

Keywords: alcoholic pancreatitis, fibrosis, inflammation, PI3K/AKT/mTOR, urolithin A
Abstract
Heavy alcohol consumption is the dominant risk factor for chronic pancreatitis (CP); however, treatment and prevention strategies for alcoholic chronic pancreatitis (ACP) remains limited. The present study demonstrates that ACP induction in C57BL/6 mice causes significant acinar cell injury, pancreatic stellate cell (PSC) activation, exocrine function insufficiency, and an increased fibroinflammatory response when compared with alcohol or CP alone. Although the withdrawal of alcohol during ACP recovery led to reversion of pancreatic damage, continued alcohol consumption with established ACP perpetuated pancreatic injury. In addition, phosphokinase array and Western blot analysis of ACP-induced mice pancreata revealed activation of the phosphatidylinositol 3 kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) and cyclic AMP response element binding protein (CREB) signaling pathways possibly orchestrating the fibroinflammatory program of ACP pathogenesis. Mice treated with urolithin A (Uro A, a gut-derived microbial metabolite) in the setting of ACP with continued alcohol intake (during the recovery period) showed suppression of AKT and P70S6K activation, and acinar damage was significantly reduced with a parallel reduction in pancreas-infiltrating macrophages and proinflammatory cytokine accumulation. These results collectively provide mechanistic insight into the impact of Uro A on attenuation of ACP severity through suppression of PI3K/AKT/mTOR signaling pathways and can be a useful therapeutic approach in patients with ACP with continuous alcohol intake.
NEW & NOTEWORTHY Our novel findings presented here demonstrate the utility of Uro A as an effective therapeutic agent in attenuating alcoholic chronic pancreatitis (ACP) severity with alcohol continuation after established disease, through suppression of the PI3K/AKT/mTOR signaling pathway.
INTRODUCTION
Chronic pancreatitis (CP) is a debilitating disease that is a significant source of morbidity in the United States (1). Excessive alcohol use is associated with 40%–70% of cases and is a dominant risk factor for the development of this disease (2, 3). Alcohol abuse is known to induce numerous pathological stress responses, the most common being endoplasmic reticulum (ER) stress, which leads to the accumulation of unfolded protein in the ER contributing to alcohol-related disorders (4). The hallmark of alcoholic CP (ACP) is persistent inflammation of the pancreas and progressive loss of the endocrine and exocrine function leading to its atrophy and fibrosis (5). Accumulating scientific evidence suggests that continued alcohol consumption, with established ACP, instigates irreversible pancreatic damage due to recurrent episodes of acute pancreatitis (AP) by fostering a continuous fibroinflammatory microenvironment within the pancreas (6–8). The molecular mechanisms involved in the pathophysiology of ACP with continuous alcohol intake remain ambiguous; treatment options and preventative care strategies are restricted due to limited experimental animal models that successfully recapitulate the human CP arising from prolonged or continued alcohol use after established pancreatic injury (7, 9, 10). Therefore, in this study, using an established ACP mice model, we have addressed two of the major unanswered questions with regards to its pathogenesis; characterization of the pancreas-specific signaling pathways in this process, and whether utilizing novel therapeutic agents can attenuate the severity of ACP progression despite continued alcohol trigger.
Several signaling pathways, including mitogen-activated protein kinases (MAPKs), JAK/STAT, and phosphatidylinositol 3 kinase (PI3K), have been identified as critical mediators of acinar cell plasticity and the fibroinflammatory microenvironment of pancreatitis (10–15). Specifically, PI3K and its downstream effector kinases, including AKT and mammalian target of rapamycin (mTOR) have also been implicated in pancreatitis progression. However, the role of the PI3K/AKT/mTOR pathway in the pathogenesis of ACP is poorly understood. In caerulein-induced CP models, PI3K pathway activation leads to premature activation of trypsinogen, accelerating acinar cell death, and triggering deleterious changes to the pancreatic microenvironment (16–18). In addition, mice lacking the PI3Kγ gene, a specific isoform of PI3K, were found to be protected from acinar cell injury/necrosis and displayed reduced severity of pancreatitis (19, 20), suggesting a promising role for PI3K inhibitors in the management and prevention of this disease.
In the context of gastrointestinal inflammation, the microbiome has been implicated as a critical mediator of overall gut health. Urolithin A (Uro A) is a natural compound synthesized by gut bacteria from ingested ellagitannins, a class of hydrolyzable tannins found mainly in pomegranates, berries, and nuts (21, 22). Previous work from our group has shown that Uro A is a potent anti-inflammatory agent in several preclinical disease models and exhibits antitumor activity in gastrointestinal cancers (23–25). Furthermore, we have shown that Uro A targets the PI3K/AKT/mTOR signaling axis, leading to a significant reduction of tumor burden, and marked improvement in overall survival in a genetic mouse model of pancreatic cancer (25, 26). Moreover, our studies have demonstrated that Uro A is well tolerated and does not elicit any adverse toxic effects at clinically relevant doses in these mice. However, despite the promising effect of Uro A in several malignancies and inflammatory disorders, the benefit of this microbial metabolite in the prevention of pancreatitis has not yet been investigated.
In the present study, using an experimental animal model, we have demonstrated that with established ACP, continued alcohol intake promotes persistent pancreatic injury, activation of pancreatic stellate cells (PSCs), and infiltration of macrophages, thereby promoting an irreversible fibroinflammatory milieu within the pancreas. Mechanistically, our results also demonstrate pancreas-specific activation of PI3K/AKT/mTOR signaling as one of the major pathways driving these changes associated with ACP. Most importantly, we found that Uro A treatment can ameliorate the characteristic hallmarks of this disease even with alcohol continuation by suppressing activation of AKT and p70S6K signaling. These results provide preclinical rationale for using this anti-inflammatory agent to reduce the severity and progression of ACP in a translational setting and can benefit patients suffering from chronic alcohol abuse.
MATERIALS AND METHODS
Animals
Wild-type C57BL/6 mice were purchased from Jackson Laboratory and maintained in the animal core facility at the University of Miami. Male and female mice (8–10 wk old, weighing 20–25 g) were used in this study and were housed in pathogen-free conditions under a 12-h light-dark diurnal cycle with a controlled temperature of (21°C–23°C) and maintained on standard rodent chow diet (Harlan Laboratories) before the onset of the experimental treatment protocol.
All animal experiments were approved and performed in compliance with the regulations and ethical guidelines for experimental and animal studies of the Institutional Animal Care (IACUC) and the University of Miami (Miami, FL; Protocol No. 15-057, 15-099, and 18-081).
Animal Model of Alcoholic Chronic Pancreatitis
C57BL/6 mice at 8–10 wk old were pair-fed with alcohol for 14 wk using Lieber-DeCarli alcohol-based liquid diet (A) (Cat. No. F1259SP; Bioserv, Inc.), containing 5% vol/vol ethanol, whereas control mice received a standard control-liquid diet containing 28% carbohydrates instead of ethanol. This feeding regimen has previously been reported to mimic pancreatic damage due to chronic alcohol use in humans (27, 28). During the last 4 wk of this alcohol exposure, chronic pancreatitis (CP) was established in C57BL/6 mice by administrating caerulein (solubilized in PBS at a final concentration of 10 µg/mL). Caerulein was given at a dose of 50 μg/kg by intraperitoneal hourly injections (6 times a day for 3 days a week) for 4 wk. With the combination of the effect of alcohol and caerulein, this group is referred to as ACP (n = 7 mice in each group). Animals were euthanized at the end of alcohol withdrawal/continuation period of 3 and 21 days, also referred to as ACP recovery phase, and peripheral blood and pancreata were collected for study. The detailed description of all the treatment groups from the induction and recovery phase is shown in Table 1.
Table 1.
List of treatment groups from induction and recovery phases
| Groups | Induction Phase | Recovery Phase | Group Abbreviation |
|---|---|---|---|
| 1 | Control diet (14 wk) |
Continued control diet | Ctrl |
| 2 | Alcohol diet (14 wk) |
Control diet (3 or 21 days) |
A |
| 3 | Control diet + caerulein injections (for 4 wk) | Continued control diet | CP |
| 4 | Alcohol diet + caerulein injections | Control diet (−A) (3 or 21 days) |
ACP (−A) (3- or 21-days recovery) = ACP−A |
| 5 | Alcohol diet + caerulein injections | Continued alcohol diet (+A) (3 or 21 days) | ACP (+A) (3- or 21-days recovery) = ACP+A |
| 6 | Alcohol diet + caerulein injections+ Uro A treatment | Control diet (−A) (3 days or 2 wk) |
ACP + Uro A (−A) (3 days or 2 wk) = (ACP+Uro A)−A |
| 7 | Alcohol diet + caerulein injections + Uro A treatment | Continued alcohol diet (+A) (3 days or 2 wk) | ACP + Uro A (+A) (3 days or 2 wk) = (ACP+Uro A)+A |
ACP, alcoholic chronic pancreatitis; CP, chronic pancreatitis; Uro A, urolithin A.
Pancreata removed were fixed in 10% neutral buffered formalin (pH 7.2–7.4) for histological analysis or snapped frozen in liquid nitrogen for molecular analyses. Blood samples harvested from mice were centrifuged at 1,500 rpm for 10 min to separate the serum. Pancreas weight for each mouse was measured and plotted relative to its corresponding body weight (as mg/g).
Serum Alcohol Estimation
Peak alcohol concentration in mouse serum was estimated using an ethanol assay kit (Abcam, ab65343) as per the manufacturer’s protocol. Alcohol oxidase oxidizes ethanol to generate H2O2 in the assay reaction, which reacted with a probe and generated color recorded at absorbance max of 570 nm using a microplate reader.
Serum Amylase Analysis
Serum amylase in the blood samples of C57BL/6 mice from different study groups was estimated using a quantitative colorimetric amylase assay kit (Cat. No. ab102523, Abcam) as per the manufacturer’s protocol. The α-amylase in the serum cleave the substrate ethylidene-pNP-G7 to produce smaller fragments that were eventually modified by α-glucosidase. This caused the release of a chromophore and was measured at optical density (O.D.) = 405 nm using a 96-well multimode plate reader.
Histological and Immunohistochemical Analysis
Pancreatic tissues obtained were fixed in 10% neutral-buffered formalin, embedded in paraffin, and sectioned at 4 µm for routine hematoxylin and eosin (H&E) examination. Histological assessment of fibrotic milieu was done using special stains, including Sirius red and Masson’s trichrome blue. Immunohistochemistry (IHC) and immunofluorescence (IF) staining analyses were done as described previously using primary antibodies listed in Supplemental Table S1 (29, 30). Immunostained tissues were imaged using DM750 Leica microscope (Leica Microsystems). IF-images were acquired using the Leica DMi8 microscope system (Leica Microsystems). Quantitative histological assessment for each stain was done by choosing multiple random, nonoverlapping fields for each mouse tissue. Image J analysis tool was used to quantify the percent positive area staining for all protein markers as described previously (25, 29, 31).
Phosphokinase Array Analysis
Screening for phosphorylation profiles of multiple effector kinases was done using phospho-kinase antibody array (Cat. No. ARY003B, R&D System). Briefly, pancreatic tissue lysates containing 200 µg of equal protein were exposed on a nitrocellulose membrane containing capture antibody spots in duplicate. Levels of phosphorylated protein were assessed using phospho-specific antibodies and detected using chemiluminescent-based reaction. The density of each spot blotted on the membrane was calculated using HL++ image analysis software.
Inflammation Antibody Array Analysis
Lysates prepared from pancreatic tissue homogenate were used for bicinchoninic acid (BCA)-based protein estimation. Pooled lysates collected from each group containing an equal amount of protein (300 µg) were subjected to inflammation antibody array profiling as per the manufacturer’s protocol (Cat. No. ab133999, Abcam).
Enzyme-Linked Immunosorbent Assay
Serum levels of cytokines were assayed using commercially available sandwich enzyme-linked immunosorbent assay (ELISA) kits specific for mouse TNF-α (MTA00B, R&D Systems), IL-1β (MLB00C, R&D Systems), IL-6 (M6000B, R&D Systems), and stromal cell derived factor-1 (SDF-1)(MCX120, R&D Systems) as per the manufacturer’s instructions.
Western Blot Analysis
Snapped frozen pancreatic tissue proteins were analyzed using Western blot as previously detailed (29, 31). Immunoreactive bands were developed using Pierce ECL Western blotting substrate (Cat. No. 32106, Thermo Scientific) or Super Signal West Pico PLUS chemiluminescent substrate (Cat. No. 32132, Thermo Scientific). Densitometric analysis for quantification of protein expression was performed using ImageJ software (NIH, Bethesda, MD).
Uro A Treatment in Mice
ACP-induced C57BL/6 mice were treated with vehicle or Uro A (20 mg/kg/day) as detailed previously (25). Mice in the Uro A arm received treatment by oral gavage 5 days/wk. Mice were euthanized at day 3 and day 14 at the end of alcohol withdrawal/continuation recovery period. Mice were euthanized and pancreatic tissues along with blood were harvested for molecular and serum analysis, respectively.
Statistical Analysis
Descriptive statistics were calculated using Prism software (GraphPad Software Inc). Results are shown as values of means ± SD unless otherwise indicated. Multiple comparisons were performed using one-way ANOVA followed by Tukey’s or Dunnett’s multiple comparisons tests where appropriate. A two-tailed Student’s t test was used for two-group comparisons. Statistical significance was defined using a cutoff of 0.05.
RESULTS
Animal Model of Alcoholic Chronic Pancreatitis
The experimental murine model of ACP was established as described in the methodology section (Fig. 1A). All mice cohorts gained weight during alcohol and CP induction, and the rate of weight gain was consistent among the experimental cohorts (Supplemental Fig. S1A). Notably, ACP mice with continued exposure to alcohol during the recovery phase showed maximum weight loss as compared with ACP-induced mice without alcohol, suggesting that the ACP induction with alcohol continuation causes heightened levels of toxicity (Supplemental Fig. S1B).
Figure 1.

Effect of alcohol continuation on mice pancreatic histology with established alcoholic chronic pancreatitis (ACP). A: schematic showing the protocol of alcoholic chronic pancreatitis (ACP) induction in C57BL/6 mice exposed to ethanol (alcohol) enriched liquid diet (A) and repetitive caerulein administration (or recurrent acute pancreatitis) to establish chronic pancreatitis (CP) model. Mice were euthanized after 3 and 21 days of recovery period after the last episode of CP induction with (+A) or without (−A) continuing alcohol intake to harvest pancreas and blood for analysis (see Table 1). B: blood alcohol concentration was measured and expressed as (mg/dL), in control, alcohol alone (A), and ACP induced (with continued alcohol for 3 days) C57BL/6 mice. C: relative pancreas weight (pancreas weight/body weight) measurements of C57BL/6 mice in A, CP, and ACP-induced with continued alcohol groups. D: representative images of mouse pancreas showing significant atrophy and loss of acinar tissue mass after ACP induction (with continued alcohol for 3 days) as compared with control mouse pancreas. E: measurements of serum amylase activity in control, A, CP, and ACP-induced C57BL/6 mice. F: H&E based histological assessment of pancreas harvested from control, A, CP, and ACP-induced C57BL/6 mice groups. Representative photomicrographs of immunofluorescence (IF)-based imaging analysis of acinar cell specific amylase expression (shown in red) in the pancreatic tissue sections of A, CP, and ACP-induced mice. Representative histological sections of pancreas showing collagen levels (trichrome blue) and PSC activation (vimentin and αSMA) immunohistochemical expression in control, A, CP, and ACP-induction with continued alcohol for 3- and 21-days during recovery phase. Scale bar, 50 μm. G: quantitative assessment of amylase expression, collagen deposition, and PSC activation markers in pancreata harvested from each experimental group is shown. H: quantitative histological assessment analysis of amylase expression and collagen deposition on pancreatic tissue sections obtained from control and ACP with 21 days of recovery phase with and without alcohol. Individual data points with means ± SD are shown and compared by one-way ANOVA. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. n = 5−7 mice/group. H&E, hematoxylin and eosin; ns, nonsignificant; PSC, pancreatic stellate cell.
ACP Induction with Prolonged Alcohol Exposure Causes Persistent Pancreatic Exocrine Insufficiency and Promotes PSC Activation and Fibrosis
Intake and metabolism of alcohol were confirmed in mice receiving an ethanol-based liquid diet by an increased concentration of blood alcohol levels compared with control mice fed with a regular diet (Fig. 1B). Furthermore, mice exposed to alcohol displayed a significant increase in pancreas weight compared with the control group. However, caerulein alone or in combination with alcohol exposure demonstrated significant atrophy of the pancreas and loss in its mass as compared with control or alcohol-exposed mice alone (Fig. 1, C and D). Overall reduction in the pancreas weight was found to be highest in mice, which continued alcohol diet for 3 and 21 days after initial ACP induction when compared with control, A, or CP alone. Conversely, withdrawal of alcohol after ACP induction resulted in the recovery of pancreas weight to normal at the end of 21 days, thus implying resolution of pancreatic damage associated with ACP after removal of alcohol (Supplemental Fig. S1C).
In addition, levels of amylase were increased in the serum of alcohol-exposed mice (Fig. 1E) suggesting acinar damage compared with control liquid diet-fed mice. ACP-induced mice showed the highest reduction in serum amylase levels compared with control, A, and CP alone (Fig. 1E), demonstrating an overall significant acinar atrophy during ACP induction.
Next, histological assessment of pancreas tissues harvested after ACP induction (both after 3 and 21 days of recovery period with continued alcohol exposure demonstrated cell death, glandular atrophy, and loss of acinar-specific amylase expression as compared with control or alcohol alone (A) exposed mice pancreata (Fig. 1, F and G). Notably, these morphological changes within the pancreas were found to be less prominent in mice exposed to A or CP alone when compared with ACP induction. Interestingly, in concordance with the previous findings by Vonlaufen et al. (7) withdrawal of alcohol after established ACP, resulted in recovery of the damaged acinar compartment and displayed histological architecture identical to control mouse pancreas when compared with ACP mice cohort fed with alcohol during the 3 and 21 days of recovery period (Fig. 1H and Supplemental Fig. S2). Taken together, these results demonstrate that ACP induction with continued alcohol triggers significant and persistent damage to the exocrine acinar cell compartment of the pancreas.
One of the hallmarks of ACP induction is the presence of parenchymal fibrosis associated with increased collagen deposition in the pancreas (32). To assess this feature, we performed Masson’s trichome staining analysis. Evaluation of fibrotic content revealed maximal collagen deposition within the pancreas after ACP induction (both after 3 and 21 days of recovery period with continued alcohol) as compared with control, A, or CP mice pancreata (Fig. 1, F and G). Interestingly, when animals were switched to a control diet for a period of 21 days of recovery after establishing ACP, we observed a drastic reduction of fibrosis after alcohol withdrawal. Conversely, continuation of an alcohol diet led to a persistent fibrotic response within the mice pancreas (Fig. 1H).
An underlying mechanism of fibrosis associated with ACP induction is the activation of PSCs toward myofibroblasts. Our immunohistochemical analysis demonstrated a significant increase in the PSC activation markers vimentin and αSMA in ACP-induced pancreatic tissues (both after 3 and 21 days of recovery period with continued alcohol) as compared with pancreata harvested from control, A, or CP mice (Fig. 1, F and G). Overall, these findings confirm that only continued alcohol intake with established ACP promotes irreversible damage to mouse pancreas and promotes fibrosis, possibly through persistent activation of PSCs, whereas withdrawal of alcohol after ACP induction leads to the reversion of these alterations during recovery.
ACP Induction Promotes Infiltration of Intrapancreatic Macrophages and Release of Proinflammatory Cytokines
Leukocytic infiltration, particularly of macrophages, has been shown to play an important role in CP fibrogenesis within the pancreatic milieu (33). However, the innate immune response during the pathogenesis of ACP with prolonged exposure to alcohol remains poorly understood. Therefore, we next assessed the changes in the immune compositions within the pancreatic tissues following ACP induction in C57BL/6 mice. Immunohistochemical analysis of immune cell macrophages (F4/80+) demonstrated a significant and maximum increase in the infiltration of these immune subsets at the site of pancreatic injury after ACP induction (with continued alcohol intake) as compared with control, A or CP alone (Fig. 2A). Next, we sought to investigate the changes in the intrapancreatic levels of inflammatory mediators, which are often associated with enhanced immune cell infiltration. Multiplex inflammation array analysis of mice pancreata obtained after ACP induction showed a significant upregulation in key inflammatory cytokines including IL-1β, IL-6, TNF-α, and SDF-1 compared with control liquid diet-fed mice (Fig. 2, B and C). Interestingly, serum-based ELISA quantification also demonstrated a significant increase in the circulating levels of these cytokines with ACP induction (3 and 21 days of recovery period with continued alcohol intake; Fig. 2D). Overall, these findings demonstrated that experimental induction of ACP with continuous alcohol exposure led to both a sustained immune response and the highest inflammatory immune response as compared with the other groups.
Figure 2.

ACP induction promotes intrapancreatic macrophage infiltration and release of proinflammatory cytokines. A: immunohistochemical localization and expression analysis of F4/80+ immune subsets within the pancreas of control, alcohol (A), CP, and ACP-induced C57BL/6 mice. For the ACP group, pancreata were harvested after 3 and 21 days of recovery period with continuation of alcohol in these mice. Quantitative analysis was done in each group. Scale bars, 50 μm. B and C: representative image of mouse inflammatory array membranes (left) and its corresponding quantification (right) obtained from pancreas harvested from control and ACP-induced with 3 days of recovery period with continuation of alcohol in C57BL/6 mice. D: ELISA-based quantitative protein estimation of proinflammatory mediators including IL-1β, IL-6, TNF-α, and SDF-1 levels in serum procured from control, A, CP, and ACP-induced (3 and 21 days of recovery period with continuation of alcohol) in C57BL/6 mice. Data points shown represent each group with means ± SD shown and compared by one-way ANOVA. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. n = 5 mice/group. ACP, alcoholic chronic pancreatitis; CP, chronic pancreatitis; ns, nonsignificant.
ACP Induction Activates PI3K/AKT and mTOR Cellular Signaling
Emerging evidence reveals that the process of pancreatic tissue damage and repair involves the activation of multiple cellular signaling networks to orchestrate inflammation, PSC activation, and subsequent fibrosis (34). Within this context, dysregulation of PI3K signaling with increased phosphorylation of RPS6, a major downstream effector of PI3K/AKT/mTOR pathway has been shown in patients with CP, highlighting the importance of this pathway in the disease (35). However, the involvement of this signaling in an inflamed pancreatic microenvironment, secondary to alcohol abuse-associated chronic pancreatitis remains unknown. Therefore, to address this, we first performed Western blot analysis of pancreatic tissue lysates obtained after alcohol exposure alone, CP, and ACP induction (Fig. 3A). As shown previously in multiple models of caerulein-induced CP, our data also showed an increase in the activation of pAKT in these mice as compared with control and A alone. Interestingly, exposure to alcohol and CP induction (ACP) together revealed the highest activation of pAKT expression (T308) as compared with the other cohorts (Fig. 3A). This further prompted us to evaluate the involvement of additional cellular kinases involved in the pathogenesis of ACP in our model. Intriguingly, phosphokinase array analysis in pancreatic tissue lysates after ACP induction in C57BL/6 mice showed top upregulated expression of the Wnt signaling component β catenin, which has been associated with pancreatitis previously (36, 37). In addition, we observed upregulated expression of several key effector kinases, including pAKT (T308), p70S6K (T421/S424), and pCREB (Ser133; Fig. 3, B and C). These signaling molecules are part of the intricate PI3K signaling network. Further validation using Western blot analysis of pancreatic tissue lysates (n = 4 mice per group) with and without ACP induction further revealed activation of AKT, p70S6K, and CREB (Fig. 3, D and E).
Figure 3.

ACP induction promotes pancreas specific activation of PI3K/AKT/mTOR/CREB signaling. A: Western blot analysis of C57BL/6 mice pancreata demonstrating highest activation of pAKT after ACP induction as compared with either control, alcohol (A), or CP-induced mice. B and C: representative image of phosphokinase array membranes (left) and its corresponding quantification (right) on pancreatic tissue lysates harvested from control, and ACP-induction with continuation of alcohol for 3 days during recovery period in C57BL/6 mice. D and E: Western blot analysis and its quantification to validate increased phosphorylated levels of pAKT, p70S6K, and CREB in mice pancreata obtained after ACP induction with continuation of alcohol for 3 days as compared with controls, n = 4 mice/group. F: representative photomicrographs of IHC based imaging analysis demonstrating acinar cell specific expression of pAKT and p70S6K in the pancreatic tissue sections of control and ACP-induced mice. Individual data points obtained from each mouse with means ± SD are reported and compared by two-tailed unpaired t test. *P < 0.05. ACP, alcoholic chronic pancreatitis; CP, chronic pancreatitis; IHC, immunohistochemistry; PI3K, phosphatidylinositol 3 kinase.
Next, we performed immunohistochemical analysis to identify the dominant cellular compartment within the pancreas expressing these signaling molecules in ACP. Our results revealed significant overexpression and localization of both pAKT (T308) and pP70 S6K(T421/S424) within the pancreatic acinar cells after ACP induction as compared with control (Fig. 3F). Taken together, these results confirmed the involvement of acinar specific PI3K/AKT/mTOR axis along with CREB activation as molecular drivers of ACP in our experimental mouse model.
Uro A Treatment in ACP-Induced Mice with Continued Alcohol Intake Suppresses the Activation of the PI3K/AKT/mTOR Signaling Axis
Uro A is a gut-derived metabolite of pomegranate phenolics that exhibits potent antioxidant and anti-inflammatory properties, and it has also been associated with multiple therapeutic benefits (23, 24). Our previous work has shown that treatment with Uro A successfully attenuated the PI3K/AKT/mTOR signaling axis in a murine model of pancreatic cancer (25). Given these properties, we hypothesized that Uro A treatment might ameliorate the progression and severity of established ACP even with continuous alcohol exposure through inhibition of the PI3K/AKT/mTOR signaling axis.
To examine this, we treated ACP-induced mice with Uro A during the last 3 wk of ACP induction (ACP + UroA). In addition, mice pancreata were harvested after day 3 and 2 wk of recovery with either continued alcohol intake or alcohol withdrawal for subsequent downstream analysis. Treatment with Uro A significantly attenuated pancreatic atrophy, as evidenced by an increase in pancreatic weight compared with untreated ACP mice exposed to continued alcohol intake during the recovery period (Fig. 4A). Interestingly, we also observed a drastic recovery in the pancreas weight (Fig. 4B), almost identical to control with Uro A treatment in ACP-induced mice with alcohol withdrawal as well, overall suggestive of the profound therapeutic benefit of this compound in ACP (Supplemental Fig. S3A).
Figure 4.

Uro A inhibits PI3K/AKT/mTOR signaling pathway in ACP-induced mouse model. A: relative pancreas weight measurements of C57BL/6 mice after ACP induction with Uro A treatment. ACP with Uro A-treated mice pancreas weight was measured after 3 days and 2 wk of recovery period with continued alcohol exposure. B: representative images of mice pancreas showing attenuation of acinar tissue atrophy with Uro A treatment in ACP-induction with continuation of alcohol for 3 days compared with untreated C57BL/6 mice. C and D: Western blot analysis along with its corresponding quantification (right) showing significant reduction in phosphorylated levels of AKT and P70S6K in pancreatic tissue lysates of Uro A-treated mice after ACP-induction compared with ACP alone. Individual data points with means ± SD are shown in each group. Statistical differences were compared by two-tailed unpaired t test. **P < 0.01; ***P < 0.001; ****P < 0.0001. n = 4–7 mice/group. ACP, alcoholic chronic pancreatitis; CP, chronic pancreatitis; ns, nonsignificant; PI3K, phosphatidylinositol 3 kinase; Uro A, urolithin A.
In addition, at the cellular level, Western blot analysis of pancreatic tissue lysates further revealed a significant and persistent reduction in the phosphorylated levels of AKT (T308), which remained reduced even after 2 wk of recovery period (with continued alcohol intake) as well as of P70S6K (T421/S424) in Uro A-treated ACP mice as compared with untreated ACP-induced mice pancreata (Fig. 4, C and D, Supplemental Fig. S3B). Overall, our results demonstrate the pivotal role of Uro A in reducing activated AKT/mTOR signaling in alcoholic pancreatitis model.
Uro A Inhibits PI3K/AKT/mTOR Signaling Axis and Attenuates the Severity of ACP in Mice
To further explore the impact of Uro A on the pancreatic architecture in alcohol-associated CP, we performed histological evaluation of these tissue sections obtained after ACP induction with Uro A treatment (after day 3 and 2 wk of recovery period with continued alcohol intake). H&E staining analysis from Uro A-treated mice revealed a drastic reduction in acinar cell death as compared with pancreata of untreated mice with ACP, which exhibited near pan-acinar cell death in analyzed tissue sections (Fig. 5A). Pancreata harvested from Uro A-treated mice demonstrated normal organization of acinar cells and were histologically identical to the pancreas of control mice, overall suggesting that this natural gut-derived metabolite promotes regression of ACP associated pancreatic injury despite continued alcohol exposure in our mice model.
Figure 5.

Uro A treatment attenuates ACP severity in C57BL/6 mice even with continuous alcohol uptake by ameliorating acinar cell damage and fibroinflammatory milieu. A: representative photomicrographs of pancreas histology in control, ACP, and ACP+Uro A treated (both after 3 days and 2 wk of recovery period with continuation of alcohol diet) C57BL/6 mice depicted through H&E based analysis, collagen levels (Sirius red and trichrome blue), and PSC activation (αSMA immunoreactivity), (left). Scale bars, 50 μm. Image J based quantitative assessment of collagen levels and αSMA expression in the pancreas (right). B: immunohistochemical staining and quantification of F4/80 expression in the pancreatic tissue sections of control, ACP, and ACP+Uro A treated with 3 and 14 days of recovery period in C57BL/6 mice. Scale bars, 50 μm. C: ELISA based estimation of serum cytokines, IL-1β, IL-6, and SDF-1, from C57BL/6 mice treated with ACP and ACP+Uro A with 3 and 14 days of recovery period. Individual data points with means ± SD are shown and compared by one-way ANOVA for multiple comparisons or by two-tailed unpaired t test for two group comparison. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. n = 5 mice/group. ACP, alcoholic chronic pancreatitis; CP, chronic pancreatitis; H&E, hematoxylin and eosin; ns, nonsignificant; Uro A, urolithin A.
We then assessed whether the treatment with Uro A impacts the overall fibrotic content and PSCs activation in ACP-induced mice. Sirius red and trichrome staining with their corresponding quantification showed a significant reduction in collagen deposition within the pancreas of ACP + Uro A compared with untreated ACP-induced mice (Fig. 5A). Interestingly, this resolution of fibrosis observed with Uro A treatment in ACP-induced mice pancreata was evident in both groups with either alcohol continuation or withdrawal during the recovery phase (Supplemental Fig. S4). In addition, this reduction in collagen levels corresponded with a marked reduction in αSMA immunoreactivity in the pancreatic tissue sections obtained from ACP + Uro A mice (after day 3 and 2 wk recovery period of alcohol continuation), (Fig. 5A), further exemplifying the therapeutic efficacy of Uro A in reducing activated PSCs as well.
Next, we examined the effects of Uro A on the infiltration of macrophages within the pancreatic microenvironment in ACP. An immunohistochemical-based assessment revealed a significant reduction in macrophage (F4/80+) populations in the pancreata of ACP + Uro (after day 3 and 2 wk of recovery period with alcohol continuation) as compared with untreated ACP mice (Fig. 5B). Interestingly, these changes in the immune subsets corresponded with a drastic reduction in the levels of soluble inflammatory mediators including IL-1β, IL-6, and SDF-1 in the serum of Uro A-treated mice (Fig. 5C) demonstrating that Uro A can suppress the systemic inflammatory response during established ACP with continued alcohol intake. Taken together, these results demonstrate Uro A inhibits PI3K/AKT/mTOR signaling, remodels the fibroinflammatory cytokine milieu, reduces infiltration of inflammatory macrophages, and decreases acinar cell injury in an experimental mouse model of ACP.
DISCUSSION
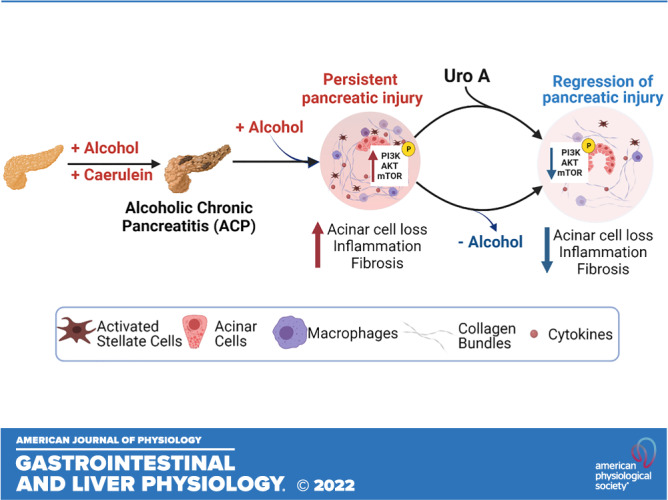
Excess alcohol consumption is one of the major etiologic factors for alcoholic pancreatitis (38), despite that, an in-depth understanding of the critical cellular drivers involved in its pathogenesis remains limited. In the present study, utilizing an experimental mouse model of ACP, we show that while continued alcohol intake perpetuates pancreatic injury, withdrawal of alcohol led to the resolution of chronic lesions with the pancreas after established ACP. The present study also identifies pancreas-specific hyperactivation of PI3K/AKT/mTOR pathways in the pathogenesis of ACP. Furthermore, we have shown that Uro A-mediated treatment substantially ameliorates fibrosis and inflammation, thereby attenuating this disease progression despite continuous alcohol trigger (Fig. 6). Therefore, these results establish a mechanistic rationale for inhibiting this signaling axis with Uro A, in the context of preventing alcohol-associated chronic pancreatitis and may benefit patients with difficulty in alcohol abstinence.
Figure 6.

Schematic demonstrating that ACP induction with continuation of alcohol intake causes severe pancreatic atrophy and promotes fibroinflammatory milieu in mice, and withdrawal of alcohol promotes regression of pancreatic injury. Treatment with Uro A attenuates ACP disease severity despite continued alcohol intake in C57BL/6 mice by inhibiting the PI3K/AKT/mTOR signaling pathway. (Image created with Bio-Render with license agreement number WO249FGLAB). ACP, alcoholic chronic pancreatitis; PI3K, phosphatidylinositol 3 kinase; Uro A, urolithin A.
Damage to the acinar cell compartment in CP is often associated with increased secretion of several inflammatory mediators. In the present study, we observed elevated levels of inflammatory cytokines including IL-1β, IL-6, TNF-α, and SDF-1 after ACP induction. These soluble inflammatory mediators have been documented to activate several cellular signaling pathways in a paracrine and autocrine manner (12). Once bound to their receptors, these cytokines activate downstream effector kinases to exert their pleiotropic effects on the various cellular compartments of the pancreas. Therefore, an in-depth understanding of the cellular complexities associated with the hallmark features of ACP is key to designing novel therapeutic strategies. We further observed activation of PI3K/AKT and mTOR signaling as a major cellular driver associated with enhanced fibroinflammatory response after ACP induction. These findings are in accordance with the previously reported role of this pathway in several phenotypic responses of pancreatitis, including modulation of cell proliferation and survival, activation of PSCs, extracellular matrix (ECM) production, and inflammation (11, 39–41).
Over the years, different relevant dietary and naturally occurring (poly) phenols have been tested as adjuvant therapeutic tools due to their high antioxidant and anti-inflammatory properties (42). The protective role of Uro A in suppressing detrimental intestinal inflammation through blocking inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 expression was previously demonstrated in an animal model of colitis (23). In addition, Singh et al. (24) showed the anti-inflammatory property of Uro A occurs via upregulation of tight junction protein claudins. Here, we have demonstrated that Uro A effectively ameliorates the fibroinflammatory milieu associated with ACP induction in C57BL/6 mice through targeted inhibition of PI3K/AKT/mTOR signaling axis. In addition, Uro A substantially prevented the systemic toxicity and weight loss that are associated with ACP induction despite continued alcohol intake in these mice. Importantly, these findings show that treatment with Uro A is well tolerated by mice and exerts its anti-inflammatory effects at a physiologically appropriate dose. These findings may have translational relevance for Uro A and related compounds in the management of established ACP disease in patients with difficulty in alcohol abstinence. However, the protective effects of this regimen are likely to be highly contingent on the correct timing and duration of therapy within the progression of acute pancreatitis to CP and should be clarified in future studies.
Similar to ACP, the fibrotic progression of CP has been characterized by infiltration of innate immune cells, predominantly macrophages, at the site of pancreatic injury in PSCs-mediated fibrosis (39). Previous studies have shown the immunomodulatory potential of Uro A on neutrophil and macrophage function by reducing oxidative stress and inflammation (43, 44). In concordance, with this, our study demonstrates Uro A treatment suppresses intrapancreatic and systemic levels of proinflammatory mediators, including IL-1β, IL-6, and SDF-1, whereas simultaneously decreasing the infiltration of F4/80+ macrophages in mice with ACP. These findings suggest that Uro A exhibits its protective, anti-inflammatory role in part by impacting the innate immune microenvironment in ACP, which has shown to be deleterious in the progression of this disease (33). Importantly, the profound protective benefits of Uro A in increasing the number of acinar cells, regression of fibroinflammatory milieu with ACP, persisted despite continuous alcohol intake in our experimental model.
Pathophysiology of pancreatitis is driven by the complex interplay of several cell types present within the inflamed microenvironment, including acinar, stellate, and immune cells. Currently, we are in the process of deciphering the compartment-specific role of PI3K/AKT/mTOR signaling axis in driving the pathogenesis of both CP and ACP with alcohol continuation. In addition, an in-depth understanding of how Uro A impacts these different cellular constituents is also underway. Overall, our present study collectively demonstrates that alcohol alone does not induce overt pancreatic damage, alcohol continuation with established pancreatitis promotes persistent acinar cell injury and fibrosis. Uro A treatment with established ACP mimics conditions of alcohol abstinence and lead to regression of pancreatic injury along with attenuation of fibroinflammatory milieu by suppressing PI3K/AKT/mTOR signaling pathway, thereby, providing substantial evidence for its potential as a therapeutic agent in the treatment of this disease.
DATA AVAILABILITY
All data generated or analyzed during this study are included in this article.
SUPPLEMENTAL DATA
Supplemental Table S1 and Supplemental Figs. S1–S4: https://doi.org/10.6084/m9.figshare.20532144.
GRANTS
This work was supported by the National Cancer Institute Grant R03 CA249401 and James Esther and King Biomedical Research Program by Florida Department of Health Grant 22K06 (to N. Nagathihalli) and the R01 CA161976 and National Institutes of Health (NIH) Grant T32 CA211034 (to N. Merchant). Histopathology Core Service was performed through the Sylvester Comprehensive Cancer Center (SCCC) support Grant by the National Cancer Institute (NCI) of the NIH under Award No. P30 CA240139.
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.M., S. Srinivasan, N.N. conceived and designed research; S.M., S. Srinivasan, Z.Z., I.D.C.S., T.M.T., X.D. performed experiments; S.M., S. Srinivasan, Z. Z., T.M.T., X.D., and N.N. analyzed data; S.M., S.S., Z.Z., T.M.T., X.D., V.R.J., M.V., and N.N. interpreted results of experiments; S.M., S.S., and N.N. prepared figures; S.M., S. Srinivasan, and S. Singh drafted manuscript; S.M., S. Srinivasan, S. Singh, Z.Z., V.G., I.D.C.S., A.R.D., R.K.D., V.R.J., C.S., J.D., M.V., N.M., and N.N. edited and revised manuscript; S.M., S. Srinivasan, S. Singh, Z.Z., V.G., I.D.C.S., T.M.T., A.R.D., X.D., R.K.D., V.R.J., C.S., J.D., M.V., N.M., and N.N. approved the final version of the manuscript.
ACKNOWLEDGMENTS
The authors thank Drs. Ifeanyichukwu Chinedu Ogobuiro, Christine Isabelle Rafie, and Ajay Dixit, for providing critical insights and editing during the preparation of the manuscript. Figure 6 was created with Bio-Render with license agreement number WO249FGLAB.
REFERENCES
- 1. Muniraj T, Aslanian HR, Farrell J, Jamidar PA. Chronic pancreatitis, a comprehensive review and update. Part I: epidemiology, etiology, risk factors, genetics, pathophysiology, and clinical features. Dis Mon 60: 530–550, 2014. doi: 10.1016/j.disamonth.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 2. Yadav D, Whitcomb DC. The role of alcohol and smoking in pancreatitis. Nat Rev Gastroenterol Hepatol 7: 131–145, 2010. doi: 10.1038/nrgastro.2010.6. [DOI] [PubMed] [Google Scholar]
- 3. Frulloni L, Gabbrielli A, Pezzilli R, Zerbi A, Cavestro GM, Marotta F, Falconi M, Gaia E, Uomo G, Maringhini A, Mutignani M, Maisonneuve P, Di Carlo V, Cavallini G. Chronic pancreatitis: report from a multicenter Italian survey (PanCroInfAISP) on 893 patients. Dig Liver Dis 41: 311–317, 2009. doi: 10.1016/j.dld.2008.07.316. [DOI] [PubMed] [Google Scholar]
- 4. Ji C. Mechanisms of alcohol-induced endoplasmic reticulum stress and organ injuries. Biochem Res Int 2012: 216450, 2012. doi: 10.1155/2012/216450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 144: 1252–1261, 2013. doi: 10.1053/j.gastro.2013.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oruc N, Whitcomb DC. Theories, mechanisms, and models of alcoholic chronic pancreatitis. Gastroenterol Clin North Am 33: 733–750, 2004. doi: 10.1016/j.gtc.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 7. Vonlaufen A, Phillips PA, Xu Z, Zhang X, Yang L, Pirola RC, Wilson JS, Apte MV. Withdrawal of alcohol promotes regression while continued alcohol intake promotes persistence of LPS-induced pancreatic injury in alcohol-fed rats. Gut 60: 238–246, 2011. doi: 10.1136/gut.2010.211250. [DOI] [PubMed] [Google Scholar]
- 8. Deng X, Wang L, Elm MS, Gabazadeh D, Diorio GJ, Eagon PK, Whitcomb DC. Chronic alcohol consumption accelerates fibrosis in response to cerulein-induced pancreatitis in rats. Am J Pathol 166: 93–106, 2005. doi: 10.1016/S0002-9440(10)62235-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhan X, Wang F, Bi Y, Ji B. Animal models of gastrointestinal and liver diseases. Animal models of acute and chronic pancreatitis. Am J Physiol Gastrointest Liver Physiol 311: G343–G355, 2016. doi: 10.1152/ajpgi.00372.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu X, Yu H, Sun L, Zheng C, Shan Y, Zhou Z, Wang C, Chen B. Adipose-derived mesenchymal stem cells ameliorate dibutyltin dichloride-induced chronic pancreatitis by inhibiting the PI3K/AKT/mTOR signaling pathway. Mol Med Rep 21: 1833–1840, 2020. doi: 10.3892/mmr.2020.10995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nishida A, Andoh A, Shioya M, Kim-Mitsuyama S, Takayanagi A, Fujiyama Y. Phosphatidylinositol 3-kinase/Akt signaling mediates interleukin-32α induction in human pancreatic periacinar myofibroblasts. Am J Physiol Gastrointest Liver Physiol 294: G831–G838, 2008. doi: 10.1152/ajpgi.00535.2007. [DOI] [PubMed] [Google Scholar]
- 12. Xu XF, Liu F, Xin JQ, Fan JW, Wu N, Zhu LJ, Duan LF, Li YY, Zhang H. Respective roles of the mitogen-activated protein kinase (MAPK) family members in pancreatic stellate cell activation induced by transforming growth factor-β1 (TGF-β1). Biochem Biophys Res Commun 501: 365–373, 2018. doi: 10.1016/j.bbrc.2018.04.176. [DOI] [PubMed] [Google Scholar]
- 13. Lesina M, Wörmann SM, Neuhöfer P, Song L, Algül H. Interleukin-6 in inflammatory and malignant diseases of the pancreas. Semin Immunol 26: 80–87, 2014. doi: 10.1016/j.smim.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 14. Collins MA, Yan W, Sebolt-Leopold JS, Pasca di Magliano M. MAPK signaling is required for dedifferentiation of acinar cells and development of pancreatic intraepithelial neoplasia in mice. Gastroenterology 146: 822–834.e7, 2014. doi: 10.1053/j.gastro.2013.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Komar HM, Serpa G, Kerscher C, Schwoegl E, Mace TA, Jin M, Yang M-C, Chen C-S, Bloomston M, Ostrowski MC, Hart PA, Conwell DL, Lesinski GB. Inhibition of Jak/STAT signaling reduces the activation of pancreatic stellate cells in vitro and limits caerulein-induced chronic pancreatitis in vivo. Sci Rep 7: 1787, 2017. doi: 10.1038/s41598-017-01973-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dienstmann R, Rodon J, Serra V, Tabernero J. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther 13: 1021–1031, 2014. doi: 10.1158/1535-7163.MCT-13-0639. [DOI] [PubMed] [Google Scholar]
- 17. Singh VP, Saluja AK, Bhagat L, van Acker GJ, Song AM, Soltoff SP, Cantley LC, Steer ML. Phosphatidylinositol 3-kinase-dependent activation of trypsinogen modulates the severity of acute pancreatitis. J Clin Invest 108: 1387–1395, 2001. doi: 10.1172/JCI12874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saluja A, Dudeja V, Dawra R, Sah RP. Early intra-acinar events in pathogenesis of pancreatitis. Gastroenterology 156: 1979–1993, 2019. doi: 10.1053/j.gastro.2019.01.268. [DOI] [PubMed] [Google Scholar]
- 19. Gukovsky I, Cheng JH, Nam KJ, Lee OT, Lugea A, Fischer L, Penninger JM, Pandol SJ, Gukovskaya AS. Phosphatidylinositide 3-kinase γ regulates key pathologic responses to cholecystokinin in pancreatic acinar cells. Gastroenterology 126: 554–566, 2004. doi: 10.1053/j.gastro.2003.11.017. [DOI] [PubMed] [Google Scholar]
- 20. Lupia E, Goffi A, De Giuli P, Azzolino O, Bosco O, Patrucco E, Vivaldo MC, Ricca M, Wymann MP, Hirsch E, Montrucchio G, Emanuelli G. Ablation of phosphoinositide 3-kinase-γ reduces the severity of acute pancreatitis. Am J Pathol 165: 2003–2011, 2004. doi: 10.1016/s0002-9440(10)63251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Heilman J, Andreux P, Tran N, Rinsch C, Blanco-Bose W. Safety assessment of urolithin A, a metabolite produced by the human gut microbiota upon dietary intake of plant derived ellagitannins and ellagic acid. Food Chem Toxicol 108: 289–297, 2017. doi: 10.1016/j.fct.2017.07.050. [DOI] [PubMed] [Google Scholar]
- 22. Espín JC, Larrosa M, García-Conesa MT, Tomás-Barberán F. Biological significance of urolithins, the gut microbial ellagic acid-derived metabolites: the evidence so far. Evid Based Complement Alternat Med 2013: 270418, 2013. doi: 10.1155/2013/270418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Larrosa M, González-Sarrías A, Yáñez-Gascón MJ, Selma MV, Azorín-Ortuño M, Toti S, Tomás-Barberán F, Dolara P, Espín JC. Anti-inflammatory properties of a pomegranate extract and its metabolite urolithin-A in a colitis rat model and the effect of colon inflammation on phenolic metabolism. J Nutr Biochem 21: 717–725, 2010. doi: 10.1016/j.jnutbio.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 24. Singh R, Chandrashekharappa S, Bodduluri SR, Baby BV, Hegde B, Kotla NG, Hiwale AA, Saiyed T, Patel P, Vijay-Kumar M, Langille MGI, Douglas GM, Cheng X, Rouchka EC, Waigel SJ, Dryden GW, Alatassi H, Zhang H-G, Haribabu B, Vemula PK, Jala VR. Enhancement of the gut barrier integrity by a microbial metabolite through the Nrf2 pathway. Nat Commun 10: 89, 2019. doi: 10.1038/s41467-018-07859-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Totiger TM, Srinivasan S, Jala VR, Lamichhane P, Dosch AR, Gaidarski AA 3rd, Joshi C, Rangappa S, Castellanos J, Vemula PK, Chen X, Kwon D, Kashikar N, VanSaun M, Merchant NB, Nagathihalli NS. Urolithin A, a novel natural compound to target PI3K/AKT/mTOR pathway in pancreatic cancer. Mol Cancer Ther 18: 301–311, 2019. doi: 10.1158/1535-7163.MCT-18-0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mehra S, Deshpande N, Nagathihalli N. Targeting PI3K pathway in pancreatic ductal adenocarcinoma: rationale and progress. Cancers (Basel) 13: 4434, 2021. doi: 10.3390/cancers13174434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. D'Souza El-Guindy NB, Kovacs EJ, De Witte P, Spies C, Littleton JM, de Villiers WJ, Lott AJ, Plackett TP, Lanzke N, Meadows GG. Laboratory models available to study alcohol-induced organ damage and immune variations: choosing the appropriate model. Alcohol Clin Exp Res 34: 1489–1511, 2010. doi: 10.1111/j.1530-0277.2010.01234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Perides G, Tao X, West N, Sharma A, Steer ML. A mouse model of ethanol dependent pancreatic fibrosis. Gut 54: 1461–1467, 2005. doi: 10.1136/gut.2004.062919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dosch AR, Dai X, Reyzer ML, Mehra S, Srinivasan S, Willobee BA, Kwon D, Kashikar N, Caprioli R, Merchant NB, Nagathihalli NS. Combined Src/EGFR inhibition targets STAT3 signaling and induces stromal remodeling to improve survival in pancreatic cancer. Mol Cancer Res 18: 623–631, 2020. doi: 10.1158/1541-7786.MCR-19-0741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Willobee BA, Gaidarski AA, Dosch AR, Castellanos JA, Dai X, Mehra S, Messaggio F, Srinivasan S, VanSaun MN, Nagathihalli NS, Merchant NB. Combined blockade of MEK and CDK4/6 pathways induces senescence to improve survival in pancreatic ductal adenocarcinoma. Mol Cancer Ther 20: 1246–1256, 2021. doi: 10.1158/1535-7163.MCT-19-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nagathihalli NS, Castellanos JA, Lamichhane P, Messaggio F, Shi C, Dai X, Rai P, Chen X, VanSaun MN, Merchant NB. Inverse correlation of STAT3 and MEK signaling mediates resistance to RAS pathway inhibition in pancreatic cancer. Cancer Res 78: 6235–6246, 2018. doi: 10.1158/0008-5472.CAN-18-0634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gukovsky I, Lugea A, Shahsahebi M, Cheng JH, Hong PP, Jung YJ, Deng QG, French BA, Lungo W, French SW, Tsukamoto H, Pandol SJ. A rat model reproducing key pathological responses of alcoholic chronic pancreatitis. Am J Physiol Gastrointest Liver Physiol 294: G68–G79, 2008. doi: 10.1152/ajpgi.00006.2007. [DOI] [PubMed] [Google Scholar]
- 33. Xue J, Sharma V, Hsieh MH, Chawla A, Murali R, Pandol SJ, Habtezion A. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat Commun 6: 7158, 2015. doi: 10.1038/ncomms8158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu J, Gao M, Nipper M, Deng J, Sharkey FE, Johnson RL, Crawford HC, Chen Y, Wang P. Activation of the intrinsic fibroinflammatory program in adult pancreatic acinar cells triggered by Hippo signaling disruption. PLoS Biol 17: e3000418, 2019. doi: 10.1371/journal.pbio.3000418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bellizzi AM, Bloomston M, Zhou X-P, Iwenofu OH, Frankel WL. The mTOR pathway is frequently activated in pancreatic ductal adenocarcinoma and chronic pancreatitis. Appl Immunohistochem Mol Morphol 18: 442–447, 2010. doi: 10.1097/PAI.0b013e3181de115b. [DOI] [PubMed] [Google Scholar]
- 36. Siveke JT, Lubeseder-Martellato C, Lee M, Mazur PK, Nakhai H, Radtke F, Schmid RM. Notch signaling is required for exocrine regeneration after acute pancreatitis. Gastroenterology 134: 544–555, 2008. doi: 10.1053/j.gastro.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 37. Eisses JF, Criscimanna A, Dionise ZR, Orabi AI, Javed TA, Sarwar S, Jin S, Zhou L, Singh S, Poddar M, Davis AW, Tosun AB, Ozolek JA, Lowe ME, Monga SP, Rohde GK, Esni F, Husain SZ. Valproic acid limits pancreatic recovery after pancreatitis by inhibiting histone deacetylases and preventing acinar redifferentiation programs. Am J Pathol 185: 3304–3315, 2015. doi: 10.1016/j.ajpath.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Whitcomb DC, LaRusch J, Krasinskas AM, Klei L, Smith JP, Brand RE, et al. Common genetic variants in the CLDN2 and PRSS1-PRSS2 loci alter risk for alcohol-related and sporadic pancreatitis. Nat Genet 44: 1349–1354, 2012. doi: 10.1038/ng.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lupia E, Pigozzi L, Goffi A, Hirsch E, Montrucchio G. Role of phosphoinositide 3-kinase in the pathogenesis of acute pancreatitis. World J Gastroenterol 20: 15190–15199, 2014. doi: 10.3748/wjg.v20.i41.15190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Birtolo C, Go VL, Ptasznik A, Eibl G, Pandol SJ. Phosphatidylinositol 3-kinase: a link between inflammation and pancreatic cancer. Pancreas 45: 21–31, 2016. doi: 10.1097/MPA.0000000000000531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McCarroll JA, Phillips PA, Kumar RK, Park S, Pirola RC, Wilson JS, Apte MV. Pancreatic stellate cell migration: role of the phosphatidylinositol 3-kinase(PI3-kinase) pathway. Biochem Pharmacol 67: 1215–1225, 2004. doi: 10.1016/j.bcp.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 42. Savi M, Bocchi L, Sala R, Frati C, Lagrasta C, Madeddu D, Falco A, Pollino S, Bresciani L, Miragoli M, Zaniboni M, Quaini F, Del Rio D, Stilli D. Parenchymal and stromal cells contribute to pro-inflammatory myocardial environment at early stages of diabetes: protective role of resveratrol. Nutrients 8: 729, 2016. doi: 10.3390/nu8110729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Komatsu W, Kishi H, Yagasaki K, Ohhira S. Urolithin A attenuates pro-inflammatory mediator production by suppressing PI3-K/Akt/NF-κB and JNK/AP-1 signaling pathways in lipopolysaccharide-stimulated RAW264 macrophages: possible involvement of NADPH oxidase-derived reactive oxygen species. Eur J Pharmacol 833: 411–424, 2018. doi: 10.1016/j.ejphar.2018.06.023. [DOI] [PubMed] [Google Scholar]
- 44. Saha P, Yeoh BS, Singh R, Chandrasekar B, Vemula PK, Haribabu B, Vijay-Kumar M, Jala VR. Gut microbiota conversion of dietary ellagic acid into bioactive phytoceutical urolithin A inhibits heme peroxidases. PLoS One 11: e0156811, 2016. doi: 10.1371/journal.pone.0156811. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1 and Supplemental Figs. S1–S4: https://doi.org/10.6084/m9.figshare.20532144.
Data Availability Statement
All data generated or analyzed during this study are included in this article.