

ABSTRACT
The epidemiology of antimicrobial resistance (AMR) is complex, with multiple interfaces (human-animal-environment). In this context, One Health surveillance is essential for understanding the distribution of microorganisms and antimicrobial resistance genes (ARGs). This report describes a multicentric study undertaken to evaluate the bacterial communities and resistomes of food-producing animals (cattle, poultry, and swine) and healthy humans sampled simultaneously from five Brazilian regions. Metagenomic analysis showed that a total of 21,029 unique species were identified in 107 rectal swabs collected from distinct hosts, the highest numbers of which belonged to the domain Bacteria, mainly Ruminiclostridium spp. and Bacteroides spp., and the order Enterobacterales. We detected 405 ARGs for 12 distinct antimicrobial classes. Genes encoding antibiotic-modifying enzymes were the most frequent, followed by genes related to target alteration and efflux systems. Interestingly, carbapenemase-encoding genes such as blaAIM-1, blaCAM-1, blaGIM-2, and blaHMB-1 were identified in distinct hosts. Our results revealed that, in general, the bacterial communities from humans were present in isolated clusters, except for the Northeastern region, where an overlap of the bacterial species from humans and food-producing animals was observed. Additionally, a large resistome was observed among all analyzed hosts, with emphasis on the presence of carbapenemase-encoding genes not previously reported in Latin America.
IMPORTANCE Humans and food production animals have been reported to be important reservoirs of antimicrobial resistance (AMR) genes (ARGs). The frequency of these multidrug-resistant (MDR) bacteria tends to be higher in low- and middle-income countries (LMICs), due mainly to a lack of public health policies. Although studies on AMR in humans or animals have been carried out in Brazil, this is the first multicenter study that simultaneously collected rectal swabs from humans and food-producing animals for metagenomics. Our results indicate high microbial diversity among all analyzed hosts, and several ARGs for different antimicrobial classes were also found. As far as we know, we have detected for the first time ARGs encoding carbapenemases, such as blaAIM-1, blaCAM-1, blaGIM-2, and blaHMB-1, in Latin America. Thus, our results support the importance of metagenomics as a tool to track the colonization of food-producing animals and humans by antimicrobial-resistant bacteria. In addition, a network surveillance system called GUARANI, created for this study, is ready to be expanded and to collect additional data.
KEYWORDS: One Health, drug resistance, surveillance, metagenomics, antimicrobial resistance genes, bacterial communities
INTRODUCTION
The consequences of commensal or unsuspected microorganisms for the health of both animals and humans are often underestimated. It is estimated that 58% of microorganisms known to be pathogenic to humans can be transmitted by animals (1). Furthermore, 73% of pathogenic species reported to be emerging or reemerging have zoonotic origins (1). To achieve a rapid response to mitigate disease, it is essential to investigate entire microbial communities, including both pathogenic and nonpathogenic microorganisms. Microbiota shared among humans and animals must be considered under a One Health approach, as continuous environmental changes and close contact with animals can impact human health (2).
Antimicrobial resistance (AMR) is considered a global public health problem (3, 4). According to the most recent CDC report, more than 2.8 million antibiotic-resistant infections occur in the United States each year, resulting in more than 35,000 deaths (5). A similar number of deaths (33,110) attributed to antimicrobial-resistant infections has been estimated in European countries using EARS-Net data collected in 2015 (6). Although a large proportion of AMR infections are health care-associated infections (7), several studies have documented the emergence and spread of antimicrobial-resistant pathogens in community settings (8–10), especially those pathogens causing foodborne and urinary tract infections (7). Overdevest et al. (11) showed that clones of Escherichia coli isolated from humans and poultry meat in the Netherlands shared the same extended-spectrum-β-lactamase (ESBL)-encoding gene. Additionally, Leverstein-van Hall et al. (12) demonstrated the presence of an IncI1 plasmid that carried blaCTX-M-1 or blaTEM-52 among E. coli isolates recovered from poultry and bloodstream and urinary tract infections.
The selective pressure exerted mainly by the massive use of antimicrobials has had an unprecedented impact on the spread of antimicrobial-resistant pathogens (13). According to the World Organization for Animal Health (OIE), 41% of 146 countries that use antimicrobials in livestock for prophylaxis or treatment allow their use as growth promoters (14). Making the situation even worse, most of these antimicrobials show broad-spectrum activity and are also prescribed for humans (14). According to a previous study by Van Boeckel et al. (15), Brazil ranks third in the consumption of antimicrobials in food animal production. Although those authors reported that China and India represent the largest sources of antimicrobial resistance genes (ARGs) in animals and food products from developing countries, the scarcity of data from South America makes it difficult to estimate the actual occurrence of ARGs in this geographic region (16). Humans and animals share the same environment, are epidemiologically related, and are directly involved in ARG acquisition and dissemination (4, 17). In this context, the need to detect ARGs in distinct ecological niches justifies the implementation of surveillance based on the One Health approach (18, 19). In addition, ARGs can be easily transferred from the environment to human-pathogenic bacteria and vice versa due to horizontal gene transfer (13). Strategies for controlling AMR dissemination have been widely discussed due to the direct and indirect impacts on global public health and the global economy (20). Metagenomic tools have been widely used in surveillance projects conducted in high-income countries to verify the compositions of different microbiomes as well as the occurrence of ARGs, providing more accurate results than conventional culture methods (21–24).
Brazil is the fifth largest country by area (3.2 million mi2) and represents 47% of the South American continent. It is also the sixth most populous country in the world (212,6 million inhabitants), the largest exporter of beef (2,359 million tons) and poultry (3,875 million tons), and the fourth largest exporter of pork (1,178 million tons) according to a 2020 report (25), with Asian countries being the main importers of these animal products (26). As part of a One Health-based surveillance study, we characterized the fecal metagenomes of food-producing animals (poultry, cattle, and swine) and humans from specimens collected in the same time frame from all five Brazilian geographic regions as well as the resistomes encountered as part of the GUARANI (One Health Brazilian Group) network. To the best of our knowledge, this is the first study of this kind conducted in South America.
RESULTS
Diversity and host microbial characterization.
The microbial compositions in 107 samples collected from poultry (n = 30), cattle (n = 30), swine (n = 15), and humans (n = 32) from all five Brazilian geographic regions were determined (Fig. 1). Each data set had three replicates, totaling 321 individual samplings. The average number of trimmed reads by sample varied from 1,474,376 to 49,909,522, leading to a total of 1.62 billion bp. Host-derived reads over all samples were poorly represented, with a frequency of <0.01% in the majority of samples. The median number of N50 contigs was 4,160, and that of coding sequences of genes was 112,267. Around 5.4 million reads had taxonomic signatures up to the species level (see Tables S1 and S2 in the supplemental material).
FIG 1.

Map of Brazil showing the geographic locations of the five participating centers.
The analyzed metagenomes showed a dominance of a few microorganisms, with the 10 most abundant species accounting for nearly 20% to 50% of the total microbial diversity (Table S3). The domain Bacteria was overrepresented compared to other domains of life (Table S3). A total of 21,029 unique bacterial species were identified, with numbers ranging from 12,388 in humans to 16,779 in poultry (Table S3). When the diversity and richness of the human bacterial composition were compared to those of animals, the Chao1 richness estimator showed that the numbers of species were significantly higher in cattle, poultry, and swine (P ≤ 0.05) (Fig. 2 and Table S4). When the number of distinct species present in each host was measured based on the Shannon index, the animals received similar diversity estimates, except for swine from the Southern region and cattle from the Midwestern region, which showed higher diversity (P ≤ 0.05). Furthermore, the bacterial composition varied among cattle herds present in the Southern and Northeastern regions (Table S5). The observed diversity was not affected by the dominance of a few species, as estimated by the Simpson index, which showed elevated evenness in all hosts, with no statistically significant difference being found (Fig. 2 and Tables S4 and S5).
FIG 2.
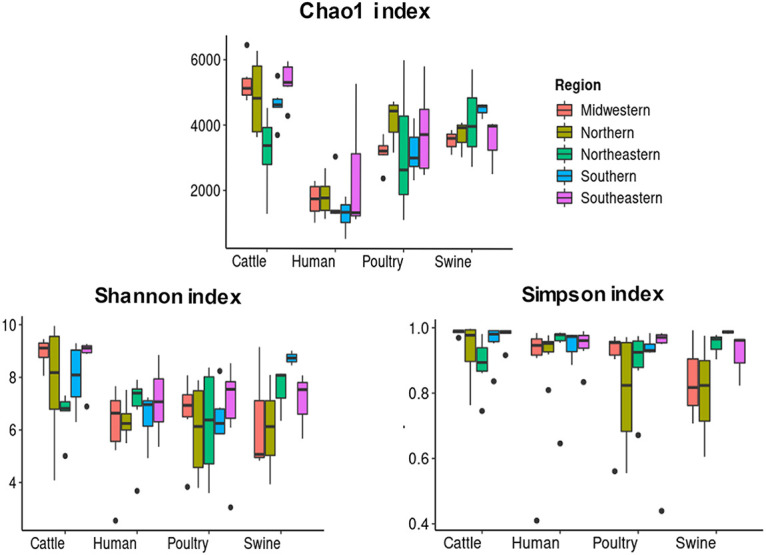
Alpha-diversity comparison of the bacterial compositions among poultry, cattle, swine, and humans, measured according to the Chao1, Shannon, and Simpson indices. Samples from five Brazilian regions are presented. Boxes represent the interquartile ranges (IQRs) between the first and third quartiles (25th and 75th percentiles, respectively), and the line inside denotes the median. Whiskers indicate the lowest and highest values within a range of 1.5-fold and the IQRs from the first and third quartiles, respectively. Dots represent outliers.
Interestingly, principal-coordinate analysis (PCoA) and hierarchical clustering methods showed that most human samples formed a single restricted cluster in all geographic regions analyzed, except for the Northeastern region (Fig. S1). The observation of nonclustering bacterial microbiota in cattle, poultry, and swine suggested that species were shared among these hosts (Fig. S1).
The domain Bacteria was composed mainly of species belonging to the phyla Firmicutes (41%), Proteobacteria (29.4%), Bacteroidetes (14.8%), and Actinobacteria (8.7%) (Fig. 3 and Table S6). Cattle, poultry, and swine harbored Firmicutes as the most abundant phylum, represented mainly by the genera Ruminiclostridium and Bacteroides. Among these bacteria, Ruminiclostridium cellobioparum, R. papyrosolvens, and R. hungatei were the most frequent, followed by Bacteroides xylanolyticus and B. graminisolvens. Additionally, in cattle, Comamonas kerstersii (Proteobacteria) and Cellulomonas persica (Actinobacteria) were very abundant (Fig. 3). Poultry also showed considerable counts of Petrimonas sp. strain IBARAKI, Sphingobacterium mizutaii (both from the phylum Bacteroidetes), and the proteobacteria E. coli and Bordetella avium. In swine, E. coli, S. mizutaii, Bacteroides paurosaccharolyticus, and C. kerstersii were representative species. The dominance of species belonging to the Proteobacteria was particularly notable in humans, especially regarding the frequencies of E. coli and C. kerstersii (Fig. 3 and Table S6). Pseudomonas aeruginosa and Prevotella copri were also observed, as were species of the genera Clostridium and Achromobacter but at lower frequencies (Fig. 3).
FIG 3.

Relative frequencies of the most common species in the microbial compositions of the sampled hosts. The top 10 microorganisms from each host are presented.
Sharing of species among humans and animals: potential scenario for cross talk colonization.
The possible cross talk of bacterial colonization between humans and animals was investigated by considering the species with the highest abundances in their respective microbiomes and those described as priority bacterial groups by the WHO, thus including species of 17 distinct genera. Among the species of these genera shared among animals and humans (Fig. 4A), approximately 17 to 24 microorganisms were detected as being the most abundant in each of the Brazilian regions studied (Fig. 4B and Tables S7 and S11).
FIG 4.

Common and exclusive species analysis among poultry, cattle, swine, and humans. (A and B) Venn diagram results for all species belonging to the 17 selected genera (comprising the 10 most abundant in host species and WHO priority groups) (A) and the predominant microorganisms from each of these genera (B). (C) Prevalence of abundant but uncommon clinical bacteria identified among the hosts within the selected genera.
In the Northern region, C. kerstersii was predominant in humans (12%) (Table 1). Pseudomonas stutzeri, Achromobacter insolitus, P. aeruginosa, E. coli, and Bordetella hinzii were also present but at lower abundances than C. kerstersii (Table 1). More precise estimated prevalences were observed for P. stutzeri, P. aeruginosa, and E. coli (Table 1). These species were also prevalent in cattle samples (Table S7). In humans of the Southern region, E. coli (8.1%), P. aeruginosa (5.9%), and P. copri (2.7%) were abundant (Table 1); the first two species were also found in cattle, poultry, and swine (Fig. 4B and Table S8). In addition to the species described previously in hosts from other Brazilian geographic regions, the occurrence of Clostridium spp. (C. botulinum, C. intestinale, and C. ventriculi) was observed in both animals and humans in the Southeastern region (Fig. 4B). Surprisingly, C. kerstersii was more frequent in humans (23.7%) from the Midwestern region (Table 1), where Acinetobacter baumannii, Salmonella enterica, Shigella sonnei, and Streptococcus pneumoniae were also among the commonly detected species (Fig. 4B). In the Northeastern region, the species present in humans and all examined animals included E. coli and P. copri (Fig. 4B).
TABLE 1.
Frequency and prevalence of predominant microorganisms belonging to 17 previously selected genera in humans from all five Brazilian geographic regions, considering species shared with animals
| Region and species detected in humans | Relative frequency (%) | Prevalence (%) | Lower CI (0.95) (%) | Upper CI (0.95) (%) |
|---|---|---|---|---|
| Northern region | ||||
| Comamonas kerstersii | 12.5479 | 50 | 19 | 81 |
| Pseudomonas stutzeri | 2.7876 | 100 | 61 | 100 |
| Achromobacter insolitus | 2.0550 | 50 | 19 | 81 |
| Pseudomonas aeruginosa | 1.8135 | 100 | 61 | 100 |
| Escherichia coli | 1.6270 | 100 | 61 | 100 |
| Bordetella hinzii | 1.0281 | 67 | 30 | 90 |
| Klebsiella pneumoniae | 0.4865 | 100 | 61 | 100 |
| Acinetobacter baumannii | 0.1403 | 100 | 61 | 100 |
| Salmonella enterica | 0.1298 | 100 | 61 | 100 |
| Streptococcus pneumoniae | 0.0666 | 100 | 61 | 100 |
| Achromobacter xylosoxidans | 0.0269 | 67 | 30 | 90 |
| Anaerospora hongkongensis | 0.0247 | 67 | 30 | 90 |
| Haemophilus parainfluenzae | 0.0200 | 67 | 30 | 90 |
| Enterococcus faecalis | 0.0138 | 83 | 44 | 99 |
| Staphylococcus aureus | 0.0084 | 100 | 61 | 100 |
| Bordetella pertussis | 0.0067 | 50 | 19 | 81 |
| Streptococcus suis | 0.0060 | 83 | 44 | 99 |
| Haemophilus influenzae | 0.0055 | 83 | 44 | 99 |
| Chryseobacterium mucoviscidosis | 0.0051 | 17 | 1 | 56 |
| Chryseobacterium spp. | 0.0006 | 17 | 1 | 56 |
| Haemophilus haemolyticus | 0.0005 | 33 | 10 | 70 |
| Southern region | ||||
| Escherichia coli | 8.1077 | 100 | 65 | 100 |
| Pseudomonas aeruginosa | 5.9414 | 100 | 65 | 100 |
| Comamonas kerstersii | 4.5719 | 14 | 1 | 51 |
| Prevotella copri | 2.7534 | 100 | 65 | 100 |
| Faecalibacterium prausnitzii | 2.0582 | 100 | 65 | 100 |
| Chryseobacterium spp. | 1.6796 | 29 | 8 | 64 |
| Bilophila wadsworthia | 1.4102 | 100 | 65 | 100 |
| Klebsiella pneumoniae | 1.0562 | 100 | 65 | 100 |
| Acinetobacter baumannii | 0.6048 | 100 | 65 | 100 |
| Enterococcus faecium | 0.1687 | 100 | 65 | 100 |
| Salmonella enterica | 0.1443 | 100 | 65 | 100 |
| Streptococcus pneumoniae | 0.0346 | 100 | 65 | 100 |
| Staphylococcus aureus | 0.0175 | 100 | 65 | 100 |
| Haemophilus influenzae | 0.0104 | 100 | 65 | 100 |
| Bordetella pertussis | 0.0046 | 29 | 8 | 64 |
| Cellulomonas hominis | 0.0003 | 14 | 1 | 51 |
| Southeastern region | ||||
| Comamonas kerstersii | 6.436 | 57 | 25 | 84 |
| Clostridium botulinum | 3.985 | 100 | 65 | 100 |
| Escherichia coli | 3.074 | 100 | 65 | 100 |
| Clostridium ventriculi | 2.816 | 57 | 25 | 84 |
| Prevotella copri | 2.085 | 100 | 65 | 100 |
| Marasmitruncus massiliensis | 2.085 | 86 | 49 | 99 |
| Achromobacter xylosoxidans | 2.057 | 71 | 36 | 92 |
| Pseudomonas stutzeri | 1.949 | 71 | 36 | 92 |
| Flavobacterium spp. | 1.848 | 71 | 36 | 92 |
| Clostridium intestinale | 1.507 | 71 | 36 | 92 |
| Streptococcus pneumoniae | 1.317 | 100 | 65 | 100 |
| Bilophila wadsworthia | 1.261 | 86 | 49 | 99 |
| Alcaligenes faecalis | 1.248 | 57 | 25 | 84 |
| Pseudomonas aeruginosa | 1.160 | 86 | 49 | 99 |
| Staphylococcus aureus | 1.143 | 100 | 65 | 100 |
| Cellulomonas hominis | 0.699 | 29 | 8 | 64 |
| Streptococcus suis | 0.604 | 100 | 65 | 100 |
| Haemophilus influenzae | 0.418 | 71 | 36 | 92 |
| Achromobacter denitrificans | 0.252 | 43 | 16 | 75 |
| Acinetobacter baumannii | 0.191 | 100 | 65 | 100 |
| Paenibacillus stellifer | 0.167 | 29 | 8 | 64 |
| Klebsiella pneumoniae | 0.158 | 100 | 65 | 100 |
| Enterococcus faecium | 0.131 | 100 | 65 | 100 |
| Paenibacillus timonensis | 0.118 | 14 | 1 | 51 |
| Enterococcus faecalis | 0.112 | 100 | 65 | 100 |
| Salmonella enterica | 0.082 | 100 | 65 | 100 |
| Midwestern region | ||||
| Comamonas kerstersii | 23.718 | 33 | 9 | 70 |
| Escherichia coli | 8.210 | 100 | 60 | 100 |
| Bilophila wadsworthia | 1.476 | 83 | 43 | 99 |
| Shigella sonnei | 1.135 | 100 | 60 | 100 |
| Klebsiella pneumoniae | 0.611 | 100 | 60 | 100 |
| Acinetobacter baumannii | 0.423 | 100 | 60 | 100 |
| Salmonella enterica | 0.140 | 100 | 60 | 100 |
| Pseudomonas aeruginosa | 0.093 | 100 | 60 | 100 |
| Enterococcus faecium | 0.053 | 100 | 60 | 100 |
| Staphylococcus aureus | 0.031 | 100 | 60 | 100 |
| Oscillibacter ruminantium | 0.017 | 83 | 43 | 99 |
| Haemophilus influenzae | 0.012 | 100 | 60 | 100 |
| Streptococcus pneumoniae | 0.007 | 100 | 60 | 100 |
| Northeastern region | ||||
| Comamonas kerstersii | 12.311 | 33 | 9 | 70 |
| Escherichia coli | 4.790 | 100 | 60 | 100 |
| Prevotella copri | 4.732 | 100 | 60 | 100 |
| Pseudomonas aeruginosa | 1.435 | 83 | 43 | 99 |
| Faecalibacterium prausnitzii | 1.363 | 100 | 60 | 100 |
| Bilophila wadsworthia | 1.083 | 100 | 60 | 100 |
| Klebsiella pneumoniae | 0.363 | 100 | 60 | 100 |
| Bacteroides fragilis | 0.313 | 100 | 60 | 100 |
| Enterococcus faecium | 0.111 | 100 | 60 | 100 |
| Acinetobacter baumannii | 0.109 | 100 | 60 | 100 |
| Salmonella enterica | 0.104 | 100 | 60 | 100 |
| Citrobacter freundii | 0.078 | 100 | 60 | 100 |
| Streptococcus pneumoniae | 0.023 | 100 | 60 | 100 |
| Staphylococcus aureus | 0.012 | 100 | 60 | 100 |
| Haemophilus influenzae | 0.009 | 83 | 43 | 99 |
| Bordetella pertussis | 0.001 | 33 | 9 | 70 |
Despite showing lower abundances, species not previously mentioned had increased prevalences in humans and some sampled animals, such as Staphylococcus aureus (from the Northern region), Faecalibacterium prausnitzii and Klebsiella pneumoniae (from the Southern region), Marasmitruncus massiliensis (from the Southeastern region), Enterococcus spp. (E. faecalis and E. faecium) (from the Midwestern region), and Bacteroides fragilis (from the Northeastern region) (Fig. 4C and Tables S7 and S11).
Variability and spread of ARGs between human and livestock resistomes.
Functional analysis detected a total of 405 ARGs for 12 distinct antimicrobial classes (Table S12). Genes encoding antibiotic-modifying enzymes (n = 231; 57%) were the most frequent, followed by genes related to target alteration (n = 95; 23.5%) and efflux pump systems (n = 79; 19.3%).
Among the genes encoding antibiotic-modifying enzymes, those encoding β-lactamases and aminoglycoside-modifying enzymes (AMEs) were the most frequent (Table S12). Intrinsic and acquired β-lactamase-encoding genes were by far the most frequent and diverse group of enzymes found (n = 122; 52.8%). According to Ambler’s classification, 42 β-lactamase-encoding genes belonged to molecular class C (n = 42; 34.4%), followed by class A (n = 39; 32.0%), class D (n = 25; 20.5%), and class B (n = 16; 13.1%) (Table S12). Interestingly, acquired carbapenemase-encoding genes such as blaAIM-1, blaCAM-1, blaHMB-1, blaGIM-2, and blaSME-1, which had not been previously reported in South American isolates, were observed (Table S12). The occurrence of blaAIM-1 was noted in cattle from the Southern region, in poultry from the Southeastern region, and in cattle and poultry from the Northeastern region (Fig. 5A). Similarly, blaSME-1 and blaSME-4 were found in cattle from the Southern and Midwestern regions and in humans from the Northern region (Fig. 5A). Curiously, some infrequent ARGs were identified in specific locations; for example, blaGIM-2 and blaCAM-1 were recovered from different hosts in the Southeastern region, and blaHMB-1 and blaVEB-9 were found in the Midwestern and Northeastern regions, respectively (Fig. S2). In addition, a total of 59 distinct AME-encoding genes were observed (Table S11), among which acetyltransferases (AACs) were the most frequent enzymes (n = 27 variants), including aac(6′)-Ib-cr, followed by adenylyltransferase (ANTs) (n = 18 variants) and phosphotransferases (APHs) (n = 14 variants) (Table S12).
FIG 5.

Gene network showing genes encoding products related to antibiotic inactivation according to the host and the geographic region. Circle sizes represent the abundance of each indicated ARG.
In terms of target alteration mechanisms, several trimethoprim-resistant dihydrofolate reductase (DFR)-encoding genes (n = 22 variants) were found (Table S12), among which dfrF was the most frequently recorded, except in metagenomes recovered from the Northern region, where dfrA1 and dfrA8 were predominant (Fig. 6). In addition, Erm 23S rRNA methyltransferase-encoding genes (n = 14 variants), which confer resistance to macrolides, lincosamides, and streptogramins, were also commonly found (Fig. 6 and Table S12), especially ermF (n = 36), ermB (n = 27), and ermG (n = 26) in humans and poultry (Fig. 5B). Finally, nine distinct tetracycline resistance ribosomal protection protein-encoding genes (tet genes) were found (Fig. 6 and Table S12), among which tetO, tetQ, and tetW were found to be widespread in all Brazilian geographic regions (Fig. 5B). Additionally, quinolone resistance protein (Qnr)-encoding genes (n = 13) (Table S12) were found in all hosts, among which qnrB10 and qnrB19 were the most frequent variants (Fig. 5B). Interestingly, qnrD1 was frequently found in poultry (n = 8), mostly from the Midwestern region, followed by humans (n = 4) and swine (n = 2) (Fig. 5B). In addition, the ciprofloxacin-modifying-enzyme-encoding gene crpP was found in all hosts, particularly humans, and was distributed in all geographic regions (Fig. 5B). Moreover, a total of nine variants of fosA, which is responsible for resistance to fosfomycin (Table S12), were also detected by the metagenome analysis and were most frequently found in humans and poultry, as were the tet37 (cattle) and tetX (human) genes that confer resistance to tetracyclines and glycylcyclines (tigecycline) (Fig. 6).
FIG 6.

Gene network showing genes encoding products related to target alteration according to the host and the geographic region. Circle sizes represent the abundance of each indicated ARG.
Genes encoding efflux pump system components were identified, most of which belonged to the major facilitator superfamily (MFS) (n = 42), especially the versatile tet group, followed by the resistance-nodulation-cell division (RND) family (n = 25). (Table S12). Curiously, the frequency of multidrug and toxic compound extrusion (MATE) efflux pump systems varied according to the region evaluated (Fig. S3). These efflux pump systems were absent in swine from the Northern region, poultry from the Southern region, and, surprisingly, cattle, poultry, and swine from the Southeastern and Midwestern regions. In addition, the the small multidrug resistance (SMR) family was not found in swine from the Southeastern region (Fig. S3).
DISCUSSION
AMR has been recognized as a serious public health concern worldwide (3, 4). The epidemiology of bacterial resistance is complex and is not restricted to humans and food-producing animals (15), as it is also associated with the environment (27) and is influenced by modern events such as international trade and travel (12, 13) and global warming (28). Because ARGs are versatile and widely distributed in different ecological niches (5, 7, 23), it is essential that AMR surveillance be based on the One Health approach (13, 17). Brazil is divided into five geographic regions, which display different sociodemographic and geographic characteristics.
In our study, cattle, poultry, and swine showed high species richness. The similar microbial structures observed in these food-producing animals, represented mainly by cellulose- and xylan-degrading Firmicutes species, seem to reflect their lifestyle conditions, as previously observed (2). Members of the phyla Firmicutes, Bacteroidetes, and Actinobacteria were found in swine and cattle, as previously reported (29, 30). In swine, Ruminococcus and Bacteroides abundance can result from early-stage animal growth, as observed previously by Han et al. (29). Jurburg et al. (31) showed that in developing chickens, the first stage was dominated by Streptococcus spp. and Escherichia spp./Shigella spp., which were displaced in the second stage by rapidly growing taxa, including Ruminococcus-like species variants (31). Beyond these taxa, distinct families and genera were identified in the animals studied in our work.
In addition to Enterobacterales, we observed that environmental or commensal proteobacteria, such as C. kerstersii (32, 33), A. insolitus (34), and B. hinzii (35), were abundant in both animals and humans and have been reported to be opportunistic agents colonizing humans. Microorganisms from distinct genera, such as P. stutzeri, Clostridium ventriculi, Faecalibacterium prausnitzii, and Bilophila wadsworthia, accounted for a proportion similar to those of some well-described antimicrobial-resistant species. As proposed previously by Woolhouse and Gowtage-Sequeria (1), small changes in animal-human interactions, such as differences in the numbers of introductions of bacteria into the host and the sizes of the susceptible populations, can lead to the spread of bacterial species, and their zoonotic potential should not be overlooked (33, 36).
In this study, our results demonstrated that resistance to β-lactams and aminoglycosides, especially that mediated by antibiotic inactivation, was the most frequent mechanism of AMR. The occurrence of antibiotic inactivation mechanisms varied slightly according to the host and geographic region and could be related to the type of feeding and handling characteristics of local livestock farming. Our results are complementary to those of previous studies (37, 38) that have evaluated environmental samples. β-Lactamase- and AME-encoding genes were also reported to be among the most frequent AMR mechanisms in water samples from Lake Bolonha, which is located in the Brazilian Amazon (37), and Brazilian mangrove regions (38).
In Brazil, the production of β-lactamases, particularly carbapenemases, by Gram-negative bacilli is the main challenge faced by physicians (39) since β-lactams have been widely used as the first line to treat serious infections (40). Interestingly, we did not observe the occurrence of blaKPC-like genes, which are the most widespread carbapenemases in Enterobacterales recovered from Brazilian hospitals, but a variety of class A ESBLs and class B carbapenemases were found (41). Similar results were observed previously by Alves et al. (37) in Lake Bolonha, where those authors found the presence of blaIMP-like, blaVIM-like, and blaCTX-M-like but not blaKPC-like genes. Curiously, we detected the occurrence of blaAIM-1 in livestock feces for the first time in South America, to the best of our knowledge. This class B carbapenemase-encoding gene was first described in 2012 in three P. aeruginosa clinical isolates recovered in Australia (42). To date, this type of gene has been reported only in K. pneumoniae recovered in 2019 from a patient with diarrhea in China (43). Other carbapenemase-encoding genes, blaCAM-1 and blaGIM-2-blaHMB-1, which have been described only in Canada and Germany, respectively (44–46), were also detected in our study. These findings might be justified by the detection of environmental/uncultured bacteria, which could be primary sources of these carbapenemase-encoding genes that have been further mobilized to generate resistant clinical isolates. The spread of bacterial species carrying carbapenemase-encoding genes by migratory birds around the globe to rural areas where humans and birds are in constant contact provides the opportunity for interspecies transmission and might give rise to new hypotheses (47). Recently, two studies reported the presence of endemic P. aeruginosa sequence type 277 (ST277) and A. baumannii ST79 clones carrying the carbapenemase-encoding genes blaSPM-1 and blaOXA-72 in the microbiota of migratory birds in Brazil (48, 49), respectively, reinforcing their role as hosts of MDR microorganisms.
Interestingly, we also observed the occurrence of the crpP gene in healthy individuals and animals for the first time in South America. crpP was recently described as a ciprofloxacin-modifying phosphotransferase carried by a plasmid in a P. aeruginosa strain isolated in Mexico (50). After it was initially reported in 2018, the presence of crpP was demonstrated in European countries (France and Switzerland) (51), Africa (Cameroon and South Africa) (52, 53), India (54), and Australia (54). It was subsequently shown that Mexican Enterobacterales isolates recovered in 1994 also carried this gene (55).
Our results allowed us to describe the bacterial communities and ARGs found in healthy humans and food-producing animals from distinct Brazilian geographic regions. The number of samples collected might be considered a limitation of this study; however, due to budget restrictions, we decided to obtain triplicate rectal swabs from the same host to obtain high depth and coverage of metagenomic results. In this manner, we were able to identify microorganisms to the species level and ARGs. Other authors have pointed out Brazil as a hot spot for the emergence of ARGs (16). The emergence of resistance in Brazil has a high chance of impacting all global regions because Brazil has been one of the largest exporters of food-producing animals. This study was also important for building a network that can be used in the future to initiate One Health surveillance at the national level, incorporating a higher number of centers and samples.
Conclusion.
To the best of our knowledge, we report the first description of the bacteriome and resistome of the feces of healthy individuals and food-producing animals (poultry, cattle, and swine) collected simultaneously within the same period of time from five Brazilian geographic regions. Our results are a snapshot of the distribution of microbial species and ARGs in humans and food-producing animals. Although in most geographic regions, the microbial diversities of animals and humans were distinct, we observed a resemblance between the species isolated from humans and those from food-producing animals collected from the center located in the Northeastern region. This may suggest the influence of regional habits favoring microbiota sharing. To the best of our knowledge, we detected for the first time carbapenemase-encoding genes such as blaAIM-1, blaCAM-1, blaGIM-2, and blaHMB-1 in Latin America. In this manner, our results corroborate the importance of metagenomics as a tool for tracking the colonization of livestock and humans by antimicrobial-resistant microorganisms. Moreover, a network surveillance program named GUARANI, which was created for this study, is ready to be scaled up. It would be important to delineate the countrywide panorama of antimicrobial resistance since Brazil plays an important role in the world scenario as one of the largest exporters of meat.
MATERIALS AND METHODS
Ethics and regulatory approval.
Ethics approval for this study was obtained from the Research Ethics Committee (CEP) and the Committee on Ethics in the Use of Animals (CEUA) of the Universidade Federal de São Paulo (UNIFESP) (process numbers 3.116.383 and 2607170119, respectively). This project was also registered by the National System for the Management of Genetic Heritage and Associated Traditional Knowledge (process number AA1668A).
One Health.
One Health is a collaborative, multisectoral, and transdisciplinary approach with the goal of achieving optimal health outcomes recognizing the interconnection among people, animals, plants, and their shared environment (2, 14, 18). In recent years, the One Health concept has gained importance in tackling AMR, one of the top 10 global public health threats facing humanity (56, 57).
Sample selection.
To perform this study, rectal swabs from cattle, swine, poultry, and humans were collected between February and April 2020 from five cities located in five Brazilian geographic regions: Castanhal (Northern region) (longitude [φ] 1°17′50″S, latitude [λ] 47°55′20″W), Blumenau (Southern region) (φ 26°55′7″S, λ 49°3′58″W), Bragança Paulista (Southeastern region) (φ 22°57′8″S, λ 46°32′33″W), Dourados (Midwestern region) (φ 22°13′16″S, λ 54°48′20″W), and Fortaleza (Northeastern region) (φ 3°43′6″S, λ 38°32′36″W), as shown in Fig. 1. The GUARANI One Health Network was established based on previous research collaboration, including one researcher from each of the five distinct Brazilian geographic regions. Three rural properties of each Brazilian geographic region were randomly selected based on two criteria: (i) they should be classified as small properties according to ordinance number 8.629, 25 February 1993, established by the Brazilian Institute of Colonization and Agrarian Reform (Instituto Nacional de Colonização e Reforma Agrária [INCRA]) (https://www.gov.br/incra/pt-br/assuntos/governanca-fundiaria/modulo-fiscal), and (ii) they should raise at the same time distinct food-producing animals (cattle, swine, and poultry) for human consumption. At each property, fecal swabs were collected from cattle (n = 2), poultry (n = 2), and swine (n = 1). In addition, fecal swabs from two healthy adults (18 to 64 years old) who lived in urban areas served by the food produced in those small properties were also collected. In total, 107 subjects were selected for swab collection, representing cattle (n = 30), poultry (n = 30), swine (n = 15), and humans (n = 32) (see Table S1 in the supplemental material). The swabs were collected in triplicate from each subject. Briefly, Copan Amies sterile transport swabs (Copan Diagnostics, Corona, CA) were inserted 1 to 1.5 in. into the rectum and gently rotated. The same swabs were placed into the tube deep enough that the medium covered the cotton tips and were transported at room temperature to the laboratory for DNA extraction.
DNA extraction and sequencing.
Total DNA extraction was performed using the ZymoBIOMICS DNA miniprep kit (Zymo, USA) according to the manufacturer’s guidelines. The extracted DNA was transported at 4°C to the Laboratório Nacional de Computação Científica (LNCC), where metagenomic library preparation and sequencing was performed. Libraries were constructed using the Nextera DNA Flex library preparation kit (Illumina, USA) according to the manufacturer’s recommendations. Library quality control (QC) and quantification procedures were performed using the high-sensitivity D5000 ScreenTape assay on a 4200 TapeStation system (Agilent, USA). For each sequencing run, 48 libraries were pooled by volume, and sequencing was conducted on a NextSeq 500 system using the NextSeq 500/550 high-output kit v2.5 (300 cycles) (Illumina, USA), with the system set to produce 2 × 150-bp reads.
Bioinformatic processing and analysis. (i) Data trimming and host sequence mapping.
Raw reads were submitted to BBduk (BBMap software v.38.81 [https://github.com/BioInfoTools/BBMap]) for quality control (i.e., the identification and filtering of low-quality reads and sequencing artifacts). Reads with a quality threshold lower than a Phred score of 20 (with a sliding window of 10 bases) and a length smaller than 50 bp, Illumina adapters, and phiX174 were removed using the following parameters: minlength=50, mink=8, qout=auto, hdist=1, k=31, trimq=10, qtrim=rl, ktrim=l, minavgquality=20, and statscolumns=5. Next, the remaining reads were mapped against NCBI reference genomes for host-associated read filtering. The mappings were performed against human (Homo sapiens, GRCh38.p13 [NCBI accession number GCF_000001405.39]), poultry (Gallus gallus, GRCg6a [accession number GCF_000002315.6]), cattle (Bos taurus, ARS-UCD1.2 [accession number GCF_002263795.1]), and swine (Sus scrofa, Sscrofa11.1 [accession number GCF_000003025.6]) genomes. All mappings were done in Bowtie 2.4.156 using the end-to-end very-sensitive option (58).
(ii) Taxonomic inference and statistical analyses.
Taxonomic analysis of the high-quality reads was performed with Kaiju software (59) (version 1.7.3) using the NR_EUK database (January 2020 version). Sequencing depth variations among samples were corrected by nonrandom library size normalization in order to make the samples comparable. For this, a factor reflecting each sample-specific library size was applied to the respective read counts [calculated as factor = (n trim reads ss/n trim reads sls) × OTU reads ss, where ss is a specific sample, sls is the smallest library sample size across all samples, and OTU is operational taxonomic units]. Species whose relative abundance was <0.001% were filtered to avoid false-positive results (60). Considering that a nomenclature revision has been proposed for some bacterial species, the data presented here are described according to the first name previously validated by the International Committee on Systematics of Prokaryotes. To investigate the community composition diversity, Shannon and Simpson indices and Chao1 richness estimators were computed under the relative abundance of bacterial species using the skbio.diversity.alpha_diversity function of a Python script written in the skbio package (61). The statistical significance of the diversity metrics was evaluated using analysis of variance (ANOVA) (P < 0.05) and Tukey’s post hoc test on the R statistical platform. Principal-coordinate analysis (PCoA) and hierarchical clustering were conducted to determine the distances or dissimilarities between the structures of the bacterial communities. PCoA matrices were analyzed using the Bray-Curtis dissimilarity metric of the Phyloseq R package (62). Multivariate analysis of agglomerative hierarchical clustering was performed using a binary distance and the Ward.D2 method in the dendextend R package (63).
(iii) Common and exclusive microbiota analyses.
The relationship among the microbial compositions of the different hosts in this study was determined using Jvenn viewer (64). The genera analyzed were selected based on two criteria. The first one included the genera of the 10 most abundant species from each host. The second criterion was the inclusion of 7 genera of 12 pathogens listed under distinct priority groups by the WHO: critical (Acinetobacter and Pseudomonas), high (Enterococcus, Staphylococcus, and Salmonella), or medium (Streptococcus and Haemophilus) (65). The order Enterobacterales was reported to be of critical priority by the WHO. However, genera of this order were observed to be abundant in some hosts and were previously included according to the first criterion. This resulted in 17 bacterial genera being selected. The predominant species from each genus were analyzed. To investigate the occurrence of species in an epidemiological context, an estimate of prevalence was inferred. For this, appropriate confidence intervals (CIs) were provided, accounting for the changes in variance metrics that arise from imperfect test sensitivity and specificity. The prevalence of each species was estimated using the epi.prev function in R (confidence level of 0.95, sensibility of 70%, and range of 90 to 95% specificity) and the Blaker method (66), based on cutoffs proposed in the literature (67, 68).
(iv) De novo assembly and gene prediction.
To maximize the identification of ARGs in the data set, the trimmed reads of each biological replicate were grouped and assembled as a unique file sample. The assemblies of reads into contigs were performed using metaSPAdes (69) software (v.3.14) with parameter settings -k 21,33,55,77. Only contigs longer than 500 bp were included in the downstream analyses. The remaining contigs were predicted in open reading frames (ORFs) with Prodigal software (70) version 2.6.3 (applying the -g 1 -p meta options).
(v) Identification of antimicrobial resistance genes.
Annotation and alignment against a functional database were conducted with ORFs of >50 amino acids. Predicted ORFs were aligned against the Comprehensive Antibiotic Resistance Database (CARD) (downloaded in August 2020) (71) for ARG identification. An E value of ≤1e−5, a minimum identity of 90%, and a minimum query length and subject coverage of 90% were applied as parameters. Analyses were done considering the gene assignments with the highest-scoring annotated hits. To avoid single nucleotide polymorphisms (SNPs) at specific loci within the ARGs, only genes with both 100% identity and 100% coverage of a match to a CARD reference sequence were discussed. Community detection analyses were performed to identify how groups of ARGs are clustered and can indicate interactions among the hosts. The network was constructed using the fast greedy algorithm implemented in the plot.igraph function (default parameters) available at the igraph R library (72).
(vi) Graphics visualization.
Bar representations of microbial abundance distributions and box plots for both richness of species and AMR genes most frequently found across the hosts, also shown as heatmaps, were generated with the ggplot2 R package (73). Pairwise correlations on scatter matrices were done using the pairs function in the R language.
Data availability.
The data sets supporting the conclusions of this article are available in the NCBI SRA (www.ncbi.nlm.nih.gov/sra) under BioProject accession number PRJNA684454.
ACKNOWLEDGMENTS
We thank all researchers from the seven participating centers belonging to the GUARANI (Grupo Brasileiro de Saúde Única) One Health Network for their commitment and hard work, even during the SARS-CoV-2 pandemic.
This study was supported by the National Council for Science and Technological Development (CNPq) and the Bill & Melinda Gates Foundation (process numbers 402659/2018-0, 443805/2018-0, and OPP1193112). We are grateful to the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) for providing grants to T.B.V., F.F.S. (PNPD), F.O.B.N., and R.G.B.D.S. and the CNPq for providing grants to R.V. and A.C.G. (process number 312066/2019-8), and A.T.R.D.V. is supported by the CNPq (307145/2021-2) and FAPERJ (E-26/201.046/2022).
A.C.G. has recently received research funding and/or consultation fees from Bayer, Cristália, InfectoPharm, Eurofarma, MSD, Pfizer, and Zambon. The other authors have nothing to declare. This study was not financially supported by any diagnostic/pharmaceutical company.
A.C.G., A.C.D.O.S., A.T.R.D.V., C.D.O.S., C.R.V.K., D.D.S.C.M.C.-B., D.M.B., E.K.A., L.F.C.F., R.C., and S.S. contributed to the study conception and design. A.C.G., A.C.D.O.S., A.T.R.D.V., C.D.O.S., D.D.S.C.M.C.-B., D.M.B., E.K.A., F.F.S., L.F.C.F., R.C., S.S., and T.B.V. supervised the assays. A.C.D.O.S., C.D.O.S., D.D.S.C.M.C.-B., D.M.B., E.K.A., L.F.C.F., S.S., and W.A.D.O.L. provided the stool samples. Data collection and analysis were performed by A.C.G., A.C.D.O.S., A.T.R.D.V., C.D.O.S., C.R.V.K., D.C.C., D.D.S.C.M.C.-B., D.M.B., E.K.A., F.F.S., F.M.D.C., F.O.B.N., G.H.D.A.S., G.M.D.M.G., L.N.L., L.F.C.F., M.D.N.M.B., M.S.M.V., R.C., R.G.B.D.S., R.V., S.S., T.B.V., and W.A.D.O.L. The first draft of the manuscript was written by A.C.G., A.T.R.D.V., F.M.D.C., F.F.S., L.N.L., R.C., and T.B.V. The final draft of the manuscript was reviewed by A.C.G., A.T.R.D.V., F.M.D.C., F.F.S., L.N.L., R.C., and T.B.V. All authors read and approved the final version of the manuscript.
Footnotes
Supplemental material is available online only.
Contributor Information
Ana Cristina Gales, Email: ana.gales@unifesp.br.
Hermine V. Mkrtchyan, University of West London
REFERENCES
- 1.Woolhouse ME, Gowtage-Sequeria S. 2005. Host range and emerging and reemerging pathogens. Emerg Infect Dis 11:1842–1847. doi: 10.3201/eid1112.050997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trinh P, Zaneveld JR, Safranek S, Rabinowitz PM. 2018. One Health relationships between human, animal, and environmental microbiomes: a mini-review. Front Public Health 6:235. doi: 10.3389/fpubh.2018.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Neill J. 2016. Tackling drug-resistant infections globally: final report and recommendations. Review on Antimicrobial Resistance, London, United Kingdom. https://amr-review.org/. [Google Scholar]
- 4.World Health Organization. 2019. No time to wait: securing the future from drug resistance infections. Report to the Secretary-General of the United Nations—April 2019. World Health Organization, Geneva, Switzerland. https://www.who.int/publications/i/item/no-time-to-wait-securing-the-future-from-drug-resistant-infections. [Google Scholar]
- 5.Centers for Disease Control and Prevention. 2019. Antibiotic resistance threats in the United States, 2019. US Department of Health and Human Services, Centers for Disease Control and Prevention, Atlanta, GA. https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf. [Google Scholar]
- 6.Cassini A, Högberg LD, Plachouras D, Quattrocchi A, Hoxha A, Simonsen GS, Colomb-Cotinat M, Kretzschmar ME, Devleesschauwer B, Cecchini M, Ouakrim DA, Oliveira TC, Struelens MJ, Suetens C, Monnet DL, Burden of AMR Collaborative Group . 2019. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: a population-level modelling analysis. Lancet Infect Dis 19:56–66. doi: 10.1016/S1473-3099(18)30605-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levy SB, Marshall B. 2004. Antibacterial resistance worldwide: causes, challenges and responses. Nat Med 10:S122–S129. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- 8.Saleem AF, Allana A, Hale L, Diaz A, Salinas R, Salinas C, Qureshi SM, Hotwani A, Rahman N, Khan A, Zaidi AK, Seed PC, Arshad M. 2020. The gut of healthy infants in the community as a reservoir of ESBL and carbapenemase-producing bacteria. Antibiotics (Basel) 9:286. doi: 10.3390/antibiotics9060286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stoesser N, Xayaheuang S, Vongsouvath M, Phommasone K, Elliott I, Del Ojo Elias C, Crook DW, Newton PN, Buisson Y, Lee SJ, Dance DA. 2015. Colonization with Enterobacteriaceae producing ESBLs in children attending pre-school childcare facilities in the Lao People’s Democratic Republic. J Antimicrob Chemother 70:1893–1897. doi: 10.1093/jac/dkv021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woerther PL, Burdet C, Chachaty E, Andremont A. 2013. Trends in human fecal carriage of extended-spectrum β-lactamases in the community: toward the globalization of CTX-M. Clin Microbiol Rev 26:744–758. doi: 10.1128/CMR.00023-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Overdevest I, Willemsen I, Rijnsburger M, Eustace A, Xu L, Hawkey P, Heck M, Savelkoul P, Vandenbroucke-Grauls C, van der Zwaluw K, Huijsdens X, Kluytmans J. 2011. Extended-spectrum β-lactamase genes of Escherichia coli in chicken meat and humans, the Netherlands. Emerg Infect Dis 17:1216–1222. doi: 10.3201/eid1707.110209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leverstein-van Hall MA, Dierikx CM, Cohen Stuart J, Voets GM, van den Munckhof MP, van Essen-Zandbergen A, Platteel T, Fluit AC, van de Sande-Bruinsma N, Scharinga J, Bonten MJM, Mevius DJ, National ESBL Surveillance Group . 2011. Dutch patients, retail chicken meat and poultry share the same ESBL genes, plasmids and strains. Clin Microbiol Infect 17:873–880. doi: 10.1111/j.1469-0691.2011.03497.x. [DOI] [PubMed] [Google Scholar]
- 13.Christaki E, Marcou M, Tofarides A. 2020. Antimicrobial resistance in bacteria: mechanisms, evolution, and persistence. J Mol Evol 88:26–40. doi: 10.1007/s00239-019-09914-3. [DOI] [PubMed] [Google Scholar]
- 14.McEwen SA, Collignon PJ. 2018. Antimicrobial resistance: a One Health perspective. Microbiol Spectr 6:ARBA-0009-2017. doi: 10.1128/microbiolspec.ARBA-0009-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Boeckel TP, Brower C, Gilbert M, Grenfell BT, Levin SA, Robinson TP, Teillant A, Laxminarayan R. 2015. Global trends in antimicrobial use in food animals. Proc Natl Acad Sci USA 112:5649–5654. doi: 10.1073/pnas.1503141112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Boeckel TP, Pires J, Silvester R, Zhao C, Song J, Criscuolo NG, Gilbert M, Bonhoeffer S, Laxminarayan R. 2019. Global trends in antimicrobial resistance in animals in low- and middle-income countries. Science 365:eaaw1944. doi: 10.1126/science.aaw1944. [DOI] [PubMed] [Google Scholar]
- 17.World Health Organization. 2015. WHO estimates of the global burden of foodborne diseases: Foodborne Disease Burden Epidemiology Reference Group 2007-2015. World Health Organization, Geneva, Switzerland. https://apps.who.int/iris/bitstream/handle/10665/199350/9789241565165_eng.pdf?sequence=1. [Google Scholar]
- 18.Walsh TR. 2018. A one-health approach to antimicrobial resistance. Nat Microbiol 3:854–855. doi: 10.1038/s41564-018-0208-5. [DOI] [PubMed] [Google Scholar]
- 19.Thanner S, Drissner D, Walsh F. 2016. Antimicrobial resistance in agriculture. mBio 7:e02227-15. doi: 10.1128/mBio.02227-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tacconelli E, Carrara E, Savoldi A, Harbarth S, Mendelson M, Monnet DL, Pulcini C, Kahlmeter G, Kluytmans J, Carmeli Y, Ouellette M, Outterson K, Patel J, Cavaleri M, Cox EM, Houchens CR, Grayson ML, Hansen P, Singh N, Theuretzbacher U, Magrini N, WHO Pathogens Priority List Working Group . 2018. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect Dis 18:318–327. doi: 10.1016/S1473-3099(17)30753-3. [DOI] [PubMed] [Google Scholar]
- 21.Yee R, Breitwieser FP, Hao S, Opene BNA, Workman RE, Tamma PD, Dien-Bard J, Timp W, Simner PJ. 2021. Metagenomic next-generation sequencing of rectal swabs for the surveillance of antimicrobial-resistant organisms on the Illumina Miseq and Oxford MinION platforms. Eur J Clin Microbiol Infect Dis 40:95–102. doi: 10.1007/s10096-020-03996-4. [DOI] [PubMed] [Google Scholar]
- 22.De R. 2019. Metagenomics: aid to combat antimicrobial resistance in diarrhea. Gut Pathog 11:47. doi: 10.1186/s13099-019-0331-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quince C, Walker AW, Simpson JT, Loman NJ, Segata N. 2017. Shotgun metagenomics, from sampling to analysis. Nat Biotechnol 35:833–844. doi: 10.1038/nbt.3935. [DOI] [PubMed] [Google Scholar]
- 24.Forsberg KJ, Patel S, Gibson MK, Lauber CL, Knight R, Fierer N, Dantas G. 2014. Bacterial phylogeny structures soil resistomes across habitats. Nature 509:612–616. doi: 10.1038/nature13377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.US Department of Agriculture Foreign Agricultural Service. 2020. Livestock and poultry: world markets and trade. US Department of Agriculture, Washington, DC. https://apps.fas.usda.gov/psdonline/circulars/livestock_poultry.pdf. [Google Scholar]
- 26.Associação Brasileira de Proteína Animal. 2022. Relatório anual. Associação Brasileira de Produção Animal, São Paulo, Brazil. https://abpa-br.org/wp-content/uploads/2022/05/Relatorio-Anual-ABPA-2022-1.pdf. [Google Scholar]
- 27.Richet H. 2012. Seasonality in Gram-negative and healthcare-associated infections. Clin Microbiol Infect 18:934–940. doi: 10.1111/j.1469-0691.2012.03954.x. [DOI] [PubMed] [Google Scholar]
- 28.Nyonyo T, Shinkai T, Mitsumori M. 2014. Improved culturability of cellulolytic rumen bacteria and phylogenetic diversity of culturable cellulolytic and xylanolytic bacteria newly isolated from the bovine rumen. FEMS Microbiol Ecol 88:528–537. doi: 10.1111/1574-6941.12318. [DOI] [PubMed] [Google Scholar]
- 29.Han GG, Lee JY, Jin GD, Park J, Choi YH, Kang SK, Chae BJ, Kim EB, Choi YJ. 2018. Tracing of the fecal microbiota of commercial pigs at five growth stages from birth to shipment. Sci Rep 8:6012. doi: 10.1038/s41598-018-24508-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sabino YNV, Santana MF, Oyama LB, Santos FG, Moreira AJS, Huws SA, Mantovani HC. 2019. Characterization of antibiotic resistance genes in the species of the rumen microbiota. Nat Commun 10:5252. doi: 10.1038/s41467-019-13118-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jurburg SD, Brouwer MSM, Ceccarelli D, van der Goot J, Jansman AJM, Bossers A. 2019. Patterns of community assembly in the developing chicken microbiome reveal rapid primary succession. Microbiologyopen 8:e00821. doi: 10.1002/mbo3.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Opota O, Ney B, Zanetti G, Jaton K, Greub G, Prod’hom G. 2014. Bacteremia caused by Comamonas kerstersii in a patient with diverticulosis. J Clin Microbiol 52:1009–1012. doi: 10.1128/JCM.02942-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang X, Liu W, Zheng B. 2018. Complete genome sequencing of Comamonas kerstersii 8943, a causative agent for peritonitis. Sci Data 5:180222. doi: 10.1038/sdata.2018.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li G, Zhang T, Yang L, Cao Y, Guo X, Qin J, Yang Q, You S, Yuan G, Jiang K, Luo J, Li Z, Gao L, Jiang K, Wu L, Zheng J. 2017. Complete genome sequence of Achromobacter insolitus type strain LMG 6003T, a pathogen isolated from leg wound. Pathog Dis 75:ftx037. doi: 10.1093/femspd/ftx037. [DOI] [PubMed] [Google Scholar]
- 35.Fabre A, Dupin C, Bénézit F, Goret J, Piau C, Jouneau S, Guillot S, Mégraud F, Kayal S, Desrues B, Le Coustumier A, Guiso N. 2015. Opportunistic pulmonary Bordetella hinzii infection after avian exposure. Emerg Infect Dis 21:2122–2126. doi: 10.3201/eid2112.150400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Filipic B, Malesevic M, Vasiljevic Z, Lukic J, Novovic K, Kojic M, Jovcic B. 2017. Uncovering differences in virulence markers associated with Achromobacter species of CF and non-CF origin. Front Cell Infect Microbiol 7:224. doi: 10.3389/fcimb.2017.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alves J, Dias L, Mateus J, Marques J, Graças D, Ramos R, Seldin L, Henriques I, Silva A, Folador A. 2020. Resistome in Lake Bolonha, Brazilian Amazon: identification of genes related to resistance to broad-spectrum antibiotics. Front Microbiol 11:67. doi: 10.3389/fmicb.2020.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Imchen M, Kumavath R. 2021. Metagenomic insights into the antibiotic resistome of mangrove sediments and their association to socioeconomic status. Environ Pollut 268:115795. doi: 10.1016/j.envpol.2020.115795. [DOI] [PubMed] [Google Scholar]
- 39.Agência Nacional de Vigilância Sanitária. 2020. Boletim Segurança do Paciente e Qualidade em Serviços de Saúde n° 18: avaliação dos indicadores nacionais das infecções relacionadas à assistência à saúde (IRAS) e resistência microbiana do ano de 2018. Agência Nacional de Vigilância Sanitária, Brasília, Brazil. https://www.gov.br/anvisa/pt-br/centraisdeconteudo/publicacoes/servicosdesaude/boletim-seguranca-do-paciente/boletim-seguranca-do-paciente-e-qualidade-em-servicos-de-saude-n-20-incidentes-relacionados-a-assistencia-a-saude-2018.pdf/view. [Google Scholar]
- 40.Van Boeckel TP, Gandra S, Ashok A, Caudron Q, Grenfell BT, Levin SA, Laxminarayan R. 2014. Global antibiotic consumption 2000 to 2010: an analysis of national pharmaceutical sales data. Lancet Infect Dis 14:742–750. doi: 10.1016/S1473-3099(14)70780-7. [DOI] [PubMed] [Google Scholar]
- 41.Sampaio JL, Gales AC. 2016. Antimicrobial resistance in Enterobacteriaceae in Brazil: focus on β-lactams and polymyxins. Braz J Microbiol 47:31–37. doi: 10.1016/j.bjm.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yong D, Toleman MA, Bell J, Ritchie B, Pratt R, Ryley H, Walsh TR. 2012. Genetic and biochemical characterization of an acquired subgroup B3 metallo-β-lactamase gene, blaAIM-1, and its unique genetic context in Pseudomonas aeruginosa from Australia. Antimicrob Agents Chemother 56:6154–6159. doi: 10.1128/AAC.05654-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou H, Guo W, Zhang J, Li Y, Zheng P, Zhang H. 2019. Draft genome sequence of a metallo-β-lactamase blaAIM-1-producing Klebsiella pneumoniae ST1916 isolated from a patient with chronic diarrhoea. J Glob Antimicrob Resist 16:165–167. doi: 10.1016/j.jgar.2019.01.010. [DOI] [PubMed] [Google Scholar]
- 44.Boyd DA, Lisboa LF, Rennie R, Zhanel GG, Dingle TC, Mulvey MR. 2019. Identification of a novel metallo-β-lactamase, CAM-1, in clinical Pseudomonas aeruginosa isolates from Canada. J Antimicrob Chemother 74:1563–1567. doi: 10.1093/jac/dkz066. [DOI] [PubMed] [Google Scholar]
- 45.Pfennigwerth N, Lange F, Belmar Campos C, Hentschke M, Gatermann SG, Kaase M. 2017. Genetic and biochemical characterization of HMB-1, a novel subclass B1 metallo-β-lactamase found in a Pseudomonas aeruginosa clinical isolate. J Antimicrob Chemother 72:1068–1073. doi: 10.1093/jac/dkw554. [DOI] [PubMed] [Google Scholar]
- 46.Wendel AF, Brodner AH, Wydra S, Ressina S, Henrich B, Pfeffer K, Toleman MA, Mackenzie CR. 2013. Genetic characterization and emergence of the metallo-β-lactamase GIM-1 in Pseudomonas spp. and Enterobacteriaceae during a long-term outbreak. Antimicrob Agents Chemother 57:5162–5165. doi: 10.1128/AAC.00118-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Casadevall A, Kontoyiannis DP, Robert V. 2019. On the emergence of Candida auris: climate change, azoles, swamps, and birds. mBio 10:e01397-19. doi: 10.1128/mBio.01397-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Narciso AC, Martins WMBS, Almeida LGP, Cayô R, Santos SV, Ramos PL, Lincopan N, Vasconcelos ATR, Gales AC. 2020. Healthcare-associated carbapenem-resistant OXA-72-producing Acinetobacter baumannii of the clonal complex CC79 colonizing migratory and captive aquatic birds in a Brazilian zoo. Sci Total Environ 726:138232. doi: 10.1016/j.scitotenv.2020.138232. [DOI] [PubMed] [Google Scholar]
- 49.Martins WMBS, Narciso AC, Cayô R, Santos SV, Fehlberg LCC, Ramos PL, da Cruz JB, Gales AC. 2018. SPM-1-producing Pseudomonas aeruginosa ST277 clone recovered from microbiota of migratory birds. Diagn Microbiol Infect Dis 90:221–227. doi: 10.1016/j.diagmicrobio.2017.11.003. [DOI] [PubMed] [Google Scholar]
- 50.Chávez-Jacobo VM, Hernández-Ramírez KC, Romo-Rodríguez P, Pérez-Gallardo RV, Campos-García J, Gutiérrez-Corona JF, García-Merinos JP, Meza-Carmen V, Silva-Sánchez J, Ramírez-Díaz MI. 2018. CrpP is a novel ciprofloxacin-modifying enzyme encoded by the Pseudomonas aeruginosa pUM505 plasmid. Antimicrob Agents Chemother 62:e02629-17. doi: 10.1128/AAC.02629-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ortiz de la Rosa JM, Nordmann P, Poirel L. 2020. Pathogenicity genomic island-associated CrpP-like fluoroquinolone-modifying enzymes among Pseudomonas aeruginosa clinical isolates in Europe. Antimicrob Agents Chemother 64:e00489-20. doi: 10.1128/AAC.00489-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Madaha EL, Mienie C, Gonsu HK, Bughe RN, Fonkoua MC, Mbacham WF, Alayande KA, Bezuidenhout CC, Ateba CN. 2020. Whole-genome sequence of multi-drug resistant Pseudomonas aeruginosa strains UY1PSABAL and UY1PSABAL2 isolated from human broncho-alveolar lavage, Yaoundé, Cameroon. PLoS One 15:e0238390. doi: 10.1371/journal.pone.0238390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Founou RC, Founou LL, Allam M, Ismail A, Essack SY. 2020. First report of a clinical multidrug-resistant Pseudomonas aeruginosa ST532 isolate harbouring a ciprofloxacin-modifying enzyme (CrpP) in South Africa. J Glob Antimicrob Resist 22:145–146. doi: 10.1016/j.jgar.2020.05.012. [DOI] [PubMed] [Google Scholar]
- 54.Khan M, Summers S, Rice SA, Stapleton F, Willcox MDP, Subedi D. 2020. Acquired fluoroquinolone resistance genes in corneal isolates of Pseudomonas aeruginosa. Infect Genet Evol 85:104574. doi: 10.1016/j.meegid.2020.104574. [DOI] [PubMed] [Google Scholar]
- 55.Chávez-Jacobo VM, Hernández-Ramírez KC, Silva-Sánchez J, Garza-Ramos U, Barrios-Camacho H, Ortiz-Alvarado R, Cervantes C, Meza-Carmen V, Ramírez-Díaz MI. 2019. Prevalence of the crpP gene conferring decreased ciprofloxacin susceptibility in enterobacterial clinical isolates from Mexican hospitals. J Antimicrob Chemother 74:1253–1259. doi: 10.1093/jac/dky562. [DOI] [PubMed] [Google Scholar]
- 56.World Health Organization. 2020. 10 global health issues to track in 2021. World Health Organization, Geneva, Switzerland. https://www.who.int/news-room/spotlight/10-global-health-issues-to-track-in-2021. [Google Scholar]
- 57.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.World Health Organization. 2018. Ten threats to global health in 2019. World Health Organization, Geneva, Switzerland. https://www.who.int/news-room/spotlight/ten-threats-to-global-health-in-2019. [Google Scholar]
- 59.Menzel P, Ng KL, Krogh A. 2016. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat Commun 7:11257. doi: 10.1038/ncomms11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ye SH, Siddle KJ, Park DJ, Sabeti PC. 2019. Benchmarking metagenomics tools for taxonomic classification. Cell 178:779–794. doi: 10.1016/j.cell.2019.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.scikit-bio Development Team. 2020. scikit-bio: a bioinformatics library for data scientists, students, and developers (version 0.5.5). http://scikit-bio.org.
- 62.McMurdie PJ, Holmes S. 2013. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Galili T. 2015. dendextend: an R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 31:3718–3720. doi: 10.1093/bioinformatics/btv428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bardou P, Mariette J, Escudié F, Djemiel C, Klopp C. 2014. jvenn: an interactive Venn diagram viewer. BMC Bioinformatics 15:293. doi: 10.1186/1471-2105-15-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.World Health Organization. 2017. Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. World Health Organization, Geneva, Switzerland. https://www.who.int/publications/i/item/WHO-EMP-IAU-2017.12. [Google Scholar]
- 66.Rogan W, Gladen B. 1978. Estimating prevalence from results of a screening test. Am J Epidemiol 107:71–76. doi: 10.1093/oxfordjournals.aje.a112510. [DOI] [PubMed] [Google Scholar]
- 67.Miller S, Naccache SN, Samayoa E, Messacar K, Arevalo S, Federman S, Stryke D, Pham E, Fung B, Bolosky WJ, Ingebrigtsen D, Lorizio W, Paff SM, Leake JA, Pesano R, DeBiasi R, Dominguez S, Chiu CY. 2019. Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res 29:831–842. doi: 10.1101/gr.238170.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Govender KN, Street TL, Sanderson ND, Eyre DW. 2021. Metagenomic sequencing as a pathogen-agnostic clinical diagnostic tool for infectious diseases: a systematic review and meta-analysis of diagnostic test accuracy studies. J Clin Microbiol 59:e02916-20. doi: 10.1128/JCM.02916-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. 2017. metaSPAdes: a new versatile metagenomic assembler. Genome Res 27:824–834. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Alcock BP, Raphenya AR, Lau TTY, Tsang KK, Bouchard M, Edalatmand A, Huynh W, Nguyen A-LV, Cheng AA, Liu S, Min SY, Miroshnichenko A, Tran H-K, Werfalli RE, Nasir JA, Oloni M, Speicher DJ, Florescu A, Singh B, Faltyn M, Hernandez-Koutoucheva A, Sharma AN, Bordeleau E, Pawlowski AC, Zubyk HL, Dooley D, Griffiths E, Maguire F, Winsor GL, Beiko RG, Brinkman FSL, Hsiao WWL, Domselaar GV, McArthur AG. 2020. CARD 2020: antibiotic resistome surveillance with the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res 48:D517–D525. doi: 10.1093/nar/gkz935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Csardi G, Nepusz T. 2006. The igraph software package for complex network research. https://www.semanticscholar.org/paper/The-igraph-software-package-for-complex-network-Cs%C3%A1rdi-Nepusz/1d2744b83519657f5f2610698a8ddd177ced4f5c.
- 73.Wickham H. 2016. ggplot2: elegant graphics for data analysis. Springer-Verlag, New York, NY. https://ggplot2.tidyverse.org. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download spectrum.00565-22-s0001.pdf, PDF file, 1.5 MB (1.5MB, pdf)
Supplemental material. Download spectrum.00565-22-s0002.xlsx, XLSX file, 0.2 MB (238.8KB, xlsx)
Data Availability Statement
The data sets supporting the conclusions of this article are available in the NCBI SRA (www.ncbi.nlm.nih.gov/sra) under BioProject accession number PRJNA684454.