

ABSTRACT
Viral hijacking of microtubule (MT)-dependent transport is well understood, but several viruses also express discrete MT-associated proteins (vMAPs), potentially to modulate MT-dependent processes in the host cell. Specific roles for vMAP-MT interactions include subversion of antiviral responses by P3, an isoform of the P protein of rabies virus (RABV; genus Lyssavirus), which mediates MT-dependent antagonism of interferon (IFN)-dependent signal transducers and activators of transcription 1 (STAT1) signaling. P3 also undergoes nucleocytoplasmic trafficking and inhibits STAT1-DNA binding, indicative of intranuclear roles in a multipronged antagonistic strategy. MT association/STAT1 antagonist functions of P3 correlate with pathogenesis, indicating potential as therapeutic targets. However, key questions remain, including whether other P protein isoforms interact with MTs, the relationship of these interactions with pathogenesis, and the extent of conservation of P3-MT interactions between diverse pathogenic lyssaviruses. Using super-resolution microscopy, live-cell imaging, and immune signaling analyses, we find that multiple P protein isoforms associate with MTs and that association correlates with pathogenesis. Furthermore, P3 proteins from different lyssaviruses exhibit variation in intracellular localization phenotypes that are associated with STAT1 antagonist function, whereby P3-MT association is conserved among lyssaviruses of phylogroup I but not phylogroup II, while nucleocytoplasmic localization varies between P3 proteins of the same phylogroup within both phylogroup I and II. Nevertheless, the divergent P3 proteins retain significant IFN antagonist function, indicative of adaptation to favor different inhibitory mechanisms, with MT interaction important to phylogroup I viruses.
IMPORTANCE Lyssaviruses, including rabies virus, cause rabies, a progressive encephalomyelitis that is almost invariably fatal. There are no effective antivirals for symptomatic infection, and effective application of current vaccines is limited in areas of endemicity, such that rabies causes ~59,000 deaths per year. Viral subversion of host cell functions, including antiviral immunity, is critical to disease, and isoforms of the lyssavirus P protein are central to the virus-host interface underpinning immune evasion. Here, we show that specific cellular interactions of P protein isoforms involved in immune evasion vary significantly between different lyssaviruses, indicative of distinct strategies to evade immune responses. These findings highlight the diversity of the virus-host interface, an important consideration in the development of pan-lyssavirus therapeutic approaches.
KEYWORDS: immune evasion, lyssavirus, microtubules, nuclear localisation, rabies virus, super-resolution microscopy
INTRODUCTION
The microtubule (MT) cytoskeleton comprises an extensive cytoplasmic filamentous network with classical functions in the transport and positioning of cellular contents and is a major component of the mitotic spindle (1, 2). In addition, MTs have regulatory roles in signal transduction, apoptosis, and innate immunity (3, 4). To carry out these roles, MTs act in concert with MT-associated proteins (MAPs), many of which regulate dynamic structural changes integral to MT function and/or act as molecular motors for cargo transport (5, 6). Viruses commonly exploit MTs/MAPs as part of their life cycle, including using motor proteins for subcellular virion/genome trafficking; several viruses also use MTs as replication platforms (7, 8). Notably, a growing number of viruses are reported to encode viral MAPs (vMAPS) that can form discrete interactions with MTs, either to facilitate viral transport/replication processes or to subvert MT-dependent functions (7, 8).
Rabies virus (RABV) is the prototypical member of the genus Lyssavirus (family Rhabdoviridae), which comprises at least 17 species, 14 of which are classified within two defined phylogroups based on antigenic properties and phylogenetic relationships (9). The remaining species are not classified in phylogroup I or II, so the genus appears to include three or more phylogroups (10). Lyssaviruses are maintained in nonhuman mammalian reservoirs, including bats and terrestrial carnivores. Zoonotic infections, including by multiple members of phylogroup I (e.g., RABV and European bat lyssavirus 2 [EBLV2]) and phylogroup II (e.g., Mokola virus [MOKV]), cause rabies, a lethal acute meningoencephalitis (10–12). RABV is considered the primary etiological agent of human rabies, which is estimated to cause ~59,000 deaths/year (13). Underreporting, misdiagnosis, and lack of discriminatory tests suggest that estimates of fatalities due to rabies are conservative and probably include a greater proportion of infections by non-RABV lyssavirus than is currently assumed (10–12).
Following detection of infection, host cells express type I interferons (IFNs; e.g., IFN-α/β) as part of the principal antiviral innate immune response (14). IFNs signal in autocrine and paracrine fashions to activate intracellular signal transducers and activators of transcription 1 (STAT1) and STAT2 by tyrosine phosphorylation, resulting in their translocation to the nucleus and activation of antiviral and immunomodulatory IFN-stimulated genes (ISGs) (14). Viruses counteract this response by expressing proteins with IFN antagonist functions, using mechanisms that can diverge significantly between different antagonist proteins and viruses (15, 16). The principal IFN antagonist of lyssaviruses is the P protein, which is expressed as the full-length P1 (an essential cofactor of the viral polymerase L protein) and several N-terminally truncated isoforms generated via ribosomal leaky scanning of the P gene mRNA (17) (Fig. 1A). Four isoforms (P2 to P5) have been detected for RABV, all of which lack N-terminal sequence necessary for L protein binding (17, 18). However, all isoforms contain a STAT1-binding site in the C-terminal domain (CTD), which enables antagonism of IFN/STAT1 signaling (19, 20). Studies to date have focused on P1 and P3 and indicate that antagonism involves several mechanisms deriving from distinct phenotypes of the isoforms (21–25). P1 is primarily cytoplasmic due to a strong N-terminal nuclear export sequence (N-NES), and so it effects nuclear exclusion/cytoplasmic accumulation of associated STAT1 to prevent ISG activation (24–26). Conservation of the STAT1-binding region and N-NES, and equivalent localization of P2 (27), suggest that P2 effects a similar mechanism but is “specialized” for antagonism due to a lack of L binding. Although this has not been directly assessed, it is consistent with studies using viruses with modified P1/P2 expression (28).
FIG 1.

RABV P3 to P5, but not P1 and P2, associate with MTs. (A) Schematic representation of RABV P1 protein; four N-terminally truncated isoforms (P2 to P5) are generated in infected cells by translation from internal in-frame AUG codons (17). Start sites are indicated by arrows, with residue numbers corresponding to position in P1. The CTD (containing the MTAS, C-NLS, C-NES, and STAT1-binding regions) and NTR (containing the S-AD, N-NLS, N-NES, and L-binding region) are indicated. (B) Residues at positions 56, 58, 66, 81, and 226 differ between P proteins from the Ni and Ni-CE strains of RABV (substitutions in Ni-CE P3 are shown in red). (C) COS-7 cells were transfected to express the indicated proteins and were treated with (+Tax) or without (no add.) paclitaxel (1 μg/mL, 4 h) before analysis by live-cell CLSM; each image is representative of the MT phenotype in cells observed in 15 fields of view sampled over two independent assays. Scale bar = 5 μm.
The N-terminal truncation generating P3 produces a profoundly different phenotype, including acquisition of MT interaction via an MT association sequence (MTAS) located within the CTD (Fig. 1A) (22). This is implicated in an MT-dependent mechanism of IFN antagonism (currently unique to RABV), whereby P3 can induce MT association of STAT1 (21, 22). IFN-induced STAT1 nuclear accumulation and ISG activation is consequently suppressed by P3, dependent on MT integrity (22). Consistent with this, inhibition of IFN/STAT1 signaling in RABV-infected cells is MT dependent (22). P3 additionally localizes to the nucleus, with molecular analyses showing that this results from truncation/deactivation of the P1/P2 N-NES as well as activation of an N-terminal nuclear localization signal (N-NLS) (27, 29). Since STAT1 binding by P proteins can interfere with STAT1-DNA interaction, this localization is thought to enable an intranuclear blockade of IFN signaling (20, 23).
Understanding of P protein function in infection and pathogenesis has been advanced by a reverse genetics system using a pathogenic RABV strain, Nishigahara (Ni), and an Ni-derivative strain, Ni-CE, that is attenuated through mechanisms such as increased IFN sensitivity (30, 31). Using this system, the P gene was shown to be a key determinant of pathogenesis due to defective STAT1 antagonism by Ni-CE P, such that substitution of the Ni P gene into Ni-CE virus is sufficient to restore resistance to IFN and enhance pathogenesis (30). The Ni-CE P proteins differ from Ni P proteins by only five amino acid substitutions (Fig. 1B), and these do not appear to impair STAT1 binding, indicating that they affect mechanisms by which P isoforms efficiently antagonize associated STAT1 (32). Analysis of Ni-CE P1 revealed that substitutions in the N-NES impair nuclear exclusion of P1 and consequently of P1-STAT1 complexes, indicating the significance of this mechanism in pathogenesis (32). MT association and IFN antagonism by Ni-CE P3 is also defective, with the single substitution in the CTD, N226-H, sufficient to impair MT association, IFN/STAT1 antagonism, and viral pathogenesis (21). Recently, nuclear localization of Ni-CE P3 was also shown to be defective due to the combined effect of the substitutions N226-H and F81-P that impact the activity of distinct NLS/NES sequences (33). Notably, phosphomimetic mutation of a protein kinase C site (residue S210) within the CTD of P3 impacts interactions of the side chain of N226 and inhibits nuclear localization and MT association, suggesting that N226 has key regulatory roles in P3 trafficking, with N226-H substitution mimicking the phosphorylated state (33–35). Thus, P1 and P3 appear to affect a multipronged strategy to shut down IFN signaling, analogous to P gene products of several paramyxoviruses (36, 37). However, the vMAP activity of other P protein isoforms, and their relationship with pathogenesis, remain unresolved. Furthermore, the conservation/divergence of MT association between RABV and other lyssaviruses are unknown.
Here, we show that, in addition to P3, the P4 and P5 isoforms of pathogenic (Ni), but not attenuated (Ni-CE), RABV associate strongly with MTs. Nuclear localization of P4 and P5 of Ni-CE is also defective, consistent with roles of both MT association and nuclear localization of multiple isoforms in immune evasion/pathogenesis. Notably, in contrast to previous data indicating a conserved subcellular localization of the P1 isoform between lyssaviruses (38), our data for P3 indicate significant diversity. Specifically, MT association of P3 is conserved between several viruses of phylogroup I, but nuclear localization differs significantly, and MT association and nuclear localization of P3 proteins of phylogroup II clearly diverged from one another and from P3 proteins of phylogroup I. Nevertheless, STAT1 antagonist function was retained, indicating that lyssaviruses use phenotypically diverse P3 proteins to effect immune evasion via differing antagonistic strategies. These data propose that the overall IFN antagonistic strategy differs between lyssaviruses due to differing phenotypes of the short P protein isoforms P3 to P5.
RESULTS AND DISCUSSION
P3, P4, and P5 form MT interactions that correlate with pathogenesis.
Previous analysis of P protein isoforms from the challenge virus standard (CVS) strain of RABV indicated that MT association of transfected P3, but not P1, is clearly detectable by conventional fluorescence microscopy, suggesting that P1 does not interact with MTs due to a suppressive activity of the extended N-terminal region (residues 1 to 52), which is removed from P3 (Fig. 1A) (22). To assess MT interaction/bundling by other isoforms, and the relationship with viral IFN antagonism/pathogenesis, we compared the localization of green fluorescent protein (GFP)-fused P1, P2, P3, P4, and P5 of Ni and Ni-CE RABV in living COS-7 (Fig. 1B and C) and HeLa (Fig. S1 in the supplemental material) cells following treatment with or without the MT-stabilizing drug paclitaxel (Tax). GFP-fused P proteins have been previously confirmed to be functional and to have phenotypes consistent with nonfused proteins (21, 39). Analysis by confocal laser scanning microscopy (CLSM) indicated diffuse cytoplasmic localization with no evident filamentous organization of Ni P1 (consistent with previous data for P1 of CVS RABV [22]) (Fig. 1C and Fig. S1). In contrast, Ni P3 interacted with filaments, the appearance of which was enhanced by Tax (as described previously [21]) (Fig. 1C and Fig. S1). The Ni P3-associated filaments were confirmed to be MTs by immunostaining of cells for β-tubulin, which indicated colocalization of filamentous P3 with MTs (Fig. S2). The P3-MT filaments were also enhanced by Tax and dispersed by nocodazole (NCZ; which causes disassembly of MT networks).
To further assess the difference in MT association by P1 and P3, we used direct stochastic optical reconstruction microscopy (dSTORM), which enables resolution and measurement of single proximally localized MTs that occupy the same diffraction-limited space and so would otherwise appear as a single MT feature by CLSM. We previously applied this technique to show that P3 induces MT bundling, which can thus be used as an indirect metric to sensitively quantify P3-MT interaction and effects thereon of substitutions between Ni and Ni-CE P3 (21, 40). The imaging resolution afforded with dSTORM provides visualization of individual MT filaments (40 to 100 nm after immunolabelling [21]) and sensitivity to detect subtle changes in MT structure, which can be analyzed by measuring MT feature diameters (MTfds). Using dSTORM, we first confirmed the effects of the MT-targeting drugs Tax and NCZ on assembly and disassembly of MTs, respectively (Fig. S3). We then analyzed the effects of P3 on MTs, confirming that expression of Ni P3 induces the formation of distinct MT bundles, some wider than 200 nm (Fig. 2A and B), with a clear increase in the median MTfd (Fig. 2C). Expression of Ni P1, while showing minimal visible changes compared to the GFP control (Fig. 2A and B), induced a small but significant level of MT bundling (Fig. 2C) and, therefore, appears to retain a low level of MT interaction that is not detectable using CLSM. Comparable expression of Ni P1 and P3 was confirmed by Western blotting (Fig. S6A). This is consistent with the fact that P1 contains the entire sequence of P3 and supports the idea that P1 has intrinsic vMAP activity, but that this is inhibited by residues 1 to 52. CLSM analysis of P2 indicated a diffuse cytoplasmic localization comparable to P1 (Fig. 1C). Thus, it appears that P2 lacks substantial capacity for MT association, such that residues 20 to 52 are sufficient to suppress the intrinsic vMAP activity.
FIG 2.

MT interaction/bundling by RABV P1 is inhibited compared with RABV P3. (A) dSTORM images of immunostained β-tubulin in COS-7 cells expressing the indicated proteins; MTfds for the indicated filaments are shown below. (B and C) Frequency distribution (B) and Tukey box plots depicting median, interquartile range, and range (C) of MTfds calculated for each protein (n = 531 [GFP-Ni-P1], 496 [GFP-Ni-P3], and 535 [GFP]; measurements are from 10 cells for each protein from two independent assays). Scale bars = 1 μm.
Phenotypes of Ni P4 and Ni P5 were similar to Ni P3, with clear cytoplasmic filaments that colocalized with MTs and were enhanced by Tax and dispersed by NCZ (Fig. 1C and Fig. S1 and S2), indicating that these shorter isoforms contribute to MT targeting by RABV. Previous analysis of P3 and truncated proteins indicated that the minimal MTAS is in the CTD, but MT association is also dependent on multimerization, which is mediated by the self-association domain (S-AD) in the N-terminal region (NTR) (22). Our finding that P5 retains association with MTs supports roles for the S-AD and MTAS as these regions are maintained in P5 (Fig. 1A). Notably, Ni-CE P4 and Ni-CE P5 showed little to no cytoplasmic filament association in the absence of Tax, and only minimal filamentous organization was observed following Tax treatment (Fig. 1C and Fig. S1). This is consistent with strongly reduced MT interaction comparable to that observed for Ni-CE P3, both here and previously (21). Thus, P4 and P5 are additional RABV vMAPs, with this MT interaction correlating with IFN antagonist function and pathogenesis of virus, similar to P3. Notably, Ni-CE P5 differs from Ni P5 by only a single substitution, N226-H (Fig. 1B), consistent with our previous findings using P3 that this is a key determinant of MT association and STAT antagonist function (21).
The CLSM images also indicated that, as previously reported (41), Ni-CE P1 is more nuclear than Ni P1. A similar effect was observed for P2, consistent with substitutions in the N-NES, suggesting that defective nuclear export of P2 also contributes to reduced IFN resistance of Ni-CE virus (41). In addition to defective vMAP function, the nuclear localization of Ni-CE P3 is defective compared with that of Ni P3 through a concerted effect of substitutions F81-P (present in P4 but absent from P5) and N226-H (present in the CTD of all isoforms) (Fig. 1A and B) (33). Consistent with the presence of N226-H, nuclear localization of Ni-CE P4 and P5 was also inhibited compared with the respective Ni proteins. Previous studies using P3 protein suggest that N226-H inhibits MT association and nuclear localization by mimicking the effects of phosphorylation of P3 at residue S210. Our findings extend these observations to shorter isoforms P4 and P3. Together, these data indicate that defects in both MT association of the truncated P3 to P5 isoforms and in the specific nucleocytoplasmic trafficking phenotype of all of the P protein isoforms are associated with altered pathogenesis of Ni and Ni-CE viruses. This is consistent with previous data implicating nuclear localization and MT association of P1 and P3 in STAT1 antagonism (21–25).
P3-MT targeting is conserved between phylogroup I, but not phylogroup II, lyssaviruses.
The steady-state localization and IFN antagonist function of P1 are largely conserved across the Lyssavirus genus due to the conserved N-NES (38), but phenotypes of other isoforms are poorly defined. CLSM analysis of COS-7 and HeLa cells expressing P3 proteins of field strains of phylogroup I viruses, silver-haired bat RABV (SHBRV) and European bat lyssavirus 2 (EBLV2), and of fixed strains of RABV, Ni, and CVS, treated without or with Tax, indicated conservation of MT association (Fig. 3A and Fig. S4). CVS and Ni P3-associated filaments were shown previously (21, 22) to correspond to MTs, and we confirmed that filaments formed by SHBRV and EBLV2 P3 proteins colocalize with MTs that are enhanced in Tax-treated cells and lost in NCZ-treated cells (Fig. S5). Although MT filament interaction was clearly detectable for CVS P3 (which was used originally to identify P3-MT association and MT-dependent IFN antagonist function [22]), it typically appeared less pronounced than that of P3 proteins of other phylogroup I viruses (Fig. 3A), perhaps relating to specific conditions in laboratory adaptation of CVS. Nevertheless, MT association was more evident for CVS P3 than for Ni-CE P3 (Fig. 1C). Intriguingly, no cytoplasmic filament association was detectable for P3 of phylogroup II viruses Lagos bat virus (LBV-P3) or Mokola virus (MOKV-P3), including in cells treated with Tax (Fig. 3A and Fig. S4). Comparable expression of the P3 proteins was observed in Western blotting analysis of cell lysates (Fig. S6).
FIG 3.

MT association and nuclear localization varies between P3 proteins of different lyssaviruses. (A) COS-7 cells were transfected to express the indicated proteins and treated with (+Tax) or without (no add.) paclitaxel (1 μg/mL, 4 h) before analysis by live-cell CLSM. Images are representative of cells observed in 13 fields of view sampled over two independent assays. Scale bars = 5 μm. (B) dSTORM images of immunostained β-tubulin in COS-7 cells expressing the indicated proteins. MTfds for filaments indicated by arrows are shown below. Scale bars = 1 μm. (C and D) Frequency distribution (C) and Tukey box plots depicting median, interquartile range, and range (D) of MTfds calculated for each protein (n = 512 [Ni], 562 [CVS], 855 [SHBRV], 617 [EBLV2], 556 [LBV], and 667 [MOKV]; measurements are from ≥7 cells for each protein from two independent assays). (E) Images such as those shown in A were analyzed to derive the ratio of nuclear-to-cytoplasmic fluorescence (Fn/c) for GFP-P3 (mean ± standard error of the mean [SEM]; n ≥ 54 cells from two independent assays).
Quantitative dSTORM analysis confirmed the results from CLSM, revealing substantial MT bundling in COS-7 cells expressing phylogroup I P3 proteins, the extent of which correlated with interactions indicated by CLSM (Fig. 3B to D). In contrast, no substantial bundling was observed in LBV P3- or MOKV P3-expressing cells, where the majority of MT features were equivalent to single MTs (30 to 90 nm) (Fig. 3C), and the median MTfds were significantly reduced compared with those for phylogroup I proteins (Fig. 3D). These data suggest that P3-MT association has conserved roles in immune evasion within phylogroup I, while the more distantly related phylogroup II viruses appear to differ in strategies associated with IFN antagonism.
vMAPs from diverse viral families are implicated in the facilitation of virion/genome trafficking or the exploitation of MTs as platforms for virus replication (7, 8). As MTs are important to the transport of RABV and maturation of RABV replication factories (42, 43), P isoform-MT interaction could conceivably have roles in facilitating or regulating these processes and so have evolved additional MT-dependent accessory functions in immune evasion. However, as replication of Ni-CE virus is not impaired compared with replication of Ni in the absence of IFN (30, 31), roles for P isoform-MT interaction in basic replication appear unlikely, with current data indicating that immune evasion is a principal function for MT interaction of P3 (21, 22) and presumably other truncated forms (this study). Our new data showing that P3-MT interaction is not conserved in phylogroup II viruses are consistent with an accessory function rather than an integral role in virus replication processes.
Nuclear localization of P3 varies significantly between lyssaviruses.
Despite the conservation of MT association between P3 of phylogroup I lyssaviruses, CLSM analysis of nuclear localization (Fig. 3A and Fig. S4), including quantitative analysis of images to calculate the ratio of nuclear-to-cytoplasmic fluorescence (Fn/c; Fig. 3E) (44, 45), indicated significant divergence between viruses. Specifically, SHBRV P3 was strongly cytoplasmic, while the P3 proteins of CVS, Ni, and EBLV2 were able to localize in the nucleus. This suggests that MT interaction is particularly important to immune evasion strategies of SHBRV P3. Phylogroup II virus P3 proteins interrogated in this work were more divergent, with LBV P3 localizing largely in the cytoplasm, while MOKV P3 (previously reported to be able to localize to the nucleus [29]) differed from the other P3 proteins in being almost exclusively nuclear (Fig. 3A and E and Fig. S4). Notably, although lyssavirus P1 proteins show comparable nuclear exclusion at steady state, inhibition of exportin 1, which mediates nuclear export via the N-NES, results in much greater nuclear localization by MOKV P1 (38), indicating that the nucleocytoplasmic shuttling of P proteins of MOKV involves greater intrinsic nuclear import activity than that of proteins of other lyssaviruses tested.
The distinct subcellular localization phenotype of MOKV P3 derives from a coordinated effect of different regulatory regions.
Subcellular localization of RABV P protein has been characterized in detail, largely by using CVS P protein/isoforms, and involves multiple sequences/domains that regulate trafficking and so contribute to different STAT-antagonistic processes (22–25, 27, 29, 34, 44). The principal sequences regulating nuclear localization include the N-NES and N-NLS; additional regulation involves a CTD-localized NLS and NES (C-NLS and C-NES, which are present in all isoforms), while the MTAS mediates MT interaction/tethering that suppresses nuclear accumulation (Fig. 1A) (22, 27, 29, 34, 44). Furthermore, phosphorylation at S210 is indicated to regulate nuclear localization and MT association (33, 35). Consistent with the presence of multiple trafficking sequences, defective nuclear localization of Ni-CE P3 involves two mutations that impact the functions of distinct NLS/NES sequences to inhibit nuclear accumulation (33). Due to the intricacy of P3 intracellular trafficking, the differing phenotypes of lyssavirus P3 proteins could result from altered activity of several sequences (Fig. S7).
To examine this, we generated chimeras of the phenotypically divergent Ni P3 and MOKV P3, in which the NTR and CTD regions were substituted (Fig. 4A). CLSM analysis indicated that the NTR of MOKV P3 is sufficient to produce a highly nuclear phenotype, as the nuclear localization of MOKVNTR-NiCTD P3 was similar to that of wild-type MOKV P3 (Fig. 3A and 4B); this chimera also did not associate with MTs. Since the N-terminal region of MOKV is enriched in basic residues, which are commonly found in NLSs (46), the nuclear phenotype of MOKV P3 is likely to involve enhanced activity of the N-NLS. In contrast, combination of the Ni NTR, which is clearly sufficient to support MT association by the C-terminal MTAS of Ni P3, with the MOKV CTD (NiNTR-MOKVCTD P3) resulted in almost exclusively cytoplasmic localization with no evident MT association, except for limited filamentous organization in some cells following Tax treatment. Thus, it appears that MOKV P3 lacks significant MTAS activity and has a stronger N-NLS than Ni P3, suggesting that it may be specifically adapted to produce strong nuclear localization through a coordinated effect on several sequences/domains. These data have potential implications for understanding of lyssavirus evolution and, together with results for Ni and Ni-CE P isoforms, highlight key regions of P3 related to pathogenic processes.
FIG 4.
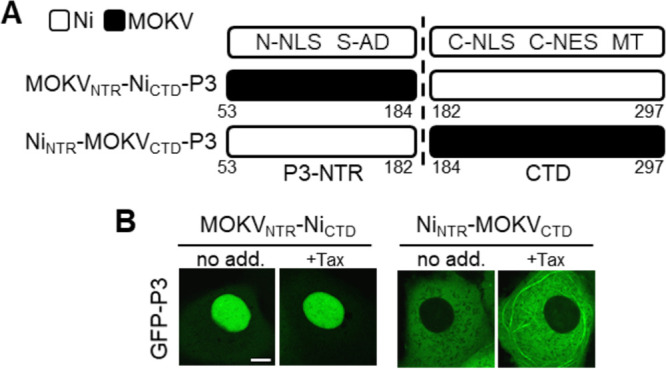
The unique subcellular localization phenotype of MOKV P3 involves the NTR and CTD. (A) Schematic representation of Ni-MOKV P3 chimeras generated. (B) Live-cell CLSM images of COS-7 cells transfected to express the indicated proteins and treated with (+Tax) or without (no add.) paclitaxel before analysis by live-cell CLSM. Images are representative of cells observed in 13 fields of view sampled over two independent assays. Scale bars = 5 μm.
Phenotypically diverse P3 proteins retain STAT1 antagonist function.
Nuclear localization and MT association of RABV P3 has been implicated in IFN/STAT1 antagonism. To examine whether P3 proteins with significant divergence in localization (cytoplasmic/filamentous/nuclear [Ni P3], cytoplasmic/filamentous/non-nuclear [SHBRV], and non-filamentous/strongly nuclear [MOKV]) retain antagonist function, we initially assessed the capacity of the proteins to target STAT1. IFN induces phosphorylation of STAT1 at residue Y701 (pY-STAT1), promoting nuclear import and ISG activation before dephosphorylation, which becomes evident from ~0.5 h after IFN treatment and ultimately deactivates STAT1 (47–49). P1 proteins use an unusual mechanism of IFN antagonism that is conserved among proteins of different lyssaviruses, whereby P1 binds efficiently to pY-STAT1 and inhibits its dephosphorylation, thus retaining inactive pY-STAT1-P1 complexes in the cytoplasm (24–26, 38). As a result, P1 expression or RABV infection results in accumulation of pY-STAT1 in IFN-treated cells (24–26, 38). Dephosphorylation of pY-STAT1 follows nuclear entry and DNA binding (to activate transcription) followed by release from the DNA (16). Thus, accumulation of pY-STAT1 is consistent with inhibition of pY-STAT1 signaling, resulting in defective recycling. Immunoblotting of lysates of cells expressing GFP-RABV N protein (GFP-N; a negative control that does not affect STAT1 [38]) following treatment with IFN-α for 0.5 or 4 h confirmed rapid induction of phosphorylation (0.5 h) followed by dephosphorylation consistent with functional signaling and recycling (Fig. 5). pY-STAT1 was also rapidly induced in cells expressing lyssavirus P3 proteins, consistent with previous reports that P1 protein expression or RABV infection does not inhibit STAT1 activation (24, 25, 38). All lyssavirus P3 proteins tested also inhibited STAT1 dephosphorylation, as indicated by similar levels of pY-STAT1 at 0.5 and 4 h posttreatment, demonstrating that isoforms other than P1 suppress STAT1 dephosphorylation/recycling. This is consistent with the localization of the STAT1 binding site in the CTD, which is common to all isoforms, and indicates an equivalent mode of binding by different isoforms. Thus, despite divergent localization of the P3 proteins tested (Fig. 3A), the data indicate that pY-STAT1 targeting is conserved.
FIG 5.
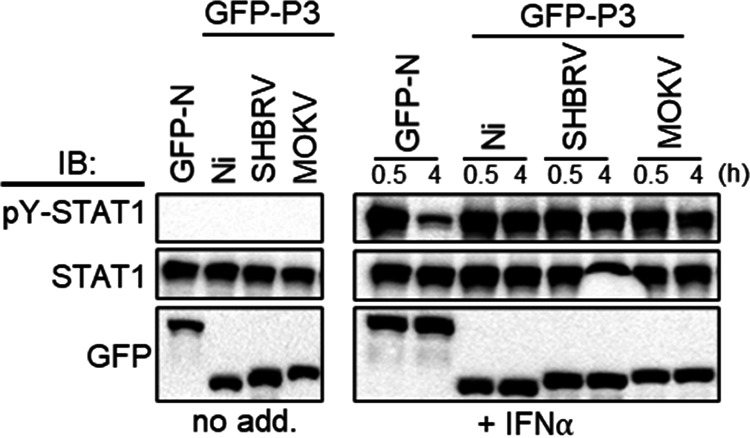
Lyssavirus P3 proteins inhibit pY-STAT1 dephosphorylation. COS-7 cells expressing the indicated proteins were treated with or without IFN-α (1,000 U/mL) for the indicated times before lysis and immunoblotting (IB) using the indicated antibodies. Results are representative of two independent assays.
Previous analysis of RABV P1 and P3 indicate that interactions with STAT1 in the cytoplasm, the nucleus, and at MTs enable efficient inhibition of signaling via different mechanisms involving MT association, inhibition of DNA interaction, and active nuclear export/cytoplasmic retention (41). Since the different lyssavirus P3 proteins similarly affected STAT1 dephosphorylation, this is indicative of conserved STAT1 targeting and antagonism. CLSM analysis of cells expressing the P3 proteins and immmunostained for STAT1 indicated no substantial difference in nuclear localization of STAT1 between non-IFN-α-treated cells expressing Ni P3, SHBRV P3, or the control protein GFP-N (Fig. 6A and B; note that the fixation conditions required for STAT1 immunostaining do not maintain MT filamentous structures). This is consistent with data for P1 isoforms showing that efficient interaction of P1 with STAT1 is dependent on STAT1 activation/phosphorylation (25, 38, 41). Intriguingly, in cells expressing MOKV P3, a degree of nuclear accumulation of STAT1 was observed in the absence of IFN-α; this observation is unique among available data for lyssavirus P proteins and probably relates to the strong nuclear localization of MOKV P3. Following IFN-α treatment, STAT1 accumulated into nuclei of cells expressing GFP-N or cells lacking expression of transfected proteins, but this was clearly inhibited in cells expressing each of the P3 proteins examined (Fig. 6C and D). Furthermore, analysis using an IFN/STAT1-dependent reporter gene assay indicated that all P3 proteins tested inhibit IFN-activated transcriptional activity (Fig. 7); thus, despite differing localization phenotypes, lyssavirus P3 proteins retain the capacity to target and modulate STAT1 responses.
FIG 6.

Lyssavirus P3 proteins inhibit IFN-α-dependent nuclear accumulation of STAT1. (A and C) COS-7 cells expressing the indicated proteins were treated with (+ IFN-α) or without (no add.) IFN-α (1,000 U/mL, 0.5h) before fixation and immunostaining for STAT1 and analysis by CLSM. (B and D) Images such as those shown in A and C were analyzed to determine the Fn/c for STAT1 (mean ± SEM; n ≥ 88 cells from four independent assays). Scale bars = 5 μm.
FIG 7.
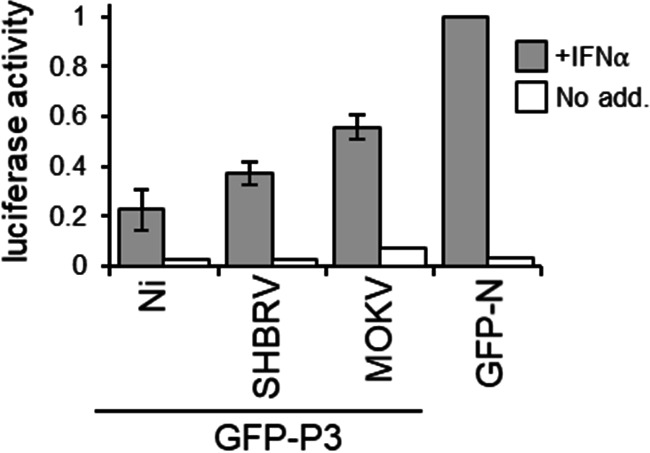
IFN-α-dependent STAT1 signaling is antagonized by lyssavirus P3 proteins. IFN-α-dependent signaling in NA cells expressing the indicated proteins was analyzed using a dual luciferase reporter gene assay. Luciferase activity is expressed as a fold change relative to that obtained for IFN-α-treated cells expressing GFP-N protein (mean relative luciferase activity ± standard deviation [SD]; n = 2 from 2 separate assays for +IFN-α samples).
Taken together, these data indicate that MT association and nucleocytoplasmic localization of P3 can vary significantly between lyssaviruses and that such differences result from coordinated effects on different trafficking sequences. Despite this divergence, the retention of STAT1 antagonist function by all P3 proteins is consistent with important roles for truncated isoforms in immune evasion, although the relative contribution of MT-dependent and other STAT1-antagonistic mechanisms likely differs between lyssaviruses such that they use distinct strategies for P protein-dependent immune evasion. This may relate to adaptation to different reservoir hosts and/or divergence in other nuclear interactions/functions, such as interactions of P3 with nucleoli or nuclear bodies (50, 51). It is also likely that alternative mechanisms involving different viral proteins might contribute to immune evasion by different lyssaviruses, resulting in altered phenotypes; notably, M and N proteins participate in IFN-antagonistic mechanisms distinct from those of P protein, and the L protein can also associate with MTs (41, 52). Thus, that the overall immune evasion “armory” of different lyssaviruses might vary significantly, with the balance of different components resulting in an altered phenotype of P protein isoforms. Nevertheless, the finding that MT association and nuclear localization of all P protein isoforms differ between Ni and Ni-CE viruses, correlating with altered IFN antagonist function and pathogenesis, supports prior studies indicating the importance of both processes to RABV immune evasion (20–23, 32, 33).
The divergence of the P3 phenotype contrasts with the conserved steady-state cytoplasmic phenotype of P1 across the genus, although some quantitative differences in P1 nuclear shuttling are observed following inhibition of nuclear export, including between different species of lyssaviruses and lineages/strains associated with different hosts, suggestive of some limited adaptation (38, 39). Greater adaptability of P3 probably relates to essential roles for P1 in genome transcription/replication through interactions with cytoplasmic N and L proteins (53), while P3 and shorter isoforms lack cofactor function and are thought to be liberated for accessory functions, including immune evasion (18, 28, 54). Similarly, the apparent capacity of the P gene of lyssaviruses and some other viruses of the order Mononegavirales to encode several phenotypically distinct isoforms, and permissiveness to variability in phenotype between strains/species (37), is likely to relate to less restricted requirements for structure/sequence of the non-catalytic P protein compared with enzymatic and structural proteins. Nevertheless, the conservation of MT association between P3 proteins of RABV strains/phylogroup I lyssaviruses and correlation with pathogenesis indicate potential for the P protein-MT interface in the development of vaccines or antivirals against the viruses that account for the majority of human cases (55). The apparent divergence of this phenotype in other lyssaviruses, however, highlights the diversity of the virus-host interface, an important consideration in the potential development of pan-lyssavirus approaches (56).
MATERIALS AND METHODS
Constructs.
Constructs were generated by PCR amplification of inserts from lyssavirus P or N gene cDNA for cloning into the mammalian expression vector pEGFP-C1 (to express GFP-fused protein) as previously described (21, 22, 38). Chimeric P3 proteins were generated via overlap PCR mutagenesis.
Cell culture, transfection, and drug treatments.
COS-7 and HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM), and mouse neuroblastoma NA cells were cultured in RPMI supplemented with 10% fetal calf serum (FCS; 37°C and 5% CO2). Cells were grown to ~80% confluence in 6-well culture dishes (for luciferase assays), on coverslips in 6-well culture dishes (for CLSM of living cells) or 12-well culture dishes (for CLSM of fixed, immunostained cells), or in 8-well Lab-Tek chambered cover glass (Nunc, Roskilde, Denmark; for dSTORM). Cells were transfected using Lipofectamine 2000 (Thermo Fisher Scientific) or FugeneHD (Promega), according to the manufacturer’s instructions. To stabilize MTs, cells were treated with paclitaxel (Sigma-Aldrich; 4 h, 1 μg/mL) (24).
Confocal laser scanning microscopy (CLSM).
For live-cell analysis, cells were imaged in phenol-free DMEM in a 37°C heated chamber. Cells immunostained for STAT1 were generated as previously described (22) by treatment with or without recombinant human IFN-α (PBL Interferon Source; 1,000 U/mL, 30 min) before fixation with 3.75% (wt/vol) formaldehyde (10 min, 37°C) in phosphate-buffered saline (PBS) and permeabilization with 90% (vol/vol) methanol (5 min, room temperature). Following incubation in 1% (wt/vol) bovine serum albumin (BSA) in PBS blocking solution, cells were immunostained with anti-STAT1 (Cell Signaling Technologies) followed by Alexa 568-labeled secondary antibodies (Molecular Probes) and were mounted in 13% (wt/vol) Mowiol solution. Immunostaining for β-tubulin was performed as previously described (21), whereby cells were fixed with 3% (wt/vol) paraformaldehyde (PFA)/0.1% (vol/vol) glutaraldehyde (GA; 10 min, 37°C), followed by blocking in 1% (wt/vol) BSA, immunostaining with anti-β-tubulin (Sigma-Aldrich) and Alexa 568-labeled antibodies, and mounting in 13% (wt/vol) Mowiol solution.
CLSM analysis used a Nikon Eclipse C1 or Leica SP5 inverted confocal laser scanning microscope with a 60× or 63× oil immersion objective, respectively. Image acquisition used Nikon NIS-Elements (Eclipse C1) or Leica LAS AF (SP5) software. Digitized confocal files (single slices) were analyzed using ImageJ 1.62 software (NIH) to determine the ratio of nuclear-to-cytoplasmic fluorescence (Fn/c) for each cell as previously described (44, 45). The Fn/c ratio was calculated (Fn-Fb/Fc-Fb, where F, Fb, and Fc are nuclear, background, and cytoplasmic fluorescence, respectively) to determine the relative nuclear accumulation of fluorescently labeled proteins.
Dual luciferase reporter assays.
NA cells were transfected with pRL-TK (encoding Renilla luciferase under the control of a constitutively active promoter) and pISRE-luc (encoding IFN-α-inducible Firefly luciferase) plasmids and with plasmid encoding GFP-fused proteins, as described previously (38, 57). Cells were then treated 21 h post-transfection with or without recombinant human IFN-α (PBL Interferon Source; 500 U/mL, 6 h) before analysis of luciferase activity, as previously described (38). Normalized luciferase activity was calculated by dividing values for Firefly luciferase by those for Renilla luciferase before determination of the fold change relative to values obtained for IFN-α-treated cells expressing control protein (GFP-N).
dSTORM.
Cells were fixed 18 h post-transfection using 2% (vol/vol) GA in cytoskeleton buffer (CB; 10 mM 2-ethanesulfonic acid, 150 mM NaCl, 5 mM ethylene glycol tetraacetic acid, 5 mM glucose, and 5 mM MgCl2) for 10 min and then permeabilized in 0.3% (vol/vol) GA/0.25% (vol/vol) Triton X-100 in CB (37°C) for 2 min. Residual glutaraldehyde was then quenched with 0.1% (wt/vol) NaBH4 in PBS for 7 min to reduce autofluorescence. Cells were blocked in 5% (wt/vol) bovine serum albumin (BSA) in PBS for 30 min and immunostained using an anti-β-tubulin primary antibody (1:100, 1 h; Sigma-Aldrich) and an Alexa Fluor-647 (1:200, 45 min; Molecular Probes) secondary antibody (Fig. 2) or F(ab′)2 antibody fragment (Fig. 3). This was followed by a secondary fixation with 4% (wt/vol) formaldehyde in PBS for 5 min. Note that the use of antibody fragment secondaries, compared to full antibodies, reduces the expected MTfd of single filaments by ~10 nm.
Cells were imaged in a switching buffer containing 10% glucose, 100 mM mercaptoethylamine, 400 μg/mL glucose oxidase, and 35 μg/mL catalase in PBS, adjusted to pH 8.5. Imaging was performed on a home-built single-molecule super-resolution microscope (Olympus IX81, 100×, 1.49-NA TIRF objective) based on a previously described setup (58). For transfected cells, epifluorescence (488-nm excitation, Toptica laser diode, ~10 to 50 W/cm2) was used to select GFP-positive cells. For dSTORM, high-power red laser excitation (638-nm excitation, Oxxius Laser Boxx laser diode, ~3 to 5 kW/cm2) was used to induce photoswitching of Alexa Fluor 647, where resulting single-molecule emissions were captured at 50 Hz for several minutes (10,000 to 20,000 frames) on an Andor iXon EM-CCD. Acquired frames were analyzed in rapidSTORM (59) using an input pixel size of 100 nm and point spread function full-width half-maximum (PSF FWHM) of 360 nm. Fitting each PSF with a two-dimensional (2D) Gaussian function produces a list of single-molecule coordinates, which is reconstructed into the super-resolved dSTORM image. MT bundling was then quantified as described in Brice et al. (21).
Western blotting.
COS-7 cells transfected to express GFP-fused proteins were treated with or without recombinant human IFN-α (PBL Assay Science, Piscataway, New Jersey; 1,000 U/mL) for 0.5 or 4 h before lysis in 50 mM Tris, pH 7.4, 150 mM NaCl, 2 mM EDTA, 10 mM NaF, and 0.5% IGEPAL (Sigma-Aldrich) supplemented with protease and phosphatase inhibitor cocktails (Roche). Isolated proteins were separated by SDS-PAGE and transferred to 0.2-μm nitrocellulose membranes before immunoblotting using anti-STAT1 (Cell Signaling Technology), anti-pY-STAT1 (Cell Signaling Technology), anti-actin (Abcam), and anti-GFP (Abcam) antibodies, followed by horseradish peroxidase (HRP)-conjugated secondary antibodies (Merck Millipore). Blots were incubated in Western Lightning ECL chemiluminescent substrate (PerkinElmer) and visualized using a ChemiDoc MP imaging system (Bio-Rad).
Statistical analysis.
Prism version 7 software (GraphPad) was used for statistical analysis to calculate P values using Student’s t test (unpaired, two tailed). If variances were determined to be significantly different (F test), a Welch’s correction was applied. If data sets failed the normality test, the alternative Mann-Whitney test was used. Significance is represented in figures using the following designations: ****, P ≤ 0.0001; ***, P ≤ 0.001; **, P ≤ 0.01; *, P ≤ 0.5; ns, not significant.
ACKNOWLEDGMENTS
We acknowledge the facilities and technical assistance of Monash Micro Imaging (Monash University) and the Biological Optical Microscopy Platform (University of Melbourne) for confocal imaging.
We declare no conflicts of interest.
This work was supported by National Health and Medical Research Council Australia project grants 1160838, 1125704, and 1079211 to G.W.M., Australian Research Council discovery project grants DP150102569 to G.W.M. and DP170104477 to T.D.M.B., and Miegunyah Trust Grimwade Fellowship to G.W.M.
Footnotes
Supplemental material is available online only.
Contributor Information
Gregory W. Moseley, Email: greg.moseley@monash.edu.
Susana López, Instituto de Biotecnologia/UNAM.
REFERENCES
- 1.Prosser SL, Pelletier L. 2017. Mitotic spindle assembly in animal cells: a fine balancing act. Nat Rev Mol Cell Biol 18:187–201. 10.1038/nrm.2016.162. [DOI] [PubMed] [Google Scholar]
- 2.de Forges H, Bouissou A, Perez F. 2012. Interplay between microtubule dynamics and intracellular organization. Int J Biochem Cell Biol 44:266–274. 10.1016/j.biocel.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 3.Povea-Cabello S, Oropesa-Ávila M, de la Cruz-Ojeda P, Villanueva-Paz M, de la Mata M, Suárez-Rivero JM, Álvarez-Córdoba M, Villalón-García I, Cotán D, Ybot-González P, Sánchez-Alcázar JA. 2017. Dynamic reorganization of the cytoskeleton during apoptosis: the two coffins hypothesis. Int J Mol Sci 18:2393. 10.3390/ijms18112393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiang H-S, Zhao Y, Song J-H, Liu S, Wang N, Terhorst C, Sharpe AH, Basavappa M, Jeffrey KL, Reinecker H-C. 2014. GEF-H1 controls microtubule-dependent sensing of nucleic acids for antiviral host defenses. Nat Immunol 15:63–71. 10.1038/ni.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sweeney HL, Holzbaur ELF. 2018. Motor proteins. Cold Spring Harb Perspect Biol 10:a021931. 10.1101/cshperspect.a021931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bodakuntla S, Jijumon AS, Villablanca C, Gonzalez-Billault C, Janke C. 2019. Microtubule-associated proteins: structuring the cytoskeleton. Trends Cell Biol 29:804–819. 10.1016/j.tcb.2019.07.004. [DOI] [PubMed] [Google Scholar]
- 7.Brice A, Moseley GW. 2013. Viral interactions with microtubules: orchestrators of host cell biology? Future Virol 8:229–243. 10.2217/fvl.12.137. [DOI] [Google Scholar]
- 8.Naghavi MH, Walsh D. 2017. Microtubule regulation and function during virus infection. J Virol 91:e00538-17. 10.1128/JVI.00538-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walker PJ, Blasdell KR, Calisher CH, Dietzgen RG, Kondo H, Kurath G, Longdon B, Stone DM, Tesh RB, Tordo N, Vasilakis N, Whitfield AE. 2018. ICTV virus taxonomy profile: Rhabdoviridae. J Gen Virol 99:447–448. 10.1099/jgv.0.001020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fisher CR, Streicker DG, Schnell MJ. 2018. The spread and evolution of rabies virus: conquering new frontiers. Nat Rev Microbiol 16:241–255. 10.1038/nrmicro.2018.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Banyard AC, Hayman DTS, Freuling CM, Müller T, Fooks AR, Johnson N. 2013. Bat rabies, p 215–267. In Jackson AC (ed), Rabies, 3rd ed. Academic Press, Cambridge, Massachusetts. [Google Scholar]
- 12.Marston DA, Banyard AC, McElhinney LM, Freuling CM, Finke S, de Lamballerie X, Müller T, Fooks AR. 2018. The lyssavirus host-specificity conundrum-rabies virus-the exception not the rule. Curr Opin Virol 28:68–73. 10.1016/j.coviro.2017.11.007. [DOI] [PubMed] [Google Scholar]
- 13.World Health Organization. 2018. WHO expert consultation on rabies: third report. https://apps.who.int/iris/handle/10665/272364. Accessed 4 October, 2022.
- 14.Bonjardim CA, Ferreira PCP, Kroon EG. 2009. Interferons: signaling, antiviral and viral evasion. Immunol Lett 122:1–11. 10.1016/j.imlet.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Versteeg GA, García-Sastre A. 2010. Viral tricks to grid-lock the type I interferon system. Curr Opin Microbiol 13:508–516. 10.1016/j.mib.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harrison AR, Moseley GW. 2020. The dynamic interface of viruses with STATs. J Virol 94:e00856-20. 10.1128/JVI.00856-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chenik M, Chebli K, Blondel D. 1995. Translation initiation at alternate in-frame AUG codons in the rabies virus phosphoprotein mRNA is mediated by a ribosomal leaky scanning mechanism. J Virol 69:707–712. 10.1128/JVI.69.2.707-712.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chenik M, Schnell M, Conzelmann KK, Blondel D. 1998. Mapping the interacting domains between the rabies virus polymerase and phosphoprotein. J Virol 72:1925–1930. 10.1128/JVI.72.3.1925-1930.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wiltzer L, Okada K, Yamaoka S, Larrous F, Kuusisto HV, Sugiyama M, Blondel D, Bourhy H, Jans DA, Ito N, Moseley GW. 2014. Interaction of rabies virus P-protein with STAT proteins is critical to lethal rabies disease. J Infect Dis 209:1744–1753. 10.1093/infdis/jit829. [DOI] [PubMed] [Google Scholar]
- 20.Hossain MA, Larrous F, Rawlinson SM, Zhan J, Sethi A, Ibrahim Y, Aloi M, Lieu KG, Mok Y-F, Griffin MDW, Ito N, Ose T, Bourhy H, Moseley GW, Gooley PR. 2019. Structural elucidation of viral antagonism of innate immunity at the STAT1 interface. Cell Rep 29:1934–1945. 10.1016/j.celrep.2019.10.020. [DOI] [PubMed] [Google Scholar]
- 21.Brice A, Whelan DR, Ito N, Shimizu K, Wiltzer-Bach L, Lo CY, Blondel D, Jans DA, Bell TDM, Moseley GW. 2016. Quantitative analysis of the microtubule interaction of rabies virus P3 protein: roles in immune evasion and pathogenesis. Sci Rep 6:33493. 10.1038/srep33493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moseley GW, Lahaye X, Roth DM, Oksayan S, Filmer RP, Rowe CL, Blondel D, Jans DA. 2009. Dual modes of rabies P-protein association with microtubules: a novel strategy to suppress the antiviral response. J Cell Sci 122:3652–3662. 10.1242/jcs.045542. [DOI] [PubMed] [Google Scholar]
- 23.Vidy A, El Bougrini J, Chelbi-Alix MK, Blondel D. 2007. The nucleocytoplasmic rabies virus P protein counteracts interferon signaling by inhibiting both nuclear accumulation and DNA binding of STAT1. J Virol 81:4255–4263. 10.1128/JVI.01930-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vidy A, Chelbi-Alix M, Blondel D. 2005. Rabies virus P protein interacts with STAT1 and inhibits interferon signal transduction pathways. J Virol 79:14411–14420. 10.1128/JVI.79.22.14411-14420.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brzózka K, Finke S, Conzelmann K-K. 2006. Inhibition of interferon signaling by rabies virus phosphoprotein P: activation-dependent binding of STAT1 and STAT2. J Virol 80:2675–2683. 10.1128/JVI.80.6.2675-2683.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manokaran G, Audsley MD, Funakoda H, David CT, Garnham KA, Rawlinson SM, Deffrasnes C, Ito N, Moseley GW. 2022. Deactivation of the antiviral state by rabies virus through targeting and accumulation of persistently phosphorylated STAT1. PLoS Pathog 18:e1010533. 10.1371/journal.ppat.1010533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oksayan S, Wiltzer L, Rowe CL, Blondel D, Jans DA, Moseley GW. 2012. A novel nuclear trafficking module regulates the nucleocytoplasmic localization of the rabies virus interferon antagonist, P protein. J Biol Chem 287:28112–28121. 10.1074/jbc.M112.374694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marschalek A, Drechsel L, Conzelmann K-K. 2012. The importance of being short: the role of rabies virus phosphoprotein isoforms assessed by differential IRES translation initiation. Eur J Cell Biol 91:17–23. 10.1016/j.ejcb.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 29.Pasdeloup D, Poisson N, Raux H, Gaudin Y, Ruigrok RWH, Blondel D. 2005. Nucleocytoplasmic shuttling of the rabies virus P protein requires a nuclear localization signal and a CRM1-dependent nuclear export signal. Virology 334:284–293. 10.1016/j.virol.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 30.Shimizu K, Ito N, Sugiyama M, Minamoto N. 2006. Sensitivity of rabies virus to type I interferon is determined by the phosphoprotein gene. Microbiol Immunol 50:975–978. 10.1111/j.1348-0421.2006.tb03875.x. [DOI] [PubMed] [Google Scholar]
- 31.Shimizu K, Ito N, Mita T, Yamada K, Hosokawa-Muto J, Sugiyama M, Minamoto N. 2007. Involvement of nucleoprotein, phosphoprotein, and matrix protein genes of rabies virus in virulence for adult mice. Virus Res 123:154–160. 10.1016/j.virusres.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 32.Ito N, Moseley GW, Blondel D, Shimizu K, Rowe CL, Ito Y, Masatani T, Nakagawa K, Jans DA, Sugiyama M. 2010. Role of interferon antagonist activity of rabies virus phosphoprotein in viral pathogenicity. J Virol 84:6699–6710. 10.1128/JVI.00011-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brice AM, Watts E, Hirst B, Jans DA, Ito N, Moseley GW. 2021. Implication of the nuclear trafficking of rabies virus P3 protein in viral pathogenicity. Traffic 22:482–489. 10.1111/tra.12821. [DOI] [PubMed] [Google Scholar]
- 34.Moseley GW, Filmer RP, DeJesus MA, Jans DA. 2007. Nucleocytoplasmic distribution of rabies virus P-protein is regulated by phosphorylation adjacent to C-terminal nuclear import and export signals. Biochemistry 46:12053–12061. 10.1021/bi700521m. [DOI] [PubMed] [Google Scholar]
- 35.Zhan J, Watts E, Brice AM, Metcalfe RD, Rozario AM, Sethi A, Yan F, Bell TDM, Griffin MDW, Moseley GW, Gooley PR. 2022. Molecular basis of functional effects of phosphorylation of the C-terminal domain of the rabies virus P protein. J Virol 96:e0011122. 10.1128/jvi.00111-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Audsley MD, Jans DA, Moseley GW. 2016. Roles of nuclear trafficking in infection by cytoplasmic negative-strand RNA viruses: paramyxoviruses and beyond. J Gen Virol 97:2463–2481. 10.1099/jgv.0.000575. [DOI] [PubMed] [Google Scholar]
- 37.Audsley MD, Moseley GW. 2013. Paramyxovirus evasion of innate immunity: diverse strategies for common targets. World J Virol 2:57–70. 10.5501/wjv.v2.i2.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wiltzer L, Larrous F, Oksayan S, Ito N, Marsh GA, Wang LF, Blondel D, Bourhy H, Jans DA, Moseley GW. 2012. Conservation of a unique mechanism of immune evasion across the lyssavirus genus. J Virol 86:10194–10199. 10.1128/JVI.01249-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deffrasnes C, Luo M-X, Wiltzer-Bach L, David CT, Lieu KG, Wang L-F, Jans DA, Marsh GA, Moseley GW. 2021. Phenotypic divergence of P proteins of Australian bat lyssavirus lineages circulating in microbats and flying foxes. Viruses 13:831. 10.3390/v13050831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rozario AM, Zwettler F, Duwé S, Hargreaves RB, Brice A, Dedecker P, Sauer M, Moseley GW, Whelan DR, Bell TDM. 2020. ‘Live and large’: super-resolution optical fluctuation imaging (SOFI) and expansion microscopy (ExM) of microtubule remodelling by rabies virus P protein. Aust J Chem 73:686. 10.1071/CH19571. [DOI] [Google Scholar]
- 41.Ito N, Moseley GW, Sugiyama M. 2016. The importance of immune evasion in the pathogenesis of rabies virus. J Vet Med Sci 78:1089–1098. 10.1292/jvms.16-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ugolini G. 2011. Rabies virus as a transneuronal tracer of neuronal connections. Adv Virus Res 79:165–202. 10.1016/B978-0-12-387040-7.00010-X. [DOI] [PubMed] [Google Scholar]
- 43.Lahaye X, Vidy A, Pomier C, Obiang L, Harper F, Gaudin Y, Blondel D. 2009. Functional characterization of Negri bodies (NBs) in rabies virus-infected cells: evidence that NBs are sites of viral transcription and replication. J Virol 83:7948–7958. 10.1128/JVI.00554-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rowe CL, Wagstaff KM, Oksayan S, Glover DJ, Jans DA, Moseley GW. 2016. Nuclear trafficking of the rabies virus interferon antagonist P-protein is regulated by an importin-binding nuclear localization sequence in the C-terminal domain. PLoS One 11:e0150477. 10.1371/journal.pone.0150477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Audsley MD, Jans DA, Moseley GW. 2016. Nucleocytoplasmic trafficking of Nipah virus W protein involves multiple discrete interactions with the nuclear import and export machinery. Biochem Biophys Res Commun 479:429–433. 10.1016/j.bbrc.2016.09.043. [DOI] [PubMed] [Google Scholar]
- 46.Soniat M, Chook YM. 2015. Nuclear localization signals for four distinct karyopherin-β nuclear import systems. Biochem J 468:353–362. 10.1042/BJ20150368. [DOI] [PubMed] [Google Scholar]
- 47.Lim CP, Cao X. 2006. Structure, function, and regulation of STAT proteins. Mol Biosyst 2:536–550. 10.1039/b606246f. [DOI] [PubMed] [Google Scholar]
- 48.Reich NC. 2013. STATs get their move on. JAKSTAT 2:e27080. 10.4161/jkst.27080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maher SG, Sheikh F, Scarzello AJ, Romero-Weaver AL, Baker DP, Donnelly RP, Gamero AM. 2008. IFNα and IFNλ differ in their antiproliferative effects and duration of JAK/STAT signaling activity. Cancer Biol Ther 7:1109–1115. 10.4161/cbt.7.7.6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blondel D, Regad T, Poisson N, Pavie B, Harper F, Pandolfi PP, De Thé H, Chelbi-Alix MK. 2002. Rabies virus P and small P products interact directly with PML and reorganize PML nuclear bodies. Oncogene 21:7957–7970. 10.1038/sj.onc.1205931. [DOI] [PubMed] [Google Scholar]
- 51.Oksayan S, Nikolic J, David CT, Blondel D, Jans DA, Moseley GW. 2015. Identification of a role for nucleolin in rabies virus infection. J Virol 89:1939–1943. 10.1128/JVI.03320-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bauer A, Nolden T, Nemitz S, Perlson E, Finke S. 2015. A dynein light chain 1 binding motif in rabies virus polymerase L protein plays a role in microtubule reorganization and viral primary transcription. J Virol 89:9591–9600. 10.1128/JVI.01298-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Albertini AAV, Ruigrok RWH, Blondel D. 2011. Rabies virus transcription and replication. Adv Virus Res 79:1–22. 10.1016/B978-0-12-387040-7.00001-9. [DOI] [PubMed] [Google Scholar]
- 54.Okada K, Ito N, Yamaoka S, Masatani T, Ebihara H, Goto H, Nakagawa K, Mitake H, Okadera K, Sugiyama M. 2016. Roles of the rabies virus phosphoprotein isoforms in pathogenesis. J Virol 90:8226–8237. 10.1128/JVI.00809-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bourhy H, Dautry-Varsat A, Hotez PJ, Salomon J. 2010. Rabies, still neglected after 125 years of vaccination. PLoS Negl Trop Dis 4:e839. 10.1371/journal.pntd.0000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Evans JS, Horton DL, Easton AJ, Fooks AR, Banyard AC. 2012. Rabies virus vaccines: is there a need for a pan-lyssavirus vaccine? Vaccine 30:7447–7454. 10.1016/j.vaccine.2012.10.015. [DOI] [PubMed] [Google Scholar]
- 57.Audsley MD, Marsh GA, Lieu KG, Tachedjian M, Joubert DA, Wang L-F, Jans DA, Moseley GW. 2016. The immune evasion function of J and Beilong virus V proteins is distinct from that of other paramyxoviruses, consistent with their inclusion in the proposed genus Jeilongvirus. J Gen Virol 97:581–592. 10.1099/jgv.0.000388. [DOI] [PubMed] [Google Scholar]
- 58.Whelan DR, Holm T, Sauer M, Bell TDM. 2014. Focus on super-resolution imaging with direct stochastic optical reconstruction microscopy (dSTORM). Aust J Chem 67:179. 10.1071/CH13499. [DOI] [Google Scholar]
- 59.Wolter S, Löschberger A, Holm T, Aufmkolk S, Dabauvalle M-C, van de Linde S, Sauer M. 2012. rapidSTORM: accurate, fast open-source software for localization microscopy. Nat Methods 9:1040–1041. 10.1038/nmeth.2224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S7. Download jvi.01396-22-s0001.pdf, PDF file, 0.9 MB (947.1KB, pdf)