

Abstract
Background
Polycystic ovary syndrome (PCOS) is the most common endocrine disorder among reproductive‐age women and has lifelong effects on health.
Methods
In this review, I discuss the pathophysiology of PCOS. First, I summarize our current understanding of the etiology and pathology of PCOS, then, discuss details of two representative environmental factors involved in the pathogenesis of PCOS. Finally, I present perspectives regarding the directions of future research.
Main findings
The pathophysiology of PCOS is heterogeneous and shaped by the interaction of reproductive dysfunction and metabolic disorders. Hyperandrogenism and insulin resistance exacerbate one another during the development of PCOS, which is also affected by dysfunction of the hypothalamus‐pituitary‐ovarian axis. PCOS is a highly heritable disorder, and exposure to certain environmental factors causes individuals with predisposing genetic factors to develop PCOS. The environmental factors that drive the development of PCOS pathophysiology make a larger contribution than the genetic factors, and may include the intrauterine environment during the prenatal period, the follicular microenvironment, and lifestyle after birth.
Conclusion
On the basis of this current understanding, three areas are proposed to be subjects for future research, with the ultimate goals of developing therapeutic and preventive strategies and providing appropriate lifelong management, including preconception care.
Keywords: delayed effects of prenatal exposure, endoplasmic reticulum stress (ER stress), follicular microenvironment, gastrointestinal microbiome, polycystic ovary syndrome (PCOS)
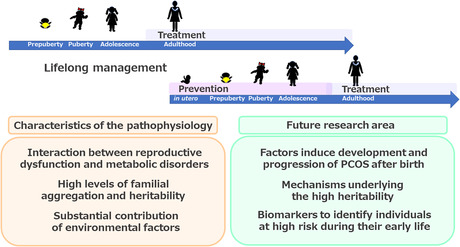
Three principal characteristics of the pathophysiology of PCOS have been identified to date: an interaction between reproductive dysfunction and metabolic disorders, high familial aggregation and heritability, and a substantial contribution of environmental factors. The following three areas represent targets for future research. 1) To identify the factors that induce the development and progression of PCOS after birth. 2) To elucidate the mechanisms underlying the high heritability of PCOS. 3) To identify biomarkers to that should be used to identify individuals at high risk during their early life. Future research should aim to develop therapeutic and preventive strategies, with the ultimate goal of achieving appropriate lifelong management, including preconception care.
1. INTRODUCTION
Polycystic ovary syndrome (PCOS) is the most common endocrine disorder among reproductive‐age women, affecting up to 15% of women in this age group. There are no significant geographical differences in prevalence when the most inclusive definition is used (the 2003 Rotterdam criteria, which are currently the most widely used). 1 , 2 , 3 PCOS manifests as various phenotypes that differ between ethnic groups. For example, high body mass index (BMI) is a feature in Caucasians, whereas lower BMI and mild hirsutism are features in East Asians, although no significant differences across ethnic groups in the prevalence of PCOS itself have been identified when diagnoses are made using the Rotterdam criteria. 1 , 3 PCOS is essentially shaped by the interaction between reproductive dysfunction and metabolic disorders, and has lifelong effects on the health of affected women.
PCOS is diagnosed according to several criteria: the 1990 National Institutes of Health (NIH) criteria, the 2003 Rotterdam criteria, the 2006 Androgen Excess & PCOS (AE‐PCOS) Society criteria (worldwide), and the 2007 Japan Society of Obstetrics and Gynecology (JSOG) criteria (Japan). 2 , 4 , 5 A comparison of these criteria is shown in Figure 1. The Rotterdam criteria, published by the European Society for Human Reproduction & Embryology (ESHRE) and the American Society of Reproductive Medicine (ASRM), require at least two out of the following three characteristics to be present for a diagnosis of PCOS to be made: clinical and/or biochemical hyperandrogenism, ovulatory dysfunction, and polycystic ovarian morphology (PCOM). In 2012, the NIH Consensus Conference Panel recommended specifying the phenotype that is identified using the Rotterdam criteria, namely, phenotype A, in which all three characteristics are present, and phenotypes B–D, in which two out of three characteristics are present: phenotype B, hyperandrogenism and ovulatory dysfunction; phenotype C, hyperandrogenism and PCOM; and phenotype D, ovulatory dysfunction and PCOM. The NIH criteria require the presence of two characteristics, hyperandrogenism and ovulatory dysfunction, while the AE‐PCOS criteria require two characteristics, hyperandrogenism and ovarian dysfunction, which may be indicated by ovulatory dysfunction or PCOM. Therefore, PCOS diagnosed using the NIH criteria corresponds to phenotype A or B, while that diagnosed using the AE‐PCOS criteria corresponds to phenotypes A, B, or C. The JSOG criteria require the presence of two characteristics, ovulatory dysfunction and PCOM, and one additional characteristic, biochemical hyperandrogenism or a high serum concentration of luteinizing hormone (LH) alongside a normal concentration of follicle‐stimulating hormone (FSH). A PCOS phenotype diagnosed using the JSOG criteria, comprising ovulatory dysfunction, PCOM, and biochemical hyperandrogenism, corresponds to phenotype A. However, it is not clear whether a PCOS phenotype diagnosed using the JSOG criteria, featuring ovulatory dysfunction, PCOM, and a high LH concentration, corresponds to some of the phenotypes defined using the Rotterdam criteria. However, this is a matter of debate, and the implementation of diagnostic criteria varies around the globe and between ethnicities, owing to variations in the prevalence of the components of PCOS, such as clinical and/or biochemical hyperandrogenism and ovulatory dysfunction.
FIGURE 1.
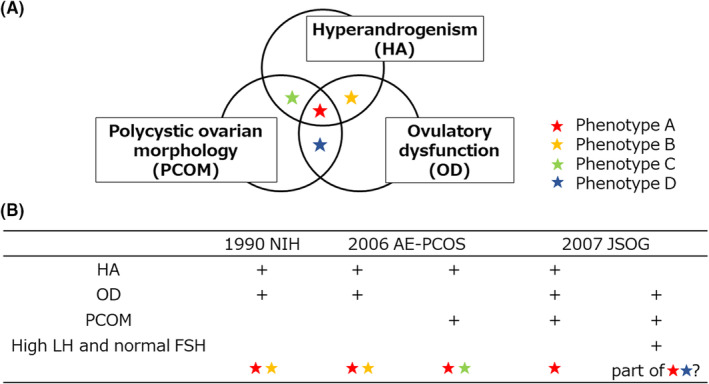
Diagnostic criteria. (A) 2003 Rotterdam criteria. Phenotype A, featuring all three characteristics of hyperandrogenism (HA), ovulatory dysfunction (OD), and polycystic ovarian morphology (PCOM); phenotype B, featuring HA and OD but not PCOM; phenotype C, featuring HA and PCOM but not OD; and phenotype D, featuring OD and PCOM but not HA. (B) Comparison of the phenotypes defined using the 2003 Rotterdam criteria and the other diagnostic criteria. Polycystic ovary syndrome (PCOS) diagnosed using the 1990 National Institutes of Health (NIH) criteria corresponds to phenotypes A or B, while a diagnosis made using the 2006 androgen excess & PCOS (AE‐PCOS) criteria corresponds to phenotypes A, B, or C. PCOS diagnosed using the 2007 Japan Society of Obstetrics and Gynecology (JSOG) criteria may correspond to components of phenotype A or D. FSH, follicle‐stimulating hormone; LH, luteinizing hormone
The first international evidence‐based guidelines, published in 2018, pose various problems for the diagnosis and understanding of the epidemiology and etiology of PCOS. 3 They also clarify the various comorbidities associated with PCOS, which include psychological problems, requiring lifelong management. These guidelines serve as a basis for our common understanding of this enigmatic disorder, and should stimulate further discussion, basic research regarding its pathophysiology, and clinical research regarding its management.
In this review, I discuss the pathophysiology of PCOS. First, I summarize current understanding regarding the etiology and pathology of PCOS. Next, I discuss in detail two representative environmental factors that are involved in the pathogenesis of PCOS, follicular endoplasmic reticulum stress (ER stress) and intrauterine hyperandrogenism, sharing recent findings from my laboratory and discussing their clinical implications. Finally, I present perspectives regarding potential directions for future research.
2. WHAT IS PCOS?
The main cause of difficulties in understanding the pathophysiology of PCOS is its heterogeneous and complex nature. Hyperandrogenism, ovulatory dysfunction, aberrant gonadotropin‐releasing hormone (GnRH) pulsation and the resulting abnormal gonadotropin secretion, and insulin resistance have been implicated in the pathophysiology of PCOS; these factors interact and exacerbate one another (Figure 2). Ovarian dysfunction involves the hypersecretion of androgens, which is associated with aberrant follicular growth and ovulatory dysfunction, causing PCOM. 4 , 6 , 7 High concentrations of anti‐Müllerian hormone (AMH), secreted by pre−/small antral follicles that accumulate in PCOS ovaries, further exacerbate the ovarian dysfunction by altering the follicular microenvironment and/or GnRH pulsation. Hyperandrogenism causes a dysregulation of the pulsatile secretion of GnRH, which can at least in part be explained by aberrant negative or positive feedback by progesterone and estrogen, causing the abnormal secretion of gonadotropins and, specifically, excess secretion of LH. 8 , 9 High concentrations of LH and the resulting imbalance in the LH/FSH ratio exacerbate the dysregulation of follicular growth, as well as causing the hypersecretion of androgens from thecal cells. 9 , 10 , 11 Insulin resistance is another key component of the pathophysiology of PCOS, although it is not included in the diagnostic criteria. 12 , 13 This insulin resistance manifests in insulin‐sensitive organs, such as liver and muscle, and is associated with visceral adiposity and adipocyte dysfunction. 11 Excess androgen secretion increases the level of insulin resistance, and hyperinsulinemia, which develops secondary to the insulin resistance, further increases androgen secretion and induces the production of sex hormone‐binding globulin (SHBG) in the liver, thereby increasing the circulating concentration of bioactive free testosterone and further aggravating the disorders associated with hyperandrogenism. 14 , 15 , 16 , 17 , 18
FIGURE 2.
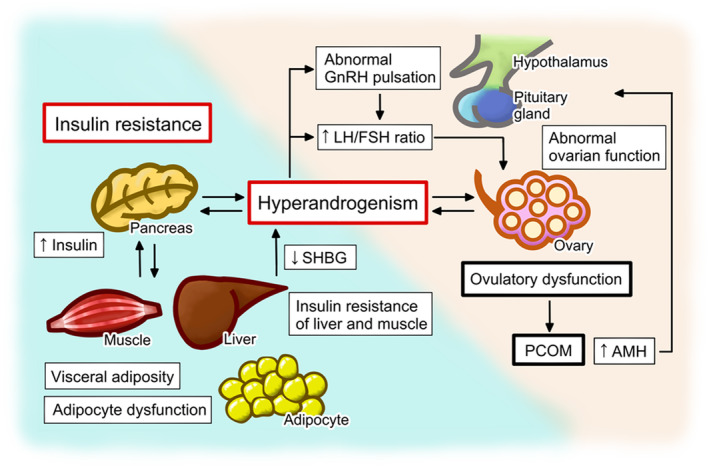
Pathophysiology of polycystic ovary syndrome (PCOS). Hyperandrogenism is a key feature and has a synergistic effect with insulin resistance to induce the development of PCOS. Individual contributions of hyperandrogenism and insulin resistance differ from patient to patient, which accounts for the heterogenous nature of PCOS and its presentation. Hyperandrogenism, ovulatory dysfunction, and polycystic ovarian morphology (PCOM) are the characteristics defined in the 2003 Rotterdam criteria. Hyperandrogenism, ovulatory dysfunction, aberrant gonadotropin‐releasing hormone (GnRH) pulsation and the resulting abnormal gonadotropin secretion, and insulin resistance comprise the vicious cycle that underpins the pathophysiology of PCOS. The abnormalities in the ovarian function of women with PCOS include the hypersecretion of androgens and ovulatory dysfunction, which causes PCOM. The hypersecretion of androgens is caused by intrinsic dysfunction of theca cells and/or the hypothalamus‐pituitary‐ovarian axis, while hyperandrogenism causes abnormal GnRH pulsation and gonadotropin secretion through the aberrant negative or positive feedback of progesterone and estrogen. The abnormal gonadotropin secretion in patients with PCOS is characterized by a high luteinizing hormone (LH)/follicle‐stimulating hormone (FSH) ratio, which induces ovarian dysfunction, including the hypersecretion of androgens. In addition, the high concentration of anti‐Müllerian hormone (AMH), which is secreted by the pre−/small antral follicles that accumulate in the ovaries of women with PCOS, further exacerbates the ovarian dysfunction by having deleterious effects on the follicular microenvironment and/or GnRH pulsation. Hyperandrogenism is further aggravated by hyperinsulinemia, which develops secondary to insulin resistance. Hyperinsulinemia causes an increase in androgen secretion by theca cells and an inhibition of the production of sex hormone‐binding globulin (SHBG) in the liver, thereby increasing the circulating concentration of bioactive free testosterone. Insulin resistance develops in tissues such as liver and muscle, and is associated with visceral adiposity and adipocyte dysfunction, which are exacerbated by hyperandrogenism
The origin of this vicious cycle of disorders that comprises the pathology of PCOS remains unknown. The simplest explanation for this complex and heterogeneous syndrome would be that hyperandrogenism is a predisposing factor, while insulin resistance triggers the development of PCOS. 4 , 6 , 7 , 18 , 19 Hyperandrogenism is caused by intrinsic dysfunction of theca cells and/or the hypothalamus‐pituitary‐ovarian axis, and is exacerbated by high AMH concentrations. 8 , 9 , 10 , 11 , 20 , 21 The hyperandrogenism and insulin resistance exacerbate one another: excess androgen secretion induces visceral adiposity and adipocyte dysfunction, while the hyperinsulinemia that develops secondary to the insulin resistance stimulates androgen secretion by theca cells and modulates the effects of gonadotropins on theca cells. 14 , 15 , 16 , 17 , 18
Health professionals, especially in East Asia, may wonder whether hyperandrogenism is as common in their patients with PCOS as in those of other ethnicities. Clinical or biochemical hyperandrogenism may not be accurately diagnosed at present, and it has not been determined whether the prevalence of phenotypes A–D among PCOS patients varies according to ethnicity. It is plausible that clinical hyperandrogenism is frequently overlooked in East Asia, where people have less dense villus hair, because appropriate cutoff values of the modified Ferriman‐Gallway (mFG) score for the definition of hirsutism in ethnic groups with less dense villus hair have not been established, nor has it even been determined whether it is appropriate to use the mFG score in these groups for the diagnosis of hirsutism. 3 , 22 Biochemical hyperandrogenism is also difficult to identify at present because assays of direct free testosterone, such as the radiometric and enzyme‐linked assays that are frequently used in the clinical setting, are not particularly sensitive or accurate. 3 Furthermore, the serum androgen concentration does not necessarily reflect local hyperandrogenism in the ovary: high testosterone concentrations in the follicular fluid were found to be present in two‐thirds of East Asian patients with PCOS and a normal serum testosterone concentration using radioimmunoassay. 23 Furthermore, body composition varies substantially among ethnic groups, with East Asian people showing less adiposity than other ethnicities. 3 However, the prevalence of impaired glucose tolerance is higher in patients with PCOS, independent of obesity, than in individuals who do not have PCOS, by five‐fold in Asia, four‐fold in the Americas, and three‐fold in Europe. 13
Hyperandrogenism and insulin resistance together contribute to the pathophysiology, but their individual contributions differ from patient to patient, which accounts for the heterogeneous nature of PCOS and its presentation.
3. WHAT DRIVES THE PATHOGENESIS OF PCOS?
A high level of familial aggregation has been observed for PCOS. A Swedish nationwide register‐based cohort study that included over 29 000 participants showed that daughters born to women with PCOS had a five‐fold higher risk of PCOS than those born to women without PCOS, 24 which is consistent with the findings described above. In addition, a cohort of twins studied in the Netherlands estimated the heritability of PCOS to be ~70%. 25 More recently, the prevalence of all three traits of PCOS, hyperandrogenism, ovulatory dysfunction, and PCOM was compared in the daughters born to women with and without PCOS several years after menarche. This was found to be 16.2% (7/43) in the daughters born to women with PCOS, while none of those born to women without PCOS (0/28) exhibited all of these three characteristics. 26
To elucidate the mechanism underlying the familial clustering of PCOS, the associations of various genes with PCOS have been studied. Candidate genes suggested by genome‐wide association studies (GWAS) include genes that regulate gonadotropin secretion and action and ovarian function, such as FSHB (follicle‐stimulating hormone B polypeptide), LHCGR (luteinizing hormone/choriogonadotropin receptor), FSHR (follicle‐stimulating hormone receptor), AMH, and DENND1A (DENN domain containing 1A); and genes associated with metabolism, such as THADA (thyroid adenoma‐associated gene) and INSR (insulin receptor). 27 , 28 , 29 , 30 Furthermore, an evaluation of the effects of 12 PCOS loci identified by GWAS in Chinese individuals in patients of Northern European descent showed similar effect sizes and directions in both ethnic groups. 31 This finding of shared susceptibility loci in individuals of Chinese and European ancestry suggests that these loci are conserved genetic susceptibility factors for PCOS, implying that PCOS was present at least 50 000–60 000 years ago, when their ancestors migrated out of Africa and then racially diverged, and further implies a common genetic risk profile across populations. 32 , 33 , 34 These findings are consistent with the absence of differences in the prevalence of PCOS in people of differing ethnicity when identical diagnostic criteria are used. 1
Although genetic factors are known to be involved in the pathogenesis of PCOS, it is estimated that the loci identified by GWAS account for less than 10% of its high heritability. 33 , 34 The precise mechanisms underlying its heritability remain to be determined, but it is currently considered that PCOS has a multifactorial etiology: the exposure of individuals with predisposing genetic factors to potent environmental factors drives the development of the various PCOS phenotypes. 19 , 34 The environmental factors involved in the pathophysiology of PCOS may include prenatal exposure to the intrauterine environment of mothers with PCOS, the follicular microenvironment, and lifestyle following birth. The intrauterine environment of mothers with PCOS features high concentrations of androgens because of high circulating androgen concentrations and functional abnormalities of the placenta. 35 , 36 Excess androgen production by the fetal ovaries in response to the intrauterine environment of mothers with PCOS may further exacerbate the androgen‐richness of the intrauterine environment. Details of the mechanisms that induce intrauterine hyperandrogenism in mothers with PCOS will be discussed in section 5. A high circulating concentration of AMH during pregnancy, along with hyperinsulinemia owing to metabolic abnormalities, in mothers with PCOS also contribute to the abnormal intrauterine environment. 21 , 37 , 38
In addition, there is plentiful evidence of an abnormal follicular microenvironment in the ovaries of women with PCOS. 39 This is also hyperandrogenic because of the abnormalities in thecal cell function and the hypothalamus‐pituitary‐ovarian axis. 8 , 9 , 10 , 11 , 20 The local concentration of AMH is high, reflecting the larger number and altered function of pre−/small antral follicles, which secrete AMH. 40 , 41 , 42 , 43 A proinflammatory state in the follicular microenvironment, as well as low‐grade systemic inflammation, is a key feature of PCOS. Furthermore, local oxidative stress and ER stress are features of PCOS. 44 , 45 , 46 , 47 , 48 Inflammation, oxidative stress, and ER stress form a vicious circle that adversely affects the follicular microenvironment of the PCOS ovary, and the local hyperandrogenic conditions further activate these pathways. In addition, metabolic abnormalities, including hyperinsulinemia, have deleterious effects on the follicular microenvironment. 45 , 49 , 50 , 51 , 52 , 53 The accumulation of several exogenous toxins has also been identified in the ovaries of women with PCOS, including endocrine‐disrupting chemicals (EDCs) and advanced glycation end products (AGEs). 54 , 55 AGEs are produced by the Maillard reaction, in which the carbonyl groups of carbohydrates react non‐enzymatically with the primary amino groups of proteins. I will further discuss the role of ER stress in the pathophysiology of PCOS in section 4.
There is no doubt that an unfavorable lifestyle, including a poor diet, which also increases the risk of metabolic disease, drives the pathogenesis of PCOS. 11 , 19 Certain lifestyle factors result in the accumulation of exogenous toxins in the follicular environment, such as the exposure to EDCs and the consumption of AGE‐rich food, may also drive the development of PCOS. AGEs can form either endogenously or exogenously, but most originate exogenously, from smoking or a high‐fat and/or high‐protein diet, especially if this includes food items cooked at high temperature and in a low‐moisture environment. 56
In the following sections, I will discuss in detail the involvement of the two environmental factors mentioned above in the pathogenesis of PCOS, namely, local ER stress in the follicular microenvironment and the androgen‐rich intrauterine environment of mothers with PCOS, and share our recent findings.
4. ROLE OF FOLLICULAR ER STRESS IN THE PATHOPHYSIOLOGY OF PCOS
4.1. ER stress
ER stress has recently been recognized to play important roles in both the pathogenesis of various diseases and the maintenance of physiological processes. The ER is the organelle responsible for the folding and assembly of secretory proteins, and ER stress is defined as a condition in which unfolded or misfolded proteins accumulate in the ER because of an imbalance in the demand for protein folding and the protein‐folding capacity of the ER. 49 , 53 , 57 , 58 ER stress results in the activation of several signal transduction cascades, collectively termed the unfolded protein response (UPR), which affect and regulate various cellular functions. In principle, the UPR exists to restore homeostasis and keep the cell alive in three ways: by reducing the translation of proteins; by increasing the synthesis of ER chaperones, thereby increasing protein‐folding capacity; and by generating ER‐associated degradation (ERAD) factors that remove irreparably misfolded proteins. However, if the ER stress cannot be resolved, the UPR induces programmed cell death. ER stress and the UPR play critical roles in various pathological conditions in humans, including diabetes, neurodegeneration, cancer, inflammatory conditions, and fibrosis. 49 , 59
4.2. ER stress in PCOS
The follicular microenvironment is regulated by gonadotrophins and intraovarian factors in a spatially and temporally coordinated manner. Intraovarian factors have regulatory roles during the entire process of follicular growth and play crucial roles in pathological conditions of the ovary, including PCOS. 39 , 53 , 60 , 61 We demonstrated for the first time that ER stress pathways are activated in the granulosa cells of both a mouse model of PCOS induced by the continuous administration of androgen and in humans, and this finding has been confirmed by other groups. 48 , 62 , 63 , 64 , 65 , 66 , 67 We also found that local hyperandrogenism in the follicular microenvironment of PCOS is an activator of ER stress in human granulosa cells, 52 and this finding has been confirmed in mouse granulosa cells. 63 Other potential activators of ER stress in the follicular microenvironment of PCOS would be local inflammation and oxidative stress, which are closely connected with ER stress and form a vicious circle, and the local hyperandrogenic conditions further activate these pathways. 45 , 49 , 50 , 51 , 52 , 53 Accumulation of AGEs and lipid observed in the follicular microenvironment of PCOS may also account for activation of ER stress. 68 , 69 Various local factors exacerbated in the follicular microenvironment of PCOS may cooperatively compromise ER function, thus activate ER stress.
The ovarian dysfunction in PCOS is characterized by aberrant follicular growth, which accelerates in the early stage and arrests at the antral stage; a failure of the selection of dominant follicles to ovulate; and an ovulatory disorder. 70 The ovarian morphology of patients with PCOS is characterized as PCOM with interstitial fibrosis. 71 We demonstrated that ER stress contributes to the pathophysiology of PCOS through multiple functional alterations in granulosa cells 48 , 52 , 53 , 72 , 73 , 74 (Figure 3). ER stress stimulates the production of transforming growth factor‐β1 (TGF‐β1), a profibrotic growth factor, in granulosa cells and accelerates interstitial fibrosis in the ovary, which is a characteristic of PCOS. 48 ER stress mediates the testosterone‐induced apoptosis of granulosa cells via the induction of the proapoptotic factor death receptor 5 (DR5) and is associated with follicular growth arrest at the antral stage, which is another characteristic of PCOS. 52 ER stress also mediates the testosterone‐induced expression of receptor for advanced glycation end products (RAGE) in granulosa cells, resulting in the accumulation of AGEs in these cells. 72 AGEs are known to accumulate in the granulosa cells of patients with PCOS and are associated with the pathology. 75 ER stress also activates aryl hydrocarbon receptor (AHR), a representative receptor for EDCs, and its downstream signaling in granulosa cells, by which means it could plausibly alter steroid metabolism in these cells. 73 Furthermore, ER stress induces the expression of multiple genes associated with cumulus oocyte‐complex (COC) expansion in granulosa cells via Notch signaling, one of the most evolutionarily highly conserved signaling systems, which regulates various cellular processes via juxtacrine cell–cell interactions. 76 Measurement of the diameters of COCs showed that the ER stress‐Notch pathway increases the size of COCs, although it remains to be determined whether such hypermaturity of COCs plays a causative role in the ovulatory dysfunction that characterizes PCOS. 74
FIGURE 3.
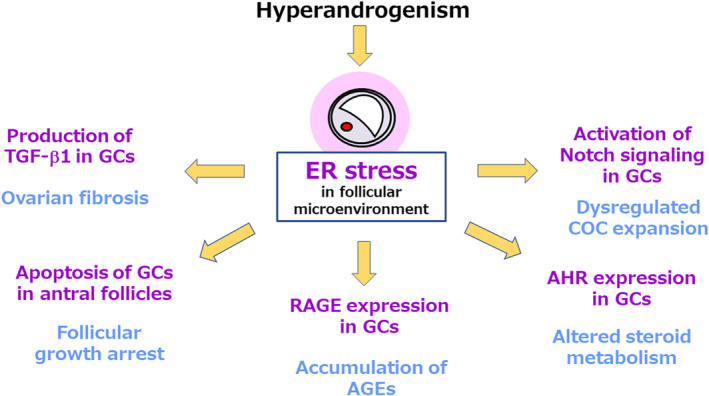
Endoplasmic reticulum stress (ER stress) develops in the follicles and forms a key component of the pathophysiology of polycystic ovary syndrome (PCOS). Local hyperandrogenism in the follicular microenvironment activates ER stress in PCOS. ER stress contributes to the pathophysiology of PCOS by affecting the function of granulosa cells (GCs) in a number of ways. ER stress stimulates the production of transforming growth factor‐β1 (TGF‐β1), a profibrotic growth factor, in GCs and accelerates interstitial fibrosis in the ovary. ER stress mediates the testosterone‐induced apoptosis of granulosa cells by inducing expression of the proapoptotic factor death receptor 5 (DR5) and is associated with follicular growth arrest at the antral follicle stage. ER stress also mediates the effects of testosterone to induce the expression of receptor for advanced glycation end products (RAGE) in GCs, which results in the accumulation of advanced glycation end products (AGEs), thereby affecting various cellular processes. ER stress also activates the aryl hydrocarbon receptor (AHR), a representative receptor for endocrine‐disrupting chemicals (EDCs), and its downstream signaling in GCs, which may alter steroid metabolism in these cells. Furthermore, ER stress induces the expression of multiple genes that are associated with cumulus oocyte‐complex (COC) expansion in GCs via notch signaling
4.3. Clinical implications
These findings suggest that ER stress and the UPR represent potential therapeutic targets for PCOS. Indeed, the treatment of dehydroepiandrosterone (DHEA)‐induced PCOS mice with ER stress inhibitors ameliorates their reproductive dysfunction; specifically, it improves the estrous cycle and PCOM by reducing the number of atretic antral follicles. 72 Histologically, treatment with ER stress inhibitors reduces the levels of interstitial fibrosis and collagen deposition in the ovary, the apoptosis of granulosa cells of the antral follicles, and the accumulation of AGEs in these cells, with a concomitant reduction in ovarian ER stress. 48 , 52 , 72
There are two means of modulating ER stress and the UPR: one is to reduce ER stress by attenuating the protein misfolding that underpins it, and the other is the targeting of specific UPR factors. 53 For the first strategy, chemical chaperones can be used as a pharmacological approach, and lifestyle interventions may also be effective. Chemical chaperones are a group of low‐molecular mass compounds that stabilize the folding of proteins and reduce abnormal protein aggregation, thereby reducing protein misfolding. 77 Two chemical chaperones, tauroursodeoxycholic acid (TUDCA) and 4‐phenylbutyrate (4‐PBA), have been used clinically to treat liver diseases and urea cycle disorders, respectively, for a long time, and recent studies have shown that they function as chemical chaperones. Lifestyle is associated with the activation of ER stress, either directly or indirectly. For instance, ER stress is activated in the granulosa cells of obese women. 78 In addition, it is closely connected with other local factors, including oxidative stress, AGEs, and inflammation, all of which are affected by lifestyle. 68 , 79 Lifestyle interventions, including the consumption of supplements, may represent valuable approaches, given that they are usually accepted as part of preconception care. In contrast, there are no small molecules in clinical use that target specific UPR factors at present. Various promising molecules are in development, in particular targeting the UPR branch activated by double‐stranded RNA‐activated protein kinase‐like ER kinase (PERK). 59
5. PRENATAL EXPOSURE TO EXCESS ANDROGENS, ALTERATIONS IN THE GUT MICROBIOME, AND THE DEVELOPMENT OF PCOS
5.1. Prenatal exposure to excess androgens in the daughters of women with PCOS and the development of PCOS in their later life
The daughters of women with PCOS are prenatally exposed to high concentrations of androgens. Daughters born to women with PCOS have a larger anogenital distance, which serves as a biomarker of intrauterine exposure to excess androgens, during the fetal and neonatal periods as well as in adulthood. 80 , 81 , 82 , 83 In addition, serum androgen concentrations are higher in women with PCOS than in those without it during pregnancy. 36 , 84 Moreover, placental tissue from women with PCOS is characterized by a higher activity of 3β‐hydroxysteroid dehydrogenase‐1 (3β‐HSD‐1) and a lower activity of P450 aromatase than that from women without PCOS, which could also explain the hyperandrogenic intrauterine environment of pregnant women with PCOS. 36 These findings are consistent with a role for prenatal exposure to excess androgen in the daughters of women with PCOS.
Several other mechanisms may contribute to the hyperandrogenic intrauterine environment of pregnant women with PCOS. Metabolic dysfunction in women with PCOS can induce fetal hyperinsulinemia, which increases androgen production from the ovaries of the fetus after midgestation. 37 , 38 In addition, pregnant women with PCOS have higher serum AMH concentrations than women without, and this high AMH may result in maternal neuroendocrine‐induced testosterone excess and less placental metabolism of testosterone to estradiol, as shown in pregnant mice treated with AMH, leading to hyperandrogenism in utero. 21
Numerous studies have shown that prenatally androgenized (PNA) animals, including rodents, sheep, and rhesus monkeys, exhibit PCOS‐like reproductive and metabolic phenotypes in adulthood, as extensively reviewed previously. 85 , 86 , 87 The investigation of the mechanism by which prenatal androgen exposure may induce the development of PCOS in adulthood has just started. One possible mechanism is the induction of epigenetic changes in fetal somatic and/or germ cells by prenatal androgen exposure. Prenatal exposure to androgens induces specific epigenetic changes in the ovary, including in granulosa and theca cells, and in the key metabolic tissues, including liver, muscle, and visceral and subcutaneous adipose tissue. 88 , 89 Prenatal androgen exposure may also induce the epigenetic modification of germ cells: PCOS‐like reproductive and metabolic traits are observed in the granddaughters of female mice that are perinatally exposed to androgens, as well as in those exposed to AMH, which causes intrauterine hyperandrogenism. 24 , 90 It is also plausible that in utero exposure to androgens might cause abnormal ovarian and early follicular development. Daughters born to mothers with PCOS exhibit high serum concentrations of AMH, which is produced by granulosa cells and reflects follicular development during infancy, prepuberty, and at the time of delivery, than those born to mothers without PCOS. 35 , 91 In addition, a high density of small preantral follicles is present in the ovaries of women with PCOS, which may be the result of a larger population of germ cells in the fetal ovary, or a lower rate of loss of oocytes during late gestation, childhood, and puberty. 92 Another interesting finding that may link prenatal androgen exposure to PCOS in later life is the dysbiosis of the gut microbiome of adult female rodents prenatally exposed to androgens. 93 , 94
5.2. Potential role of the gut microbiome in the pathogenesis of PCOS
The links between the gut microbiome and various physiological and pathological conditions have been receiving an increasing amount of attention during the past decade. Metabolic disorders are of particular interest in this research field, and mounting evidence is consistent with the important and causative roles of dysbiosis of the gut microbiome in metabolic disorders, such as obesity and type 2 diabetes. 95 , 96 Given that sex steroid hormone concentrations are related to gut microbial composition, 97 , 98 it is conceivable that changes in gut microbiome may play a role in the pathophysiology of PCOS. Recent studies have demonstrated that the gut microbiomes of adult patients with PCOS and in various models of PCOS differs from those of healthy individuals and control animals, respectively, as summarized in a recent review. 99 Moreover, the transplantation of feces from or cohousing with healthy rodents ameliorates the PCOS‐like phenotypes of such models. These findings imply that the gut microbiome plays a causative role in the pathogenesis of PCOS. 100 , 101
5.3. Effects of prenatal exposure to high androgen concentrations on the gut microbiome and the development of the PCOS phenotype
We hypothesized that prenatal exposure to high concentrations of androgens would induce dysbiosis of the gut microbiome early in life and thus lead to the development of PCOS in later life. As a first step in testing this hypothesis, we compared the gut microbiomes of PNA mice induced by the injection of dihydrotestosterone (DHT) into pregnant dams and control mice when they were prepubertal (4 weeks of age); at puberty (6 weeks); and during their adolescence (8 weeks), young adulthood (12 weeks), and adulthood (16 weeks). We then studied the temporal relationship between the alterations in the gut microbiome and the development of PCOS‐like phenotypes. 102 The PCOS‐like reproductive phenotype was identified through the assessment of estrous cyclicity, ovarian histology, and serum testosterone concentration, and the metabolic phenotype was assessed using the measurement of body mass, the size of visceral adipocytes, insulin tolerance, and fasting blood glucose (FBG) concentration. We performed next‐generation sequencing (NGS) of fecal bacterial 16S rRNA genes to characterize the gut microbiomes of mice. The α‐ and β‐diversities, which reflect the richness of the microbial species and the similarity between the groups, respectively, were calculated, and the differences in the bacterial taxa between the control and PNA offspring were also identified.
We found that abnormalities appear in the gut microbiome as early as or even before PCOS‐like phenotypes manifest in PNA offspring 102 (Figure 4). Specifically, the reproductive phenotype of PCOS became apparent at puberty in the PNA offspring, which was followed by the appearance of the metabolic features of PCOS in young adulthood. By contrast, alterations to the composition of the gut microbiome of female PNA offspring were apparent as early as before puberty and continued throughout the study, with a lower richness of bacterial taxa after young adulthood in PNA offspring, and significant differences in the microbial communities of the PNA and control groups during adolescence. Eleven bacterial taxa were consistently more or less abundant in the PNA group across multiple time points, and the abundances of nine of these taxa were lower in the PNA group. In addition, 5 and 10 of these 11 taxa were already less abundant before and at puberty, respectively.
FIGURE 4.

Temporal relationship between alterations in the gut microbiome and the development of polycystic ovary syndrome (PCOS)‐like phenotypes in prenatally androgenized (PNA) mice. PNA mice were generated by injecting dihydrotestosterone (DHT) into pregnant dams. The gut microbiomes of PNA and control mice were analyzed when they were prepubertal (4 weeks of age); at puberty (6 weeks); and during their adolescence (8 weeks), young adulthood (12 weeks), and adulthood (16 weeks). Then, the temporal relationship between the alterations in the gut microbiome and the development of the PCOS‐like phenotype was evaluated. The reproductive phenotype of PCOS, involving changes in estrous cyclicity, ovarian histology, and serum testosterone concentration, became apparent at puberty in the PNA offspring. This was followed by the appearance of the metabolic characteristics of PCOS in young adulthood, featuring differences in body mass, the size of visceral adipocytes, insulin tolerance, and fasting blood glucose (FBG) concentration. By contrast, alterations to the gut microbiome of female PNA offspring were apparent as early as before puberty and were present throughout the study
5.4. Clinical implications
Our findings that abnormalities appear in the gut microbiome as early as or even before PCOS‐like phenotypes manifest in PNA offspring, and that these mimic the in utero environment of women with PCOS, imply that female PNA offspring that are susceptible to PCOS already have an abnormal gut microbial composition soon after weaning, and this may be amplified by the consumption of certain foods and sex steroid action after puberty, contributing to the development of the reproductive and metabolic phenotypes of PCOS.
Importantly, given that there is currently no preventive strategy for use in girls at a high risk of subsequently developing PCOS, including for the daughters of mothers with PCOS, our findings suggest a novel strategy for the prevention of PCOS in later life. To clarify the role of alterations of gut microbiome in the development of PCOS in later life, intervention studies are needed. In addition, the identification of the mechanism by which dysbiosis is induced in PNA offspring as early as just after weaning should provide insight into whether interventions in pregnant women with PCOS or in daughters born to such mothers would be more effective. Alterations to the intrauterine environment of PNA dams might affect the formation of gut microbiome in their pups. Alternatively, the gut, skin, and/or milk microbiome of PNA dams might be altered by DHT treatment, and this altered microbial population might be transmitted to their pups during delivery and feeding. Furthermore, the identification of fecal metabolites that are affected by dysbiosis in PNA offspring would be helpful in guiding the selection of novel pre‐/pro‐/postbiotics that might help prevent the development of PCOS.
6. FUTURE PERSPECTIVES
The pathophysiology of PCOS is heterogeneous in nature and is further complicated by the variations in presentation between ethnicities. The pathogenesis is shaped by interactions between reproductive dysfunction and metabolic disorders: hyperandrogenism and insulin resistance exacerbate one another in the development of PCOS, which is also affected by altered function of hypothalamus‐pituitary‐ovarian axis. PCOS is highly heritable, and genetic factors are certainly involved in its pathogenesis, but the loci identified using GWAS appear to only account for a small proportion of this heritability. It is currently considered that PCOS is a multifactorial disorder, like type 2 diabetes, with the exposure to environmental factors causing individuals with predisposing genetic factors to develop PCOS. However, environmental factors play a much larger role than genetic factors. These environmental factors may include the intrauterine environment during the prenatal period, the follicular microenvironment, and lifestyle following birth.
On the basis of this current understanding, the following three areas represent targets for future research (Figure 5). The first is to identify the factors that induce the development and progression of PCOS, and to elucidate the underlying mechanisms. In particular, the mechanism connecting the reproductive and metabolic abnormalities that comprise the pathophysiology of PCOS is of great interest because such knowledge could be used to target the interruption of the vicious cycle involved, but research in this area to date has been limited. In addition, further investigation of the environmental factors that are known to drive the development of PCOS through effects after birth, such as the follicular microenvironment and lifestyle, is necessary. Furthermore, the identification of novel factors, which may include psychosocial factors, would add to our understanding of the pathophysiology.
FIGURE 5.
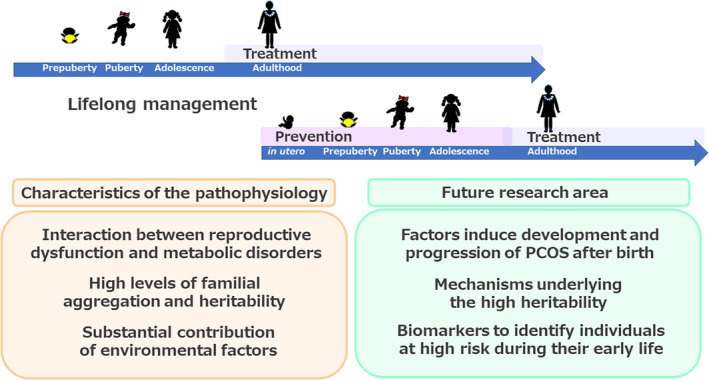
Summary and future directions. Three principal characteristics of the pathophysiology of polycystic ovary syndrome (PCOS) have been identified to date: an interaction between reproductive dysfunction and metabolic disorders, high familial aggregation and heritability, and a substantial contribution of environmental factors. On the basis of this current understanding, the following three areas represent targets for future research. (1) To identify the factors that induce the development and progression of PCOS after birth and elucidate the underlying mechanisms. (2) To elucidate the mechanisms underlying the high heritability of PCOS. (3) To identify biomarkers that could be used to identify individuals at high risk of developing PCOS during their early life. Future research should aim to develop therapeutic and preventive strategies, with the ultimate goal of achieving appropriate lifelong management, including preconception care
The second area for further research is the elucidation of the mechanisms underlying the high heritability of PCOS. The research performed to date may have underestimated the contribution of genetic factors. The limited contribution of the genetic loci identified by GWAS may be at least in part attributable to the ‘common disease, common variants’ rationale of GWAS, which means that the etiology of common and complex diseases cannot be explained by the limited number of common variants of moderate‐to‐low effect size that can be detected using this method. 103 The use of novel strategies in combination with GWAS, including the investigation of gene–environment interactions, may help elucidate the actual contributions of genetic factors to the high familial aggregation and heritability of PCOS.
Investigation of the mechanisms by which the intrauterine environment during the perinatal period predisposes toward the development of PCOS in later life has only just started, and this is definitely a promising area of research, given that accumulating evidence implies that this phenomenon is important in humans rather than just in experimental animals. As discussed, the induction of epigenetic changes in fetal somatic and/or germ cells, and of dysbiosis of the offspring's gut microbiome, requires investigation. With respect to the gut microbiome, the identification of the fecal metabolite concentrations that are affected by dysbiosis and an exploration of the functional links between the gut and other organs should improve our understanding of the pathophysiology of PCOS as a systemic disorder. Other environmental factors present after birth that may be involved in the high familial aggregation and heritability, the follicular microenvironment and lifestyle, overlap with the first area for future research described above.
The third area for further research is the identification of biomarkers that could be used to identify individuals early in life who are at high risk of developing PCOS in later life. As discussed, women born to mothers with PCOS are at high risk of developing PCOS, but a Swedish nationwide register‐based cohort study showed that only 3.3% (78/2275) of daughters born to mothers with PCOS were subsequently diagnosed as having PCOS, while the remaining 96.7% were not. 24 The identification of biomarkers capable of distinguishing these two groups early in life would help them receive appropriate care. However, the mothers of women diagnosed with PCOS were not necessarily diagnosed as having PCOS themselves: in the Swedish cohort, 67.1% (159/237) of all daughters diagnosed with PCOS were born to mothers who did not have PCOS. 24 Biomarkers that can be used to identify women who are predisposed toward the development of PCOS in groups that are theoretically at low risk, such as those who were born to mothers without PCOS, would also be helpful. Evaluation of maternal metabolic and/or hormonal status during pregnancy may be helpful for distinguishing daughters at high and low risk. Alternatively, metabolic and/or hormonal status of daughters themselves at their early stage of life, that is at infancy or childhood, may serve as a biomarker identifying those who may develop PCOS in later life. Totally novel biomarkers, including fecal metabolites or gut microbial composition, would be of special interest for future research.
These areas for future research should enhance our understanding of this complex and enigmatic disorder, facilitate the development of therapeutic and preventive strategies that would supersede the current approach that targets individual symptoms, and help us achieve the eventual goal of achieving appropriate lifelong management of PCOS, including preconception care.
CONFLICT OF INTEREST
Miyuki Harada declares that she has no conflict of interest.
APPROVAL BY ETHICS COMMITTEE
Not applicable
HUMAN RIGHTS STATEMENTS AND INFORMED CONSENT
Not applicable
ANIMAL RIGHTS
Not applicable
ACKNOWLEDGMENTS
The author thanks the members of her laboratory, Nozomi Takahashi, Chisato Kunitomi, Akari Kusamoto, and Hiroshi Koike (The University of Tokyo), and Jerilee MK Azhary (The University of Tokyo and University of Malaya), for their contribution to the papers cited; Akari Kusamoto and Hiroshi Koike for their support in drawing schematic diagrams; and all the members of her laboratory for their critical reading of the manuscript and valuable comments. The author also thanks Prof. Yutaka Osuga (The University of Tokyo) for his supervision. This study was supported by Japan Society for the Promotion of Science (JSPS; 22 K09614).
A summary of the content of this paper was presented during Educational Lecture 4 at the 66th Japan Society of Reproductive Medicine (JSRM) meeting in 2021, hosted by Prof. Tasuku Harada at Tottori University. The author expresses her sincere appreciation to Prof. Harada for giving her the opportunity to present this work. Finally, the author expresses special thanks to the Editorial committee of Reproductive Medicine and Biology for inviting her to contribute to the journal.
Harada M. Pathophysiology of polycystic ovary syndrome revisited: Current understanding and perspectives regarding future research. Reprod Med Biol. 2022;21:e12487. doi: 10.1002/rmb2.12487
Clinical trial registry: Not applicable.
REFERENCES
- 1. Bozdag G, Mumusoglu S, Zengin D, Karabulut E, Yildiz BO. The prevalence and phenotypic features of polycystic ovary syndrome: a systematic review and meta‐analysis. Hum Reprod. 2016;31:2841–55. [DOI] [PubMed] [Google Scholar]
- 2. Rotterdam ESHRE/ASRM‐Sponsored PCOS Consensus Workshop Group . Revised 2003 consensus on diagnostic criteria and long‐term health risks related to polycystic ovary syndrome (PCOS). Hum Reprod. 2004;19:41–7. [DOI] [PubMed] [Google Scholar]
- 3. International PCOS Network in Collaboration with Funding Partner and Collaborating Partners. International evidence‐based guideline for the assessment and management of polycystic ovary syndrome 2018. https://www.monash.edu/__data/assets/pdf_file/0004/1412644/PCOS_Evidence‐Based‐Guidelines_20181009.pdf. 2018. Accessed 17 Jun 2022.
- 4. Azziz R, Carmina E, Dewailly D, Diamanti‐Kandarakis E, Escobar‐Morreale HF, Futterweit W, et al. Positions statement: criteria for defining polycystic ovary syndrome as a predominantly hyperandrogenic syndrome: an androgen excess society guideline. J Clin Endocrinol Metab. 2006;91:4237–45. [DOI] [PubMed] [Google Scholar]
- 5. Kubota T. Update in polycystic ovary syndrome: new criteria of diagnosis and treatment in Japan. Reprod Med Biol. 2013;12:71–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Legro RS, Driscoll D, Strauss JF 3rd, Fox J, Dunaif A. Evidence for a genetic basis for hyperandrogenemia in polycystic ovary syndrome. Proc Natl Acad Sci U S A. 1998;95:14956–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McAllister JM, Legro RS, Modi BP, Strauss JF 3rd. Functional genomics of PCOS: from GWAS to molecular mechanisms. Trends Endocrinol Metab. 2015;26:118–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Taylor AE, McCourt B, Martin KA, Anderson EJ, Adams JM, Schoenfeld D, et al. Determinants of abnormal gonadotropin secretion in clinically defined women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1997;82:2248–56. [DOI] [PubMed] [Google Scholar]
- 9. Burt Solorzano CM, McCartney CR, Blank SK, Knudsen KL, Marshall JC. Hyperandrogenaemia in adolescent girls: origins of abnormal gonadotropin‐releasing hormone secretion. BJOG. 2010;117:143–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Visser JA, Durlinger AL, Peters IJ, van den Heuvel ER, Rose UM, Kramer P, et al. Increased oocyte degeneration and follicular atresia during the estrous cycle in anti‐Müllerian hormone null mice. Endocrinology. 2007;148:2301–8. [DOI] [PubMed] [Google Scholar]
- 11. Azziz R, Carmina E, Chen Z, Dunaif A, Laven JS, Legro RS, et al. Polycystic ovary syndrome. Nat Rev Dis Primers. 2016;2:16057. [DOI] [PubMed] [Google Scholar]
- 12. Cassar S, Misso ML, Hopkins WG, Shaw CS, Teede HJ, Stepto NK. Insulin resistance in polycystic ovary syndrome: a systematic review and meta‐analysis of euglycaemic‐hyperinsulinaemic clamp studies. Hum Reprod. 2016;31:2619–31. [DOI] [PubMed] [Google Scholar]
- 13. Kakoly NS, Khomami MB, Joham AE, Cooray SD, Misso ML, Norman RJ, et al. Ethnicity, obesity and the prevalence of impaired glucose tolerance and type 2 diabetes in PCOS: a systematic review and meta‐regression. Hum Reprod Update. 2018;24:455–67. [DOI] [PubMed] [Google Scholar]
- 14. Nestler JE, Powers LP, Matt DW, Steingold KA, Plymate SR, Rittmaster RS, et al. A direct effect of hyperinsulinemia on serum sex hormone‐binding globulin levels in obese women with the polycystic ovary syndrome. J Clin Endocrinol Metab. 1991;72:83–9. [DOI] [PubMed] [Google Scholar]
- 15. Nestler JE, Jakubowicz DJ, de Vargas AF, Brik C, Quintero N, Medina F. Insulin stimulates testosterone biosynthesis by human thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. J Clin Endocrinol Metab. 1998;83:2001–5. [DOI] [PubMed] [Google Scholar]
- 16. Escobar‐Morreale HF, San Millán JL. Abdominal adiposity and the polycystic ovary syndrome. Trends Endocrinol Metab. 2007;18:266–72. [DOI] [PubMed] [Google Scholar]
- 17. Dumesic DA, Akopians AL, Madrigal VK, Ramirez E, Margolis DJ, Sarma MK, et al. Hyperandrogenism accompanies increased intra‐abdominal fat storage in Normal weight polycystic ovary syndrome women. J Clin Endocrinol Metab. 2016;101:4178–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thong EP, Codner E, Laven JSE, Teede H. Diabetes: a metabolic and reproductive disorder in women. Lancet Diabetes Endocrinol. 2020;8:134–49. [DOI] [PubMed] [Google Scholar]
- 19. Escobar‐Morreale HF. Polycystic ovary syndrome: definition, aetiology, diagnosis and treatment. Nat Rev Endocrinol. 2018;14:270–84. [DOI] [PubMed] [Google Scholar]
- 20. Gilling‐Smith C, Story H, Rogers V, Franks S. Evidence for a primary abnormality of thecal cell steroidogenesis in the polycystic ovary syndrome. Clin Endocrinol (Oxf). 1997;47:93–9. [DOI] [PubMed] [Google Scholar]
- 21. Tata B, Mimouni NEH, Barbotin AL, Malone SA, Loyens A, Pigny P, et al. Elevated prenatal anti‐Müllerian hormone reprograms the fetus and induces polycystic ovary syndrome in adulthood. Nat Med. 2018;24:834–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yildiz BO, Bolour S, Woods K, Moore A, Azziz R. Visually scoring hirsutism. Hum Reprod Update. 2010;16:51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li A, Zhang L, Jiang J, Yang N, Liu Y, Cai L, et al. Follicular hyperandrogenism and insulin resistance in polycystic ovary syndrome patients with normal circulating testosterone levels. J Biomed Res. 2017;32:208–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Risal S, Pei Y, Lu H, Manti M, Fornes R, Pui HP, et al. Prenatal androgen exposure and transgenerational susceptibility to polycystic ovary syndrome. Nat Med. 2019;25:1894–904. [DOI] [PubMed] [Google Scholar]
- 25. Vink JM, Sadrzadeh S, Lambalk CB, Boomsma DI. Heritability of polycystic ovary syndrome in a Dutch twin‐family study. J Clin Endocrinol Metab. 2006;91:2100–4. [DOI] [PubMed] [Google Scholar]
- 26. Crisosto N, Ladrón de Guevara A, Echiburú B, Maliqueo M, Cavada G, Codner E, et al. Higher luteinizing hormone levels associated with antimüllerian hormone in postmenarchal daughters of women with polycystic ovary syndrome. Fertil Steril. 2019;111:381–8. [DOI] [PubMed] [Google Scholar]
- 27. Chen ZJ, Zhao H, He L, Shi Y, Qin Y, Shi Y, et al. Genome‐wide association study identifies susceptibility loci for polycystic ovary syndrome on chromosome 2p16.3, 2p21 and 9q33.3. Nat Genet. 2011;43:55–9. [DOI] [PubMed] [Google Scholar]
- 28. Shi Y, Zhao H, Shi Y, Cao Y, Yang D, Li Z, et al. Genome‐wide association study identifies eight new risk loci for polycystic ovary syndrome. Nat Genet. 2012;44:1020–5. [DOI] [PubMed] [Google Scholar]
- 29. Hayes MG, Urbanek M, Ehrmann DA, Armstrong LL, Lee JY, Sisk R, et al. Genome‐wide association of polycystic ovary syndrome implicates alterations in gonadotropin secretion in European ancestry populations. Nat Comm. 2015;6:7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Day FR, Hinds DA, Tung JY, Stolk L, Styrkarsdottir U, Saxena R, et al. Causal mechanisms and balancing selection inferred from genetic associations with polycystic ovary syndrome. Nat Comm. 2015;6:8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Louwers YV, Stolk L, Uitterlinden AG, Laven JS. Cross‐ethnic meta‐analysis of genetic variants for polycystic ovary syndrome. J Clin Endocrinol Metab. 2013;98:E2006–12. [DOI] [PubMed] [Google Scholar]
- 32. Ünlütürk U, Sezgin E, Yildiz BO. Evolutionary determinants of polycystic ovary syndrome: part 1. Fertil Steril. 2016;106:33–41. [DOI] [PubMed] [Google Scholar]
- 33. Dunaif A. Perspectives in polycystic ovary syndrome: from hair to eternity. J Clin Endocrinol Metab. 2016;101:759–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Azziz R. Polycystic ovary syndrome. Obstet Gynecol. 2018;132:321–36. [DOI] [PubMed] [Google Scholar]
- 35. Sir‐Petermann T, Codner E, Maliqueo M, Echiburú B, Hitschfeld C, Crisosto N, et al. Increased anti‐Müllerian hormone serum concentrations in prepubertal daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91:3105–9. [DOI] [PubMed] [Google Scholar]
- 36. Maliqueo M, Lara HE, Sánchez F, Echiburú B, Crisosto N, Sir‐Petermann T. Placental steroidogenesis in pregnant women with polycystic ovary syndrome. Eur J Obstet Gynecol Reprod Biol. 2013;166:151–5. [DOI] [PubMed] [Google Scholar]
- 37. Shifren JL, Osathanondh R, Yeh J. Human fetal ovaries and uteri: developmental expression of genes encoding the insulin, insulin‐like growth factor I, and insulin‐like growth factor II receptors. Fertil Steril. 1993;59:1036–40. [DOI] [PubMed] [Google Scholar]
- 38. Fowler PA, Anderson RA, Saunders PT, Kinnell H, Mason JI, Evans DB, et al. Development of steroid signaling pathways during primordial follicle formation in the human fetal ovary. J Clin Endocrinol Metab. 2011;96:1754–62. [DOI] [PubMed] [Google Scholar]
- 39. Dumesic DA, Meldrum DR, Katz‐Jaffe MG, Krisher RL, Schoolcraft WB. Oocyte environment: follicular fluid and cumulus cells are critical for oocyte health. Fertil Steril. 2015;103:303–16. [DOI] [PubMed] [Google Scholar]
- 40. Laven JS, Mulders AG, Visser JA, Themmen AP, De Jong FH, Fauser BC. Anti‐Müllerian hormone serum concentrations in normoovulatory and anovulatory women of reproductive age. J Clin Endocrinol Metab. 2004;89:318–23. [DOI] [PubMed] [Google Scholar]
- 41. Homburg R, Ray A, Bhide P, Gudi A, Shah A, Timms P, et al. The relationship of serum anti‐Mullerian hormone with polycystic ovarian morphology and polycystic ovary syndrome: a prospective cohort study. Hum Reprod. 2013;28:1077–83. [DOI] [PubMed] [Google Scholar]
- 42. Pellatt L, Hanna L, Brincat M, Galea R, Brain H, Whitehead S, et al. Granulosa cell production of anti‐Müllerian hormone is increased in polycystic ovaries. J Clin Endocrinol Metab. 2007;92:240–5. [DOI] [PubMed] [Google Scholar]
- 43. Bhide P, Dilgil M, Gudi A, Shah A, Akwaa C, Homburg R. Each small antral follicle in ovaries of women with polycystic ovary syndrome produces more antimüllerian hormone than its counterpart in a normal ovary: an observational cross‐sectional study. Fertil Steril. 2015;103:537–41. [DOI] [PubMed] [Google Scholar]
- 44. Schmidt J, Weijdegård B, Mikkelsen AL, Lindenberg S, Nilsson L, Brännström M. Differential expression of inflammation‐related genes in the ovarian stroma and granulosa cells of PCOS women. Mol Hum Reprod. 2014;20:49–58. [DOI] [PubMed] [Google Scholar]
- 45. Adams J, Liu Z, Ren YA, Wun WS, Zhou W, Kenigsberg S, et al. Enhanced inflammatory transcriptome in the granulosa cells of women with polycystic ovarian syndrome. J Clin Endocrinol Metab. 2016;101:3459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chattopadhayay R, Ganesh A, Samanta J, Jana SK, Chakravarty BN, Chaudhury K. Effect of follicular fluid oxidative stress on meiotic spindle formation in infertile women with polycystic ovarian syndrome. Gynecol Obstet Invest. 2010;69:197–202. [DOI] [PubMed] [Google Scholar]
- 47. González F, Rote NS, Minium J, Kirwan JP. Reactive oxygen species‐induced oxidative stress in the development of insulin resistance and hyperandrogenism in polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91:336–40. [DOI] [PubMed] [Google Scholar]
- 48. Takahashi N, Harada M, Hirota Y, Nose E, Azhary JM, Koike H, et al. Activation of endoplasmic reticulum stress in granulosa cells from patients with polycystic ovary syndrome contributes to ovarian fibrosis. Sci Rep. 2017;7:10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rutkowski DT, Kaufman RJ. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem Sci. 2007;32:469–76. [DOI] [PubMed] [Google Scholar]
- 50. González F. Inflammation in polycystic ovary syndrome: underpinning of insulin resistance and ovarian dysfunction. Steroids. 2012;77:300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hasnain SZ, Lourie R, Das I, Chen AC, McGuckin MA. The interplay between endoplasmic reticulum stress and inflammation. Immunol Cell Biol. 2012;90:260–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Azhary JMK, Harada M, Takahashi N, Nose E, Kunitomi C, Koike H, et al. Endoplasmic reticulum stress activated by androgen enhances apoptosis of granulosa cells via induction of death receptor 5 in PCOS. Endocrinology. 2019;160:119–32. [DOI] [PubMed] [Google Scholar]
- 53. Harada M, Takahashi N, Azhary JM, Kunitomi C, Fujii T, Osuga Y. Endoplasmic reticulum stress: a key regulator of the follicular microenvironment in the ovary. Mol Hum Reprod. 2021;27:gaaa088. [DOI] [PubMed] [Google Scholar]
- 54. Rutkowska AZ, Diamanti‐Kandarakis E. Polycystic ovary syndrome and environmental toxins. Fertil Steril. 2016;106:948–58. [DOI] [PubMed] [Google Scholar]
- 55. Diamanti‐Kandarakis E, Piperi C, Patsouris E, Korkolopoulou P, Panidis D, Pawelczyk L, et al. Immunohistochemical localization of advanced glycation end‐products (AGEs) and their receptor (RAGE) in polycystic and normal ovaries. Histochem Cell Biol. 2007;127:581–9. [DOI] [PubMed] [Google Scholar]
- 56. Garg D, Merhi Z. Advanced glycation end products: link between diet and ovulatory dysfunction in PCOS? Nutrients. 2015;7:10129–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–6. [DOI] [PubMed] [Google Scholar]
- 58. Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21:421–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hetz C, Axten JM, Patterson JB. Pharmacological targeting of the unfolded protein response for disease intervention. Nat Chem Biol. 2019;15:764–75. [DOI] [PubMed] [Google Scholar]
- 60. Harada M, Nose E, Takahashi N, Hirota Y, Hirata T, Yoshino O, et al. Evidence of the activation of unfolded protein response in granulosa and cumulus cells during follicular growth and maturation. Gynecol Endocrinol. 2015;31:783–7. [DOI] [PubMed] [Google Scholar]
- 61. Takahashi N, Harada M, Hirota Y, Zhao L, Yoshino O, Urata Y, et al. A potential role of endoplasmic reticulum stress in development of ovarian hyperstimulation syndrome. Mol Cell Endocrinol. 2016;428:161–9. [DOI] [PubMed] [Google Scholar]
- 62. Peng Y, Guo L, Gu A, Shi B, Ren Y, Cong J, et al. Electroacupuncture alleviates polycystic ovary syndrome‐like symptoms through improving insulin resistance, mitochondrial dysfunction, and endoplasmic reticulum stress via enhancing autophagy in rats. Mol Med. 2020;26:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jin J, Ma Y, Tong X, Yang W, Dai Y, Pan Y, et al. Metformin inhibits testosterone‐induced endoplasmic reticulum stress in ovarian granulosa cells via inactivation of p38 MAPK. Hum Reprod. 2020;35:1145–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Brenjian S, Moini A, Yamini N, Kashani L, Faridmojtahedi M, Bahramrezaie M, et al. Resveratrol treatment in patients with polycystic ovary syndrome decreased pro‐inflammatory and endoplasmic reticulum stress markers. Am J Reprod Immunol. 2020;83:e13186. [DOI] [PubMed] [Google Scholar]
- 65. Zhang Y, Weng Y, Wang D, Wang R, Wang L, Zhou J, et al. Curcumin in combination with aerobic exercise improves follicular dysfunction via inhibition of the Hyperandrogen‐induced IRE1α/XBP1 endoplasmic reticulum stress pathway in PCOS‐like rats. Oxid Med Cell Longev. 2021;2021:7382900–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sun HL, Tian MM, Jiang JX, Liu CJ, Zhai QL, Wang CY, et al. Does endoplasmic reticulum stress stimulate the apoptosis of granulosa cells in polycystic ovary syndrome? J Physiol Pharmacol. 2021;72:785–92. [DOI] [PubMed] [Google Scholar]
- 67. El‐Saka MH, Barhoma RA, Ibrahim RR, Elsaadany A, Alghazaly GM, Elshwaikh S, et al. Potential effect of adrenomedullin on metabolic and endocrinal dysfunctions in the experimentally induced polycystic ovary: targeting implication of endoplasmic reticulum stress. J Biochem Mol Toxicol. 2021;35:e22725. [DOI] [PubMed] [Google Scholar]
- 68. Takahashi N, Harada M, Azhary JMK, Kunitomi C, Nose E, Terao H, et al. Accumulation of advanced glycation end products in follicles is associated with poor oocyte developmental competence. Mol Hum Reprod. 2019;25:684–94. [DOI] [PubMed] [Google Scholar]
- 69. Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res. 2006;47:2726–37. [DOI] [PubMed] [Google Scholar]
- 70. Jonard S, Dewailly D. The follicular excess in polycystic ovaries, due to intra‐ovarian hyperandrogenism, may be the main culprit for the follicular arrest. Hum Reprod Update. 2004;10:107–17. [DOI] [PubMed] [Google Scholar]
- 71. Hughesdon PE. Morphology and morphogenesis of the stein‐Leventhal ovary and of so‐called "hyperthecosis". Obstet Gynecol Surv. 1982;37:59–77. [DOI] [PubMed] [Google Scholar]
- 72. Azhary JMK, Harada M, Kunitomi C, Kusamoto A, Takahashi N, Nose E, et al. Androgens increase accumulation of advanced glycation end products in granulosa cells by activating ER stress in PCOS. Endocrinology. 2020;161:bqaa015. [DOI] [PubMed] [Google Scholar]
- 73. Kunitomi C, Harada M, Kusamoto A, Azhary JM, Nose E, Koike H, et al. Induction of aryl hydrocarbon receptor in granulosa cells by endoplasmic reticulum stress contributes to pathology of polycystic ovary syndrome. Mol Hum Reprod. 2021;27:gaab003. [DOI] [PubMed] [Google Scholar]
- 74. Koike H, Harada M, Kusamoto A, Kunitomi C, Xu Z, Tanaka T, et al. Notch signaling induced by endoplasmic reticulum stress regulates cumulus‐oocyte complex expansion in polycystic ovary syndrome. Biomolecules. 2022;12:1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Merhi Z, Kandaraki EA, Diamanti‐Kandarakis E. Implications and future perspectives of AGEs in PCOS pathophysiology. Trends Endocrinol Metabol. 2019;30:150–62. [DOI] [PubMed] [Google Scholar]
- 76. Vanorny DA, Mayo KE. The role of notch signaling in the mammalian ovary. Reproduction. 2017;153:R187–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discov. 2013;12:703–19. [DOI] [PubMed] [Google Scholar]
- 78. Takahashi N, Harada M, Hirota Y, Zhao L, Azhary JM, Yoshino O, et al. A potential role for endoplasmic reticulum stress in progesterone deficiency in obese women. Endocrinology. 2017;158:84–97. [DOI] [PubMed] [Google Scholar]
- 79. Kunitomi C, Harada M, Takahashi N, Azhary JMK, Kusamoto A, Nose E, et al. Activation of endoplasmic reticulum stress mediates oxidative stress‐induced apoptosis of granulosa cells in ovaries affected by endometrioma. Mol Hum Reprod. 2020;26:40–52. [DOI] [PubMed] [Google Scholar]
- 80. Barrett ES, Hoeger KM, Sathyanarayana S, Abbott DH, Redmon JB, Nguyen RHN, et al. Anogenital distance in newborn daughters of women with polycystic ovary syndrome indicates fetal testosterone exposure. J Dev Orig Health Dis. 2018;9:307–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Perlman S, Toledano Y, Kivilevitch Z, Halevy N, Rubin E, Gilboa Y. Foetal sonographic anogenital distance is longer in polycystic ovary syndrome mothers. J Clin Med. 2020;9:2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sánchez‐Ferrer ML, Mendiola J, Hernández‐Peñalver AI, Corbalán‐Biyang S, Carmona‐Barnosi A, Prieto‐Sánchez MT, et al. Presence of polycystic ovary syndrome is associated with longer anogenital distance in adult Mediterranean women. Hum Reprod. 2017;32:2315–23. [DOI] [PubMed] [Google Scholar]
- 83. Wu Y, Zhong G, Chen S, Zheng C, Liao D, Xie M. Polycystic ovary syndrome is associated with anogenital distance, a marker of prenatal androgen exposure. Hum Reprod. 2017;32:937–43. [DOI] [PubMed] [Google Scholar]
- 84. Sir‐Petermann T, Maliqueo M, Angel B, Lara HE, Pérez‐Bravo F, Recabarren SE. Maternal serum androgens in pregnant women with polycystic ovarian syndrome: possible implications in prenatal androgenization. Hum Reprod. 2002;17:2573–9. [DOI] [PubMed] [Google Scholar]
- 85. Abbott DH, Dumesic DA, Levine JE. Hyperandrogenic origins of polycystic ovary syndrome ‐ implications for pathophysiology and therapy. Expert Rev Endocrinol Metab. 2019;14:131–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Stener‐Victorin E, Padmanabhan V, Walters KA, Campbell RE, Benrick A, Giacobini P, et al. Animal models to understand the etiology and pathophysiology of polycystic ovary syndrome. Endocr Rev. 2020;41:bnaa010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Dumesic DA, Hoyos LR, Chazenbalk GD, Naik R, Padmanabhan V, Abbott DH. Mechanisms of intergenerational transmission of polycystic ovary syndrome. Reproduction. 2020;159:R1–R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Guo X, Puttabyatappa M, Domino SE, Padmanabhan V. Developmental programming: prenatal testosterone‐induced changes in epigenetic modulators and gene expression in metabolic tissues of female sheep. Mol Cell Endocrinol. 2020;514:110913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Salinas I, Sinha N, Sen A. Androgen‐induced epigenetic modulations in the ovary. J Endocrinol. 2021;249:R53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Mimouni NEH, Paiva I, Barbotin AL, Timzoura FE, Plassard D, Le Gras S, et al. Polycystic ovary syndrome is transmitted via a transgenerational epigenetic process. Cell Metab. 2021;33:513–530.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Detti L, Christiansen ME, Francillon L, Ikuwezunma G, Diamond MP, Mari G, et al. Serum anti‐Müllerian hormone (AMH) in mothers with polycystic ovary syndrome (PCOS) and their term fetuses. System Biol Reprod Med. 2019;65:147–54. [DOI] [PubMed] [Google Scholar]
- 92. Webber LJ, Stubbs S, Stark J, Trew GH, Margara R, Hardy K, et al. Formation and early development of follicles in the polycystic ovary. Lancet. 2003;362:1017–21. [DOI] [PubMed] [Google Scholar]
- 93. Lindheim L, Manti M, Fornes R, Bashir M, Czarnewski P, Diaz OE, et al. Reproductive and behavior dysfunction induced by maternal androgen exposure and obesity is likely not gut microbiome‐mediated. J Endocr Soc. 2018;2:1363–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Sherman SB, Sarsour N, Salehi M, Schroering A, Mell B, Joe B, et al. Prenatal androgen exposure causes hypertension and gut microbiota dysbiosis. Gut Microbes. 2018;9:400–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Tremaroli V, Bäckhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489:242–9. [DOI] [PubMed] [Google Scholar]
- 96. Sonnenburg JL, Bäckhed F. Diet‐microbiota interactions as moderators of human metabolism. Nature. 2016;535:56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez‐Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Flak MB, Neves JF, Blumberg RS. Immunology. Welcome to the microgenderome. Science. 2013;339:1044–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Rizk MG, Thackray VG. Intersection of polycystic ovary syndrome and the gut microbiome. J Endocr Soc. 2021;5:bvaa177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Guo Y, Qi Y, Yang X, Zhao L, Wen S, Liu Y, et al. Association between polycystic ovary syndrome and gut microbiota. PloS One. 2016;11:e0153196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Torres PJ, Ho BS, Arroyo P, Sau L, Chen A, Kelley ST, et al. Exposure to a healthy gut microbiome protects against reproductive and metabolic dysregulation in a PCOS mouse model. Endocrinology. 2019;160:1193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kusamoto A, Harada M, Azhary JMK, Kunitomi C, Nose E, Koike H, et al. Temporal relationship between alterations in the gut microbiome and the development of polycystic ovary syndrome‐like phenotypes in prenatally androgenized female mice. FASEB J. 2021;35:e21971. [DOI] [PubMed] [Google Scholar]
- 103. Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–53. [DOI] [PMC free article] [PubMed] [Google Scholar]