

ABSTRACT
Mutations at spike protein L452 are recurrently observed in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants of concern (VOC), including omicron lineages. It remains elusive how amino acid substitutions at L452 are selected in VOC. Here, we characterized all 19 possible mutations at this site and revealed that five mutants expressing the amino acids Q, K, H, M, and R gained greater fusogenicity and pseudovirus infectivity, whereas other mutants failed to maintain steady-state expression levels and/or pseudovirus infectivity. Moreover, the five mutants showed decreased sensitivity toward neutralization by vaccine-induced antisera and conferred escape from T cell recognition. Contrary to expectations, sequence data retrieved from the Global Initiative on Sharing All Influenza Data (GISAID) revealed that the naturally occurring L452 mutations were limited to Q, M, and R, all of which can arise from a single nucleotide change. Collectively, these findings highlight that the codon base change mutational barrier is a prerequisite for amino acid substitutions at L452, in addition to the phenotypic advantages of viral fitness and decreased sensitivity to host immunity.
IMPORTANCE In a span of less than 3 years since the declaration of the coronavirus pandemic, numerous SARS-CoV-2 variants of concern have emerged all around the globe, fueling a surge in the number of cases and deaths that caused severe strain on the health care system. A major concern is whether viral evolution eventually promotes greater fitness advantages, transmissibility, and immune escape. In this study, we addressed the differential effect of amino acid substitutions at a frequent mutation site, L452 of SARS-CoV-2 spike, on viral antigenic and immunological profiles and demonstrated how the virus evolves to select one amino acid over the others to ensure better viral infectivity and immune evasion. Identifying such virus mutation signatures could be crucial for the preparedness of future interventions to control COVID-19.
KEYWORDS: SARS-CoV-2, L452, spike, substitution, fitness, coronavirus, mutational studies, spike protein
INTRODUCTION
Since the emergence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in the Wuhan province in China, this fast-evolving virus has acquired a significant number of mutations across its genome. As a result, many variants have surfaced in different parts of the world, such as the D614G variant, which first emerged in Europe during mid-2020 (1), and three major variants of concern (VOC) that emerged near the end of 2020, namely, the UK/alpha variant (B.1.1.7), the South Africa/beta variant (B.1.351), and the Brazil/gamma variant (P.1) (2–4). In the middle of 2021, the world was plagued with the fast-spreading delta variant (B.1.617.2) (5) before it was replaced with the currently circulating VOC, the omicron variant (B.1.1.529) (6).
The L452R mutation located within the receptor-binding domain (RBD) of the SARS-CoV-2 spike, first garnered attention during multiple outbreaks in California, United States, where a sharp rise of sequences harboring L452R was detected from September 2020 to January 2021 in the genome sequences deposited in the Global Initiative on Sharing All Influenza Data (GISAID) (7–9). Prior to that, mink-associated L452M variant was found in several mink farms in the Netherlands, where few cases of two-way transmission from human to mink and back to human were reported (10–12). Thereafter, similar L452-associated mutations, including L452Q-harboring C.37 lineage (lambda variant) (13) and L452R-harboring B.1.617.1/2 lineage (kappa and delta variants) (14) and A.27 lineage (15) were also reported. Several reports suggested that L452R contributed to the enhanced infectivity and reduced susceptibility to antibody neutralization that is observed in these SARS-CoV-2 lineages (8, 13, 16–19). In addition, the L452 is located within a 9-mer immunodominant cytotoxic T lymphocyte (CTL) epitope presented by HLA-A*24:02 (448NYNYLYRLF456) (20–22), where L452R mutation confers escape from HLA-A*24:02-restricted CTL recognition (23).
In this study, we devised to clarify why certain amino acids are preferentially selected over the others in the light of global mutation landscape of the L452 variants. We constructed a panel of L452 spike mutants substituted with all 19 canonical amino acids and investigated their phenotypical, functional and immunological impacts. Our study showed that single nucleotide accessible mutation (SNAM) is the prerequisite factor for amino acid selection in L452 mutation, which was ultimately shaped by viral fitness and sensitivity to host immunity.
RESULTS
Recurrent mutations at spike L452 in multiple lineages of SARS-CoV-2.
We constructed a phylogenetic tree based on major SARS-CoV-2 genotypes (n = 25,796) extracted from GISAID databases as of 27 April 2022, where 10,113 of them (~39%) had the spike L452 substituted with either arginine (R), glutamine (Q), or methionine (M) (Fig. 1A). These strains were derived from multiple SARS-CoV-2 lineages isolated from over 35 countries across all six continents. When we analyzed the timeline of the emergence of SARS-CoV-2 variants of interest (VOI) or VOC, the L452 mutation was first reported in VOI Epsilon (L452R) expanded in California (17, 24) from November 2020 but peaked in March 2021 (Fig. 1B). Following that, VOI Lambda (L452Q), which was first reported in Peru (25) in December 2020, then went on to spread in the South America countries in the first half of 2021 before declining (Fig. 1B). Meanwhile, VOI Kappa (L452R) (14), and VOC Delta (L452R) (5) were identified in India in October 2020. VOC Delta ended up becoming the predominant variant circulating globally from the second half of 2021 before it was replaced by the omicron variant in November 2021 (Fig. 1B). Very recently, several lineages of omicron acquired additional mutations, including at position L452. These omicron subvariants have since been assigned by the World Health Organization (WHO) to the newly added category, termed omicron VOC lineages under monitoring (VOC-LUMs) (26). We collected the metadata of deposited sequences from GISAID and depicted the trend of these omicron VOC-LUMs in different continents across the globe from January until the end of April 2022 (Fig. 1C). During this period, the BA.4 and BA.5 variants (harboring L452R) dominated South Africa, while the BA.2.12.1 subvariant (harboring L452Q), which was first detected in New York, outcompeted the BA.2 variant to become the dominant subvariant all over the United States. Other VOC-LUMs, including BA.2.13 (harboring L452M), BA.2.9.1 (harboring L452M), and BA.2.11 (harboring L452R), were showing signs of expansion in Europe (27) (Fig. 1C). The global emergence of L452 mutations in multiple lineages of SARS-CoV-2 demonstrated a pattern of convergent evolution, suggesting a common beneficial trait for the emerging variants.
FIG 1.

Recurrent mutations at spike L452 in multiple SARS-CoV-2 lineages. (A) Maximum-likelihood phylogenetic tree based on full-length genome sequences of a total of 25,796 major genotypes of SARS-CoV-2. The sequences were examined for amino acid mutations at spike position 452, and their numbers are shown in red. (B) Global epidemic dynamics of the L452Q-harboring lambda, as well as the L452R-harboring epsilon, kappa, and delta variants spanning from October 2020 to April 2022. (C) Recent emergence of the omicron lineages, including L452R-harboring BA.4, BA.5, and BA.2.11, L452Q-harboring BA2.12.1, and L452M-harboring BA.2.13 and BA.2.9.1 from January to April 2022. All data were retrieved from the GISAID database as of 27 April 2022.
Codon base change as a genetic barrier in the global occurrences of spike L452 mutation.
When we aligned the part of nucleotide sequences encompassing the region encoding position 452, we noticed that all nonsynonymous mutations at this site in epsilon, delta, kappa, lambda, and the omicron VOC-LUMs resulted from a single nucleotide substitution within the CUG codon of the ancestral Wuhan-Hu-1 strain (Fig. 2A). In light of this, we calculated the probability of occurrences of each canonical amino acid substitution at this position when leucine encoded by CUG as the prototype sequence, with the single (P = 0.25), doublet (P = 0.0625), or triplet (P = 0.0156) substitution required to achieve a nonsynonymous mutation and the number of codons available for a particular amino acid. We used a neutral model of codon substitution probability estimation without weighing in on codon preferences or the mutation rate of the virus because SARS-CoV-2 is known to have a relatively low codon usage bias (28). The analysis showed that the substitutions with the highest probability of occurrence are L452R (P = 0.516), followed by L452P and L452V (both P = 0.438), L452Q (P = 0.313), and L452M (P = 0.25) (Fig. 2B), suggesting that newly arising variants (e.g., Q, M, and R) have a low genetic barrier toward the codon change at this position. This is in agreement with a prior global mutational analysis study of SARS-CoV-2, which demonstrated that SNAMs are present at much higher frequencies (29). However, some exceptions on the basis of selection pressure exist, for instance, certain L452 mutations despite having a lower genetic barrier (e.g., P and V) showed very low global frequency (Fig. 2C), suggesting phenotypic constraints in these mutations.
FIG 2.

Preference of SNAMs at spike L452. (A) Alignment of spike sequence showing the single base change at nucleotide numbers 22916/22917 that leads to nonsynonymous L452 mutation among circulating SARS-CoV-2 variants. (B) Likelihood for each amino acid substitution from leucine (encoded by CUG) based on a neutral probability estimation model. (C) Likelihood versus actual frequency of L452 amino acid substitutions. Actual global frequency of each amino acid was analyzed using the Table of Mutation Sites tool (https://cov.lanl.gov/content/sequence/TBLS_MUT_SITES/tbls_mut_sites.html; updated on 27 April 2022). Amino acids characterized by a SNAM are highlighted in red.
Changes in spike-mediated fusion and pseudovirus entry by L452 mutations.
By constructing a set of spike mutants substituted with 19 different canonical amino acids at position L452, we first tested the effect on the steady-state expression of the spike protein in 293T cells after transfection of spike-encoding plasmids. Immunoblot analyses showed that wild-type (WT) spike was detected both in the full-length (180 kDa) and the cleaved forms (90 kDa) (Fig. 3A). While 14 of 19 mutants showed expression levels comparable to that of WT for both the full-length and cleaved forms, expression of the cleaved form was substantially reduced in L452 mutations to P and C and moderately reduced in I, N, and D mutations. However, the expression level of the full-length forms was not reduced in these mutants, suggesting either inefficient cleavage by a host protease or instability of the cleaved forms following the mutations (Fig. 3A). The introduction of P and C mutations at position 452 may trigger conformational defect of the spike protein (30, 31) and may lead to altered protein processing by host proteases and/or enhanced degradation by host machinery (32).
FIG 3.

Biochemical and virological characterization of spike L452 variants. (A) Immunoblots showing total cellular expression and virion incorporation of spike L452 mutants in producer cell and virion (left panel). The spike incorporation level was quantified and normalized to p24 Gag levels and is expressed relative to the WT in three independent replicates (right panel). Statistical significance was determined by one-way ANOVA with multiple-comparison tests (*, P < 0.05; **, P < 0.01; ***, P < 0.001). (B) Titers (ng/mL) of pseudoviruses collected from the culture supernatant at 48 h posttransfection. (C) Infectivity of reporter lentiviruses pseudotyped by SARS-CoV-2 spike L452-mutants and WT D614G. The indicated titers of pseudoviruses (1, 3, and 5 ng) were exposed to 293T cells expressing ACE2 and TMPRSS2. The amount of pseudoviruses successfully infected into target cells was determined from the luciferase activity, and the relative infectivity is expressed as the percentage normalized to the WT (infection at 5 ng). (D) Infectivity of reporter lentiviruses pseudotyped with SARS-CoV-2 BA.2 spike and its L452 mutants (infection at 5 ng). (E) Fusion formation between the spike-expressing cells and ACE2-TMPRSS2-expressing cells was continuously monitored at intervals of 3, 6, 12, 24, and 48 h. The fusion activity was expressed relative to WT at 3 h after coincubation. (F) Correlation between infectivity of pseudoviruses (infection at 5 ng) and cell-to-cell fusion activity (at 48 h). The statistical significance was determined by using the Pearson’s correlation coefficient test. (G) Structure of SARS-CoV-2 spike trimer with single RBD up-conformation (PDB 7KRR). Each protomer is differently colored. The L452 residues are shown as spheres, while glycans are indicated as sticks. In the RBD with the up-conformation, the L452 residue is exposed (yellow box). In contrast, in the RBD with the down-conformation, the L452 residue is buried at the interface with the NTD of another protomer (orange box). The data shown are means ± the SD of triplicate determinations.
We then prepared lentiviral particles pseudotyped with the spike mutants. In addition to the P and C mutations, L452 mutations to I, N, and D showed substantial impairment both in spike incorporation into the nascent virions (Fig. 3A) and in production of the progeny viral particles (Fig. 3B), most likely reflecting the reduced cleaved form of the spike protein observed in these mutants. The rest of the mutants showed spike incorporation to virions and the subsequent production of progeny virions at levels comparable to WT. Of note, although the L452 mutations to I, P, and C exhibited substantial impairment in the incorporation of spike into lentiviral particles, I and L are considered to have relatively similar chemical properties compared to the side chain moieties of P and C (Fig. 3A).
Next, we tested the effect of the mutations on the infectivity of pseudotyped viral particles toward 293T target cells transiently expressing human ACE2 receptor and serine protease TMPRSS2 (33). As expected from these data, the L452 mutations to P, C, I, and D diminished the infectivity potential of the pseudoviruses to less than 50% of the WT level (Fig. 3C). The L452 mutations to Q, K, H, M, and R showed a >2-fold increase in infectivity compared to the WT (Fig. 3C), indicating that not only naturally arising mutants (i.e., Q, M, and R) but also naturally absent mutants (i.e., K and H) can enhance infectivity. In contrast, compared to WT, the L452 mutations to A, V, F, W, G, Y, S, T, and E retained infectivity (within a 2-fold increase) (Fig. 3C). Because L452-M, -Q, and -R mutations recently appeared in multiple lineages derived from omicron BA.2 (Fig. 1C), we tested the effect of these naturally arising mutations on pseudovirus infectivity in the context of the omicron spike backbone. Notably, we observed that introduction of M, Q, and R mutations to the parental BA.2 spike demonstrated a pronounced effect on enhancing infectivity (3- to 5-fold) (Fig. 3D), suggesting a common role of L452 mutations in the context of multiple variant lineages.
Fusogenicity between spike and host membranes via its interaction with the ACE2 receptor is known to be important for SARS-CoV-2 infectivity and transmissivity (34). We therefore analyzed the spike-mediated cell-to-cell fusion using a dual-split reporter protein system (DSP) (35). Time course experiments showed that the cell-to-cell fusion commenced immediately after initiating coculture of spike and ACE2/TMPRSS2-expressing cells and continued for at least 48 h. The L452 substitutions to amino acid residues harboring basic side chains (i.e., K, R, and H) exhibited cell-to-cell fusion most efficiently (Fig. 3E). In contrast, fusogenicity of the L452P mutant was the lowest, most likely because of impaired cleavage of the spike protein in the cells (Fig. 3A). Of interest, the L452D mutant exhibited only approximately 15% reduction in fusion activity compared to WT; although it showed much more impairment in the virion incorporation of the spike protein (Fig. 3A) and in infectivity of the pseudovirus particles (Fig. 3C). It remains unclear whether incorporation of the L452D spike is similarly abrogated in authentic SARS-CoV-2. Overall, the fusion activity and the infectivity of pseudoviruses were well correlated (r = 0.9030, P < 0.0001) (Fig. 3F), confirming the importance of fusogenicity between spike and host membrane via its interaction with the ACE2 receptor in the viral entry to the target cells.
Previously reported cryo-electron microscopy structure of full-length spike trimer shows that the L452 amino acid residue in RBD at down conformation is buried at the interface with the N-terminal domain (NTD) of another protomer (PDB 7KRR) (36) (Fig. 3G). It was also demonstrated that the delta variant bearing spike L452R has an up-conformation more frequently than that of D614G variant (37). In parallel, our mutagenesis study demonstrated that amino acids with positive charge (e.g., R, K, and H) and/or long side chain (e.g., R, K, Q, and M) at position 452 of SARS-CoV-2 spike have a positive effect on viral entry. Hence, it is electrostatic repulsion or steric hindrance of the 452nd residue with its neighboring NTD that may induce RBD up-conformation which is more accessible to ACE2 entry receptor (37).
Changes in immune recognition of spike proteins by L452 mutations.
We obtained a panel of antisera from nine healthy volunteers who had received the two doses of BNT162b2 (Pfizer-BioNTech) vaccine (Table 1) and tested for neutralizing activity against the lentiviral reporters pseudotyped with the 14 L452 spike variants that showed expression and virion incorporation levels comparable to the WT D614G (Fig. 3A). Half of the L452 variants (7 of 14) maintained the sensitivity to the antisera with <20% difference in NT50. On the other hand, L452R and L452Q showed a >20% decrease in sensitivity (Fig. 4A), which in good agreement with previous studies demonstrating that L452 mutations play some roles in escape from neutralizing antibodies in epsilon, delta, and lambda variants (13, 16, 18, 24, 38). Interestingly, we found that the L452T mutant significantly (>60%) enhanced the sensitivity to neutralization by vaccine-induced antisera (Fig. 4A).
TABLE 1.
Vaccinated donors in this study
| Donor no. | Sex | Age (yrs) | No. of doses | Doses |
Time since last shot (days) | History of COVID-19 | pNT50a against: |
|||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | WTb | BA.2c | ||||||
| 1 | F | 18 | 2 | BNT162b2 | BNT162b2 | NAd | 21 | No | 3685.391 | NA |
| 2 | F | 24 | 2 | BNT162b2 | BNT162b2 | NA | 22 | No | 858.9506 | NA |
| 3 | F | 25 | 2 | BNT162b2 | BNT162b2 | NA | 23 | No | 585.8983 | NA |
| 4 | M | 23 | 2 | BNT162b2 | BNT162b2 | NA | 22 | No | 824.0571 | NA |
| 5 | M | 34 | 2 | BNT162b2 | BNT162b2 | NA | 14 | No | 779.7973 | NA |
| 6 | M | 55 | 2 | BNT162b2 | BNT162b2 | NA | 14 | No | 1037.38 | NA |
| 7 | M | 35 | 2 | BNT162b2 | BNT162b2 | NA | 14 | No | 1821.565 | NA |
| 8 | M | 33 | 2 | BNT162b2 | BNT162b2 | NA | 13 | No | 506.6158 | NA |
| 9 | F | 57 | 2 | BNT162b2 | BNT162b2 | NA | 10 | No | 1504.086 | NA |
| 10 | M | 23 | 3 | BNT162b2 | BNT162b2 | mRNA-1273 | 44 | No | NA | 525.8267 |
| 11 | F | 29 | 3 | BNT162b2 | BNT162b2 | mRNA-1273 | 40 | No | NA | 228.7586 |
| 12 | F | 24 | 3 | BNT162b2 | BNT162b2 | mRNA-1273 | 42 | No | NA | 167.915 |
| 13 | M | 22 | 3 | BNT162b2 | BNT162b2 | mRNA-1273 | 42 | No | NA | 336.4254 |
| 14 | F | 23 | 3 | BNT162b2 | BNT162b2 | mRNA-1273 | 43 | No | NA | 201.9978 |
| 15 | M | 23 | 3 | BNT162b2 | BNT162b2 | mRNA-1273 | 44 | No | NA | 155.8458 |
| 16 | M | 56 | 3 | BNT162b2 | BNT162b2 | mRNA-1273 | 24 | No | NA | 191.1671 |
| 17 | M | 39 | 3 | BNT162b2 | BNT162b2 | mRNA-1273 | 24 | No | NA | 133.8658 |
| 18 | F | 67 | 3 | BNT162b2 | BNT162b2 | BNT162b2 | 15 | No | NA | 124.8006 |
pNT50, pseudovirus 50% neutralization titer (serum dilution).
WT, wild-type D614G (B.1 lineage).
BA.2, Omicron BA.2 (B.1.1.529 lineage).
NA, not applicable.
FIG 4.
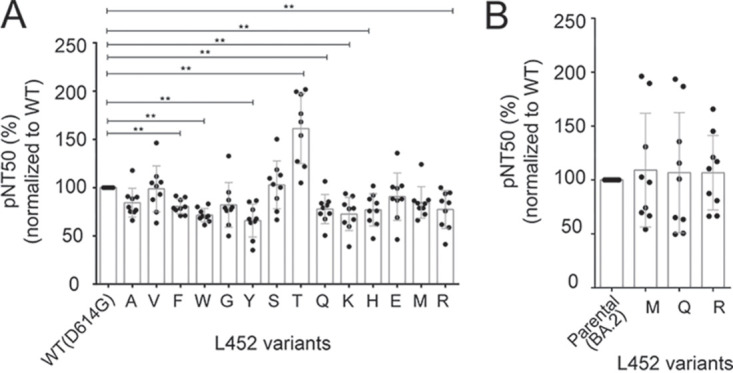
Changes in neutralizing sensitivity by L452 spike variants. (A and B) Neutralizing sensitivity expressed as the pseudovirus neutralizing titer (pNT50) of sera from nine donors receiving two and three doses of the vaccines (Table 1) against a panel of lentiviruses pseudotyped with L452 spike mutants in the context of WT D614G (A) and omicron BA.2 (B), respectively. The relative pNT50 (%) values normalized to their parental pairs are shown. The data shown are means ± the SD of triplicate determinations. Statistical significance was determined by Wilcoxon paired signed rank test (*, P < 0.05; **, P < 0.01).
Next, we wanted to test the neutralization sensitivity when the naturally arising L452 mutations were introduced into BA.2 spike. In this assay, we used another panel of antisera from nine healthy volunteers who had received the three doses of BNT162b2 (Pfizer-BioNTech) or mRNA-1273 (Moderna) (Table 1) as BA.2 acquired substantial resistance against vaccinated antisera obtained from a two-dose regimen (39). No difference was observed in neutralization sensitivity between BA.2 spike and its L452 mutations to M, Q, and R (Fig. 4B), suggesting that none of the L452 mutations on the omicron BA.2 backbone was selected due to their reduced sensitivity to neutralization.
A nonamer peptide 448-NYNYLYRLF-456 (here “NF9”), which spans the spike L452 residue at its fifth position, is an immunodominant CTL epitope presented by HLA-A*24:02, a prevalent HLA allele in Asia (20–22, 40), and the spike L452R mutation confers escape from recognition by the cognate CTLs (23). Three NF9-specific TCR clones were isolated from three donors (23) and determined their TCR gene usages (Table 2). TCR clonotypes were found to be very similar. Specifically, in the TCR α chain, the identical V segment was shared by T1 and T3, and the same J segment was shared by T1 and T2. In the TCR β chain, all harbored the same J segment and only one amino acid difference at the complementary determining region 3 (CDR3) (Table 2). To analyze their reactivity toward the panel of NF9 and NF9_5X peptide (where “X” represents any amino acid substitution at the fifth position of the peptide), we used previously established reporter T cell assay (41). The A549 cells expressing HLA-A*24:02 as target cells were pulsed with a set of the synthetic peptides, followed by coculture with Jurkat cells expressing the cognate TCRs as effector cells (Fig. 5A). As expected from similar TCR clonotypes (Table 2), all three TCRs showed very similar cross-reactivity profiles (Fig. 5B). Specifically, all TCRs recognized variant peptides NF9_5V, 5I and 5T comparably to NF9, and peptide NF9_5W to a lesser extent. In contrast, while TCR 1 and TCR 3 showed minimal cross-reactivity to the peptide NF9_ 5N and 5M, TCR 2 showed none. No TCR could cross-recognize the other 13 variant peptides (Fig. 5B). Furthermore, we examined the TCR cross-reactivity of L452 variants in the context of the omicron BA.2 spike. DNAs encoding the omicron BA.2 spike and its L452-M, -Q, and -R mutants were transfected to HLA-A*24:02-expressing A542 cells and tested for recognition by the same TCRs (Fig. 5A). The results showed that none of the naturally arising L452 variants (i.e., L452-M, -Q and -R) retained sensitivity to TCR recognition (Fig. 5C). Of interest, the L452T mutant, which is naturally absent, showed greater sensitivity to neutralization by antisera and TCRs isolated from vaccine recipients (Fig. 4A and 5B).
TABLE 2.
Clonotypes of NF9-specific TCRs
| TCR | Donor ID | TRAa |
TRBb |
|||||
|---|---|---|---|---|---|---|---|---|
| Vc | Jd | CDR3e | V | J | Df | CDR3 | ||
| T1 | GV34#3-4 | TRAV12-1*01 | TRAJ33*01 | CVVNLFDSNYQLIW | TRBV2*01 | TRBJ2-7*01 | TRBD1*01 | CASSEGAGYEQYF |
| T2 | GV36#16-2 | TRAV12-2*02 | TRAJ33*01 | CAVNGLKDSNYQLIW | TRBV6-1*01 | TRBJ2-7*01 | TRBD2*01 | CASSEGGGYEQYF |
| T3 | KK08#14-1 | TRAV12-1*01 | TRAJ12*01 | CVVNIIMDSSYKLIF | TRBV6-4*01 | TRBJ2-7*01 | TRBD2*02 | CASSEGEGYEQYF |
TRA, T cell receptor alpha chain.
TRB, T cell receptor beta chain.
V, V segment.
J, J segment.
CDR3, complementary determining region 3.
D, D segment.
FIG 5.

Changes in TCR recognition by L452 spike variants. (A) Schematic diagram of TCR recognition assay. NFAT-luciferase Jurkat T reporter cells (TCRαβ–/–) expressing the defined αβ TCR clonotypes were used as effector cells. A549 cells expressing human ACE2 receptor and HLA-A*A24:02 (A549/hACE2/HLA-A*24:02) were loaded with a panel of synthetic peptides or transfected with a panel of genes expressing spike proteins and used as target cells. (B and C) TCR activation level as measured by luciferase activity. A549/hACE2/HLA-A*24:02 target cells loaded with a panel of NF9 and the indicated mutant peptides (B) and transfected by DNAs encoding BA.2 spike and its L452 variants (C) were cocultured with the NFAT-luciferase Jurkat T reporter cells that had been engineered to stably express the indicated NF9-specific TCR clones. The data shown are means ± the SD of triplicate determinations.
DISCUSSION
In the present study, by employing a systematic and comprehensive analysis of recurrent mutations at SARS-CoV-2 spike L452, we demonstrated that the naturally arising L452 mutations in SARS-CoV-2 VOCs were characterized by (i) SNAM amino acid changes, (ii) amino acid residues that maintain a steady-state expression level and downstream intracellular processing, (iii) enhancement of spike fusogenicity, (iv) enhancement of pseudovirus infectivity, and (v) amino acid mutations associated with less susceptibility to neutralization and TCR recognition.
Structural analysis shows that amino acids with longer and/or positively charged side chains at the position 452 tend to demonstrate better viral entry probably due to a steric clash with neighboring residues that leads to RBD up-conformation, which is known to be more accessible to the ACE2 receptor (37). Interestingly, glutamic acid (E) which contains a long side chain showed a rather modestly enhanced entry, probably due to additional restraint in conformation by its negative charge. Further detailed structural analysis is needed to elucidate the molecular basis of L452 mutations.
Of additional interest, the L452T mutant, which is naturally absent, showed greater sensitivity to neutralization by antisera and TCRs isolated from vaccine recipients. One could infer that the L452 mutation results in forming a classical N-x-S/T motif for N-linked glycosylation because the amino acid residue at position 450 was asparagine. The addition of glycan at N450 could influence the RBD conformation and sensitivity to immune recognition. However, such enhanced immune recognition was observed in L452T, but not in L452S. Whether the L452T mutant could be more immunogenic when introduced into vaccine platforms warrants further evaluation.
Our study employed a lentiviral pseudotyping system for the phenotypic analysis. It is important to note that the mutational effects of spike proteins on the infectivity of the authentic SARS-CoV-2 may have different profiles. Thus, further studies are needed to clarify this issue by creating recombinant SARS-CoV-2 particles harboring defined L452 mutations with reverse genetic technology, as demonstrated in a previous study (23). Despite this limitation, our findings suggest that the codon base change mutational barrier is a prerequisite for amino acid substitutions at L452, in addition to the phenotypic advantages of viral fitness and decreased sensitivity to host immunity, highlighting a convergent evolutional pathway of SARS-CoV-2 spike protein in emerging variants.
MATERIALS AND METHODS
Phylogenetic analysis.
A maximum-likelihood phylogenetic tree of the coding regions (266 to 29,674 nucleotides [nt] in the reference sequence, EPI_ISL_402124) of the representative SARS-CoV-2 genomes was constructed. First, publicly available SARS-CoV-2 genome sequences (approximately 10.5 million) were downloaded from the GISAID EpiCoV database on 27 April 2022 (https://www.gisaid.org/). Sequences were excluded if (i) they carried any ambiguous bases (i.e., genetic codes other than A, U, G, and C) or (ii) they were shorter than 29,000 nt, as previously described (42). Next, the coding region of each filtered sequence was extracted, and a genotype was estimated by using a set of existing nucleotide mutations for the respective coding sequence, as previously described (42). A total of 25,796 major genotypes shared among ≥10 sequences (except recombinant lineages) were selected, and one sequence for each major genotype was applied for the phylogenetic analysis. Of note, no genotypes within the BA.4 lineage were shared among ≥10 sequences at the time of analysis. An approximate maximum-likelihood phylogenetic tree was constructed using FastTree (v2.1.11) with the GTR+CAT model (43). The tree was drawn using FigTree (v1.4.4; http://tree.bio.ed.ac.uk/software/figtree/). Selected sequences were also examined to detect amino acid mutations at spike position 452.
Probability estimation model.
The probability of occurrences for each canonical amino acid substitution at position L452 was estimated by the number of nucleotide substitutions required for nonsynonymous mutation, together with the codonicity of each amino acid (i.e., the number of codons available for a particular amino acid). Substitution of one nucleotide (A/U/G/C) in a codon is computed at a probability of 0.25, two simultaneous nucleotides change at a probability of 0.252 and three simultaneous nucleotides change at a probability of 0.253. In the event of amino acid substitution of leucine-452 (original codon CUG, as in ancestral SARS-CoV-2; GenBank MN908947.3) by each of the canonical amino acids, the final probability of the event was calculated by adding up the nucleotide change probabilities of each codon coded for that particular amino acid.
Plasmid constructs.
The firefly luciferase-expressing lentiviral transfer plasmid pWPI-Luc2, the HiBiT-tagged lentiviral packaging plasmid psPAX2-IN/HiBiT, the ACE2-expressing plasmid pC-ACE2, the TMPRSS2-expressing plasmid pC-TMPRSS2, and the SARS-CoV-2 spike D614G-expressing plasmid pC-SARS2-S-D614G (generous gifts from Kenzo Tokunaga) have previously been described elsewhere (33). The SARS-CoV-2 spike L452 mutants in which the position L452 was mutated from leucine to other canonical amino acids, including alanine (L452A), valine (L452V), isoleucine (L452I), phenylalanine (L452F), tryptophan (L452W), proline (L452P), glycine (L452G), tyrosine (L452Y), serine (L452S), threonine (L452T), cysteine (L452C), asparagine (L452N), glutamine (L452Q), lysine (L452K), histidine (L452H), aspartic acid (L452D), glutamic acid (L452E), methionine (L452M), and arginine (L452R) were prepared by PCR-based site-directed mutagenesis using pC-SARS-S-D614G as a template. The BA.2 L452 variants, including pC-SARS-S-BA.2-L452M, pC-SARS-S-BA.2-L452Q, and pC-SARS-S-BA.2-L452R, were constructed using pC-SARS2-S-BA.2 (39) (a generous gift from Kei Sato) as a template. The resultant PCR fragment was digested with KpnI and NotI and inserted into the corresponding site of the pCAGGS mammalian expression vector (44). Nucleotide sequences of the spike were determined by Genetic Analyzer 3500xL (Applied Biosystems), followed by analysis using GENETYX software (v12 622; GENETYX Corporation).
Pseudovirus assay.
SARS-CoV-2 pseudoviruses were prepared by cotransfecting 1 μg of pWPI-LUC2, 1 μg of psPAX2-IN/HiBiT, and 400 ng of various spike-encoding plasmids in 293T cells (6 × 105) by using a polyethylenimine (PEI) Max transfection system (Polysciences, Inc., catalog no. 24765-1). The secreted viral particles were harvested from the culture supernatant, clarified by centrifugation at 3,000 × g for 3 min at 4°C, and quantified as assessed by NanoLuc expression that was proportional to the level of the p24 antigen, as described previously (45). To prepare 293T/ACE2/TMPRSS2-expressing target cells, equal amounts (500 ng) of pC-ACE2 and pC-TMPRSS2 were cotransfected in 293T cells (6 × 105). After 48 h, the cells were split and seeded at 2.2 × 104 per well on a 96-well plate and then exposed to equal amounts of viral inoculum at titers of 1, 3, and 5 ng of the p24 antigen level. The single-round infectivity was determined 48 h later by measuring the luciferase activity with a One-Glo luciferase assay system (Promega, catalog no. E6130) in a Centro XS3 LB960 luminometer (Berthold Technologies). Data were normalized to WT D614G (5 ng) or parental BA.2 as 100%.
Cell-to-cell fusion assay.
The SARS-CoV-2 spike-based fusion assay was performed based on a dual-split protein (DSP), DSP1-7 and DSP8-11, which together encode a chimera of Renilla luciferase (RL) and GFP reporter proteins (46, 47). To prepare spike-expressing cells, HEK293T cells (3 × 105) seeded on a 24-well plate were cotransfected with equal amounts (200 ng) of pDSP1-7 and various spike-expressing plasmids, as indicated above. To prepare ACE2- and TMPRSS2-expressing target cells, HEK293 cells (8 × 105) seeded on a six-well plates were cotransfected with 500 ng of pC-ACE2, 250 ng of pC-TMPRSS2, and 500 ng of pDSP8-11. At 48 h posttransfection, the ACE2- and TMPRSS2-expressing cells were incubated with EnduRen live cell substrate (Promega, catalog no. E6481) for 3 h. Then, the spike-expressing cells (1.6 × 104 per well) and ACE2- and TMPRSS2-expressing cells (3.2 × 104 per well) were mixed in a 96-well black plate (Falcon, catalog no. 353376), and the RL activity was measured at the indicated time points by using a Centro XS3 LB960 luminometer (Berthold Technologies).
Protein structure.
The cryo-electron microscopy structure of a full-length spike trimer (PDB 7KRR) (36) was drawn using PyMOL v2.3 (https://pymol.org/2/).
Western blotting.
The samples for immunoblotting were prepared as described previously (48). Briefly, transfected cells were lysed directly on ice for 15 min in a buffer (100 mM NaCl, 1 mM TCEP [Tris(2-carboxyethyl)phosphine hydrochloride], 2× protease inhibitor, and 10 mM HEPES; pH 7.5) containing 1% n-dodecyl-β-d-maltoside (DDM; Nacalai Tesque, catalog no. 14239-41). For the spike incorporation study, the virus-containing culture supernatant was clarified by filtering through a 0.45-μm-pore-size filter, layered onto a 20% (wt/vol) sucrose cushion, pelleted by ultracentrifugation at 50,000 rpm for 30 min at 4°C (Beckman Coulter Optima-TLX), and then lysed in 1% DDM buffer. The resultant samples were resuspended in 1× Laemmli buffer containing 5% β-mercaptoethanol (Bio-Rad, catalog no. 1610710), boiled for 10 min, and subjected to protein separation by SDS-PAGE in 4 to 20% Mini-Protean TGX precast gels (Bio-Rad, catalog no. 4561096) before transfer to nitrocellulose membranes (Wako, catalog no. 032-22663). The membranes were incubated in a blocking buffer (Nacalai Tesque, catalog no. 03953-95) for 1 h at room temperature and then mixed with primary antibodies, including mouse anti-SARS-CoV-2 Spike (S2 subunit) monoclonal antibody (1:4,000; GeneTex, catalog no. GTX632604), rabbit anti-HIV Gag p24 monoclonal antibody (1:5,000; Bioacademia, catalog no. 65-004), and mouse anti-β-actin monoclonal antibody (1:5,000; Wako, catalog no. 281-98721), followed by staining with the appropriate secondary antibodies, horseradish peroxidase-conjugated anti-mouse (1:25,000; GE Healthcare, catalog no. NA931VS) or anti-rabbit (1:50,000; GE Healthcare, catalog no. NA94VS) IgG antibodies. The membrane was developed with the ImmunoStar LD enhanced chemiluminescence reagents (Wako, catalog no. 290-69904) and visualized using ImageQuant LAS 400 (GE Healthcare). Band intensities were analyzed using ImageJ software (49).
Pseudovirus neutralization assay.
Serum samples were collected from nine donors with two doses of BNT162b2 (Pfizer-BioNTech) and another group of nine donors with three doses of vaccines, including BNT162b2 or mRNAQ-123 (Moderna) (Table 1). The serum samples were heat inactivated at 56°C for 30 min and stored at −80°C until use. To compare the neutralizing activity against different spike-pseudotyped lentiviruses, 2-fold serially diluted sera were mixed with an equal volume of pseudoviruses (5 ng of p24 level per well) and incubated at 37°C for 1 h. Finally, 2.2 × 104 293T cells transiently expressing ACE2 and TMPRSS2 were added into each well in a 96-well plate and incubated further for 48 h prior to quantification of luciferase expression using a One-Glo luciferase assay system in Centro XS3 LB960 luminometer (Berthold Technologies). The neutralization level was determined by the percent decrease in luminescence relative to that obtained in the absence of sera. Neutralization curves were plotted, and 50% pseudovirus neutralization titer (pNT50) values (in serum dilution factor) were inferred using GraphPad Prism, and expressed in percentage normalized to parental WT D614G or BA.2. Prepandemic sera isolated from three donors were simultaneously tested and always yielded negative results for neutralization.
T cell receptor recognition assay.
Construction of NF9-specific effector T cells and TCR activation assays were performed as previously described (23, 41, 50). In brief, TCR clones were reconstituted and stably expressed in an NFAT-luciferase Jurkat T reporter cell line (TCRαβ −/−) that allows detection of T cell activation. Also, HLA-A*A24:02 was stably transduced in A549-expressing human ACE2 receptor, giving rise to A549/hACE2/HLA-A*24:02 (23, 50). A549/hACE/HLA-A*24:02 target cells were loaded with no peptide or a panel of NF9 and NF9_5X peptides (where “X” represents any amino acid substitution at the fifth position of the peptide) in a final concentration of 50 nM. Alternatively, the same target cells were transfected or not with 2 μg of various spike-expressing plasmids, pC-BA.2, pC-BA.2-L452M, pC-BA.2-L452Q, and pC-BA.2-L452R for 48 h. In both cases, target cells (0.5 × 105 per well) and NFAT-luciferase Jurkat T reporter cells expressing NF9-specific TCR clones (1 × 105 per well) were cocultured in a 96-well plate for 8 h. The T cell activation level was measured by detecting the luciferase activity using a Steady-Glo assay system (Promega, catalog no. E2550) in a Centro XS3 LB960 luminometer.
Statistical analysis.
Statistical tests were performed by using GraphPad Prism 6. Standard deviations (SD) were calculated to estimate the variance. Statistical comparisons were made as described for each data set and figure. A P value of <0.05 was considered significant.
ACKNOWLEDGMENTS
We thank Kenzo Tokunaga (National Institute of Infectious Disease, Tokyo, Japan) and Kei Sato and Jin Gohda (University of Tokyo, Tokyo, Japan) for providing the necessary reagents.
This study was supported in part by AMED Research Program on Emerging and Re-emerging Infectious Diseases 20fk0108539h0001 and 20fk0108451s0101 (to T.U.) and AMED Research Program on HIV/AIDS 21fk0410046 (to C.M.), JSPS KAKENHI Grants-in-Aid for Scientific Research B 19H03703 and 22H03119 (to T.U.) and 22H02877 (to C.M.), Scientific Research C grants 19K07623 (to C.M.) and 22K07089 (to M.T.), Takeda Science Foundation grants (to C.M. and M.T.) and an intramural grant from Kumamoto University COVID-19 Research Projects (AMABIE) (to C.M.), IMAI Memorial Trust for AIDS Research (to M.T.), and Shin-Nihon Foundation of Advanced Medical Research (to M.T.). This study was also supported in part by JSPS Bilateral Open Partnership Joint Research Project, JPJSBP120219933 and JSPS Core-to-Core Program, JPJSCCB20220010 (to T.U.).
The authors declare there are no competing interests.
Conceptualization: T.S.T., M.T., C.M., and T.U.; reagents and specimens: M.T., G.B., H.H., M.K., H.K., and C.M.; data collection: T.S.T., M.T., H.O., C.M., and Y.I.; manuscript writing: T.S.T. and T.U.; supervision: T.U.; funding acquisition: M.T., C.M., and T.U.
Contributor Information
Takamasa Ueno, Email: uenotaka@kumamoto-u.ac.jp.
Mark T. Heise, University of North Carolina at Chapel Hill
REFERENCES
- 1.Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L, Wang X. 2020. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581:215–220. 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 2.Iacobucci G. 2021. Covid-19: New UK variant may be linked to increased death rate, early data indicate. BMJ 372:n230. 10.1136/bmj.n230. [DOI] [PubMed] [Google Scholar]
- 3.Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Fonseca V, Giandhari J, Doolabh D, Pillay S, San EJ, Msomi N, Mlisana K, von Gottberg A, Walaza S, Allam M, Ismail A, Mohale T, Glass AJ, Engelbrecht S, Van Zyl G, Preiser W, Petruccione F, Sigal A, Hardie D, Marais G, Hsiao NY, Korsman S, Davies MA, Tyers L, Mudau I, York D, Maslo C, Goedhals D, Abrahams S, Laguda-Akingba O, Alisoltani-Dehkordi A, Godzik A, Wibmer CK, Sewell BT, Lourenço J, Alcantara LCJ, Kosakovsky Pond SL, Weaver S, Martin D, Lessells RJ, Bhiman JN, Williamson C, de Oliveira T. 2021. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 592:438–443. 10.1038/s41586-021-03402-9. [DOI] [PubMed] [Google Scholar]
- 4.Voloch CM, Silva F, de Almeida LGP, Cardoso CC, Brustolini OJ, Gerber AL, Guimarães A, Mariani D, Costa R, Ferreira OC, Cavalcanti AC, Frauches TS, de Mello CMB, Galliez RM, Faffe DS, Castiñeiras TMPP, Tanuri A, de Vasconcelos ATR. 2020. Genomic characterization of a novel SARS-CoV-2 lineage from Rio de Janeiro, Brazil. medRxiv. https://www.medrxiv.org/content/10.1101/2020.12.23.20248598v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mlcochova P, Kemp SA, Dhar MS, Papa G, Meng B, Ferreira IATM, Datir R, Collier DA, Albecka A, Singh S, Pandey R, Brown J, Zhou J, Goonawardane N, Mishra S, Whittaker C, Mellan T, Marwal R, Datta M, Sengupta S, Ponnusamy K, Radhakrishnan VS, Abdullahi A, Charles O, Chattopadhyay P, Devi P, Caputo D, Peacock T, Wattal C, Goel N, Satwik A, Vaishya R, Agarwal M, Mavousian A, Lee JH, Bassi J, Silacci-Fegni C, Saliba C, Pinto D, Irie T, Yoshida I, Hamilton WL, Sato K, Bhatt S, Flaxman S, James LC, Corti D, Piccoli L, Barclay WS, Rakshit P, CITIID-NIHR BioResource COVID-19 Collaboration , et al. 2021. SARS-CoV-2 B.1.617.2 delta variant replication and immune evasion. Nature 599:114–119. 10.1038/s41586-021-03944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meng B, Abdullahi A, Ferreira IATM, Goonawardane N, Saito A, Kimura I, Yamasoba D, Gerber PP, Fatihi S, Rathore S, Zepeda SK, Papa G, Kemp SA, Ikeda T, Toyoda M, Tan TS, Kuramochi J, Mitsunaga S, Ueno T, Shirakawa K, Takaori-Kondo A, Brevini T, Mallery DL, Charles OJ, Baker S, Dougan G, Hess C, Kingston N, Lehner PJ, Lyons PA, Matheson NJ, Ouwehand WH, Saunders C, Summers C, Thaventhiran JED, Toshner M, Weekes MP, Maxwell P, Shaw A, Bucke A, Calder J, Canna L, Domingo J, Elmer A, Fuller S, Harris J, Hewitt S, Kennet J, Jose S, Kourampa J, The CITIID-NIHR BioResource COVID-19 Collaboration ., et al. 2022. Altered TMPRSS2 usage by SARS-CoV-2 omicron impacts tropism and fusogenicity. Nature 603:706–714. 10.1038/s41586-022-04474-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tchesnokova V, Kulasekara H, Larson L, Bowers V, Rechkina E, Kisiela D, Sledneva Y, Choudhury D, Maslova I, Deng K, Kutumbaka K, Geng H, Fowler C, Greene D, Ralston J, Samadpour M, Sokurenko E. 2021. Acquisition of the L452R mutation in the ACE2-binding interface of Spike protein triggers recent massive expansion of SARS-CoV-2 variants. J Clin Microbiol 59:e00921-21. 10.1128/JCM.00921-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng X, Garcia-Knight MA, Khalid MM, Servellita V, Wang C, Morris MK, Sotomayor-González A, Glasner DR, Reyes KR, Gliwa AS, Reddy NP, Sanchez San Martin C, Federman S, Cheng J, Balcerek J, Taylor J, Streithorst JA, Miller S, Sreekumar B, Chen P-Y, Schulze-Gahmen U, Taha TY, Hayashi JM, Simoneau CR, Kumar GR, McMahon S, Lidsky PV, Xiao Y, Hemarajata P, Green NM, Espinosa A, Kath C, Haw M, Bell J, Hacker JK, Hanson C, Wadford DA, Anaya C, Ferguson D, Frankino PA, Shivram H, Lareau LF, Wyman SK, Ott M, Andino R, Chiu CY. 2021. Transmission, infectivity, and neutralization of a spike L452R SARS-CoV-2 variant. Cell 184:3426–3437.e8. 10.1016/j.cell.2021.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang W, Davis BD, Chen SS, Sincuir Martinez JM, Plummer JT, Vail E. 2021. Emergence of a novel SARS-CoV-2 variant in southern California. JAMA 325:1324–1326. 10.1001/jama.2021.1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu L, Sikkema RS, Velkers FC, Nieuwenhuijse DF, Fischer EAJ, Meijer PA, Bouwmeester-Vincken N, Rietveld A, Wegdam-Blans MCA, Tolsma P, Koppelman M, Smit LAM, Hakze-van der Honing RW, van der Poel WHM, van der Spek AN, Spierenburg MAH, Molenaar RJ, Rond J, Augustijn M, Woolhouse M, Stegeman JA, Lycett S, Oude Munnink BB, Koopmans MPG. 2021. Adaptation, spread and transmission of SARS-CoV-2 in farmed minks and associated humans in the Netherlands. Nat Commun 12:6802. 10.1038/s41467-021-27096-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Munnink BBO, Sikkema RS, Nieuwenhuijse DF, Molenaar RJ, Munger E, Molenkamp R, Spek A, Tolsma P, Rietveld A, Brouwer M, Bouwmeester-Vincken N, Harders F, Honing RH-v, Wegdam-Blans MCA, Bouwstra RJ, GeurtsvanKessel C, Eijk A, Velkers FC, Smit LAM, Stegeman A, Poel W, Koopmans MPG. 2021. Transmission of SARS-CoV-2 on mink farms between humans and mink and back to humans. Science 371:172–177. 10.1126/science.abe5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoffmann M, Zhang L, Krüger N, Graichen L, Kleine-Weber H, Hofmann-Winkler H, Kempf A, Nessler S, Riggert J, Winkler MS, Schulz S, Jäck HM, Pöhlmann S. 2021. SARS-CoV-2 mutations acquired in mink reduce antibody-mediated neutralization. Cell Rep 35:109017. 10.1016/j.celrep.2021.109017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kimura I, Kosugi Y, Wu J, Zahradnik J, Yamasoba D, Butlertanaka EP, Tanaka YL, Uriu K, Liu Y, Morizako N, Shirakawa K, Kazuma Y, Nomura R, Horisawa Y, Tokunaga K, Ueno T, Takaori-Kondo A, Schreiber G, Arase H, Motozono C, Saito A, Nakagawa S, Sato K. 2022. The SARS-CoV-2 lambda variant exhibits enhanced infectivity and immune resistance. Cell Rep 38:110218. 10.1016/j.celrep.2021.110218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCallum M, Walls AC, Sprouse KR, Bowen JE, Rosen LE, Dang HV, Marco AD, Franko N, Tilles SW, Logue J, Miranda MC, Ahlrichs M, Carter L, Snell G, Pizzuto MS, Chu HY, Voorhis WCV, Corti D, Veesler D. 2021. Molecular basis of immune evasion by the delta and kappa SARS-CoV-2 variants. Science 374:1621–1626. 10.1126/science.abl8506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaleta T, Kern L, Hong SL, Hölzer M, Kochs G, Beer J, Schnepf D, Schwemmle M, Bollen N, Kolb P, Huber M, Ulferts S, Weigang S, Dudas G, Wittig A, Jaki L, Padane A, Lagare A, Salou M, Ozer EA, Nnaemeka N, Odoom JK, Rutayisire R, Benkahla A, Akoua-Koffi C, Ouedraogo A-S, Simon-Lorière E, Enouf V, Kröger S, Calvignac-Spencer S, Baele G, Panning M, Fuchs J. 2022. Antibody escape and global spread of SARS-CoV-2 lineage A.27. Nat Commun 13:1152. 10.1038/s41467-022-28766-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Planas D, Veyer D, Baidaliuk A, Staropoli I, Guivel-Benhassine F, Rajah MM, Planchais C, Porrot F, Robillard N, Puech J, Prot M, Gallais F, Gantner P, Velay A, Le Guen J, Kassis-Chikhani N, Edriss D, Belec L, Seve A, Courtellemont L, Péré H, Hocqueloux L, Fafi-Kremer S, Prazuck T, Mouquet H, Bruel T, Simon-Lorière E, Rey FA, Schwartz O. 2021. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 596:276–280. 10.1038/s41586-021-03777-9. [DOI] [PubMed] [Google Scholar]
- 17.Shen X, Tang H, Pajon R, Smith G, Glenn GM, Shi W, Korber B, Montefiori DC. 2021. Neutralization of SARS-CoV-2 variants B.1.429 and B.1.351. N Engl J Med 384:2352–2354. 10.1056/NEJMc2103740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tada T, Zhou H, Dcosta BM, Samanovic MI, Mulligan MJ, Landau NR. 2021. Partial resistance of SARS-CoV-2 Delta variants to vaccine-elicited antibodies and convalescent sera. iScience 24:103341. 10.1016/j.isci.2021.103341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Q, Wu J, Nie J, Zhang L, Hao H, Liu S, Zhao C, Zhang Q, Liu H, Nie L, Qin H, Wang M, Lu Q, Li X, Sun Q, Liu J, Zhang L, Li X, Huang W, Wang Y. 2020. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 182:1284–1294.e9. 10.1016/j.cell.2020.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao A, Chen Z, Amitai A, Doelger J, Mallajosyula V, Sundquist E, Pereyra Segal F, Carrington M, Davis MM, Streeck H, Chakraborty AK, Julg B. 2021. Learning from HIV-1 to predict the immunogenicity of T cell epitopes in SARS-CoV-2. iScience 24:102311. 10.1016/j.isci.2021.102311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kared H, Redd AD, Bloch EM, Bonny TS, Sumatoh H, Kairi F, Carbajo D, Abel B, Newell EW, Bettinotti MP, Benner SE, Patel EU, Littlefield K, Laeyendecker O, Shoham S, Sullivan D, Casadevall A, Pekosz A, Nardin A, Fehlings M, Tobian AA, Quinn TC. 2021. SARS-CoV-2-specific CD8+ T cell responses in convalescent COVID-19 individuals. J Clin Invest 131:e145476. 10.1172/JCI145476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu C, Shen M, Han X, Chen Q, Li L, Chen S, Zhang J, Gao F, Wang W, Wang Y, Li T, Li S, Huang J, Wang J, Zhu J, Chen D, Wu Q, Tao K, Pang D, Jin A. 2022. Identification of cross-reactive CD8+ T cell receptors with high functional avidity to a SARS-CoV-2 immunodominant epitope and its natural mutant variants. Genes Dis 9:216–229. 10.1016/j.gendis.2021.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Motozono C, Toyoda M, Zahradnik J, Saito A, Nasser H, Tan TS, Ngare I, Kimura I, Uriu K, Kosugi Y, Yue Y, Shimizu R, Ito J, Torii S, Yonekawa A, Shimono N, Nagasaki Y, Minami R, Toya T, Sekiya N, Fukuhara T, Matsuura Y, Schreiber G, Ikeda T, Nakagawa S, Ueno T, Sato K, Genotype to Phenotype Japan (G2P-Japan) Consortium . 2021. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 29:1124–1136.e11. 10.1016/j.chom.2021.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou Z, Du P, Yu M, Baptista-Hon DT, Miao M, Xiang AP, Lau JY-N, Li N, Xiong X, Huang H, Liu Z, Dai Q, Zhu J, Wu S, Li G, Zhang K, Group C-II, COVID-19 Immunity Investigation Group . 2021. Assessment of infectivity and the impact on neutralizing activity of immune sera of the COVID-19 variant, CAL.20C. Signal Transduction and Targeted Therapy 6:285. 10.1038/s41392-021-00695-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vargas-Herrera N, Araujo-Castillo RV, Mestanza O, Galarza M, Rojas-Serrano N, Solari-Zerpa L. 2022. SARS-CoV-2 Lambda and Gamma variants competition in Peru, a country with high seroprevalence. Lancet Reg Health Am 6:100112. 10.1016/j.lana.2021.100112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.World Health Organization. 2022. Tracking SARS-CoV-2 variants. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 27.Cao Y, Yisimayi A, Jian F, Song W, Xiao T, Wang L, Du S, Wang J, Li Q, Chen X, Wang P, Zhang Z, Liu P, An R, Hao X, Wang Y, Feng R, Sun H, Zhao L, Zhang W, Zhao D, Zheng J, Yu L, Li C, Zhang N, Wang R, Niu X, Yang S, Song X, Zheng L, Li Z, Gu Q, Shao F, Huang W, Jin R, Shen Z, Wang Y, Wang X, Xiao J, Xie XS. 2022. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. bioRxiv. https://www.biorxiv.org/content/10.1101/2022.04.30.489997v2. [DOI] [PMC free article] [PubMed]
- 28.Hou W. 2020. Characterization of codon usage pattern in SARS-CoV-2. Virol J 17:138. 10.1186/s12985-020-01395-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Balasco N, Damaggio G, Esposito L, Villani F, Berisio R, Colonna V, Vitagliano L. 2021. A global analysis of conservative and non-conservative mutations in SARS-CoV-2 detected in the first year of the COVID-19 world-wide diffusion. Sci Rep 11:24495. 10.1038/s41598-021-04147-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morgan AA, Rubenstein E. 2013. Proline: the distribution, frequency, positioning, and common functional roles of proline and polyproline sequences in the human proteome. PLoS One 8:e53785. 10.1371/journal.pone.0053785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wedemeyer WJ, Welker E, Narayan M, Scheraga HA. 2000. Disulfide bonds and protein folding. Biochemistry 39:4207–4216. 10.1021/bi992922o. [DOI] [PubMed] [Google Scholar]
- 32.Bass J, Turck C, Rouard M, Steiner DF. 2000. Furin-mediated processing in the early secretory pathway: sequential cleavage and degradation of misfolded insulin receptors. Proc Natl Acad Sci USA 97:11905–11909. 10.1073/pnas.97.22.11905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ozono S, Zhang Y, Ode H, Sano K, Tan TS, Imai K, Miyoshi K, Kishigami S, Ueno T, Iwatani Y, Suzuki T, Tokunaga K. 2021. SARS-CoV-2 D614G spike mutation increases entry efficiency with enhanced ACE2-binding affinity. Nat Commun 12:848. 10.1038/s41467-021-21118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pal D. 2021. Spike protein fusion loop controls SARS-CoV-2 fusogenicity and infectivity. J Struct Biol 213:107713. 10.1016/j.jsb.2021.107713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teeranaipong P, Hosoya N, Kawana-Tachikawa A, Fujii T, Koibuchi T, Nakamura H, Koga M, Kondo N, Gao GF, Hoshino H, Matsuda Z, Iwamoto A. 2013. Development of a rapid cell-fusion-based phenotypic HIV-1 tropism assay. J Int AIDS Soc 16:18723–18723. 10.7448/IAS.16.1.18723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang J, Cai Y, Xiao T, Lu J, Peng H, Sterling SM, Walsh RM, Rits-Volloch S, Zhu H, Woosley AN, Yang W, Sliz P, Chen B. 2021. Structural impact on SARS-CoV-2 spike protein by D614G substitution. Science 372:525–530. 10.1126/science.abf2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Liu C, Zhang C, Wang Y, Hong Q, Xu S, Li Z, Yang Y, Huang Z, Cong Y. 2022. Structural basis for SARS-CoV-2 Delta variant recognition of ACE2 receptor and broadly neutralizing antibodies. Nat Commun 13:871. 10.1038/s41467-022-28528-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang M, Zhang L, Li Q, Wang B, Liang Z, Sun Y, Nie J, Wu J, Su X, Qu X, Y L, Wang Y, Huang W. 2022. Reduced sensitivity of the SARS-CoV-2 Lambda variant to monoclonal antibodies and neutralizing antibodies induced by infection and vaccination. Emerg Microbes Infect 11:18–29. 10.1080/22221751.2021.2008775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamasoba D, Kimura I, Nasser H, Morioka Y, Nao N, Ito J, Uriu K, Tsuda M, Zahradnik J, Shirakawa K, Suzuki R, Kishimoto M, Kosugi Y, Kobiyama K, Hara T, Toyoda M, Tanaka YL, Butlertanaka EP, Shimizu R, Ito H, Wang L, Oda Y, Orba Y, Sasaki M, Nagata K, Yoshimatsu K, Asakura H, Nagashima M, Sadamasu K, Yoshimura K, Kuramochi J, Seki M, Fujiki R, Kaneda A, Shimada T, Nakada T-a, Sakao S, Suzuki T, Ueno T, Takaori-Kondo A, Ishii KJ, Schreiber G, Sawa H, Saito A, Irie T, Tanaka S, Matsuno K, Fukuhara T, Ikeda T, Sato K. 2022. Virological characteristics of the SARS-CoV-2 Omicron BA.2 spike. Cell 185:2103–2115. 10.1016/j.cell.2022.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kiyotani K, Toyoshima Y, Nemoto K, Nakamura Y. 2020. Bioinformatic prediction of potential T cell epitopes for SARS-Cov-2. J Hum Genet 65:569–575. 10.1038/s10038-020-0771-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mahiti M, Toyoda M, Jia X, Kuang XT, Mwimanzi F, Mwimanzi P, Walker BD, Xiong Y, Brumme ZL, Brockman MA, Ueno T. 2016. Relative Resistance of HLA-B to Downregulation by Naturally Occurring HIV-1 Nef Sequences. mBio 7:e01516-15–e01515. 10.1128/mBio.01516-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ode H, Nakata Y, Nagashima M, Hayashi M, Yamazaki T, Asakura H, Suzuki J, Kubota M, Matsuoka K, Matsuda M, Mori M, Sugimoto A, Imahashi M, Yokomaku Y, Sadamasu K, Iwatani Y. 2022. Molecular epidemiological features of SARS-CoV-2 in Japan. Virus Evol 8:veac034. 10.1093/ve/veac034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2: approximately maximum-likelihood trees for large alignments. PLoS One 5:e0009490. 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199. 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 45.Ozono S, Zhang Y, Tobiume M, Kishigami S, Tokunaga K. 2020. Super-rapid quantitation of the production of HIV-1 harboring a luminescent peptide tag. J Biol Chem 295:13023–13030. 10.1074/jbc.RA120.013887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ikeda T, Symeonides M, Albin JS, Li M, Thali M, Harris RS. 2018. HIV-1 adaptation studies reveal a novel Env-mediated homeostasis mechanism for evading lethal hypermutation by APOBEC3G. PLoS Pathog 14:e1007010. 10.1371/journal.ppat.1007010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kondo N, Miyauchi K, Matsuda Z. 2011. Monitoring viral-mediated membrane fusion using fluorescent reporter methods. Curr Protoc Cell Biol Chapter 26:Unit 26.9. 10.1002/0471143030.cb2609s50. [DOI] [PubMed] [Google Scholar]
- 48.Tan TS, Toyoda M, Tokunaga K, Ueno T. 2021. Aromatic side chain at position 412 of SERINC5 exerts restriction activity toward HIV-1 and other retroviruses. J Virol 95:e00634-21. 10.1128/JVI.00634-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Motozono C, Toyoda M, Tan TS, Hamana H, Goto Y, Aritsu Y, Miyashita Y, Oshiumi H, Nakamura K, Okada S, Udaka K, Kitamatsu M, Kishi H, Ueno T. 2022. The SARS-CoV-2 Omicron BA.1 spike G446S mutation potentiates antiviral T-cell recognition. Nat Commun 13:5440. 10.1038/s41467-022-33068-4. [DOI] [PMC free article] [PubMed] [Google Scholar]