


Keywords: AT1-AA blockade, fetal growth restriction, intrauterine growth restriction, microRNAs, 'n7AAc', preeclampsia
Abstract
Placenta ischemia, the initiating event in preeclampsia (PE), is associated with fetal growth restriction. Inhibition of the agonistic autoantibody against the angiotensin type 1 receptor AT1-AA, using an epitope-binding inhibitory peptide ('n7AAc') attenuates increased blood pressure at gestational day (G)19 in the clinically relevant reduced uterine perfusion pressure (RUPP) model of PE. Thus we tested the hypothesis that maternal administration of 'n7AAc' does not transfer to the fetus, improves uterine blood flow and fetal growth, and attenuates elevated placental expression of miRNAs implicated in PE and FGR. Sham or RUPP surgery was performed at G14 with vehicle or 'n7AAc' (144 µg/day) administered via an osmotic pump from G14 to G20. Maternal plasma levels of the peptide on G20 were 16.28 ± 4.4 nM, and fetal plasma levels were significantly lower at 1.15 ± 1.7 nM (P = 0.0007). The uterine artery resistance index was significantly elevated in RUPP (P < 0.0001) but was not increased in 'n7AAc'-RUPP or 'n7AAc'-Sham versus Sham. A significant reduction in fetal weight at G20 in RUPP (P = 0.003) was not observed in 'n7AAc'-RUPP. Yet, percent survival was reduced in RUPP (P = 0.0007) and 'n7AAc'-RUPP (P < 0.0002). Correlation analysis indicated the reduction in percent survival during gestation was specific to the RUPP (r = 0.5342, P = 0.043) and independent of 'n7AAc'. Placental miR-155 (P = 0.0091) and miR-181a (P = 0.0384) expression was upregulated in RUPP at G20 but was not elevated in 'n7AAc'-RUPP. Collectively, our results suggest that maternal administration of 'n7AAc' does not alter fetal growth in the RUPP implicating its potential as a therapeutic for the treatment of PE.
NEW & NOTEWORTHY The seven amino acid inhibitory peptide to the AT1-AA ('n7AAc') has limited transfer to the fetus at gestational day 20, improves uterine blood flow and fetal growth in the reduced uterine perfusion pressure model of preeclampsia (PE), and does not impair fetal survival during gestation in sham-operated or placental ischemic rats. Collectively, these findings suggest that maternal administration of 'n7AAc' as an effective strategy for the treatment of PE is associated with improved outcomes in the fetus.
INTRODUCTION
The renin angiotensin system (RAS) plays a significant role in fluid homeostasis and the long-term control of blood pressure (BP) regulation (1, 2). Overactivation of the RAS system is an essential contributor to the pathophysiology of hypertension (3). Sex influences the expression and activity of the RAS in cardiovascular control (4). The RAS is also altered during pregnancy (5). In normal pregnancy, plasma angiotensin II (ANG II) levels are increased and remain elevated throughout gestation, whereas ANG II is decreased in late gestation in women with preeclampsia (PE) (5, 6). Yet, despite the drop in ANG II in PE, sensitivity to ANG II is increased (7). Administration of an angiotensin-converting enzyme (ACE) inhibitor or an angiotensin II receptor blocker (ARB) is a common standard of care for hypertension and cardiovascular disease (8). However, the RAS is critical for proper fetal development and nephrogenesis (9), and the use of these drugs during pregnancy is contraindicated due to increases in fetal morbidity (10). Other therapeutic options to improve or prevent PE are lacking, and to date, delivery of the fetus remains the only option for intervention in PE (11).
In 1999, using a bioassay, Wallukat et al. (12) reported that an agonistic autoantibody against the angiotensin type 1 receptor (AT1R), the AT1-AA, is elevated in women with PE. Furthermore, this study demonstrated that increased bioactivity of the AT1R by the AT1R agonistic antibody was abolished by a seven amino acid peptide that consists of the second extracellular loop of the AT1 R (13), implicating agonistic activity by the AT1-AA. In 2008, LaMarca and colleagues (14) reported that circulating AT1-AA levels are increased in association with increased BP and fetal growth restriction (FGR) at gestational day 19 (G19) in the well-characterized and clinically relevant reduced uterine perfusion pressure (RUPP) rat model of PE induced by placental insufficiency. In 2009, LaMarca et al. (15) demonstrated that BP is elevated at G19 in pregnant rats treated chronically with AT1-AAs collected from RUPP. A later study by Cunningham et al. (16) reported that chronic infusion of the AT1-AA in late gestation augments ANG II-mediated increases in renal vascular resistance in the pregnant rat, highlighting a role for the AT1-AA as a mediator of increased BP and enhanced ANG II sensitivity in PE. To determine the direct importance of the AT1-AA in the etiology of PE. Lamarca and colleagues (17) developed a novel inhibitory peptide that contains the seven amino acid sequence, AFHYESQ that corresponds to the second extracellular loop of the AT1R. This novel seven amino acid (7aa) peptide consists of NH2-terminal acetyl and COOH-terminal amide caps ('n7AAc') to increase the half-life of the inhibitory peptide and protect against degradation (17). Moreover, this novel inhibitory peptide binds to circulating AT1-AAs inhibiting the binding of the AT1-AA to the AT1R and preventing AT1-AA-induced activation of the AT1R (17). Maternal administration of 'n7AAc' from gestational day 14 (G14) to G19 in the RUPP is associated with a significant reduction in BP, cytolytic natural killer cells, and mitochondrial oxidative stress (18, 19). Collectively, these studies implicate an important role for the AT1-AA as a mediator of increased BP and ANG II sensitivity in the RUPP and indicate the use of the 'n7AAc' as a potential intervention for the treatment and management of PE. Yet, benefit to the mother must also occur without harm to the developing fetus. Moreover, the molecular mechanisms that contribute to the initiation of increased AT1-AAs remain unknown.
Epigenetic mechanisms are involved in the regulation of gene expression during development and in differentiated tissues including the placenta (20, 21). Epigenetic mechanisms include microRNAs (miRNAs) that regulate target mRNA transcripts involved in a variety of regulatory processes (21, 22). In 2007, Pineles et al. (23) reported that a number of placental miRNAs are upregulated in PE including miR-155, miR-182, and miR-210. Recent studies report upregulation of placental and circulating miR-16, miR-181, and miR-222 and indicate that aberrant expression of these miRNAs can serve as potential biomarkers for PE and PE plus FGR (24–28). Ishibashi et al. (29) reported that the expression of miR-210 is upregulated by hypoxia in trophoblastic cell lines. Wang et al. (30) reported that overexpression of miR-16 inhibits proliferation and migration of decidua-derived mesenchymal stem cells. Taken together, these studies indicate that the expression of placental miRNAs responds to hypoxia, a contributing factor to PE, and that changes in placental miRNA expression are associated with disruption of angiogenesis and vascular integrity. Yet, the direct importance of miRNAs in the etiology and pathogenesis of PE and FGR remains controversial. Moreover, very few studies have examined whether placental miRNA expression is altered in well-established, preclinical models of PE reflecting the clinical relevance of miRNAs to the pathophysiology of this disease.
Therefore, one aim of this study was to determine if the novel inhibitory peptide 'n7AAc' crosses the uteroplacental barrier and to determine whether maternal administration of 'n7AAc' is associated with an improvement in uteroplacental perfusion and mitigation of impaired fetal growth in the RUPP model of PE, a model our laboratory has used extensively to study the developmental origins of chronic disease. A second aim was to determine if expression of placental miRNAs implicated in the pathophysiology of PE is associated with placental ischemia and FGR in the RUPP model of PE and whether chronic maternal administration of 'n7AAc' alters expression.
METHODS
Animals
All experimental procedures were conducted in accordance with the National Institute of Health (NIH) guidelines. All animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Mississippi Medical Center. Primiparous, timed pregnant Sprague-Dawley rats (225–250 g) were purchased from Envigo, Inc. (Indianapolis, IN). All animals upon arrival were placed on 2020x diet, a nonsoy-based breeding and lactation-specific diet (Envigo, Inc). Food and water were available to all animals ad libitum, and rats were housed in a temperature-controlled environment (23°C) with a 12:12-h light-dark cycle. All animals undergoing surgical procedures were anesthetized using 2–5% isoflurane by inhalation. Analgesics (Rodent MD’s, Rimadyln, 2 mg/tablet, Bioserve, Flemington, NJ) were provided before, then twice per day for 3 days after sham operation (Sham) or RUPP surgery. At the study end on gestational day 20 (G20), all rats were anesthetized with isoflurane for collection of fetal and placental tissue and maternal blood via abdominal butterfly catheter.
For pharmacokinetic and biodistribution studies, timed pregnant rats at G14 were randomly allocated for treatment with saline or the seven amino acid AT1-AA inhibitory peptide carrying an NH2-terminal rhodamine label [5(6)-carboxytetramethyrhodamine (Genscript)] (n = 4). At G14, 2ML1 osmotic pumps (Alzet, Corp. Cupertino, CA) with saline or the fluorescently labeled seven amino acid AT1-AA inhibitory peptide 7aa were placed in the abdomen of a timed pregnant rat to deliver a dose of 144 µg/day. Blood samples were collected twice a day up to G20. Plasma fluorescence was determined by direct measurement of the fluorescence intensity of 2-μL plasma samples with 535-nm excitation and 580-nm emission using a fluorescence plate reader and a Take3 Microvolume Plate (Biotek). Plasma fluorescence intensity was fit to standard curves generated from known quantities of the labeled peptide to determine molar plasma concentrations at each time point, and data were averaged for all rats. Whole organ and pup ex vivo fluorescence imaging was preformed using the IVIS spectrum (Caliper Life Sciences, Perkin Elmer) with 535-nm excitation, 580-nm emission, and autoexposure in the Animal Imaging Core Facility at the University of Mississippi Medical Center. The mean fluorescence radiant efficiency was measured for maternal organs and fetal pups with placenta attached, after removal from the amniotic sack, using Living Image Software (Caliper). Background autofluorescence from tissues from saline-treated animals was subtracted, and the mean fluorescence radiant efficiency of all organs and fetal pups was fit to a standard curve to determine concentration in the fetal tissue. Data were averaged for all rats.
For maternal administration of 'n7AAc' to study the effect of inhibition of the AT1-AA on placental ischemia and fetal growth, timed pregnant rats at G14 were randomly allocated into four experimental groups: vehicle-treated sham (Sham) (n = 8), vehicle-treated RUPP (RUPP) (n = 10), 'n7AAc'-treated sham ('n7AAc'-Sham) (n = 9), and 'n7AAc'-treated RUPP ('n7AAc'-RUPP) (n = 10). Osmotic pumps (model 2002, Alzet, Corp. Cupertino, CA) were implanted at G14 with saline or the modified capped seven amino acid inhibitory peptide ('n7AAc'; 144 µg/day ip, Thermo Fisher Scientific, Waltham, MA) administered until G20. Uterine artery resistance index (UARI) was performed on gestation day 20 (G20) followed by collection of maternal blood and fetal and placental tissues. Placental location on the uterine horn was recorded, and placental, fetal body, liver, and brain weights were obtained from each viable offspring. The viability of each fetus was determined based on the active breathing and movement of pups at G20 by a trained investigator blinded to dam identity. Placentas were sliced in half and frozen at −80°C for molecular analysis of miRNA expression. Placentas for assessments were selected based on representative average placental weights and median location on the uterine horn, and only placentas from viable pups were used in the analysis. In an additional group, a half dose of 'n7AAc' (½'n7AAc') was utilized to determine efficacy. Timed pregnancy rats were randomly allocated into four experimental groups: vehicle-treated sham (Sham) (n = 4), vehicle-treated RUPP (RUPP) (n = 4), ½'n7AAc'-treated sham ('n7AAc'-Sham) (n = 5), and ½'n7AAc'-treated RUPP ('n7AAc'-RUPP) (n = 6) and were administered 'n7AAc' (72 µg/day ip, Thermo Fisher Scientific, Waltham, MA) from G14 to G20.
Sham or Reduced Uterine Perfusion Pressure in the Pregnant Rat
At G14, animals were randomly allocated to undergo the Sham or the RUPP procedure. In brief, the RUPP procedure was performed by placing a sliver clip (0.203 mm) around the abdominal aorta directly rostral to the iliac bifurcation. A sliver clip was also placed around both branches of the uterine artery (0.100 mm) to prevent a compensatory increase in flow to the uteroplacental units. The Sham procedure involved complete visualization of the uterine horns to ensure both exposure to anesthesia and comparable surgical procedure as the RUPP, all as previously described (31).
Uterine Artery Resistance Index
The uterine artery resistance index (UARI) was measured by Doppler sonography at G20, the morning before euthanasia. Investigators were blinded to dam identity during measurement and analysis. Dams were anesthetized by isoflurane and placed fixed on the platform of the Vevo 3100 unit (FUJIFLIM, Visualsonics, Inc. Toronto, Canada). Doppler velocimetry measurements were taken on uterine arteries from each uterine horn. Waveform images representing the peak systolic velocity (PSV) and end-diastolic velocity (EDV) were captured. Three waveforms were measured from one frame. UARI was calculated using the formula: UARI = (PSV – EDV)/PSV. An average of three measurements (three different frames) was reported as the final UARI for each animal.
Fetal Weight, Placental Weight, Fetal Survival Ratio, and Litter Size
At G14, the number of total implanted embryos was counted per uterine horn and recorded during the Sham or RUPP procedure. At G20, fetal and placental weights of viable pups and litter size were determined for all viable pups. To determine the fetal survival ratio, the number of viable pups at G20 was compared with the number of implanted embryos at G14: fetal survival ratio = (number of viable offspring at G20/number of implanted embryos at G14) × 100.
Placental miRNA Expression Analysis
Total RNA was isolated from rat placenta using TRIzol Reagent (Invitrogen) and was purified using the Pure-link RNA isolation kit (Invitrogen), all according to the manufacturer’s protocol. DNase-treated total RNA (5 ng/μL) was used for the first-strand cDNA synthesis using miRCURY LNA RT Kit (Qiagen) per the manufacturer’s protocol. Quantitative PCR was performed using QuantStudio3 Real-Time PCR cycler (Life Technologies, ThermoFisher Scientific) using a miRCURY LNA SYBR Green PCR Kit (Qiagen) with miRNA expression quantitated using validated miRNA primer-specific HAS-MIR MIRCUY LNA MIR PCR assay kits (cat. nos. YP00206010, YP02119304, YP00204755, P00206081, YP00205702, YP00204333, YP00204306, YP02119303, YP00204551, and YP00204532, Qiagen). The miRNA expression data were normalized with miR-191, and the changes in gene expression were calculated by the ΔΔCt method; data are expressed as fold change relative to control.
Statistical Analysis
Data were analyzed with GraphPad Prism 9 software utilizing the Student’s t test for two-group comparison and two-way ANOVA followed by two-way ANOVA with Dunnett’s multiple comparison test for comparison or Pearson’s test for regression as appropriate. Statistical significance was set at a P value of <0.05 and a 95% confidence interval, and power analysis was set at a minimum of 0.80 to calculate the sample size for each group. Figures were generated with GraphPad Prism software (version 9; GraphPad Software, Inc., La Jolla, CA). Data are presented as means ± SE.
RESULTS
Passage of the AT1-AA Inhibitory Peptide 7aa across the Placental-Fetal Barrier
A steady-state concentration of the AT1-AA inhibitory peptide 7aa, carrying an NH2-terminal rhodamine label [5(6)-carboxytetramethyrhodamine], in the maternal plasma was present on G15 and remained constant until G20 (Fig. 1A). Terminal paired maternal and fetal blood samples confirmed a significantly lower fetal plasma concentration (1.15 ± 1.7 nM) relative to maternal plasma concentration (16.28 ± 4.4 nM) after 6 days of continuous infusion at the therapeutic dose (Fig. 1B, P = 0.0007), equating to a placental transfer percentage of 6.4 ± 9.4%. Moreover, accumulation of the 7aa peptide was visible in the placental tissue of pregnant rats by ex vivo whole organ fluorescence imaging at G20 (Fig. 1C) with significantly decreased levels in the pups as assessed by quantification of the ex vivo fluorescence images (13.89 ± 6.07 nM vs. 3.35 ± 1.59 nM, respectively; P = 0.015).
Figure 1.

Biodistribution of the 7 amino acid inhibitory peptide. A: plasma levels of a NH2-terminal rhodamine-labeled [5(6)-carboxytetramethyrhodamine] seven amino acid (7aa) inhibitory peptide to the angiotensin type II receptor agonistic antibody (AT1-AA) from gestational day 14 (G14) to gestational day 20 (G20) in infused pregnant rats (n = 4). B: maternal and fetal plasma concentration of the rhodamine-labeled AT1-AA 7aa inhibitory peptide from infused pregnant rats (n = 4) euthanized on G20. C: representative ex vivo fluorescence images of the placenta and fetal pups euthanized on G20 from saline or rhodamine-labeled AT1-AA 7aa inhibitory peptide (n = 4)-infused pregnant rats (top, full litter; bottom, close up of representative placenta and fetus) at G20. For A and B, each scatter plot represents means ± SD. P = 0.0007, statistical significance.
Effect of Maternal Intervention with 'n7AAc' and Uterine Artery Resistance Index at G20
UARI, a noninvasive procedure to confirm placental ischemia, was performed at G20. Representative UARI images for each experimental group are shown in (Fig. 2, A–D). The presence of an early diastolic notch in the flow velocity waveform in the RUPP (Fig. 2B, smaller white arrow) was significantly improved in 'n7AAc'-RUPP (Fig. 2D). At G20, UARI was significantly elevated in RUPP compared with Sham (P < 0.0001) (Fig. 2E). However, UARI was not significantly different upon comparison of 'n7AAc'-RUPP to Sham, 'n7AAc'-RUPP to RUPP, or 'n7AAc'-Sham to Sham (Fig. 2E).
Figure 2.

Uterine artery resistance index (UARI) measurement by Doppler ultrasound. UARI was measured in sham-operated and reduced uterine perfusion pressure (RUPP)-pregnant rats treated with saline or the novel seven amino acid inhibitory peptide against the agonistic autoantibody to the angiotensin type II receptor ('n7AAc') on gestational day 20 (G20) by Doppler sonography. A–D: representative UARI figures. Thin white arrow in B represents an early diastolic notch in the flow velocity waveform. Sham, n = 8; RUPP, n = 10; 'n7AAc'-Sham, n = 9; 'n7AAc'-RUPP, n = 10. For E, each scatter plot represents mean ± SE. EDV, end-diastolic velocity; PSV, peak systolic velocity. Two-way ANOVA with Dunnett’s multiple comparison test was used for comparisons. P < 0.05, statistical significance.
Effect of Maternal Intervention with 'n7AAc' on Viable Placental Weight and Fetal Morphometrics
Average viable fetal weight per litter was significantly reduced in viable offspring from RUPP compared with Sham at G20 (P = 0.0036) (Fig. 3A). However, average viable fetal weight per litter did not differ in 'n7AAc'-RUPP compared with Sham (Fig. 3A). Moreover, average viable fetal weight at G20 did not differ in 'n7AAc'-RUPP compared with RUPP or 'n7AAc'-Sham compared with Sham (Fig. 3A). Placental weights for viable offspring did not differ upon comparison of Sham to RUPP offspring regardless of treatment (Fig. 3B); brain (Fig. 3C) and liver (Fig. 3D) weights were also not significantly different.
Figure 3.

Average fetal weight and morphometrics of viable pups at G20. A–D: average fetal weight (A), average placental weight (B), average brain weight (C), and average liver weight (D) of viable pups per litter at gestational day 20 (G20) in sham-operated and reduced uterine perfusion pressure (RUPP) dams treated with saline or the novel seven amino acid inhibitory peptide against the agonistic autoantibody to the angiotensin type II receptor ('n7AAc'). Sham, n = 8; RUPP, n = 10; 'n7AAc'-Sham, n = 9; 'n7AAc'-RUPP, n = 10. Each data point represents an average of viable pups per litter. Scatter plots represent means ± SE. Two-way ANOVA with Dunnett’s multiple comparison test was used for comparisons. P < 0.05, statistical significance.
Effect of Maternal Intervention with 'n7AAc' on Litter Size and Survival Ratio at G20
The number of viable pups at G20 did not differ upon comparison of Sham and RUPP, Sham and 'n7AAc'-Sham, or RUPP and 'n7AAc'-RUPP (Fig. 4A); however, there was a significant reduction in viable pups on the comparison of Sham to 'n7AAc' to RUPP (P = 0.0055) (Fig. 4A). The percent survival between G14 and G19 did not differ between Sham and 'n7AAc'-Sham (Fig. 4B). However, the percent survival was significantly decreased in RUPP (P = 0.007) and in 'n7AAc'-RUPP compared with Sham (P = 0.001) (Fig. 4B), and RUPP compared with 'n7AAc'-RUPP (P = 0.002).
Figure 4.

Number of viable pups and percentage of survival. A: number of viable pups per litter at gestational day 20 (G20). B: percent survival of viable pups [percent survival = number of viable pups at G20/number of viable embryos at gestational day 14 (G14) × 100]. Groups include Sham-operated and reduced uterine perfusion pressure (RUPP) dams treated with saline or the novel seven amino acid inhibitory peptide against the agonistic autoantibody to the angiotensin type II receptor ('n7AAc'). Sham, n = 8; RUPP, n = 10; 'n7AAc'-Sham, n = 9; 'n7AAc'-RUPP, n = 10 on G20. Each data point represents an average of viable pups per litter. Scatter plots represent means ± SE. Two-way ANOVA with Dunnett’s multiple comparison test was used for comparisons. P < 0.05, statistical significance.
Correlation between Fetal Weight of Viable Pups at G20 and Percent Survival
A significant correlation was found between fetal weight of viable pups at G20 and percent survival in 'n7AAc'-RUPP and RUPP (r = 0.5342, P = 0.043) but not in 'n7AAc'-Sham and Sham (r = 0.3097, P = 0.211) (Fig. 5, A and B).
Figure 5.

Correlation of fetal weight of viable pups at gestational day 20 (G20) and percent survival. Relationship between fetal weight of viable offspring at G20 and percent survival at G20. Sham-operated (A) or reduced uterine perfusion pressure (RUPP) (B) dams treated with saline or the novel seven amino acid inhibitory peptide against the agonistic autoantibody to the angiotensin type II receptor ('n7AAc'). Pearson’s correlation analysis: r = 0.3097, P= 0.2110 (A); r = 0.5342, P = 0.0403 (B). P < 0.05, statistical significance.
Efficacy of a Half Dose of 'n7AAc'
At G20, UARI was significantly elevated in RUPP compared with Sham (P < 0.0003) and in ½'n7AAc'-RUPP compared with Sham (P < 0.0026) (Supplemental Fig. S1A: all Supplemental material is available at https://doi.org/10.6084/m9.figshare.20498862.v1). In this study group, pup weight did not differ at G20 (Supplemental Fig. S1B). However, the number of viable pups was significantly decreased in RUPP compared with ½'n7AAc'-Sham (P < 0.023) (Supplemental Fig. 1C); percent survival was only significantly increased in Sham compared with RUPP (P < 0.03) (Supplemental Fig. S1D).
Placental microRNA Expression in the RUPP Model of PE
When compared with Sham, RUPP had a significantly higher expression of placental miR-181a (P = 0.0384) (Fig. 6A). However, placental expression of miR-181a was not significantly elevated in 'n7AAc'-RUPP compared with Sham or RUPP (Fig. 6A), and placental miR-181a levels remained unchanged on comparison of Sham to 'n7AAc'-Sham (Fig. 6A). Placental miR-155 expression was also significantly increased in RUPP compared with Sham (P = 0.0091) (Fig. 6B). Placental expression of miR-155 was also not significantly elevated in 'n7AAc'-RUPP compared with Sham or RUPP (Fig. 6B), and placental miR-151 levels remained unchanged upon comparison of Sham to 'n7AAc'-Sham (Fig. 6A). Placental expression of miR-126 was only significantly different between sham and 'n7AAc'-RUPP (P = 0.0418) (Table 1). However, no difference was observed in placental expression of miR-16, miR-17, miR-210, miR-221, and miR-222 upon comparison of Sham and RUPP regardless of treatment (Table 1).
Figure 6.
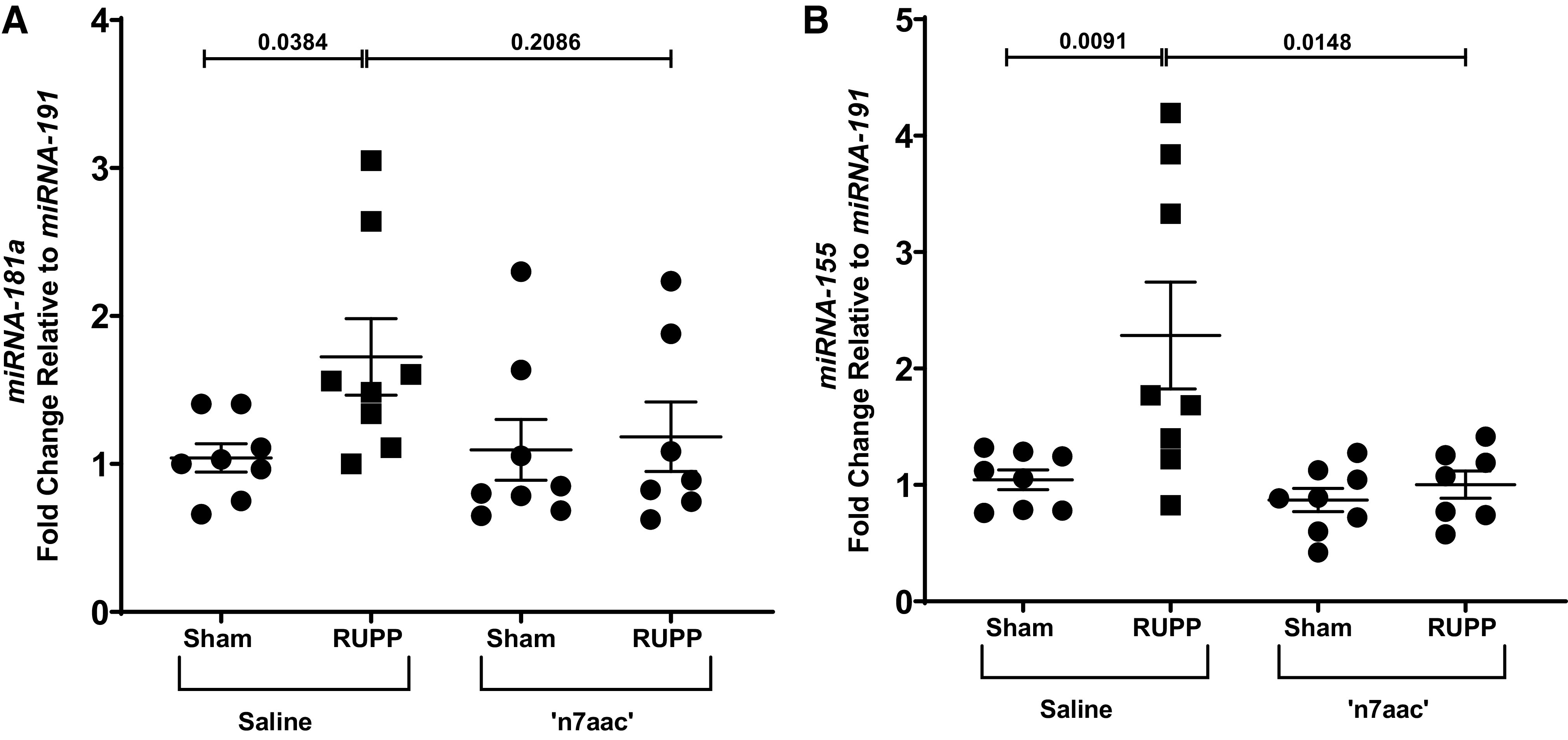
miRNA-181a and miRNA-155 expression. Total RNA extracted from the placenta of viable pups from sham-operated and reduced uterine perfusion pressure (RUPP) dams at gestational day 20 (G20) that were treated with saline or the novel seven amino acid inhibitory peptide against the agonistic autoantibody to the angiotensin type II receptor ('n7AAc') was used to quantify miRNA expression. Sham, n = 8; RUPP, n = 10; 'n7AAc'-Sham, n = 9; 'n7AAc'-RUPP, n = 10. Placental expression levels for miRNA 181a (A) and miRNA-155 (B) were measured using quantitative RT-PCR. Scatter plots represent means ± SE. Two-way ANOVA with Dunnett’s multiple comparison test was used for comparisons. P < 0.05, statistical significance.
Table 1.
Placental miRNA expression in sham-operated and reduced uterine perfusion pressure treated with saline of the novel seven amino acid inhibitory peptide against the agonistic autoantibody to the angiotensin type II receptor
| miRNA/Group | Relative Expression | P Value vs. Sham |
|---|---|---|
| miR-16 | ||
| Sham | 1.041 | |
| RUPP | 1.783 | 0.0853 |
| 'n7AAc'-Sham | 1.403 | 0.5627 |
| 'n7AAc'-RUPP | 1.556 | 0.2627 |
| miR-17 | ||
| Sham | 1.071 | |
| RUPP | 1.679 | 0.3781 |
| 'n7AAc'-Sham | 1.490 | 0.6529 |
| 'n7AAc'-RUPP | 2.049 | 0.0801 |
| miR-126 | ||
| Sham | 1.066 | |
| RUPP | 2.108 | 0.2594 |
| 'n7AAc'-Sham | 1.390 | 0.9187 |
| 'n7AAc'-RUPP | 2.699 | 0.0418* |
| miR-210 | ||
| Sham | 1.048 | |
| RUPP | 1.425 | 0.3007 |
| 'n7AAc'-Sham | 1.214 | 0.8341 |
| 'n7AAc'-RUPP | 1.172 | 0.8561 |
| miR-221 | ||
| Sham | 1.011 | |
| RUPP | 1.616 | 0.2403 |
| 'n7AAc'-Sham | 1.289 | 0.7764 |
| 'n7AAc'-RUPP | 1.446 | 0.5354 |
| miR-222 | ||
| Sham | 1.063 | |
| RUPP | 1.781 | 0.3211 |
| 'n7AAc'-Sham | 1.708 | 0.4030 |
| 'n7AAc'-RUPP | 1.229 | 0.9363 |
Values represent means ± SE; n, number of rats. miRNA, microRNA; 'n7AAc', epitope-binding inhibitory peptide; RUPP, reduced uterine perfusion pressure. Sham, n = 8; RUPP, n = 10; 'n7AAc'-treated sham, n = 9; 'n7AAc'-treated RUPP, n = 10. Two-way ANOVA with Dunnett’s multiple comparison test was used for comparisons. *P < 0.05, statistical significance. P value shown represents the P value vs. Sham.
DISCUSSION
This study tested the hypothesis that administration of the novel inhibitory peptide 'n7AAc' against the AT1-AA during late gestation (G14 until G20) in a rat model of PE improved fetal growth in association with mitigation of increased expression of miRNA-181a and miRNA-155, which are miRNAs associated with FGR. A major discovery from this study demonstrated that the novel seven amino acid inhibitory peptide against the AT1-AA exhibited a low level of passage across the placental-fetal barrier. As reported previously at G19 (17), UARI was significantly increased in RUPP compared with Sham at G20; UARI was not significantly elevated in 'n7AAc'-RUPP compared with Sham or RUPP. The reduction in pup weight in RUPP compared with Sham was abolished in n7aac’-RUPP. Yet, percent survival was reduced in RUPP and 'n7AAc'-RUPP compared with Sham. To determine the significance of the maternal treatment versus the RUPP procedure on percent survival during gestation, correlation analysis revealed that percent survival was significantly correlated to viable fetal weight at G20 and was independent of the maternal treatment with the inhibitory peptide 'n7AAc'. Collectively, these findings indicate that maternal administration of 'n7AAc' is associated with improved uteroplacental perfusion, limited transfer of peptide to the fetus, restored fetal weight, and no impact on fetal survival in late gestation in the RUPP model of PE. However, maternal administration of a half dose of 'n7AAc' was not associated with attenuation of elevated UARI in ½'n7AAc' RUPP. To initiate studies into epigenetic mechanisms involved in the pathophysiology of PE and FGR, we showed that placental miR-155 and miR-181a expression was significantly elevated in RUPP at G20 compared with Sham but did not differ in 'n7AAc'-treated RUPP compared with Sham or RUPP. Therefore, these studies indicate 'n7AAc' as a potential therapeutic for use in PE that will not impair fetal development during gestation.
Currently, there are no effective therapeutic interventions to treat PE (11, 32) or alleviate FGR, implicating the importance of continued drug discovery. Clinically, the cornerstone of therapy in PE remains stabilization of the mother and early delivery of the fetus, which results in preterm birth or low birth weight. Yet, like FGR, preterm birth and low birth weight are also associated with increased cardiorenal and metabolic risk in offspring (33, 34), indicating that PE remains a significant risk factor for chronic disease in the offspring. Any therapeutic intervention administered to the mother must provide benefit to the developing fetus without compromising uteroplacental perfusion, fetal development and viability, and subsequent long-term chronic health in the offspring.
Low placental transfer of a drug including peptides is critical to prevent adverse fetal effects. ACE inhibitors easily cross the placental barrier and are associated with serious fetal complications such as FGR, fetal neonatal hypotension, renal failure, limb deformities, and an increased rate of fetal loss (35, 36). ARBs are also reported to cross the placental barrier impairing renal development in the offspring resulting in a reduction in renal function associated with an increase in BP in the offspring (37–39). Placental passage of magnesium sulfate, a drug used for the treatment of seizures in PE, is virtually 100% (40). Peptides of considerable size (e.g., oxytocin or cyanocobalamin) are also capable of crossing the placental barrier by active transport or passive diffusion. The molecular mass of the 7aa peptide is less than 1 kDa. The low level of placental transfer observed in this study could be due to diffusion across the placental barrier or to active transport (including, possibly, transport via direct binding to the AT1-AA via active shuttling of the antibody by the neonatal Fc receptor). Tocolytic drugs, or small-molecule drugs that prevent preterm labor and immature birth by suppressing uterine contractions, display low rates of placental transfer (41). For example, the degree of transfer of barusiban, a selective oxytocin antagonist, across the placental barrier is limited to less than 5% in the rabbit and 11% in humans (42); atosiban also demonstrates limited passage (43). Whether placental sequestering, binding of tocolytic drugs to placenta tissue (44), limits placental transfer is unknown but indicates another potential mechanism that may prevent the placental passage of the 7aa peptide. Constant levels of 7aa were maintained from G15 to the end of the study at G20 in the maternal circulation, suggesting that a reduction in 7aa in late gestation was not a rate-limiting factor for limited placental transfer. Whether longer exposure to 7aa, and/or the transition for use of this inhibitory peptide in PE will be associated with the increased passage of 7aa remains to be determined. The mechanism and dose dependence of the peptide’s placental transport will be the subject of future studies.
Ultrasonography in the second trimester demonstrated an increase in UARI (45), and the presence of an early diastolic notch in the flow velocity waveform at 24 to 26 wk of gestation (46) is indicative of abnormal placental blood flow and is predictive of subsequent PE and/or FGR. As previously reported at G19 (19), the increase in UARI observed in RUPP at G20 was absent in 'n7AAc'-RUPP in association with improved fetal weight. In this study, the presence of an early diastolic notch in the flow velocity waveform in RUPP was also absent in 'n7AAc'-RUPP implicating improvement in placental blood flow. Yet, UARI was significantly increased in ½'n7AAc'-treated RUPP suggesting that the benefit from this therapeutic approach may be dose dependent. Only studies exploring benefit versus harm at birth and beyond will be able to determine the efficacy of dose on offspring survival and chronic health.
For this study, we explored the potential role of miRNAs that are linked to PE via their association with factors critical to PE and FGR. These included miRNAs that associate with angiogenesis, which is critical for proper placental development; hypoxia, which is critical in the initiation of PE; and immunology, another important mediator in the etiology of PE (47). Based on relevance to PE, we determined if the placental expression of miRNAs relevant to PE was increased in the RUPP and whether maternal administration of ‘n7aaac’ altered their expression.
Nitric oxide (NO), synthesized from l-arginine by endothelial NO synthase (eNOS), plays a pivotal role in the regulation of vascular resistance and hemodynamic changes during normal pregnancy (48, 49) with alterations in the NO pathway associated with PE (50, 51). Placental eNOS expression is downregulated in PE, resulting in placental tissue damage caused by an increase in reactive oxygen species (ROS) (52–54). Many reports suggest that an increase in placental ROS in response to placental ischemia contributes to FGR in PE (55–57). Maternal administration of l-arginine (58), or the antioxidant tempol (59), attenuates increased BP and FGR in the RUPP implicating an important role for a deficiency in NO and increased ROS in the etiology of increased BP and FGR in this model of PE. However, the molecular mechanisms that govern the balance of NO and ROS in PE and in response to placental ischemia are not clear. miRNA-155 targets the 3′-untranslated region of eNOS. Overexpression of miRNA-155 in human umbilical vein endothelial cells and the immortalized trophoblast cell line HTR8/Svneo decreases eNOS expression and NO production, whereas inhibition of miRNA-155 increases eNOS expression and NO production implicating a direct role of miRNA-155 on NO production (53). Moreover, Xu et al. (60) report that miRNA-155 reduces inducible nitric oxide synthase (iNOS) expression by targeting iNOS upstream regulators. Placenta and circulating levels of miRNA-155 are increased in PE (23, 61), suggesting that downregulation of placental eNOS by miRNA-155 may contribute to decreased placental NO production and increased placental ROS production contributing to impaired placental perfusion and fetal growth and the subsequent development of increase cardiovascular risk in the FGR offspring. Our study showed that the placental expression of miRNA-155 was significantly increased at G20 in RUPP. However, placental expression of miRNA-155 was not increased in 'n7AAc'-RUPP compared with Sham suggesting that placental miRNA-155 may be a potential target for future studies directed at the development of novel targets including the 'n7AAc' for improvement of maternal health and fetal development in PE.
Circulating and placental miRNA-181a is upregulated in PE (62, 63). Li et al. showed that upregulation of miR-181a in mesenchymal stem cells (MSCs) blocks activation of the transforming growth factor-β signaling pathway preventing MSC proliferation and enhancing expression of IL-6 (62), a proinflammatory cytokine that is elevated in PE (64). Thus the study by Li and colleagues indicates an important role for miR-181a as a potential indirect mediator of inflammation in PE. In PE, increases in IL-6 are associated with elevated AT1-AAs (65, 66). Circulating IL-6 in the RUPP is also associated with elevated AT1-AAs (67). Maternal AT1-AAs collected from women with PE induce AT1R-specific increases in IL-6 in primary human mesangial cells demonstrating an important role for the AT1-AA as a mediator of increased IL-6 and inflammation in PE (66). Vaka et al. (18) showed that the activity of circulating AT1-AA is decreased in 'n7AAc'-RUPP at G19. In our study, placental miRNA-181a expression was significantly upregulated in the RUPP at G20 but was not elevated in 'n7AAc'-RUPP compared with Sham, suggesting that placental miRNA-181a may be a potential target for future studies directed at the improvement of maternal health and fetal development in PE. Benyo et al. (68) suggested that tissues other than the placenta may be a source of increased circulating levels of IL-6. Yet, in our study, we observed that other miRNAs reported to be upregulated in PE were not upregulated in the placenta from RUPP. These included miR-16, miR-17, miR-126, miR-210, miR-221, and miR-222. These differential findings could be due to the polygenic origins of PE, variation in the gestational age at the time of study in human PE compared with the RUPP model of PE in the rat and, as stated above, the study of circulating versus placental specific miRNAs. Additionally, our study does not rule out the potential importance of other miRNAs in PE. However, this study highlights that placental ischemia alters placental expression of a potential target miRNA, which was reversed by inhibition of the AT1R activation by the AT1-AA. Understanding the importance of miRNAs in the regulation of gene expression in the PE placenta will help to identify novel targets to improve and manage PE, consequently preventing or attenuating FGR and programmed cardiovascular risk.
Limitations to this study include identification of the sex of the fetus at the time of study and further investigation into downstream mediators of identified miRNAs associated with placental ischemia and FGR in the RUPP. An additional limitation is the small number of animals used for the half-dose analysis. Future studies will explore the sex-specific benefit of chronic maternal administration of the novel inhibitory peptide 'n7AAc' on fetal growth and chronic health in the offspring. Another limitation involved the use of an uncapped version of the 7aa inhibitory peptide in the biodistribution study. Capping of 7aa provides stability not observed in the 7aa peptide alone.
Perspectives and Significance
PE is a leading cause of maternal and fetal morbidity and mortality (69). Cardiovascular risk is increased in offspring of women with PE (70–76), demonstrating that PE has a long-term adverse effect on the developing fetus. Yet, no therapeutics are available to treat PE beyond early delivery of the fetus, which is also associated with increased cardiovascular risk in the offspring. Our laboratory has extensively studied the mechanisms that contribute to increased BP in FGR offspring from RUPP dams (31, 77–82). However, the identification of therapeutic approaches to treat PE that limit fetal exposure and sustain fetal development is critical for improving the long-term cardiovascular health of the offspring. This study demonstrated low placental transfer of the novel AT1-AA inhibitory peptide, suggesting a limited risk for off-target effects on the fetus. Improvement in fetal weight was associated with no adverse effects on viability during gestation further suggesting the potential benefit without harm to the fetus. Future studies will determine the effect of this novel treatment strategy during gestation on the long-term cardiovascular health of the offspring. We will also determine the importance of placental miRNAs in the etiology of FGR in PE and their role in the benefit gained by this novel therapeutic intervention and further explore the influence of fetal sex.
SUPPLEMENTAL DATA
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.20498862.v1.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants HL143459 (to B.T.A.) with additional funding provided by NIH Grants HL51971, P20-GM104357, HD067541, and P20-GM121334 (to B.T.A.). J.P.W. was supported by NIH F31HL151180. U.M.A. was supported by NIH T32-HL105324.
DISCLAIMERS
The content of the manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
U.M.A. B.T.A. J.P.W., and G.L.B. conceived and designed research; U.M.A. D.L.H. J.P.W. A.Z.R. K.C., and G.L.B. performed experiments; U.M.A. N.B.O. B.L. B.T.A., and G.L.B. analyzed data; U.M.A. N.B.O. B.L. B.T.A. N.C. D.S. G.L.B., and D.G.R. interpreted results of experiments; U.M.A. B.T.A., and G.L.B. prepared figures; U.M.A. and B.T.A. drafted manuscript; U.M.A. N.B.O. B.L. B.T.A. D.L.H. N.C. J.P.W. A.Z.R. D.S. G.L.B., and D.G.R. edited and revised manuscript; U.M.A. N.B.O. B.L. B.T.A. D.L.H. N.C. J.P.W. A.Z.R. D.S. K.C. G.L.B., and D.G.R. approved final version of manuscript.
REFERENCES
- 1. Te Riet L, van Esch JH, Roks AJ, van den Meiracker AH, Danser AH. Hypertension: renin-angiotensin-aldosterone system alterations. Circ Res 116: 960–975, 2015. doi: 10.1161/CIRCRESAHA.116.303587. [DOI] [PubMed] [Google Scholar]
- 2. Hall JE, Brands MW, Henegar JR. Angiotensin II and long-term arterial pressure regulation: the overriding dominance of the kidney. J Am Soc Nephrol 10, Suppl 12: S258–265, 1999. [PubMed] [Google Scholar]
- 3. Navar LG, Prieto MC, Satou R, Kobori H. Intrarenal angiotensin II and its contribution to the genesis of chronic hypertension. Curr Opin Pharmacol 11: 180–186, 2011. doi: 10.1016/j.coph.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Medina D, Mehay D, Arnold AC. Sex differences in cardiovascular actions of the renin-angiotensin system. Clin Auton Res 30: 393–408, 2020. doi: 10.1007/s10286-020-00720-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brosnihan KB, Merrill DC, Yamaleyeva LM, Chen K, Neves L, Joyner J, Givner C, Lanier K, Moorefield C, Westwood B. Longitudinal study of angiotensin peptides in normal and pre-eclamptic pregnancy. Endocrine 69: 410–419, 2020. doi: 10.1007/s12020-020-02296-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Langer B, Grima M, Coquard C, Bader AM, Schlaeder G, Imbs JL. Plasma active renin, angiotensin I, and angiotensin II during pregnancy and in preeclampsia. Obstet Gynecol 91: 196–202, 1998. doi: 10.1016/S0029-7844(97)00660-1. [DOI] [PubMed] [Google Scholar]
- 7. Gant NF, Daley GL, Chand S, Whalley PJ, MacDonald PC. A study of angiotensin II pressor response throughout primigravid pregnancy. J Clin Invest 52: 2682–2689, 1973. doi: 10.1172/JCI107462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li EC, Heran BS, Wright JM. Angiotensin converting enzyme (ACE) inhibitors versus angiotensin receptor blockers for primary hypertension. Cochrane Database Syst Rev 8: 9096, 2014. doi: 10.1002/14651858.cd009096.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gomez RA. Role of angiotensin in renal vascular development. Kidney Int Suppl 67: S12–S16, 1998. doi: 10.1046/j.1523-1755.1998.06703.x. [DOI] [PubMed] [Google Scholar]
- 10. Moretti ME, Caprara D, Drehuta I, Yeung E, Cheung S, Federico L, Koren G. The fetal safety of angiotensin converting enzyme inhibitors and angiotensin II receptor blockers. Obstet Gynecol Int 2012:658310, 2012. doi: 10.1155/2012/658310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Amaral LM, Wallace K, Owens M, LaMarca B. Pathophysiology and current clinical management of preeclampsia. Curr Hypertens Rep 19: 61, 2017. doi: 10.1007/s11906-017-0757-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, Baur E, Nissen E, Vetter K, Neichel D, Dudenhausen JW, Haller H, Luft FC. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest 103: 945–952, 1999. doi: 10.1172/JCI4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu C, Kellems RE, Xia Y. Inflammation, autoimmunity, and hypertension: the essential role of tissue transglutaminase. Am J Hypertens 30: 756–764, 2017. doi: 10.1093/ajh/hpx027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. LaMarca B, Wallukat G, Llinas M, Herse F, Dechend R, Granger JP. Autoantibodies to the angiotensin type I receptor in response to placental ischemia and tumor necrosis factor alpha in pregnant rats. Hypertension 52: 1168–1172, 2008. doi: 10.1161/HYPERTENSIONAHA.108.120576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. LaMarca B, Parrish M, Ray LF, Murphy SR, Roberts L, Glover P, Wallukat G, Wenzel K, Cockrell K, Martin JN Jr, Ryan MJ, Dechend R. Hypertension in response to autoantibodies to the angiotensin II type I receptor (AT1-AA) in pregnant rats: role of endothelin-1. Hypertension 54: 905–909, 2009. doi: 10.1161/HYPERTENSIONAHA.109.137935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cunningham MW Jr, Williams JM, Amaral L, Usry N, Wallukat G, Dechend R, LaMarca B. Agonistic autoantibodies to the angiotensin II type 1 receptor enhance angiotensin II-induced renal vascular sensitivity and reduce renal function during pregnancy. Hypertension 68: 1308–1313, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cunningham MW Jr, Castillo J, Ibrahim T, Cornelius DC, Campbell N, Amaral L, Vaka VR, Usry N, Williams JM, LaMarca B. AT1-AA (angiotensin II type 1 receptor agonistic autoantibody) blockade prevents preeclamptic symptoms in placental ischemic rats. Hypertension 71: 886–893, 2018. doi: 10.1161/HYPERTENSIONAHA.117.10681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vaka VR, Cunningham MW, Deer E, Franks M, Ibrahim T, Amaral LM, Usry N, Cornelius DC, Dechend R, Wallukat G, LaMarca BD. Blockade of endogenous angiotensin II type I receptor agonistic autoantibody activity improves mitochondrial reactive oxygen species and hypertension in a rat model of preeclampsia. Am J Physiol Regul Integr Comp Physiol 318: R256–R262, 2020. doi: 10.1152/ajpregu.00179.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deer E, Vaka VR, McMaster KM, Wallace K, Cornelius DC, Amaral LM, Cunningham MW, LaMarca B. Vascular endothelial mitochondrial oxidative stress in response to preeclampsia: a role for angiotension II type 1 autoantibodies. Am J Obstet Gynecol MFM 3: 100275, 2021. doi: 10.1016/j.ajogmf.2020.100275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Skinner MK. Role of epigenetics in developmental biology and transgenerational inheritance. Birth Defects Res C Embryo Today 93: 51–55, 2011. doi: 10.1002/bdrc.20199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Apicella C, Ruano CS, Mehats C, Miralles F, Vaiman D. The role of epigenetics in placental development and the etiology of preeclampsia. Int J Mol Sci 20: 2837, 2019. doi: 10.3390/ijms20112837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Butler AA, Webb WM, Lubin FD. Regulatory RNAs and control of epigenetic mechanisms: expectations for cognition and cognitive dysfunction. Epigenomics 8: 135–151, 2016. doi: 10.2217/epi.15.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pineles BL, Romero R, Montenegro D, Tarca AL, Han YM, Kim YM, Draghici S, Espinoza J, Kusanovic JP, Mittal P, Hassan SS, Kim CJ. Distinct subsets of microRNAs are expressed differentially in the human placentas of patients with preeclampsia. Am J Obstet Gynecol 196: 261-e1-6, 2007. doi: 10.1016/j.ajog.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 24. Srinivasan S, Treacy R, Herrero T, Olsen R, Leonardo TR, Zhang X, DeHoff P, To C, Poling LG, Fernando A, Leon-Garcia S, Knepper K, Tran V, Meads M, Tasarz J, Vuppala A, Park S, Laurent CD, Bui T, Cheah PS, Overcash RT, Ramos GA, Roeder H, Ghiran I, Parast M, PAPR Study Consortium, Breakefield XO, Lueth AJ, Rust SR, Dufford MT, Fox AC, Hickok DE, Burchard J, Boniface JJ, Laurent LC. Discovery and verification of extracellular mirna biomarkers for non-invasive prediction of pre-eclampsia in asymptomatic women. Cell Rep Med 1: 100013, 2020. doi: 10.1016/j.xcrm.2020.100013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Awamleh Z, Gloor GB, Han VK. Placental microRNAs in pregnancies with early onset intrauterine growth restriction and preeclampsia: potential impact on gene expression and pathophysiology. BMC Med Genomics 12: 91, 2019. doi: 10.1186/s12920-019-0548-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li L, Huang X, He Z, Xiong Y, Fang Q. miRNA-210-3p regulates trophoblast proliferation and invasiveness through fibroblast growth factor 1 in selective intrauterine growth restriction. J Cell Mol Med 23: 4422–4433, 2019. doi: 10.1111/jcmm.14335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guo L, Tsai SQ, Hardison NE, James AH, Motsinger-Reif AA, Thames B, Stone EA, Deng C, Piedrahita JA. Differentially expressed microRNAs and affected biological pathways revealed by modulated modularity clustering (MMC) analysis of human preeclamptic and IUGR placentas. Placenta 34: 599–605, 2013. doi: 10.1016/j.placenta.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zou Z, He Z, Cai J, Huang L, Zhu H, Luo Y. Potential role of microRNA-424 in regulating ERRgamma to suppress trophoblast proliferation and invasion in fetal growth restriction. Placenta 83: 57–62, 2019. doi: 10.1016/j.placenta.2019.07.001. [DOI] [PubMed] [Google Scholar]
- 29. Ishibashi O, Ohkuchi A, Ali MM, Kurashina R, Luo SS, Ishikawa T, Takizawa T, Hirashima C, Takahashi K, Migita M, Ishikawa G, Yoneyama K, Asakura H, Izumi A, Matsubara S, Takeshita T, Takizawa T. Hydroxysteroid (17-β) dehydrogenase 1 is dysregulated by miR-210 and miR-518c that are aberrantly expressed in preeclamptic placentas: a novel marker for predicting preeclampsia. Hypertension 59: 265–273, 2012. doi: 10.1161/HYPERTENSIONAHA.111.180232. [DOI] [PubMed] [Google Scholar]
- 30. Wang Y, Fan H, Zhao G, Liu D, Du L, Wang Z, Hu Y, Hou Y. miR-16 inhibits the proliferation and angiogenesis-regulating potential of mesenchymal stem cells in severe pre-eclampsia. FEBS J 279: 4510–4524, 2012. doi: 10.1111/febs.12037. [DOI] [PubMed] [Google Scholar]
- 31. Alexander BT. Placental insufficiency leads to development of hypertension in growth-restricted offspring. Hypertension 41: 457–462, 2003. doi: 10.1161/01.HYP.0000053448.95913.3D. [DOI] [PubMed] [Google Scholar]
- 32. Zhang N, Tan J, Yang H, Khalil RA. Comparative risks and predictors of preeclamptic pregnancy in the Eastern, Western and developing world. Biochem Pharmacol 182: 114247, 2020. doi: 10.1016/j.bcp.2020.114247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Davis EF, Lewandowski AJ, Aye C, Williamson W, Boardman H, Huang RC, Mori TA, Newnham J, Beilin LJ, Leeson P. Clinical cardiovascular risk during young adulthood in offspring of hypertensive pregnancies: insights from a 20-year prospective follow-up birth cohort. BMJ Open 5: e008136, 2015. doi: 10.1136/bmjopen-2015-008136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Andraweera PH, Condon B, Collett G, Gentilcore S, Lassi ZS. Cardiovascular risk factors in those born preterm - systematic review and meta-analysis. J Dev Orig Health Dis 12: 539–554, 2021. doi: 10.1017/S2040174420000914. [DOI] [PubMed] [Google Scholar]
- 35. Pryde PG, Sedman AB, Nugent CE, Barr M Jr.. Angiotensin-converting enzyme inhibitor fetopathy. J Am Soc Nephrol 3: 1575–1582, 1993. doi: 10.1681/ASN.V391575. [DOI] [PubMed] [Google Scholar]
- 36. Quan A. Fetopathy associated with exposure to angiotensin converting enzyme inhibitors and angiotensin receptor antagonists. Early Hum Dev 82: 23–28, 2006. doi: 10.1016/j.earlhumdev.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 37. Wolf G. Angiotensin II and tubular development. Nephrol Dial Transplant 17, Suppl 9: 48–51, 2002. doi: 10.1093/ndt/17.suppl_9.48. [DOI] [PubMed] [Google Scholar]
- 38. Spence SG, Allen HL, Cukierski MA, Manson JM, Robertson RT, Eydelloth RS. Defining the susceptible period of developmental toxicity for the AT1-selective angiotensin II receptor antagonist losartan in rats. Teratology 51: 367–382, 1995. doi: 10.1002/tera.1420510603. [DOI] [PubMed] [Google Scholar]
- 39. Woods LL, Rasch R. Perinatal ANG II programs adult blood pressure, glomerular number, and renal function in rats. Am J Physiol Regul Integr Comp Physiol 275: R1593–R1599, 1998. doi: 10.1152/ajpregu.1998.275.5.R1593. [DOI] [PubMed] [Google Scholar]
- 40. Duley L, Neilson JP. Magnesium sulphate and pre-eclampsia. Trial needed to see whether it’s as valuable in pre-eclampsia as in eclampsia. BMJ 319: 3–4, 1999. doi: 10.1136/bmj.319.7201.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Green KW, Key TC, Coen R, Resnik R. The effects of maternally administered magnesium sulfate on the neonate. Am J Obstet Gynecol 146: 29–33, 1983. doi: 10.1016/0002-9378(83)90922-5. [DOI] [PubMed] [Google Scholar]
- 42. Helmer H, Saleh L, Petricevic L, Knofler M, Reinheimer TM. Barusiban, a selective oxytocin receptor antagonist: placental transfer in rabbit, monkey, and human. Biol Reprod 103: 135–143, 2020. doi: 10.1093/biolre/ioaa048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Valenzuela GJ, Craig J, Bernhardt MD, Holland ML. Placental passage of the oxytocin antagonist atosiban. Am J Obstet Gynecol 172: 1304–1306, 1995. doi: 10.1016/0002-9378(95)91497-8. [DOI] [PubMed] [Google Scholar]
- 44. Syme MR, Paxton JW, Keelan JA. Drug transfer and metabolism by the human placenta. Clin Pharmacokinet 43: 487–514, 2004. doi: 10.2165/00003088-200443080-00001. [DOI] [PubMed] [Google Scholar]
- 45. Neto RM, Ramos JG. 3D power Doppler ultrasound in early diagnosis of preeclampsia. Pregnancy Hypertens 6: 10–16, 2016. doi: 10.1016/j.preghy.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 46. Mose JC. The role of maternal & fetal doppler in pre-eclampsia. Pregnancy Hypertens 4: 242, 2014. doi: 10.1016/j.preghy.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 47. Sheikh AM, Small HY, Currie G, Delles C. Systematic review of micro-RNA expression in pre-eclampsia identifies a number of common pathways associated with the disease. PLoS One 11: e0160808, 2016. doi: 10.1371/journal.pone.0160808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu VW, Huang PL. Cardiovascular roles of nitric oxide: a review of insights from nitric oxide synthase gene disrupted mice. Cardiovasc Res 77: 19–29, 2008. doi: 10.1016/j.cardiores.2007.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ortega MA, Romero B, Asunsolo A, Martinez-Vivero C, Sainz F, Bravo C, De León-Luis J, Alvarez-Mon M, Bujan J, Garcia-Honduvilla N. Pregnancy-associated venous insufficiency course with placental and systemic oxidative stress. J Cell Mol Med 24: 4157–4170, 2020. doi: 10.1111/jcmm.15077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shaheen G, Jahan S, Ain QU, Ullah A, Afsar T, Almajwal A, Alam I, Razak S. Placental endothelial nitric oxide synthase expression and role of oxidative stress in susceptibility to preeclampsia in Pakistani women. Mol Genet Genomic Med 8: e1019, 2020. [Erratum in Mol Genet Genomic Med 8: e1191, 2020]. doi: 10.1002/mgg3.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Choi JW, Im MW, Pai SH. Nitric oxide production increases during normal pregnancy and decreases in preeclampsia. Ann Clin Lab Sci 32: 257–263, 2002. [PubMed] [Google Scholar]
- 52. Xu Y, Sui L, Qiu B, Yin X, Liu J, Zhang X. ANXA4 promotes trophoblast invasion via the PI3K/Akt/eNOS pathway in preeclampsia. Am J Physiol Cell Physiol 316: C481–C491, 2019. doi: 10.1152/ajpcell.00404.2018. [DOI] [PubMed] [Google Scholar]
- 53. Kim J, Lee KS, Kim JH, Lee DK, Park M, Choi S, Park W, Kim S, Choi YK, Hwang JY, Choe J, Won MH, Jeoung D, Lee H, Ryoo S, Ha KS, Kwon YG, Kim YM. Aspirin prevents TNF-alpha-induced endothelial cell dysfunction by regulating the NF-kappaB-dependent miR-155/eNOS pathway: role of a miR-155/eNOS axis in preeclampsia. Free Radic Biol Med 104: 185–198, 2017. doi: 10.1016/j.freeradbiomed.2017.01.010. [DOI] [PubMed] [Google Scholar]
- 54. Du L, He F, Kuang L, Tang W, Li Y, Chen D. eNOS/iNOS and endoplasmic reticulum stress-induced apoptosis in the placentas of patients with preeclampsia. J Hum Hypertens 31: 49–55, 2017. doi: 10.1038/jhh.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Aouache R, Biquard L, Vaiman D, Miralles F. Oxidative stress in preeclampsia and placental diseases. Int J Mol Sci 19: 1496, 2018. doi: 10.3390/ijms19051496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. LaMarca B, Amaral LM, Harmon AC, Cornelius DC, Faulkner JL, Cunningham MW Jr.. Placental ischemia and resultant phenotype in animal models of preeclampsia. Curr Hypertens Rep 18: 38, 2016. doi: 10.1007/s11906-016-0633-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schoots MH, Gordijn SJ, Scherjon SA, van Goor H, Hillebrands JL. Oxidative stress in placental pathology. Placenta 69: 153–161, 2018. doi: 10.1016/j.placenta.2018.03.003. [DOI] [PubMed] [Google Scholar]
- 58. Alexander BT, Llinas MT, Kruckeberg WC, Granger JP. L-arginine attenuates hypertension in pregnant rats with reduced uterine perfusion pressure. Hypertension 43: 832–836, 2004. doi: 10.1161/01.HYP.0000119192.32360.a9. [DOI] [PubMed] [Google Scholar]
- 59. Sedeek MH, Llinas MT, Drummond H, Fortepiani L, Abram SR, Alexander BT, Reckelhoff JF, Granger JP. Role of reactive oxygen species in endothelin-induced hypertension. Hypertension 42: 806–810, 2003. doi: 10.1161/01.HYP.0000084372.91932.BA. [DOI] [PubMed] [Google Scholar]
- 60. Xu C, Ren G, Cao G, Chen Q, Shou P, Zheng C, Du L, Han X, Jiang M, Yang Q, Lin L, Wang G, Yu P, Zhang X, Cao W, Brewer G, Wang Y, Shi Y. miR-155 regulates immune modulatory properties of mesenchymal stem cells by targeting TAK1-binding protein 2. J Biol Chem 288: 11074–11079, 2013. doi: 10.1074/jbc.M112.414862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cheng W, Liu T, Jiang F, Liu C, Zhao X, Gao Y, Wang H, Liu Z. microRNA-155 regulates angiotensin II type 1 receptor expression in umbilical vein endothelial cells from severely pre-eclamptic pregnant women. Int J Mol Med 27: 393–399, 2011. doi: 10.3892/ijmm.2011.598. [DOI] [PubMed] [Google Scholar]
- 62. Lui L, Wang Y, Fan H, Zhao X, Liu D, Hu Y, Kidd AR, Bao J, Hou Y. MicroRNA-181a regulates local immune balance by inhibiting proliferation and immunosuppressive properties of mesenchymal stem cells. Stem Cells 30: 1756–1770, 2012. doi: 10.1002/stem.1156. [DOI] [PubMed] [Google Scholar]
- 63. Wu L, Zhou H, Lin H, Qi J, Zhu C, Gao Z, Wang H. Circulating microRNAs are elevated in plasma from severe preeclamptic pregnancies. Reproduction 143: 389–397, 2012. doi: 10.1530/REP-11-0304. [DOI] [PubMed] [Google Scholar]
- 64. Gadonski G, LaMarca BB, Sullivan E, Bennett W, Chandler D, Granger JP. Hypertension produced by reductions in uterine perfusion in the pregnant rat: role of interleukin 6. Hypertension 48: 711–716, 2006. doi: 10.1161/01.HYP.0000238442.33463.94. [DOI] [PubMed] [Google Scholar]
- 65. Lamarca B, Speed J, Ray LF, Cockrell K, Wallukat G, Dechend R, Granger J. Hypertension in response to IL-6 during pregnancy: role of AT1-receptor activation. Int J Interferon Cytokine Mediat Res 2011: 65–70, 2011. doi: 10.2147/IJICMR.S22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bobst SM, Day MC, Gilstrap LC 3rd, Xia Y, Kellems RE. Maternal autoantibodies from preeclamptic patients activate angiotensin receptors on human mesangial cells and induce interleukin-6 and plasminogen activator inhibitor-1 secretion. Am J Hypertens 18: 330–336, 2005. doi: 10.1016/j.amjhyper.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 67. Cornelius DC, Amaral LM, Harmon A, Wallace K, Thomas AJ, Campbell N, Scott J, Herse F, Haase N, Moseley J, Wallukat G, Dechend R, LaMarca B. An increased population of regulatory T cells improves the pathophysiology of placental ischemia in a rat model of preeclampsia. Am J Physiol Regul Integr Comp Physiol 309: R884–R891, 2015. doi: 10.1152/ajpregu.00154.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Benyo DF, A Smarason A, Redman CW, Sims C, Conrad KP. Expression of inflammatory cytokines in placentas from women with preeclampsia. J Clin Endocrinol Metab 86: 2505–2512, 2001. doi: 10.1210/jcem.86.6.7585. [DOI] [PubMed] [Google Scholar]
- 69. Rana S, Lemoine E, Granger JP, Karumanchi SA. Preeclampsia: pathophysiology, challenges, and perspectives. Circ Res 124: 1094–1112, 2019. [Erratum in Circ Res 126: e8, 2020]. doi: 10.1161/CIRCRESAHA.118.313276. [DOI] [PubMed] [Google Scholar]
- 70. Davis EF, Lazdam M, Lewandowski AJ, Worton SA, Kelly B, Kenworthy Y, Adwani S, Wilkinson AR, McCormick K, Sargent I, Redman C, Leeson P. Cardiovascular risk factors in children and young adults born to preeclamptic pregnancies: a systematic review. Pediatrics 129: e1552-61, 2012. doi: 10.1542/peds.2011-3093. [DOI] [PubMed] [Google Scholar]
- 71. Staley JR, Bradley J, Silverwood RJ, Howe LD, Tilling K, Lawlor DA, Macdonald-Wallis C. Associations of blood pressure in pregnancy with offspring blood pressure trajectories during childhood and adolescence: findings from a prospective study. J Am Heart Assoc 4: e001422, 2015. doi: 10.1161/JAHA.114.001422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Geelhoed JJ, Fraser A, Tilling K, Benfield L, Davey Smith G, Sattar N, Nelson SM, Lawlor DA. Preeclampsia and gestational hypertension are associated with childhood blood pressure independently of family adiposity measures: the Avon Longitudinal Study of Parents and Children. Circulation 122: 1192–1199, 2010. doi: 10.1161/CIRCULATIONAHA.110.936674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lazdam M, de la Horra A, Diesch J, Kenworthy Y, Davis E, Lewandowski AJ, Szmigielski C, Shore A, Mackillop L, Kharbanda R, Alp N, Redman C, Kelly B, Leeson P. Unique blood pressure characteristics in mother and offspring after early onset preeclampsia. Hypertension 60: 1338–1345, 2012. doi: 10.1161/HYPERTENSIONAHA.112.198366. [DOI] [PubMed] [Google Scholar]
- 74. Seidman DS, Laor A, Gale R, Stevenson DK, Mashiach S, Danon YL. Pre-eclampsia and offspring’s blood pressure, cognitive ability and physical development at 17-years-of-age. Br J Obstet Gynaecol 98: 1009–1014, 1991. doi: 10.1111/j.1471-0528.1991.tb15339.x. [DOI] [PubMed] [Google Scholar]
- 75. Alsnes IV, Vatten LJ, Fraser A, Bjorngaard JH, Rich-Edwards J, Romundstad PR, Asvold BO. Hypertension in pregnancy and offspring cardiovascular risk in young adulthood: prospective and sibling studies in the HUNT Study (Nord-Trondelag Health Study) in Norway. Hypertension 69: 591–598, 2017. doi: 10.1161/HYPERTENSIONAHA.116.08414. [DOI] [PubMed] [Google Scholar]
- 76. Tenhola S, Rahiala E, Halonen P, Vanninen E, Voutilainen R. Maternal preeclampsia predicts elevated blood pressure in 12-year-old children: evaluation by ambulatory blood pressure monitoring. Pediatr Res 59: 320–324, 2006. doi: 10.1203/01.pdr.0000196734.54473.e3. [DOI] [PubMed] [Google Scholar]
- 77. Payne JA, Alexander BT, Khalil RA. Reduced endothelial vascular relaxation in growth-restricted offspring of pregnant rats with reduced uterine perfusion. Hypertension 42: 768–774, 2003. doi: 10.1161/01.HYP.0000084990.88147.0C. [DOI] [PubMed] [Google Scholar]
- 78. Alexander BT, Hendon AE, Ferril G, Dwyer TM. Renal denervation abolishes hypertension in low-birth-weight offspring from pregnant rats with reduced uterine perfusion. Hypertension 45: 754–758, 2005. doi: 10.1161/01.HYP.0000153319.20340.2a. [DOI] [PubMed] [Google Scholar]
- 79. Grigore D, Ojeda NB, Robertson EB, Dawson AS, Huffman CA, Bourassa EA, Speth RC, Brosnihan KB, Alexander BT. Placental insufficiency results in temporal alterations in the renin angiotensin system in male hypertensive growth restricted offspring. Am J Physiol Regul Integr Comp Physiol 293: R804–811, 2007. doi: 10.1152/ajpregu.00725.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Dasinger JH, Intapad S, Backstrom MA, Carter AJ, Alexander BT. Intrauterine growth restriction programs an accelerated age-related increase in cardiovascular risk in male offspring. Am J Physiol Renal Physiol 311: F312–F319, 2016. doi: 10.1152/ajprenal.00123.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ojeda NB, Hennington BS, Williamson DT, Hill ML, Betson NE, Sartori-Valinotti JC, Reckelhoff JF, Royals TP, Alexander BT. Oxidative stress contributes to sex differences in blood pressure in adult growth-restricted offspring. Hypertension 60: 114–122, 2012. doi: 10.1161/HYPERTENSIONAHA.112.192955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Coats LE, Bamrick-Fernandez DR, Ariatti AM, Bakrania BA, Rawls AZ, Ojeda NB, Alexander BT. Stimulation of soluble guanylate cyclase diminishes intrauterine growth restriction in a rat model of placental ischemia. Am J Physiol Regul Integr Comp Physiol 320: R149–R161, 2021. doi: 10.1152/ajpregu.00234.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.20498862.v1.