

Abstract
The stabilization of natural proteins is a long‐standing desired goal in protein engineering. Optimizing the hydrophobicity of the protein core often results in extensive stability enhancements. However, the presence of totally or partially buried catalytic charged residues, essential for protein function, has limited the applicability of this strategy. Here, focusing on the thioredoxin, we aimed to augment protein stability by removing buried charged residues in the active site without loss of catalytic activity. To this end, we performed a charged‐to‐hydrophobic substitution of a buried and functional group, resulting in a significant stability increase yet abolishing catalytic activity. Then, to simulate the catalytic role of the buried ionizable group, we designed a combinatorial library of variants targeting a set of seven surface residues adjacent to the active site. Notably, more than 50% of the library variants restored, to some extent, the catalytic activity. The combination of experimental study of 2% of the library with the prediction of the whole mutational space by partial least squares regression revealed that a single point mutation at the protein surface is sufficient to fully restore the catalytic activity without thermostability cost. As a result, we engineered one of the highest thermal stabilities reported for a protein with a natural occurring fold (137°C). Further, our hyperstable variant preserves the catalytic activity both in vitro and in vivo.
Keywords: genotype–phenotype mapping, partial least squares reconstruction, protein design, protein evolution, protein stability, thioredoxin
1. INTRODUCTION
Most proteins exhibit nearly balanced free energy profiles for folded and unfolded states 1 which hampers their biotechnological applications. Furthermore, as most mutations are destabilizing, 2 marginal stability becomes a significant bottleneck for the laboratory evolution 3 , 4 and computational design of proteins. 5 , 6 In consequence, many efforts have been directed to increase protein stability. 1 , 7 , 8 , 9 , 10 Notably, de novo design has been revealed as one of the most successful strategies to achieve protein hyperstability, partly attributed to the well‐packed and exclusively hydrophobic cores of its designs. 11 Indeed, optimizing natural proteins' core to maximize its buried hydrophobic surface area often results in extensive stability enhancements. 12 , 13 , 14 , 15 , 16 , 17 However, it is frequently impossible to fully implement such a strategy in natural proteins, as buried ionizable residues are often part of active sites and, thus, linked to function. 18 , 19 , 20 Therefore, when applying this stability strategy to natural proteins, functional buried ionizable residues typically remain untouched.
This trade‐off between stability and function is exemplified by the electron transfer protein thioredoxin. The thioredoxin 21 (Trx) is a small and well‐characterized protein with an active site center partially exposed. 22 It comprises a tryptophan followed by two vicinal cysteines spaced by a glycine‐proline segment (Trp‐Cys32‐Gly‐Pro‐Cys35), enumerated according to the sequence of the E. coli‐Trx, and a conserved and buried aspartic residue (Asp26). 21 , 22 , 23 Mutating the buried Asp26 to a hydrophobic residue dramatically drops the catalytic activity 23 , 24 yet increases the protein stability. 14 , 21
We aim to increase protein thermostability by replacing a buried and functional charged group with a hydrophobic residue without activity cost. It has been previously shown how the rational redesign of the surface charge distribution can modulate the pK a and the ionization state of a buried residue. 25 In this work, we go a step further and aim to simulate a buried ionizable group's functional role, comprised in the active site center of a protein, by redesigning the surface charge distribution adjacent to the active site. Such a strategy may allow maximizing the buried hydrophobic area of proteins while preserving their catalytic capabilities.
Here, we optimized the thioredoxin's protein core by mutating its buried and functional Asp26 to Ile. Then, based on structural criteria, we selected a set of solvent‐exposed positions that might balance the absence of the buried aspartic residue. After that, we constructed a combinatorial library of thioredoxin variants where the previously selected positions were mutated to lysine, glutamate, or kept the wild‐type residue. We tested the thermostability and catalytic activity of 39 variants accounting for 1.8% of the total library of 2,187 variants. We used partial least squares (PLS) reconstruction to predict the whole mutational space of the library. Lastly, we selected those variants with predicted higher redox activity and experimentally validated their thermostability and activity in vitro and in vivo.
Notably, our results show that about half of the library's variants restored the catalytic activity of the thioredoxin to some degree. Strikingly, introducing a sole lysine on the protein surface was enough to completely restore the activity of the conserved and buried aspartic residue in vitro. This redesigned active site variant was functional in vivo and displayed one of the highest thermal stabilities ever reported. 26 These findings support that neutral‐to‐charged mutations of solvent‐exposed residues can mimic the select physicochemical properties of catalytic buried ionizable groups. Furthermore, this potential might be harnessed to stabilize natural proteins by substituting functional yet destabilizing buried charged groups without compromising their catalytic activities.
2. RESULTS
2.1. Surface mutations simulate the catalytic role of a buried charged residue in thioredoxin in vitro
We want to increase protein stability maximizing the protein core's hydrophobicity. An established approach consists in substituting buried charged residues with hydrophobics. 11 , 12 , 13 , 14 , 15 , 16 However, as buried charged groups are often engaged in functional roles, this strategy risks augmenting stability at the expense of function. Therefore, first, we aim to fashion the functional role of a buried charged residue by surface mutations directed to redesign the charge surface distribution nearby. This strategy profits from the plasticity of the protein's surface, which allows rearranging the protein, minimizing the stability effect of the mutations. 27 , 28
To meet this goal, we focused on thioredoxin. Thioredoxins are proteins present in all organisms that catalyze disulfide bond reductions. 29 Their canonical active site center hosts the segment Trp‐Cys32‐Gly‐Pro‐Cys35. Close to the disulfide and part of the active site center, a buried carboxylic residue (Asp26) is utterly conserved among thioredoxins (Figure 1a). The reaction catalyzed by Trx is a bimolecular nucleophilic substitution that involves shuttling two electrons from the Trx to the substrate. Firstly, the hydrophobic active site residues interact with the substrate protein. Then, in the hydrophobic environment of the complex, Cys32 nucleophilically attacks the substrate's disulfide bond, resulting in an intermediate disulfide bond between Trx and the substrate. Finally, the deprotonated Cys35 nucleophilically attacks the mixed disulfide bond, resulting in the oxidation of Trx and the reduction of the substrate 30 (Figure 1b). The buried Asp26 deprotonates and activates the Cys35, 24 , 31 , 32 guiding the reaction's molecular mechanism and removing the Asp26 results in a dramatic decrease in the redox capacity of Trx. 23 To test whether we can mimic the contribution to the redox activity of Asp26, we used as the library starting point a modified version of an ancestral Trx from the last universal common ancestor of the Cyanobacterial, Deinococcus, and Thermus groups, 33 called from now on trx0, that has a melting temperature (T m) of 128°C. 21 Trx0 will likely accept several destabilizing mutations, allowing to explore a more expansive mutational space and, therefore, increasing the chances to accommodate catalytic residues at positions different than those found in all‐natural thioredoxins.
FIGURE 1.
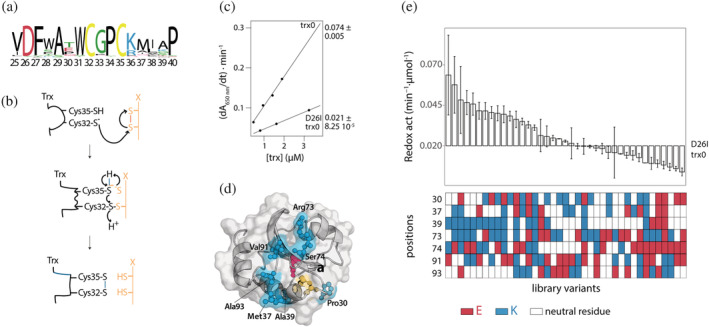
Redesign of the conserved active site of the thioredoxin. (a) Sequence logo showing the conservation of the thioredoxin active site. The logo was inferred from an alignment of 120 thioredoxin sequences which shared at least 30% of identity with E. coli Trx. (b) Molecular mechanism of the redox activity of the thioredoxin. Reduced thioredoxin binds, through its hydrophobic active site area, to the substrate. Then, the thiolate of Cys32 attacks the substrate and forms a transient mixed disulfide. After that, the thiolate of Cys35 attacks this mixed disulfide, generating the oxidized thioredoxin and the reduced substrate protein. 30 (c) The redox activity of the thioredoxins was determined testing their capacity to reduce insulin disulfide bridges. The activity values are determined from the slope of the linear relationship between the maximum rate of the insulin reduction curves versus thioredoxin concentration. The errors shown correspond to the standard deviation associated with the slope. The mutation D26I significantly drops the redox activity of trx0. (d) Crystallographic structure of the ancestral thioredoxin from the last common ancestor of the Cyanobacterial, Deinococcus and Thermus groups (LPBCA, pdb code: 2YJ7). 77 The catalytic, partially buried cysteines are shown in yellow, and the catalytic buried aspartic in red. Those solvent‐exposed residues selected to modulate the functional role of the buried Asp26 are shown in cyan. (e) Experimental redox activity of the 39 randomly selected variants of the library. The activity values are determined from the slope of the linear relationship between the maximum rate of the insulin reduction curves versus thioredoxin concentration. The error is the standard deviation associated with the slope. As a color code, below is shown the type of residues found at the seven positions targeted in the library. White when there is a neutral residue, blue when there is lysine, and red when there is glutamate
First, we measured the effect of the D26I substitution on trx0 activity. The D26I substitution reduced the redox capacity by 72% (Figure 1c), consistent with the previously described functional role of the buried aspartic residue in Escherichia coli Trx. 23 , 24 Then, we measured the thermostability of D26I‐trx0. The D26I substitution increases by 10°C the protein stability, scaling the T m up to 138°C, in agreement with the previously reported stability effect of the D26I mutation in E. coli Trx. 14 Thus, D26I‐trx0 is a hyperstable protein with a residual capacity to catalyze the reduction of disulfide bonds.
We selected a set of residues that, upon mutation, might simulate the buried aspartic's functional role based on structural criteria: (a) We selected those residues close to the active site center. For this purpose, we picked all residues in a radius of 11 Å centered in the sulfur atom of the catalytic Cys32. (b) Among those residues, we discarded the buried ones, that is, we only considered residues with an accessible surface area (ASA) above 0.3. (c) Lastly, we excluded exposed residues directly involved in the redox activity, such as Trp31, Asp61, Pro76, and Lys57, 34 and those located adjacent to the sulfur atom of the Cys32—closer than 7 Å. With these criteria, seven positions were selected as promising candidates to change the surface charge distribution and simulate the catalytic role of the buried Asp26: Pro30, Met37, Ala39, Arg73, Ser74, Val91, and Ala93 (Figure 1d and Table 1). We then designed a combinatorial library of variants where each position could be occupied by a neutral, a positive, or a negative charged residue. Positions 30, 37, 39, 74, 91, and 93 could be hosted by their wild‐type neutral residue or mutated either to lysine or glutamic acid. However, the wild‐type residue found in position 73 is already positively charged; therefore, in the library, this position could be occupied by its wild‐type residue or mutated either to glutamic acid or to methionine (since methionine is often found in this position in an alignment of thioredoxins).
TABLE 1.
Selected solvent‐exposed positions to simulate the functional properties of the buried Asp26
| Residue | Position |
|
ASA | |
|---|---|---|---|---|
| Proline | 30 | 7.0 | 0.58 | |
| Methionine | 37 | 9.4 | 0.73 | |
| Alanine | 39 | 9.5 | 0.33 | |
| Arginine | 73 | 10.1 | 0.93 | |
| Serine | 74 | 7.5 | 0.58 | |
| Valine | 91 | 10.2 | 0.56 | |
| Alanine | 93 | 8.4 | 0.33 |
Note: In the first column is annotated the amino acids found in the position shown in the second column. In the third column it is written the distance from the sulfur atom of the catalytic Cys32 to the closest atom of the amino acid at each position, and in the fourth column, the accessible surface area.
We constructed the combinatorial library and transformed it into E. coli cells. We randomly selected 104 colonies, from which 43 expressed soluble thioredoxin variants. Sequencing of the soluble variants revealed 39 unique mutants. We purified these mutants and studied their redox activity in vitro using insulin as a substrate. 35 Remarkably, more than half of them showed enhanced redox activity compared to D26I‐trx0. Indeed, some mutants displayed up to three‐fold redox activity than D26I‐trx0, almost fully compensating for the lack of the buried Asp26 (Figure 1e, Figure S1). The results showed that redesigning the solvent‐exposed charge distribution adjacent to the active site might allow the simulation of the catalytic properties of buried charged residues.
2.2. Partial least squares allows the phenotypic prediction of the entire mutational space
Complete experimental characterization of the whole library is infeasible. We still did not determine most variants' activity (more than 98% of the library). Hence, we applied a computational method that allows the whole library's phenotypic prediction from the 1.8% experimentally studied. We fit the experimental data to the equation:
where A is the redox activity; is a vector of values (1, 0, 0) when the position is occupied by a neutral residue, (0, 1, 0) when a positively charged residue occupies it, and (0, 0, 1) when a negatively charged residue occupies it; is the effect of the residue in the position on the redox activity; takes different values depending on the type of residue found in position and (Table S1) and describes the coupling effect between residues at positions and in the redox activity. Hence, the first term of the equation describes the effect of the individual mutations on the activity and the second, the coupling effect of the mutations on the activity, considering a possible nonadditive effect. The fitting requires 231 output parameters (21 parameters and 210 parameters). At the same time, the number of experimental values to be fitted is only 39 (values of the redox activity of the screened library variants). We addressed the fitting using partial least squares, an established algorithm to describe the relationship between protein sequence and function. 36 , 37 , 38 This approach has been used to predict the phenotypic outcome of the presence or absence of mutations. 36 , 37 Here, we further increase the mutational space to explore three different alternatives per position (a neutral, a positive, and a negatively charged amino acid) and consider the synergistic effect among mutations. PLS can predict a significant number of fitting parameters using very few experimental data because it first reduces the number of independent variables ( and ) to fewer latent variables. 39 These latent variables are orthogonal combinations of the original independent variables that define most of their variance and explain the correlation between the independent and dependent variables (A, the redox activity). Thus, PLS uses the two data sets' information, the dependent and independent variables, to define latent variables that maximize prediction capacity.
We bootstrapped the experimental data set to assess the prediction's uncertainty, that is, we repeated the PLS fitting using 10 different data sets by resampling the 39 initial experimental values. Full‐library reconstructions of such 10 replicas are shown in Figure 2a and their mean values are shown in Figure 2b. The reconstruction displayed an unexpected scenario, as the redox activity might reach wild‐type levels with very few mutations. We validated the reconstruction by selecting the best‐predicted variants with one or two mutations (A39K, A39E, P30K/A39K/S74K, A39K/K73M, and A39K/P30K) and experimentally examined their redox activity in vitro (Figure 2c). Remarkably, a single point mutation alanine‐to‐lysine in a solvent‐exposed position (A39K) almost fully compensates for the functional properties of the buried Asp26.
FIGURE 2.
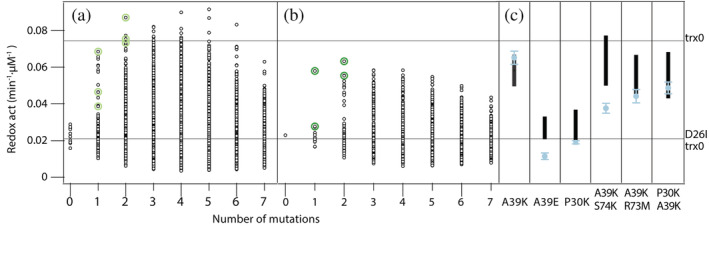
Partial least squares reconstruction of the redox activity of all the library variants. (a) Full‐library reconstruction of the 10 data sets, obtained by resampling the 39 initial experimental values, are shown in black. In light green are highlighted the highest predictions for those variants with one or two mutations. (b) In black, mean values of the predicted redox activity for each library variant. In green, mean values of the predicted redox activity for the best variants with one or two mutations. (c) For each selected variant, in black is shown the range of predicted values and in blue the experimental value for the redox activity
We attempted to rationalize this result on a molecular basis. For the thioredoxin to be active, the pK a of the catalytic Cys32 must be sufficiently low to provide a convenient population of negatively charged thiolate at neutral pH. 40 In fact, removing the Asp26 increases the pK a of Cys32 (from 7.1–7.4 to 7.8–9.0), 40 reducing its reactivity. We used PROPKA 3 41 to assess, based on the 3D structure, how the identity of the residue in position 39 influences the Cys32's pK a. To this end, we simulated the structures and performed energy minimization with Chimera 2. 42 As expected, a decrease in the Cys32's pK a was observed when a lysine is introduced in position 39. On the contrary, an increase in the Cys32's pK a was predicted when a glutamate is placed in position 39 (Table S2). This data must be interpreted cautiously because the predicted shift on the pK a is relatively small and differs from the experimental value. However, it qualitatively proposes an explanation for the catalytic role of the Lys39. We further use PROPKA3 to assess the Cys32's pK a of some inactive variants (S74E/D26I‐trx0 and P30E/D26I‐trx0 since they are overrepresented among the inactive variants experimentally tested, and P30E/R73E/S4E/V91E/D26I‐trx0 and P30E/R73M/S74E/V91E/D26I‐trx0 since they are the two variants less active). As expected, for all these inactive variants, PROPKA 3 predicted an increase in the Cys32's pK a. These predictions may be limited since: (a) pK a values are challenging to reproduce based on structure‐based calculations 43 ; (b) We lack the experimental 3D structures of the studied mutants and the structure of the catalytic cysteines in the thiolate form. Accordingly, we simulated the structures and performed energy minimization with Chimera 2. 42
A deeper analysis of the pK a prediction allows us to further unravel how the Lys39 may shape the Cys32's properties. According to PROPKA 3's prediction, a coulombic interaction between Lys39 and Cys32 is one of the main contributors to the decrease in the pK a of the latter (Table S2), stabilizing the thiolate form. Further, when placing a glutamate in position 39, PROPKA 3 predicted a change in the hydrogen bond pattern between the catalytic Cys32 and Cys35 that causes an increase in the Cys32's pK a (Table S2) that destabilizes the thiolate form. Interestingly, a lysine at position 39 is highly conserved in thioredoxins from mammalian organisms, suggesting a potential key role of this residue in modulating the pK a of Cys32 in mammals. 34
The fact that a single point mutation alanine‐to‐lysine in a solvent‐exposed position (A39K) almost fully compensates for the functional properties of the buried Asp26 suggests a remarkable plasticity of the thioredoxin's active site center. Further, the A39K mutation involves the accommodation of an ionizable residue in the protein surface, and thus, it likely implies a minor stability effect.
2.3. Simultaneous enhancement of redox activity and stability in thioredoxin
We showed that surface mutations could reproduce the role of a buried charge on the redox activity of thioredoxin in vitro. However, the mutations' stability effect and the interplay between stability and activity remain to be tested.
To understand the energetic landscape confined by the library variants, we studied their thermostability by differential scanning calorimetry (DSC) (Figure 3a and S2). Few variants displayed thermal stabilities equivalent to trx0. Remarkably, the A39K substitution, with similar catalytic activity to trx0, presented a Tm of 137.1°C (Figure 3b, c, and d). Therefore, we wondered whether the redesigned active site center allows the multi‐feature optimization of the enzyme, that is, whether simultaneously improving stability and activity is feasible.
FIGURE 3.
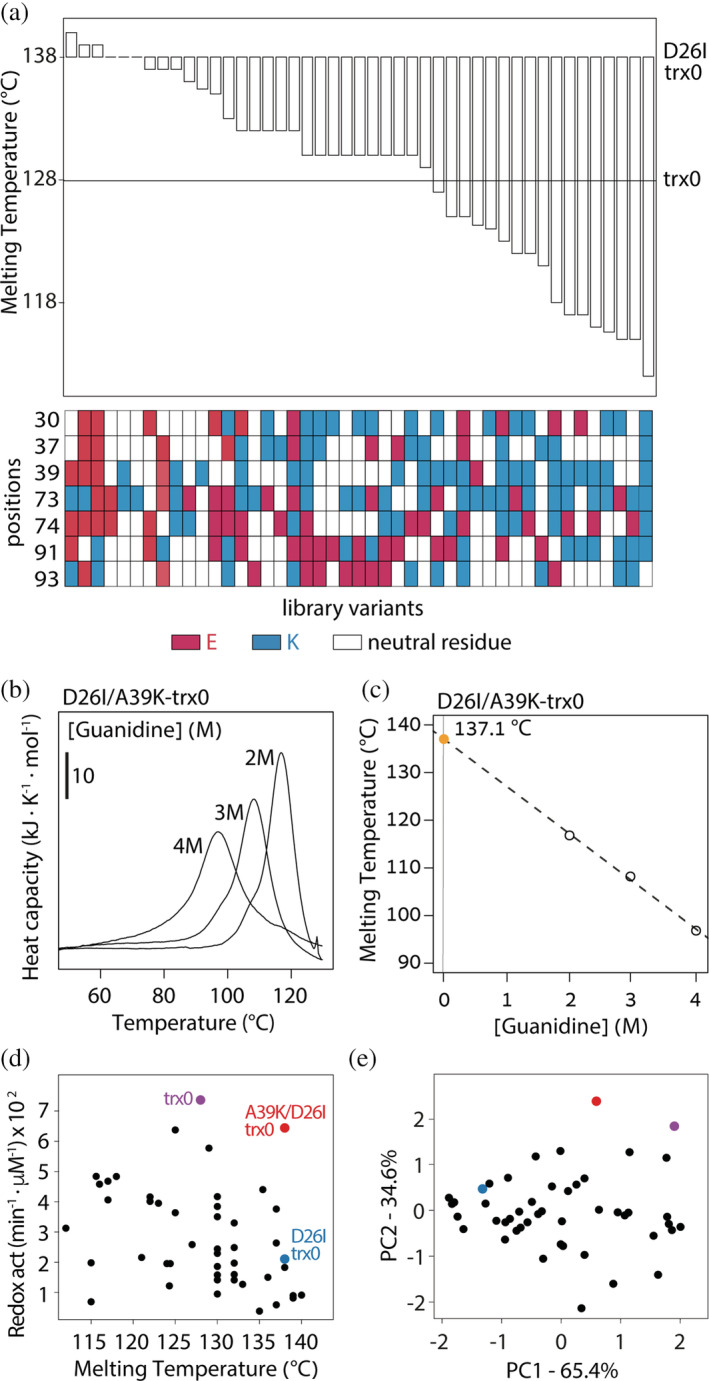
Redesigning the conserved active site center allows the multi‐feature optimization of the enzyme. (a) Most of the variants decrease the thermostability of the library's background—D26I‐trx0. However, compared with trx0, half of the variants have improved stability. (b) Scanning calorimetry profiles of the variant D26I/A39K‐trx0 measured in HEPES 50 mM pH 7 at three different concentrations of guanidinium hydrochloride (guanidine). Melting temperatures correspond to the maximum heat capacity of the calorimetric traces. (c) Melting temperatures of D26I/A39K‐trx0 at different guanidine concentrations (open circles). Closed orange circle corresponds to the extrapolation to zero concentration of guanidine. (d) Plotting the redox activity versus the thermostability of each variant displays a spread‐out distribution of the experimental dataset. (e) The PCA analysis displays two components with significant size. PC1 explained the 65% of the variance, whereas PC2 explained the 35%
To analyze whether stability and activity trade‐off, we studied the stability‐activity correlation among the experimentally studied variants. Plotting both properties shows, at a glance, a spread‐out distribution of the dataset (Figure 3d). To quantify the degree of interplay between properties, we carried out a Principal Component Analysis (PCA). 44 PCA is a mathematical algorithm that reduces the dataset's dimensionality, preserving as much statistical information as possible. It identifies directions, called Principal Components, along which the variation in the data is maximal. If two properties correlate, the dataset can be reduced to only one component. Hence, PCA displays a major component and a very minor one. On the contrary, if two properties are independent, two components are needed to explain the data variation. Then, the PCA analysis displays two components with significant size. Two components contributed significantly to the variance of the stability‐activity dataset. PC1 explained 65% of the variance, whereas PC2 explained 35% (Figure 3e). Thus, the PCA analysis revealed a weak correlation and an efficient independent modulation of both properties. Overall, the modulation range achieved for the catalysis and stability was broad and, since both parameters weakly correlate, a simultaneous improvement of activity and stability seems possible. Indeed, the mutant A39K, whose redox activity is comparable to trx0 and higher than that of E. coli and human thioredoxins, 33 has a T m of 137°C.
Taken together, the results show that the small impact of surface mutations on protein stability permits improving the catalytic activity without compromising the stability.
2.4. Synthetic thioredoxin functions in vivo
The mutation A39K in thioredoxin reproduces the catalytic role of the buried Asp26 in vitro and stabilizes the protein scaffold. We wondered whether our redesigned variant is active in a cellular context. In the cell, thioredoxin must catalyze the reduction of disulfide bridges of numerous protein substrates, and multiple factors such as crowding and viscosity are at play. To assess whether the redesigned thioredoxin (D26I/A39K‐trx0) functions in vivo, we genome‐integrated selected thioredoxin variants (trx0, D26I‐trx0, and D26I/A39K‐trx0) in a Saccharomyces cerevisiae strain lacking the TRX2 gene. 45 TRX2 encodes the protein thioredoxin II, which maintains thiol peroxidases in the reduced state. Consequently, the TRX2 deletion mutant (trx2Δ) is hypersensitive to oxidative stress. 46 Thus, if a thioredoxin variant is active in vivo, it should, to some extent, complement the function of trx2 under oxidative stress. As a positive control, we also genome‐integrated the yeast TRX2. As thioredoxin is a moonlighting protein 47 with multiple interaction partners, 48 , 49 , 50 a possible increase in fitness could be partially attributed to specific interactions or functionalities independent from the active site. To minimize this effect, we conduct the experiments in yeast, an organism whose thioredoxin is evolutively distant from trx0, which is derived from an ancestral thioredoxin corresponding to an internal bacterial node. 21 Indeed, while trx0 and E. coli's thioredoxin share an identity of 58.3%, the identity shared by trx0 and yeast's trx2 is only 38.3%.
We studied the growth of these mutants in normal growing conditions and under oxidative stress. The lag time of the trx2Δ strain increased 3.3‐fold and 6.3‐fold in the presence of 3 mM H2O2 and 4 mM H2O2 respectively (Figures 4a, S3, and Table S3). We observed a slightly reduced lag‐phase under H2O2 exposure for the D26I‐trx0 variant (2.9‐fold increase at 3 mM H2O2 and 5.8‐fold at 4 mM H2O2), and a more reduced for trx0 (2.7‐fold and 5.2‐fold increase at 3 and 4 mM H2O2) and even more for the redesigned thioredoxin D26I/A39K‐trx0 (2.6‐fold and 5.0‐fold at 3 and 4 mM H2O2; Figure 4a and Table S3). Notably, the expression of D26I/A39K‐trx0 decreased the lag‐phase in 3.2 hr at 3 mM H2O2 and in 9.1 hr at 4 mM H2O2 with respect to D26I‐trx0. While D26I/A39K‐trx0 was not able to suppress the peroxide sensitivity to the same degree than trx2 (Figure 4b and Table S3; 1.8‐fold and 2.5‐fold increase in the lag‐phase at 3 and 4 mM H2O compared with the lag‐phase at 0 mM H2O), it displayed similar positive complementation as trx0 and significantly improved compared with D26I‐trx0. The trx0 is an ancestral thioredoxin variant that was supposed to exist in the Precambrian era. 21 , 33 The coevolution of the thioredoxin with the organism's protein network is fundamental to optimizing its performance. This is reflected in the superior capacity of endogenous thioredoxin (trx2) compared with D26I/A39K‐trx0. However, D26I/A39K‐trx0 significantly raises the redox activity in vivo with respect to trx2Δ and D26I‐trx0.
FIGURE 4.
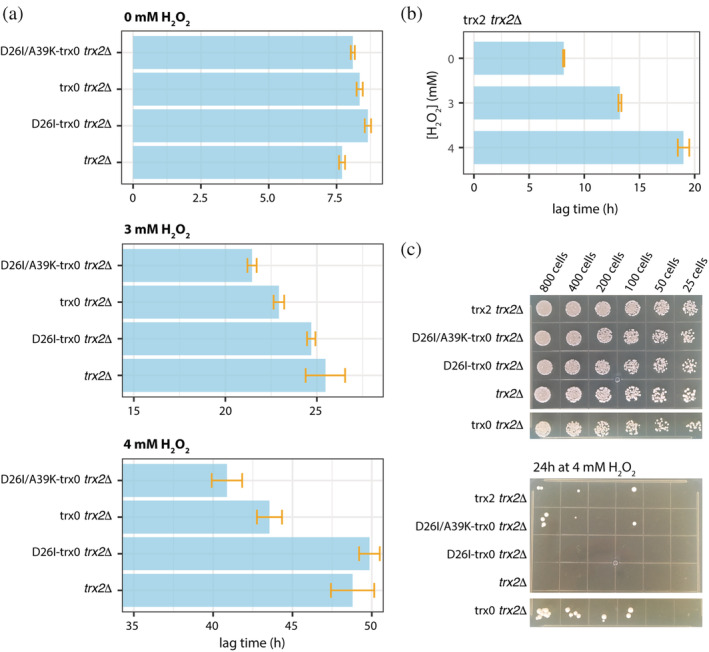
The redesigned thioredoxin, D26I/A39K‐trx0, can catalyze the reduction of disulfide bonds in vivo. (a) In vivo complementation assays in yeast displayed the following scenario. The growth of trx2Δ presents a long lag‐phase when stressing with H2O2. This lag‐phase is maintained when overexpressing D26I‐trx0. However, the lag‐phase at the highest H2O2 concentration is shortened in 5 hr and 8 hr with the overexpression of trx0 and A39K/D26I‐trx0, respectively. (b) Yeast strain trx2Δ complemented with yeast trx2 was grown at different H2O2 concentrations. The lag‐phase extends as the H2O2 concentration grows higher, but significantly less than the rest of the complemented strains. (c) Plate‐based spot assay to visualize yeast cells' survival after treatment with 4 H2O2 for 24 hr at 30°C. trx2Δ and D26I‐trx0 trx2Δ could not survive the peroxide stress. Overexpression of trx0, D26I/A39K‐trx0, and trx2 allows surviving the peroxide treatment. In the upper panel is shown the number of colonies before the treatment with 4 mM H2O2 and below after 24 hr at 30°C with YPD supplemented with 4 mM H2O2
The growth of the trx2Δ yeast constructs presented a lag‐phase phenotype in the presence of H2O2 while no phenotype in the exponential or stationary phase (Figure S3). However, stress response assays can be highly variable depending on cell growth conditions, 51 as observed by the elongated error bars, especially at the exponential and stationary phases (Figure S3). Thus, we performed a second assay for oxidative stress induced by hydrogen peroxide treatment, optimized to obtain highly reproducible results. 52 This plate‐based spot assay allows the visualization of yeast cells' survival after treatment with 4 mM H2O2 for 24 hr at 30°C. The results showed that the knocked‐out trx2Δ could not survive the peroxide exposure. Although the endogenous thioredoxin's expression allows surviving the peroxide treatment, the viability, measured as the percentage of live cells, was compromised. Similarly, expression of trx0 and D26I/A39K‐trx0 complemented the lack of endogenous thioredoxin while D26I‐trx0 was not able to complement (Figure 4c). Taken together, these experiments suggested that the engineered active site center of the thioredoxin could catalyze the reduction of disulfide bonds in vivo.
3. DISCUSSION
Our results illustrate that optimizing the protein's core hydrophobicity combined with surface charge distribution's rearrangement results in an extensive stability enhancement while maintaining the functional properties of the thioredoxin. We showed that, for a previously designed hyperstable thioredoxin, 21 mutating a buried ionizable group results in a dramatic decrease in the redox capacity and, at the same time, increases protein stability by 10 degrees. Combining the experimental screening of a library of mutations at selected positions with partial least squares reconstruction of the whole mutational space, we found out that a sole alanine‐to‐lysine mutation in a solvent‐exposed position is enough to restore the functional properties of the buried charge. Further, we found a very weak stability and redox activity trade‐off that allows thioredoxin's multi‐feature optimization. Thus, we engineered thioredoxin's active site to efficiently catalyze the reduction of disulfide bonds in vitro and stabilize the protein scaffold 10 degrees. Lastly, we showed that the new thioredoxin could also function in vivo.
Protein engineering studies often seek improved stabilities. Several evolutionary‐based strategies, such as back‐to‐consensus mutations 53 , 54 , 55 and ancestral protein reconstruction, 21 , 56 , 57 have been well used to achieve protein stabilization. Also, directed evolution techniques have shown great promise in stabilizing proteins. 10 , 58 Overall, these methods have proven successful in stabilizing natural proteins. However, engineered proteins with the highest stabilities mostly come from de novo design. 16 , 59 , 60 , 61 , 62 Some of them display exceptionally high thermal stabilities; for example, Top7 and DRNN have melting temperatures of 133°C 60 , 61 and 142°C, 16 respectively. One of the reasons for such impressive stabilities could be explained by the exclusively hydrophobic core of the designs. 11 However, natural proteins often display functional ionizable residues in their hydrophobic cores 25 , 63 achieving functionality at the expense of thermodynamic stability. 18 , 64 , 65 Redesigning active sites with more stable configuration without compromising function is, thus, a challenge. Here, we have shown for the thioredoxin that redesigning its surface charge distribution nearby the active site can simulate the selected catalytic properties of a conserved and buried ionizable group. This approach allowed us to design one of the enzymes with the highest thermal stabilities ever reported. In fact, out of more than 32,000 entries in the 2021 version of the ProTherm database (https://web.iitm.ac.in/bioinfo2/prothermdb), 26 only six entries corresponding to four natural proteins have similar or higher reported thermal stability (Figure S4). Overall, this approach optimizes the thioredoxin's hydrophobic core while preserving its enzymatic activity and, therefore, opens a new strategy to stabilize enzymes.
We also showed that the redesigned thioredoxin can function in vivo yet does not fully compensate for the lack of the endogenous thioredoxin. This could be due to the multifunctional nature of the thioredoxin. 47 It reduces oxidized cysteine residues of several biological substrates, such as hydrogen peroxide, transcription factors, 66 unfolded proteins, 67 and kinases, 68 among others. Thus, thioredoxin is essential to keep intracellular protein disulfides reduced, prevent oxidative stress, regulate transcription factors' activity, and assist other proteins' folding. The active site center of the thioredoxin is plausibly the outcome of satisfying many of these functions. Here, we demonstrated that an alanine‐to‐lysine mutation in a solvent‐exposed position is enough to restore the properties of the buried Asp26 to catalyze the reduction of disulfide bridges. However, we did not consider the role of the Asp26 in other functionalities and specific interactions with other proteins. Nevertheless, in vivo assays showed a slightly better complementation of D26I/A39K‐trx0 relative to trx0, suggesting the nonessentiality of the Asp26. Another reason to explain the failure to fully complement the endogenous thioredoxin could be that the endogenous thioredoxin has coevolved with the organism's protein network. However, the D26I/A39K‐trx0 has not.
The design of new active sites from scratch is a grand challenge. For this purpose, a general approach implies burying, totally or partially, ionizable groups in the hydrophobic protein core 5 , 69 , 70 , 71 because such buried groups often have perturbed physicochemical properties essential for their catalytic roles. 25 , 63 , 71 However, placing a charge in a hydrophobic pocket occurs at a thermodynamic cost. 18 , 64 , 65 As a result, a significant limitation to design enzymes with tailored functionalities resides in the thermostability of the protein scaffold. 8 Here, we have shown for the thioredoxin that modulating the surface charge distribution nearby its active site can simulate the catalytic properties of a buried charged group. Thus, our results open a new strategy that might help design new enzymes.
In summary, this work suggests that it is possible to introduce functional innovation in a protein scaffold targeting those structural features more plastic. With this approach, we redesigned the thioredoxin active site center against conservation, expanding the evolutionary mutational space's boundaries. We think this strategy can be generalized and exploited to stabilize natural proteins.
4. METHODS
4.1. Sequence alignment and conservation analysis
We have carried out a BLAST (https://www.ebi.ac.uk/Tools/sss/ncbiblast/) search using as query the E. coli thioredoxin sequence (pdb:2trx). The Swissprot database of November 2020 and the default options of the search were used. The retrieved sequences were aligned using the Smith‐Waterman algorithm, and those sharing a minimum of 30% of identity and with entry names corresponding to thioredoxins (a total of 120) were retained. The consensus logo was generated with Weblogo (https://weblogo.berkeley.edu/logo.cgi).
4.2. Gene library construction and protein expression and purification
The combinatorial library was constructed by gene assembly mutagenesis 37 and cloned into the plasmid pQE‐80L with a 6xHis tag at the N‐terminal to facilitate protein purification. The thioredoxin variants were expressed and purified as previously described. 72 Purity of the protein fractions was assessed by SDS‐PAGE.
4.3. In vitro activity assay
In vitro redox activity of the thioredoxin variants was determined by a turbidimetric assay using insulin as a substrate. 35 Briefly, we prepared protein solutions at pH 6.5 in phosphate buffer 0.1 M, 2 mM EDTA, and 0.5 mg/ml of insulin. The addition of DTT until 1 mM final concentration initiated the reaction that was monitored by measuring the absorbance at 650 nm over time. Then, the maximum value of the derivative of the kinetics was calculated. For each variant, 3 to 4 concentrations were assayed. The redox activity was quantified from the linear fit of the maximum derivative versus protein concentration (Figure 1c and S1).
4.4. Partial least squares analysis
PLS analyses were carried out with the program Unscrambler X from CAMO software using the NIPALS algorithm (Figure 2). The dependent variable was the logarithm value of the redox activity and was auto‐scaled before the analysis. We used leave‐one‐out cross‐validation. The number of latent variables retained was the Unscrambler program's optimum value based on the mean square error of cross‐validation. The PLS analyses were carried with 10 replica sets constructed from the original set through random resampling (bootstrapping). The number of latent variables retained depends on the replica set used. Typical values were much smaller than the numbers of dependent and independent variables involved. Illustrative plots of experimental versus predicted activities are given in Figure 2c.
4.5. Thermostability experiments
Differential scanning calorimetry experiments were performed using an automatic VP‐DSC microcalorimeter (VP‐Capillary DSC; MicroCal, Malvern Panalytical) with a 0.133 ml cell volume. Solutions of 0.5 mg/ml protein concentration in 50 mM HEPES at pH 7 were warmed up from 25°C until 130°C or 140°C at 3.5 K/min scan rate. Heat up to 140°C requires manipulating the calorimeter settings under the supervision of MicroCal. For technical reasons, some experiments were performed warming up the samples to 130°C. To prevent boiling above 100°C, overpressure was implemented. All thermograms were dynamically corrected using the software provided by MicroCal. The melting temperature (T m) was estimated at the maximum heat capacity of the calorimetric curve. The calorimetry curves of D26I/A39K‐trx0 do not show the peak of the scan in the studied range of temperatures. Its T m was, thus, obtained heating up the protein until 130°C at a concentration of 1.6 mg/ml in 50 mM HEPES pH 7 at 2, 3, and 4 M Guanidine and extrapolating to 0 M Guanidine. The latter experiments were performed in a VP‐DSC microcalorimeter (MicroCal, General Electric) at a scan rate of 1.5 K/min.
4.6. Principal component analysis
Principal Component Analysis was performed using R 73 prior to the analysis both variables (Redox activity and Thermal stability) were auto‐scaled.
4.7. Genome integration of thioredoxins in yeast
Genes encoding the ancestral thioredoxin variants trx0, D26I‐trx0, D26I/A39K‐trx0, and S. cerevisiae trx2 were subcloned downstream of the yeast GPD promoter into an M3925 plasmid (trp1::KanMX3) with a cassette for genome integration. 74 For genome integration, the cassette was amplified by PCR and transformed into BY4741 cells. Transformants were selected by G418 resistance, and correct integration was assessed by sequencing.
4.8. Growth assays of yeast strains
Yeast strains were grown for 2 days in YPD containing 200 μg/ml of G418 at 30°C until reaching the stationary phase. Then, strains were diluted 1:100 in YPD with concentrations of 0, 3, and 4 mM H2O2 in a 96‐well plate for a final volume of 150 μl per well. Three replicates of each strain were done for each condition. Strains were grown under constant shaking for 60 hr at 30°C in a plate reader. Absorbance at 600 nm was continuously monitored throughout the experiment. Analysis of the growth curves (Figure 4 and Table S3) was done with the growth rate R package. 75
4.9. Plate‐based spot viability assay
Yeast cultures of 2 ml of YPD supplemented with G418 200 μg/ml were grown in a 96‐well plate, at 30°C with saturated humidity until saturation. Then, from this preculture, two new cultures were prepared at 0.1 OD, the control and the oxidatively stressed culture. The control culture was prepared in YPD, and from this one, five more cultures were prepared by two times dilution. In total, for each yeast strain, six solutions in YPD with an absorbance at 600 nm of 0.1, 0.05, 0.025, 0.0125, 0.006, and 0.003, respectively. We spotted 5 μl of each culture in a YPD‐agar plate, let it dry, and left it at 30°C for 2 days. Since absorbance 1 at 600 nm correspond around 0.5 × 107 cells/ml, we spotted 2,500, 1,250, 625, 312, 160, and 80 cells in each drop (Figure 4c). For the oxidative stress shock, we prepared six solutions in YPD supplemented with 4 mM H2O2 with an absorbance at 600 nm of 0.1, 0.05, 0.025, 0.0125, 0.006, and 0.003. The cultures were incubated 24 hr at 30°C with saturated humidity. Then, we spotted 5 μL of each culture in a YPD‐agar plate, let it dry, and left it at 30°C for 2 days (Figure 4c).
4.10. pK a prediction of ionizable groups
Structure of the mutants (D26I, A39K/D26I, A39E/D26I, S74E/D26I, P30E/D26I, P30E/R73E/S74E/V91E/D26I, and P30E/R73M/S74E/V91E/D26I) was modeled on the V55F/Q69K/D93Q‐LPBCA (trx0) model structure with the Cys 32 and 35 in the reduced form. The latter was modeled using the LPBCA thioredoxin structure (pdb code: 2YJ7) with Chimera 2. 42 For each mutation we chose the rotamer with the highest probability and a subsequent energy minimization was performed. Prediction of ionizable group's pK a was done using the PROPKA 3.2 software 41 integrated in the APBS‐PDB2PQR web server (https://server.poissonboltzmann.org/).
In PROPKA 3, 76 pK a values are modeled as follows:
where is the pK a of the residue in water and it is a fixed value. is the contribution to the pK a from the residue's environment and it is predicted on the basis of the supplied structure as:
where is the contribution due to desolvation effects, due to hydrogen‐bond interactions, due to unfavorable electrostatic reorganization energies, and due to Coulombic interactions. and are generic parameters and and are calculated.
AUTHOR CONTRIBUTIONS
Maria Luisa Romero: Conceptualization (lead); formal analysis (lead); investigation (lead); methodology (lead); project administration (lead); supervision (lead); visualization (lead); writing – original draft (lead); writing – review and editing (lead). Hector Garcia Seisdedos: Formal analysis (equal); investigation (equal); methodology (equal); visualization (equal); writing – original draft (equal); writing – review and editing (equal). Beatriz Ibarra‐Molero: Formal analysis (supporting); funding acquisition (lead); investigation (supporting); visualization (supporting); writing – original draft (supporting); writing – review and editing (equal).
Supporting information
Appendix S1 Supporting Information
ACKNOWLEDGEMENTS
Maria Luisa Romero Romero thanks the late Dr Dan Tawfik for his passionate discussions with long‐lasting effect on her scientific thinking. We thank Dr Jose Manuel Sanchez Ruiz for invaluable input throughout the realization of this work, Maria Del Carmen Tejero Extremera for helping with protein purification, Dr Agnes Toth‐Petrozcy for useful discussions on the manuscript, Dr Emmanuel D. Levy for sharing the yeast strains and the plasmids with the cassettes for genome integration, and Dr Paola Laurino and Dr Benjamin Clifton for feedback and comments that greatly improved the manuscript. This work was supported by European Regional Development Fund funds and grants (CSD2009‐00088 and BIO2012‐34937), by the Spanish Ministry of Economy and Competitiveness (grant BIO2015‐66426‐R), and by the “Junta de Andalucia” (grant P09‐CVI‐5073). Maria Luisa Romero Romero and Hector Garcia Seisdedos received support from the Koshland Foundation and a McDonald‐Leapman Grant. Open Access funding enabled and organized by Projekt DEAL.
Romero ML, Garcia Seisdedos H, Ibarra‐Molero B. Active site center redesign increases protein stability preserving catalysis in thioredoxin. Protein Science. 2022;31(9):e4417. 10.1002/pro.4417
Review editor: John Kuriyan
Funding information European Regional Development Fund funds and grants, Grant/Award Numbers: BIO2012‐34937, CSD2009‐00088; Junta de Andalucia, Grant/Award Number: P09‐CVI‐5073; Spanish Ministry of Economy and Competitiveness, Grant/Award Number: BIO2015‐66426‐R
REFERENCES
- 1. Magliery TJ. Protein stability: Computation, sequence statistics, and new experimental methods. Curr Opin Struct Biol. 2015;33:161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tokuriki N, Stricher F, Serrano L, Tawfik DS. How protein stability and new functions trade off. PLoS Comput Biol. 2008;4:e1000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Khersonsky O, Kiss G, Röthlisberger D, et al. Bridging the gaps in design methodologies by evolutionary optimization of the stability and proficiency of designed Kemp eliminase KE59. Proc Natl Acad Sci USA. 2012;109:10358–10363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bloom JD, Labthavikul ST, Otey CR, Arnold FH. Protein stability promotes evolvability. Proc Natl Acad Sci USA. 2006;103:5869–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Röthlisberger D, Khersonsky O, Wollacott AM, et al. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453:190–195. [DOI] [PubMed] [Google Scholar]
- 6. Fleishman SJ, Whitehead TA, Ekiert DC, et al. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science. 2011;332:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wijma HJ, Floor RJ, Janssen DB. Structure‐ and sequence‐analysis inspired engineering of proteins for enhanced thermostability. Curr Opin Struct Biol. 2013;23:588–594. 10.1016/j.sbi.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 8. Goldenzweig A, Fleishman SJ. Principles of protein stability and their application in computational design. Annu Rev Biochem. 2018;87:105–129. [DOI] [PubMed] [Google Scholar]
- 9. Goldenzweig A, Goldsmith M, Hill SE, et al. Automated structure‐ and sequence‐based design of proteins for high bacterial expression and stability. Mol Cell. 2018;70:380. 10.1016/j.molcel.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chandler PG, Broendum SS, Riley BT, et al. Strategies for increasing protein stability. Methods Mol Biol. 2020;2073:163–181. [DOI] [PubMed] [Google Scholar]
- 11. Baker D. What has de novo protein design taught us about protein folding and biophysics? Protein Sci. 2019;28:678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Munson M, Balasubramanian S, Fleming KG, et al. What makes a protein a protein? Hydrophobic core designs that specify stability and structural properties. Protein Sci. 1996;5:1584–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Malakauskas SM, Mayo SL. Design, structure and stability of a hyperthermophilic protein variant. Nat Struct Biol. 1998;5:470–475. [DOI] [PubMed] [Google Scholar]
- 14. Bolon DN, Mayo SL. Polar residues in the protein core of Escherichia coli thioredoxin are important for fold specificity. Biochemistry. 2001;40:10047–10053. [DOI] [PubMed] [Google Scholar]
- 15. Borgo B, Havranek JJ. Automated selection of stabilizing mutations in designed and natural proteins. Proc Natl Acad Sci USA. 2012;109:1494–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Murphy GS, Mills JL, Miley MJ, Machius M, Szyperski T, Kuhlman B. Increasing sequence diversity with flexible backbone protein design: The complete redesign of a protein hydrophobic core. Structure. 2012;20:1086–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rocklin GJ, Chidyausiku TM, Goreshnik I, et al. Global analysis of protein folding using massively parallel design, synthesis, and testing. Science. 2017;357:168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Isom DG, Castañeda CA, Cannon BR, Velu PD, García‐Moreno EB. Charges in the hydrophobic interior of proteins. Proc Natl Acad Sci USA. 2010;107:16096–16100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu J, Swails J, Zhang JZH, He X, Roitberg AE. A coupled ionization‐conformational equilibrium is required to understand the properties of ionizable residues in the hydrophobic interior of staphylococcal nuclease. J Am Chem Soc. 2018;140:1639–1648. [DOI] [PubMed] [Google Scholar]
- 20. Narayan A, Naganathan AN. Switching protein conformational substates by protonation and mutation. J Phys Chem B. 2018;122:11039–11047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Romero‐Romero ML, Risso VA, Martinez‐Rodriguez S, Ibarra‐Molero B, Sanchez‐Ruiz JM. Engineering ancestral protein hyperstability. Biochem J. 2016;473:3611–3620. [DOI] [PubMed] [Google Scholar]
- 22. Holmgren A, Söderberg BO, Eklund H, Brändén CI. Three‐dimensional structure of Escherichia coli thioredoxin‐S2 to 2.8 A resolution. Proc Natl Acad Sci USA. 1975;72:2305–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gleason FK. Mutation of conserved residues in Escherichia coli thioredoxin: Effects on stability and function. Protein Sci. 1992;1:609–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chivers PT, Raines RT. General acid/base catalysis in the active site of Escherichia coli thioredoxin. Biochemistry. 1997;36:15810–15816. [DOI] [PubMed] [Google Scholar]
- 25. Pey AL, Rodriguez‐Larrea D, Gavira JA, Garcia‐Moreno B, Sanchez‐Ruiz JM. Modulation of buried ionizable groups in proteins with engineered surface charge. J Am Chem Soc. 2010;132:1218–1219. [DOI] [PubMed] [Google Scholar]
- 26. Nikam R, Kulandaisamy A, Harini K, Sharma D, Gromiha MM. ProThermDB: Thermodynamic database for proteins and mutants revisited after 15 years. Nucleic Acids Res. 2021;49:D420–D424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tóth‐Petróczy Á, Tawfik DS. Slow protein evolutionary rates are dictated by surface–core association. Proc Natl Acad Sci USA. 2011;108:11151–11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tokuriki N, Stricher F, Schymkowitz J, Serrano L, Tawfik DS. The stability effects of protein mutations appear to be universally distributed. J Mol Biol. 2007;369:1318–1332. [DOI] [PubMed] [Google Scholar]
- 29. Holmgren A. Thioredoxin. Annu Rev Biochem. 1985;54:237–271. [DOI] [PubMed] [Google Scholar]
- 30. Holmgren A. Thioredoxin structure and mechanism: Conformational changes on oxidation of the active‐site sulfhydryls to a disulfide. Structure. 1995;3:239–243. [DOI] [PubMed] [Google Scholar]
- 31. LeMaster DM, Springer PA, Unkefer CJ. The role of the buried aspartate of Escherichia coli Thioredoxin in the activation of the mixed disulfide intermediate. J Biol Chem. 1997;272:29998–30001. 10.1074/jbc.272.48.29998. [DOI] [PubMed] [Google Scholar]
- 32. Menchise V, Corbier C, Didierjean C, et al. Crystal structure of the wild‐type and D30A mutant thioredoxin h of Chlamydomonas reinhardtii and implications for the catalytic mechanism. Biochem J. 2001;359:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Perez‐Jimenez R, Inglés‐Prieto A, Zhao Z‐M, et al. Single‐molecule paleoenzymology probes the chemistry of resurrected enzymes. Nat Struct Mol Biol. 2011;18:592–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eklund H, Gleason FK, Holmgren A. Structural and functional relations among thioredoxins of different species. Proteins. 1991;11:13–28. [DOI] [PubMed] [Google Scholar]
- 35. Holmgren A. Thioredoxin catalyzes the reduction of insulin disulfides by dithiothreitol and dihydrolipoamide. J Biol Chem. 1979;254:9627–9632. [PubMed] [Google Scholar]
- 36. Fox RJ, Davis SC, Mundorff EC, et al. Improving catalytic function by ProSAR‐driven enzyme evolution. Nat Biotechnol. 2007;25:338–344. [DOI] [PubMed] [Google Scholar]
- 37. Garcia‐Seisdedos H, Ibarra‐Molero B, Sanchez‐Ruiz JM. Probing the mutational interplay between primary and promiscuous protein functions: A computational‐experimental approach. PLoS Comput Biol. 2012;8:e1002558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Berland M, Offmann B, André I, Remaud‐Siméon M, Charton P. A web‐based tool for rational screening of mutants libraries using ProSAR. Protein Eng Des Sel. 2014;27:375–381. [DOI] [PubMed] [Google Scholar]
- 39. Anon . Partial least squares regression. Available from: http://methods.sagepub.com/reference/the-sage-encyclopedia-of-social-science-research-methods/n690.xml.
- 40. Dyson HJ, Jeng MF, Tennant LL, et al. Effects of buried charged groups on cysteine thiol ionization and reactivity in Escherichia coli thioredoxin: Structural and functional characterization of mutants of Asp 26 and Lys 57. Biochemistry. 1997;36:2622–2636. [DOI] [PubMed] [Google Scholar]
- 41. Olsson MHM, Søndergaard CR, Rostkowski M, Jensen JH. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J Chem Theory Comput. 2011;7:525–537. [DOI] [PubMed] [Google Scholar]
- 42. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 43. Kougentakis CM, Grasso EM, Robinson AC, et al. Anomalous properties of Lys residues buried in the hydrophobic interior of a protein revealed with 15N‐detect NMR spectroscopy. J Phys Chem Lett. 2018;9:383–387. [DOI] [PubMed] [Google Scholar]
- 44. Lever J, Krzywinski M, Altman N. Principal component analysis. Nat Methods. 2017;14:641–642. 10.1038/nmeth.4346. [DOI] [Google Scholar]
- 45. Giaever G, Chu AM, Ni L, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. [DOI] [PubMed] [Google Scholar]
- 46. Kuge S, Jones N. YAP1 dependent activation of TRX2 is essential for the response of Saccharomyces cerevisiae to oxidative stress by hydroperoxides. EMBO J. 1994;13:655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Arnér ES, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem. 2000;267:6102–6109. [DOI] [PubMed] [Google Scholar]
- 48. Kumar JK, Tabor S, Richardson CC. Proteomic analysis of thioredoxin‐targeted proteins in Escherichia coli. Proc Natl Acad Sci USA. 2004;101:3759–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Arts IS, Vertommen D, Baldin F, Laloux G, Collet J‐F. Comprehensively characterizing the thioredoxin interactome in vivo highlights the central role played by this ubiquitous oxidoreductase in redox control. Mol Cell Proteomics. 2016;15:2125–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Oughtred R, Rust J, Chang C, et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021;30:187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tran K, Green EM. Assessing yeast cell survival following hydrogen peroxide exposure. Bio Protoc. 2019;9:e3149. 10.21769/BioProtoc.3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kwolek‐Mirek M, Zadrag‐Tecza R. Comparison of methods used for assessing the viability and vitality of yeast cells. FEMS Yeast Res. 2014;14:1068–1079. [DOI] [PubMed] [Google Scholar]
- 53. Oda K, Lee Y, Wiriyasermkul P, et al. Consensus mutagenesis approach improves the thermal stability of system xc—transporter, xCT, and enables cryo‐EM analyses. Protein Sci. 2020;29:2398–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Georgoulis A, Louka M, Mylonas S, Stavros P, Nounesis G, Vorgias CE. Consensus protein engineering on the thermostable histone‐like bacterial protein HUs significantly improves stability and DNA binding affinity. Extremophiles. 2020;24:293–306. [DOI] [PubMed] [Google Scholar]
- 55. Yao H, Cai H, Li D. Thermostabilization of membrane proteins by consensus mutation: A case study for a fungal Δ8‐7 sterol isomerase. J Mol Biol. 2020;432:5162–5183. [DOI] [PubMed] [Google Scholar]
- 56. Risso VA, Gavira JA, Mejia‐Carmona DF, Gaucher EA, Sanchez‐Ruiz JM. Hyperstability and substrate promiscuity in laboratory resurrections of Precambrian β‐lactamases. J Am Chem Soc. 2013;135:2899–2902. [DOI] [PubMed] [Google Scholar]
- 57. Risso VA, Sanchez‐Ruiz JM, Ozkan SB. Biotechnological and protein‐engineering implications of ancestral protein resurrection. Curr Opin Struct Biol. 2018;51:106–115. [DOI] [PubMed] [Google Scholar]
- 58. Höcker B. Directed evolution of (betaalpha)(8)‐barrel enzymes. Biomol Eng. 2005;22:31–38. [DOI] [PubMed] [Google Scholar]
- 59. Lin Y‐R, Koga N, Tatsumi‐Koga R, et al. Control over overall shape and size in de novo designed proteins. Proc Natl Acad Sci U S A. 2015;112:E5478–E5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Koga R, Yamamoto M, Kosugi T, et al. Robust folding of a de novo designed ideal protein even with most of the core mutated to valine. Proc Natl Acad Sci USA. 2020;117:31149–31156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Design of a novel globular protein fold with atomic‐level accuracy. Science. 2003;302:1364–1368. [DOI] [PubMed] [Google Scholar]
- 62. Marcos E, Chidyausiku TM, McShan AC, et al. De novo design of a non‐local β‐sheet protein with high stability and accuracy. Nat Struct Mol Biol. 2018;25:1028–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Isom DG, Castañeda CA, Cannon BR, García‐Moreno B. Large shifts in pKa values of lysine residues buried inside a protein. Proc Natl Acad Sci USA. 2011;108:5260–5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Garcia‐Seisdedos H, Ibarra‐Molero B, Sanchez‐Ruiz JM. How many ionizable groups can sit on a protein hydrophobic core? Proteins: Struct Funct Bioinf. 2012;80:1–7. [DOI] [PubMed] [Google Scholar]
- 65. Pace CN, Grimsley GR, Scholtz JM. Protein ionizable groups: pK values and their contribution to protein stability and solubility. J Biol Chem. 2009;284:13285–13289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Matthews JR, Wakasugi N, Virelizier J‐L, Yodoi J, Hay RT. Thiordoxin regulates the DNA binding activity of NF‐χB by reduction of a disulphid bond involving cysteine 62. Nucleic Acids Res. 1992;20:3821–3830. 10.1093/nar/20.15.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kern R, Malki A, Holmgren A, Richarme G. Chaperone properties of Escherichia coli thioredoxin and thioredoxin reductase. Biochem J. 2003;371:965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Saitoh M, Nishitoh H, Fujii M, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal‐regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Korendovych IV, Kulp DW, Wu Y, Cheng H, Roder H, DeGrado WF. Design of a switchable eliminase. Proc Natl Acad Sci USA. 2011;108:6823–6827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Burton AJ, Thomson AR, Dawson WM, Brady RL, Woolfson DN. Installing hydrolytic activity into a completely de novo protein framework. Nat Chem. 2016;8:837–844. [DOI] [PubMed] [Google Scholar]
- 71. Risso VA, Martinez‐Rodriguez S, Candel AM, et al. De novo active sites for resurrected Precambrian enzymes. Nat Commun. 2017;8:16113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Romero‐Romero ML, Inglés‐Prieto A, Ibarra‐Molero B, Sanchez‐Ruiz JM. Highly anomalous energetics of protein cold denaturation linked to folding‐unfolding kinetics. PLoS One. 2011;6:e23050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. R Core Team . RA language and environment for statistical computing. Vienna: R Foundation of Statistical Computing, 2019. Available from: http://www.R-project.org/Cited5. [Google Scholar]
- 74. Garcia‐Seisdedos H, Levin T, Shapira G, Freud S. Mutants libraries reveal negative design shielding proteins from mis‐assembly and re‐localization in cells. Proc Natl Acad Sci USA. 2021;119(5):e2101117119. 10.1101/2021.01.20.427404v1.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hall BG, Acar H, Nandipati A, Barlow M. Growth rates made easy. Mol Biol Evol. 2014;31:232–238. [DOI] [PubMed] [Google Scholar]
- 76. Søndergaard CR, Olsson MHM, Rostkowski M, Jensen JH. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J Chem Theory Comput. 2011;7:2284–2295. [DOI] [PubMed] [Google Scholar]
- 77. Ingles‐Prieto A, Ibarra‐Molero B, Delgado‐Delgado A, et al. Conservation of protein structure over four billion years. Structure. 2013;21:1690–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information