

Keywords: biglycan, cancer, decorin, Met, proteoglycan
Abstract
Decorin, a small leucine-rich proteoglycan with multiple biological functions, is known to evoke autophagy and mitophagy in both endothelial and cancer cells. Here, we investigated the effects of soluble decorin on mitochondrial homeostasis using live cell imaging and ex vivo angiogenic assays. We discovered that decorin triggers mitochondrial depolarization in triple-negative breast carcinoma, HeLa, and endothelial cells. This bioactivity was mediated by the protein core in a time- and dose-dependent manner and was specific for decorin insofar as biglycan, the closest homolog, failed to trigger depolarization. Mechanistically, we found that the bioactivity of decorin to promote depolarization required the MET receptor and its tyrosine kinase. Moreover, two mitochondrial interacting proteins, mitostatin and mitofusin 2, were essential for downstream decorin effects. Finally, we found that decorin relied on the canonical mitochondrial permeability transition pore to trigger tumor cell mitochondrial depolarization. Collectively, our study implicates decorin as a soluble outside-in regulator of mitochondrial dynamics.
INTRODUCTION
Soluble decorin was the first small leucine-rich proteoglycan (SLRP) to be shown to inhibit cell growth in Chinese hamster ovary cells (1), primarily by binding to and blocking transforming growth factor beta (TGFβ) (2). However, we reasoned that TGFβ could not be the main reason for growth inhibition in cancer cells as most of the primary and metastatic carcinomas do not respond to nor are dependent on TGFβ. Thus, we investigated the role of decorin in cancer growth and found that de novo decorin gene expression suppressed the malignant phenotype in human colon cancer cells (3). In addition, we found that ectopic expression of decorin protein core caused a generalized growth suppression in neoplastic cells of various histogenetic origins (4), and this bioactivity required endogenous p21 (4, 5), an inhibitor of cyclin-dependent kinases. As decorin is a secreted proteoglycan, we reasoned that cell-surface receptors should be involved in mediating its outside-in signaling cascade (6). We discovered that decorin bound with relatively high affinity to epidermal growth factor receptor (EGFR) in squamous carcinoma cells overexpressing this receptor (7, 8). In general, soluble decorin or its protein core in a monomeric form (9) binds to a narrow region of EGFR partially overlapping with but distinct from the epidermal growth factor (EGF)-binding epitope (10). This interaction leads to a transient EGFR activation that includes intracellular mobilization of Ca2+ (11), followed by a protracted downregulation of the receptor (12) that is internalized via caveosomes and ultimately degraded in lysosomes (13). The discovery of decorin/EGFR interaction has opened this field of research and has led to the discovery of multiple receptor tyrosine kinase interacting with decorin including ErbB2 (14), Met (15, 16), insulin-like growth factor receptor 1 (IGF-IR) (17, 18), vascular endothelial growth factor receptor 2 (VEGFR2) (19, 20) and platelet-derived growth factor receptor (PDGFR) (21). Indeed, the protein interacting network of decorin is quite large, now encompassing over 75 proteins, including growth factors, receptor tyrosine kinases (RTKs), and immune receptors (22). There is strong genetic evidence supporting a role for decorin as an oncosuppressive proteoglycan. For example, cooperative action of germ-line targeting mutations of decorin and p53 accelerates lymphoma tumorigenesis (23). In line with these observations, a genetic background lacking decorin promotes hepatic carcinogenesis (24), favors spontaneous colon tumor formation (25–27), and promotes epithelial-mesenchymal transition and colon cancer metastasis (28). Some of these activities could be pharmacologically targeted (29). Notably, decorin has been implicated in inflammation and fibrosis (30, 31) and in the pathophysiology of several organs and tissues including the heart (32, 33), bone (34), tendon and ligaments (35, 36), cervical remodeling (37), ocular pathology (38), osteoarthritis and cartilage homeostasis (39, 40), and matrix remodeling (41).
Another important function of decorin is its ability to evoke autophagy in endothelial cells (20, 42, 43). Indeed, decorin itself is an autophagy-inducible proteoglycan and is required for proper in vivo autophagy (44). Decorin-evoked signaling through VEGFR2 leads to activation of the energy sensor kinase AMP-activated protein kinase (AMPK) (45), and also to the autophagic degradation of vascular endothelial growth factor A (VEGFA) (46), a mechanism additional to those previously found in support of decorin angiostatic activity (47–51). Notably, biglycan, the closest homolog of decorin, does not cause autophagy in endothelial cells (20); however, it evokes autophagy in macrophages via a novel CD44/Toll-like receptor 4 (TLR4) signaling axis (52, 53). Moreover, biglycan-triggered TLR2 and TLR4 signaling exacerbates the pathophysiology of ischemic acute renal injury (54).
As a mechanistic corollary to decorin to evoked endothelial cell autophagy (42, 51, 55), we found that decorin evokes tumor cell mitochondrial autophagy (mitophagy) (56, 57). This process relies on decorin binding to the Met receptor and initiating a complex signaling cascade involving peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α)-mediated stabilization of mitostatin mRNA and protein (43). Mitostatin has been characterized as a putative tumor suppressor (58, 59) and is required for decorin-evoked tumor cell mitophagy (56, 60). Indeed, loss of mitostatin prevented decorin from inducing mitophagy including loss of mitochondrial fragmentation, mtDNA loss, and voltage-dependent anion channel 1 (VDAC1) clearance. As the most direct result of future mitophagy, mitochondrial depolarization is critical (61–63). Thus, we sought to mechanistically decipher the role of decorin in coordinating this early event as an initiating signal for downstream mitophagy in cancer.
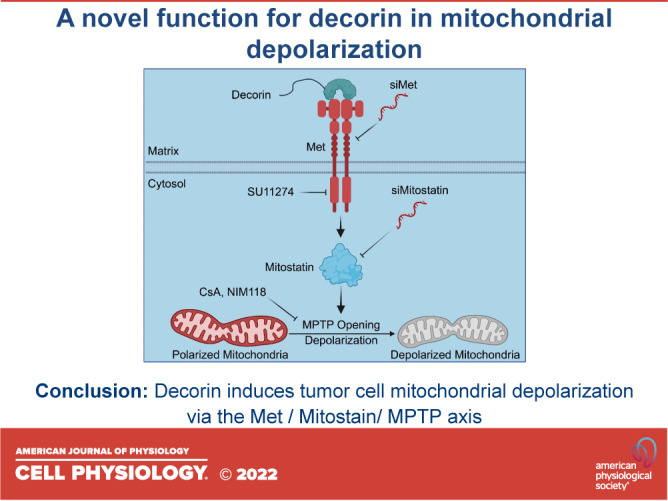
Here, we report on a novel signaling pathway that mechanistically connects decorin-dependent RTK signaling to mitochondrial depolarization in various cancer and vascular models. These findings redefine the stage at which mitostatin functions in the mitophagic cascade and broaden our understanding on the extent of the effect decorin has on tumor cell mitophagy.
METHODS
Cells, Chemicals, and General Reagents
The human triple-negative breast cancer cell line MDA-MB-231 and cervical cancer cell line HeLa were obtained from American Type Cell Culture. Cells were grown at 37°C in a 5% CO2 atmosphere in Dulbecco’s modified Eagle’s medium (DMEM) containing 4.5 g/L glucose, l-glutamine, and sodium pyruvate from Life Technologies and supplemented with 5% fetal bovine serum (FBS) from ThermoFisher Scientific and 100 U/mL penicillin/streptomycin from Life Technologies. Primary human umbilical vein endothelial cells (HUVECs) were obtained from Lifeline Cell Technology, grown in basal media supplemented with the VascuLife EnGS LifeFactors Kit (from Lifeline Cell Technology), and used within the first five passages. HUVECs were also grown at 37°C in a 5% CO2 atmosphere. Dimethyl sulfoxide (DMSO) was obtained from ThermoFisher Scientific. Rabbit monoclonal antibodies against mitofusin 2 (64) and GAPDH (65) were obtained from Cell Signaling Technologies. The rabbit polyclonal antibody against Met (66) and α-Tubulin (67) were obtained from Santa Cruz Biotechnology. The rabbit anti-mitostatin affinity-purified antibody was described elsewhere (68). The validation for the custom mitostatin antibody comes from our siRNA experiments (please consult the mitostatin immunoblots in Figs. 4A and 6, A and D). Briefly, relative to siScramble controls, use of a siRNA specific for mitostatin results in the depletion of mitostatin immunoreactivity, thereby validating the mitostatin antibody. Moreover, this same biochemical check is valid for the commercially available mitofusin 2 antibody. Please consult the immunoblots in Figs. 5A and 6, A and D. Recombinant human hepatocyte growth factor (HGF), NIM811, AG1478, and SU11274 were purchased from Sigma. The mitochondrial dyes JC-1, JC-10, TMRE, and TMRM were purchased from Abcam. The mitochondrial dyes used here are cell-permeant, positively charged dyes that readily accumulate in active mitochondria due to their relative negative charge. Depolarized or inactive mitochondria have decreased membrane potential and fail to sequester TMRE and TMRM, resulting in a lower intensity signal. For JC-1 and JC-10, this results in a color change from red (J-aggregates sequestered within the mitochondria) to green (J-monomers) following depolarization. The plasmids encoding myc-Met and 3×FLAG-mitostatin were purchased from Origene. DMSO was used as vehicle where appropriate for all experiments. All primary antibodies were used at 1:1,000 diluted in 1% BSA-TBST except for GAPDH, which was used at 1:10,000. Secondary antibodies for chemiluminescence were used at 1:5,000 in the same buffer as above. The SuperSignal West Pico-enhanced chemiluminescence substrate was purchased from ThermoFisher Scientific.
Figure 4.

Mitostatin, a key downstream effector of decorin, is required for depolarization. A: representative immunoblots confirming the efficacy of mitostatin depletion in 231 (left) or HeLa (right) via siRNA with densitometry values below the loading control. Amounts of siRNA for siScr and siMitostatin were kept constant throughout (100 pM). B: gallery of live cell images depicting 231 (top series of images) or HeLa (bottom series of images) cells following transient knockdown of mitostatin, challenged with decorin (6 h, 100 nM), and then stained with JC-10. C and D: quantification of the JC-10 signal in 231 (C) or HeLa (D) from data in B. E and F: quantification of the resultant TMRE (E) or TMRM (F) signal in 231 cells as per the same experimental conditions as in B. G and H: quantification of the resultant TMRE (G) or TMRM (H) signal in HeLa cells as per the same experimental conditions as in B. I and J: mitostatin rescue experiment done in 231 (I) or HeLa (J) and stained with TMRE as per the conditions listed. Vehicle (pcDNA3.1) and active plasmid (FLAG-mitostatin) were used at equimolar amounts (4 µg). For immunoblots (A), GAPDH served as the loading control. PBS served as vehicle for all experiments. All data presented are expressed as n = 4 (unless otherwise noted) independent biological replicates. Data are expressed as means ± SE. Statistics calculated via one-way ANOVA (***P < 0.001).
Figure 6.

Mitostatin and mitofusin 2 are in the same pathway and required for depolarization. A: representative immunoblots confirming the efficacy of mitostatin and/or mitofusin 2 depletion via siRNA in 231 cells in combination with decorin (6 h, 100 nM) as noted. B: gallery of live cell images depicting JC-10 staining in 231 cells under a variety of experimental conditions as labeled. C: quantification of the JC-10 signal in B. D: representative immunoblots confirming the efficacy of mitostatin and/or mitofusin 2 depletion via siRNA in HeLa cells in combination with decorin (6 h, 100 nM) as noted. E: gallery of live cell images depicting JC-10 staining in HeLa cells under a variety of experimental conditions as indicated. F: quantification of the JC-10 signal in E. For immunoblots (A and D), GAPDH served as the loading control. PBS served as vehicle for all experiments. All data presented are expressed as n = 4 independent biological replicates. Data are expressed as means ± SE. Statistics calculated via one-way ANOVA (***P < 0.001).
Transient DNA Expression and RNAi-Mediated Silencing
We transiently transfected the appropriate cell line with empty vector (pcDNA3.1) or plasmids encoding myc-Met or 3xFLAG-mitostatin using Lipofectamine 2000 (Life Technologies) in OptiMEM reduced serum media (Gibco). Expression was verified by immunoblot using target-specific antibodies. A description of DNA transfection has been described (46). Cells were transiently transfected using Lipofectamine RNAiMAX (Life Technologies) mixed with siRNA against Met, mitostatin, and/or mitofusin 2 that were purchased from Santa Cruz Biotechnology. The siRNA used represents a validated cocktail of 3–5 targeting oligonucleotides for each gene of interest. Scrambled siRNA (sc-37007, Santa Cruz Biotechnology) served as a control for all siRNA experiments presented herein. The full protocol is described in Ref. 69.
Mitochondrial Membrane Potential
At least four individual assays were performed in the appropriate cell line and experimental condition using the mitochondrial dyes JC-1, JC-10, TMRE, or TMRM. Cells were treated as per the experimental conditions described herein. The TMRE and TMRM signals were normalized to FCCP (carbonylcyanide 4-triflouromethoxy phenylhydrazone) for 10 min following treatment to completely collapse the mitochondrial membrane potential. Each chamber or well of cells was incubated with the appropriate dye for 20–30 min. The cells were washed three times with PBS and imaged live using a Leica DM5500B microscope. All the images were procured using the same exposure, gain, and intensity.
Mitochondrial Membrane Potential Assay in Ex Vivo Aortic Rings
All of the animal experiments were performed according to the Guide for Care and Use of Laboratory Animals and the Institutional Animal Care and Use Committee of Thomas Jefferson University.
We initially coated 35-mm plastic bottom plates with a 3D collagen type I, rat tail (Corning) solution. After polymerization of the collagen, we selected male or female C57BL/6 mice between 1 and 3 mo of age and anesthetized the mice with isoflurane. We dissected the mice by removing organs to expose the aorta. We carefully removed the aorta from the spine with a scalpel under a stereoscopic microscope. After trimming excess fat, we then sectioned the aorta into rings that were ∼1 mm in width and placed these rings into the 35-mm dishes, 4 rings per dish. We then covered the rings with a top layer of collagen and VascuLife Basal Medium (Lifeline). Minimal sprouting should be apparent after 2 days, and after 4 days, sprouting should be both dense and widespread.
Mitochondrial depolarization was visualized via a Mitochondrial Membrane Potential Assay Kit (Abcam), which used TMRE (tetramethylrhodamine, ethyl ester) to label active mitochondria with a red fluorescence. TMRE staining was done for 30 min at a concentration of 150 nM (the volume of the collagen was included in the total volume, ∼2.5 mL). For the treatment groups, FCCP was used at 10 μM with a 15-min pretreatment to TMRE staining. Decorin treatment was done at 1 μM with overnight pretreatment to TMRE staining. This higher concentration of decorin was used to adjust for the loss of decorin bound to collagen. Four images of each ring were taken at ×10 magnification and 200 ms exposure time. A Nikon Eclipse Ts2 inverted microscope was used for image capture. Fluorescent intensity was measured using ImageJ, in which the background was subtracted with the Subtract background function with a rolling ball radius of 700 pixels. The mean gray value of sprouting was measured by splitting the channel by color and using the red color channel. Then we selected Analyze → Set Measurements → Mean gray value as well as Limit to threshold. By adjusting the threshold via Image → Adjust → Threshold to highlight the sprouting without picking up any background, we measured the fluorescence intensity of areas with dense sprouting. The FCCP negative control was used to subtract from vehicle- and decorin-treated values as a background fluorescence.
Statistical Analysis
Immunoblots were quantified by scanning densitometry using Scion Image software (NIH). Graphs were generated using Sigma Stat 3.10. Experiments with three or more comparison groups were subjected to one-way ANOVA followed by a Bonferroni post hoc test using the Systat Package of SigmaPlot 13.0. Differences were considered significant at two-sided P < 0.05.
RESULTS
Decorin Triggers Mitochondrial Depolarization in Tumor Cells
To investigate in-depth role of soluble decorin on mitochondrial homeostasis, we used live cell imaging with fluorescence microscopy and TMRE in triple-negative MDA-MB-231 breast carcinoma cells (231 hereafter). We discovered that decorin triggered depolarization in a dose- (Fig. 1A) and time-dependent manner (Fig. 1C). Starting with as little as 1 nM of decorin (Fig. 1, A and B), mitochondria began depolarizing as demonstrated by a visible loss of the TMRE signal. Mitochondrial depolarization progressed with increasing concentrations of decorin until 100 nM where decorin achieved maximal depolarization and was stable up to 1 µM (Fig. 1, A and B). Therefore, unless otherwise noted, we used decorin at 100 nM. Next, we evaluated the temporal effects of decorin and found that as early as 1 h, decorin already significantly decreased the TMRE signal, indicating depolarization (Fig. 1, C and D). Depolarization became more pronounced over time, became maximal at 6 h, and was sustained for up to 24 h (Fig. 1, C and D). Based on these data, unless otherwise noted, we exposed the breast carcinoma cells to soluble decorin for 6 h.
Figure 1.

Decorin triggers depolarization in tumor cells. A: gallery of live cell images depicting 231 cells treated with increasing concentrations of decorin, as noted, for a fixed time (6 h) and stained with tetramethyl rhodamine ethyl ester (TMRE; 150 nM). B: quantification of the normalized TMRE signal as in A. C: gallery of live cell images depicting 231 cells treated over time with decorin, as noted, at a fixed concentration of decorin (100 nM) and stained with TMRE (150 nM). D: quantification of the normalized TMRE signal as seen in C. E: identical experiment as shown in A, but stained with JC-10 (1 µM). F: quantification of the JC-10 signal as seen in E. G: identical experiment as shown in C, but stained with JC-10. H: quantification of the JC-10 signal as seen in G. I and J: gallery of images depicting the depolarization recovery experiments done in 231 (I) or HeLa (J) and stained with JC-10. K and L: quantification of the JC-10 signal from the recovery experiments for 231 (K) or HeLa (L). PBS served as vehicle for all experiments. All data presented are reflective of several (n = 4) independent biological replicates. Data are expressed as means ± SE. Statistics calculated via one-way ANOVA (***P < 0.001; **P < 0.01).
To corroborate these findings, we performed additional experiments using the cationic ratiometric dye, JC-1 (70). We corroborated the temporal dynamics of decorin to induce mitochondrial depolarization over time with this dye (Supplemental Fig. S1, A and B). We then switched to JC-10 for further validation of our model and kinetics of decorin-evoked ΔΨm. JC-10 is a much more soluble and more robust variant of the JC-1 ratiometric dye to measure mitochondrial permeability (71, 72). Using this more vigorous approach, we discovered very comparable effects. Decorin evoked ΔΨm in 231 cells with as little as 1 nM (Fig. 1, E and F) in as quickly as 2 h (Fig. 1, G and H). In both sets of experiments, depolarization was progressive and was maintained up to 1 µM and 24 h, respectively. By using HeLa cells and JC-10, we found that decorin triggered depolarization at 1 nM (Supplemental Fig. S1, C and D) and within 2 h (Supplemental Fig. S1, E and F). Similarly, these effects were progressive and maintained to the endpoints of 1 µM and 24 h (Supplemental Fig. S1, C–F), respectively.
It is important to note that we used highly purified decorin proteoglycan expressed in 293-EBNA cells as a His6-protein using a Celligen Plus bioreactor and affinity purified on a Ni-NTA column (73). The purity of our decorin preparations was assessed via SDS-PAGE. As colloidal Coomassie has a detection threshold of ∼5-ng protein, and as no additional bands were detectable at 5 µg (Supplemental Fig. S2A), we conclude that our protein-core preparations are >99.9% pure (<5 ng/5,000 ng).
We next evaluated whether decorin required the covalently attached dermatan/chondroitin sulfate glycosaminoglycan chain (74) to trigger depolarization in tumor cells. We found that in either 231 (Supplemental Fig. S2, B–E) or HeLa cells (Supplemental Fig. S2, F–I), in the presence of TMRE or JC-10, the GAG chain was dispensable for mitochondrial depolarization. Quantification of the TMRE or JC-10 signals for either 231 or HeLa revealed no statistically significant differences in the magnitude of depolarization between decorin proteoglycan or decorin protein core. These data are congruent with past and recent findings where the GAG chain is dispensable for most of the biological effects of decorin (46, 75).
To establish specificity of action, we next evaluated the effects of biglycan on triggering depolarization. Biglycan is a class I SLRP and closest relative of decorin with >65% homology at the protein level (76). Biglycan was synthesized in a manner similar to decorin and was found to be contaminant-free via colloidal Coomassie staining (Supplemental Fig. S2J). Interestingly, biglycan had no discernable effect on depolarization in either 231 (Supplemental Fig. S2K) or HeLa (Supplemental Fig. S2L) when assessed via JC-10 (Supplemental Fig. S2M).
To test whether decorin effects were species-specific, we used mouse E0771 cells. These are triple-negative, basal-like tumor cells (77, 78) with a molecular profile similar to that of 231 breast carcinoma cells. Importantly, the E0711 cells responded to exogenous human decorin by triggering depolarization via a significant loss of TMRE signal, and this effect was similar to FCCP (Supplemental Fig. S2N). Collectively, our results demonstrate that soluble decorin specifically triggers depolarization in both human and murine breast carcinoma cells, in a time- and dose-dependent manner. Moreover, this bioactivity is specific for this Class I SLRP as biglycan failed to trigger depolarization.
Decorin-Dependent Mitochondrial Depolarization is Reversible in Carcinoma Cells
It is widely accepted that decorin binds to cell-surface RTKs and triggers internalization with subsequent degradation of the decorin/RTK complex within lysosomes (43, 79, 80). This process is the cornerstone of the mechanism of action for tumorigenic suppression via decorin for protracted biological effects (20, 45, 46, 56, 81). Empirical experimental evidence clearly demonstrates decorin-mediated internalization of RTK complexes following high-affinity binding for EGFR (13) and Met (16). However, it remains unknown whether removal of soluble decorin would lead to a reversal of the downstream biological effects. Therefore, we treated 231 or HeLa cells with decorin (100 nM) for 4 h to trigger depolarization (prewash, Fig. 1, I and J) as determined by JC-10 staining (Fig. 1, K and L). We then washed the cells and monitored depolarization over time in the absence of exogenous decorin. Strikingly, within 30 min of removing the decorin-laden media (e.g., postwash), mitochondrial membrane potential began to recover in both 231 (Fig. 1, I and K) and HeLa cells (Fig. 1, J and L). Within 1 h and by 2 h, mitochondrial membrane potential fully recovered and stabilized in 231 and HeLa cells (Fig. 1, I–L). Therefore, these data indicate that the continued presence of decorin is required to sustain protracted depolarization. Furthermore, these data began to substantiate that once internalized, the putative decorin/RTK complex is not capable of intracellular signaling, consistent with a wholly degradative role for decorin to silence oncogenic signals and curb tumorigenesis.
Decorin Requires the Met Receptor Kinase to Trigger Mitochondrial Depolarization in Carcinoma Cells
Given that decorin predominantly uses RTKs (60) to transduce its biological signals (51, 82), we assessed the requirement of Met or EGFR. As measured by JC-10 staining, decorin triggered depolarization in 231 (Fig. 2, B and I) and HeLa cells (Fig. 2, K and R) vis-à-vis their respective vehicles (Fig. 2, A and J). Treatment with a SU11274, a tyrosine kinase inhibitor (TKi) of the Met tyrosine kinase (83), alone had no discernable effect on depolarization at basal conditions (Fig. 2, C, L, I, and R); however, in combination with decorin SU11274 significantly blocked (Fig. 2, D and M) decorin-triggered depolarization (Fig. 2, I and R). Next, using an EGFR-specific inhibitor, AG1478 (84, 85), we discovered that blocking EGFR under basal conditions had no effect on depolarization (Fig. 2, E, N, I, and R) akin to the Met Tki. Similarly, combination treatment of AG1478 and decorin results in a significant abrogation of mitochondrial depolarization in 231 Fig. 2, F and I) and HeLa cells (Fig. 2, O and R).
Figure 2.

Decorin requires the Met receptor kinase to trigger depolarization in carcinoma cells. A–H: gallery of live cell images of 231 cells treated in various combinations of decorin (100 nM), SU11274 (1 µM), or AG1478 (1 µM) and stained with JC-10. I: quantification of the JC-10 signal from A–H. J–Q: identical experiment as in A–H performed in HeLa cells. R: quantification of the JC-10 signal from J–Q. S and T: identical experiments as performed above with combinations of decorin and TKi but stained with TMRE in 231 (S) or HeLa cells (T). DMSO served as vehicle to account for SU11274 and AG1478 for all experiments. All data presented are reflective of several (n = 4) independent biological replicates. Data presented are expressed as means ± SE. Statistics calculated via one-way ANOVA (***P < 0.001).
Next, we sought to rule out any possible contributions from other RTKs that decorin might use to trigger depolarization, as decorin uses Met and EGFR to achieve the majority of antitumorigenic outcomes. Therefore, we used both inhibitors either in combination with or without decorin. We found that treatment with both inhibitors alone had no effect on basal mitochondrial membrane potential in 231 (Fig. 2, G and I) or HeLa (Fig. 2, P and R). Adding both inhibitors, in the presence of decorin, blocked depolarization in a magnitude akin to single-agent alone (Fig. 2, H, I, Q, and R). Furthermore, there was no observed compensation or redundant receptor for decorin to evoke ΔΨm in 231 or HeLa cells.
We repeated these experiments in 231 cells with JC-1 and obtained identical results. Decorin-mediated depolarization was significantly blocked with single-agent Tki with no significant change in magnitude or additional redundancy found with dual TKi (Supplemental Fig. S3, A and B).
We used TMRE to validate our findings with the ratiometric dyes (e.g., JC-10, JC-1). Importantly, we found highly similar patterns using TMRE. Blocking either Met or EGFR individually significantly blocked decorin-mediated depolarization in 231 (Fig. 2S) and HeLa (Fig. 2T). Lastly, dual agent in the presence of decorin recapitulated the concept that there was no further change in the magnitude of depolarization when both receptors were blocked and no additional redundancy conveyed by a different receptor complex (Fig. 2, S and T).
Collectively, our findings indicate that decorin used Met and/or EGFR to evoke an intracellular signaling cascade that leads to a reversible but profound mitochondrial depolarization.
The Met Receptor is Required for Decorin-Mediated Tumor Cell Mitochondrial Depolarization
We decided to sharpen our studies on only the Met receptor as it represents the primary receptor (15) for a multitude of antioncogenic activities imbued by decorin (86) against cancer. To this end, we used siRNA and significantly depleted Met from 231 (approximately eightfold, Fig. 3A) and HeLa cells (∼3.6 folds, Fig. 3B). Using JC-1, we discovered that physical depletion of Met completely blocked the ability of decorin to induce depolarization in both cell types (Fig. 3, C–E).
Figure 3.

The Met receptor is required for decorin-dependent tumor cell depolarization. Representative immunoblots confirming the efficacy of Met depletion in 231 (A) or HeLa (B) cells via siRNA with densitometry values below the loading control. Amounts of siRNA for siScr and siMet were kept constant throughout (100 pM). C: gallery of live cell images depicting 231 or HeLa cells following transient knockdown of the Met receptor, challenged with decorin (6 h, 100 nM), and then stained with either JC-1 (top) or JC-10 (bottom). D and E: quantification of the JC-1 signal in 231 (D) or HeLa (E) from data in C. F and G: quantification of the JC-10 signal in 231 (F) or HeLa (G) from data in C. H and I: identical Met knockdown experiment as in C, but stained with TMRE in 231 (H) or HeLa (I). J and K: quantification of the resultant TMRE signal in 231 (J) or HeLa (K) after being stimulated with a combination, as noted, of decorin (100 nM) or HGF (50 ng/mL) for 6 h. L and M: Met rescue experiment done in 231 (L) or HeLa (M) and stained with TMRE as per the conditions listed. Vehicle (pcDNA3.1) and active plasmid (myc-Met) were used at equimolar amounts (4 µg). For immunoblots A and B, α-tubulin served as the loading control. PBS served as vehicle for all experiments. All data presented are reflective of several (n = 4, unless otherwise noted) independent biological replicates. Data are expressed as means ± SE. Statistics calculated via one-way ANOVA (***P < 0.001).
Next, we complemented the JC-1 data with a more robust mitochondria dye (JC-10) and found similar patterns for 231 and HeLa (Fig. 3, C, F and G). Finally, we used TMRE as an additional verification of decorin-driven depolarization. We found that loss of decorin and staining with TMRE revealed a similar mechanism whereby depolarization was abrogated following Met silencing in 231 (Fig. 3H) and HeLa cells (Fig. 3I). Interestingly, similar to TKi alone, depletion of Met itself had no observable effects on depolarization in neither 231 nor HeLa cells.
Next, we evaluated whether decorin could interfere with Met signaling conveyed by hepatocyte growth factor (HGF), the endogenous agonist of Met (87), widely considered to be pro-oncogenic and proangiogenic (88–94). Mechanistically, decorin displaces HGF from binding the Met ectodomain (15) resulting in decreased breast cancer metastasis (95) and decreased angiogenesis (48). Moreover, prooncogenic growth factors can increase mitochondrial polarization (96–98). Assaying with TMRE, we found that HGF was indeed capable of mitochondrial hyperpolarization in 231 (Fig. 3J) and HeLa (Fig. 3K) cells. As expected, decorin-induced depolarization, and strikingly, was able to significantly blunt HGF-evoked mitochondrial hyperpolarization (Fig. 3, J and K) in both cell types. These data reinforce the role of decorin as a critical antagonist of HGF in cancer.
We wanted to further establish the sufficiency of Met-dependent signaling to induce mitochondrial depolarization in response to decorin. Therefore, we performed rescue experiments by reexpressing siRNA-resistant MET cDNA (which has an N-terminal myc-tag) in 231 and HeLa cells followed by TMRE staining. We reproduced our results insofar as that loss of Met via siRNA prevented decorin-induced mitochondrial depolarization (Fig. 3, L and M) in the presence of empty vector (pcDNA3.1). Notably, supertransfecting 231 or HeLa with myc-Met alone did not have an adverse effect on basal mitochondria membrane potential, as the resting magnitude is comparable with experiments reported herein.
We observed two important effects after reintroducing myc-Met into cells that have been depleted of the receptor. First, overexpressing Met in cells that only received scramble siRNA enhanced the effect of decorin-mediated depolarization in 231 and HeLa (compare condition 2 with condition 5, Fig. 3, L and M). Second, restoring decorin in Met-depleted cells significantly restored the effect of decorin to induce depolarization (compare condition 4 with condition 8, Fig. 3, L and M). Akin to TKi and siRNA, overexpression of Met had negligible effects on basal mitochondrial membrane potential (Fig. 3, L and M).
Collectively, we have demonstrated a mechanistic dependency of Met to convey signal required for decorin-mediated depolarization. Moreover, Met is both necessary and sufficient for this process and complements the pharmacological approach underscoring the importance of the decorin/Met interaction in cancer cells.
Mitostatin, a Key Downstream Effector of Decorin, is Required for Mitochondrial Depolarization
We next turned our attention to potential downstream effectors that could be critical for decorin-mediated depolarization. A key modulator of decorin bioactivity is mitostatin. We reasoned that mitostatin may have a functional role in the early stages of mitophagy (63, 99) and potentially in decorin-mediated depolarization. To this end, we used siRNA and significantly depleted mitostatin in both 231 and HeLa (Fig. 4A) cells. Using JC-10, we discovered that loss of mitostatin severely abrogated the ability of decorin to evoke ΔΨm in either cell type (P < 0.001 for both, Fig. 4, B–D). These data were recapitulated using JC-1 in 231 whereupon mitostatin silencing significantly prevented decorin-mediated depolarization (Supplemental Fig. S4, A and B).
We then sought to further authenticate our findings. Using 231 and HeLa and different mitochondrial membrane potential dyes [TMRE and TMRM (100)], we found a strikingly similar pattern in decorin-mediated depolarization following mitostatin depletion. In 231 cells loaded with either TMRE (Fig. 4E) or TMRM (Fig. 4F), loss of mitostatin significantly impaired decorin-mediated depolarization. In HeLa cells, we found an identical pattern where cells loaded with TMRE (Fig. 4G) or TMRM (Fig. 4H) following mitostatin silencing significantly abrogated decorin-mediated depolarization. These sets of data corroborate the JC-10 and JC-1 staining above and illustrate a critical role of mitostatin in conveying information for decorin-mediated depolarization. Interestingly, in all cases independent of staining method or cell type, loss of mitostatin alone did not perturb basal mitochondrial membrane potential (Fig. 4B). These data indicate that mitostatin is an active molecule in transducing signals given the appropriate stimulus (e.g., decorin) in tumor cells.
As we did with Met (cf. Fig. 3, L and M), we wanted to establish the sufficiency of mitostatin-dependent depolarization in the presence or absence of decorin. Thus, we executed rescue experiments by reexpressing siRNA-resistant mitostatin cDNA (which harbors an N-terminal 3xFLAG tag) in 231 and HeLa cells followed by TMRE staining. We were able to reproduce our depolarization results to the degree that loss of mitostatin prevented depolarization in cells supertransfected with equal amounts of empty plasmid (231, Fig. 4I; HeLa, Fig. 4J).
Surprisingly, supertransfecting 231 or HeLa cells with 3xFLAG-mitostatin alone was sufficient to evoke significant mitochondrial depolarization (fifth and seventh condition, Fig. 4, I and J). Depolarization occurred without the use of any external stimulant (e.g., decorin) and seemingly independent of whether endogenous mitostatin was present. Concurrent treatment with decorin and 3x-FLAG-mitostatin had a marginally combined effect on depolarization (sixth and eighth condition, Fig. 4, I and J). These data substantiate and confirm that mitostatin alone is capable of mediating depolarization in tumor cells.
Collectively, these data provide a mechanistic basis for the role of mitostatin in acting at the earliest step in the mitophagic process by evoking mitochondrial depolarization. Moreover, decorin relies on mitostatin for depolarization to occur.
Mitostatin and Mitofusin 2 Are in the Same Pathway and Required for Mitochondrial Depolarization
A key binding partner for mitostatin was identified as mitofusin 2 (101) as a regulator of mitochondria-associated membranes. Mitofusin 2 is a critical mediator of overall mitochondrial health (61, 101), ER/mitochondrial calcium homeostasis (102, 103), and mitophagy (104–106). The fact that mitostatin binds mitofusin 2 (a finding we recapitulated in 231 and HeLa cells, data not shown) provided a rationale to test two hypotheses: 1) does decorin-mediated depolarization also require mitofusin 2? and 2) do mitofusin 2 and mitostatin reside in the same pathway to promote depolarization?
To begin testing these possibilities, we depleted mitofusin 2 from 231 and HeLa (Fig. 5A) cells via siRNA. Following authentication that mitofusin 2 was substantially depleted, we loaded 231 and HeLa with JC-10. We discovered that loss of mitofusin 2 phenocopied the loss of mitostatin insofar as blocking the ability of decorin to mediate depolarization in 231 and HeLa cells (Fig. 5B) (Fig. 5, D and F). We also performed this experiment with JC-1 and found that it operates identically (Supplemental Fig. S4C). Loss of mitofusin 2 blocked decorin when stained with JC-1 (Supplemental Fig. S4D). Next, we loaded 231 or HeLa cells with TMRE and found a very similar response that loss of mitofusin 2 rendered decorin unable to mediate depolarization (Fig. 5, C and D). These results mechanistically link decorin to a critical and well-established mitochondrial mediator.
Figure 5.

Decorin requires mitofusin 2 for depolarization in tumor cells. A: representative immunoblots confirming the efficacy of mitofusin 2 depletion in 231 (left) or HeLa (right) via siRNA with densitometry values below the loading control. Amounts of siRNA for siScr and siMitostatin were kept constant throughout (100 pM). B: gallery of live cell images depicting 231 (top series of images) or HeLa (bottom series of images) cells following transient knockdown of mitostatin, challenged with decorin (6 h, 100 nM) and then stained with JC-10. C and D: quantification of the JC-10 signal in 231 (C) or HeLa (D) from data in B. E and F: quantification of the resultant TMRE signal in 231 (E) or HeLa (F) cells as per the same experimental conditions as in B. For immunoblots (A), GAPDH served as the loading control. PBS served as vehicle for all experiments. All data presented are expressed as n = 4 (unless otherwise noted) independent biological replicates. Data are expressed as means ± SE. Statistics calculated via one-way ANOVA.
Next, we tested whether mitostatin and mitofusin, two physically interacting proteins (107) (and unpublished results), could be in the same signaling pathway that leads to depolarization. We approached this by performing single- or double-agent siRNA against mitostatin, mitofusin 2, or both in the presence or absence of decorin. First, we documented proper and significant knockdown of both proteins in 231 cells (Fig. 6A and Supplemental Fig. S5, A and B). Importantly, we found that decorin increased mitostatin levels in 231 cells (Fig. 6A and Supplemental Fig. S5A) in line with our previous findings (56, 58). We found that decorin significantly increased mitofusin 2 levels (Fig. 6A and Supplemental Fig. S5B). Using JC-10, we independently and fully reproduced our earlier findings (cf. Figs. 4 and 5) that loss of either mitostatin or mitofusin 2 alone blocked decorin-mediated depolarization (Fig. 6, B and C). Further loss of single agent alone without decorin had no adverse effect on basal depolarization (Fig. 6, B and C). Interestingly, siRNA-mediated depletion of both mitostatin and mitofusin 2 did not perturb basal mitochondrial membrane potential (Fig. 6, B and C). Notably, dual depletion of mitostatin and mitofusin 2 in the presence of decorin did not result in either an attenuated blockade or a more severe blockade of decorin activity (Fig. 6, B and C). Indeed, the magnitude of preventing decorin from driving depolarization was visually and statistically similar to either single agent knockdown alone (Fig. 6, B and C).
We then fully recapitulated these critical set of experiments in HeLa cells. We determined that single and dual knockdown of mitostatin and/or mitofusin 2 was robust and significant (Fig. 6D and Supplemental Fig. S5, C and D). We found that akin to 231 cells, decorin increases mitostatin and mitofusin 2 levels (Fig. 6D and Supplemental Fig. S5 C and D). Importantly, in a manner that mimics the situation in 231 cells, we independently reproduced that loss of either mitostatin or mitofusin 2 singly abrogated the prodepolarization effect of decorin (Fig. 6, E and F). Moreover, dual knockdown of mitostatin and mitofusin 2 did not overtly affect basal mitochondrial membrane potential, which is in line with single knockdown as we have demonstrated in Fig. 6, E and F. Dual depletion of mitostatin and mitofusin 2 did not alter the effects of decorin (Fig. 6, E and F). Collectively, these data implicate mitofusin 2 as a key mediator of decorin-evoked ΔΨm in two different cancer cell lines. Mechanistically, it appears that mitostatin and mitofusin 2 are functionally within the same signaling pathway as there were neither additive nor synergistic effects when single versus dual knockdowns were performed. Because we used only one concentration of decorin (100 nM) and one time point (6 h) for these studies, as empirically determined (cf. Fig. 1), it is possible that differential combinations of concentrations and time points may promote a different response.
Decorin Relies on the Canonical mPTP to Evoke Tumor Cell Mitochondrial Depolarization
Canonical mitochondrial depolarization proceeds via a specialized pore widely known as the mitochondrial permeability transition pore (mPTP) (108). The formation of the highly conductive and long-lasting mPTP is often the result of a damaging stimulus or tightly regulated signal such as an overload of Ca2+ (108) or reactive oxygen species (ROS) (109). Indeed, long-term opening of the mPTP leads to oxidative phosphorylation (OXPHOS) decoupling, mitochondrial injury, and cellular death (109). Although the molecular composition of the mPTP is still hotly contested (110, 111), there remains critical physiological relevance for its function (112).
One vital molecular component that aids in the opening of the mPTP is cyclophilin D (CypD) (110). Inhibition of cyclophilin D with cyclosporine A (CsA) (113, 114) provides a useful tool to investigate the molecular function of the mPTP (115). We, therefore, investigated whether decorin uses the canonical mPTP to mediate depolarization in tumor cells.
To this end, we treated 231 cells with decorin in the presence or absence of CsA. As before, decorin potently mediated depolarization when stained with JC-10 relative to vehicle-treated controls (Fig. 7, A and B) as quantified (Fig. 7J). The magnitude of depolarization was similar to that of FCCP (Fig. 7, C and J). Strikingly, we found that application of CsA in combination with decorin significantly blocked decorin-mediated depolarization (Fig. 7, E and J). Interestingly, CsA alone did not significantly affect mitochondrial membrane polarization (Fig. 7, D and J). Consistent, however, with the mechanism of action for an uncoupler such as FCCP (116), CsA did not block the ability of FCCP to induce mitochondrial depolarization (Fig. 7, F and J). These data are important as it provides a point of inflection between the RTK (and mPTP)-dependent mechanism used by decorin and that of a potent protonophore.
Figure 7.

Decorin relies on the canonical mPTP to trigger tumor cell depolarization. A–I: gallery of live cell images depicting 231 cells treated in a variety of combinations with decorin (100 nM), carbonylcyanide 4-triflouromethoxy phenylhydrazone (FCCP; 1 µM), cyclosporin A (5 µM), or NIM811 (5 µM) and stained with JC-10. J: quantification of the JC-10 signal as in (A–I). K–S: gallery of live cell images depicting an identical experiment as in (A–I) carried out in HeLa cells. T: quantification of the JC-10 signal as in K–S. U and V: quantification of the resultant tetramethyl rhodamine ethyl ester (TMRE) signal under identical conditions (e.g., A–I; K–S) performed in 231 (U) or HeLa (V). Dimethyl sulfoxide (DMSO) served as vehicle for all experiments to account for cyclosporin A and NIM811. All data presented are expressed as n = 4 independent biological replicates. Data are expressed as means ± SE. Statistics calculated via one-way ANOVA (***P < 0.001).
Next, we used a more specific CypD inhibitor with less potential off-target effects, NIM811 (117). NIM811 functions similarly to CypD in which it inhibits the transition of the mPTP but does not bind to calcineurin, making it nonimmunosupressive (117). Akin to CsA, NIM811 alone had no effects on basal mitochondrial membrane polarization (Fig. 7, G and J), but did significantly blunt the ability of decorin to trigger depolarization (Fig. 7, H and J). Moreover, NIM811 did not have any effect on FCCP-mediated uncoupling and resulting depolarization (Fig. 7, I and J).
To provide additional mechanistic robustness to this mechanism, we recapitulated these experiments in HeLa cells and stained with JC-10. As shown, decorin evoked a strong depolarization relative to vehicle treatments (Fig. 7, K, L, and T) that was comparable in responsiveness to FCCP (Fig. 7, M and T). Similar to 231, CsA had no substantive effect on resting mitochondrial membrane potential (Fig. 7, N and T), but potently blocked decorin-evoked ΔΨm (Fig. 7, O and T). Furthermore, CsA was functionally unable to block FCCP (Fig. 7, P and T). We then applied NIM811 and treated it in combination with decorin or FCCP in HeLa cells. We observed similar results. NIM811 had no effect on basal mitochondrial membrane potential (Fig. 7, Q and T), but completely abrogated decorin-mediated depolarization (Fig. 7, R and T). As for CsA, NIM811 did not impair FCCP from evoking potent depolarization in HeLa cells (Fig. 7, S and T).
We next verified these findings with TMRE in 231 (Fig. 7U) and HeLa cells (Fig. 7V) insofar as that CsA and NIM811 potently block decorin-mediated depolarization in both cell types without compromising FCCP-mediated depolarization. These independently obtained data corroborate our JC-10 staining.
Collectively, these data substantiate a mechanistic role for the mPTP in significantly and specifically mediating decorin-mediated depolarization. This mechanism is different from that of a well-established uncoupler (FCCP) in triggering depolarization. Therefore, an undefined signaling pathway involving communication from a cell-surface RTK exists that serves as the link between decorin and mitochondrial depolarization.
Decorin Triggers Mitochondrial Depolarization in Vascular Endothelial Cells and in Ex Vivo Aortic Ring Assays
In an effort to validate our profound mitochondrial depolarization findings, we used an ex vivo approach vis-à-vis aortic ring assays that have recently been improved upon in our laboratory (118). The aortic rings are carefully harvested and embedded in a 3D collagen type I matrix.
Therefore, TMRE staining in aortic rings isolated from C57BL/6 mice revealed that an 18-h decorin treatment (1 µM) significantly induced depolarization in the sprouts relative to vehicle conditions (Fig. 8, A and B). Interestingly decorin used at half this concentration (0.5 µM) had no discernable effect on sprout mitochondrial depolarization (Fig. 8, A and B).
Figure 8.

Decorin induces depolarization in ex vivo aortic ring assays in 3D collagen I and in vascular endothelial cells. A: serialized aortic rings treated overnight with the indicated concentrations of decorin followed by tetramethyl rhodamine ethyl ester (TMRE) staining (150 nM). Live bright field images were acquired to demonstrate the presence of sprouts from the aortic rings. All images were captured with a 200-ms exposure at ×10 magnification. Bar ∼ 90 µm. B: quantification of the TMRE signal from A. C: live cell images of human umbilical vein endothelial cells (HUVECs) treated with decorin (6 h, 200 nM) and stained with JC-1. D: quantification of the resultant TMRE signal in HUVEC following decorin or CCCP (1 µM). PBS served as control vehicle for all experiments. All data presented are reflective of several (n = 4, unless otherwise indicated here or in the text) independent biological replicates. Data are expressed as means ± SE. Statistics calculated via one-way ANOVA (***P < 0.001).
Considering the cellular similarity between aortic endothelial cells (comprising the aortic ring sprouts) and HUVECs, which are a target of decorin (20, 46), we assayed depolarization in this system. Akin to the robust depolarization we discovered in tumor cells and aortic rings, decorin triggered significant depolarization in HUVECs as per JC-1 staining (Fig. 8C) and TMRE (Fig. 8D).
Taken together, these data provide the first evidence that decorin can evoke mitochondrial depolarization in an ex vivo aortic ring system as well as in HUVECs. These data expand on the already exciting mechanism of RTK-mediated mitochondrial depolarization seen in tumor cells.
DISCUSSION
One of the earliest, if not the earliest, step in the mitophagic cascade is the rapid and widespread loss of the mitochondrial membrane potential (57, 63). We previously identified that decorin evokes mitochondrial depolarization in 231 cells immediately before mitochondrial network fragmentation (42, 56). However, despite the early evidence that decorin triggers mitochondrial depolarization, the mechanism, molecular factors, general applicability, and biological function were unknown.
Therefore, in this study, we delved deeper into the mechanism of action that governs decorin-dependent depolarization in multiple tumor cell types and increased scientific robustness surrounding this biological process. We have demonstrated that there is a dose- and time-dependent pattern in which decorin promotes extensive depolarization of the tumor mitochondrial network. The depolarization evoked by decorin is also conserved among various types of human and mouse tumor cells including MDA-231, HeLa, and E0771. This effect is also seen in normal, genomically stable HUVECs, and ex vivo murine aortic ring assays. Moreover, using a combination of up to four different mitochondrial-specific dyes (TMRE, TMRE, JC-1, and JC-10), we validated that decorin is capable of mediated depolarization under a variety of detection methods. Interestingly, biglycan, the closest relative of decorin, was unable to initiate depolarization in our tumor models, most likely owing to the lack of a specific receptor.
Starting from the outside-in signaling paradigm (51), decorin takes on average ∼1–2 h to trigger mitochondrial depolarization. This is consistent with decorin-transducing signals via a cell-surface receptor tyrosine kinase. In this study, we found that both EGFR and Met are equally capable to communicate the signal for depolarization. This lends additional credence to decorin integrating the signal among several receptors for biological effects (43). Considering that Met represents the primary biological receptor for the observed antiangiogenic and antioncogenic activities of decorin (15), we pursued more in-depth studies with the decorin/Met interaction under the construct of depolarization. We discovered the physical presence of Met is not only necessary but also sufficient for decorin bioactivity.
As we found that mitostatin was crucial for executing decorin-evoked mitophagy (56), we redefined the role of mitostatin that it carries out in the mitophagic program. Indeed, mitostatin is not only required early for decorin-mediated depolarization but also it is capable of triggering depolarization alone (cf. Fig. 4, I and J), without external stimulation. This opens new avenues for understanding mitostatin biology as a tumor suppressor and regulation of mitochondrial health and dynamics. Furthermore, in studies done by our group in endothelial cells (119) and unpublished data in tumor cells, and others (107), mitostatin physically binds mitofusin 2. Mitofusin 2 is a key regulator of mitochondrial Ca2+ signaling and overall homeostasis (101, 103, 106). We discovered a functional role for mitofusin 2 in coordinating decorin-mediated depolarization in tumor cells. This is important given the role of calcium flux into and out of the mitochondria. Moreover, via dual knockdown experiments, we discovered that mitostatin and mitofusin 2 are within the same signaling pathway, giving more robustness and functionally corroborating the biochemical data of these two proteins physically interacting with each other. It will be interesting to determine if the innate GTPase activity of mitofusin 2 (102, 105) is required for either mitostatin binding and downstream depolarization and/or coordinating external signals (such as those conveyed by decorin) to depolarize the mitochondria.
Considering that decorin engages a Met/mitostatin/mitofusin 2 signaling axis to affect depolarization, the connections to PINK1/Parkin-mediated mitophagy are substantially strengthened. Damaged mitochondria (99, 104), mitochondria in highly metabolic tissues (120), or mitochondria that have received a signal for depolarization (62, 121) are “marked” to undergo a disconnection from the tubular mitochondrial network and are eventually degraded by mitophagosomes (63, 122–126). Deciphering this mechanism would be directly relevant to fully comprehending decorin-evoked tumor cell mitophagy.
Using various inhibitors, we have determined that decorin engages and “opens” the mitochondria permeability transition pore (mPTP) for extracellular signal-dependent depolarization. Although the responsible members of signaling apparatus currently remain unknown, several candidates may be involved. There is evidence that energy-sensing kinases such as AMPK and ULK1 may be responsible in coordinating mitophagy (127, 128). It is not known, however, if these kinases can also induce ΔΨm. Furthermore, it is also unknown where these kinases, if suitable for mediating depolarization, fall in the decorin/Met/mitostatin/mitofusin 2/mPTP axis. Whether there is a connection between these kinases and the molecular components of the mPTP is unknown as well but is currently the subject of future studies.
It has been shown before that a cocktail of three chicken brain hyalectans (aggrecan, phosphacan, and versican) mediates depolarization in embryonic avian axons (129). In this work, the mixture of the three proteoglycans was primarily bound to the substrate. In another study, soluble aggrecan used at double the concentration was able to induce a higher magnitude of mitochondrial depolarization in sensory neurons (130). Thus, although the mechanism of action was not delineated in these abovementioned studies, it is plausible to infer that minimal aggrecan harbors the depolarizing effect on growing axons.
It has been previously shown in an ex vivo assay using cartilage explants that following a single impact load, calcium is released from the endoplasmic reticulum via the ryanodine receptor and is taken up by the mitochondria via the uniport transporter, causing depolarization and caspase 9 activation (131). Notably, soluble decorin is capable of increasing intracellular calcium, [Ca2+]I, in EGFR-overexpressing squamous carcinoma cells (11). Decorin effects persisted in the absence of extracellular Ca2+ but were blocked by specific inhibitors of EGFR tyrosine kinase, by physical downregulation of the EGFR, and were not mimicked by biglycan (11), as in the present report for depolarization.
Finally, we previously found that decorin-evoked mitophagy is required for decreased VEGFA (56). This is consistent with recent findings that decorin catabolizes intracellular endothelial VEGFA via autophagy (46) and in this study that decorin mediates depolarization in ex vivo aortic rings and HUVEC (cf. Fig. 8). This lays the foundational framework to investigate whether ΔΨm is needed for decorin-mediated angiostasis. Using either Ca2+ release antagonists or mPTP inhibitors (pharmacological or genetic) will help us understand the underlying connection to this important aspect of decorin biology in cancer.
The potential future use of decorin as a next generation protein therapy against human cancer is bright. This assertion is based on and reinforced by an overwhelming number of studies conducted over the past couple of decades. The supporting data and rationale demonstrate that reduced DCN expression is significantly associated with a poor clinical outcome (132–136). Conversely, high DCN expression within the stromal compartment is a positive prognosticator for patients with breast cancer (137). These clinical relations are further buttressed by a plethora of data documenting the antioncogenic properties of decorin in preclinical and various genetic studies (23–28, 138–148).
In conclusion, we have delineated a molecular axis focused on delivering prodepolarization signals to the mitochondrial network in tumor cells. These findings constitute a framework by which decorin could potentially exert promitophagic programs in an effort to curb tumorigenesis.
SUPPLEMENTAL DATA
Supplemental Figs. S1–S5: https://doi.org/10.6084/m9.figshare.20352915.
GRANTS
This original research was supported in part by the National Institutes of Health Grants RO1 CA39481 and RO1 CA245311 (to R.V.I).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTION
T.N. and R.V.I. conceived and designed research; T.N., C.X., and R.V.I. performed experiments; T.N., C.X., and R.V.I. analyzed data; T.N., C.X., and R.V.I. interpreted results of experiments; T.N., C.X., and R.V.I. prepared figures; T.N. and R.V.I. drafted manuscript; T.N., C.X., and R.V.I. edited and revised manuscript; T.N., C.X., and R.V.I. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank all the members of the Iozzo laboratory for helpful scientific input, Dr. Rick Owens for providing recombinant decorin and biglycan, and Dr. Gianluca Gallo for valuable information. Graphical abstract was created with BioRender.com and published with permission.
REFERENCES
- 1. Yamaguchi Y, Ruoslahti E. Expression of human proteoglycan in Chinese hamster ovary cells inhibits cell proliferation. Nature 336: 244–246, 1988. doi: 10.1038/336244a0. [DOI] [PubMed] [Google Scholar]
- 2. Yamaguchi Y, Mann DM, Ruoslahti E. Negative regulation of transforming growth factor-b by the proteoglycan decorin. Nature 346: 281–284, 1990. doi: 10.1038/346281a0. [DOI] [PubMed] [Google Scholar]
- 3. Santra M, Skorski T, Calabretta B, Lattime EC, Iozzo RV. De novo decorin gene expression suppresses the malignant phenotype in human colon cancer cells. Proc Natl Acad Sci U S A 92: 7016–7020, 1995. doi: 10.1073/pnas.92.15.7016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Santra M, Mann DM, Mercer EW, Skorski T, Calabretta B, Iozzo RV. Ectopic expression of decorin protein core causes a generalized growth suppression in neoplastic cells of various histogenetic origin and requires endogenous p21, an inhibitor of cyclin-dependent kinases. J Clin Invest 100: 149–157, 1997. doi: 10.1172/JCI119507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. De Luca A, Santra M, Baldi A, Giordano A, Iozzo RV. Decorin-induced growth suppression is associated with upregulation of p21, an inhibitor of cyclin-dependent kinases. J Biol Chem 271: 18961–18965, 1996. doi: 10.1074/jbc.271.31.18961. [DOI] [PubMed] [Google Scholar]
- 6. Iozzo RV, Cohen I. Altered proteoglycan gene expression and the tumor stroma. Experientia 49: 447–455, 1993. doi: 10.1007/BF01923588. [DOI] [PubMed] [Google Scholar]
- 7. Moscatello DK, Santra M, Mann DM, McQuillan DJ, Wong AJ, Iozzo RV. Decorin suppresses tumor cell growth by activating the epidermal growth factor receptor. J Clin Invest 101: 406–412, 1998. doi: 10.1172/JCI846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Iozzo RV, Moscatello D, McQuillan DJ, Eichstetter I. Decorin is a biological ligand for the epidermal growth factor receptor. J Biol Chem 274: 4489–4492, 1999. doi: 10.1074/jbc.274.8.4489. [DOI] [PubMed] [Google Scholar]
- 9. Goldoni S, Owens RT, McQuillan DJ, Shriver Z, Sasisekharan R, Birk DE, Campbell S, Iozzo RV. Biologically active decorin is a monomer in solution. J Biol Chem 279: 6606–6612, 2004. doi: 10.1074/jbc.M310342200. [DOI] [PubMed] [Google Scholar]
- 10. Santra M, Reed CC, Iozzo RV. Decorin binds to a narrow region of the epidermal growth factor (EGF) receptor, partially overlapping with but distinct from the EGF-binding epitope. J Biol Chem 277: 35671–35681, 2002. doi: 10.1074/jbc.M205317200. [DOI] [PubMed] [Google Scholar]
- 11. Patel S, Santra M, McQuillan DJ, Iozzo RV, Thomas AP. Decorin activates the epidermal growth factor receptor and elevates cytosolic Ca2+ in A431 cells. J Biol Chem 273: 3121–3124, 1998. doi: 10.1074/jbc.273.6.3121. [DOI] [PubMed] [Google Scholar]
- 12. Csordás G, Santra M, Reed CC, Eichstetter I, McQuillan DJ, Gross D, Nugent MA, Hajnóczky G, Iozzo RV. Sustained down-regulation of the epidermal growth factor receptor by decorin. A mechanism for controlling tumor growth in vivo. J Biol Chem 275: 32879–32887, 2000. doi: 10.1074/jbc.M005609200. [DOI] [PubMed] [Google Scholar]
- 13. Zhu J-X, Goldoni S, Bix G, Owens RA, McQuillan D, Reed CC, Iozzo RV. Decorin evokes protracted internalization and degradation of the EGF receptor via caveolar endocytosis. J Biol Chem 280: 32468–32479, 2005. doi: 10.1074/jbc.M503833200. [DOI] [PubMed] [Google Scholar]
- 14. Santra M, Eichstetter I, Iozzo RV. An anti-oncogenic role for decorin: downregulation of ErbB2 leads to growth suppression and cytodifferentiation of mammary carcinoma cells. J Biol Chem 275: 35153–35161, 2000. [DOI] [PubMed] [Google Scholar]
- 15. Goldoni S, Humphries A, Nyström A, Sattar S, Owens RT, McQuillan DJ, Ireton K, Iozzo RV. Decorin is a novel antagonistic ligand of the Met receptor. J Cell Biol 185: 743–754, 2009. doi: 10.1083/jcb.200901129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Buraschi S, Pal N, Tyler-Rubinstein N, Owens RT, Neill T, Iozzo RV. Decorin antagonizes Met receptor activity and downregulates β-catenin and Myc levels. J Biol Chem 285: 42075–42085, 2010. doi: 10.1074/jbc.M110.172841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schönherr E, Sunderkötter C, Iozzo RV, Schaefer L. Decorin, a novel player in the insulin-like growth factor system. J Biol Chem 280: 15767–15772, 2005. doi: 10.1074/jbc.M500451200. [DOI] [PubMed] [Google Scholar]
- 18. Iozzo RV, Buraschi S, Genua M, Xu S-Q, Solomides CC, Peiper SC, Gomella LG, Owens RT, Morrione A. Decorin antagonizes IGF receptor I (IGF-IR) function by interfering with IGF-IR activity and attenuating downstream signaling. J Biol Chem 286: 34712–34721, 2011. doi: 10.1074/jbc.M111.262766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Khan GA, Girish GV, Lala N, Di Guglielmo GM, Lala PK. Decorin is a novel VEGFR-2-binding antagonist for the human extravillous trophoblast. Mol Endocrinol 25: 1431–1443, 2011. doi: 10.1210/me.2010-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Buraschi S, Neill T, Goyal A, Poluzzi C, Smythies J, Owens RT, Schaefer L, Torres A, Iozzo RV. Decorin causes autophagy in endothelial cells via Peg3. Proc Natl Acad Sci U S A 110: E2582–E2591, 2013. doi: 10.1073/pnas.1305732110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baghy K, Horváth Z, Regős E, Kiss K, Schaff Z, Iozzo RV, Kovalszky I. Decorin interferes with platelet-derived growth factor receptor signaling in experimental hepatocarcinogenesis. FEBS J 280: 2150–2164, 2013. doi: 10.1111/febs.12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gubbiotti MA, Vallet SD, Ricard-Blum S, Iozzo RV. Decorin interacting network: a comprehensive analysis of decorin-binding partners and their versatile functions. Matrix Biol 55: 7–21, 2016. doi: 10.1016/j.matbio.2016.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Iozzo RV, Chakrani F, Perrotti D, McQuillan DJ, Skorski T, Calabretta B, Eichstetter I. Cooperative action of germline mutations in decorin and p53 accelerates lymphoma tumorigenesis. Proc Natl Acad Sci U S A 96: 3092–3097, 1999. doi: 10.1073/pnas.96.6.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Horvath Z, Kovalszky I, Fullar A, Kiss K, Schaff Z, Iozzo RV, Baghy K. Decorin deficiency promotes hepatic carcinogenesis. Matrix Biol 35: 194–205, 2014. doi: 10.1016/j.matbio.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bi X, Tong C, Dockendorff A, Bancroft L, Gallagher L, Guzman G, Iozzo RV, Augenlicht LH, Yang W. Genetic deficiency of decorin causes intestinal tumor formation through disruption of intestinal cell maturation. Carcinogenesis 29: 1435–1440, 2008. doi: 10.1093/carcin/bgn141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bi X, Pohl NM, Qian Z, Yang GR, Gou Y, Guzman G, Kajdacsy-Balla A, Iozzo RV, Yang W. Decorin-mediated inhibition of colorectal cancer growth and migration is associated with E-cadherin in vitro and in mice. Carcinogenesis 33: 326–330, 2012. doi: 10.1093/carcin/bgr293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bi X, Xia X, Fan D, Mu T, Zhang Q, Iozzo RV, Yang W. Oncogenic activin C interacts with decorin in colorectal cancer in vivo and in vitro. Mol Carcinog 55: 1786–1795, 2016. doi: 10.1002/mc.22427. [DOI] [PubMed] [Google Scholar]
- 28. Mao L, Yang J, Yue J, Chen Y, Zhou H, Fan D, Zhang Q, Buraschi S, Iozzo RV, Bi X. Decorin deficiency promotes epithelial-mesenchymal transition and colon cancer metastasis. Matrix Biol 95: 1–14, 2021. doi: 10.1016/j.matbio.2020.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Theocharis AD, Skandalis SS, Tzanakakis GN, Karamanos NK. Proteoglycans in health and disease: novel proteoglycan roles in malignancy and their pharmacological targeting. FEBS J 277: 3904–3923, 2010. doi: 10.1111/j.1742-4658.2010.07800.x. [DOI] [PubMed] [Google Scholar]
- 30. Baghy K, Dezsó K, László V, Fullár A, Péterfia B, Paku S, Nagy P, Schaff Z, Iozzo RV, Kovalszky I. Ablation of the decorin gene enhances experimental hepatic fibrosis and impairs hepatic healing in mice. Lab Invest 91: 439–451, 2011. doi: 10.1038/labinvest.2010.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Baghy K, Iozzo RV, Kovalszky I. Decorin-TGFb axis in hepatic fibrosis and cirrhosis. J Histochem Cytochem 60: 262–268, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weis SM, Zimmerman SD, Shah M, Covell JW, Omens JH, Ross J, Jr, Dalton N, Jones Y, Reed CC, Iozzo RV, McCulloch AD. A role for decorin in the remodeling of myocardial infarction. Matrix Biol 24: 313–324, 2005. doi: 10.1016/j.matbio.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 33. Barallobre-Barreiro J, Gupta SK, Zoccarato A, Kitazume-Taneike R, Fava M, Yin X, Werner T, Hirt MN, Zampetaki A, Viviano A, Chong M, Bern M, Kourliouros A, Domenech N, Willeit P, Shah AM, Jahangiri M, Schaefer L, Fischer JW, Iozzo RV, Viner R, Thum T, Heineke J, Kichler A, Otsu K, Mayr M. Glycoproteomics reveals decorin peptides with anti-myostatin activity in human atrial fibrillation. Circulation 134: 817–832, 2016. doi: 10.1161/CIRCULATIONAHA.115.016423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nikitovic D, Aggelidakis J, Young MF, Iozzo RV, Karamanos NK, Tzanakakis GN. The biology of small leucine-rich proteoglycans in bone pathophysiology. J Biol Chem 287: 33926–33933, 2012. doi: 10.1074/jbc.R112.379602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Häkkinen L, Strassburger S, Kähäri VM, Scott PG, Eichstetter I, Lozzo RV, Larjava H. A role for decorin in the structural organization of periodontal ligament. Lab Invest 80: 1869–1880, 2000. doi: 10.1038/labinvest.3780197. [DOI] [PubMed] [Google Scholar]
- 36. Robinson KA, Sun M, Barnum CE, Weiss SN, Huegel J, Shetye SS, Lin L, Saez D, Adams SM, Iozzo RV, Soslowsky LJ, Birk DE. Decorin and biglycan are necessary for maintaining collagen fibril structure, fiber realignment, and mechanical properties of mature tendons. Matrix Biol 64: 81–93, 2017. doi: 10.1016/j.matbio.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Colon-Caraballo M, Lee N, Nallasamy S, Myers K, Hudson D, Iozzo RV, Mahendroo M. Novel regulatory roles of small leucine-rich proteoglycans in remodeling of the uterine cervix in pregnancy. Matrix Biol 105: 53–71, 2022. doi: 10.1016/j.matbio.2021.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38. Balne PK, Gupta S, Zhang J, Bristow D, Faubion M, Heil SD, Sinha PR, Green SL, Iozzo RV, Mohan RR. The functional role of decorin in corneal neovascularization in vivo. Exp Eye Res 207: 108610, 2021. doi: 10.1016/j.exer.2021.108610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li Q, Han B, Wang C, Tong W, Wei Y, Tseng WJ, Han LH, Liu XS, Enomoto-Iwamoto M, Mauck RL, Qin L, Iozzo RV, Birk DE, Han L. Mediation of cartilage matrix degeneration and fibrillation by decorin in post-traumatic osteoarthritis. Arthritis Rheumatol 72: 1266–1277, 2020. doi: 10.1002/art.41254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Han B, Li Q, Wang C, Patel P, Adams SM, Doyran B, Nia HT, Oftadeh R, Zhou S, Li CY, Liu XS, Lu XL, Enomoto-Iwamoto M, Qin L, Mauck RL, Iozzo RV, Birk DE, Han L. Decorin regulates the aggrecan network integrity and biomechanical functions of cartilage extracellular matrix. ACS Nano 13: 11320–11333, 2019. doi: 10.1021/acsnano.9b04477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Karamanos NK, Theocharis AD, Neill T, Iozzo RV. Matrix modeling and remodeling: a biological interplay regulating tissue homeostasis and diseases. Matrix Biol 75-76: 1–11, 2019. doi: 10.1016/j.matbio.2018.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Neill T, Schaefer L, Iozzo RV. Instructive roles of extracellular matrix on autophagy. Am J Pathol 184: 2146–2153, 2014. doi: 10.1016/j.ajpath.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Neill T, Schaefer L, Iozzo RV. Decoding the matrix: instructive roles of proteoglycan receptors. Biochemistry 54: 4583–4598, 2015. doi: 10.1021/acs.biochem.5b00653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gubbiotti MA, Neill T, Frey H, Schaefer L, Iozzo RV. Decorin is an autophagy-inducible proteoglycan and is required for proper in vivo autophagy. Matrix Biol 48: 14–25, 2015. doi: 10.1016/j.matbio.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Goyal A, Neill T, Owens RT, Schaefer L, Iozzo RV. Decorin activates AMPK, an energy sensor kinase, to induce autophagy in endothelial cells. Matrix Biol 34: 46–54, 2014. doi: 10.1016/j.matbio.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Neill T, Chen CG, Buraschi S, Iozzo RV. Catabolic degradation of endothelial VEGFA via autophagy. J Biol Chem 295: 6064–6079, 2020. doi: 10.1074/jbc.RA120.012593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Grant DS, Yenisey C, Rose RW, Tootell M, Santra M, Iozzo RV. Decorin suppresses tumor cell-mediated angiogenesis. Oncogene 21: 4765–4777, 2002. doi: 10.1038/sj.onc.1205595. [DOI] [PubMed] [Google Scholar]
- 48. Neill T, Painter H, Buraschi S, Owens RT, Lisanti MP, Schaefer L, Iozzo RV. Decorin antagonizes the angiogenic network. Concurrent inhibition of Met, hypoxia inducible factor-1a and vascular endothelial growth factor A and induction of thrombospondin-1 and TIMP3. J Biol Chem 287: 5492–5506, 2012. doi: 10.1074/jbc.M111.283499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Neill T, Jones HR, Crane-Smith Z, Owens RT, Schaefer L, Iozzo RV. Decorin induces rapid secretion of thrombospondin-1 in basal breast carcinoma cells via inhibition of Ras homolog gene family, member A/Rho-associated coiled-coil containing protein kinase 1. FEBS J 280: 2353–2368, 2013. doi: 10.1111/febs.12148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Neill T, Schaefer L, Iozzo RV. Decorin as a multivalent therapeutic agent against cancer. Adv Drug Deliv Rev 97: 174–185, 2016. doi: 10.1016/j.addr.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Neill T, Kapoor A, Xie C, Buraschi S, Iozzo RV. A functional outside-in signaling network of proteoglycans and matrix molecules regulating autophagy. Matrix Biol 100-101: 118–149, 2021. doi: 10.1016/j.matbio.2021.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Poluzzi C, Nastase MV, Zeng-Brouwers J, Roedig H, Hsieh LT, Michaelis JB, Buhl EM, Rezende F, Manavski Y, Bleich A, Boor P, Brandes RP, Pfeilschifter J, Stelzer EHK, Munch C, Dikic I, Brandts C, Iozzo RV, Wygrecka M, Schaefer L. Biglycan evokes autophagy in macrophages via a novel CD44/Toll-like receptor 4 signaling axis in ischemia/reperfusion injury. Kidney Int 95: 540–562, 2019. doi: 10.1016/j.kint.2018.10.037. [DOI] [PubMed] [Google Scholar]
- 53. Diehl V, Huber LS, Trebicka J, Wygrecka M, Iozzo RV, Schaefer L. The role of decorin and biglycan signaling in tumorigenesis. Front Oncol 11: 801801, 2021. doi: 10.3389/fonc.2021.801801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Moreth K, Frey H, Hubo M, Zeng-Brouwers J, Nastase MV, Hsieh LT, Haceni R, Pfeilschifter J, Iozzo RV, Schaefer L. Biglycan-triggered TLR-2- and TLR-4-signaling exacerbates the pathophysiology of ischemic acute kidney injury. Matrix Biol 35: 143–151, 2014. doi: 10.1016/j.matbio.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Neill T, Buraschi S, Kapoor A, Iozzo RV. Proteoglycan-driven autophagy: a nutrient-independent mechanism to control intracellular catabolism. J Histochem Cytochem 68: 733–746, 2020. doi: 10.1369/0022155420937370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Neill T, Torres A, Buraschi S, Owens RT, Hoek J, Baffa R, Iozzo RV. Decorin induces mitophagy in breast carcinoma cells via peroxisome proliferator-activated receptor g coactivator-1a (PGC-1a) and mitostatin. J Biol Chem 289: 4952–4968, 2014. doi: 10.1074/jbc.M113.512566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Neill T, Iozzo RV. The role of decorin proteoglycan in mitophagy. Cancers (Basel) 14: 804, 2022. doi: 10.3390/cancers14030804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vecchione A, Fassan M, Anesti V, Morrione A, Goldoni S, Baldassarre G, Byrne D, D'Arca D, Palazzo JP, Lloyd J, Scorrano L, Gomella LG, Iozzo RV, Baffa R. MITOSTATIN, a putative tumor suppressor on chromosome 12q24.1, is downregulated in human bladder and breast cancer. Oncogene 28: 257–269, 2009. doi: 10.1038/onc.2008.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fassan M, D'Arca D, Letko J, Vecchione A, Gardiman MP, McCue P, Wildemore B, Rugge M, Shupp-Byrne D, Gomella LG, Morrione A, Iozzo RV, Baffa R. Mitostatin is down-regulated in human prostate cancer and suppresses the invasive phenotype of prostate cancer cells. PLoS ONE 6: e19771, 2011. doi: 10.1371/journal.pone.0019771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Buraschi S, Neill T, Iozzo RV. Decorin is a devouring proteoglycan: remodeling of intracellular catabolism via autophagy and mitophagy. Matrix Biol 75-76: 260–270, 2019. doi: 10.1016/j.matbio.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chourasia AH, Boland ML, Macleod KF. Mitophagy and cancer. Cancer Metab 3: 4, 2015. doi: 10.1186/s40170-015-0130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dagda R, Cherra SJI, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 284: 13843–13855, 2009. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Durcan TM, Fon EA. The three 'P's of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev 29: 989–999, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chung KP, Hsu CL, Fan LC, Huang Z, Bhatia D, Chen YJ, Hisata S, Cho SJ, Nakahira K, Imamura M, Choi ME, Yu CJ, Cloonan SM, Choi AMK. Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nat Commun 10: 3390, 2019. doi: 10.1038/s41467-019-11327-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Heard A, Landmann JH, Hansen AR, Papadopolou A, Hsu Y-S, Selli ME, Warrington JM, Lattin J, Chang J, Ha H, Haug-Kroeper M, Doray B, Gill S, Ruella M, Hayer KE, Weitzman MD, Green AM, Fluhrer R, Singh N. Antigen glycosylation regulates efficacy of CAR T cells targeting CD19. Nat Commun 13: 3367, 2022. doi: 10.1038/s41467-022-31035-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kühbacher A, Dambournet D, Echard A, Cossart P, Pizarro-Cerdá J. Phosphatidylinositol 5-phosphatase oculocerebrorenal syndrome of Lowe protein (OCRL) controls actin dynamics during early steps of Listeria monocytogenes infection. J Biol Chem 287: 13128–13136, 2012. doi: 10.1074/jbc.M111.315788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. McKinzey DR, Gomathinayagam S, Griffin WC, Klinzing KN, Jeffries EP, Rajkovic A, Trakselis MA. Motifs of the C-terminal domain of MCM9 direct localization to sites of mitomycin-C damage for RAD51 recruitment. J Biol Chem 296: 100355, 2021. doi: 10.1016/j.jbc.2021.100355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nicoli S, DeSena G, Presta M. Fibroblast Growth Factor 2-induced angiognesis in zebrafish: the zebrafish yok membrane (ZFYM) angiogenesis assay. J Cell Mol Med 13: 2061–2068, 2009. doi: 10.1111/j.1582-4934.2008.00432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Poluzzi C, Casulli J, Goyal A, Mercer TJ, Neill T, Iozzo RV. Endorepellin evokes autophagy in endothelial cells. J Biol Chem 289: 16114–16128, 2014. doi: 10.1074/jbc.M114.556530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cossarizza A, Baccarani-Contri M, Kalashnikova G, Franceschi C. A new method for the cytofluorimetric analysis of mitochondrial membrane potential using the J-aggregate forming lipophilic cation 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1). Biochem Biophys Res Commun 197: 40–45, 1993. doi: 10.1006/bbrc.1993.2438. [DOI] [PubMed] [Google Scholar]
- 71. Jiang P, Wang M, Xue L, Xiao Y, Yu J, Wang H, Yao J, Liu H, Peng Y, Liu H, Li H, Chen Y, Guan MX. A Hypertension-Associated tRNAAla Mutation Alters tRNA Metabolism and Mitochondrial Function. Mol Cell Biol 36: 1920–1930, 2016. doi: 10.1128/MCB.00199-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Soldevila-Barreda JJ, Romero-Canelon I, Habtemariam A, Sadler PJ. Transfer hydrogenation catalysis in cells as a new approach to anticancer drug design. Nat Commun 6: 6582, 2015. doi: 10.1038/ncomms7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dickeson SK, Walsh JJ, Santoro SA. Contribution of the I and EF hand domains to the divalent cation-dependent collagen binding activity of the α2β1 integrin. J Biol Chem 272: 7661–7668, 1997. doi: 10.1074/jbc.272.12.7661. [DOI] [PubMed] [Google Scholar]
- 74. Karamanos NK, Piperigkou Z, Theocharis AD, Watanabe H, Franchi M, Baud S, Brezillon S, Gotte M, Passi A, Vigetti D, Ricard-Blum S, Sanderson RD, Neill T, Iozzo RV. Proteoglycan chemical diversity drives multifunctional cell regulation and therapeutics. Chem Rev 118: 9152–9232, 2018. doi: 10.1021/acs.chemrev.8b00354. [DOI] [PubMed] [Google Scholar]
- 75. Reed CC, Waterhouse A, Kirby S, Kay P, Owens RA, McQuillan DJ, Iozzo RV. Decorin prevents metastatic spreading of breast cancer. Oncogene 24: 1104–1110, 2005. doi: 10.1038/sj.onc.1208329. [DOI] [PubMed] [Google Scholar]
- 76. Schulz M, Diehl V, Trebicka J, Wygrecka M, Schaefer L. Biglycan: a regulator of hepatorenal inflammation and autophagy. Matrix Biol 100–101: 150–161, 2021. doi: 10.1016/j.matbio.2021.06.001. [DOI] [PubMed] [Google Scholar]
- 77. Johnstone CN, Smith YE, Cao Y, Burrows AD, Cross RSN, Ling X, Redvers RP, Doherty JP, Eckhardt BL, Natoli AL, Restall CM, Lucas E, Pearson HB, Deb S, Britt KL, Rizzitelli A, Li J, Harmey JH, Pouliot N, Anderson RL. Functional and molecular characterisation of EO771.LMB tumours, a new C57BL/6-mouse-derived model of spontaneously metastatic mammary cancer. Dis Model Mech 8: 237–251, 2015. doi: 10.1242/dmm.017830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hiraga T, Ninomiya T. Establishment and characterization of a C57BL/6 mouse model of bone metastasis of breast cancer. J Bone Miner Metab 37: 235–242, 2019. doi: 10.1007/s00774-018-0927-y. [DOI] [PubMed] [Google Scholar]
- 79. Iozzo RV, Schaefer L. Proteoglycans in health and disease: novel regulatory signaling mechanisms evoked by the small leucine-rich proteoglycans. FEBS J 277: 3864–3875, 2010. doi: 10.1111/j.1742-4658.2010.07797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Merline R, Nastase MV, Iozzo RV, Schaefer L. Small Leucine-rich proteoglycans: multifunctional signaling effectors. In: Extracellular Matrix: Pathobiology and signaling, edited by Karamanos N. Berlin: Walter de Gruytier Gmbh and Co., 2012. [Google Scholar]
- 81. Buraschi S, Neill T, Owens RT, Iniguez LA, Purkins G, Vadigepalli R, Evans B, Schaefer L, Peiper SC, Wang Z, Iozzo RV. Decorin protein core affects the global gene expression profile of the tumor microenvironment in a triple-negative orthotopic breast carcinoma xenograft model. PLoS ONE 7: e45559, 2012. doi: 10.1371/journal.pone.0045559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gubbiotti MA, Iozzo RV. Proteoglycans regulate autophagy via outside-in signaling: an emerging new concept. Matrix Biol 48: 6–13, 2015. doi: 10.1016/j.matbio.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Berthou S, Aebersold DM, Schmidt LS, Stroka D, Heigl C, Streit B, Stalder D, Gruber G, Liang C, Howlett AR, Candinas D, Greiner RH, Lipson KE, Zimmer Y. The Met kinase inhibitor SU11274 exhibits a selective inhibition pattern toward different receptor mutated variants. Oncogene 23: 5387–5393, 2004. doi: 10.1038/sj.onc.1207691. [DOI] [PubMed] [Google Scholar]
- 84. Zhou Y, Brattain MG. Synergy of epidermal growth factor receptor kinase inhibitor AG1478 and ErbB2 kinase inhibitor AG879 in human colon carcinoma cells is associated with induction of apoptosis. Cancer Res 65: 5848–5856, 2005. doi: 10.1158/0008-5472.CAN-04-3509. [DOI] [PubMed] [Google Scholar]
- 85. Ellis AG, Doherty MM, Walker F, Weinstock J, Nerrie M, Vitali A, Murphy R, Johns TG, Scott AM, Levitzki A, McLachlan G, Webster LK, Burgess AW, Nice EC. Preclinical analysis of the analinoquinazoline AG1478, a specific small molecule inhibitor of EGF receptor tyrosine kinase. Biochem Pharmacol 71: 1422–1434, 2006. doi: 10.1016/j.bcp.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 86. Mongiat M, Buraschi S, Andreuzzi E, Neill T, Iozzo RV. Extracellular matrix: the gatekeeper of tumor angiogenesis. Biochem Soc Trans 47: 1543–1555, 2019. doi: 10.1042/BST20190653. [DOI] [PubMed] [Google Scholar]
- 87. Stamos J, Lazarus RA, Yao X, Kirchhofer D, Wiesmann C. Crystal structure of the HGF β-chain in complex with the Sema domain of the Met receptor. EMBO J 23: 2325–2335, 2004. doi: 10.1038/sj.emboj.7600243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Date K, Matsumoto K, Kuba K, Shimura H, Tanaka M, Nakamura T. Inhibition of tumor growth and invasion by a four-kringle antagonist (HGF/NK4) for hepatocyte growth factor. Oncogene 17: 3045–3054, 1998. doi: 10.1038/sj.onc.1202231. [DOI] [PubMed] [Google Scholar]
- 89. Cao B, Su Y, Oskarsson M, Zhao P, Kort EJ, Fisher RJ, Wang LM, Vande Woude GF. Neutralizing monoclonal antibodies to hepatocyte growth factor/scatter factor (HGF/SF) display antitumor activity in animal models. Proc Natl Acad Sci U S A 98: 7443–7448, 2001. doi: 10.1073/pnas.131200498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wang X, Le P, Liang C, Chan J, Kiewlich D, Miller T, Harris D, Sun L, Rice A, Vasile S, Blake RA, Howlett AR, Patel N, McMahon G, Lipson KE. Potent and selective inhibitors of Met [hepatocyte growth factor/scatter factor (HGF/SF) receptor] tyrosyne kinase block HGF/SF-induced tumor cell growth. Mol Cancer Ther 2: 1085–1092, 2003. [PubMed] [Google Scholar]
- 91. Prat M, Crepaldi T, Pennacchietti S, Bussolino F, Comoglio PM. Agonistic monoclonal antibodies against the Met receptor dissect the biological responses to HGF. J Cell Sci 111: 237–247, 1998. doi: 10.1242/jcs.111.2.237. [DOI] [PubMed] [Google Scholar]
- 92. Shojaei F, Lee JH, Simmons BH, Wong A, Esparza CO, Plumlee PA, Feng J, Stewart AE, Hu-Lowe DD, Christensen JG. HGF/c-Met acts as an alternative angiogenic pathway in sunitinib-resistant tumors. Cancer Res 70: 10090–10100, 2010. doi: 10.1158/0008-5472.CAN-10-0489. [DOI] [PubMed] [Google Scholar]
- 93. Holland JD, Gyorffy B, Vogel R, Eckert K, Valenti G, Fang L, Lohneis P, Elezkurtaj S, Ziebold U, Birchmeier W. Combined Wnt/beta-catenin, Met, and CXCL12/CXCR4 signals characterize basal breast cancer and predict disease outcome. Cell Rep 5: 1214–1227, 2013. doi: 10.1016/j.celrep.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 94. Leung E, Xue A, Wang Y, Rougerie P, Sharma VP, Eddy R, Cox D, Condeelis J. Blood vessel endothelium-directed tumor cell streaming in breast tumors requires the HGF/C-Met signaling pathway. Oncogene 36: 2680–2692, 2017. doi: 10.1038/onc.2016.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Goldoni S, Seidler DG, Heath J, Fassan M, Baffa R, Thakur ML, Owens RA, McQuillan DJ, Iozzo RV. An anti-metastatic role for decorin in breast cancer. Am J Pathol 173: 844–855, 2008. doi: 10.2353/ajpath.2008.080275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Verburg J, Hollenbeck PJ. Mitochondrial membrane potential in axons increases with local nerve growth factor or semaphorin signaling. J Neurosci 28: 8306–8315, 2008. doi: 10.1523/JNEUROSCI.2614-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lyons A, Coleman M, Riis S, Favre C, O'Flanagan CH, Zhdanov AV, Papkovsky DB, Hursting SD, O'Connor R. Insulin-like growth factor 1 signaling is essential for mitochondrial biogenesis and mitophagy in cancer cells. J Biol Chem 292: 16983–16998, 2017. doi: 10.1074/jbc.M117.792838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Martorana F, Gaglio D, Bianco MR, Aprea F, Virtuoso A, Bonanomi M, Alberghina L, Papa M, Colangelo AM. Differentiation by nerve growth factor (NGF) involves mechanisms of crosstalk between energy homeostasis and mitochondrial remodeling. Cell Death Dis 9: 391, 2018. doi: 10.1038/s41419-018-0429-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189: 211–221, 2010. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ishigaki M, Iketani M, Sugaya M, Takahashi M, Tanaka M, Hattori S, Ohsawa I. STED super-resolution imaging of mitochondria labeled with TMRM in living cells. Mitochondrion 28: 79–87, 2016. doi: 10.1016/j.mito.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 101. Filadi R, Pendin D, Pizzo P. Mitofusin 2: from functions to disease. Cell Death Dis 9: 330, 2018. doi: 10.1038/s41419-017-0023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456: 605–610, 2008. [Erratum in Nature 513: 266, 2014]. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 103. de Brito OM, Scorrano L. Mitofusin 2: a mitochondria-shaping protein with signaling roles beyond fusion. Antioxid Redox Signal 10: 621–633, 2008. doi: 10.1089/ars.2007.1934. [DOI] [PubMed] [Google Scholar]
- 104. Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 20: 1726–1737, 2011. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Chen Y, Dorn GW. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340: 471–475, 2013. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Pallanck L. Mitophagy: mitofusin recruits a mitochondrial killer. Curr Biol 23: R570–R572, 2013. [DOI] [PubMed] [Google Scholar]
- 107. Cerqua C, Anesti V, Pyakurel A, Liu D, Naon D, Wiche G, Baffa R, Dimmer KS, Scorrano L. Trichoplein/mitostatin regulates endoplasmic reticulum-mitochondria juxtaposition. EMBO Rep 11: 854–860, 2010. doi: 10.1038/embor.2010.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Antony AN, Paillard M, Moffat C, Juskeviciute E, Correnti J, Bolon B, Rubin E, Csordas G, Seifert EL, Hoek JB, Hajnoczky G. MICU1 regulation of mitochondrial Ca(2+) uptake dictates survival and tissue regeneration. Nat Commun 7: 10955, 2016. doi: 10.1038/ncomms10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Zorova LD, Popkov VA, Plotnikov EY, Silachev DN, Pevzner IB, Jankauskas SS, Babenko VA, Zorov SD, Balakireva AV, Juhaszova M, Sollott SJ, Zorov DB. Mitochondrial membrane potential. Anal Biochem 552: 50–59, 2018. doi: 10.1016/j.ab.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Hurst S, Hoek J, Sheu SS. Mitochondrial Ca(2+) and regulation of the permeability transition pore. J Bioenerg Biomembr 49: 27–47, 2017. doi: 10.1007/s10863-016-9672-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Siemen D, Ziemer M. What is the nature of the mitochondrial permeability transition pore and what is it not? IUBMB Life 65: 255–262, 2013. doi: 10.1002/iub.1130. [DOI] [PubMed] [Google Scholar]
- 112. Bonora M, Pinton P. The mitochondrial permeability transition pore and cancer: molecular mechanisms involved in cell death. Front Oncol 4: 302, 2014. doi: 10.3389/fonc.2014.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Liu J, Farmer JDJr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 66: 807–815, 1991. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 114. Halestrap AP, Connern CP, Griffiths EJ, Kerr PM. Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol Cell Biochem 174: 167–172, 1997. [PubMed] [Google Scholar]
- 115. Kruglov AG, Kharechkina ES, Nikiforova AB, Odinokova IV, Kruglova SA. Dynamics of the permeability transition pore size in isolated mitochondria and mitoplasts. FASEB J 35: e21764, 2021. doi: 10.1096/fj.202100596R. [DOI] [PubMed] [Google Scholar]
- 116. Samartsev VN, Semenova AA, Dubinin MV. A comparative sttudy of the action of protonophore uncouplers and decoupling agents as inducers of free respiration in mitochondria in states 3 and 4: theoretical and experimental approaches. Cell Biochem Biophys 78: 203–216, 2020. doi: 10.1007/s12013-020-00914-5. [DOI] [PubMed] [Google Scholar]
- 117. Huang ZL, Pandya D, Banta DK, Ansari MS, Oh U. Cyclophilin inhibitor NIM811 ameliorates experimental allergic encephalomyelitis. J Neuroimmunol 311: 40–48, 2017. doi: 10.1016/j.jneuroim.2017.07.016. [DOI] [PubMed] [Google Scholar]
- 118. Kapoor A, Chen CG, Iozzo RV. A simplified aortic ring assay: a useful ex vivo method to assess biochemical and functional parameters of angiogenesis. Matrix Biol Plus 6–7: 100025, 2020. doi: 10.1016/j.mbplus.2020.100025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Neill T, Andreuzzi E, Wang Z-X, Peiper SC, Mongiat M, Iozzo RV. Endorepellin remodels the endothelial transcriptome toward a pro-autophagic and pro-mitophagic gene signature. J Biol Chem 293: 12137–12148, 2018. doi: 10.1074/jbc.RA118.002934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. McWilliams TG, Prescott AR, Montava-Garriga L, Ball G, Singh F, Barini E, Muqit MMK, Brooks SP, Ganley IG. Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab 27: 439–449.e5, 2018. doi: 10.1016/j.cmet.2017.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Biel TG, Rao VA. Mitochondrial dysfunction activates lysosomal-dependent mitophagy selectively in cancer cells. Oncotarget 9: 995–1011, 2018. doi: 10.18632/oncotarget.23171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6: 1090–1106, 2010. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Vincow ES, Merrihew G, Thomas RE, Shulman NJ, Beyer RP, MacCoss MJ, Pallanck LJ. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci U S A 110: 6400–6405, 2013. doi: 10.1073/pnas.1221132110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12: 119–131, 2010. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 125. Durcan TM, Fon EA. USP8 and PARK2/parkin-mediated mitophagy. Autophagy 11: 428–429, 2015. doi: 10.1080/15548627.2015.1009794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. McWilliams TG, Prescott AR, Allen GF, Tamjar J, Munson MJ, Thomson C, Muqit MM, Ganley IG. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J Cell Biol 214: 333–345, 2016. doi: 10.1083/jcb.201603039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331: 456–461, 2011. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Laker RC, Drake JC, Wilson RJ, Lira VA, Lewellen BM, Ryall KA, Fisher CC, Zhang M, Saucerman JJ, Goodyear LJ, Kundu M, Yan Z. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun 8: 548, 2017. doi: 10.1038/s41467-017-00520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Sainath R, Ketschek A, Grandi L, Gallo G. CSPGs inhibit axon branching by impairing mitochondria-dependent regulation of actin dynamics and axonal translation. Dev Neurobiol 77: 454–473, 2017. doi: 10.1002/dneu.22420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Sainath R, Armijo-Weingart L, Ketscheck A, Xu Z, Li S, Gallo G. Chondroitin sulfate proteoglycans negatively regulate the positioning of mitochondria and endoplasmic reticulum to distal axons. Dev Neurobiol 77: 1351–1370, 2017. doi: 10.1002/dneu.22535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Huser CA, Davies ME. Calcium signaling leads to mitochondrial depolarization in impact-induced chondrocyte death in equine articular cartilage explants. Arthritis Rheum 56: 2322–2334, 2007. doi: 10.1002/art.22717. [DOI] [PubMed] [Google Scholar]
- 132. Mihály Z, Kormos M, Lánczky A, Dank M, Budczies J, Szász MA, Győrffy B. A meta-analysis of gene expression-based biomarkers predicting outcome after tamoxifen treatment in breast cancer. Breast Cancer Res Treat 140: 219–232, 2013. doi: 10.1007/s10549-013-2622-y. [DOI] [PubMed] [Google Scholar]
- 133. Troup S, Njue C, Kliewer EV, Parisien M, Roskelley C, Chakravarti S, Roughley PJ, Murphy LC, Watson PH. Reduced expression of the small leucine-rich proteoglycans, lumican, and decorin is associated with poor outcome in node-negative invasive breast cancer. Clin Cancer Res 9: 207–214, 2003. [PubMed] [Google Scholar]
- 134. Leygue E, Snell L, Dotzlaw H, Troup S, Hiller-Hitchcock T, Murphy LC, Roughley PJ, Watson PH. Lumican and decorin are differentially expressed in human breast carcinoma. J Pathol 192: 313–320, 2000. doi:. [DOI] [PubMed] [Google Scholar]
- 135. Oda G, Sato T, Ishikawa T, Kawachi H, Nakagawa T, Kuwayama T, Ishiguro M, Iida S, Uetake H, Sugihara K. Significance of stromal decorin expression during the progression of breast cancer. Oncol Rep 28: 2003–2008, 2012. doi: 10.3892/or.2012.2040. [DOI] [PubMed] [Google Scholar]
- 136. Hosoya T, Oda G, Nakagawa T, Onishi I, Hosoya T, Ishiguro M, Ishikawa T, Uetake H. Plasma levels of decorin increased in patients during the progression of breast cancer. J Clin Med 10: 5530, 2021.doi: 10.3390/jcm10235530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Li SJ, Chen DL, Zhang WB, Shen C, Che GW. Prognostic value of stromal decorin expression in patients with breast cancer: a meta-analysis. J Thorac Dis 7: 1939–1950, 2015. doi: 10.3978/j.issn.2072-1439.2015.11.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Reed CC, Gauldie J, Iozzo RV. Suppression of tumorigenicity by adenovirus-mediated gene transfer of decorin. Oncogene 21: 3688–3695, 2002. doi: 10.1038/sj.onc.1205470. [DOI] [PubMed] [Google Scholar]
- 139. Tralhão JG, Schaefer L, Micegova M, Evaristo C, Schönherr E, Kayal S, Veiga-Fernandes H, Danel C, Iozzo RV, Kresse H, Lemarchand P. In vivo selective and distant killing of cancer cells using adenovirus-mediated decorin gene transfer. FASEB J 17: 464–466, 2003. doi: 10.1096/fj.02-0534fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Li X, Pennisi A, Yaccoby S. Role of decorin in the antimyeloma effects of osteoblasts. Blood 112: 159–168, 2008. doi: 10.1182/blood-2007-11-124164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Hu Y, Sun H, Owens RT, Wu J, Chen YQ, Berquin IM, Perry D, O'Flaherty JT, Edwards IJ. Decorin suppresses prostate tumor growth through inhibition of epidermal growth factor and androgen receptor pathways. Neoplasia 11: 1042–1053, 2009. doi: 10.1593/neo.09760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Bozoky B, Savchenko A, Guven H, Ponten F, Klein G, Szekely L. Decreased decorin expression in the tumor microenvironment. Cancer Med 3: 485–491, 2014. doi: 10.1002/cam4.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Sainio A, Nyman M, Lund R, Vuorikoski S, Boström P, Laato M, Boström PJ, Järveläinen H. Lack of decorin expression by human bladder cancer cells offers new tools in the therapy of urothelial malignancies. PLoS ONE 8: e76190, 2013. doi: 10.1371/journal.pone.0076190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Jackson DG. Hyaluronan in the lymphatics: the key role of the hyaluronan receptor LYVE-1 in leucocyte trafficking. Matrix Biol 78-79: 219–235, 2019. doi: 10.1016/j.matbio.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 145. Xu W, Neill T, Yang Y, Hu Z, Cleveland E, Wu Y, Hutten R, Xiao X, Stock SR, Shevrin D, Kaul K, Brendler C, Iozzo RV, Seth P. The systemic delivery of an oncolytic adenovirus expressing decorin inhibits bone metastasis in a mouse model of human prostate cancer. Gene Ther 22: 247–256, 2015. doi: 10.1038/gt.2014.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Yang Y, Xu WW, Neill T, Hu Z, Wang CH, Xiao X, Stock S, Guise T, Yun CO, Brendler CB, Iozzo RV, Seth P. Systemic delivery of an oncolytic adenovirus expressing decorin for the treatment of breast cancer bone metastases. Hum Gene Ther 26: 813–825, 2015. doi: 10.1089/hum.2015.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Reszegi A, Horváth Z, Karászi K, Regős E, Postniková V, Tátrai P, Kiss A, Schaff Z, Kovalszky I, Baghy K. The protective role of decorin in hepatic metastasis of colorectal carcinoma. Biomolecules 10: 1199, 2020. doi: 10.3390/biom10081199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Zhang Y, Liu C, Wang T, Kong F, Zhang H, Yi J, Dong X, Duan H, Tao N, Yang Y, Wang H. Therapeutic effects of mesenchymal stem cells loaded with oncolytic adenovirus carrying decorin on a breast cancer lung metastatic mouse model. Mol Ther Oncolytics 24: 486–496, 2022. doi: 10.1016/j.omto.2022.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figs. S1–S5: https://doi.org/10.6084/m9.figshare.20352915.