

ABSTRACT
Saccharomyces cerevisiae, whose evolutionary past includes a whole-genome duplication event, is characterized by a mosaic genome configuration with substantial apparent genetic redundancy. This apparent redundancy raises questions about the evolutionary driving force for genomic fixation of “minor” paralogs and complicates modular and combinatorial metabolic engineering strategies. While isoenzymes might be important in specific environments, they could be dispensable in controlled laboratory or industrial contexts. The present study explores the extent to which the genetic complexity of the central carbon metabolism (CCM) in S. cerevisiae, here defined as the combination of glycolysis, the pentose phosphate pathway, the tricarboxylic acid cycle, and a limited number of related pathways and reactions, can be reduced by elimination of (iso)enzymes without major negative impacts on strain physiology. Cas9-mediated, groupwise deletion of 35 of the 111 genes yielded a “minimal CCM” strain which, despite the elimination of 32% of CCM-related proteins, showed only a minimal change in phenotype on glucose-containing synthetic medium in controlled bioreactor cultures relative to a congenic reference strain. Analysis under a wide range of other growth and stress conditions revealed remarkably few phenotypic changes from the reduction of genetic complexity. Still, a well-documented context-dependent role of GPD1 in osmotolerance was confirmed. The minimal CCM strain provides a model system for further research into genetic redundancy of yeast genes and a platform for strategies aimed at large-scale, combinatorial remodeling of yeast CCM.
KEYWORDS: Saccharomyces cerevisiae, central carbon metabolism, minimal genome, genetic redundancy
INTRODUCTION
The fundamental challenge of defining the minimum complement of genes required for life has been addressed by theoretical as well as experimental approaches. Bottom-up and top-down strategies have mainly focused on bacteria with small genomes (1–9). Their larger genome sizes might appear to make eukaryotic microorganisms less relevant for this type of research. However, they do offer attractive models to explore the biological significance of (apparent) genetic redundancy. Different evolutionary advantages have been proposed for the fixation of duplicated genes in genomes, including provision of a molecular landscape for functional (minor or major) innovation (e.g., neo- and subfunctionalization), a functional backup, gene dosage effects, or increased buffering to respond to environmental cues (10, 11). Systematically identifying the physiological significance underlying gene fixation presents a daunting challenge.
With its relatively small genome (12 Mb), tractability, and high genetic accessibility, the yeast Saccharomyces cerevisiae is a valuable model for fundamental research on minimal genetic requirements. S. cerevisiae underwent a whole-genome duplication (WGD) approximately 100 million years ago, as well as smaller-scale duplication (SSD) events. While 90% of the WGD genes were lost during evolution, some duplicates remain (11). As observed in humans, a substantial fraction of the total gene duplicates in S. cerevisiae originates from a WGD (approximately 63% of duplicates in the S. cerevisiae genome and 62% in the human genome), while a smaller fraction originates from SSD events (approximately 37% of duplicates in the S. cerevisiae genome and 38% in the human genome) (12, 13). While systematic, large-scale studies like the construction of the yeast deletion collections (14–17), the synthetic genetic array projects (18–20), or the recent SCRaMbLE-based genome compaction (21), have provided valuable information on the dispensability of (a combination of) genes, the physiological roles of many of these paralogous genes remain poorly defined.
In addition to the fundamental scientific questions raised by genetic redundancy, it also complicates genome engineering of S. cerevisiae. The conversion of substrate into product via native or engineered pathways relies on the microbial host core pathways for the supply of metabolic precursors, energy-rich molecules, and redox equivalents. These biochemical reactions are catalyzed by sets of “metabolic” genes that are characterized by a high genetic redundancy in eukaryotes (11). Not only has the physiological role of many paralogous genes not been fully elucidated, but also the manipulation of specific biochemical reactions is hindered by the presence of multiple paralogous genes with poorly known functions that are scattered over the 12-Mb, mosaic yeast genome and its 16 chromosomes. Additionally, expression of these redundant genes dissipates cellular resources (e.g., carbon, energy) that might be better invested in industrially relevant properties, such as high product yield or cellular robustness to the stressful environment of large-scale fermentation.
To tackle these fundamental and applied challenges, taking glycolysis and ethanolic fermentation as starting points, Solis-Escalante and colleagues pioneered the genetic reduction of central carbon metabolism in S. cerevisiae. The set of 26 genes encoding the (iso)enzymes catalyzing 12 reactions was reduced to 13 genes (22). Remarkably, this 50% genetic reduction did not result in any visible phenotypic effect, although a wide range of growth conditions was tested. These observations argued against gene dosage being a strong driving force in the evolution of Crabtree-positive yeasts (11, 22) and raised questions on the mechanisms involved in the fixation of these gene duplicates in the S. cerevisiae genome. A recent study suggested that the role of the redundant paralogs might be highly context dependent and that some relevant conditions were not tested by Solis-Escalante and coworkers (22) (e.g., the role of pyruvate kinase 2 in the utilization of three-carbon substrates such as dihydroxyacetone [23]). The surprising lack of phenotype of the “minimal glycolysis” yeast strain (called MG) enabled the construction of a genetically simplified version of the glycolytic pathway, which was subsequently relocalized to a single chromosomal locus (24). The resulting yeast strain with a single locus for glycolysis presents a powerful tool to remodel the glycolytic pathway in two single steps into any redesigned (heterologous) version.
Glycolysis is an important but small part of central carbon metabolism (CCM), a set of reactions required for the conversion of carbon feedstocks into any industrially relevant product (Fig. 1). For cells, CCM is primarily the set of reactions that convert carbon sources into the 12 building blocks required for the synthesis of cellular components (25). CCM encompasses ~111 genes, with 66% duplicates (Fig. 2). Reducing its genetic complexity would be the first step in an attempt to construct a modular, designer yeast genome, with a single-locus CCM, as previously achieved for glycolysis and fermentation. Modular, specialized synthetic chromosomes could be ideal platforms for the centralization of the CCM genes (26).
FIG 1.
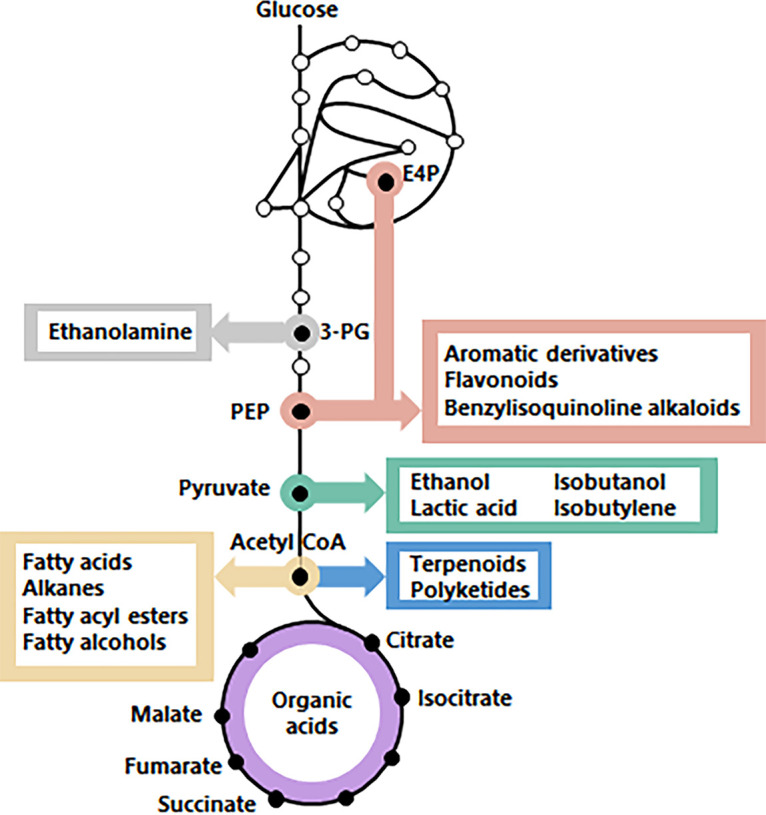
CCM precursors of industrially relevant chemicals. 3-PG, 3-phosphoglycerate; PEP, phosphoenolpyruvate; E4P, erythrose-4-phosphate.
FIG 2.

Reactions of the CCM of S. cerevisiae considered for genetic reduction in this study, including subprocesses and pathways in CCM metabolic pathways. Enzyme-catalyzed conversions (black lines) and transport processes (dotted lines) are shown between intermediates and through mitochondrial transporters (circles and ovals), respectively. Directionality and reversibility of reactions were based on information from the Yeast Pathways database (https://pathway.yeastgenome.org/). Enzyme localization was based on literature information. Genes retained in the genetic reduction strategy are shown in black, and genes selected for deletion in the minimal CCM strain are indicated in bold and underlined. Occurrence of pathways in different cellular compartments is shown by gray borders. Simplifications have been made for visualization reasons, for example, H2O and inorganic phosphate are not shown. 6P-GLCN-lac, 6-phosphogluconolactone; 6P-GLCN, 6-phosphoglucononate; RL5P, ribulose 5-phosphate; R5P, ribose 5-phosphate; XUL-5P, xylulose 5-phosphate; Fru-6P, fructose-6-phosphate; Ery-4P, erythrose 4-phosphate; S7P, sedoheptulose 7-phosphate; GAP, glyceraldehyde-3-phosphate; Glc, glucose; Glc-6P, glucose-6-phosphate; Fru-1,6-BP, fructose-1,6-bisphosphate; DHAP, dihydroxyacetone phosphate; 1,3-BPG, 1,3-bisphosphoglycerate; 3-PG, 3-phosphoglycerate; 2-PG, 2-phosphoglycerate; PEP, phosphoenolpyruvate; Pyr, pyruvate; AcAl, acetaldehyde; EtOH, ethanol; OAA, oxaloacetate; Cit, citrate; Cis-Aco, cis-aconitate; Isocit, isocitrate; α-KG, α-ketoglutarate; Suc-CoA, succinyl-CoA; Suc, succinate; Fum, fumarate; Mal, malate; Glyox, glyoxylate; Ace, acetate; Ac-CoA, acetyl-CoA; Glyc-3P, glycerol-3-phosphate.
The main goal of this study was to explore the extent to which the number of genes encoding CCM enzymes in S. cerevisiae can be reduced without substantially affecting fitness under a set of chosen growth conditions. To this end, redundancies were first predicted based on data in the literature on gene expression, enzyme activities, and phenotypes of (single) deletion mutants. Subsequently, phenotypes of mutants with mutations in sets of genes encoding CCM enzymes were tested under a wider range of growth conditions. In this first attempt of genetic reduction of yeast CCM at this scale, special attention was given to possible synergistic effects of mutations that were previously studied in separate strains.
RESULTS
Genetic reduction strategy.
In this study, the CCM of S. cerevisiae was defined as the set of biochemical reactions encompassed by glycolysis, ethanolic fermentation, pentose-phosphate pathway (PPP), acetyl-coenzyme A (CoA) synthesis, tricarboxylic acid cycle (TCA), anaplerosis, gluconeogenesis, glyoxylate cycle, and glycerol metabolism. As CCM reactions occur in multiple compartments, mitochondrial transporters were also considered (Fig. 2). Transport through the peroxisomal membrane was not considered, as this phenomenon is poorly studied (27). For the construction of a minimal CCM strain, decisions to remove or retain genes were based on (i) transcript levels from an expression compendium encompassing 170 different cultivation conditions (28), (ii) enzyme activities in cell extracts of mutant strains when data were available, and (iii) reported phenotypes of null mutants. Genes encoding proteins with reported secondary (“moonlighting”) functions or proteins known to cause auxotrophy upon deletion were retained (29).
Genes were classified as functionally redundant when at least 75% of the specific growth rate of the congenic reference strains CEN.PK113-7D (Ura+) or IMX581 (Ura−) was retained during aerobic batch cultivation on synthetic medium supplied with either glucose or ethanol. Ethanol-grown cultures were included because, in contrast to glucose, ethanol can only be dissimilated by respiration and because its metabolism involves different sets of CCM enzymes and transporters. In addition, testing for growth on ethanol ensured that the intensive engineering undergone by the strains, including removal of several mitochondrial proteins, did not cause respiratory deficiency.
The previously constructed MG strain, in which 13 out of the 26 existing paralogs of genes encoding glycolytic enzymes and fermentation enzymes were deleted without the detection of major phenotypes, was used as a starting point of the present CCM reduction endeavor. To identify any synergistic effects between the newly introduced deletions and the 13 deletions already present in the MG strain, a congenic naive reference strain (IMX581) with a full complement of glycolytic and fermentation genes was also used in parallel to MG for serial deletions. To accelerate the deletion workflow, genes involved in individual pathways or processes were deleted in sets of two to four (Fig. 3). When a substantial loss of fitness was observed, the contribution of individual deletions was dissected by constructing additional strains with various combinations and numbers of deletions.
FIG 3.

Deletion strategy and specific growth rates of resulting strains. (A) Workflow for construction of relevant S. cerevisiae strains. (B) The strains’ respective specific growth rates, measured as shake flask growth rate on synthetic medium with glucose (SMD) or ethanol (SME) as carbon source, supplemented with uracil. Specific growth rates represent averages and standard deviations of measurements on independent duplicate cultures for each strain and are expressed as a percentage of specific growth rate of the naive uracil-auxotrophic reference strain S. cerevisiae IMX581 or the naive uracil-prototrophic reference strain S. cerevisiae CEN.PK113-7D. Significant differences in specific growth rates relative to the control strain are indicated with an asterisk (P < 0.05; two-tailed paired homoscedastic t test).
Deletion of 35 CCM genes had minimal impact on specific growth rates on chemically defined glucose medium.
Pentose-phosphate pathway.
The pentose-phosphate pathway reduces cellular NADP+, generates ribose-5-phosphate and erythrose-4-phosphate for nucleic acid and amino acid synthesis and, in strains engineered for pentose fermentation, acts as a dissimilatory pathway (30).
Four of the seven reactions in the PPP are catalyzed by pairs of isoenzymes encoded by WGD paralogs, for which sequence similarities ranged from 47% (SOL3 and SOL4) to 87% (GND1 and GND2) (see Table S1 in the supplemental material). Based on transcript levels across a wide range of cultivation conditions (28), SOL4, GND2, TKL2, and NQM1 were considered minor paralogs. Moreover, deletion of TKL2 and NQM1 was previously reported not to affect growth on glucose synthetic medium (31–33). While similar in vitro enzyme activities were reported for Sol3 and Sol4 in cell extracts (34), SOL4 was deleted based on its consistently lower transcript level (28). Simultaneous deletion of GND2, TKL2, SOL4, and NQM1 in the naive reference strain or in the MG strain, while retaining SOL3, GND1, TKL1, and TAL1, did not significantly affect growth rate on either synthetic medium with 2% (wt/vol) glucose (SMD) or 2% (vol/vol) ethanol (SME) as carbon source (strains IMX1592 [pppmin] and IMX1591 [CCMin1, glycmin fermin pppmin]) (pppmin, minimized pentose phosphate pathway; glycmin, minimized glycolysis; fermin, minimized ethanolic fermentation) (Fig. 3). Previously reported extended lag phase and slower growth on ethanol of sol4 null mutants (35) were not observed. This difference may have been related to the use of different S. cerevisiae strain backgrounds.
Genetic characteristics of the paralogs considered for deletion. Download Table S1, PDF file, 0.1 MB (144.9KB, pdf) .
Copyright © 2022 Postma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Tricarboxylic acid cycle, anaplerotic reactions, and gluconeogenesis.
In addition to its dissimilatory role in oxidizing acetyl-CoA units to CO2, the TCA cycle supplies precursors, NADH, FADH2, and ATP (36). During growth on fermentable sugars, the TCA cycle is a mitochondrial pathway, with acetyl-CoA resulting from oxidative decarboxylation of pyruvate by the pyruvate dehydrogenase complex. To replenish use of TCA cycle intermediates for biosynthesis, the cycle’s acceptor molecule, oxaloacetate, can be imported from the cytosol, where it is produced by carboxylation of pyruvate. The nine biochemical reactions of the TCA cycle involve 22 mitochondrial enzymes, which show little genetic redundancy. Two reactions are catalyzed by single enzymes, Mdh1 and Fum1, while three steps are catalyzed by complexes of two to five proteins. Deletion of genes encoding individual subunits of the α-ketoglutarate and succinyl-CoA synthetase complexes renders the complexes dysfunctional (37–42). In contrast, the succinate dehydrogenase complex, in which four functions are performed by seven proteins, does show some redundancy. SDH1, SDH3, and SDH4 proteins have the genes SDH1b, SHH3, and SHH4, respectively, as homologs originating from the WGD, while SDH2 is a unique gene (43–48). Deletion of SDH1b, SHH3, and SHH4 has a minor or no effect on complex integrity and yeast physiology, and these genes are considered functionally redundant (45, 48, 49). Citrate synthase (Cit1 and Cit3) and isocitrate dehydrogenase (Idh1, Idh2, and Idp1) have functionally redundant mitochondrial enzymes. Based on expression data and lack of a phenotypic difference during fermentative and respiratory growth, Cit3 and the NADP+-dependent Idp1 are considered redundant (28, 50–52). Single deletion of ACO1 or ACO2, which encode aconitase isoenzymes, causes amino acid auxotrophies (53, 54). Idh1 and Idh2 are part of a complex and are both required for isocitrate dehydrogenase activity (37, 38). Based on this information, only 5 of the 22 TCA cycle mitochondrial proteins were considered functionally redundant and, therefore, selected as candidates for elimination: Cit3, Idp1, Sdh1b, Shh3, and Shh4. IDP1 was targeted in a later deletion round, along with extramitochondrial paralogs of the TCA cycle that are part of the glyoxylate cycle.
There are several enzymes that form an interface between the TCA cycle and glycolysis. The WGD paralog pair PYC1 and PYC2 encodes isoenzymes of the anaplerotic enzyme pyruvate carboxylase. Transcript levels of these two highly similar genes (92%) (see Table S1) are condition dependent, and despite some conflicting reports on the physiological impact of PYC1 and PYC2 deletion (28, 55–57), one study showed that only deletion of PYC1 leads to aspartate auxotrophy (58). PYC2 was therefore deleted. Deletion of MAE1, which encodes a mitochondrial malic enzyme catalyzing the oxidative decarboxylation of malate to pyruvate, does not show a clear phenotype. However, double deletion of MAE1 and PYK2 reduces the specific growth rate on ethanol by 62% (58). As PYK2 was deleted in the MG strain, MAE1 was retained. The gluconeogenic enzymes phosphoenolpyruvate carboxykinase (Pck1) and fructose-1,6-bisphosphatase (Fbp1) are essential for bypassing the irreversible pyruvate kinase and phosphofructokinase reactions, respectively, during growth on nonfermentable carbon sources (59, 60).
CIT3, SDH1b, SHH3, SHH4, and PYC2 were deleted in two consecutive transformation rounds in the naive reference strain and the CCMin1 strain (glycmin fermin pppmin), resulting in IMX1805 (tcamin) and IMX1806 (CCMin2, glycmin fermin pppmin tcamin), respectively. Both strains grew as well as their parental strains in chemically defined medium supplemented with glucose or ethanol (Fig. 3).
Mitochondrial carriers.
The 35 nuclearly encoded mitochondrial carriers (MCs) mediate transport of numerous metabolites, nucleotides, cofactors, and inorganic anions between the mitochondrial matrix and cytosol (61). Based on extensive functional analysis studies (62, 63), 19 MCs involved in transport of pyruvate, TCA cycle intermediates CoA, ADP, ATP, Pi, NAD+, FAD+, and thiamine pyrophosphate (a cofactor of pyruvate dehydrogenase and α-ketoglutarate dehydrogenase) were considered part of CCM (Fig. 2; see also Table S2). Potential genetic redundancy was identified for 10 of these MCs, with protein sequence similarity varying between 51 and 87% (see Table S1). In addition to genetic redundancy, functional redundancy has to be considered, since several genetically distinct transporters can transport the same solutes, as exemplified by the antiport of ADP and ATP across the mitochondrial membrane by three Aac isoforms as well as by Sal1. Aac2 and Aac3 originate from WGD, while Aac1 does not. Sal1 shares no homology with the Aac carriers and harbors an additional Ca2+-binding domain (64, 65). Several studies indicate Aac2 as a major paralog whose presence suffices to sustain adenine nucleotide transport during respiratory growth (28, 64–69). AAC1, AAC3, and SAL1 were therefore all candidates for deletion. NAD+, synthesized in the cytosol and required for the NAD+-dependent mitochondrial dehydrogenases in CCM, is imported by two MCs encoded by NDT1 and NDT2, paralogs with 51% similarity at the protein level (see Table S1). NDT1 and NDT2 are individually dispensable for growth on glucose or ethanol, but deletion of both precludes growth on nonfermentable carbon sources (70). Therefore, only one of the paralogs, NDT2, was chosen for deletion. Since import of FAD+, CoA, and thiamine pyrophosphate is crucial for mitochondrial activity, the corresponding unique genes (FLX1, LEU5, and TPC1) were retained (71–74).
Mitochondrial carrier proteins. Download Table S2, PDF file, 0.1 MB (148.9KB, pdf) .
Copyright © 2022 Postma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Pyruvate is located at the interface of glycolysis and the TCA cycle and, in addition, mitochondrial pyruvate is required for synthesis of branched-chain amino acids (BCAA). Pyruvate import into mitochondria is mediated by three isoforms: Mpc1, Mpc2, and Mpc3. Mpc1 is constitutively expressed and forms complexes with either of the highly homologous Mpc2 or Mpc3 (75). MPC2 is expressed during fermentative growth, while MPC3 is expressed during respiratory growth. Deletion of MPC2 leads to a severe growth defect, even in glucose-containing medium supplied with BCAA, while MPC3 deletion leads to a modest (20%) decrease of specific growth rates on nonfermentable carbon sources (76, 77). Based on these literature data, it was decided to delete MPC3.
Sfc1 and Dic1 employ different mechanisms to import succinate into mitochondria and, since both are essential for growth on ethanol (78–80), neither was eliminated. Oxaloacetate is mainly transported by Oac1, whose removal only has a minor impact on specific growth rate on glucose medium, which is linked to its secondary function as an exporter of α-isopropylmalate for leucine biosynthesis (81, 82). Since we observed a 26% reduction of the specific growth rate on glucose upon deletion of OAC1 in the CEN.PK genetic background used in this study (see Fig. S1), it was retained in strain construction.
Growth rate of the OAC1 deletion mutant. Maximum specific growth rate of S. cerevisiae CEN.PK113-7D (naive reference strain) and the OAC1 deletion mutant, S. cerevisiae IMK588, tested in shake flasks on selective SMD medium. Growth rates represent averages and standard deviations of two biological duplicates. *, P < 0.05; IMK588 had a 26% slower growth rate with respect to CEN.PK113-7D (two-tailed paired homoscedastic t test). Download FIG S1, PDF file, 0.2 MB (154KB, pdf) .
Copyright © 2022 Postma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Four additional and partially functionally redundant MCs with different transport mechanisms and affinities mediate organic acid transport. Ctp1 is a citrate-malate antiporter, the paralogous carriers Odc1 and Odc2 are α-ketoglutarate and oxodicarboxylate antiporters, and Yhm2 exchanges α-ketoglutarate and citrate, thereby enabling NADPH shuttling between cytosol and mitochondria (involving isocitrate dehydrogenase and aconitase) (83–89). Deletion of CTP1 or double deletion of ODC1 and ODC2 does not affect growth, while triple deletion of YHM2, ODC1, and ODC2 does (84, 85). Based on these literature data, CTP1, ODC1, and ODC2 were selected for deletion, with the realization that their combined deletion might affect di- and tricarboxylic acid trafficking.
In total, 8 MCs were targeted for elimination. First, AAC1, AAC3, SAL1, and MPC3 were simultaneously deleted, followed by simultaneous deletion of NDT2, CTP1, ODC1, and ODC2. Deletion of AAC1, AAC3, SAL1, and MPC3 in the naive reference strain, resulting in strain IMX2360, only marginally affected the specific growth rate on SMD (3 to 5% decrease) but had a stronger impact on growth on SME (14 to 18% slower growth) (Fig. 3). These results are in agreement with the reported roles of these MCs in respiratory growth. When introduced in CCMin2 (glycmin fermin pppmin tcamin), resulting in strain IMX1984, the same set of deletions did not affect the specific growth rate on either SMD or SME.
Combined deletion of NDT2, CTP1, ODC1, and ODC2 reduced the specific growth rate on SMD by 23% in the naive reference strain (resulting in strain IMX2230) and by 30% in IMX1984 (glycmin fermin pppmin tcamin aac1Δ aac3Δ sal1Δ mpc3Δ; resulting in strain IMX2231) (Fig. 3). When instead only NDT2, CTP1, and ODC1 were deleted in the naive reference strain (resulting in strain IMX2404) or in engineered background strain IMX1984 (resulting in strain IMX2407, called CCMin3: glycmin fermin pppmin tcamin mcmin; mcmin is an abbreviation for minimized mitochondrial carriers), the specific growth rate on SMD was not affected and only a small (3 to 7%) reduction of growth rate was observed on SME (Fig. 3; see also Fig. S2). CCMin3 (glycmin fermin pppmin tcamin mcmin) retained 96% of the specific growth rate of the reference strain on SMD and 82% of its specific growth rate on SME.
Growth rates of deletion strains. Maximum specific growth rates of all S. cerevisiae strains tested in shake flasks on selective SMD (A and C) or SME (B and D) supplemented with uracil. Represented are deletion strains in the naive background (A and B) and in the engineered background (C and D). Color coding matches that used in Fig. 2 in the main article. Growth rates represent averages and standard deviations of two biological duplicates. Growth rates of the deletion strains are expressed as percentages of the control strain IMX581 growth rate. Significant changes in growth rate with respect to the control strain are indicated (*, P < 0.05, two-tailed paired homoscedastic t test). Download FIG S2, PDF file, 0.3 MB (266.1KB, pdf) .
Copyright © 2022 Postma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Fumarate reductases, acetyl-CoA synthesis, and glyoxylate cycle.
Cytosolic (Frds1) and mitochondrial (Osm1) fumarate reductases reoxidize FADH2, which has been proposed to be important for protein folding under anaerobic conditions (90–93). Double deletion of FRDS1 and OSM1 has no phenotypic effect on complex glucose medium under aerobic conditions (94). However, Osm1 has a moonlighting function outside CCM, as it contains two translation sites, leading to the targeting to the endoplasmic reticulum of an Osm1 variant. Therefore, only FRDS1 was considered for deletion in the design of a minimal CCM strain.
The glyoxylate cycle, which is essential for providing biosynthetic precursors with more than 2 carbon atoms during growth on fatty acids and two-carbon compounds, encompasses reactions in the peroxisome and cytosol (95, 96) and uses acetyl-CoA as substrate, which is made by the acetyl-CoA synthesis pathway. Ethanol is converted into to acetyl-CoA via alcohol dehydrogenase (already reduced in the MG strain), acetaldehyde dehydrogenases, and acetyl-CoA synthetases. Five acetaldehyde dehydrogenase isoenzymes, Ald2 to Ald6, oxidize acetaldehyde to acetate with either NADP+ or NAD+ as cofactor. The mitochondrial isoenzymes Ald4 and Ald5, required for growth on ethanol (97, 98) and for maintenance of a functional respiratory chain (98), were both retained. Ald6 is the major cytosolic isoenzyme whose elimination strongly affects growth on fermentable and nonfermentable carbon sources (99). The other two cytosolic acetaldehyde dehydrogenases, Ald2 and Ald3, are involved in conversion of 3-aminopropanal to β-alanine for pantothenic acid biosynthesis (100, 101). As single deletion of ALD2 or ALD3 does not affect growth on ethanol or glucose and ALD2 is the major paralog in pantothenic acid production (100, 101), ALD3 was considered for deletion.
Acetate is then converted to acetyl-CoA via Acs1, whose localization is under debate. Acs1 has been reported to occur in the cytosol, nucleus, and peroxisomes, depending on growth conditions (102, 103). Acs1 and its isoenzyme Acs2 are essential for growth on nonfermentable and fermentable carbon sources, respectively (103). The mitochondrial acetyl-CoA hydrolase Ach1 is also able to convert acetate into acetyl-CoA but uses succinyl-CoA as CoA donor. Deletion of ACH1 leads to reduced chronological life span, severe mitochondrial damage, and accumulation of reactive oxygen species (104, 105). ACS1, ACS2, and ACH1 were therefore retained.
The glyoxylate cycle is initiated by Cit2, an extramitochondrial isoenzyme of the mitochondrial Cit1 and Cit3 citrate synthases, whose localization is under debate and has been reported in the cytosol and peroxisome (103, 106). Citrate is then converted into isocitrate in the cytosol by the dually localized enzyme Aco1 in cytosol and mitochondria (107). Via a series of cytosolic and peroxisomal reactions (some localizations under debate), including the isocitrate lyase Icl1 (cytosol) (108, 109), the malate synthase (Mls1 or Mls2, cytosol and peroxisome) (103, 110, 111), and malate dehydrogenase (Mdh2 in cytosol and Mdh3 occurs in the peroxisome) (111), the net synthesis of TCA cycle intermediates is enabled from acetyl-CoA.
Possible redundancies of glyoxylate enzymes also involved in the TCA cycle were discussed above, with only Cit3 selected for elimination in a minimal CCM strain. Since the glyoxylate cycle enzymes Cit2, Mls1, Icl1, and Mdh2 (cytosolic isoenzyme of the mitochondrial Mdh1) are either essential for growth on C2 compounds or their elimination leads to strong reductions in growth rate, they were retained in the minimal CCM design (95, 103, 112–116). The proteins Icl2, Mls2, and Mdh3 are homologous to Icl1, Mls1, and Mdh1 or Mdh2, respectively, but have (additional) functions outside the CCM (113, 117, 118) and were therefore also retained.
The peroxisomes harbor the NADP+-dependent isocitrate dehydrogenase Idp3. Deletion of its mitochondrial homologs Idp1 and Idp2 does not affect growth on ethanol or glucose (51, 52). Idp1 and Idp2 were therefore the only genes considered for elimination in the minimal CCM design.
Triple deletion of FRDS1, IDP1, and IDP2 in the naive reference strain (resulting in strain IMX2470, fummin glyoxmin) (fummin, minimized fumarate reductases; glyoxmin, minimized glyoxylate cycle) did not affect the specific growth rate on SMD and caused a 7% lower growth rate on SME (Fig. 3; see also Fig. S2). Deletion in CCMin3 (IMX2407, glycmin fermin pppmin tcamin mcmin) did not affect the specific growth rate on either SMD or SME (Fig. 3). The resulting strain, CCMin4 (IMX2475, glycmin fermin pppmin tcamin mcmin fummin glyoxmin), retained 94% and 79% of the specific growth rate of the naive reference strain IMX581 on SMD and SME, respectively. For reasons of experimental efficiency, ALD3 was removed in the final deletion round (see below).
Glycerol synthesis.
Glycerol production is essential for redox balancing in anaerobic S. cerevisiae cultures (119). In addition, glycerol plays a key role in osmotolerance and maintenance of cellular volume and turgor pressure during growth under hypertonic conditions (120, 121). The conversion of dihydroxyacetone phosphate to glycerol-3-phosphate is catalyzed by the isoenzymes Gpd1 and Gpd2. gpd1 deletion mutants are osmosensitive but show no growth defects in the absence of stress (122–124). In contrast, gpd2 null mutants show a mild reduction of aerobic growth rates and strongly decreased growth rates under anaerobic conditions (125, 126). Glycerol-3-phosphate is converted into glycerol by the redundant Gpp1 and Gpp2 isoenzymes. Single deletion of either enzyme affected neither osmotolerance nor growth on glucose or ethanol, while gpp1 mutants have been reported to show extended lag phases in anaerobic cultures (127, 128). Therefore, GPD1 and GPP2 were chosen for deletion.
Triple deletion of ALD3, GPD1, and GPP2 did not significantly affect the specific growth rate on SMD, while a small growth rate reduction was observed on SME in both the naive reference strain and the engineered background (IMX2509 [acemin glycerolmin] and IMX2519 [CCMin5, glycmin fermin pppmin tcamin mcmin fummin glyoxmin acemin glycerolmin], respectively) (acemin, minimized acetyl-CoA synthesis; glycerolmin, minimized glycerol synthesis) (Fig. 3). The lower specific growth rate on SME could be attributed to the double deletion of GPD1 and GPP2 (strain IMX2612) (Fig. 3; see also Fig. S2).
The auxotrophic 35-deletion strain IMX2519 (glycmin fermin pppmin tcamin mcmin fummin glyoxmin acemin glycerolmin) grew at 93% and 71% of the specific growth rate of the control strain IMX581 on uracil-supplemented SMD and SME, respectively. Integration of a URA3 cassette yielded the uracil-prototrophic 35-deletion strain IMX2538, which was labeled the minimal CCM strain. This prototrophic strain grew at 94% of the rate of the prototrophic control strain with a full set of CCM genes, CEN.PK113-7D, on SMD and at 76% of the control on SME (Fig. 3). These values were within the 25% boundary that were initially set, and the physiology of the minimal CCM strain was further explored.
An S. cerevisiae strain with minimalized CCM shows only mild growth defects on synthetic media.
The genome sequence of the minimal CCM strain was analyzed by short-read and long-read techniques. Long-read sequencing revealed that 9 transformation rounds and deletion of 22 genes from the MG strain had not led to chromosomal rearrangements or deletions. Previously reported duplicated regions on chromosomes 3 and 5 of the MG strain, based on karyotyping and short read sequencing (22), were also observed in this study with long-read sequencing. Sequence analysis confirmed that all 22 targeted CCM genes were correctly deleted from the MG strain. The genome of the minimal CCM strain showed 45 single-nucleotide polymorphisms (SNPs) relative to the MG strain, of which 8 were located in genes and only 4 led to an amino acid change (Table 1), none of which affected proteins involved in CCM.
TABLE 1.
Single-nucleotide mutations identified in coding regions of the prototrophic minimal CCM strain IMX2538a
| Systematic name | Gene name | Typeb | Amino acid change |
|---|---|---|---|
| YBR114W | RAD16 | NS | Ile-202-Thr |
| YDL035C | GPR1 | S | Asn-523-Asn |
| YDR098C | GRX3 | NS | Glu-239-Asp |
| YFL062W | COS4 | S | Cys-151-Cys |
| YLR002C | NOC3 | NS | Asp-526-Glu |
| YML058W | SML1 | NS | Gly-52-Ser |
| YMR154C | RIM13 | S | Lys-265-Lys |
| YNL273W | TOF1 | S | Asn-117-Asn |
Single-nucleotide changes in S. cerevisiae IMX2538 (prototrophic minimal CCM) relative to the genome sequence of S. cerevisiae IMX372 (prototrophic minimal glycolysis (MG) (22) are shown.
S, synonymous; NS, nonsynonymous.
The physiology of the minimal CCM strain was compared to that of the congenic reference strain CEN.PK113-7D, which has a full complement of CCM genes, in pH-controlled aerobic bioreactor cultures on SMD. Consistent with the analyses in shake flasks, the specific growth rate of the minimal CCM strain in these cultures was 8% lower than the level of the reference strain CEN.PK113-7D (Table 2). During the glucose consumption phase, biomass-specific glucose and oxygen consumption rates of the two strains, as well as their ethanol and CO2 production rates and their biomass and ethanol yields on glucose, were also similar. The minimal CCM strain did exhibit a higher acetate production rate and yield (63% and 71% higher, respectively) than the reference strain, a difference already observed for the MG strain (22). Similarly, a lower glycerol production rate and glycerol yield on glucose (27% and 23% lower, respectively) was in line with data reported for a gpd1 deletion mutant (122). After the diauxic shift, growth of the minimal CCM strain on ethanol, glycerol, and organic acids produced during the glucose phase proceeded at a 17% lower rate than observed for the reference strain (Table 2). As a macroscopic characterization based on extracellular products might mask subtle differences of intracellular fluxes, intracellular concentrations of CCM intermediates were measured during the mid-exponential growth phase on glucose. Despite higher T6P and NAD+ concentrations than previously reported (129, 130), the concentrations of these metabolites hardly differed between the minimal CCM and the control strains (Table 3). These results indicated that a 32% reduction of the complement of genes encoding CCM enzymes of S. cerevisiae had only a small impact on its physiology under standard laboratory conditions.
TABLE 2.
Physiological characterization of a 35-deletion, minimal CCM prototrophic S. cerevisiae strain in aerobic bioreactor batch culturesa
| Growth phase and factor | CEN.PK113-7D (naive reference) | IMX372 (minimal glycolysis) | IMX2538 (minimal CCM) |
|---|---|---|---|
| Glucose phase (μmax, h−1) | 0.37 ± 0.00 | 0.38 ± 0.01* | 0.34 ± 0.00* |
| qs (mmol gDW−1 h−1) | −16.2 ± 0.2 | −15.7 ± 0.7 | −15.4 ± 0.5 |
| qEthanol (mmol gDW−1 h−1) | 23.5 ± 1.5 | 23.1 ± 1.1 | 23.2 ± 2.1 |
| qGlycerol (mmol gDW−1 h−1) | 1.52 ± 0.05 | 1.40 ± 0.02 | 1.11 ± 0.05* |
| qAcetate (mmol gDW−1 h−1) | 0.44 ± 0.03 | 0.82 ± 0.04* | 0.71 ± 0.02* |
| qCO2 (mmol gDW−1 h−1) | 23.4 ± 0.2 | 22.6 ± 0.5 | |
| qO2 (mmol gDW−1 h−1) | −6.8 ± 0.4 | −7.0 ± 0.2 | |
| Ybiomass/glucose (gDW gglucose−1) | 0.13 ± 0.00 | 0.13 ± 0.00 | 0.12 ± 0.00 |
| Yethanol/glucose (mol mol−1) | 1.45 ± 0.09 | 1.48 ± 0.01 | 1.51 ± 0.09 |
| Yglycerol/glucose (mol mol−1) | 0.09 ± 0.00 | 0.09 ± 0.01 | 0.07 ± 0.00* |
| Yacetate/glucose (mol mol−1) | 0.03 ± 0.00 | 0.05 ± 0.00 | 0.05 ± 0.00* |
| Postdiauxic phase | |||
| μmax (h−1) | 0.10 ± 0.00 | 0.12 ± 0.00* | 0.08 ± 0.01* |
| qEthanol (mmol gDW−1 h−1) | −3.10 ± 0.19 | −3.93 ± 0.04* | −3.07 ± 0.31 |
S. cerevisiae strains were grown at pH 5.0 and at 30°C in aerobic bioreactors on synthetic medium with glucose as sole carbon source. Data are presented as averages and standard deviations of 3 biological replicates for S. cerevisiae strains CEN.PK113-7D (naive reference) and IMX2538 (minimal CCM). DW, dry weight. Data for S. cerevisiae IMX372 (minimal glycolysis) were recalculated from the raw data of Solis-Escalante et al. (22) and were obtained with two biological replicates. Statistical significance with respect to CEN.PK113-7D is indicated in boldface with an asterisk (P < 0.05, two-tailed t test, equal variances).
TABLE 3.
Intracellular metabolite profiles of a 35-deletion, minimal CCM prototrophic S. cerevisiae strain in aerobic bioreactor batch culturesa
| Pathway and metabolite | Amount of metabolite (μmol/g biomass [dry wt]−1) |
Fold difference | |
|---|---|---|---|
| CEN.PK113-7D (naive reference) | IMX2538 (minimal CCM) | ||
| Glycolysis | |||
| Glucose 6-phosphate | 4.13 ± 0.49 | 5.01 ± 0.38 | 1.2 |
| Fructose 6-phosphate | 0.38 ± 0.06 | 0.59 ± 0.08 | 1.5* |
| Fructose 1,6-bisphosphate | 17.53 ± 0.82 | 20.52 ± 0.66 | 1.2* |
| Glyceraldehyde 3-phosphate | 0.15 ± 0.01 | 0.19 ± 0.01 | 1.3* |
| Dihydroxyacetone phosphate | 0.79 ± 0.13 | 2.08 ± 0.66 | 2.6 |
| 3-Phosphoglycerate | 1.30 ± 0.12 | 2.52 ± 0.28 | 1.9* |
| 2-Phosphoglycerate | 0.16 ± 0.01 | 0.17 ± 0.01 | 1.0 |
| Phosphoenolpyruvate | 0.25 ± 0.01 | 0.12 ± 0.01 | 0.5* |
| Trehalose synthesis | |||
| Trehalose 6-phosphate | 9.34 ± 1.18 | 14.48 ± 1.67 | 1.6* |
| Trehalose | 0.20 ± 0.04 | 0.41 ± 0.09 | 2.1* |
| Pentose phosphate pathway | |||
| 6-Phosphoglucononate | 1.01 ± 0.02 | 1.11 ± 0.07 | 1.1 |
| Ribose 5-phosphate | 0.40 ± 0.02 | 0.51 ± 0.03 | 1.3* |
| Ribulose 5-phosphate | 0.20 ± 0.03 | 0.33 ± 0.06 | 1.6* |
| Xylulose 5-phosphate | 0.43 ± 0.07 | 0.72 ± 0.13 | 1.7 |
| Sedoheptulose 7-phosphate | 0.50 ± 0.06 | 0.54 ± 0.04 | 1.1 |
| Erythrose 4-phosphate | 0.004 ± 0.000 | 0.005 ± 0.000 | 1.3* |
| TCA cycle and glyoxylate cycle | |||
| Citrate | 4.56 ± 0.65 | 5.80 ± 0.98 | 1.3 |
| Isocitrate | 0.02 ± 0.00 | 0.03 ± 0.00 | 1.7* |
| α-Ketoglutarate | 0.29 ± 0.02 | 0.67 ± 0.24 | 2.3 |
| Succinate | 0.37 ± 0.03 | 0.37 ± 0.12 | 1.0 |
| Fumarate | 0.10 ± 0.02 | 0.18 ± 0.06 | 1.8 |
| Malate | 0.60 ± 0.08 | 1.03 ± 0.19 | 1.7* |
| Nucleotides and cofactors | |||
| Energy charge | 0.86 ± 0.00 | 0.88 ± 0.01 | 1.0 |
| AMP | 0.13 ± 0.01 | 0.17 ± 0.01 | 1.3* |
| ADP | 1.55 ± 0.08 | 1.43 ± 0.11 | 0.9 |
| ATP | 5.50 ± 0.16 | 5.58 ± 0.21 | 1.0 |
| UDP | 0.23 ± 0.01 | 0.22 ± 0.05 | 0.9 |
| UTP | 1.61 ± 0.06 | 1.57 ± 0.10 | 1.0 |
| GDP | 0.27 ± 0.02 | 0.21 ± 0.07 | 0.8 |
| GTP | 1.58 ± 0.06 | 1.49 ± 0.15 | 0.9 |
| CDP | 0.13 ± 0.02 | 0.15 ± 0.01 | 1.1 |
| CTP | 0.84 ± 0.02 | 0.82 ± 0.03 | 1.0 |
| NADH | 0.18 ± 0.02 | 0.15 ± 0.02 | 0.8 |
| NAD+ | 85.43 ± 1.90 | 91.82 ± 2.64 | 1.1* |
| NADP+ | 8.49 ± 0.67 | 6.87 ± 0.25 | 0.8* |
| Acetyl-CoA | 5.80 ± 0.18 | 5.38 ± 0.34 | 0.9 |
| FAD+ | 0.87 ± 0.12 | 0.54 ± 0.18 | 0.6 |
Intracellular metabolite contents were measured during the mid-exponential glucose phase of aerobic bioreactor batch cultures of S. cerevisiae CEN.PK113-7D (naive reference) and IMX2538 (minimal CCM) (see Table 2 for other physiological data). Data represent averages and standard deviations of data from analyses of three independent cultures for each strain. Fold differences that are statistically significant are indicated in bold with an asterisk (two-tailed t test, equal variances, P < 0.05).
Dissecting individual from synergistic responses to growth under a range of conditions.
To explore genetic redundancy of CCM genes, the minimal CCM strain and the congenic reference strain CEN.PK113-7D were grown under a broad range of conditions. Some of these were chosen based on previously reported phenotypes (e.g., high osmolarity) or connection to CCM (e.g., growth on various carbon sources), while others subjected the strains to adverse conditions (e.g., acidic or alkaline pH).
Consistent with reports that deletion of GPD1 causes decreased osmotolerance (123, 125), the minimal CCM strain grew 13% to 25% slower than the reference strain exposed to high osmolarity, which was imposed by adding high concentrations of sorbitol (1 M and 2 M) or glucose (10% and 20% [wt/vol]). Construction and analysis of strains with different combinations of deletions in reference and CCM minimization backgrounds confirmed that this growth reduction specifically resulted from GPD1 deletion, rather than from synergistic effects (Fig. 4; see also Fig. S3). MPC3 deletion has been reported to cause lower growth rates on glycerol or lactate as sole carbon source (77). Since Mpc3 is a pyruvate transporter, growth was assessed directly on chemically defined medium with pyruvate as sole carbon source. The minimal CCM strain grew 79% slower than the control strain (Fig. 4); however, this could not be attributed to the MPC3 deletion, surprisingly. Indeed, reintroduction of MPC3 in IMX2519 (CCMin 5, glycmin fermin pppmin tcamin mcmin fummin glyoxmin Acemin glycerolmin), resulting in strain IMX2641, did not restore the growth rate on pyruvate (see Fig. S3G).
FIG 4.

Specific growth rates of the 35-deletion, prototrophic minimal CCM strain under a broad range of growth conditions. Specific growth rates of the prototrophic S. cerevisiae strains CEN.PK113-7D (naive reference strain), IMX372 (minimal glycolysis (MG)), and IMX2538 (minimal CCM) under different growth conditions are shown. Specific growth rates were measured in triplicate cultures using a growth profiler, except for those in the SMPyr and SMD-anaerobic cultures, which were measured in independent duplicate shake flask cultures. Abbreviations indicate the following growth conditions: SM, synthetic medium; SMD, synthetic medium with glucose; Gal, galactose as carbon source; Mal, maltose as carbon source; Suc, sucrose as carbon source; Fruc, fructose as carbon source; Pyr, pyruvate as carbon source; YPD, complex medium with glucose. Significant changes in growth rates of IMX372 and IMX2538 with respect to that of CEN.PK113-7D are indicated with an asterisk (P < 0.05, two-tailed paired homoscedastic t test).
Growth rates in specific environments. (A to D) Specific growth rate determinations under osmotic stress conditions (supplemented with uracil), measured with the growth profiler in biological triplicates. (E) Specific growth rates of FRDS1 and AAC3 deletion mutants on SMD supplemented with Tween, ergosterol, and uracil measured under anaerobic conditions in shake flasks in biological duplicates. (F) Specific growth rates of MPC3 complementation strains on SME supplemented with uracil in shake flasks in biological duplicates. (G) Specific growth rate of MPC3 complementation strain on SM with 83.3 mM pyruvate and supplemented with uracil in shake flasks in biological duplicates. Significant changes in growth rates with respect to the parental strain are indicated (*, P < 0.05, two-tailed paired homoscedastic t test). Download FIG S3, PDF file, 0.2 MB (200KB, pdf) .
Copyright © 2022 Postma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Combined deletion of AAC1, AAC3, MPC3, and SAL1 caused a 3 to 5% and a 14 to 18% decrease of growth rates on SMD and SME, respectively, thus marking their importance on these carbon sources. According to previous reports, individual deletion of these four genes does not affect growth on glucose and individual deletion of AAC1, AAC3, and SAL1 does not affect growth on ethanol (64, 65, 77, 131). Deletion of MPC3 has been reported to cause a decrease in growth rate on glycerol and lactate (77) and may therefore also be responsible for the lower growth rate on ethanol. Reintroduction of MPC3 in strain IMX1984 (glycmin fermin pppmin tcamin aac1Δ aac3Δ sal1Δ mpc3Δ) increased the specific growth rate on SME by only 3%, while reintroduction in IMX2519 (CCMin 5, glycmin fermin pppmin tcamin mcmin fummin glyoxmin acemin glycerolmin) did not affect growth rate (see Fig. S3). These results suggest that the observed impact of quadruple deletion of AAC1, AAC3, MPC3, and SAL1 on ethanol growth was caused by synergistic effects.
Some paralogs have been reported to be specifically important under anaerobic conditions (AAC3, FRDS1) (94, 131). In line with these reports, while the MG strain showed the same growth rate as CEN.PK113-7D under anaerobic conditions, the minimal CCM strain showed a 25% lower anaerobic growth rate (Fig. 4). However, testing of deletions in the reference background indicated that this difference was not caused by the deletion of AAC3 or FRDS1 (see Fig. S3).
Over the broad range of conditions tested, including several stress conditions, few differences in specific growth rate were observed (Fig. 4). Although lag phases were observed for some conditions, as expected from cells transitioning between different growth environments (the inoculum was consistently prepared using SM glucose as medium and under standard conditions regarding pH, temperature, etc.), no difference in lag phase was observed between the final IMX2519 mutant and the control strain. Combining the phenotypes of strains with individual and clustered deletions enabled us to identify synergistic interactions between minor paralogs.
DISCUSSION
Genetic reduction has been applied in several microorganisms (1), including Bacillus subtilis (3), Escherichia coli (2), Lactococcus lactis (6), Corynebacterium glutamicum (4), Streptomyces species (7), Pseudomonas species (5) and Schizosaccharomyces pombe (132), with the purpose of discovering a minimal genome content and/or for engineering efficient cell factories. In S. cerevisiae, Marakami et al. reduced genome content by 5% by deleting 15 terminal chromosomal regions (133). Moreover, the creation of a synthetic yeast genome in the Sc2.0 project was accompanied by an 8% genome reduction by deletion of long terminal repeats, retrotransposons, and introns; in addition, engineering of a single-chromosome yeast strain was characterized by a 9% decrease in DNA (134, 135). Genome reduction studies typically target two types of DNA sequences, nonexpressed DNA (cryptic genes, mobile DNA) and irrelevant or nonessential genes. These DNA elements can be targeted by random strategies for which little knowledge is required, such as transposon mutagenesis or the elegant SCRaMbLE technique used for the recent reduction of the left synthetic chromosome arm XII in S. cerevisiae (21). In the present study, knowledge-based reduction of the gene complement for CCM in S. cerevisiae was informed by gene expression data and previous phenotypic analyses on single-knockout mutants (21, 133–135).
In this study, we reduced genetic complexity of CCM in S. cerevisiae by deletion of 35 genes encoding enzymes and transporters. This reduction corresponded to elimination of 32% of the (iso)enzymes and transporters involved in the included processes, without major impacts on strain physiology, which was tested under a broad range of conditions (Fig. 2 and 4). The present study built on earlier work by Solis-Escalante et al. (22), who eliminated 50% of isoenzymes involved in glycolysis and ethanolic fermentation with a similar small impact on physiology. The attainable reduction of gene sets for enzymes and transporters involved in other CCM pathways differed, with 50% for fumarate reduction and glycerol synthesis, 37% for the mitochondrial carriers, 36% for the pentose-phosphate pathway, 23% for the TCA cycle, 14% for the glyoxylate cycle, 20% for the glycolysis-TCA cycle interface plus gluconeogenesis, and 8% for acetyl-CoA metabolism. The lower attainable genetic reduction of the four latter pathways can be largely attributed to neofunctionalization and relocalization of enzymes during evolution.
Our results showed that yeast CCM is remarkably robust to genetic reduction, in particular during growth on glucose, yeast’s favorite carbon source, but also when challenged by a broad range of growth conditions. Notable exceptions were growth on pyruvate (79% growth rate reduction), anaerobic growth on glucose (25% reduction), growth on ethanol (24% reduction), and growth at high osmolarity (between 13% and 25% lower specific growth rates). Growth rate reductions on ethanol and at high osmolarity could be attributed to specific genes or gene combinations, while for growth on pyruvate and anaerobic growth, some hypothetical targets could be excluded. The physiological role of most deleted paralogs remains elusive. Such a situation is exemplified by TKL2 and NQM1, which are paralogs of the major PPP genes TKL1 and TAL1, respectively. In S. cerevisiae strains engineered for l-arabinose utilization, their deletion was shown to lead to lower growth rates on this pentose sugar (33, 136). Clearly, as pentoses are not natural carbon sources for S. cerevisiae, this role cannot have provided an evolutionary driving force for fixation of these paralogs in its genome, but it does indicate potential contributions to fitness under other, as-yet-unidentified growth conditions. Testing the minimal CCM strain under an even wider variety of environmental conditions, including dynamics in nutrient availability and other environmental parameters, may reveal physiological roles of these and other paralogs. Alternatively, the mechanisms that fixed some paralogs during evolution may have been disrupted by relatively recent mutations or gene loss (137). Following this reasoning, absence of a clear phenotype of knockout mutants may have captured a stage in the evolutionary trajectory of S. cerevisiae that will eventually lead to loss of a paralog, evolution toward complete subfunctionalization, or retention of functional overlap with asymmetric divergence (138).
In this first step toward the genetic minimization of CCM in yeast, choices had to be made on which pathways and genes were considered part of the CCM and on criteria for redundancy. For instance, transport of NAD+, FAD+, ADP/ATP, and Pi across the mitochondrial membrane was considered, while transport of NAD(P)H, which requires more complex shuttle systems (139, 140), was not yet included. In addition, as S. cerevisiae cannot synthetize carnitine (141), the carnitine shuttle system transporting acetyl-CoA across compartments was not considered. Since CRC1, CAT1, YAT1, and YAT2 involved in this shuttle are dispensable for growth in the absence of carnitine (141), they can be considered for further genetic reduction of the CCM. Some genes required for anaerobic growth, such as ADH3 (139, 142), were also retained but could be removed if fast anaerobic growth is excluded as a criterion. Several other processes and pathways are of particular interest for development of strain platforms for modular engineering of yeast CCM. In this context, glucose uptake, which involves a set of 20 hexose transporters (143), provides an interesting target for future experiments, whose minimization can benefit from a recently constructed Hxt0 CRISPR kit (144). Another logical target for minimization is uptake and assimilation of (alternative) carbon sources and especially of maltose, whose metabolism is enabled by highly redundant subtelomeric genes (145, 146).
Genetic reduction presents a first, indispensable step toward the construction of modular yeast strains for extensive remodeling of CCM. Current demands for economically competitive cell factories, with optimized titer, rate, and yield, requires extensive remodeling of the CCM for the supply of precursors, (redox) cofactors and energy-rich molecules (147–149). For instance, the extensive remodeling of the native Entner-Doudoroff glycolytic pathway into the Embden-Meyerhof-Parnas pathway improved carotenoid synthesis in Pseudomonas putida (150). Similarly, substantial efforts have been invested in remodeling yeast CCM in S. cerevisiae to increase the supply of cytosolic acetyl-CoA, a precursor for a wide array of attractive biomolecules (Fig. 1) (151). Also, production of complicated chemical structures, like plant natural products in S. cerevisiae, requires extensive remodeling of the entire central carbon metabolism (152–154). As demonstrated by Kuijpers and coworkers (24), genetic reduction facilitates the colocalization of sets of genes in “pathway clusters” and strongly accelerates the genetic remodeling of these pathways. With this strategy, the 12 steps of glycolysis and ethanolic fermentation were rapidly and efficiently swapped with heterologous variants and enabled the implementation of an innocuous DNA and RNA watermarking method (155). A similar strategy can be considered for remodeling CCM, with the minimal CCM strain as starting point. As recently demonstrated, 44 transcriptional unit-sized DNA fragments can be assembled in S. cerevisiae into specialized, synthetic supernumerary chromosomes (26). Since the capacity of homologous recombination was not reached, assembly of synthetic chromosomes containing the set of 76 genes encoding the minimal CCM has now become a realistic objective. Subsequent CRISPR-Cas-assisted removal of the duplicate CCM genes from their native locations could then generate powerful platforms for chromosome swapping and combinatorial CCM remodeling studies. The reduction of genetic complexity demonstrated in the present study therefore not only provides new insights in genetic redundancy of CCM but also contributes to the eventual localization of all genes required for a minimized CCM on specialized, synthetic supernumerary chromosomes that allow for extensive, combinatorial remodeling of yeast metabolism for industrial applications.
MATERIALS AND METHODS
Strains, media and maintenance.
The Saccharomyces cerevisiae strains used in this study were all derived from the CEN.PK family (156, 157) (see Table S3). The naive, uracil-auxotrophic, and Cas9-containing strain, IMX581 (158), and the uracil-auxotrophic MG (minimal glycolysis) strain IMX370 (22) were used for deletion of genes encoding enzymes or transporters involved in CCM. The naive uracil-prototrophic strain, CEN.PK113-7D, was used for physiological comparison. Complex medium used for propagation of yeast strains consisted of 10 g liter−1 Bacto yeast extract, 20 g liter−1 Bacto peptone, and 20 g liter−1 glucose (YPD), autoclaved at 110°C for 20 min. After transformation, yeast strains were selected in synthetic medium (SM) (159) containing 3.0 g liter−1 KH2PO4, 0.5 g liter−1 MgSO4·7H2O, 5.0 g liter−1 (NH4)2SO4, and 1.0 mL liter−1 trace elements autoclaved at 121°C for 20 min, whereafter 1.0 mL liter−1 of filter-sterilized vitamin solution was added. Before autoclaving, media were set to pH 6 by 1 M KOH addition. SM was supplemented with 20 g liter−1 glucose (SMD) or 2% ethanol (vol/vol) (SME) for propagation and growth characterization. Synthetic medium was supplemented with 150 mg liter−1 uracil for uracil-auxotrophic strains. For selection of transformants carrying the amdS selection marker (160), ammonium sulfate was replaced as nitrogen source with 10 mM acetamide. For experiments on SM in the growth profiler and under anaerobic conditions, ammonium sulfate was replaced by 2.3 g liter−1 urea. For both media in which ammonium sulfate was replaced, 6.6 g liter−1 K2SO4 was added. Growth was performed in 500-mL shake flasks containing 100 mL medium or in 100-mL shake flasks containing 20 mL medium at 30°C and 200 rpm in an Innova 44 incubator shaker (New Brunswick Scientific, Edison, NJ). Cultures on solid media were incubated for 3 to 5 days at 30°C.
Strains. Download Table S3, PDF file, 0.2 MB (204.1KB, pdf) .
Copyright © 2022 Postma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
CEN.PK113-7D, IMX372, IMX2538, and several intermediate strains were tested in the growth profiler on a variety of liquid media containing SM (urea) plus 2% glucose (SMD), SMD at pH 3.0, 3.5, or 7.5, SM plus 10% glucose (SMD [10%]), SM plus 20% glucose (SMD [20%]), SMD plus 1 or 2 M sorbitol, SMD plus 200 or 500 mM NaCl, SM plus 111 mM galactose (SMGal), SM plus 55 mM maltose (SMMal), SM plus 55 mM sucrose (SMSuc), SM plus 111 mM fructose (SMFruc), SM plus 125 mM acetic acid, SMD plus 1 mM paraquat and YPD (2% glucose). Growth on SM plus 83.3 mM pyruvic acid was performed in shake flasks. For anaerobic growth in shake flasks, SMD was supplemented with 0.01 g liter−1 ergosterol and 0.42 g liter−1 Tween 80 dissolved in ethanol (SMD [2%] anaerobic) (159).
Plasmids were propagated in and isolated from chemically competent Escherichia coli XL1-Blue cells, which were cultivated in lysogeny broth containing 10 g liter−1 Bacto tryptone, 5.0 g liter−1 Bacto yeast extract, and 5 g liter−1 NaCl supplemented with 100 mg liter−1 ampicillin (LB-amp) when required. E. coli was cultivated in 15-mL Greiner tubes containing 5 mL medium at 37°C and 200 rpm in an Innova 4000 incubator shaker (New Brunswick Scientific). Bacterial cultures on solid medium were incubated overnight at 37°C.
For solid medium, 20 g liter−1 of agar was added before autoclaving. All S. cerevisiae and E. coli strains were stored at −80°C in 1-mL aliquots containing 30% (vol/vol) glycerol in appropriate medium.
Molecular biology techniques.
Plasmids were isolated from E. coli using the GenElute plasmid miniprep kit (Sigma-Aldrich, St. Louis, MO) or the GeneJET plasmid miniprep kit (Thermo Fisher Scientific, Waltham, MA) according to the provided protocols. DNA fragments for plasmid construction or integrative DNA fragments used in yeast transformation were amplified using Phusion high-fidelity DNA polymerase (Thermo Fisher Scientific) according to the manufacturer’s instructions, using PAGE-purified or desalted oligonucleotides (Sigma-Aldrich) depending on the application. Purification of genomic PCR-amplified DNA was performed with the GenElute PCR Clean-Up kit (Sigma-Aldrich) or the GeneJET PCR purification kit (Thermo Fisher Scientific) if no aspecific products were present. When aspecific products were present or when DNA was amplified from plasmids, the DNA was purified by separation using electrophoresis on a 1% (wt/vol) agarose gel (TopVision agarose, Thermo Fisher Scientific) in 1× Tris-acetate-EDTA buffer (Thermo Fisher Scientific) or on a 2% (wt/vol) agarose gel (TopVision agarose) in 1× Tris-borate-EDTA buffer (Thermo Fisher Scientific) with subsequent purification with the Zymoclean gel DNA recovery kit (Zymo Research). Chemical transformation of E. coli XL1-Blue was performed by thawing of competent cells on ice, addition of DNA, and heat shock for 40 s at 42°C. Subsequently, cells were incubated on ice for 2 min and plated immediately on selective LB-amp plates and grown overnight at 37°C. Transformation of S. cerevisiae was performed using the lithium acetate–single-stranded carrier DNA–polyethylene glycol method (161). Single-cell lines were obtained by three consecutive restreaks on selective solid medium. Yeast genomic DNA was extracted according to the methods of Looke et al. (162), with the YeaStar genomic DNA kit (Zymo Research, Irvine, CA) according to Protocol I supplied by the manufacturer, or the Qiagen blood and cell culture kit with 100-G or 20-G genomic tips (Qiagen, Hilden, Germany) following the manufacturer’s recommendations. Verification of the accurate genotypes of engineered S. cerevisiae strains and E. coli plasmids was done by diagnostic PCR before strain storage at −80°C. These diagnostic PCRs were performed using desalted oligonucleotides and the DreamTaq PCR master mix (Thermo Fisher Scientific) according to the manufacturer’s protocol.
Plasmid and strain construction.
Deletions were performed using CRISPR/Cas9. CRISPR/Cas9-based genome editing of S. cerevisiae was performed as described by Mans et al.(158) with minor alterations. Plasmids containing a single guide RNA (gRNA) (see Table S4) were constructed via Gibson assembly with a backbone containing the marker cassette and one insert fragment containing the gRNA and the 2μm plasmid fragment. The backbone was amplified from a pMEL plasmid (158) with primers 5980 and 5792, and the insert fragment was amplified with a gRNA-specific primer designed with the yeastriction tool (158) and primer 5979 (primers are listed in Table S5). Plasmids containing two gRNAs were constructed using one backbone fragment and two insert fragments, each containing one gRNA and one half of the 2μm fragment. Backbones were PCR amplified from the pROS plasmids (158) with the double-binding primer 6005. Insert fragments were obtained with the gRNA-specific primers together with either primer 5974 or primer 5975 (see Table S5). The backbone and gRNA insert fragment(s) were gel purified, DpnI digested (Thermo Fisher Scientific), and Gibson assembled in a final volume of 5 μL using NEBuilder HiFi DNA assembly master mix (NEB, Ipswich, MA), according to the manufacturer’s instructions. Assembled plasmids were transformed and subsequently isolated from LB-amp-grown E. coli. Correct assembly was checked using diagnostic PCR (see Table S5).
Plasmids. Download Table S4, PDF file, 0.2 MB (193.4KB, pdf) .
Copyright © 2022 Postma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Primers used in this study. Download Table S5, PDF file, 0.2 MB (199.6KB, pdf) .
Copyright © 2022 Postma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
IMK588 was constructed by integrating the KanMX marker at the OAC1 locus of CEN.PK113-7D. The KanMX marker with homologous flanks to OAC1 was amplified with primers 6358 and 6359 from pUG6 (see Tables S4 and S5).
In order to perform CRISPR editing in the MG strain (IMX370), Cas9 was integrated by transforming a Cas9 and natNT2 DNA fragment, which could assemble by homologous recombination at the CAN1 locus. The Cas9 fragment (can1 flank-Cas9 expression cassette-SHR A) was PCR amplified with primers 2873 and 4653 from plasmid p414-TEF1p-Cas9-CYC1t (see Tables S4 and S5). The natNT2 fragment (SHR A-NatNT2 marker cassette-can 1 flank) was amplified with primers 3093 and 5542 from plasmid pUG-natNT2 (see Tables S4 and S5). The Cas9 containing MG strain was stocked as IMX1331.
For genome editing using CRISPR, S. cerevisiae strains were transformed with 1 μg of each gRNA plasmid and 1 μg of each 120-bp double-stranded DNA repair fragment. These repair fragments were made by annealing of complimentary oligonucleotides listed in Table S5 and consisted of 60-bp homology sequences immediately upstream of the start codon and downstream of the stop codon of the targeted gene, unless stated otherwise. Transformants were plated on selective medium. Gene deletion was verified by diagnostic colony PCR on randomly picked colonies by using the primers which bind outside of the targeted open reading frame (ORF) (see Table S5). gRNA plasmids were removed by growing the colonies in liquid YPD medium and subsequent plating on solid YPD medium. Plasmid removal was confirmed by growth on selective and nonselective solid media after which the strains were stored. For all transformations, the corresponding gRNA plasmids and repair fragment are summarized in Table S6.
Strain transformations. Download Table S6, PDF file, 0.2 MB (180.1KB, pdf) .
Copyright © 2022 Postma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To obtain a prototrophic strain with 35 deletions, a URA3 transcriptional unit amplified from CEN.PK113-7D DNA (primers 17752 and 17753 [see Table S5]) was integrated at the GPP2 locus of strain IMX2520 (34 deletions). The flanks of the URA3 repair fragment were homologous to the 60 bp immediately upstream and downstream of the GPP2 ORF. The prototrophic 35-deletion strain IMX2538 was checked by diagnostic PCR, short-read sequencing, and long-read nanopore sequencing. Integration of the MPC3 transcriptional unit at the X2 locus of IMX1984 and IMX2519 was achieved by amplifying the corresponding fragment from CEN.PK113-7D genomic DNA (primers 18025 and 18026 [see Table S5]) and integration by CRISPR-Cas9 using gRNA plasmid pUDR376, resulting in strains IMX2640 and IMX2641, respectively. Correct integration was verified by diagnostic PCR (see Table S5).
Sequencing.
High-quality genomic DNA of yeast for sequencing was extracted using the Qiagen blood and cell culture kit with 100-G or 20-G genomic tips (Qiagen) according to the manufacturer’s instructions. DNA concentration was measured using the BR ds DNA kit (Invitrogen, Carlsbad, CA) and a Qubit 2.0 fluorometer (Thermo Fisher Scientific). The purity was verified with a Nanodrop 2000 UV-Vis spectrophotometer (Thermo Fisher Scientific).
Short-read sequencing.
IMX2538 (35-deletions prototrophic strain) was sequenced using 300-bp paired-end sequencing reads prepared with the MiSeq reagent kit v3 on an Illumina MiSeq sequencer (Illumina, San Diego, CA). To this end, extracted DNA was mechanically sheared to 550 bp with the M220 ultrasonicator (Covaris, Wolburn, MA) and subsequently the TruSeq DNA PCR-Free Library preparation kit (Illumina) was employed to make a six-strain library. The samples were quantified by quantitative PCR on a Rotor-Gene Q PCR cycler (Qiagen) using the KAPA library quantification kit (Kapa Biosystems, Wilmington, MA). Library integrity and fragment size were determined with a Tapestation 2200 system (Agilent Technologies). Sequencing reads were mapped onto the CEN.PK113-7D (163) reference genome using the Burrows-Wheeler alignment (BWA) tool (version 0.7.15) (164) and further processed using SAMtools (version 1.3.1) (165) and Pilon (with -vcf setting; version 1.18) (166) to identify SNPs. The sequence was analyzed by visualizing the generated .bam files in the Integrative Genomics Viewer (IGV) software (version 2.4.0) (167). Chromosomal copy number was estimated by the Magnolya algorithm (version 0.15) (168).
Long-read sequencing.
High-quality DNA of IMX2538 was isolated and checked for quantity and quality as described above. Furthermore, quality and integrity of DNA was checked with a TapeStation 2200 system (Agilent Technologies, Santa Clara, CA). IMX2538 was sequenced in-house on a single R10 flow cell (FLO-MIN111) using the SQK-LSK109 sequencing kit (Oxford Nanopore Technologies, Oxford, United Kingdom), according to the manufacturer’s instructions. With MinKnow (version 3.6.5, Oxford Nanopore Technologies), raw signal files were generated. Base-calling was performed by Guppy (version 4.0.11, Oxford Nanopore Technologies), followed by de novo assembly with Canu (version 2.0) (169).
Growth rate measurements in shake flasks.
The growth rate of the constructed strains was determined in 500-mL shake flasks containing 100 mL of SMD or SME medium. Wake-up cultures were inoculated with a 1-mL aliquot of a strain stored at −80°C and grown until late exponential phase. Precultures were inoculated from the wake-up cultures and grown to mid-exponential phase. Finally, measuring cultures were inoculated in biological duplicates from the preculture at an initial optical density at 660 nm (OD660) of 0.3. Cultures were monitored based on the OD660 with a Jenway 7200 spectrophotometer in technical duplicate (Cole-Parmer, Vernon Hills, IL). A maximum specific growth rate (μmax) was calculated from at least five data points in the exponential phase with at least 2 doublings.
Anaerobic shake flask-based experiments were performed at 30°C in a Bactron anaerobic chamber (Sheldon Manufacturing Inc., Cornelius, OR) with an atmosphere of 5% (vol/vol) H2, 6% (vol/vol) CO2, and 89% (vol/vol) N2, on an IKA KS 260 basic shaker at 200 rpm, using 50-mL shake flasks containing 30 mL SMD (2%) anaerobic medium.
Growth rate measurements in microtiter plates.
Growth measurements of strains in microtiter plates with a Growth Profiler 960 system (EnzyScreen BV, Heemstede, The Netherlands) were performed as described by Postma et al. (26). Briefly, strains from glycerol freezer stocks were inoculated and grown overnight in 10 mL SMD medium in a 50-mL shake flask. This culture was used to inoculate a preculture in 10 mL SMD medium in a 50-mL shake flask, which was cultivated until mid-exponential growth. To prepare the inoculum, the cells were then spun down and resuspended in SM without carbon source. The growth study was performed in 96-well microtiter plates (EnzyScreen type CR1496dl), with a final working volumes of 250 μL and an approximate starting OD660 of 0.3. Microtiter plates were closed with a sandwich cover (EnzyScreen type CR1296). Images of cultures were made at 30-min intervals. Growth rates were calculated in the time frame where the calculated OD was between 2 and 10. Each experimental condition was analyzed in biological triplicates.
Physiological characterization of CEN.PK113-7D and IMX2538 in bioreactor cultures.
Aerobic batch bioreactor cultures were performed in 2-liter bioreactors (Applikon, Delft, The Netherlands). Bioreactors were filled with synthetic medium containing 5.0 g liter−1 (NH4)2SO4, 3.0 g liter−1 KH2PO4, 0.5 g liter−1 MgSO4·7H2O, and 1.0 mL liter−1 trace elements. After heat sterilization, 20 g liter−1 glucose, 0.2 g liter−1 antifoam emulsion C (Sigma-Aldrich, St. Louis, MA), and filter-sterilized vitamins were added to complete the medium. Upon inoculation, bioreactors contained a working volume 1.4 liters and the culture pH was maintained at 5.0 by automated addition of 2 M H2SO4 or 2 M KOH. Temperature was kept stable at 30°C, and mixing of the medium was performed at 800 rpm. The gas flow was set to 700 mL of air per minute to supply oxygen and remove produced carbon dioxide, and an overpressure of 0.3 × 105 Pa was applied to the reactor. Dissolved oxygen tension was thus maintained for all reactors above 59% over the whole duration of the batch cultivation. Off-gas was cooled to 2°C in a condenser on the bioreactor to prevent water evaporation and further dried with a Permapure MD-110-48P-4 filter dryer (Permapure, Lakewood, NJ) for subsequent analysis of carbon dioxide and oxygen percentages by a MultiExact 4100 gas analyzer (Servomex, Zoetermeer, The Netherlands). For both CEN.PK113-7D and IMX2538, reactors were run in biological triplicates and inoculated from exponentially growing shake flask cultures.
Optical densities were measured in technical triplicates with a Jenway 7200 spectrophotometer (Cole-Parmer) at 660 nm, while cell dry weights were determined by filtration of 10 mL of well-mixed sample over dried polyethersulfone (PES) membrane filters with a pore size of 0.45 μm (Pall Corporation, Port Washington, NY). Filters were washed with demineralized water and dried in a microwave oven for 20 min at 360 W.
Extracellular organic acids, sugars, and ethanol were determined by high-performance liquid chromatography analysis using an Aminex HPX-87H ion-exchange column (Agilent, Santa Clara) with 5 mM H2SO4 as mobile phase and a flow rate of 0.6 mL min−1 at 60°C. Glucose, glycerol, and ethanol were detected by a refractive index detector (Agilent G1362A), and organic acids were detected by a dual-wavelength absorbance detector (Agilent G1314F).
During mid-exponential growth in the glucose consumption phase, intracellular metabolite samples were taken with a filtration-based washing method according to that of Douma et al. (170) with some modifications. Briefly, approximately 3 mL of cell culture was sampled in 15 mL of 100% methanol at −40°C. Biomass was washed with cooled 100% methanol on a PES membrane with a pore size of 0.45 μm (Pall Corporation), which was precooled and wetted with 100% methanol at −40°C. Finally, metabolites were extracted with 75% boiling ethanol. A 100-μL volume of 13C-labeled cell extract was added to each tube as an internal standard for metabolite quantification (171). The intracellular CCM metabolites, cofactors, and nucleotides were derivatized and quantified as described by de Jonge et al. (172) and Niedenführ et al. (173).
Data availability.
Short- and long-read sequencing data are available at NCBI under BioProject PRJNA757356.
ACKNOWLEDGMENTS
We thank Pilar de la Torre and Marcel van den Broek for sequencing and bioinformatic analysis, Roel Sarelse, Ilse Pardijs, and Lycka Kamoen for strain construction, Koen Verhagen, Patricia van Dam, and Martin Pabst for analysis of intracellular metabolites, Erik de Hulster for assistance with sampling of the bioreactors, Marijke Luttik for the growth experiment on pyruvate medium, Sofia Dashko and Jean-Marc Daran for input during the literature analysis, and Jack Pronk for feedback on an advanced version of the manuscript. This work was funded by the AdLibYeast ERC consolidator grant number 648141 attributed to P.D.-L.
Contributor Information
Pascale Daran-Lapujade, Email: p.a.s.daran-lapujade@tudelft.nl.
Jean-Paul Latge, IMBB-FORTH.
REFERENCES
- 1.Lara AR, Gosset G (ed). 2019. Minimal cells: design, construction, biotechnological applications. Springer International Publishing, Berlin, Germany. [Google Scholar]
- 2.Hashimoto M, Ichimura T, Mizoguchi H, Tanaka K, Fujimitsu K, Keyamura K, Ote T, Yamakawa T, Yamazaki Y, Mori H, Katayama T, Kato J. 2005. Cell size and nucleoid organization of engineered Escherichia coli cells with a reduced genome. Mol Microbiol 55:137–149. doi: 10.1111/j.1365-2958.2004.04386.x. [DOI] [PubMed] [Google Scholar]
- 3.Morimoto T, Kadoya R, Endo K, Tohata M, Sawada K, Liu S, Ozawa T, Kodama T, Kakeshita H, Kageyama Y, Manabe K, Kanaya S, Ara K, Ozaki K, Ogasawara N. 2008. Enhanced recombinant protein productivity by genome reduction in Bacillus subtilis. DNA Res 15:73–81. doi: 10.1093/dnares/dsn002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suzuki N, Nonaka H, Tsuge Y, Inui M, Yukawa H. 2005. New multiple-deletion method for the Corynebacterium glutamicum genome, using a mutant lox sequence. Appl Environ Microbiol 71:8472–8480. doi: 10.1128/AEM.71.12.8472-8480.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lieder S, Nikel PI, de Lorenzo V, Takors R. 2015. Genome reduction boosts heterologous gene expression in Pseudomonas putida. Microb Cell Fact 14:23. doi: 10.1186/s12934-015-0207-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu D, Fu Y, Liu F, Xu H, Saris PEJ, Qiao M. 2017. Enhanced heterologous protein productivity by genome reduction in Lactococcus lactis NZ9000. Microb Cell Fact 16:1. doi: 10.1186/s12934-016-0616-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Komatsu M, Uchiyama T, Omura S, Cane DE, Ikeda H. 2010. Genome-minimized Streptomyces host for the heterologous expression of secondary metabolism. Proc Natl Acad Sci USA 107:2646–2651. doi: 10.1073/pnas.0914833107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hutchison CA, Chuang R-Y, Noskov VN, Assad-Garcia N, Deerinck TJ, Ellisman MH, Gill J, Kannan K, Karas BJ, Ma L, Pelletier JF, Qi Z-Q, Richter RA, Strychalski EA, Sun L, Suzuki Y, Tsvetanova B, Wise KS, Smith HO, Glass JI, Merryman C, Gibson DG, Venter JC. 2016. Design and synthesis of a minimal bacterial genome. Science 351:aad6253. doi: 10.1126/science.aad6253. [DOI] [PubMed] [Google Scholar]
- 9.Mutschler H, Robinson T, Tang TD, Wegner S. 2019. Special issue on bottom-up synthetic biology. Chembiochem 20:2533–2534. doi: 10.1002/cbic.201900507. [DOI] [PubMed] [Google Scholar]
- 10.Conant GC, Wolfe KH. 2008. Turning a hobby into a job: how duplicated genes find new functions. Nat Rev Genet 9:938–950. doi: 10.1038/nrg2482. [DOI] [PubMed] [Google Scholar]
- 11.Escalera-Fanjul X, Quezada H, Riego-Ruiz L, Gonzalez A. 2019. Whole-genome duplication and yeast's fruitful way of life. Trends Genet 35:42–54. doi: 10.1016/j.tig.2018.09.008. [DOI] [PubMed] [Google Scholar]
- 12.Acharya D, Ghosh TC. 2016. Global analysis of human duplicated genes reveals the relative importance of whole-genome duplicates originated in the early vertebrate evolution. BMC Genomics 17:1–14. doi: 10.1186/s12864-016-2392-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fares MA, Keane OM, Toft C, Carretero-Paulet L, Jones GW. 2013. The roles of whole-genome and small-scale duplications in the functional specialization of Saccharomyces cerevisiae genes. PLoS Genet 9:e1003176. doi: 10.1371/journal.pgen.1003176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M'Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Véronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, et al. 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 15.Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau-Danila A, Anderson K, André B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Güldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kötter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, et al. 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 16.Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, Tan G, Wang W, Usaj M, Hanchard J, Lee SD, Pelechano V, Styles EB, Billmann M, van Leeuwen J, van Dyk N, Lin Z-Y, Kuzmin E, Nelson J, Piotrowski JS, Srikumar T, Bahr S, Chen Y, Deshpande R, Kurat CF, Li SC, Li Z, Usaj MM, Okada H, Pascoe N, San Luis B-J, Sharifpoor S, Shuteriqi E, Simpkins SW, Snider J, Suresh HG, Tan Y, Zhu H, Malod-Dognin N, Janjic V, Przulj N, Troyanskaya OG, Stagljar I, Xia T, Ohya Y, Gingras A-C, Raught B, Boutros M, Steinmetz LM, Moore CL, Rosebrock AP, et al. 2016. A global genetic interaction network maps a wiring diagram of cellular function. Science 353:aaf1420. doi: 10.1126/science.aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuzmin E, VanderSluis B, Wang W, Tan G, Deshpande R, Chen Y, Usaj M, Balint A, Mattiazzi Usaj M, van Leeuwen J, Koch EN, Pons C, Dagilis AJ, Pryszlak M, Wang JZY, Hanchard J, Riggi M, Xu K, Heydari H, San Luis B-J, Shuteriqi E, Zhu H, Van Dyk N, Sharifpoor S, Costanzo M, Loewith R, Caudy A, Bolnick D, Brown GW, Andrews BJ, Boone C, Myers CL. 2018. Systematic analysis of complex genetic interactions. Science 360:eaao1729. doi: 10.1126/science.aao1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tong AHY, Evangelista M, Parsons AB, Xu H, Bader GD, Pagé N, Robinson M, Raghibizadeh S, Hogue CWV, Bussey H, Andrews B, Tyers M, Boone C. 2001. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- 19.Tong AHY, Lesage G, Bader GD, Ding H, Xu H, Xin X, Young J, Berriz GF, Brost RL, Chang M, Chen Y, Cheng X, Chua G, Friesen H, Goldberg DS, Haynes J, Humphries C, He G, Hussein S, Ke L, Krogan N, Li Z, Levinson JN, Lu H, Ménard P, Munyana C, Parsons AB, Ryan O, Tonikian R, Roberts T, Sdicu A-M, Shapiro J, Sheikh B, Suter B, Wong SL, Zhang LV, Zhu H, Burd CG, Munro S, Sander C, Rine J, Greenblatt J, Peter M, Bretscher A, Bell G, Roth FP, Brown GW, Andrews B, Bussey H, Boone C. 2004. Global mapping of the yeast genetic interaction network. Science 303:808–813. doi: 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- 20.Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JLY, Toufighi K, Mostafavi S, Prinz J, St Onge RP, VanderSluis B, Makhnevych T, Vizeacoumar FJ, Alizadeh S, Bahr S, Brost RL, Chen Y, Cokol M, Deshpande R, Li Z, Lin Z-Y, Liang W, Marback M, Paw J, San Luis B-J, Shuteriqi E, Tong AHY, van Dyk N, Wallace IM, Whitney JA, Weirauch MT, Zhong G, Zhu H, Houry WA, Brudno M, Ragibizadeh S, Papp B, Pál C, Roth FP, Giaever G, Nislow C, Troyanskaya OG, Bussey H, Bader GD, Gingras A-C, Morris QD, Kim PM, Kaiser CA, et al. 2010. The genetic landscape of a cell. Science 327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo Z, Yu K, Xie S, Monti M, Schindler D, Fang Y, Zhao S, Liang Z, Jiang S, Luan M, Xiao C, Cai Y, Dai J. 2021. Compacting a synthetic yeast chromosome arm. Genome Biol 22:5. doi: 10.1186/s13059-020-02232-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Solis-Escalante D, Kuijpers NG, Barrajon-Simancas N, van den Broek M, Pronk JT, Daran J-M, Daran-Lapujade P. 2015. A minimal set of glycolytic genes reveals strong redundancies in Saccharomyces cerevisiae central metabolism. Eukaryot Cell 14:804–816. doi: 10.1128/EC.00064-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bradley PH, Gibney PA, Botstein D, Troyanskaya OG, Rabinowitz JD. 2019. Minor isozymes tailor yeast metabolism to carbon availability. mSystems 4. doi: 10.1128/mSystems.00170-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuijpers NG, Solis-Escalante D, Luttik MA, Bisschops MM, Boonekamp FJ, van den Broek M, Pronk JT, Daran J-M, Daran-Lapujade P. 2016. Pathway swapping: toward modular engineering of essential cellular processes. Proc Natl Acad Sci USA 113:15060–15065. doi: 10.1073/pnas.1606701113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nielsen J, Keasling JD. 2016. Engineering cellular metabolism. Cell 164:1185–1197. doi: 10.1016/j.cell.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 26.Postma ED, Dashko S, van Breemen L, Taylor Parkins SK, van den Broek M, Daran J-M, Daran-Lapujade P. 2021. A supernumerary designer chromosome for modular in vivo pathway assembly in Saccharomyces cerevisiae. Nucleic Acids Res 49:1769–1783. doi: 10.1093/nar/gkaa1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeLoache WC, Russ ZN, Dueber JE. 2016. Towards repurposing the yeast peroxisome for compartmentalizing heterologous metabolic pathways. Nat Commun 7:11152. doi: 10.1038/ncomms11152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knijnenburg TA, Daran J-MG, van den Broek MA, Daran-Lapujade PA, de Winde JH, Pronk JT, Reinders MJ, Wessels LF. 2009. Combinatorial effects of environmental parameters on transcriptional regulation in Saccharomyces cerevisiae: a quantitative analysis of a compendium of chemostat-based transcriptome data. BMC Genomics 10:53. doi: 10.1186/1471-2164-10-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gancedo C, Flores CL. 2008. Moonlighting proteins in yeasts. Microbiol Mol Biol Rev 72:197–210. doi: 10.1128/MMBR.00036-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jansen MLA, Bracher JM, Papapetridis I, Verhoeven MD, de Bruijn H, de Waal PP, van Maris AJA, Klaassen P, Pronk JT. 2017. Saccharomyces cerevisiae strains for second-generation ethanol production: from academic exploration to industrial implementation. FEMS Yeast Res 17:fox044. doi: 10.1093/femsyr/fox044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lobo Z, Maitra PK. 1982. Pentose phosphate pathway mutants of yeast. Mol Gen Genet 185:367–368. doi: 10.1007/BF00330815. [DOI] [PubMed] [Google Scholar]
- 32.Schaaff-Gerstenschläger I, Mannhaupt G, Vetter I, Zimmermann FK, Feldmann H. 1993. TKL2, a second transketolase gene of Saccharomyces cerevisiae. Eur J Biochem 217:487–492. doi: 10.1111/j.1432-1033.1993.tb18268.x. [DOI] [PubMed] [Google Scholar]
- 33.Matsushika A, Goshima T, Fujii T, Inoue H, Sawayama S, Yano S. 2012. Characterization of non-oxidative transaldolase and transketolase enzymes in the pentose phosphate pathway with regard to xylose utilization by recombinant Saccharomyces cerevisiae. Enzyme Microb Technol 51:16–25. doi: 10.1016/j.enzmictec.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 34.Stanford D, Whitney M, Hurto R, Eisaman D, Shen W-C, Hopper A. 2004. Division of labor among the yeast Sol proteins implicated in tRNA nuclear export and carbohydrate metabolism. Genetics 168:117–127. doi: 10.1534/genetics.104.030452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Castelli LM, Lui J, Campbell SG, Rowe W, Zeef LA, Holmes LE, Hoyle NP, Bone J, Selley JN, Sims PF, Ashe MP. 2011. Glucose depletion inhibits translation initiation via eIF4A loss and subsequent 48S preinitiation complex accumulation, while the pentose phosphate pathway is coordinately up-regulated. Mol Biol Cell 22:3379–3393. doi: 10.1091/mbc.E11-02-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barnett JA. 2003. A history of research on yeasts 6: the main respiratory pathway. Yeast 20:1015–1044. doi: 10.1002/yea.1021. [DOI] [PubMed] [Google Scholar]
- 37.Cupp J, McAlister-Henn L. 1991. NAD(+)-dependent isocitrate dehydrogenase. Cloning, nucleotide sequence, and disruption of the IDH2 gene from Saccharomyces cerevisiae. J Biol Chem 266:22199–22205. doi: 10.1016/S0021-9258(18)54554-3. [DOI] [PubMed] [Google Scholar]
- 38.Cupp JR, McAlister-Henn L. 1992. Cloning and characterization of the gene encoding the IDH1 subunit of NAD(+)-dependent isocitrate dehydrogenase from Saccharomyces cerevisiae. J Biol Chem 267:16417–16423. [PubMed] [Google Scholar]
- 39.Dickinson JR, Roy DJ, Dawes IW. 1986. A mutation affecting lipoamide dehydrogenase, pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase activities in Saccharomyces cerevisiae. Mol Gen Genet 204:103–107. doi: 10.1007/BF00330195. [DOI] [PubMed] [Google Scholar]
- 40.Repetto B, Tzagoloff A. 1989. Structure and regulation of KGD1, the structural gene for yeast alpha-ketoglutarate dehydrogenase. Mol Cell Biol 9:2695–2705. doi: 10.1128/mcb.9.6.2695-2705.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Repetto B, Tzagoloff A. 1990. Structure and regulation of KGD2, the structural gene for yeast dihydrolipoyl transsuccinylase. Mol Cell Biol 10:4221–4232. doi: 10.1128/mcb.10.8.4221-4232.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Przybyla‐Zawislak B, Dennis RA, Zakharkin SO, McCammon MT. 1998. Genes of succinyl‐CoA ligase from Saccharomyces cerevisiae. Eur J Biochem 258:736–743. doi: 10.1046/j.1432-1327.1998.2580736.x. [DOI] [PubMed] [Google Scholar]
- 43.Bullis BL, Lemire BD. 1994. Isolation and characterization of the Saccharomyces cerevisiae SDH4 gene encoding a membrane anchor subunit of succinate dehydrogenase. J Biol Chem 269:6543–6549. [PubMed] [Google Scholar]
- 44.Chapman KB, Solomon SD, Boeke JD. 1992. SDH1, the gene encoding the succinate dehydrogenase flavoprotein subunit from Saccharomyces cerevisiae. Gene 118:131–136. doi: 10.1016/0378-1119(92)90260-v. [DOI] [PubMed] [Google Scholar]
- 45.Colby G, Ishii Y, Tzagoloff A. 1998. Suppression of sdh1 mutations by the SDH1b gene of Saccharomyces cerevisiae. Yeast 14:1001–1006. doi: 10.1002/(SICI)1097-0061(199808)14:11<1001::AID-YEA304>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 46.Gebert N, Gebert M, Oeljeklaus S, von der Malsburg K, Stroud DA, Kulawiak B, Wirth C, Zahedi RP, Dolezal P, Wiese S, Simon O, Schulze-Specking A, Truscott KN, Sickmann A, Rehling P, Guiard B, Hunte C, Warscheid B, van der Laan M, Pfanner N, Wiedemann N. 2011. Dual function of Sdh3 in the respiratory chain and TIM22 protein translocase of the mitochondrial inner membrane. Mol Cell 44:811–818. doi: 10.1016/j.molcel.2011.09.025. [DOI] [PubMed] [Google Scholar]
- 47.Lombardo A, Carine K, Scheffler IE. 1990. Cloning and characterization of the iron-sulfur subunit gene of succinate dehydrogenase from Saccharomyces cerevisiae. J Biol Chem 265:10419–10423. doi: 10.1016/S0021-9258(18)86962-9. [DOI] [PubMed] [Google Scholar]
- 48.Szeto SS, Reinke SN, Oyedotun KS, Sykes BD, Lemire BD. 2012. Expression of Saccharomyces cerevisiae Sdh3p and Sdh4p paralogs results in catalytically active succinate dehydrogenase isoenzymes. J Biol Chem 287:22509–22520. doi: 10.1074/jbc.M112.344275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chang YL, Hsieh MH, Chang WW, Wang HY, Lin MC, Wang CP, Lou PJ, Teng SC. 2015. Instability of succinate dehydrogenase in SDHD polymorphism connects reactive oxygen species production to nuclear and mitochondrial genomic mutations in yeast. Antioxid Redox Signal 22:587–602. doi: 10.1089/ars.2014.5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jia YK, Bécam AM, Herbert C. 1997. The CIT3 gene of Saccharomyces cerevisiae encodes a second mitochondrial isoform of citrate synthase. Mol Microbiol 24:53–59. doi: 10.1046/j.1365-2958.1997.3011669.x. [DOI] [PubMed] [Google Scholar]
- 51.Haselbeck RJ, Colman RF, McAlister-Henn L. 1992. Isolation and sequence of a cDNA encoding porcine mitochondrial NADP-specific isocitrate dehydrogenase. Biochemistry 31:6219–6223. doi: 10.1021/bi00142a007. [DOI] [PubMed] [Google Scholar]
- 52.Zhao WN, McAlister-Henn L. 1996. Expression and gene disruption analysis of the isocitrate dehydrogenase family in yeast. Biochemistry 35:7873–7878. doi: 10.1021/bi9605189. [DOI] [PubMed] [Google Scholar]
- 53.Gangloff SP, Marguet D, Lauquin G. 1990. Molecular cloning of the yeast mitochondrial aconitase gene (ACO1) and evidence of a synergistic regulation of expression by glucose plus glutamate. Mol Cell Biol 10:3551–3561. doi: 10.1128/mcb.10.7.3551-3561.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fazius F, Shelest E, Gebhardt P, Brock M. 2012. The fungal α‐aminoadipate pathway for lysine biosynthesis requires two enzymes of the aconitase family for the isomerization of homocitrate to homoisocitrate. Mol Microbiol 86:1508–1530. doi: 10.1111/mmi.12076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pronk JT, Yde Steensma H, Van Dijken JP. 1996. Pyruvate metabolism in Saccharomyces cerevisiae. Yeast 12:1607–1633. doi: 10.1002/(SICI)1097-0061(199612)12:16<1607::AID-YEA70>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 56.Stucka R, Dequin S, Salmon J-M, Gancedo C. 1991. DNA sequences in chromosomes 11 and VII code for pyruvate carboxylase isoenzymes in Saccharomyces cerevisiae: analysis of pyruvate carboxylase-deficient strains. Mol Gen Genet 229:307–315. doi: 10.1007/BF00272171. [DOI] [PubMed] [Google Scholar]
- 57.Brewster NK, Val DL, Walker ME, Wallace JC. 1994. Regulation of pyruvate carboxylase isozyme (PYC1, PYC2) gene expression in Saccharomyces cerevisiae during fermentative and nonfermentative growth. Arch Biochem Biophys 311:62–71. doi: 10.1006/abbi.1994.1209. [DOI] [PubMed] [Google Scholar]
- 58.Boles E, de Jong-Gubbels P, Pronk JT. 1998. Identification and characterization of MAE1, the Saccharomyces cerevisiae structural gene encoding mitochondrial malic enzyme. J Bacteriol 180:2875–2882. doi: 10.1128/JB.180.11.2875-2882.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Valdés-Hevia MD, de la Guerra R, Gancedo C. 1989. Isolation and characterization of the gene encoding phosphoenolpyruvate carboxykinase from Saccharomyces cerevisiae. FEBS Lett 258:313–316. doi: 10.1016/0014-5793(89)81682-5. [DOI] [PubMed] [Google Scholar]
- 60.Soontorngun N, Larochelle M, Drouin S, Robert F, Turcotte B. 2007. Regulation of gluconeogenesis in Saccharomyces cerevisiae is mediated by activator and repressor functions of Rds2. Mol Cell Biol 27:7895–7905. doi: 10.1128/MCB.01055-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Palmieri F, Agrimi G, Blanco E, Castegna A, Di Noia MA, Iacobazzi V, Lasorsa FM, Marobbio CM, Palmieri L, Scarcia P, Todisco S, Vozza A, Walker J. 2006. Identification of mitochondrial carriers in Saccharomyces cerevisiae by transport assay of reconstituted recombinant proteins. Biochim Biophys Acta 1757:1249–1262. doi: 10.1016/j.bbabio.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 62.Palmieri L, Palmieri F, Runswick MJ, Walker JE. 1996. Identification by bacterial expression and functional reconstitution of the yeast genomic sequence encoding the mitochondrial dicarboxylate carrier protein. FEBS Lett 399:299–302. doi: 10.1016/s0014-5793(96)01350-6. [DOI] [PubMed] [Google Scholar]
- 63.Palmieri F, Monné M. 2016. Discoveries, metabolic roles and diseases of mitochondrial carriers: a review. Biochim Biophys Acta 1863:2362–2378. doi: 10.1016/j.bbamcr.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 64.Cavero S, Traba J, Del Arco A, Satrústegui J. 2005. The calcium-dependent ATP-Mg/Pi mitochondrial carrier is a target of glucose-induced calcium signalling in Saccharomyces cerevisiae. Biochem J 392:537–544. doi: 10.1042/BJ20050806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smith CP, Thorsness PE. 2008. The molecular basis for relative physiological functionality of the ADP/ATP carrier isoforms in Saccharomyces cerevisiae. Genetics 179:1285–1299. doi: 10.1534/genetics.108.087700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Adrian GS, McCammon MT, Montgomery DL, Douglas MG. 1986. Sequences required for delivery and localization of the ADP/ATP translocator to the mitochondrial inner membrane. Mol Cell Biol 6:626–634. doi: 10.1128/mcb.6.2.626-634.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gawaz M, Douglas MG, Klingenberg M. 1990. Structure-function studies of adenine nucleotide transport in mitochondria. II. Biochemical analysis of distinct AAC1 and AAC2 proteins in yeast. J Biol Chem 265:14202–14208. [PubMed] [Google Scholar]
- 68.Chen XJ. 2004. Sal1p, a calcium-dependent carrier protein that suppresses an essential cellular function associated with the Aac2 isoform of ADP/ATP translocase in Saccharomyces cerevisiae. Genetics 167:607–617. doi: 10.1534/genetics.103.023655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Laco J, Zeman I, Pevala V, Polcic P, Kolarov J. 2010. Adenine nucleotide transport via Sal1 carrier compensates for the essential function of the mitochondrial ADP/ATP carrier. FEMS Yeast Res 10:290–296. doi: 10.1111/j.1567-1364.2010.00606.x. [DOI] [PubMed] [Google Scholar]
- 70.Todisco S, Agrimi G, Castegna A, Palmieri F. 2006. Identification of the mitochondrial NAD+ transporter in Saccharomyces cerevisiae. J Biol Chem 281:1524–1531. doi: 10.1074/jbc.M510425200. [DOI] [PubMed] [Google Scholar]
- 71.Marobbio CM, Vozza A, Harding M, Bisaccia F, Palmieri F, Walker JE. 2002. Identification and reconstitution of the yeast mitochondrial transporter for thiamine pyrophosphate. EMBO J 21:5653–5661. doi: 10.1093/emboj/cdf583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bafunno V, Giancaspero TA, Brizio C, Bufano D, Passarella S, Boles E, Barile M. 2004. Riboflavin uptake and FAD synthesis in Saccharomyces cerevisiae mitochondria: involvement of the Flx1p carrier in FAD export. J Biol Chem 279:95–102. doi: 10.1074/jbc.M308230200. [DOI] [PubMed] [Google Scholar]
- 73.Tzagoloff A, Jang J, Glerum DM, Wu M. 1996. FLX1 codes for a carrier protein involved in maintaining a proper balance of flavin nucleotides in yeast mitochondria. J Biol Chem 271:7392–7397. doi: 10.1074/jbc.271.13.7392. [DOI] [PubMed] [Google Scholar]
- 74.Prohl C, Pelzer W, Diekert K, Kmita H, Bedekovics T, Kispal G, Lill R. 2001. The yeast mitochondrial carrier Leu5p and its human homologue Graves' disease protein are required for accumulation of coenzyme A in the matrix. Mol Cell Biol 21:1089–1097. doi: 10.1128/MCB.21.4.1089-1097.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bender T, Pena G, Martinou JC. 2015. Regulation of mitochondrial pyruvate uptake by alternative pyruvate carrier complexes. EMBO J 34:911–924. doi: 10.15252/embj.201490197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, Kunji ER, Martinou JC. 2012. Identification and functional expression of the mitochondrial pyruvate carrier. Science 337:93–96. doi: 10.1126/science.1218530. [DOI] [PubMed] [Google Scholar]
- 77.Timón-Gómez A, Proft M, Pascual-Ahuir A. 2013. Differential regulation of mitochondrial pyruvate carrier genes modulates respiratory capacity and stress tolerance in yeast. PLoS One 8:e79405. doi: 10.1371/journal.pone.0079405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Palmieri L, Lasorsa FM, De Palma A, Palmieri F, Runswick MJ, Walker JE. 1997. Identification of the yeast ACR1 gene product as a succinate-fumarate transporter essential for growth on ethanol or acetate. FEBS Lett 417:114–118. doi: 10.1016/s0014-5793(97)01269-6. [DOI] [PubMed] [Google Scholar]
- 79.Palmieri L, Vozza A, Hönlinger A, Dietmeier K, Palmisano A, Zara V, Palmieri F. 1999. The mitochondrial dicarboxylate carrier is essential for the growth of Saccharomyces cerevisiae on ethanol or acetate as the sole carbon source. Mol Microbiol 31:569–577. doi: 10.1046/j.1365-2958.1999.01197.x. [DOI] [PubMed] [Google Scholar]
- 80.Fernández M, Fernández E, Rodicio R. 1994. ACR1, a gene encoding a protein related to mitochondrial carriers, is essential for acetyl-CoA synthetase activity in Saccharomyces cerevisiae. Mol Gen Genet 242:727–735. doi: 10.1007/BF00283428. [DOI] [PubMed] [Google Scholar]
- 81.Palmieri L, Vozza A, Agrimi G, De Marco V, Runswick MJ, Palmieri F, Walker JE. 1999. Identification of the yeast mitochondrial transporter for oxaloacetate and sulfate. J Biol Chem 274:22184–22190. doi: 10.1074/jbc.274.32.22184. [DOI] [PubMed] [Google Scholar]
- 82.Marobbio CMT, Giannuzzi G, Paradies E, Pierri CL, Palmieri F. 2008. α-Isopropylmalate, a leucine biosynthesis intermediate in yeast, is transported by the mitochondrial oxalacetate carrier. J Biol Chem 283:28445–28453. doi: 10.1074/jbc.M804637200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Castegna A, Scarcia P, Agrimi G, Palmieri L, Rottensteiner H, Spera I, Germinario L, Palmieri F. 2010. Identification and functional characterization of a novel mitochondrial carrier for citrate and oxoglutarate in Saccharomyces cerevisiae. J Biol Chem 285:17359–17370. doi: 10.1074/jbc.M109.097188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tibbetts AS, Sun Y, Lyon NA, Ghrist AC, Trotter PJ. 2002. Yeast mitochondrial oxodicarboxylate transporters are important for growth on oleic acid. Arch Biochem Biophys 406:96–104. doi: 10.1016/s0003-9861(02)00419-8. [DOI] [PubMed] [Google Scholar]
- 85.Scarcia P, Palmieri L, Agrimi G, Palmieri F, Rottensteiner H. 2017. Three mitochondrial transporters of Saccharomyces cerevisiae are essential for ammonium fixation and lysine biosynthesis in synthetic minimal medium. Mol Genet Metab 122:54–60. doi: 10.1016/j.ymgme.2017.07.004. [DOI] [PubMed] [Google Scholar]
- 86.Kaplan RS, Mayor JA, Gremse DA, Wood DO. 1995. High level expression and characterization of the mitochondrial citrate transport protein from the yeast Saccharomyces cerevisiae. J Biol Chem 270:4108–4114. doi: 10.1074/jbc.270.8.4108. [DOI] [PubMed] [Google Scholar]
- 87.Kaplan RS, Mayor JA, Kakhniashvili D, Gremse DA, Wood DO, Nelson DR. 1996. Deletion of the nuclear gene encoding the mitochondrial citrate transport protein from Saccharomyces cerevisiae. Biochem Biophys Res Commun 226:657–662. doi: 10.1006/bbrc.1996.1411. [DOI] [PubMed] [Google Scholar]
- 88.Cho JH, Ha SJ, Kao LR, Megraw TL, Chae CB. 1998. A novel DNA-binding protein bound to the mitochondrial inner membrane restores the null mutation of mitochondrial histone Abf2p in Saccharomyces cerevisiae. Mol Cell Biol 18:5712–5723. doi: 10.1128/MCB.18.10.5712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Palmieri L, Agrimi G, Runswick MJ, Fearnley IM, Palmieri F, Walker JE. 2001. Identification in Saccharomyces cerevisiae of two isoforms of a novel mitochondrial transporter for 2-oxoadipate and 2-oxoglutarate. J Biol Chem 276:1916–1922. doi: 10.1074/jbc.M004332200. [DOI] [PubMed] [Google Scholar]
- 90.Camarasa C, Faucet V, Dequin S. 2007. Role in anaerobiosis of the isoenzymes for Saccharomyces cerevisiae fumarate reductase encoded by OSM1 and FRDS1. Yeast 24:391–401. doi: 10.1002/yea.1467. [DOI] [PubMed] [Google Scholar]
- 91.Jouhten P, Penttilä M. 2014. Anaerobic carbon metabolism of Saccharomyces cerevisiae, p 57–82. In Piškur J, Compagno C (ed), Molecular mechanisms in yeast carbon metabolism. Springer, Berlin, Germany. [Google Scholar]
- 92.Liu Z, Österlund T, Hou J, Petranovic D, Nielsen J. 2013. Anaerobic α-amylase production and secretion with fumarate as the final electron acceptor in Saccharomyces cerevisiae. Appl Environ Microbiol 79:2962–2967. doi: 10.1128/AEM.03207-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Neal SE, Dabir DV, Wijaya J, Boon C, Koehler CM. 2017. Osm1 facilitates the transfer of electrons from Erv1 to fumarate in the redox-regulated import pathway in the mitochondrial intermembrane space. Mol Biol Cell 28:2773–2785. doi: 10.1091/mbc.E16-10-0712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Arikawa Y, Enomoto K, Muratsubaki H, Okazaki M. 1998. Soluble fumarate reductase isoenzymes from Saccharomyces cerevisiae are required for anaerobic growth. FEMS Microbiol Lett 165:111–116. doi: 10.1111/j.1574-6968.1998.tb13134.x. [DOI] [PubMed] [Google Scholar]
- 95.Kunze M, Pracharoenwattana I, Smith SM, Hartig A. 2006. A central role for the peroxisomal membrane in glyoxylate cycle function. Biochim Biophys Acta 1763:1441–1452. doi: 10.1016/j.bbamcr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 96.Xiberras J, Klein M, Nevoigt E. 2019. Glycerol as a substrate for Saccharomyces cerevisiae based bioprocesses: knowledge gaps regarding the central carbon catabolism of this 'non-fermentable' carbon source. Biotechnol Adv 37:107378. doi: 10.1016/j.biotechadv.2019.03.017. [DOI] [PubMed] [Google Scholar]
- 97.Boubekeur S, Camougrand N, Bunoust O, Rigoulet M, Guérin B. 2001. Participation of acetaldehyde dehydrogenases in ethanol and pyruvate metabolism of the yeast Saccharomyces cerevisiae. Eur J Biochem 268:5057–5065. doi: 10.1046/j.1432-1033.2001.02418.x. [DOI] [PubMed] [Google Scholar]
- 98.Kurita O, Nishida Y. 1999. Involvement of mitochondrial aldehyde dehydrogenase ALD5 in maintenance of the mitochondrial electron transport chain in Saccharomyces cerevisiae. FEMS Microbiol Lett 181:281–287. doi: 10.1111/j.1574-6968.1999.tb08856.x. [DOI] [PubMed] [Google Scholar]
- 99.Meaden PG, Dickinson FM, Mifsud A, Tessier W, Westwater J, Bussey H, Midgley M. 1997. The ALD6 gene of Saccharomyces cerevisiae encodes a cytosolic, Mg(2+)-activated acetaldehyde dehydrogenase. Yeast 13:1319–1327. doi: 10.1002/(SICI)1097-0061(199711)13:14<1319::AID-YEA183>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 100.White WH, Skatrud PL, Xue Z, Toyn JH. 2003. Specialization of function among aldehyde dehydrogenases: the ALD2 and ALD3 genes are required for beta-alanine biosynthesis in Saccharomyces cerevisiae. Genetics 163:69–77. doi: 10.1093/genetics/163.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Navarro-Aviño JP, Prasad R, Miralles VJ, Benito RM, Serrano R. 1999. A proposal for nomenclature of aldehyde dehydrogenases in Saccharomyces cerevisiae and characterization of the stress-inducible ALD2 and ALD3 genes. Yeast 15:829–842. doi: 10.1002/(SICI)1097-0061(199907)15:10A<829::AID-YEA423>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 102.Krivoruchko A, Zhang Y, Siewers V, Chen Y, Nielsen J. 2015. Microbial acetyl-CoA metabolism and metabolic engineering. Metab Eng 28:28–42. doi: 10.1016/j.ymben.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 103.Chen Y, Siewers V, Nielsen J. 2012. Profiling of cytosolic and peroxisomal acetyl-CoA metabolism in Saccharomyces cerevisiae. PLoS One 7:e42475. doi: 10.1371/journal.pone.0042475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Orlandi I, Casatta N, Vai M. 2012. Lack of Ach1 CoA-transferase triggers apoptosis and decreases chronological lifespan in yeast. Front Oncol 2:67. doi: 10.3389/fonc.2012.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Takahashi H, McCaffery JM, Irizarry RA, Boeke JD. 2006. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol Cell 23:207–217. doi: 10.1016/j.molcel.2006.05.040. [DOI] [PubMed] [Google Scholar]
- 106.van Rossum HM, Kozak BU, Niemeijer MS, Duine HJ, Luttik MAH, Boer VM, Kötter P, Daran J-MG, van Maris AJA, Pronk JT. 2016. Alternative reactions at the interface of glycolysis and citric acid cycle in Saccharomyces cerevisiae. FEMS Yeast Res 16:fow017. doi: 10.1093/femsyr/fow017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Regev-Rudzki N, Karniely S, Ben-Haim NN, Pines O. 2005. Yeast aconitase in two locations and two metabolic pathways: seeing small amounts is believing. Mol Biol Cell 16:4163–4171. doi: 10.1091/mbc.e04-11-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Taylor KM, Kaplan CP, Gao X, Baker A. 1996. Localization and targeting of isocitrate lyases in Saccharomyces cerevisiae. Biochem J 319:255–262. doi: 10.1042/bj3190255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chaves RS, Herrero P, Ordiz I, Angeles del Brio M, Moreno F. 1997. Isocitrate lyase localisation in Saccharomyces cerevisiae cells. Gene 198:165–169. doi: 10.1016/s0378-1119(97)00311-9. [DOI] [PubMed] [Google Scholar]
- 110.Kunze M, Kragler F, Binder M, Hartig A, Gurvitz A. 2002. Targeting of malate synthase 1 to the peroxisomes of Saccharomyces cerevisiae cells depends on growth on oleic acid medium. Eur J Biochem 269:915–922. doi: 10.1046/j.0014-2956.2001.02727.x. [DOI] [PubMed] [Google Scholar]
- 111.Hiltunen JK, Mursula AM, Rottensteiner H, Wierenga RK, Kastaniotis AJ, Gurvitz A. 2003. The biochemistry of peroxisomal beta-oxidation in the yeast Saccharomyces cerevisiae. FEMS Microbiol Rev 27:35–64. doi: 10.1016/S0168-6445(03)00017-2. [DOI] [PubMed] [Google Scholar]
- 112.Kim KS, Rosenkrantz MS, Guarente L. 1986. Saccharomyces cerevisiae contains two functional citrate synthase genes. Mol Cell Biol 6:1936–1942. doi: 10.1128/mcb.6.6.1936-1942.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hartig A, Simon MM, Schuster T, Daugherty JR, Yoo HS, Cooper TG. 1992. Differentially regulated malate synthase genes participate in carbon and nitrogen metabolism of S. cerevisiae. Nucleic Acids Res 20:5677–5686. doi: 10.1093/nar/20.21.5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lee YJ, Jang JW, Kim KJ, Maeng PJ. 2011. TCA cycle-independent acetate metabolism via the glyoxylate cycle in Saccharomyces cerevisiae. Yeast 28:153–166. doi: 10.1002/yea.1828. [DOI] [PubMed] [Google Scholar]
- 115.Fernández E, Moreno F, Rodicio R. 1992. The ICL1 gene from Saccharomyces cerevisiae. Eur J Biochem 204:983–990. doi: 10.1111/j.1432-1033.1992.tb16720.x. [DOI] [PubMed] [Google Scholar]
- 116.Minard KI, McAlister-Henn L. 1991. Isolation, nucleotide sequence analysis, and disruption of the MDH2 gene from Saccharomyces cerevisiae: evidence for three isozymes of yeast malate dehydrogenase. Mol Cell Biol 11:370–380. doi: 10.1128/mcb.11.1.370-380.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Luttik MA, Kötter P, Salomons FA, van der Klei IJ, van Dijken JP, Pronk JT. 2000. The Saccharomyces cerevisiae ICL2 gene encodes a mitochondrial 2-methylisocitrate lyase involved in propionyl-coenzyme A metabolism. J Bacteriol 182:7007–7013. doi: 10.1128/JB.182.24.7007-7013.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fernández E, Fernández M, Rodicio R. 1993. Two structural genes are encoding malate synthase isoenzymes in Saccharomyces cerevisiae. FEBS Lett 320:271–275. doi: 10.1016/0014-5793(93)80601-p. [DOI] [PubMed] [Google Scholar]
- 119.Dijken JP, Scheffers W. 1986. Redox balances in the metabolism of sugars by yeasts. FEMS Microbiology Rev 32:199–224. doi: 10.1111/j.1574-6968.1986.tb01194.x. [DOI] [Google Scholar]
- 120.Ansell R, Granath K, Hohmann S, Thevelein JM, Adler L. 1997. The two isoenzymes for yeast NAD+-dependent glycerol 3-phosphate dehydrogenase encoded by GPD1 and GPD2 have distinct roles in osmoadaptation and redox regulation. EMBO J 16:2179–2187. doi: 10.1093/emboj/16.9.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nevoigt E, Stahl U. 1997. Osmoregulation and glycerol metabolism in the yeast Saccharomyces cerevisiae. FEMS Microbiol Rev 21:231–241. doi: 10.1111/j.1574-6976.1997.tb00352.x. [DOI] [PubMed] [Google Scholar]
- 122.Björkqvist S, Ansell R, Adler L, Lidén G. 1997. Physiological response to anaerobicity of glycerol-3-phosphate dehydrogenase mutants of Saccharomyces cerevisiae. Appl Environ Microbiol 63:128–132. doi: 10.1128/aem.63.1.128-132.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Albertyn J, Hohmann S, Thevelein JM, Prior BA. 1994. GPD1, which encodes glycerol-3-phosphate dehydrogenase, is essential for growth under osmotic stress in Saccharomyces cerevisiae, and its expression is regulated by the high-osmolarity glycerol response pathway. Mol Cell Biol 14:4135–4144. doi: 10.1128/mcb.14.6.4135-4144.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Al-Saryi NA, Al-Hejjaj MY, van Roermund CWT, Hulmes GE, Ekal L, Payton C, Wanders RJA, Hettema EH. 2017. Two NAD-linked redox shuttles maintain the peroxisomal redox balance in Saccharomyces cerevisiae. Sci Rep 7:11868. doi: 10.1038/s41598-017-11942-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Eriksson P, André L, Ansell R, Blomberg A, Adler L. 1995. Cloning and characterization of GPD2, a second gene encoding sn-glycerol 3-phosphate dehydrogenase (NAD+) in Saccharomyces cerevisiae, and its comparison with GPD1. Mol Microbiol 17:95–107. doi: 10.1111/j.1365-2958.1995.mmi_17010095.x. [DOI] [PubMed] [Google Scholar]
- 126.Nissen TL, Hamann CW, Kielland-Brandt MC, Nielsen J, Villadsen J. 2000. Anaerobic and aerobic batch cultivations of Saccharomyces cerevisiae mutants impaired in glycerol synthesis. Yeast 16:463–474. doi: 10.1002/(SICI)1097-0061(20000330)16:5<463::AID-YEA535>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 127.Norbeck J, Pâhlman AK, Akhtar N, Blomberg A, Adler L. 1996. Purification and characterization of two isoenzymes of DL-glycerol-3-phosphatase from Saccharomyces cerevisiae. Identification of the corresponding GPP1 and GPP2 genes and evidence for osmotic regulation of Gpp2p expression by the osmosensing mitogen-activated protein kinase signal transduction pathway. J Biol Chem 271:13875–13881. doi: 10.1074/jbc.271.23.13875. [DOI] [PubMed] [Google Scholar]
- 128.Pahlman AK, Granath K, Ansell R, Hohmann S, Adler L. 2001. The yeast glycerol 3-phosphatases Gpp1p and Gpp2p are required for glycerol biosynthesis and differentially involved in the cellular responses to osmotic, anaerobic, and oxidative stress. J Biol Chem 276:3555–3563. doi: 10.1074/jbc.M007164200. [DOI] [PubMed] [Google Scholar]
- 129.Canelas AB, van Gulik WM, Heijnen JJ. 2008. Determination of the cytosolic free NAD/NADH ratio in Saccharomyces cerevisiae under steady-state and highly dynamic conditions. Biotechnol Bioeng 100:734–743. doi: 10.1002/bit.21813. [DOI] [PubMed] [Google Scholar]
- 130.Walther T, Mtimet N, Alkim C, Vax A, Loret MO, Ullah A, Gancedo C, Smits GJ, Francois JM. 2013. Metabolic phenotypes of Saccharomyces cerevisiae mutants with altered trehalose 6-phosphate dynamics. Biochem J 454:227–237. doi: 10.1042/BJ20130587. [DOI] [PubMed] [Google Scholar]
- 131.Kolarov J, Kolarova N, Nelson N. 1990. A third ADP/ATP translocator gene in yeast. J Biol Chem 265:12711–12716. [PubMed] [Google Scholar]
- 132.Sasaki M, Kumagai H, Takegawa K, Tohda H. 2013. Characterization of genome-reduced fission yeast strains. Nucleic Acids Res 41:5382–5399. doi: 10.1093/nar/gkt233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Murakami K, Tao E, Ito Y, Sugiyama M, Kaneko Y, Harashima S, Sumiya T, Nakamura A, Nishizawa M. 2007. Large scale deletions in the Saccharomyces cerevisiae genome create strains with altered regulation of carbon metabolism. Appl Microbiol Biotechnol 75:589–597. doi: 10.1007/s00253-007-0859-2. [DOI] [PubMed] [Google Scholar]
- 134.Richardson SM, Mitchell LA, Stracquadanio G, Yang K, Dymond JS, DiCarlo JE, Lee D, Huang CLV, Chandrasegaran S, Cai Y, Boeke JD, Bader JS. 2017. Design of a synthetic yeast genome. Science 355:1040–1044. doi: 10.1126/science.aaf4557. [DOI] [PubMed] [Google Scholar]
- 135.Shao Y, Lu N, Wu Z, Cai C, Wang S, Zhang L-L, Zhou F, Xiao S, Liu L, Zeng X, Zheng H, Yang C, Zhao Z, Zhao G, Zhou J-Q, Xue X, Qin Z. 2018. Creating a functional single-chromosome yeast. Nature 560:331–335. doi: 10.1038/s41586-018-0382-x. [DOI] [PubMed] [Google Scholar]
- 136.Wisselink HW, Cipollina C, Oud B, Crimi B, Heijnen JJ, Pronk JT, van Maris AJ. 2010. Metabolome, transcriptome and metabolic flux analysis of arabinose fermentation by engineered Saccharomyces cerevisiae. Metab Eng 12:537–551. doi: 10.1016/j.ymben.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 137.Shen X-X, Opulente DA, Kominek J, Zhou X, Steenwyk JL, Buh KV, Haase MAB, Wisecaver JH, Wang M, Doering DT, Boudouris JT, Schneider RM, Langdon QK, Ohkuma M, Endoh R, Takashima M, Manabe R-i, Čadež N, Libkind D, Rosa CA, DeVirgilio J, Hulfachor AB, Groenewald M, Kurtzman CP, Hittinger CT, Rokas A. 2018. Tempo and mode of genome evolution in the budding yeast subphylum. Cell 175:1533–1545.e20. doi: 10.1016/j.cell.2018.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kuzmin E, VanderSluis B, Nguyen Ba AN, Wang W, Koch EN, Usaj M, Khmelinskii A, Usaj MM, van Leeuwen J, Kraus O, Tresenrider A, Pryszlak M, Hu M-C, Varriano B, Costanzo M, Knop M, Moses A, Myers CL, Andrews BJ, Boone C. 2020. Exploring whole-genome duplicate gene retention with complex genetic interaction analysis. Science 368:eaaz5667. doi: 10.1126/science.aaz5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bakker BM, Overkamp KM, van Maris AJ, Kötter P, Luttik MA, van Dijken JP, Pronk JT. 2001. Stoichiometry and compartmentation of NADH metabolism in Saccharomyces cerevisiae. FEMS Microbiol Rev 25:15–37. doi: 10.1111/j.1574-6976.2001.tb00570.x. [DOI] [PubMed] [Google Scholar]
- 140.Miyagi H, Kawai S, Murata K. 2009. Two sources of mitochondrial NADPH in the yeast Saccharomyces cerevisiae. J Biol Chem 284:7553–7560. doi: 10.1074/jbc.M804100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Swiegers JH, Dippenaar N, Pretorius IS, Bauer FF. 2001. Carnitine-dependent metabolic activities in Saccharomyces cerevisiae: three carnitine acetyltransferases are essential in a carnitine-dependent strain. Yeast 18:585–595. doi: 10.1002/yea.712. [DOI] [PubMed] [Google Scholar]
- 142.Bakker BM, Bro C, Kötter P, Luttik MA, van Dijken JP, Pronk JT. 2000. The mitochondrial alcohol dehydrogenase Adh3p is involved in a redox shuttle in Saccharomyces cerevisiae. J Bacteriol 182:4730–4737. doi: 10.1128/JB.182.17.4730-4737.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wieczorke R, Krampe S, Weierstall T, Freidel K, Hollenberg CP, Boles E. 1999. Concurrent knock-out of at least 20 transporter genes is required to block uptake of hexoses in Saccharomyces cerevisiae. FEBS Lett 464:123–128. doi: 10.1016/S0014-5793(99)01698-1. [DOI] [PubMed] [Google Scholar]
- 144.Wijsman M, Swiat MA, Marques WL, Hettinga JK, van den Broek M, Torre CP, Mans R, Pronk JT, Daran JM, Daran-Lapujade P. 2019. A toolkit for rapid CRISPR-SpCas9 assisted construction of hexose-transport-deficient Saccharomyces cerevisiae strains. FEMS Yeast Res 19:foy107. doi: 10.1093/femsyr/foy107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Brickwedde A, Brouwers N, van den Broek M, Gallego MJ, Fraiture JL, Pronk JT, Daran J-MG. 2018. Structural, physiological and regulatory analysis of maltose transporter genes in Saccharomyces eubayanus CBS 12357T. Front Microbiol 9 doi: 10.3389/fmicb.2018.01786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Brown CA, Murray AW, Verstrepen KJ. 2010. Rapid expansion and functional divergence of subtelomeric gene families in yeasts. Curr Biol 20:895–903. doi: 10.1016/j.cub.2010.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Aslan S, Noor E, Bar-Even A. 2017. Holistic bioengineering: rewiring central metabolism for enhanced bioproduction. Biochem J 474:3935–3950. doi: 10.1042/BCJ20170377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Papagianni M. 2012. Recent advances in engineering the central carbon metabolism of industrially important bacteria. Microb Cell Fact 11:50. doi: 10.1186/1475-2859-11-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.François JM, Lachaux C, Morin N. 2019. Synthetic biology applied to carbon conservative and carbon dioxide recycling pathways. Front Bioeng Biotechnol 7:446. doi: 10.3389/fbioe.2019.00446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sánchez-Pascuala A, Fernández-Cabezón L, de Lorenzo V, Nikel PI. 2019. Functional implementation of a linear glycolysis for sugar catabolism in Pseudomonas putida. Metab Eng 54:200–211. doi: 10.1016/j.ymben.2019.04.005. [DOI] [PubMed] [Google Scholar]
- 151.van Rossum HM, Kozak BU, Pronk JT, van Maris AJA. 2016. Engineering cytosolic acetyl-coenzyme A supply in Saccharomyces cerevisiae: pathway stoichiometry, free-energy conservation and redox-cofactor balancing. Metab Eng 36:99–115. doi: 10.1016/j.ymben.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 152.Liu Q, Yu T, Li X, Chen Y, Campbell K, Nielsen J, Chen Y. 2019. Rewiring carbon metabolism in yeast for high level production of aromatic chemicals. Nat Commun 10:4976. doi: 10.1038/s41467-019-12961-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Meadows AL, Hawkins KM, Tsegaye Y, Antipov E, Kim Y, Raetz L, Dahl RH, Tai A, Mahatdejkul-Meadows T, Xu L, Zhao L, Dasika MS, Murarka A, Lenihan J, Eng D, Leng JS, Liu CL, Wenger JW, Jiang H, Chao L, Westfall P, Lai J, Ganesan S, Jackson P, Mans R, Platt D, Reeves CD, Saija PR, Wichmann G, Holmes VF, Benjamin K, Hill PW, Gardner TS, Tsong AE. 2016. Rewriting yeast central carbon metabolism for industrial isoprenoid production. Nature 537:694–697. doi: 10.1038/nature19769. [DOI] [PubMed] [Google Scholar]
- 154.Koopman F, Beekwilder J, Crimi B, van Houwelingen A, Hall RD, Bosch D, van Maris AJ, Pronk JT, Daran JM. 2012. De novo production of the flavonoid naringenin in engineered Saccharomyces cerevisiae. Microb Cell Fact 11:155. doi: 10.1186/1475-2859-11-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Boonekamp FJ, Dashko S, Duiker D, Gehrmann T, van den Broek M, den Ridder M, Pabst M, Robert V, Abeel T, Postma ED, Daran JM, Daran-Lapujade P. 2020. Design and experimental evaluation of a minimal, innocuous watermarking strategy to distinguish near-identical DNA and RNA sequences. ACS Synth Biol 9:1361–1375. doi: 10.1021/acssynbio.0c00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Entian K-D, Kötter P. 2007. 25 yeast genetic strain and plasmid collections, p 629–666. In Stansfield I, Stark MJR (ed), Methods in microbiology, vol 36. Academic Press, Amsterdam, Netherlands. [Google Scholar]
- 157.Nijkamp JF, van den Broek M, Datema E, de Kok S, Bosman L, Luttik MA, Daran-Lapujade P, Vongsangnak W, Nielsen J, Heijne WH, Klaassen P, Paddon CJ, Platt D, Kötter P, van Ham RC, Reinders MJ, Pronk JT, de Ridder D, Daran J-M. 2012. De novo sequencing, assembly and analysis of the genome of the laboratory strain Saccharomyces cerevisiae CEN.PK113-7D, a model for modern industrial biotechnology. Microb Cell Fact 11:36. doi: 10.1186/1475-2859-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mans R, van Rossum HM, Wijsman M, Backx A, Kuijpers NG, van den Broek M, Daran-Lapujade P, Pronk JT, van Maris AJ, Daran JM. 2015. CRISPR/Cas9: a molecular Swiss army knife for simultaneous introduction of multiple genetic modifications in Saccharomyces cerevisiae. FEMS Yeast Res 15:1–15. doi: 10.1093/femsyr/fov004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Verduyn C, Postma E, Scheffers WA, Van Dijken JP. 1992. Effect of benzoic acid on metabolic fluxes in yeasts: a continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast 8:501–517. doi: 10.1002/yea.320080703. [DOI] [PubMed] [Google Scholar]
- 160.Solis-Escalante D, Kuijpers NG, Bongaerts N, Bolat I, Bosman L, Pronk JT, Daran JM, Daran-Lapujade P. 2013. amdSYM, a new dominant recyclable marker cassette for Saccharomyces cerevisiae. FEMS Yeast Res 13:126–139. doi: 10.1111/1567-1364.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Gietz RD, Woods RA. 2002. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol 350:87–96. doi: 10.1016/s0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- 162.Looke M, Kristjuhan K, Kristjuhan A. 2011. Extraction of genomic DNA from yeasts for PCR-based applications. Biotechniques 50:325–328. doi: 10.2144/000113672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Salazar AN, Gorter de Vries AR, van den Broek M, Wijsman M, de la Torre Cortes P, Brickwedde A, Brouwers N, Daran JG, Abeel T. 2017. Nanopore sequencing enables near-complete de novo assembly of Saccharomyces cerevisiae reference strain CEN.PK113-7D. FEMS Yeast Res 17. doi: 10.1093/femsyr/fox074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford) 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The sequence alignment/map format and SAMtools. Bioinformatics (Oxford) 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, Earl AM. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Thorvaldsdottir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Nijkamp JF, van den Broek MA, Geertman JM, Reinders MJ, Daran JM, de Ridder D. 2012. De novo detection of copy number variation by co-assembly. Bioinformatics 28:3195–3202. doi: 10.1093/bioinformatics/bts601. [DOI] [PubMed] [Google Scholar]
- 169.Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Douma RD, de Jonge LP, Jonker CT, Seifar RM, Heijnen JJ, van Gulik WM. 2010. Intracellular metabolite determination in the presence of extracellular abundance: application to the penicillin biosynthesis pathway in Penicillium chrysogenum. Biotechnol Bioeng 107:105–115. doi: 10.1002/bit.22786. [DOI] [PubMed] [Google Scholar]
- 171.Wu L, Mashego MR, van Dam JC, Proell AM, Vinke JL, Ras C, van Winden WA, van Gulik WM, Heijnen JJ. 2005. Quantitative analysis of the microbial metabolome by isotope dilution mass spectrometry using uniformly 13C-labeled cell extracts as internal standards. Anal Biochem 336:164–171. doi: 10.1016/j.ab.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 172.de Jonge LP, Buijs NA, ten Pierick A, Deshmukh A, Zhao Z, Kiel JA, Heijnen JJ, van Gulik WM. 2011. Scale-down of penicillin production in Penicillium chrysogenum. Biotechnol J 6:944–958. doi: 10.1002/biot.201000409. [DOI] [PubMed] [Google Scholar]
- 173.Niedenführ S, ten Pierick A, van Dam PT, Suarez-Mendez CA, Nöh K, Wahl SA. 2016. Natural isotope correction of MS/MS measurements for metabolomics and (13)C fluxomics. Biotechnol Bioeng 113:1137–1147. doi: 10.1002/bit.25859. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genetic characteristics of the paralogs considered for deletion. Download Table S1, PDF file, 0.1 MB (144.9KB, pdf) .
Copyright © 2022 Postma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Mitochondrial carrier proteins. Download Table S2, PDF file, 0.1 MB (148.9KB, pdf) .
Copyright © 2022 Postma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Growth rate of the OAC1 deletion mutant. Maximum specific growth rate of S. cerevisiae CEN.PK113-7D (naive reference strain) and the OAC1 deletion mutant, S. cerevisiae IMK588, tested in shake flasks on selective SMD medium. Growth rates represent averages and standard deviations of two biological duplicates. *, P < 0.05; IMK588 had a 26% slower growth rate with respect to CEN.PK113-7D (two-tailed paired homoscedastic t test). Download FIG S1, PDF file, 0.2 MB (154KB, pdf) .
Copyright © 2022 Postma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Growth rates of deletion strains. Maximum specific growth rates of all S. cerevisiae strains tested in shake flasks on selective SMD (A and C) or SME (B and D) supplemented with uracil. Represented are deletion strains in the naive background (A and B) and in the engineered background (C and D). Color coding matches that used in Fig. 2 in the main article. Growth rates represent averages and standard deviations of two biological duplicates. Growth rates of the deletion strains are expressed as percentages of the control strain IMX581 growth rate. Significant changes in growth rate with respect to the control strain are indicated (*, P < 0.05, two-tailed paired homoscedastic t test). Download FIG S2, PDF file, 0.3 MB (266.1KB, pdf) .
Copyright © 2022 Postma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Growth rates in specific environments. (A to D) Specific growth rate determinations under osmotic stress conditions (supplemented with uracil), measured with the growth profiler in biological triplicates. (E) Specific growth rates of FRDS1 and AAC3 deletion mutants on SMD supplemented with Tween, ergosterol, and uracil measured under anaerobic conditions in shake flasks in biological duplicates. (F) Specific growth rates of MPC3 complementation strains on SME supplemented with uracil in shake flasks in biological duplicates. (G) Specific growth rate of MPC3 complementation strain on SM with 83.3 mM pyruvate and supplemented with uracil in shake flasks in biological duplicates. Significant changes in growth rates with respect to the parental strain are indicated (*, P < 0.05, two-tailed paired homoscedastic t test). Download FIG S3, PDF file, 0.2 MB (200KB, pdf) .
Copyright © 2022 Postma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Strains. Download Table S3, PDF file, 0.2 MB (204.1KB, pdf) .
Copyright © 2022 Postma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Plasmids. Download Table S4, PDF file, 0.2 MB (193.4KB, pdf) .
Copyright © 2022 Postma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Primers used in this study. Download Table S5, PDF file, 0.2 MB (199.6KB, pdf) .
Copyright © 2022 Postma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Strain transformations. Download Table S6, PDF file, 0.2 MB (180.1KB, pdf) .
Copyright © 2022 Postma et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
Short- and long-read sequencing data are available at NCBI under BioProject PRJNA757356.