

Abstract
Technological progress over the past 15 years has fueled an explosion in genome‐wide chromatin profiling tools that take advantage of low‐cost short‐read sequencing technologies to map particular chromatin features. Here, we survey the recent development of epigenomic tools that provide precise positions of chromatin proteins genome‐wide in intact cells or nuclei. Some profiling tools are based on tethering Micrococcal Nuclease to chromatin proteins of interest in situ, whereas others similarly tether Tn5 transposase to integrate DNA sequencing adapters (tagmentation) and so eliminate the need for library preparation. These in situ cleavage and tagmentation tools have gained in popularity over the past few years, with many protocol enhancements and adaptations for single‐cell and spatial chromatin profiling. The application of experimental and computational tools to address problems in gene regulation, eukaryotic development, and human disease are helping to define the emerging field of chromatin structural epigenomics.
Keywords: Micrococcal Nuclease, single‐cell CUT&Tag, spatial profiling, Tn5 transposase
1. INTRODUCTION
Completion of the draft sequence of the human genome in 2001 1 , 2 ushered in the post‐genomic era, during which attention turned to mapping the protein constituents and chromatin modifications onto the human and other eukaryotic genomes. At the outset of the post‐genomic era, the most popular tool for chromatin analysis was chromatin immunoprecipitation (ChIP), a method dating from the mid‐1980s. 3 , 4 With ChIP, chromatin from cells or nuclei is fragmented and solubilized, usually by sonication after cross‐linking, then targeted chromatin epitopes are pulled down using an antibody, whereupon the DNA is extracted. Genome‐wide read‐out for ChIP at first consisted of various DNA microarray platforms, collectively referred to as ChIP‐chip, which at its peak was capable of genome‐wide profiling from as few as 10,000 cells. 5 However, beginning in 2007, short‐read DNA sequencing made ChIP‐seq possible, 6 , 7 which rapidly replaced ChIP‐chip as the cost of Illumina sequencing dramatically decreased. Although ChIP‐seq remains the most common method for chromatin profiling, its dominance is being challenged by Assay for Transposase‐Accessible Chromatin using sequencing (ATAC‐seq) 8 and by in situ chromatin profiling methods, which is the subject of this survey (Figure 1).
FIGURE 1.
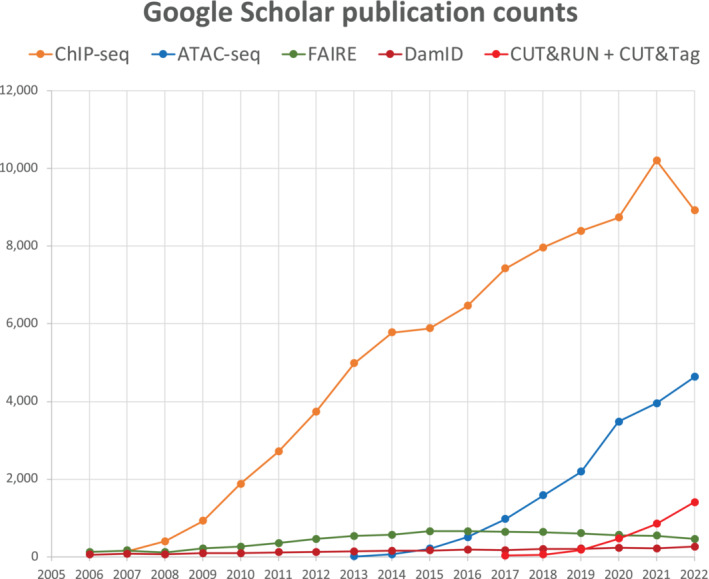
Assay for Transposase‐Accessible Chromatin using sequencing (ATAC‐seq) and in situ chromatin profiling methods are rapidly growing in popularity. Graph shows the number of Google Scholar citations each year using each of the indicated terms (together with “chromatin” to reduce false‐positives), where the number for 2022 has been linearly extrapolated based on statistics through September 17, 2022. Since its introduction in 2007, ChIP‐seq has dominated chromatin profiling, currently with ~10,000 citations per year, but the recent leveling off is likely attributable to the rapidly growing popularity of ATAC‐seq. Based on this metric, CUT&RUN and CUT&Tag, respectively introduced in 2017 and 2019, are growing in popularity at about the same rate as ATAC‐seq during its first 5 years. ATAC‐seq and in situ chromatin profiling are suitable for single‐cell platforms and so are likely to further chip away at the dominance of ChIP‐seq in the future. FAIRE and DamID are respectively alternative chromatin accessibility and chromatin profiling methods.
For ChIP the entire chromatin content of a cell is fragmented and solubilized, usually by sonication, which requires cross‐linking to prevent loss of protein from the DNA. Cross‐linking can result in epitope masking, and sonication introduces variability, limits resolution and contributes to genome‐wide background. In contrast, for in situ chromatin profiling methods, no cross‐linking is required, and only the chromatin‐bound target is solubilized for DNA purification (Figure 2). As a result, backgrounds and sequencing costs are lower with in situ profiling, and orders‐of‐magnitude less material is required to distinguish a chromatin feature from untargeted background. These advantages have led to the rapid development of single‐cell chromatin profiling methods, and recent adaptation for multimodal, multifactorial, and spatial applications (Table 1).
FIGURE 2.
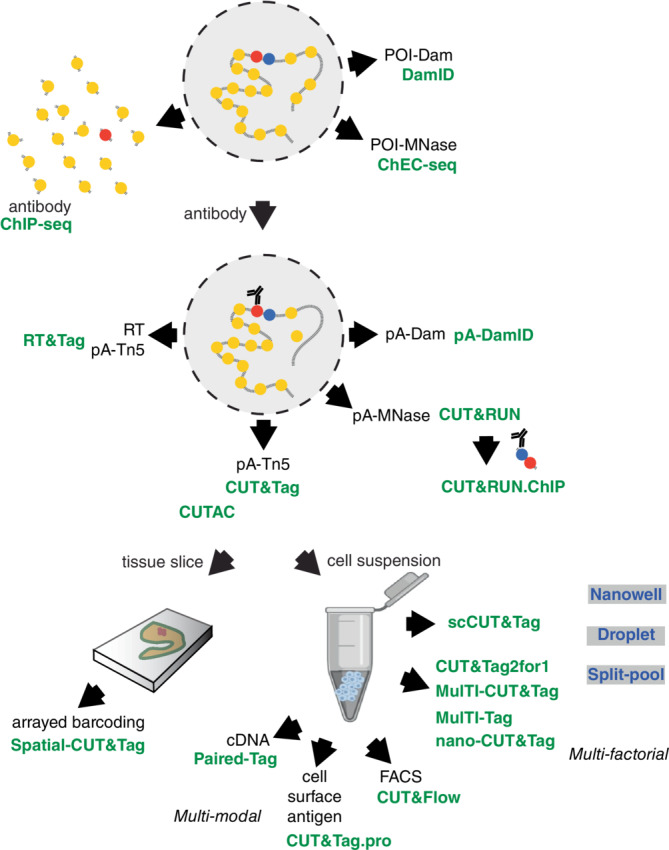
Schematic of the in situ chromatin profiling methods described in this survey. Methods are indicated in green, with their characteristic reagents or features in black. POI, protein of interest. Chromatin features within a permeabilized nucleus (dashed circle) are indicated in yellow, red, and blue. Platforms that have been used for single‐cell scCUT&Tag and multifactorial methods are shown in gray boxes.
TABLE 1.
Genome‐wide in situ chromatin profiling methods
| Method | Defining feature | Refs |
|---|---|---|
| ChEC‐seq | Micrococcal Nuclease (MNase) fused to protein of interest (POI) | [9] |
| CUT&RUN | MNase fused to Protein A bound to antibody to POI | [10, 11] |
| pA‐DamID | Dam DNA methyltransferase fused to Protein A | [12] |
| CUT&RUN.ChIP | Supernatant from CUT&RUN used as input for ChIP | [13] |
| CUT&Tag | Tn5 fused to Protein A bound to antibody to POI | [14, 15, 16, 17] |
| CUTAC | Low‐salt tagmentation of promoter epitope for accessibility | [18, 19] |
| Tip‐seq | CUT&Tag with in vitro transcription‐based amplification | [20] |
| HiCuT | Antibody‐targeted HiC with a CUT&Tag readout | [21] |
| CUT&Flow | CUT&Tag with FACS for cell cycle‐dependent profiling | [22] |
| Paired‐Tag | Joint single‐cell CUT&Tag and RNA‐seq | [23, 24] |
| Epi‐DamID | Joint single‐cell DamID and RNA‐seq | [25] |
| CUT&Tag.pro | Joint single‐cell CUT&Tag and cell‐surface antigen profiling | [26] |
| CUT&Tag2for1 | Joint single‐cell deconvolution CUT&Tag | [19] |
| Multi‐CUT&Tag | Multifactorial single‐cell CUT&Tag | [27, 28] |
| Nano‐CUT&Tag | Multifactorial single‐chain antibodies fused to Tn5 | [29, 30] |
| Spatial‐CUT&Tag | Tissue single‐cell CUT&Tag | [31] |
| RT&Tag | Proximity labeling of chromatin‐associated RNAs | [32] |
2. IN SITU CHROMATIN PROFILING
2.1. MNase‐based in situ chromatin mapping
Micrococcal Nuclease (MNase) has been used for chromatin mapping since the 1970s. 33 MNase is an endonuclease that cleaves single‐stranded nucleic acids and double‐stranded DNA one strand at a time, preferring AT‐rich regions. Cleavage of both strands results in rapid progressive “end‐nibbling” by MNase, which halts when it encounters a nucleosome or DNA‐binding protein. MNase‐seq has been the preferred method for base‐pair resolution genome‐wide mapping of nucleosomes, 34 although small molecule cleavage reagents have also been used in place of MNase with the advantage that they lack the AT‐bias and aggressive end‐nibbling activity of MNase. 35 , 36 MNase‐seq has also been used for base‐pair resolution mapping of transcription factor (TF) binding sites, 37 , 38 similar to the use of DNaseI for footprinting by the ENCODE project, 39 and MNase digestion has been used to fragment chromatin for native ChIP‐seq. 40 , 41
Alternatives to ChIP are based on targeting chromatin proteins within intact cells, originally using Escherichia coli Dam methyltransferase fused to the protein of interest (POI) for in vivo DNA methylation of chromatin targets, termed DamID. 42 Similarly, MNase can be tethered to a POI for chromatin profiling: In a pioneering study, Laemmli and colleagues used MNase for in situ chromatin mapping. 43 Their Chromatin Endogenous Cleavage (ChEC) method took advantage of the fact that MNase requires Ca++ ions for activity and there is relatively little Ca++ in the eukaryotic nucleus. By expressing a chimeric protein that fused a POI to MNase, they could target MNase cleavages to the sites of protein‐DNA binding by adding Ca++ to digitonin‐permeabilized cells. Using Southern blots with indirect end‐labeling to detect fragments cleaved from the genomic region being probed, the authors were able to demonstrate that the resolution was much higher than that obtained by ChIP or DamID chromatin profiling methods. Only much later was ChEC developed into a genome‐wide sequencing based method, ChEC‐seq. 9 Given the simplicity of ChEC‐seq and the relative ease of producing MNase fusion proteins in yeast, the method has been applied to study diverse problems in yeast chromatin biology. 44 , 45 , 46 , 47 , 48 , 49
2.2. Antibody‐targeted MNase‐based in situ chromatin mapping
In the same landmark publication that introduced ChEC, the Laemmli group introduced antibody‐directed targeting of MNase, a method termed Chromatin Immunocleavage (ChIC). 43 Crude fixed yeast nuclei were incubated with an antibody to an epitope‐tagged chromatin POI. After collecting the nuclei by centrifugation and subjecting them to washing and centrifugation steps, a protein fusion between Protein A and MNase (pA‐MN) was added. Protein A binds avidly to many Immunoglobulin Gs, such as those from rabbit, although for others a compatible secondary antibody binding step was included. Addition of Ca++ activated the pA‐MN for cleavage, after which DNA was extracted and used for Southern blot analysis. The use of antibodies with ChIC as opposed to MNase fusion proteins with ChEC extended the utility of in situ chromatin profiling by making it potentially adaptable for use with untransformed cells and tissues and for profiling histone modifications.
Thirteen years later, several modifications were applied to the ChIC strategy to adapt it for genome‐wide profiling and extend its applicability beyond yeast. 10 , 11 Native nuclei or digitonin‐permeabilized mammalian cells were immobilized on paramagnetic Concanavalin A beads to streamline the protocol and allow for stringent but gentle washes. Importantly, fragments released following pA‐MN cleavage were allowed to diffuse out of the intact nuclei or cells into the supernatant, leaving behind the untargeted DNA with the beads. DNA sequencing libraries were prepared by end‐polishing of fragments purified from the supernatant and ligation to adapters following protocols used for preparing ChIP‐seq libraries. This Cleavage Under Targets and Release Using Nuclease (CUT&RUN) method provided much lower background levels than ChIP‐seq, because the pA‐MN cleaves DNA only in the immediate vicinity of the antibody, releasing fragments into the supernatant, whereas in ChIP‐seq, the entire genome is fragmented, which results in a general untargeted background. A similar strategy has been applied to genome‐wide chromatin profiling of lamin‐associated chromatin in vivo using a fusion between Protein A and Dam methyltransferase (pA‐DamID). 12
As a chromatin profiling method, CUT&RUN has multiple advantages over standard ChIP‐seq: (a) The low backgrounds obtained with CUT&RUN relative to ChIP‐seq translate into fewer cells and less sequencing required with the in situ method. Whereas ChIP‐seq is typically done with ≥1 million mammalian cells, CUT&RUN can be performed with as few as 100 cells, making it more appropriate where sample size is limiting. 11 (b) Sequencing depths are much lower for CUT&RUN, typically 3–5 million paired‐end reads versus ≥20 million for ChIP‐seq, 50 which reduces sequencing and data handling and storage costs. (c) Whereas the resolution of standard ChIP‐seq is a few hundred base pairs, CUT&RUN achieves near base‐pair resolution allowing TF mapping with high precision, 51 and nucleosome‐bound (“pioneer”) TF binding to be distinguished from direct binding to accessible DNA. 52 (d) Full automation starting with samples of intact unfixed cells or tissues is possible with CUT&RUN, 53 whereas sonication makes full automation unfeasible for ChIP‐seq. (e) CUT&RUN supernatant can be used as input for efficient native ChIP to identify epitopes bound to the same DNA, 13 whereas sequential ChIP‐seq requires large amounts of starting material 54 and cross‐linking may complicate interpretation of co‐immunoprecipitated proteins. (f) CUT&RUN spike‐in calibration options include using carry‐over E. coli DNA left behind during pA‐Tn5 purification. 55 (g) CUT&RUN profiles epitopes on chromatin surfaces exposed in the nucleus, whereas ChIP‐seq profiles proteins that are cross‐linked to chromatin and survive sonication, so that specificity is often reduced.
Some of the problems with ChIP‐seq that in situ methods overcome have also been addressed by improvements to the traditional ChIP‐seq protocol. Among them is the use of MNase for genome‐wide fragmentation, which avoids cross‐linking and achieves high resolution for histone marks and TFs, 40 and the use of microfluidic devices for low cell numbers 56 and single cells. 57 Antibody availability is a serious limiting factor for all antibody‐directed methods, and there is currently a very large selection of ChIP‐seq grade antibodies, although as the popularity of CUT&RUN increases, antibody producers are testing their antibodies for use with this protocol. Other problems with CUT&RUN are similar to problems with MNase‐seq, which include high backgrounds due to over‐digestion, although the use of magnetic bead immobilization and stringent washes minimizes overdigestion.
2.3. Tagmentation‐based in situ chromatin mapping
The cut‐and‐paste transposase Tn5 has been used to create DNA sequencing libraries 58 and later adapted for accessibility mapping by ATAC‐seq. 8 These methods employ tagmentation, in which Tn5 is loaded with DNA sequencing adapters, 14 Mg++ is added to activate the Tn5, whereupon it performs a cut‐and‐paste integration reaction to produce DNA ends capped by barcoded adapters for Illumina sequencing. 58 The simplicity of ATAC‐seq for chromatin accessibility mapping led to its rapid acceptance by the broad epigenomics community and encouraged the adoption of tagmentation for in situ antibody‐targeted chromatin profiling. Cleavage Under Targets and Tagmentation (CUT&Tag) follows the CUT&RUN workflow, replacing pA‐MN with Protein A‐Tn5 (pA‐Tn5). 14 After incubation and binding of Tn5 to the primary and secondary antibodies and washes to remove unbound pA‐Tn5, Mg++ is added to activate the cut‐and‐paste reaction, resulting in tagmentation of targeted chromatin. Unlike ChEC‐seq and CUT&RUN, where controlling the time and temperature of MNase is critical, tagmentation is “one‐and‐done,” so that CUT&Tag integrations may proceed to completion at 37°C with little if any increase in background tagmentation.
CUT&Tag can be performed in single PCR tubes from live cells to amplified sequencing‐ready libraries in a single day 59 and has been adapted for full automation. 60 To prevent accessible site tagmentation by pA‐Tn5, CUT&Tag includes 300 mM NaCl during incubation, washes and tagmentation, where the elevated salt competes for binding of Tn5 to DNA but also limits its use for TFs, which are partially destabilized under such conditions. Three other methods using the same general strategy were introduced at about the same time. 15 , 16 , 17 Although some of the protocols were found to suffer from accessible site tagmentation, 59 it was later shown that leaving out the NaCl during tagmentation for the promoter and enhancer mark H3K4me2 resulted in high‐resolution chromatin accessibility mapping, identifying the same spectrum of promoters and enhancers as ATAC‐seq and DNAseI‐seq. 18 This Cleavage Under Targeted Accessible Chromatin (CUTAC) variation of CUT&Tag was shown to also provide sensitive high‐quality chromatin accessibility mapping using an antibody to paused RNA Polymerase II marked by Serine‐5 phosphate (RNAPIIS5p). 19 This correspondence between accessible sites and adjacent paused RNAPII at regulatory elements confirmed inferences based global run‐on sequencing that promoters and enhancers share the same chromatin architecture. 61 This finding also provided for the first time ground‐truth biological validation of accessibility mapping first introduced for mapping regulatory elements >40 years ago, 62 , 63 but previously defined only on the basis of hyper‐sensitivity to DNA cleavage, DNA modification enzymes or physical breakage by sonication.
CUT&Tag has also been adapted for 3‐dimensional contact mapping: Hi‐C Coupled chromatin cleavage and Tagmentation (HiCuT). 21 HiCuT adopts the efficient and sensitive Hi‐C 3.0 protocol, using short‐ and long‐range crosslinking, optimized restriction endonuclease digestion and proximity ligation to capture contact sites. This is followed by a standard CUT&Tag workflow using antibodies to the boundary TF CTCF. The advantages of HiCuT over second‐generation Hi‐C immunoprecipitation‐based methods such as HiChIP and PLAC‐seq parallel those of CUT&Tag over ChIP‐seq in requiring 1–2 orders of magnitude fewer cells (100,000) and an order of magnitude fewer reads (10–12 million), and can be accomplished in less time (1.5 days).
3. SINGLE‐CELL CHROMATIN PROFILING
3.1. Scalable single‐cell CUT&Tag
Performing in situ chromatin profiling on tissues presents challenges, but methods for cell‐type separation, including fluorescence activated cell sorting (FACS) 64 and Isolation of Nuclei Tagged in Specific Cell Types (INTACT) 65 may often suffice. CUT&Tag followed by FACS (CUT&Flow) has been used to follow chromatin dynamics over the cell cycle. 22 However, cell separation methods are limited as to the number and types of cellular material that can be obtained, and single‐cell approaches are increasingly becoming standard, where pseudotime analysis can define developmental trajectories. 66 Although ChIP‐seq and CUT&RUN have been successfully adapted for single‐cell chromatin profiling, 57 , 67 , 68 , 69 scalability is limited, because the targeted chromatin fragment is in solution. In contrast, the tight binding of Tn5 to both DNA ends of an antibody‐targeted fragment retains the fragment within chromatin, so that all steps in the process from intact cells or nuclei through to tagmentation can be performed in bulk. Indeed, all four 2019 publications describing versions of CUT&Tag included application to single cells. 14 , 15 , 16 , 17
Scalability is critical for single‐cell epigenomics, because single‐cell read‐out technologies are advancing at an exponential rate such that thousands to hundreds of thousands of single cells can profiled by RNA‐seq. 70 The same technologies have been applied to ATAC‐seq and CUT&Tag, where nanowell and droplet‐based CUT&Tag use the same barcoding strategies, implementations and analysis software that had been developed previously for ATAC‐seq. 23 , 24 Combinatorial barcoding, where populations of cells are arrayed and barcoded in 96‐well plates, pooled, then rearrayed in plates and a second barcode is added, and so on, 71 , 72 has also been applied to single‐cell CUT&Tag, increasing the number of cells in a single experiment and allowing for multiple experiments to be performed in parallel to minimize batch effects. 28
Although dramatic scaling up of single‐cell technologies has been achieved by increasing the number of cells in an experiment, this only partially overcomes the limitations owing to the low information content per cell inherent to single‐cell epigenomics relative to bulk chromatin profiling. Whereas single‐cell RNA‐seq takes advantage of abundant transcripts expressed from highly and moderately expressed genes, epigenomic profiling is limited to at most two copies of a chromatin feature in each diploid cell. This disadvantage relative to RNA‐seq is in part offset by the regulatory functions and the much larger number of features potentially detectable by both ATAC‐seq and CUT&Tag than by RNA‐seq. However, the inherent sparseness of regulatory information puts a premium on overall efficiency to squeeze out more cell‐type discriminatory information per cell. One method that has been shown to increase efficiency is linear amplification, for example where a T7 promoter is placed upstream of the barcode and single adapter sequence for in vitro transcription. 15 In the Targeted Insertion of Promoters method the T7 transcripts are reverse transcribed and cDNA is used for sequencing, a procedure that provides abundant sequencing templates while requiring only a single Tn5‐mediated integration event, increasing the number of fragments per cell 10‐fold relative to PCR‐based CUT&Tag. 20
3.2. Multimodal in situ chromatin profiling
Another way to gain more useful cell‐type information from each single cell is multimodal single‐cell CUT&Tag, which borrows technologies developed initially for RNA‐seq and ATAC‐seq. For example, Paired‐Tag is a combinatorial barcoding strategy for CUT&Tag and RNA‐seq in the same cell. 73 , 74 In Paired‐Tag, successive antibody, pA‐Tn5 binding and tagmentation steps are followed by reverse transcription (RT) in wells of a microtiter plate using a well‐specific barcode for both the CUT&Tag adapter and the RT primer. Cells are then pooled and dispensed with two rounds of ligation‐based split‐pooling in 96‐well plates. 75 When applied to ~10,000 single cells per mouse brain sample, all of five different histone modifications showed much improved resolution and separation of cell‐types when RNA‐seq data were jointly clustered. A similar strategy has been applied to in vivo chromatin profiling, where histone reader domains are fused to Dam methyltransferase and both methylated DNA and mRNAs are profiled (single‐cell EpiDamID 25 ).
Another multimodal strategy, scCUT&Tag‐pro, 26 is a droplet‐based strategy modeled on the Cellular Indexing of Transcriptomes and Epitopes by sequencing (CITE‐seq) method, which combines cell surface antibody multiplexing with RNA‐seq. 76 In scCUT&Tag‐pro a mixture of oligonucleotide‐conjugated antibodies to cell surface markers bound to cells, then blocked followed by permeabilization for CUT&Tag. After tagmentation, cells are subjected to 10X Genomics single‐cell indexing and DNA sequencing so that the oligonucleotides for the cell‐surface markers and the integrated adapters for the chromatin epitope have the same index. scCUT&Tag‐pro facilitates data integration of multiple histone modifications and data from CITE‐seq and ASAC‐seq (ATAC with Select Antigen Profiling by sequencing 77 ) by clustering and annotating cell types based on their cell surface markers. In this way, single‐cell CUT&Tag data from multiple experiments with multiple epitopes can be integrated with one another and with experiments using other modalities, with definitive annotation of cell types.
3.3. Multifactorial CUT&Tag
Integration of multiple histone modifications and potentially other chromatin epitopes can also be accomplished using Multifactorial single‐cell CUT&Tag approaches. CUT&Tag2for1 follows exactly the same workflow as ordinary CUT&Tag for bulk and single‐cell profiling, except that two antibodies are mixed before being added to permeabilized cells. 19 One antibody is to RNAPIIS5p and the other is to histone H3K27me3, which marks silenced Polycomb domains. Under CUTAC tagmentation conditions Tn5s anchored via RNAPIIS5p tagment active enhancers and promoters, releasing subnucleosome‐sized fragments whereas Tn5s anchored via H3K27me3 tagment Polycomb domains release nucleosomal fragments. Fragment size differences taken together with differences in the breadth of the feature (narrow peaks for RNAPIIS5p and broad domains for H3K27me3) are used to computationally deconvolve the two signals, resulting in simultaneous profiling of the active and silenced regulomes in single cells.
Other multifactorial methods based on CUT&Tag involve loading differentially barcoded adapters into pA‐Tn5. 27 , 28 Multifactorial CUT&Tag has been used to profile up to three different epitopes in single cells, with no fixed upper limit. An important recent advance in in situ chromatin profiling has been the use of nanobodies, single‐chain camelid antibodies, fused to MNase 78 or Tn5, 29 , 30 where higher efficiencies are achieved by leaving out the Protein A “middleman.” Multifactorial single‐cell CUT&Tag results from the addition of two differentially barcoded nanobody‐Tn5 fusions, one specific for rabbit primary antibodies and the other for mouse. In one study, multifactorial nanobody CUT&Tag was preceded by ATAC‐seq with pA‐Tn5 loaded with different barcoded adapters. 30 We expect that nanobody‐based single‐cell CUT&Tag will in the near future be combined with other modalities to take even fuller advantage of the possibilities of capturing diverse features and increasing the cell‐type discriminatory information that can be obtained from each single cell.
3.4. Single‐cell spatial CUT&Tag
Perhaps the most exciting advance in single‐cell chromatin profiling is the adaptation of CUT&Tag for in situ spatial profiling. 79 Spatial RNA‐seq performed on thin tissue slices on microscope slides is becoming increasing standard for research and clinical diagnosis thanks to commercial platforms such as Visium (10X Genomics) and GeoMx (Nanostring). These platforms are not yet capable of achieving single‐cell resolution, 79 however, new spatial barcoding platforms have achieved higher resolution for RNA‐seq. 31 , 80 , 81 One of these platforms has been adapted for single‐cell Spatial‐CUT&Tag, 31 whereby a fixed thin tissue section on a glass microscope slide is successively incubated with a primary antibody, a secondary antibody and pA‐Tn5, and then a 20 μm resolution grid of DNA barcodes is printed onto the slice by microchannel‐guided delivery. Although it remains to be seen which platform(s) will become commercially available for routine spatial single‐cell chromatin profiling, we expect that the advances in multifactorial, multimodal and nanobody‐based CUT&Tag technologies will soon be adaptable for the spatial single‐cell realm.
4. ANALYTICS
New genomic technologies fuel the development of new analytical tools. The long lag in development of CUT&RUN from its origin in ChIC meant that sophisticated analytical tools developed for ChIP‐seq could be immediately adapted to in situ chromatin profiling. However, the near base‐pair resolution and the ability to distinguish TFs from nucleosomes by fragment length not possible with standard ChIP‐seq motivated the development of CUT&RUN‐specific analytical tools for inferring TF and chromatin dynamics. 52 Likewise, the low backgrounds inherent to in situ chromatin profiling motivated the development of the Sparse Enrichment Analysis for CUT&RUN (SEACR) 82 and GoPeaks 83 peak callers to complement peak callers such as MACS2, 84 which were developed for ChIP‐seq. The growing popularity of in situ chromatin profiling has also motivated the development of custom analytical pipelines 85 , 86 and the CUT&Tag Data Processing and Analysis Tutorial, 87 where users can get questions answered and provide feedback to developers.
Single‐cell analytical tools first developed for single‐cell RNA‐seq and ATAC‐seq require relatively little adaptation to be applied to single‐cell CUT&Tag. 88 , 89 , 90 , 91 , 92 For example, gene activity scores calculated for RNA‐seq can be repurposed for single‐cell chromatin profiling even though features such as silencing histone modifications do not typically correspond to genes, and when they do may be interpreted as gene silencing scores. Alternatively, the recently introduced Version 2.0 of CUT&RUNTools now provides seamless analytical capabilities that include single‐cell in situ chromatin profiling. Other analytical concepts that have been applied to RNA‐seq, such as RNA velocity, show promise for adaptation to in situ chromatin profiling. 93 As multifactorial, multimodal, and spatial in situ chromatin profiling methods proliferate, they create challenges and opportunities for new approaches to data integration.
5. CONCLUSIONS
This is an exciting time for the emerging field of epigenomics, where the cost of short‐read sequencing continues to drop and single‐cell profiling is becoming increasingly routine. The tools of the trade are rapidly becoming more accessible to the broad community of cell and developmental biologists, including commercial availability of pA‐MN and pA‐Tn5 and kits for CUT&RUN and CUT&Tag that have helped accelerate the adoption of in situ chromatin profiling methods (Figure 1). While we have focused on protein‐based in situ chromatin profiling tools in this review, these tools are readily adaptable to non‐protein features of the chromatin landscape. For example, multiple groups have adapted CUT&RUN or CUT&Tag for profiling RNA‐DNA hybrids (R‐loops) and/or DNA G‐quadruplexes, 94 , 95 , 96 including in single cells. 96 In addition, a proximity dependent RNA profiling method based on CUT&Tag (RT&Tag) has recently been applied to the genome‐wide mapping of chromatin‐associated RNAs and adenine‐methylated RNAs. 32
In situ chromatin profiling technologies also have the potential of providing gene regulatory information that complements RNA‐seq and ATAC‐seq, and in the case of Paired‐Tag and nano‐CUT&Tag, combines them into a single workflow. Such multimodal, multifactorial variations of CUT&Tag are potentially suitable for adaptation to spatial single‐cell platforms and integration with imaging‐based technologies. These recently developed tools only await commercialization to be applied to large‐scale infrastructural projects such as the Human Developmental Cell Atlas 97 and made available to the large community of biologists and clinicians.
AUTHOR CONTRIBUTIONS
Steven Henikoff: Conceptualization (lead); writing – original draft (lead); writing – review and editing (equal). Kami Ahmad: Writing – review and editing (equal).
CONFLICT OF INTEREST
Steven Henikoff and Kami Ahmad have filed patents related to some of the work described in this review.
ACKNOWLEDGMENTS
This work was supported by Howard Hughes Medical Institute (Steven Henikoff) and a grant from the National Institutes of Health (R01 HG010492). We thank Paul Talbert for comments on the manuscript.
Henikoff S, Ahmad K. In situ tools for chromatin structural epigenomics. Protein Science. 2022;31(11):e4458. 10.1002/pro.4458
Review Editor: Nir Ben‐Tal
Funding information Howard Hughes Medical Institute, Grant/Award Number: Henikoff; National Institutes of Health, Grant/Award Number: R01 HG010492
REFERENCES
- 1. Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. [DOI] [PubMed] [Google Scholar]
- 2. Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291:1304–1351. [DOI] [PubMed] [Google Scholar]
- 3. Solomon MJ, Varshavsky A. Formaldehyde‐mediated DNA‐protein crosslinking: A probe for in vivo chromatin structures. Proc Natl Acad Sci U S A. 1985;82:6470–6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gilmour DS, Lis JT. In vivo interactions of RNA polymerase II with genes of Drosophila melanogaster . Mol Cell Biol. 1985;5:2009–2018. 10.1128/mcb.5.8.2009-2018.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Acevedo L, Iniguez AL, Holster HL, Zhang X, Green R, Farnham PJ. Genome‐scale ChIP‐chip analysis using 10,000 human cells. Biotechniques. 2007;43:791–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barski A, Cuddapah S, Cui K, et al. High‐resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. [DOI] [PubMed] [Google Scholar]
- 7. Robertson G, Hirst M, Bainbridge M, et al. Genome‐wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4:651–657. [DOI] [PubMed] [Google Scholar]
- 8. Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA‐binding proteins and nucleosome position. Nat Methods. 2013;10:1213–1218. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zentner GE, Kasinathan S, Xin B, Rohs R, Henikoff S. ChEC‐seq kinetics discriminate transcription factor binding sites by DNA sequence and shape in vivo. Nat Commun. 2015;6:8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Skene PJ, Henikoff S. An efficient targeted nuclease strategy for high‐resolution mapping of DNA binding sites. eLife. 2017;6:e21856. 10.7554/eLife.21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Skene PJ, Henikoff JG, Henikoff S. Targeted in situ genome‐wide profiling with high efficiency for low cell numbers. Nat Protoc. 2018;13:1006–1019. 10.1038/nprot.2018.015. [DOI] [PubMed] [Google Scholar]
- 12. van Schaik T, Vos M, Peric‐Hupkes D, Hn Celie P, van Steensel B. Cell cycle dynamics of lamina‐associated DNA. EMBO Rep. 2020;21:e50636. 10.15252/embr.202050636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brahma S, Henikoff S. RSC‐associated subnucleosomes define MNase‐sensitive promoters in yeast. Mol Cell. 2019;73:238–249. 10.1016/j.molcel.2018.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kaya‐Okur HS, Wu SJ, Codomo CA, et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun. 2019;10:1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harada A, Maehara K, Handa T, et al. A chromatin integration labelling method enables epigenomic profiling with lower input. Nat Cell Biol. 2019;21:287–296. 10.1038/s41556-018-0248-3. [DOI] [PubMed] [Google Scholar]
- 16. Carter B, Ku WL, Kang JY, et al. Mapping histone modifications in low cell number and single cells using antibody‐guided chromatin tagmentation (ACT‐seq). Nat Commun. 2019;10:3747. 10.1038/s41467-019-11559-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang Q, Xiong H, Ai S, et al. CoBATCH for high‐throughput single‐cell epigenomic profiling. Mol Cell. 2019;76:206–216.e7. 10.1016/j.molcel.2019.07.015. [DOI] [PubMed] [Google Scholar]
- 18. Henikoff S, Henikoff JG, Kaya‐Okur HS, Ahmad K. Efficient chromatin accessibility mapping in situ by nucleosome‐tethered tagmentation. Elife. 2020;9:e63274. 10.7554/eLife.63274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Janssens DH, Otto DJ, Meers MP, Setty M, Ahmad K, Henikoff S. CUT&Tag2for1: A modified method for simultaneous profiling of the accessible and silenced regulome in single cells. Genome Biol. 2022;23:81. 10.1186/s13059-022-02642-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bartlett DA, Dileep V, Henikoff S, Gilbert DM. High throughput genome‐wide single cell protein:DNA binding site mapping by targeted insertion of promoters (TIP‐seq). J Cell Biol. 2021;220:e202103078. 10.1101/2021.03.17.435909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sati S, Jones P, Kim HS, Zhou LA, Rapp‐Reyes E, Leung TH. HiCuT: An efficient and low input method to identify protein‐directed chromatin interactions. PLoS Genet. 2022;18:e1010121. 10.1371/journal.pgen.1010121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Veronezi G, Ramachandran S. Nucleation and spreading rejuvenate polycomb domains every cell cycle. biorxiv. 2022. 10.1101/2022.08.02.502476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu SJ, Furlan SN, Mihalas AB, et al. Single‐cell CUT&Tag analysis of chromatin modifications in differentiation and tumor progression. Nat Biotechnol. 2021;39:819–824. 10.1038/s41587-021-00865-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bartosovic M, Kabbe M, Castelo‐Branco G. Single‐cell CUT&Tag profiles histone modifications and transcription factors in complex tissues. Nat Biotechnol. 2021;39:825–835. 10.1038/s41587-021-00869-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rang FJ, de Luca KL, de Vries SS, et al. Single‐cell profiling of transcriptome and histone modifications with EpiDamID. Mol Cell. 2022;82:1956–1970.e14. 10.1016/j.molcel.2022.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang B, Srivastava A, Mimitou E, et al. Characterizing cellular heterogeneity in chromatin state with scCUT&tag‐pro. Nat Biotechnol. 2022;40:1220–1230. 10.1038/s41587-022-01250-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gopalan S, Wang Y, Harper NW, Garber M, Fazzio TG. Simultaneous profiling of multiple chromatin proteins in the same cells. Mol Cell. 2021;81:4736–4746.e5. 10.1016/j.molcel.2021.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meers MP, Llagas G, Janssens DH, Codomo CA, Henikoff S. Multifactorial chromatin regulatory landscapes at single cell resolution. Nat Biotechnol. 2021; In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stuart T, Hao S, Zhang B, et al. Nanobody‐tethered transposition allows for multifactorial chromatin profiling at single‐cell resolution. biorxiv. 2022. 10.1101/2022.03.08.483436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bartosovic M, Castelo‐Branco G. Multimodal chromatin profiling using nanobody‐based single‐cell CUT&tag. biorxiv. 2022. 10.1101/2022.03.08.483459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deng Y, Bartosovic M, Kukanja P, et al. Spatial‐CUT&tag: Spatially resolved chromatin modification profiling at the cellular level. Science. 2022;375:681–686. 10.1126/science.abg7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Khyzha N, Ahmad K, Henikoff S. Profiling RNA at chromatin targets in situ by antibody‐targeted tagmentation. Nat Methods. 2022. 10.1038/s41592-022-01618-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reeves R. Nucleosome structure of Xenopus oocyte amplified ribosomal genes. Biochemistry (Mosc). 1978;17:4908–4916. 10.1021/bi00616a008. [DOI] [PubMed] [Google Scholar]
- 34. Ramachandran S, Ahmad K, Henikoff S. Transcription and remodeling produce asymmetrically unwrapped nucleosomal intermediates. Mol Cell. 2017;68:1038–1053.e4. 10.1016/j.molcel.2017.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brogaard K, Xi L, Wang JP, Widom J. A map of nucleosome positions in yeast at base‐pair resolution. Nature. 2012;486:496–501. 10.1038/nature11142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ishii H, Kadonaga JT, Ren B. MPE‐seq, a new method for the genome‐wide analysis of chromatin structure. Proc Natl Acad Sci U S A. 2015;112:E3457–E3465. 10.1073/pnas.1424804112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kent NA, Adams S, Moorhouse A, Paszkiewicz K. Chromatin particle spectrum analysis: A method for comparative chromatin structure analysis using paired‐end mode next‐generation DNA sequencing. Nucleic Acids Res. 2011;39:e26. 10.1093/nar/gkq1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Henikoff JG, Belsky JA, Krassovsky K, Macalpine DM, Henikoff S. Epigenome characterization at single base‐pair resolution. Proc Natl Acad Sci U S A. 2011;108:18318–18323. 10.1073/pnas.1110731108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Neph S, Vierstra J, Stergachis AB, et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012;489:83–90. 10.1038/nature11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kasinathan S, Orsi GA, Zentner GE, Ahmad K, Henikoff S. High‐resolution mapping of transcription factor binding sites on native chromatin. Nat Methods. 2014;11:203–209. 10.1038/nmeth.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lorzadeh A, Bilenky M, Hammond C, et al. Nucleosome density ChIP‐Seq identifies distinct chromatin modification signatures associated with MNase accessibility. Cell Rep. 2016;17:2112–2124. 10.1016/j.celrep.2016.10.055. [DOI] [PubMed] [Google Scholar]
- 42. van Steensel B, Henikoff S. Identification of in vivo DNA targets of chromatin proteins using tethered dam methyltransferase. Nat Biotechnol. 2000;18:424–428. [DOI] [PubMed] [Google Scholar]
- 43. Schmid M, Durussel T, Laemmli UK. ChIC and ChEC; genomic mapping of chromatin proteins. Mol Cell. 2004;16:147–157. 10.1016/j.molcel.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 44. Zentner GE, Policastro RA, Henikoff S. ChEC‐seq produces robust and specific maps of transcriptional regulators. biorxiv. 2021. 10.1101/2021.02.11.430831. [DOI] [Google Scholar]
- 45. Donczew R, Lalou A, Devys D, Tora L, Hahn S. An improved ChEC‐seq method accurately maps the genome‐wide binding of transcription coactivators and sequence‐specific transcription factors. biorxiv. 2021. 10.1101/2021.02.12.430999. [DOI] [Google Scholar]
- 46. Bruzzone MJ, Albert B, Hafner L, et al. ChEC‐seq: A robust method to identify protein‐DNA interactions genome‐wide. biorxiv. 2021. 10.1101/2021.02.18.431798. [DOI] [Google Scholar]
- 47. Brodsky S, Jana T, Mittelman K, et al. Intrinsically disordered regions direct transcription factor in vivo binding specificity. Mol Cell. 2020;79:459–471.e4. 10.1016/j.molcel.2020.05.032. [DOI] [PubMed] [Google Scholar]
- 48. Policastro RA, Zentner GE. Enzymatic methods for genome‐wide profiling of protein binding sites. Brief Funct Genomics. 2018;17:138–145. 10.1093/bfgp/elx030. [DOI] [PubMed] [Google Scholar]
- 49. Saleh MM, Tourigny JP, Zentner GE. Genome‐wide profiling of protein‐DNA interactions with chromatin endogenous cleavage and high‐throughput sequencing (ChEC‐Seq). Methods Mol Biol. 2021;2351:289–303. 10.1007/978-1-0716-1597-3_16. [DOI] [PubMed] [Google Scholar]
- 50. Landt SG, Marinov GK, Kundaje A, et al. ChIP‐seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu N, Hargreaves VV, Zhu Q, et al. Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell. 2018;173:430–442. 10.1016/j.cell.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Meers MP, Janssens DH, Henikoff S. Pioneer factor‐nucleosome binding events during differentiation are motif encoded. Mol Cell. 2019;75:562–575. 10.1016/j.molcel.2019.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Janssens DH, Wu SJ, Sarthy JF, et al. Automated in situ chromatin profiling efficiently resolves cell types and gene regulatory programs. Epigenetics Chromatin. 2018;11:74. 10.1186/s13072-018-0243-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nekrasov M, Tremethick DJ. Sequential chromatin immunoprecipitation to identify heterotypic nucleosomes. Methods Mol Biol. 2021;2351:147–161. 10.1007/978-1-0716-1597-3_8. [DOI] [PubMed] [Google Scholar]
- 55. Meers MP, Bryson TD, Henikoff JG, Henikoff S. Improved CUT&RUN chromatin profiling tools. eLife. 2019;8:e46314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhu B, Hsieh YP, Murphy TW, Zhang Q, Naler LB, Lu C. MOWChIP‐seq for low‐input and multiplexed profiling of genome‐wide histone modifications. Nat Protoc. 2019;14:3366–3394. 10.1038/s41596-019-0223-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Grosselin K, Durand A, Marsolier J, et al. High‐throughput single‐cell ChIP‐seq identifies heterogeneity of chromatin states in breast cancer. Nat Genet. 2019;51:1060–1066. 10.1038/s41588-019-0424-9. [DOI] [PubMed] [Google Scholar]
- 58. Adey A, Morrison HG, (no last name) A, et al. Rapid, low‐input, low‐bias construction of shotgun fragment libraries by high‐density in vitro transposition. Genome Biol. 2010;11:R119. 10.1186/gb-2010-11-12-r119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kaya‐Okur HS, Janssens DH, Henikoff JG, Ahmad K, Henikoff S. Efficient low‐cost chromatin profiling with CUT&Tag. Nat Protoc. 2020;15:3264–3283. 10.1038/s41596-020-0373-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Janssens DH, Meers MP, Wu SJ, et al. Automated CUT&Tag profiling of chromatin heterogeneity in mixed‐lineage leukemia. Nat Genet. 2021;53:1586–1596. 10.1038/s41588-021-00941-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Andersson R, Sandelin A, Danko CG. A unified architecture of transcriptional regulatory elements. Trends Genet. 2015;31:426–433. 10.1016/j.tig.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 62. Stalder J, Larsen A, Engel JD, Dolan M, Groudine M, Weintraub H. Tissue‐specific DNA cleavages in the globin chromatin domain introduced by DNAase I. Cell. 1980;20:451–460. 10.1016/0092-8674(80)90631-5. [DOI] [PubMed] [Google Scholar]
- 63. Wu C. The 5′ ends of Drosophila heat shock genes in chromatin are hypersensitive to DNase I. Nature. 1980;286:854–860. 10.1038/286854a0. [DOI] [PubMed] [Google Scholar]
- 64. Whetstine JR, Van Rechem C. A cell‐sorting‐based protocol for cell cycle small‐scale ChIP sequencing. STAR Protoc. 2022;3:101243. 10.1016/j.xpro.2022.101243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mo A, Mukamel EA, Davis FP, et al. Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron. 2015;86:1369–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Trapnell C, Cacchiarelli D, Grimsby J, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–386. 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yeung J, Florescu M, Zeller P, Barbanson BA, Oudenaarden A. Deconvolving multiplexed histone modifications in single cells. biorxiv. 2021. 10.1101/2021.04.26.440629. [DOI] [Google Scholar]
- 68. Ai S, Xiong H, Li CC, et al. Profiling chromatin states using single‐cell itChIP‐seq. Nat Cell Biol. 2019;21:1164–1172. 10.1038/s41556-019-0383-5. [DOI] [PubMed] [Google Scholar]
- 69. Hainer SJ, Boškovic A, McCannell KN, Rando OJ, Fazzio TG. Profiling of pluripotency factors in individual stem cells and early embryos. Cell. 2019;177:1319–1329. 10.1101/286351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kharchenko PV. The triumphs and limitations of computational methods for scRNA‐seq. Nat Methods. 2021;18:723–732. 10.1038/s41592-021-01171-x. [DOI] [PubMed] [Google Scholar]
- 71. Cusanovich DA, Daza R, Adey A, et al. Epigenetics. Multiplex single‐cell profiling of chromatin accessibility by combinatorial cellular indexing. Science. 2015;348:910–914. 10.1126/science.aab1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cao J, Packer JS, Ramani V, et al. Comprehensive single‐cell transcriptional profiling of a multicellular organism. Science. 2017;357:661–667. 10.1126/science.aam8940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhu C, Zhang Y, Li YE, Lucero J, Behrens MM, Ren B. Joint profiling of histone modifications and transcriptome in single cells from mouse brain. Nat Methods. 2021;18:283–292. 10.1038/s41592-021-01060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Xiong H, Luo Y, Wang Q, Yu X, He A. Single‐cell joint detection of chromatin occupancy and transcriptome enables higher‐dimensional epigenomic reconstructions. Nat Methods. 2021;18:652–660. 10.1038/s41592-021-01129-z. [DOI] [PubMed] [Google Scholar]
- 75. Rosenberg AB, Roco CM, Muscat RA, et al. Single‐cell profiling of the developing mouse brain and spinal cord with split‐pool barcoding. Science. 2018;360:176–182. 10.1126/science.aam8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Stoeckius M, Hafemeister C, Stephenson W, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14:865–868. 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mimitou EP, Lareau CA, Chen KY, et al. Scalable, multimodal profiling of chromatin accessibility, gene expression and protein levels in single cells. Nat Biotechnol. 2021;39:1246–1258. 10.1038/s41587-021-00927-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Koidl S, Timmers HTM. greenCUT&RUN: Efficient genomic profiling of GFP‐tagged transcription factors and chromatin regulators. Curr Protoc. 2021;1:e266. 10.1002/cpz1.266. [DOI] [PubMed] [Google Scholar]
- 79. Moffitt JR, Lundberg E, Heyn H. The emerging landscape of spatial profiling technologies. Nat Rev Genet. 2022. 10.1038/s41576-022-00515-3. [DOI] [PubMed] [Google Scholar]
- 80. Srivatsan SR, Regier MC, Barkan E, et al. Embryo‐scale, single‐cell spatial transcriptomics. Science. 2021;373:111–117. 10.1126/science.abb9536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Stickels RR, Murray E, Kumar P, et al. Highly sensitive spatial transcriptomics at near‐cellular resolution with Slide‐seqV2. Nat Biotechnol. 2021;39:313–319. 10.1038/s41587-020-0739-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Meers MP, Tenenbaum D, Henikoff S. Peak calling by sparse enrichment analysis for CUT&RUN chromatin profiling. Epigenetics Chromatin. 2019;12:42. 10.1186/s13072-019-0287-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yashar WM, Kong G, VanCampen J, et al. GoPeaks: Histone modification peak calling for CUT&Tag. Genome Biol. 2022;23:144. 10.1186/s13059-022-02707-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhang Y, Liu T, Meyer CA, et al. Model‐based analysis of ChIP‐Seq (MACS). Genome Biol. 2008;9:R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhu Q, Liu N, Orkin SH, Yuan GC. CUT&RUNTools: A flexible pipeline for CUT&RUN processing and footprint analysis. Genome Biol. 2019;20:192. 10.1186/s13059-019-1802-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Yu F, Sankaran VG, Yuan GC. CUT&RUNTools 2.0: A pipeline for single‐cell and bulk‐level CUT&RUN and CUT&Tag data analysis. Bioinformatics. 2021;38:252–254. 10.1093/bioinformatics/btab507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zheng Y, Ahmad K, Henikoff S. CUT&Tag data processing and analysis tutorial. 2020. Available from: https://www.protocols.io/view/cut‐amp‐tag‐data‐processing‐and‐analysis‐tutorial‐e6nvw93x7gmk/v1. 10.17504/protocols.io.bjk2kkye. [DOI]
- 88. Granja JM, Corces MR, Pierce SE, et al. ArchR is a scalable software package for integrative single‐cell chromatin accessibility analysis. Nat Genet. 2021;53:403–411. 10.1038/s41588-021-00790-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single‐cell gene expression data. Nat Biotechnol. 2015;33:495–502. 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tarbell ED, Liu T. HMMRATAC: a Hidden Markov ModeleR for ATAC‐seq. Nucleic Acids Res. 2019;47:e91. 10.1093/nar/gkz533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Pliner HA, Packer JS, McFaline‐Figueroa JL, et al. Cicero predicts cis‐regulatory DNA interactions from single‐cell chromatin accessibility data. Mol Cell. 2018;71:858–871.e8. 10.1016/j.molcel.2018.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Schep AN, Wu B, Buenrostro JD, Greenleaf WJ. chromVAR: Inferring transcription‐factor‐associated accessibility from single‐cell epigenomic data. Nat Methods. 2017;14:975–978. 10.1038/nmeth.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tedesco M, Giannese F, Lazarević D, et al. Chromatin velocity reveals epigenetic dynamics by single‐cell profiling of heterochromatin and euchromatin. Nat Biotechnol. 2022;40:235–244. 10.1038/s41587-021-01031-1. [DOI] [PubMed] [Google Scholar]
- 94. Lyu J, Shao R, Kwong Yung PY, Elsasser SJ. Genome‐wide mapping of G‐quadruplex structures with CUT&Tag. Nucleic Acids Res. 2022;50:e13. 10.1093/nar/gkab1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Yan Q, Shields EJ, Bonasio R, Sarma K. Mapping native R‐loops genome‐wide using a targeted nuclease approach. Cell Rep. 2019;29:1369–1380.e5. 10.1016/j.celrep.2019.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Hui WWI, Simeone A, Zyner KG, Tannahill D, Balasubramanian S. Single‐cell mapping of DNA G‐quadruplex structures in human cancer cells. Sci Rep. 2021;11:23641. 10.1038/s41598-021-02943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Haniffa M, Taylor D, Linnarsson S, et al. A roadmap for the human developmental cell atlas. Nature. 2021;597:196–205. 10.1038/s41586-021-03620-1. [DOI] [PMC free article] [PubMed] [Google Scholar]