sensitized TiO2. It is possible that the PMAA-Ag nanocrystals help to promote interactions with the enzyme surface near the distal [4Fe-4S] cluster (209). Further divulging how specific residues on the protein promote favorable interactions with nanocrystalline components could reveal valuable information about the mechanism of CODH activation in these systems, along with how modifications may be made to produce yet better-performing assemblies.
5.2. Biomolecular Catalysts for CO2 Reduction
The complicated structure of the CODH active site has been modeled with inorganic catalysts (205). Close structural mimics have not yet been reported to have activity toward CO2 reduction, but simpler functional mimics have been reported. One of the most successful is nickel(II)-cyclam [Fig. 15], a molecular catalyst for CO2 reduction to CO that functions under mild conditions in water (200,210,211). Though active, modifications that can be made to Ni(II)-cyclam are limited and the catalyst is not selective for CO production, producing H2 as a byproduct.
Fig. 15.
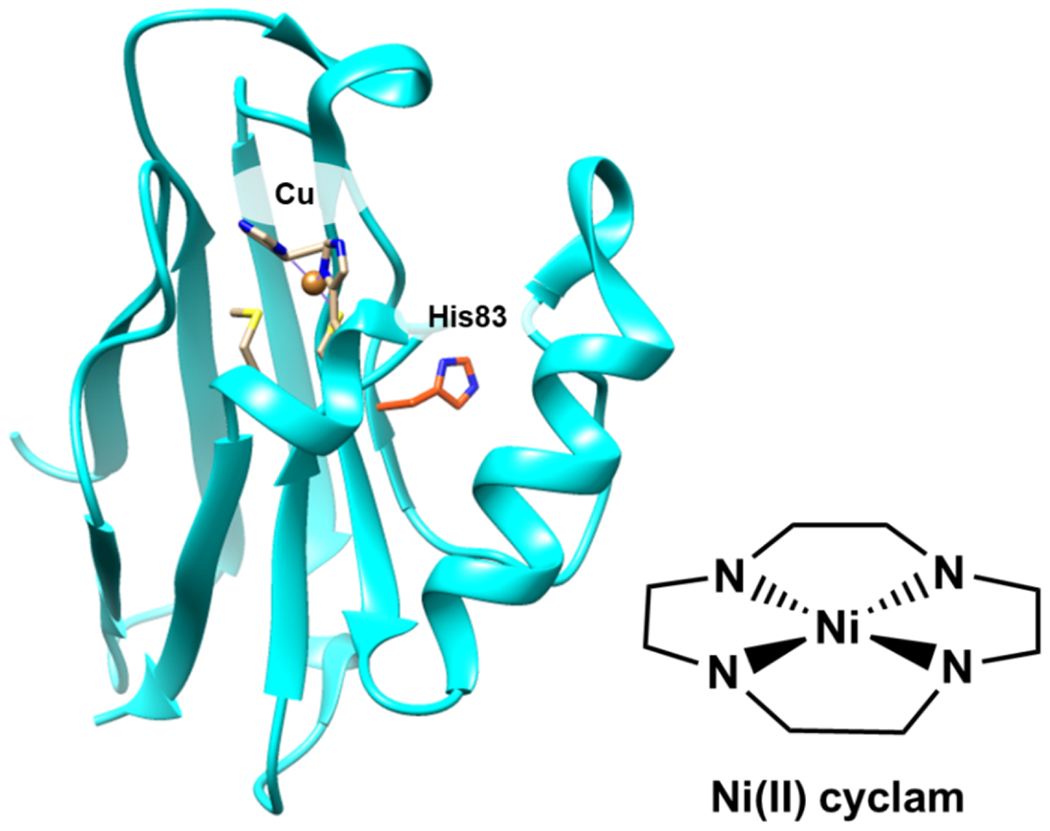
Copper containing azurin (PDB 4AZU) with His83, the binding site for Ni(II) cyclam, highlighted. A drawing of Ni(II) cyclam is shown.
To guide activity and selectivity, Ni(II)-cyclam was introduced to a protein scaffold to yield an artificial CODH enzyme (212–214). Azurin, a robust electron-transfer blue copper protein with a surface histidine residue (His83) available to bind Ni(II)-cyclam, was used for this work. An azurin mutant, H83Q/Q107H, was prepared to facilitate coordination of the catalyst with greater solvent exposure (212). Azurin with either Cu or Zn in its native Cu-bonding site were used as scaffolds for Ni(II)-cyclam. When combined with [Ru(bpy)3]2+ as a sensitizer and ascorbate as an electron donor, the CuAz-Ni(II)-cyclam variant catalyzes the highest TON for CO production among these variants. Accounting for less than 100% efficiency of catalyst incorporation into CuAz, the CuAz outperforms free Ni(II)-cyclam in terms of TONs (mol CO/mol catalyst), demonstrating an increase from approximately 15 to 35. Importantly, the protein scaffold enhances selectivity for CO2 reduction relative to H+ reduction. Interestingly, the location of catalyst attachment does not change performance in photocatalysis, but the choice of metal in the native metal-binding site does. The Zn-containing variants of azurin produce less hydrogen, suggesting that Cu(II/I) plays a role in the redox chemistry of the process. In further work, a ruthenium sensitizer was attached directly to azurin complexed with Ni(II)-cyclam (214). S78C-Ru-CuAz, the mutant where the sensitizer was in closest proximity to the catalyst, is the most active variant under visible light irradiation at pH 7.25. Remarkably, the selectivity displayed by all prepared constructs is exclusively for CO production. On the other hand, the free cyclam has poor selectivity, yielding TONsCO/TONsH2 of only ~0.1. These results set a benchmark for fully selective homogeneous CO2 catalysts functioning in water while demonstrating the advantages a protein scaffold can convey.
Another nickel-centered catalyst, nickel(II)-terpyridine (2,2′:6′,2″-terpyridine), is highly selective for CO2 reduction to CO in organic solvent (215,216). The catalyst operates at low electrochemical overpotential and has also been driven photochemically. The terpyridine ligand is flexible to modification, which allows anchoring of Ni(II)-terpy to CdS quantum dots (217). While the catalyst-sensitizer assembly notably maintains high selectivity in aqueous solution, activity is insensitive to enzyme-to-nanocrystal ratios, and instability of the tether is detrimental to long-term activity.
Ni(II)-terpyridine (Ni(II)-terpy) was incorporated into a photosensitizing mini-protein to provide more tunable CO2-reduction activity [Fig. 16] (218). A naturally fluorescent protein, superfolder yellow fluorescent protein (sfYFP), was genetically modified to incorporate benzophenone–alanine. While fluorescent proteins typically have luminescence lifetimes too short to facilitate charge transfer, rational engineering of the sfYFP yielded mutant photosensitizer proteins (PSPs) with long-lived excited states capable of fueling redox reactions. One variant that includes mutations near the chromophore, PSP2, was found to be reducing enough to pass electrons to a CO2 reduction catalyst like Ni(II)-terpy. Using a PSP to drive catalysis allows for modification of not only the chromophore site, but also the tethering location for Ni(II)-terpy and the active-site environment. An iodacetamide derivative of Ni(II)terpy was prepared for direct coordination to a cysteine residue of PSP2. The PSP2-95C mutant, when paired with BIH (4-(2,3-dihydro-1H-benzo[d] imidazol-2-yl)benzene-1,2-diol) as a sacrificial electron donor, reduces CO2 to CO upon irradiation [Fig 16]. Hydrogen evolution was dependent on the distance between the chromophore and catalyst, and the Cys95 mutant was identified as the optimal distance. In addition, the installation of tyrosine residues near Cys95 to facilitate PCET reactions boosts the activity, yielding a best quantum yield of 2.6% for photocatalytic reduction of CO2 to CO, an improvement over the CdS-Ni(II)-terpy hybrids (0.28%). The ability to engineer sensitizer and active-site environment precisely is an opportunity afforded by the choice of a mini protein catalyst, and the innovation of genetically engineering photosensitizing proteins is broadly useful at the interface of photocatalysis and biocatalysis.
Fig. 16.
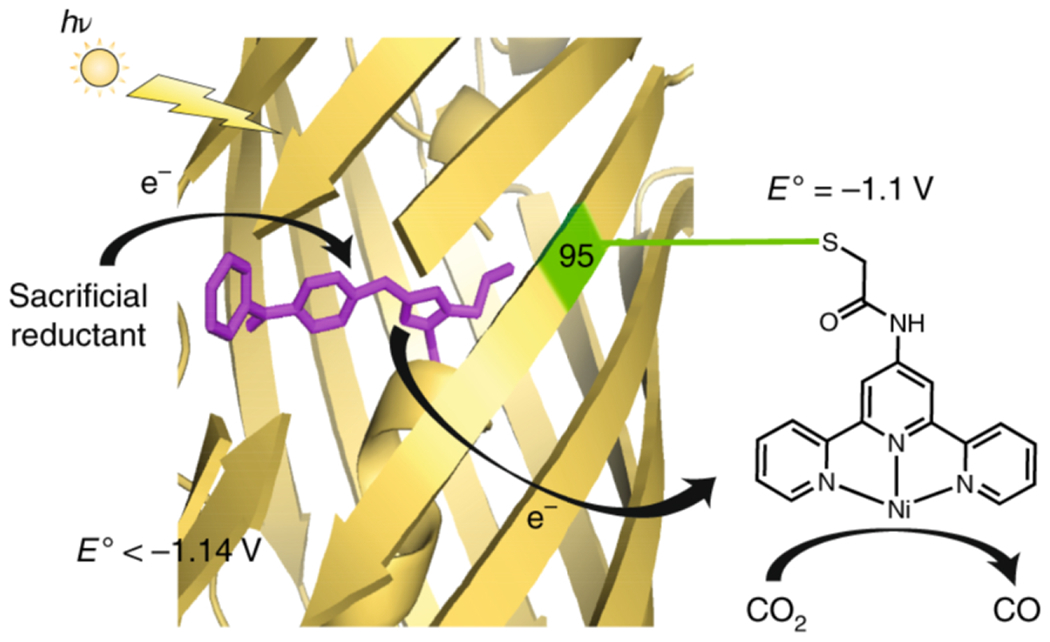
A scheme demonstrating the mechanism of CO2 reduction by Ni(II)-terpy coordinated to PSP2-95C. Reprinted with permission from Springer Nature Customer Service Centre GmbH: Springer, Nature Chemistry, Liu X, Kang F, Hu C, Wang L, Xu Z, Zheng D, Gong W, Lu Y, Ma Y, Wang J. (2018) Nature Chemistry 10, 1201-1206, copyright 2018.
Work continues on incorporating biocatalysts into photocatalytic systems for CO2 reduction. Although it is outside the scope of discussion here, many systems have focused on recycling electron donors rather than directly reducing enzymes with electrons (190,219). Recently, a graphene-based photocatalyst was reported that works with a series of coupled enzymes, formate dehydrogenase, formaldehyde dehydrogenase, and alcohol dehydrogenase, for selective reduction of CO2 to methanol (220). In addition, ongoing work looks to build structural and functional models of enzymes like CODH (221,222).
6. N2 Reduction
The Haber-Bosch nitrogen-fixation reaction has transformed life on earth (14,15). As aforementioned, the process requires harsh conditions and uses hydrogen generated from fossil fuels (15). The process consumes about 1% of the world’s total energy, and is crucial for producing the quantities of ammonia needed to sustain current human industry and agriculture. Although the Haber-Bosch process is now a significant contributor to the nitrogen cycle on earth, for millions of years, nitrogen fixation was carried out primarily by nitrogenases in few microorganisms (13).
Nitrogenase enzymes catalyze the reduction of N2 to NH3 in nitrogen-fixing bacteria [Equation 8] (13). Compared to the industrial process, these enzymes carry out the reaction under mild conditions. Molybdenum nitrogenase is the most well-studied variety. It catalyzes nitrogen fixation with a two-component system composed of an MoFe protein (dinitrogenase) and an electron-transfer Fe protein (dinitrogenase reductase). Typically, a reducing source and MgATP are required for the normal function. To accomplish nitrogen fixation on a large scale as nitrogen ase does, under relatively mild conditions without the need to input H2, would be a huge accomplishment for industry. As with previously discussed processes, though, the potential use of nitrogenase is limited by the need for complex systems to deliver reducing equivalents. In nitrogen fixation by molybdenum nitrogenase, there are eight sequential electron transfer and ATP hydrolysis reactions to generate one mole of NH3. Molybdenum nitrogenase also contains a MoFe cofactor, which is proposed to form Fe-hydrides to participate in reduction of N2, producing H2 as part of the reaction.
| (Equation 8) |
To replace the need for ATP-coupled electron transfers and dinitrogen reductase, a photosensitizer could be used to provide electrons directly to the protein. To accomplish this, molybdenum-containing nitrogenase from Azotobacter vinelandii has been coupled to CdS nanorods as photosensitizers [Fig. 17] (223). While there was some precedent for coupling nitrogenase to ruthenium photosensitizers, the enzymes had been unable to catalyze full reduction to NH3 in that system, and had catalyzed the reduction of nonphysiological substrates relatively slowly (224,225). Unlike previous instances, the pairing of CdS nanorods with molybdenum nitrogenase yielded full conversion of N2 to NH3. Reactions containing a sacrificial donor (HEPES) and self-assembled complexes of CdS-MoFe protein achieved TONs up to of 1.1 × 104 (mol NH3/mol enzyme) upon radiation with 405-nm light. As in the physiological system, catalysis is expected to occur at the MoFe-cofactor; this hypothesis is supported by studies of catalytic inhibition. The CdS nanorod thus replaces the function of the Fe protein [Fig. 17]. The structure and choice of sensitizer is crucial to driving the chemistry. While various sensitizers could potentially be chosen, the authors found that the ability of the nanorod to quickly deliver successive equivalents of electrons was important for the full reduction to NH3.
Fig. 17.
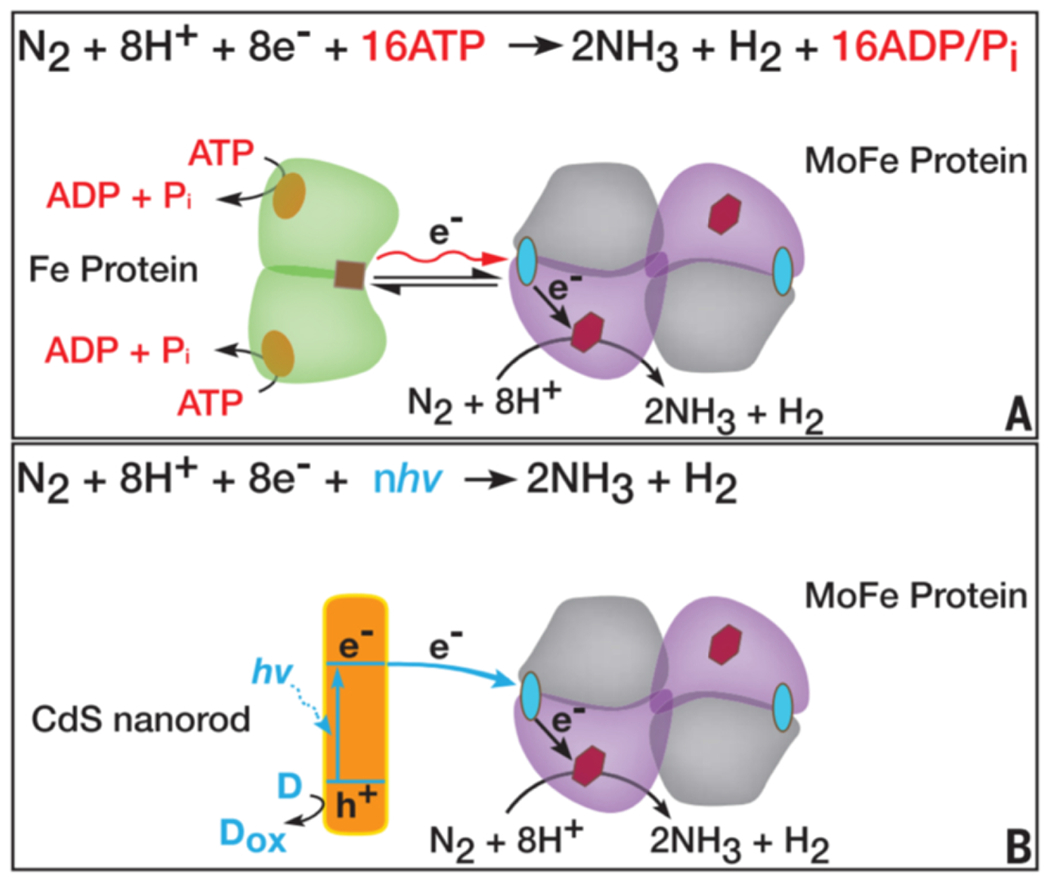
Schematic of the typical reaction catalyzed by molybdenum nitrogenase (A) demonstrating how CdS nanorods replace necessary electron transfer proteins and cofactors (B). Reprinted with permission from Brown KA, Harris DF, Wilker MB, Rasmussen A, Khadka N, Hamby H, Keable S, Dukovic G, Peters JW, Seefeldt LC, King PW. (2016) Science 352, 448-450. Copyright (2016), American Association for the Advancement of Science.
The field of N2 conversion to ammonia via light-driven reactions has gained increasing traction over the last few decades. Although largely dominated by heterogeneous catalysis (226–230), the pioneering work coupling nitrogenase to CdS nanostructures is a promising demonstration of light-driven biological ammonia production. Others have begun to follow suit, bridging the gap between bioinspired homogeneous catalysis and heterogeneous catalysis (231,232). Biomimetic chalcogels containing mimics of the MoFe protein active site can catalyze the full reduction of N2 to NH3 under ambient conditions and visible-light irradiation. Pairing enzymes with nanocrystalline photosensitizers also provides an opportunity to study fast electron transfer steps that activate enzymes.
7. Summary and Outlook
Significant strides toward developing light-driven biocatalysis have been made in the past decade. These steps forward have been enabled by advances in enzyme engineering and in photochemistry. Looking ahead on the biocatalyst side, broader uses of directed evolution methods in this field are expected to further enable the engineering and application of biomolecular catalysts for a range of reactions including those not found in nature. On the side of the photosensitizers, the development and application of semiconductor nanocrystals has yielded photosensitizers that are amenable to assembly with complex biomolecules. Putting these components together takes advantage of the best aspects of nanomaterials (high photostability and tunability) with those of enzymes and biocatalysts (efficiency, selectivity). Nevertheless, significant challenges remain. For one, while using light to initiate reactions opens doors to time-resolved studies of mechanism, in general little is known beyond the first electron transfer. The development of approaches that facilitate more detailed study of photocatalytic mechanism is vital for advancing our understanding of how these systems work. Second, the nature of the biomolecule-nanocrystal interface is poorly understood, and control of the assembly of these systems is poor. The use of molecular photosensitizers yields more well-defined photocatalytic systems, but photodegradation of these sensitizers is often a limiting factor. Furthermore, despite the high stability of nanocrystalline photosensitizers, hybrids with biocatalysts nevertheless often do not exhibit the desired longevity, and the basis for loss of activity is not always understood. Finally, the development of photo-bio systems for important and challenging multi-electron reactions such as the reduction of CO2 to alcohols and of N2 to ammonia is still in its infancy. With the rapid pace of the fields of photocatalysis as well as enzyme engineering, we can look forward to exciting developments in these areas in coming years.
Synopsis.
Efforts to drive catalytic reactions with light, inspired by natural processes like photosynthesis, have a long history and have seen significant recent growth. Successfully engineering systems using biomolecular and bioinspired catalysts to carry out light-driven chemical reactions capitalizes on advantages offered from the fields of biocatalysis and photocatalysis. In particular, driving reactions under mild conditions and in water, in which enzymes are operative, using sunlight as a renewable energy source yields environmentally friendly systems. Furthermore, using enzymes and bioinspired systems can take advantage of the high efficiency and specificity of biocatalysts. There are many challenges to overcome to fully capitalize on the potential of light driven biocatalysis. In this mini-review, we discuss examples of enzymes and engineered biomolecular catalysts that are activated via electron transfer from a photosensitizer in a photocatalytic system. We place an emphasis on selected forefront chemical reactions of high interest, including C-H oxidation, proton reduction, water oxidation, CO2 reduction, and N2 reduction.
Highlights.
Biomolecular catalysts are being developed for a wide range of reactions in the areas of synthesis and energy conversion.
Driving biocatalytic reactions with light circumvents the need to consume expensive biological electron donors like NAD(P)H.
Some light-driven biocatalytic reactions store light energy in the form of a fuel.
Acknowledgments
E.H.E.’s research in the Bren lab focuses on light-driven hydrogen production catalyzed by biomolecular catalysts and is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award DE-FG02-09ER16121. E.H.E. also acknowledges support from the N.I.H. Chemistry-Biology Interface Training Program T32-GM118283. K.L.B.’s work on this review was supported by the National Science Foundation grant CHE-1708256, which supports research on synthetic metallopeptide catalysts. The authors declare no conflict of interest.
References
- [1].Sheldon RA, Pereira PC (2017) Chem. Soc. Rev 46, 2678–2691. [DOI] [PubMed] [Google Scholar]
- [2].Cornish-Bowden A. (2011) History of Enzyme Chemistry. In: eLS, (Ed.). eLS. [Google Scholar]
- [3].Cuesta SM, Rahman SA, Furnham N, Thornton JM (2015) Biophys. J 109, 1082–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hughes G, Lewis JC (2018) Chem. Rev 118, 1–3. [DOI] [PubMed] [Google Scholar]
- [5].Devine PN, Howard RM, Kumar R, Thompson MP, Truppo MD, Turner NJ (2018) Nat. Rev. Chem 2, 409–421. [Google Scholar]
- [6].Turner NJ, Kumar R (2018) Curr. Opin. Biotechnol 43, A1–A3. [DOI] [PubMed] [Google Scholar]
- [7].Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K (2012) Nature 485, 185–194. [DOI] [PubMed] [Google Scholar]
- [8].DiCosimo R, McAuliffe J, Poulose AJ, Bohlmann G (2013) Chem. Soc. Rev 42, 6437–6474. [DOI] [PubMed] [Google Scholar]
- [9].Rozzell JD (1999) Bioorg. Med. Chem 7, 2253–2261. [DOI] [PubMed] [Google Scholar]
- [10].Truppo MD (2017) ACS Med. Chem. Lett 8, 476–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen K, Arnold FH (2020) Nat. Catal 3, 203–213. [Google Scholar]
- [12].Lewis JC, Coelho PS, Arnold FH (2011) Chem. Soc. Rev 40, 2003–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hoffman BM, Lukoyanov D, Yang Z-Y, Dean DR, Seefeldt LC (2014) Chem. Rev 114, 4041–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cherkasov N, Ibhadon AO, Fitzpatrick P (2015) Chem. Eng. Process. Process Intensif 90, 24–33. [Google Scholar]
- [15].Chen JG, Crooks RM, Seefeldt LC, Bren KL, Bullock RM, Darensbourg MY, Holland PL, Hoffman B, Janik MJ, Jones AK, Kanatzidis MG, King P, Lancaster KM, Lymar SV, Pfromm P, Schneider WF, Schrock RR (2018) Science 360, eaar6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Desai AA (2011) Angew. Chem. Int. Ed 50, 1974–1976. [DOI] [PubMed] [Google Scholar]
- [17].Savile CK, Janey JM, Mundorff EC, Moore JC, Tam S, Jarvis WR, Colbeck JC, Krebber A, Fleitz FJ, Brands J, Devine PN, Huisman GW, Hughes GJ (2010) Science 329, 305–309. [DOI] [PubMed] [Google Scholar]
- [18].Shalan H, Kato M, Cheruzel L (2018) Biochim. Biophys. Acta, Proteins Proteomics 1866, 80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Serpone N, Emeline AV, Horikoshi S, Kuznetsov VN, Ryabchuk VK (2012) Photochem. Photobiol. Sci 11, 1121–1150. [DOI] [PubMed] [Google Scholar]
- [20].Zhu S, Wang D (2017). Adv. Energy Mater 7, 1700841. [Google Scholar]
- [21].Zhang B, Sun L (2019) Chem. Soc. Rev 48, 2216–2264. [DOI] [PubMed] [Google Scholar]
- [22].Romero NA, Nicewicz DA (2016) Chem. Rev 116, 10075–10166. [DOI] [PubMed] [Google Scholar]
- [23].Prier CK, Rankic DA, MacMillan DWC (2013) Chem. Rev 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xuan J, Xiao W-J (2012) Angew. Chem. Int. Ed 51, 6828–6838. [DOI] [PubMed] [Google Scholar]
- [25].Graetzel M. (1981) Acc. Chem. Res 14, 376–384. [Google Scholar]
- [26].Meyer TJ (1989) Acc. Chem. Res 22, 163–170. [Google Scholar]
- [27].Ulmer U, Dingle T, Duchesne PN, Morris RH, Tavasoli A, Wood T, Ozin GA (2019) Nat. Commun 10, 3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Schultz DM, Yoon TP (2014) Science 343, 1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Michelin C, Hoffmann N (2018) ACS Catal. 8, 12046–12055. [Google Scholar]
- [30].Yang X, Wang D (2018) ACS Appl. Energy Mater 1, 6657–6693. [Google Scholar]
- [31].Caputo JA, Frenette LC, Zhao N, Sowers KL, Krauss TD, Weix DJ (2017) J. Am. Chem. Soc 139, 4250–4253. [DOI] [PubMed] [Google Scholar]
- [32].Ischay MA, Yoon TP (2012) Eur. J. Org. Chem 2012, 3359–3372. [Google Scholar]
- [33].Ciamician G. (1912) Science 36, 385–394. [DOI] [PubMed] [Google Scholar]
- [34].Whang DR, Apaydin DH (2018) ChemPhotoChem 2, 148–160. [Google Scholar]
- [35].Bard AJ, Fox MA (1995) Acc. Chem. Res 28, 141–145. [Google Scholar]
- [36].Bren KL (2015) Interface Focus 5, 20140091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Alstrum-Acevedo JH, Brennaman MK, Meyer TJ (2005) Inorg. Chem 44, 6802–6827. [DOI] [PubMed] [Google Scholar]
- [38].Cox N, Pantazis DA, Neese F, Lubitz W (2015) Interface Focus 5, 20150009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mirkovic T, Ostroumov EE, Anna JM, van Grondelle R, Govindjee, Scholes GD (2017) Chem. Rev 117, 249–293. [DOI] [PubMed] [Google Scholar]
- [40].Balzani V, Credi A, Venturi M (2008) ChemSusChem 1, 26–58. [DOI] [PubMed] [Google Scholar]
- [41].Seel CJ, Gulder T (2019) ChemBioChem 20, 1871–1897. [DOI] [PubMed] [Google Scholar]
- [42].Maciá-Agulló JA, Corma A, Garcia H (2015) Chem. Eur. J 21, 10940–10959. [DOI] [PubMed] [Google Scholar]
- [43].Schmermund L, Jurkaţ V, Özgen FF, Barone GD, Büchsenschütz HC, Winkler CK, Schmidt S, Kourist R, Kroutil W (2019) ACS Catal. 9, 4115–4144. [Google Scholar]
- [44].Lee SH, Choi DS, Kuk SK, Park CB (2018) Angew. Chem. Int. Ed 57, 7958–7985. [DOI] [PubMed] [Google Scholar]
- [45].Biegasiewicz KF, Cooper SJ, Gao X, Oblinsky DG, Kim JH, Garfinkle SE, Joyce LA, Sandoval BA, Scholes GD, Hyster TK (2019) Science 364, 1166–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Black MJ, Biegasiewicz KF, Meichan AJ, Oblinsky DG, Kudisch B, Scholes GD, Hyster TK (2020) Nat. Chem 12, 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Arndtsen BA, Bergman RG, Mobley TA, Peterson TH (1995) Acc. Chem. Res 28, 154–162. [Google Scholar]
- [48].Fasan R. (2012) Tuning P450 Enzymes as Oxidation Catalysts. ACS Catal. 2, 647–666. [Google Scholar]
- [49].Abrams DJ, Provencher PA, Sorensen EJ (2018) Chem. Soc. Rev 47, 8925–8967. [DOI] [PubMed] [Google Scholar]
- [50].Davies HML, Du Bois J, Yu J-Q (2011) Chem. Soc. Rev 40, 1855–1856. [DOI] [PubMed] [Google Scholar]
- [51].Gutekunst WR, Baran PS (2011) Chem. Soc. Rev 40, 1976–1991. [DOI] [PubMed] [Google Scholar]
- [52].Yamaguchi J, Yamaguchi AD, Itami K (2012) Angew. Chem. Int. Ed 51, 8960–9009. [DOI] [PubMed] [Google Scholar]
- [53].Dick AR, Hull KL, Sanford MS (2004) J. Am. Chem. Soc 126, 2300–2301. [DOI] [PubMed] [Google Scholar]
- [54].Brodsky BH, Du Bois J (2005) J. Am. Chem. Soc 127, 15391–15393. [DOI] [PubMed] [Google Scholar]
- [55].Crabtree RH, Lei A (2017) Chem. Rev 117, 8481–8482. [DOI] [PubMed] [Google Scholar]
- [56].Gandeepan P, Müller T, Zell D, Cera G, Warratz S, Ackermann L (2019) Chem. Rev 119, 2192–2452. [DOI] [PubMed] [Google Scholar]
- [57].Woźniak Ł, Cramer N (2019) Trends Chem. 1, 471–484. [Google Scholar]
- [58].Kille S, Zilly FE, Acevedo JP, Reetz MT (2011) Nat. Chem 3, 738–743. [DOI] [PubMed] [Google Scholar]
- [59].Rentmeister A, Arnold FH, Fasan R (2009) Nat. Chem. Biol 5, 26–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Guengerich FP (2018) ACS Catal. 8, 10964–10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ortiz de Montellano PR (2010) Chem. Rev 110, 932–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Groves JT (2014) Nat. Chem 6, 89–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ener ME, Lee Y-T, Winkler JR, Gray HB, Cheruzel L (2010) PNAS 107, 18783–18786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Tran N-H, Huynh N, Chavez G, Nguyen A, Dwaraknath S, Nguyen T-A, Nguyen M, Cheruzel L (2012) J. Inorg. Biochem 115, 50–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Tran N-H, Nguyen D, Dwaraknath S, Mahadevan S, Chavez G, Nguyen A, Dao T, Mullen S, Nguyen T-A, Cheruzel LE (2013) J. Am. Chem. Soc 135, 14484–14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Kato M, Lam Q, Bhandarkar M, Banh T, Heredia JUA, Cheruzel L (2017). C.R. Chim 20, 237–242. [Google Scholar]
- [67].Sosa V, Melkie M, Sulca C, Li J, Tang L, Li J, Faris J, Foley B, Banh T, Kato M, Cheruzel LE (2018) ACS Catal. 8, 2225–2229. [Google Scholar]
- [68].Kato M, Nguyen D, Gonzalez M, Cortez A, Mullen SE, Cheruzel LE (2014) Bioorg. Med. Chem 22, 5687–5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Whitehouse CJC, Bell SG, Wong L-L (2012) Chem. Soc. Rev 41, 1218–1260. [DOI] [PubMed] [Google Scholar]
- [70].Tran N-H, Huynh N, Bui T, Nguyen Y, Huynh P, Cooper ME, Cheruzel LE (2011) Chem. Comm 47, 11936–11938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Cowart LA, Falck JR, Capdevila JH (2001) Arch. Biochem. Biophys 387, 117–124. [DOI] [PubMed] [Google Scholar]
- [72].Zilly FE, Taglieber A, Schulz F, Hollmann F, Reetz MT (2009) Chem. Comm 7152–7154. [DOI] [PubMed] [Google Scholar]
- [73].Estabrook RW, Faulkner KM, Shet MS, Fisher CW (1996) Application of electrochemistry for P450-catalyzed reactions. In: Johnson EF, Waterman MR (eds) Methods in Enzymology, vol 272. Academic Press, pp 44–51. [DOI] [PubMed] [Google Scholar]
- [74].Lam Q, Cortez A, Nguyen TT, Kato M, Cheruzel L (2016) J. Inorg. Biochem 158, 86–91. [DOI] [PubMed] [Google Scholar]
- [75].Shalan H, Colbert A, Nguyen TT, Kato M, Cheruzel L (2017) Inorg. Chem 56, 6558–6564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ipe BI, Niemeyer CM (2006) Angew. Chem. Int. Ed 45, 504–507. [DOI] [PubMed] [Google Scholar]
- [77].Gandubert VJ, Torres E, Niemeyer CM (2008) J. Mater. Chem 18, 3824–3830. [Google Scholar]
- [78].Harris RD, Bettis Homan S, Kodaimati M, He C, Nepomnyashchii AB, Swenson NK, Lian S, Calzada R, Weiss EA (2016) Electronic Processes within Quantum Dot-Molecule Complexes. Chem. Rev 116, 12865–12919. [DOI] [PubMed] [Google Scholar]
- [79].Kodaimati MS, McClelland KP, He C, Lian S, Jiang Y, Zhang Z, Weiss EA (2018) Inorg. Chem 57, 3659–3670. [DOI] [PubMed] [Google Scholar]
- [80].Matsunaga I, Ueda A, Fujiwara N, Sumimoto T, Ichihara K (1999) Lipids 34, 841–846. [DOI] [PubMed] [Google Scholar]
- [81].Ipe BI, Lehnig M, Niemeyer CM (2005) Small 1, 706–709. [DOI] [PubMed] [Google Scholar]
- [82].Rajendran V, König A, Rabe KS, Niemeyer CM (2010) Small 6, 2035–2040. [DOI] [PubMed] [Google Scholar]
- [83].Nielsen AZ, Ziersen B, Jensen K, Lassen LM, Olsen CE, Müller BL, Jensen PE (2013) ACS Synth. Biol 2, 308–315. [DOI] [PubMed] [Google Scholar]
- [84].Nelson N, Ben-Shem A (2004) Nat. Rev. Mol. Cell Biol 5, 971–982. [DOI] [PubMed] [Google Scholar]
- [85].Ordonio R, Ito Y, Morinaka Y, Sazuka T, Matsuoka M (2016) Chapter Five: Molecular Breeding of Sorghum bicolor, A Novel Energy Crop. In: Jeon KW (ed) International Review of Cell and Molecular Biology, vol 321. Academic Press, pp 221–257. [DOI] [PubMed] [Google Scholar]
- [86].Rosati VC, Blomstedt CK, Møller BL, Garnett T, Gleadow R (2019) Front. Plant Sci 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Kahn RA, Bak S, Svendsen I, Halkier BA, Moller BL (1997) Plant Physiol. 115, 1661–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Mellor SB, Nielsen AZ, Burow M, Motawia MS, Jakubauskas D, Møller BL, Jensen PE (2016) ACS Chem. Biol 11, 1862–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Renault H, Bassard J-E, Hamberger B, Werck-Reichhart D (2014) Curr. Opin. Plant Biol 19, 27–34. [DOI] [PubMed] [Google Scholar]
- [90].Zhang W, Fernández-Fueyo E, Ni Y, van Schie M, Gacs J, Renirie R, Wever R, Mutti FG, Rother D, Alcalde M, Hollmann F (2018) Nat. Catal 1, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Willot SJP, Fernández-Fueyo E, Tieves F, Pesic M, Alcalde M, Arends IWCE, Park CB, Hollmann F (2019) ACS Catal. 9, 890–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Jung ST, Lauchli R, Arnold FH (2011) Curr. Opin. Biotechnol 22, 809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Groves J T, Nemo TE, Myers RS (1979) J. Am. Chem. Soc 101, 1032–1033. [Google Scholar]
- [94].Che C-M, Huang J-S (2009) Chem. Commun 27, 3996–4015. [DOI] [PubMed] [Google Scholar]
- [95].Rohde J-U, In J-H, Lim MH, Brennessel WW, Bukowski MR, Stubna A, Münck E, Nam W, Que L (2003) Science 299, 1037–1039. [DOI] [PubMed] [Google Scholar]
- [96].Usharani D, Lacy DC, Borovik AS, Shaik S (2013) J. Am. Chem. Soc 135, 17090–17104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].de Visser SP, Rohde J-U, Lee Y-M, Cho J, Nam W (2013) Coord. Chem. Rev 257, 381–393. [Google Scholar]
- [98].Cho K, Leeladee P, McGown AJ, DeBeer S, Goldberg DP (2012) J. Am. Chem. Soc 134, 7392–7399. [DOI] [PubMed] [Google Scholar]
- [99].Nam W, Lee Y-M, Fukuzumi S (2014) Acc. Chem. Res 47, 1146–1154. [DOI] [PubMed] [Google Scholar]
- [100].Dubouis N, Grimaud A (2019) Chem. Sci 10, 9165–9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Navarro RM, Peña MA, Fierro JLG (2007) Chem. Rev 107, 3952–3991. [DOI] [PubMed] [Google Scholar]
- [102].Bockris JOM (2002) Int. J. Hydrogen Energy 27, 731–740. [Google Scholar]
- [103].Nocera DG (2017) Acc. Chem. Res 50, 616–619. [DOI] [PubMed] [Google Scholar]
- [104].Wang W, Chen J, Li C, Tian W (2014) Nat. Commun 5, 4647. [DOI] [PubMed] [Google Scholar]
- [105].Santhanam KSVP, Roman J; Miri Massoud J.; Bailey Alla V.; Takacs Gerald A. (2017) Introduction to Hydrogen Technology, 2nd Edition. 2 edn. [Google Scholar]
- [106].Thoi VS, Sun Y, Long JR, Chang CJ (2013) Chem. Soc. Rev 42, 2388–2400. [DOI] [PubMed] [Google Scholar]
- [107].Eckenhoff WT, Eisenberg R (2012) Dalton Trans. 41, 13004–13021. [DOI] [PubMed] [Google Scholar]
- [108].Debe MK (2012) Nature 486, 43–51. [DOI] [PubMed] [Google Scholar]
- [109].Zeng M, Li Y (2015) J. Mater. Chem. A 3, 14942–14962. [Google Scholar]
- [110].Dalle KE, Warnan J, Leung JJ, Reuillard B, Karmel IS, Reisner E (2019) Chem. Rev 119, 2752–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Tong L, Duan L, Zhou A, Thummel RP (2020) Coord. Chem. Rev 402, 213079. [Google Scholar]
- [112].Walter MG, Warren EL, McKone JR, Boettcher SW, Mi Q, Santori EA, Lewis NS (2010) Chem. Rev 110, 6446–6473. [DOI] [PubMed] [Google Scholar]
- [113].McKone JR, Marinescu SC, Brunschwig BS, Winkler JR, Gray HB (2014) Chem. Sci 5, 865–878. [Google Scholar]
- [114].Simmons TR, Berggren G, Bacchi M, Fontecave M, Artero V (2014) Coord. Chem. Rev 270-271, 127–150. [Google Scholar]
- [115].Barton BE, Olsen MT, Rauchfuss TB (2010) Curr. Opin. Biotechnol 21, 292–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Reisner E, Powell DJ, Cavazza C, Fontecilla-Camps JC, Armstrong FA (2009) J. Am. Chem. Soc 131, 18457–18466. [DOI] [PubMed] [Google Scholar]
- [117].Lubitz W, Ogata H, Rüdiger O, Reijerse E (2014) Chem. Rev 114, 4081–4148. [DOI] [PubMed] [Google Scholar]
- [118].Melis A, Happe T (2001) Plant Physiol. 127, 740–748. [PMC free article] [PubMed] [Google Scholar]
- [119].Nandi R, Sengupta S (1998) Crit. Rev. Microbiol 24, 61–84. [DOI] [PubMed] [Google Scholar]
- [120].Ghirardi ML, Togasaki RK, Seibert M (1997) Appl. Biochem. Biotechnol 63, 141–151. [DOI] [PubMed] [Google Scholar]
- [121].McTavish H (1998) J. Biochem 123, 644–649. [DOI] [PubMed] [Google Scholar]
- [122].Ihara M, Nishihara H, Yoon K-S, Lenz O, Friedrich B, Nakamoto H, Kojima K, Honma D, Kamachi T, Okura I (2006) Photochem. Photobiol 82, 676–682. [DOI] [PubMed] [Google Scholar]
- [123].Krassen H, Schwarze A, Friedrich B, Ataka K, Lenz O, Heberle J (2009) ACS Nano 3, 4055–4061. [DOI] [PubMed] [Google Scholar]
- [124].Lubner CE, Knörzer P, Silva PJN, Vincent KA, Happe T, Bryant DA, Golbeck JH (2010) Biochemistry 49, 10264–10266. [DOI] [PubMed] [Google Scholar]
- [125].Reisner E, Fontecilla-Camps JC, Armstrong FA (2009) Chem. Comm 5, 550–552. [DOI] [PubMed] [Google Scholar]
- [126].Morra S, Valetti F, Sadeghi SJ, King PW, Meyer T, Gilardi G (2011) Chem. Comm 47, 10566–10568. [DOI] [PubMed] [Google Scholar]
- [127].Morra S, Valetti F, Sarasso V, Castrignanò S, Sadeghi SJ, Gilardi G (2015) Bioelectrochemistry 106, 258–262. [DOI] [PubMed] [Google Scholar]
- [128].Brown KA, Dayal S, Ai X, Rumbles G, King PW (2010) J. Am. Chem. Soc 132, 9672–9680. [DOI] [PubMed] [Google Scholar]
- [129].Brown KA, Wilker MB, Boehm M, Dukovic G, King PW (2012) J. Am. Chem. Soc 134, 5627–5636. [DOI] [PubMed] [Google Scholar]
- [130].Greene BL, Joseph CA, Maroney MJ, Dyer RB (2012) J. Am. Chem. Soc 134, 11108–11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Greene BL, Wu C-H, McTernan PM, Adams MWW, Dyer RB (2015) J. Am. Chem. Soc 137, 4558–4566. [DOI] [PubMed] [Google Scholar]
- [132].Chica B, Wu C-H, Liu Y, Adams MWW, Lian T, Dyer RB (2017) Energy Environ. Sci 10, 2245–2255. [Google Scholar]
- [133].Sanchez MLK, Wu C-H, Adams MWW, Dyer RB (2019) Chem. Comm 55, 5579–5582. [DOI] [PubMed] [Google Scholar]
- [134].Wilker MB, Utterback JK, Greene S, Brown KA, Mulder DW, King PW, Dukovic G (2018) J. Phys. Chem. C 122, 741–750. [Google Scholar]
- [135].Utterback JK, Wilker MB, Brown KA, King PW, Eaves JD, Dukovic G (2015) Phys. Chem. Chem. Phys 17, 5538–5542. [DOI] [PubMed] [Google Scholar]
- [136].Zhang L, Beaton SE, Carr SB, Armstrong FA (2018) Energy Environ. Sci 11, 3342–3348. [Google Scholar]
- [137].Beaton SE, Evans RM, Finney AJ, Lamont CM, Armstrong FA, Sargent F, Carr SB (2018) Biochem. J 475, 1353–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Oohora K, Onoda A, Hayashi T (2019) Acc. Chem. Res 52, 945–954. [DOI] [PubMed] [Google Scholar]
- [139].Le JM, Bren KL (2019) ACS Energy Lett. 4, 2168–2180. [Google Scholar]
- [140].Kuchenreuther JM, Grady-Smith CS, Bingham AS, George SJ, Cramer SP, Swartz JR (2010) PLoS One 5, e15491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Tard C, Pickett CJ (2009) Chem. Rev 109, 2245–2274. [DOI] [PubMed] [Google Scholar]
- [142].Artero V, Fontecave M (2005) Coord. Chem. Rev 249, 1518–1535. [Google Scholar]
- [143].Tschierlei S, Ott S, Lomoth R (2011) Energy Environ. Sci 4, 2340–2352. [Google Scholar]
- [144].Jones AK, Lichtenstein BR, Dutta A, Gordon G, Dutton PL (2007) J. Am. Chem. Soc 129, 14844–14845. [DOI] [PubMed] [Google Scholar]
- [145].Sano Y, Onoda A, Hayashi T (2011) Chem. Comm 47, 8229–8231. [DOI] [PubMed] [Google Scholar]
- [146].Na Y, Wang M, Pan J, Zhang P, Åkermark B, Sun L (2008) Inorg. Chem 47, 2805–2810. [DOI] [PubMed] [Google Scholar]
- [147].Na Y, Pan J, Wang M, Sun L (2007) Inorg. Chem 46, 3813–3815. [DOI] [PubMed] [Google Scholar]
- [148].Streich D, Astuti Y, Orlandi M, Schwartz L, Lomoth R, Hammarström L, Ott S (2010) Chem. Eur. J 16, 60–63. [DOI] [PubMed] [Google Scholar]
- [149].Zhang P, Wang M, Na Y, Li X, Jiang Y, Sun L (2010) Dalton Trans. 39, 1204–1206. [DOI] [PubMed] [Google Scholar]
- [150].Onoda A, Kihara Y, Fukumoto K, Sano Y, Hayashi T (2014) ACS Catal. 4, 2645–2648. [Google Scholar]
- [151].Roy A, Madden C, Ghirlanda G (2012) Chem. Comm 48, 9816–9818. [DOI] [PubMed] [Google Scholar]
- [152].Sano Y, Onoda A, Hayashi T (2012) J. Inorg. Biochem 108, 159–162. [DOI] [PubMed] [Google Scholar]
- [153].Slater JW, Shafaat HS (2015) J. Phys. Chem. Lett 6, 3731–3736. [DOI] [PubMed] [Google Scholar]
- [154].Slater JW, Marguet SC, Cirino SL, Maugeri PT, Shafaat HS (2017) Inorg. Chem 56, 3926–3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Stevenson MJ, Marguet SC, Schneider CR, Shafaat HS (2017) ChemSusChem 10, 4424–4429. [DOI] [PubMed] [Google Scholar]
- [156].Slater JW, Marguet SC, Monaco HA, Shafaat HS (2018) J. Am. Chem. Soc 140, 10250–10262. [DOI] [PubMed] [Google Scholar]
- [157].Marguet SC, Stevenson MJ, Shafaat HS (2019) J. Phys. Chem. B 123, 9792–9800. [DOI] [PubMed] [Google Scholar]
- [158].Slater JW, Marguet SC, Gray ME, Monaco HA, Sotomayor M, Shafaat HS (2019) ACS Catalysis 9, 8928–8942. [Google Scholar]
- [159].Saint-Martin P, Lespinat PA, Fauque G, Berlier Y, LeGall J, Moura I, Teixeira M, Xavier AV, Moura JJG (1988) PNAS 85, 9378–9380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Queyriaux N, Jane RT, Massin J, Artero V, Chavarot-Kerlidou M (2015) Coord. Chem. Rev 304-305, 3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Eckenhoff WT, McNamara WR, Du P, Eisenberg R (2013) Biochim. Biophys. Acta, Bioenerg 1827, 958–973. [DOI] [PubMed] [Google Scholar]
- [162].Rakowski DuBois M, DuBois DL (2009) Chem. Soc. Rev 38, 62–72. [DOI] [PubMed] [Google Scholar]
- [163].Du P, Schneider J, Luo G, Brennessel WW, Eisenberg R (2009) Inorg. Chem 48, 4952–4962. [DOI] [PubMed] [Google Scholar]
- [164].Dempsey JL, Brunschwig BS, Winkler JR, Gray HB (2009) Acc. Chem. Res 42, 1995–2004. [DOI] [PubMed] [Google Scholar]
- [165].Bacchi M, Berggren G, Niklas J, Veinberg E, Mara MW, Shelby ML, Poluektov OG, Chen LX, Tiede DM, Cavazza C, Field MJ, Fontecave M, Artero V (2014) Inorg. Chem 53, 8071–8082. [DOI] [PubMed] [Google Scholar]
- [166].Bacchi M, Veinberg E, Field MJ, Niklas J, Matsui T, Tiede DM, Poluektov OG, Ikeda-Saito M, Fontecave M, Artero V (2016). ChemPlusChem 81, 1083–1089. [DOI] [PubMed] [Google Scholar]
- [167].Soltau SR, Niklas J, Dahlberg PD, Poluektov OG., Tiede DM, Mulfort KL, Utschig LM (2015) Chem. Comm 51, 10628–10631. [DOI] [PubMed] [Google Scholar]
- [168].Soltau SR Dahlberg PD, Niklas J, Poluektov OG, Mulfort KL, Utschig LM (2016) Chem. Sci 7, 7068–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Brahmachari U, Pokkuluri PR, Tiede DM, Niklas J, Poluektov OG, Mulfort KL, Utschig LM (2020) Photosynth. Res 143, 183–192. [DOI] [PubMed] [Google Scholar]
- [170].Sommer DJ, Vaughn MD, Ghirlanda G (2014) Chem. Commun 50, 15852–15855. [DOI] [PubMed] [Google Scholar]
- [171].Kleingardner JG, Kandemir B, Bren KL (2014) J. Am. Chem. Soc 136, 4–7. [DOI] [PubMed] [Google Scholar]
- [172].Kandemir B, Kubie L, Guo Y, Sheldon B, Bren KL (2016) Inorg. Chem 55, 1355–1357. [DOI] [PubMed] [Google Scholar]
- [173].Chakraborty S, Edwards EH, Kandemir B, Bren KL (2019) Inorg. Chem 58, 16402–16410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Wilson AD, Newell RH, McNevin MJ, Muckerman JT, Rakowski DuBois M, DuBois DL (2006) J. Am. Chem. Soc 128, 358–366. [DOI] [PubMed] [Google Scholar]
- [175].McLaughlin MP, McCormick TM, Eisenberg R, Holland PL (2011) Chem. Comm 47, 7989–7991. [DOI] [PubMed] [Google Scholar]
- [176].Silver SC, Niklas J, Du P, Poluektov OG, Tiede DM, Utschig LM (2013) J. Am. Chem. Soc 135, 13246–13249. [DOI] [PubMed] [Google Scholar]
- [177].Dutta A, Hamilton GA, Hartnett HE, Jones AK (2012) Inorg. Chem 51, 9580–9588. [DOI] [PubMed] [Google Scholar]
- [178].Dutta A, Ginovska B, Raugei S, Roberts JAS, Shaw WJ (2016) Dalton Trans. 45, 9786–9793. [DOI] [PubMed] [Google Scholar]
- [179].Reback ML, Buchko GW, Kier BL, Ginovska-Pangovska B, Xiong Y, Lense S, Hou J, Roberts JAS, Sorensen CM, Raugei S, Squier TC, Shaw WJ (2014) Chem. Eur. J 20, 1510–1514. [DOI] [PubMed] [Google Scholar]
- [180].Reback ML, Ginovska B, Buchko GW, Dutta A, Priyadarshani N, Kier BL, Helm ML, Raugei S, Shaw WJ (2016) J. Coord. Chem 69, 1730–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Lee CH, Dogutan DK, Nocera DG (2011) J. Am. Chem. Soc 133, 8775–8777. [DOI] [PubMed] [Google Scholar]
- [182].Najafpour MM, Moghaddam AN, Allakhverdiev SI, Govindjee. (2012) Biochim. Biophys. Acta, Bioenerg 1817, 1110–1121. [DOI] [PubMed] [Google Scholar]
- [183].Kong D, Zheng Y, Kobielusz M, Wang Y, Bai Z, Macyk W, Wang X, Tang J (2018) Mater. Today 21, 897–924. [Google Scholar]
- [184].Berardi S, La Ganga G, Natali M, Bazzan I, Puntoriero F, Sartorel A, Scandola F, Campagna S, Bonchio M (2012) J. Am. Chem. Soc 134, 11104–11107. [DOI] [PubMed] [Google Scholar]
- [185].Maayan G, Gluz N, Christou G (2018) Nat. Catal 1, 48–54. [Google Scholar]
- [186].Kato M, Cardona T, Rutherford AW, Reisner E (2012) J. Am. Chem. Soc 134, 8332–8335. [DOI] [PubMed] [Google Scholar]
- [187].Utschig LM, Soltau SR, Mulfort KL, Niklas J, Poluektov OG (2018) Chem. Sci 9, 8504–8512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Allen MR, Frame DJ, Huntingford C, Jones CD, Lowe JA, Meinshausen M, Meinshausen N (2009) Nature 458, 1163–1166. [DOI] [PubMed] [Google Scholar]
- [189].Knutti R, Rogelj J (2015) Clim. Change 133, 361–373. [Google Scholar]
- [190].Morris AJ, Meyer GJ, Fujita E (2009) Acc. Chem. Res 42, 1983–1994. [DOI] [PubMed] [Google Scholar]
- [191].Nitopi S, Bertheussen E, Scott SB, Liu X, Engstfeld AK, Horch S, Seger B, Stephens IEL, Chan K, Hahn C, Nørskov JK, Jaramillo TF, Chorkendorff I (2019) Chem. Rev 119, 7610–7672. [DOI] [PubMed] [Google Scholar]
- [192].Yin Z, Palmore GTR, Sun S (2019) Trends Chem. 1, 739–750. [Google Scholar]
- [193].Low J, Cheng B, Yu J (2017) Appl. Surf. Sci 392, 658–686. [Google Scholar]
- [194].Aresta M, Dibenedetto A (2007) Dalton Trans. 28, 2975–2992. [DOI] [PubMed] [Google Scholar]
- [195].Ola O, Maroto-Valer MM (2015) J. Photochem. Photobiol. C, 24, 16–42. [Google Scholar]
- [196].Alkhatib II, Garlisi C, Pagliaro M, Al-Ali K, Palmisano G (2020) Catal. Today 340, 209–224. [Google Scholar]
- [197].Wang C-C, Zhang Y-Q, Li J, Wang P (2015) J. Mol. Struct 1083, 127–136. [Google Scholar]
- [198].Elouarzaki K, Kannan V, Jose V, Sabharwal HS, Lee J-M (2019) Adv. Energy Mater 9, 1900090. [Google Scholar]
- [199].Laitar DS, Müller P, Sadighi JP (2005) J. Am. Chem. Soc 127, 17196–17197. [DOI] [PubMed] [Google Scholar]
- [200].Beley M, Collin JP, Ruppert R, Sauvage JP (1986) J. Am. Chem. Soc 108, 7461–7467. [DOI] [PubMed] [Google Scholar]
- [201].Branduardi P, Sauer M (2017) FEMS Microbiol. Lett 365, 1–7. [DOI] [PubMed] [Google Scholar]
- [202].Ducat DC, Silver PA (2012) Curr. Opin. Chem. Biol 16, 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Fuchs G (1986) FEMS Microbiol. Rev 2, 181–213. [Google Scholar]
- [204].Ragsdale SW (1991) Crit. Rev. Biochem. Mol. Biol 26, 261–300. [DOI] [PubMed] [Google Scholar]
- [205].Can M, Armstrong F A, Ragsdale SW (2014) Chem. Rev 114, 4149–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Woolerton TW, Sheard S, Pierce E, Ragsdale SW, Armstrong FA (2011) Energy Environ. Sci 4, 2393–2399. [Google Scholar]
- [207].Woolerton TW, Sheard S, Reisner E, Pierce E, Ragsdale SW, Armstrong FA (2010) J. Am. Chem. Soc 132, 2132–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Chaudhary YS, Woolerton TW, Allen CS, Warner JH, Pierce E, Ragsdale SW, Armstrong FA (2012) Chem. Comm 48, 58–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Zhang L, Can M, Ragsdale SW, Armstrong FA (2018) ACS Catal. 8, 2789–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Song J, Klein EL, Neese F, Ye S (2014) Inorg. Chem 53, 7500–7507. [DOI] [PubMed] [Google Scholar]
- [211].Froehlich JD, Kubiak CP (2012) Inorg. Chem 51, 3932–3934. [DOI] [PubMed] [Google Scholar]
- [212].Schneider CR, Shafaat HS (2016) Chem. Comm 52, 9889–9892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Behnke SL, Manesis AC, Shafaat HS (2018) Dalton Trans. 47, 15206–15216. [DOI] [PubMed] [Google Scholar]
- [214].Schneider CR, Manesis AC, Stevenson MJ, Shafaat HS (2018) Chem. Comm 54, 4681–4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Arana C, Yan S, Keshavarz KM, Potts KT, Abruna HD (1992) Inorg. Chem 31, 3680–3682. [Google Scholar]
- [216].Elgrishi N, Chambers MB, Wang X, Fontecave M (2017) Chem. Soc. Rev 46, 761–796. [DOI] [PubMed] [Google Scholar]
- [217].Kuehnel MF, Orchard KL, Dalle KE, Reisner E (2017) J. Am. Chem. Soc 139, 7217–7223. [DOI] [PubMed] [Google Scholar]
- [218].Liu X, Kang F, Hu C, Wang L, Xu Z, Zheng D, Gong W, Lu Y, Ma Y, Wang J (2018) Nat. Chem 10, 1201–1206. [DOI] [PubMed] [Google Scholar]
- [219].Sultana S, Chandra Sahoo P, Martha S, Parida K (2016) RSC Adv. 6, 44170–44194. [Google Scholar]
- [220].Yadav RK, Oh GH, Park N-J, Kumar A, Kong K.-j., Baeg J-O (2014). J. Am. Chem. Soc 136, 16728–16731. [DOI] [PubMed] [Google Scholar]
- [221].Panda R, Berlinguette CP, Zhang Y, Holm RH (2005) J. Am. Chem. Soc 127, 11092–11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Sun J, Tessier C, Holm RH (2007) Inorg. Chem 46, 2691–2699. [DOI] [PubMed] [Google Scholar]
- [223].Brown KA, Harris DF, Wilker MB, Rasmussen A, Khadka N, Hamby H, Keable S, Dukovic G, Peters JW, Seefeldt LC, King PW (2016) Science 352, 448–450. [DOI] [PubMed] [Google Scholar]
- [224].Roth LE, Nguyen JC, Tezcan FA (2010) J. Am. Chem. Soc 132, 13672–13674. [DOI] [PubMed] [Google Scholar]
- [225].Roth LE, Tezcan FA (2012) J. Am. Chem. Soc 134, 8416–8419. [DOI] [PubMed] [Google Scholar]
- [226].Li H, Shang J, Ai Z, Zhang L (2015) J. Am. Chem. Soc 137, 6393–6399. [DOI] [PubMed] [Google Scholar]
- [227].Ali M, Zhou F, Chen K, Kotzur C, Xiao C, Bourgeois L, Zhang X, MacFarlane DR (2016) Nat. Commun 7, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Yuan S-J, Chen J-J, Lin Z-Q, Li W-W, Sheng G-P, Yu H-Q (2013) Nat. Commun 4, 2249. [DOI] [PubMed] [Google Scholar]
- [229].Medford AJ, Hatzell MC (2017) ACS Catal. 7, 2624–2643. [Google Scholar]
- [230].Guo C, Ran J, Vasileff A, Qiao S-Z (2018) Energy Environ. Sci 11, 45–56. [Google Scholar]
- [231].Banerjee A, Yuhas BD, Margulies EA, Zhang Y, Shim Y, Wasielewski MR, Kanatzidis MG (2015) J. Am. Chem. Soc 137, 2030–2034. [DOI] [PubMed] [Google Scholar]
- [232].Liu J, Kelley MS, Wu W, Banerjee A, Douvalis AP, Wu J, Zhang Y, Schatz GC, Kanatzidis MG (2016) PNAS 113, 5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Ogata H, Hirota S, Nakahara A, Komori H, Shibata N, Kato T, Kano K, Higuchi Y (2005) Structure 13, 1635–1642. [DOI] [PubMed] [Google Scholar]
- [234].Nicolet Y, Piras C, Legrand P, Hatchikian CE, Fontecilla-Camps JC (1999) Structure 7, 13–23. [DOI] [PubMed] [Google Scholar]