

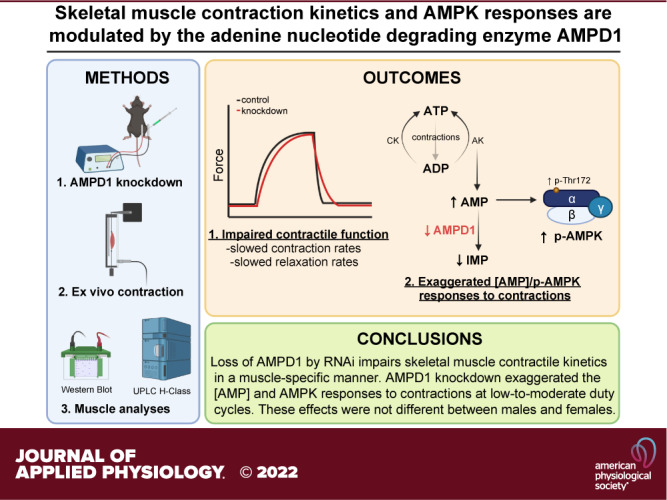
Keywords: AMP-activated protein kinase, AMP deaminase, contraction, relaxation, sex differences
Abstract
AMP deaminase 1 (AMPD1; AMP → IMP + NH3) deficiency in skeletal muscle results in an inordinate accumulation of AMP during strenuous exercise, with some but not all studies reporting premature fatigue and reduced work capacity. To further explore these inconsistencies, we investigated the extent to which AMPD1 deficiency impacts skeletal muscle contractile function of different muscles and the [AMP]/AMPK responses to different intensities of fatiguing contractions. To reduce AMPD1 protein, we electroporated either an inhibitory AMPD1-specific miRNA encoding plasmid or a control plasmid, into contralateral EDL and SOL muscles of C57BL/6J mice (n = 48 males, 24 females). After 10 days, isolated muscles were assessed for isometric twitch, tetanic, and repeated fatiguing contraction characteristics using one of four (None, LOW, MOD, and HIGH) duty cycles. AMPD1 knockdown (∼35%) had no effect on twitch force or twitch contraction/relaxation kinetics. However, during maximal tetanic contractions, AMPD1 knockdown impaired both time-to-peak tension (TPT) and half-relaxation time (½ RT) in EDL, but not SOL muscle. In addition, AMPD1 knockdown in EDL exaggerated the AMP response to contractions at LOW (+100%) and MOD (+54%) duty cycles, but not at HIGH duty cycle. This accumulation of AMP was accompanied by increased AMPK phosphorylation (Thr-172; LOW +25%, MOD +34%) and downstream substrate phosphorylation (LOW +15%, MOD +17%). These responses to AMPD1 knockdown were not different between males and females. Our findings demonstrate that AMPD1 plays a role in maintaining skeletal muscle contractile function and regulating the energetic responses associated with repeated contractions in a muscle- but not sex-specific manner.
NEW & NOTEWORTHY AMP deaminase 1 (AMPD1) deficiency has been associated with premature muscle fatigue and reduced work capacity, but this finding has been inconsistent. Herein, we report that although AMPD1 knockdown in mouse skeletal muscle does not change maximal isometric force, it negatively impacts muscle function by slowing contraction and relaxation kinetics in EDL muscle but not SOL muscle. Furthermore, AMPD1 knockdown differentially affects the [AMP]/AMPK responses to fatiguing contractions in an intensity-dependent manner in EDL muscle.
INTRODUCTION
During exercise, repeated contractions of skeletal muscle require a tremendous amount of chemical energy, with rates of ATP hydrolysis increasing up to 100-fold from rest (1). As ATP is rapidly hydrolyzed to ADP, the ATP/ADP levels are buffered primarily by the near equilibrium reactions of two enzymes: creatine kinase (PCr + ADP ↔ ATP + Cr) and adenylate kinase (ADP + ADP ↔ ATP + AMP). Thus, the breakdown of ATP to ADP shifts both reactions toward ATP resynthesis. In the case of adenylate kinase, this also results in the formation of adenosine monophosphate (AMP). In order for the adenylate kinase reaction to continue proceeding toward ATP formation, the enzyme AMP deaminase (AMPD) catalyzes the thermodynamically irreversible deamination of AMP to inosine monophosphate (AMP → IMP + NH3). In individuals with AMPD deficiency, this results in an inordinate accumulation of AMP during exercise, with reports of premature fatigue, cramping, and myalgia in some (2–6) but not in all studies (7–11). Interestingly, deficiency of the skeletal muscle AMPD isoform 1 (AMPD1) is among the most common genetic abnormalities of human skeletal muscle, with an allele mutation frequency of 12%–14% and complete loss of AMPD1 in 2% of the general population (6, 12–16). Despite the high prevalence of AMPD1 deficiency, the link between AMPD1 and skeletal muscle function has not been fully established, and the variable impact of AMPD1 deficiency on exercise performance remains a topic of debate.
The discrepant reports on the effect of reduced AMPD1 activity on physical performance may be the result of differences in exercise intensity or muscle fiber type. For example, while mutant allele frequency does tend to be lower in endurance athletes (17) than in the general population, it does not seem to impair elite endurance performance (18, 19). In addition, submaximal endurance exercise testing has not uncovered any negative physiological or performance outcomes related to AMPD1 deficiency (6). However, as exercise intensity increases toward maximal, the resulting accumulation of AMP (7) is accompanied by reductions in maximal power output (6, 20, 21). Not coincidentally, AMPD1 deficiency is even more infrequent in elite sprint and power-oriented athletes than in endurance athletes and is characterized by inferior anaerobic exercise performance (19, 22). Such observations align with reports of higher AMPD1 content in fast-twitch/glycolytic, compared with slow-twitch/oxidative muscle (23–25). Therefore, AMPD1 may play a more prominent role in regulating muscle contractile function during high-intensity contractions rather than low-intensity contractions, in a muscle-dependent manner. However, this contribution across intensities and between different muscles has not been defined. Alternatively, the preclusion of AMPD1 deficient athletes from elite power-oriented sports might not be due to direct alterations in the energetic responses to exercise, but rather the potential adaptations mediated by those responses over time (i.e., AMPK inhibition of protein synthesis and activation of mitochondrial biogenesis).
AMPD1 has gained considerable interest as a potential target for regulating AMP-activated protein kinase (AMPK) and improving insulin sensitivity in skeletal muscle (26, 27). However, its role in the regulation of nucleotides and cellular AMPK responses in contracting skeletal muscle has only been studied at high contraction workloads (23, 28). Although it is well established that the energetic demands of skeletal muscle contractions lead to increases in [AMP] and AMPK phosphorylation in a workload-dependent manner (29, 30), how AMPD1 deficiency impacts the [AMP]/p-AMPK responses to different workloads in different muscles has not been characterized.
Therefore, our main objectives were 1) to define the impact of AMPD1 deficiency on contractile function of different muscles composed of either mostly fast-twitch glycolytic [extensor digitorum longus (EDL); ∼88% type IIb and ∼10% type IIa (31, 32)] or more slow-twitch oxidative fiber-types [soleus (SOL); ∼46% type I and ∼54% type IIa (31, 32)] and 2) to characterize the effect of AMPD1 deficiency on the [AMP]/p-AMPK responses to contractions of different intensities within these muscles. Recently, AMPD1 has been found to localize in areas that experience high ATP turnover during contractions—the sarcomeric I-band (33) and the sarcoplasmic reticulum (34, 35). Since AMPD1 is localized at the sarcomere and the sarcoplasmic reticulum, and both actin-myosin binding (36, 37) and Ca2+ kinetics (38–40) in skeletal muscle are dependent upon the adenine nucleotides, we hypothesized that AMPD1 knockdown in skeletal muscle would impair contraction and relaxation kinetics. Furthermore, although lower exercise workloads are traditionally associated with lower [AMP]/p-AMPK responses in skeletal muscle, they still lead to increased IMP and NH3 (41, 42)—suggesting active AMP deamination. Therefore, we also hypothesized that AMPD1 knockdown in skeletal muscle would exaggerate the relative [AMP]/p-AMPK responses to fatiguing contractions in a workload (duty cycle)-dependent manner.
METHODS
Seventy-two (48 males, 24 females) adult C57BL/6J mice (∼16–20 wk) were acquired from Jackson Laboratories, housed in groups of two to five per cage with free access to food and water, and kept on a 12-h light/dark cycle. Prior to beginning our investigations, we obtained approval for these studies from the Indiana University School of Medicine (IUSM) Animal Care and Use Committee (IACUC Protocol No. 19149). All procedures complied with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines.
Plasmid Electroporation into Skeletal Muscle
We electroporated an inhibitory microRNA (RNAi) encoding plasmid into skeletal muscle to knock down AMPD1 protein expression. To this end, we engineered a mouse AMPD1-specific miRNA oligo using Invitrogen’s web-based RNAi Designer (Thermo Fisher):
5′- TGCTGTAATGAAGCGCAGCAGGTGCTGTTTTGGCCACTGACTGACAGCACCTGGCGCTTCATTA-3′.
The oligo was annealed, cloned, and transformed into chemically competent E. coli using the BLOCK-iT Pol II MiR RNAi Expression Vector Kit (Invitrogen). As part of our study design, we also used the negative control miRNA encoding plasmid included with the vector kit, as it was designed to not target any known vertebrate gene:
5′- GAAATGTACTGCGCGTGGAGACGTTTTGGCCACTGACTGACGTCTCCACGCAGTACATTT-3′.
We electroporated the encoding plasmids into the EDL and SOL muscles according to our established protocols (43–45). To start, we anesthetized the mice using 2% vaporized isoflurane (Piramal Critical Care) and provided a subcutaneous injection of sustained-release buprenorphine (3.25 mg/kg body mass; EthiqaXR, Fidelis Pharmaceuticals) for analgesia. The fur was shaved from the lower hindlimbs, and the skin was scrubbed with a 10% povidone-iodine solution and cleaned with 70% isopropyl alcohol. We then made a ∼1 cm incision longitudinally in the skin over the belly of the tibialis anterior (TA) muscle. We slid a plate electrode underneath the EDL muscle and injected either the AMPD1 RNAi encoding plasmid or negative control plasmid (5 µg in a 0.9% NaCl saline solution, at a final volume of 8 µL) using a 30-gauge Hamilton syringe. The opposing electrode was placed on top of the EDL, and five electrical pulses (5 V, 20 ms duration, 200 ms delay interval) were delivered using an ECM 830 Electroporator (BTX Harvard Apparatus). The soleus muscle was electroporated through the same incision using the same plasmid amount/volume and electrical pulse conditions. The incision was closed using a 5-0 Prolene suture (FS-2, Ethicon). We repeated the electroporation protocol for the contralateral EDL and SOL muscles to ensure that an EDL and SOL of each mouse received either the AMPD1 RNAi plasmid or negative control. All four muscles were collected 10 days after electroporation. Since plasmid electroporation does not result in plasmid delivery to all muscle fibers (46), it is likely that our whole muscle approach to investigating the effect of AMPD1 knockdown via RNAi plasmid electroporation underestimates the magnitude of the effect that would be observed in individual fibers.
Ex Vivo Muscle Contractions
Mice were anesthetized using a ketamine (90 mg/kg) and xylazine (10 mg/kg) cocktail, delivered intraperitoneally. As xylazine is a central nervous system (CNS) depressant and reduces heart rate and blood oxygenation (47, 48), we performed the muscle collection procedure while providing supplemental O2 through a nose cone. To isolate and remove the EDL, we carefully pulled away the skin of the lower hindlimb, cut out the TA, and sutured (4-0 Braided Silk Suture, Teleflex Medical) both the proximal and distal tendons. To isolate and remove the SOL, we tied off the distal Achilles tendon, peeled back the gastrocnemius/plantaris/SOL complex, and sutured the proximal tendon. We were then able to cut the proximal tendon above the knot and pull back the SOL, carefully isolating it from the rest of the complex. We kept the muscles in Krebs–Henseleit buffer (25 mM NaHCO3, 118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 1.2 mM CaCl2) with added glucose (5 mM) and sodium pyruvate (0.15 mM). Upon completion of the muscle collection procedure, death was confirmed by cervical dislocation.
Muscles were suspended vertically in a warmed (37°C) tissue bath (Aurora Scientific) containing Krebs–Henseleit buffer solution and gassed continuously with a 95% O2/5% CO2 mixture to a pH of 7.4. Muscles were suspended at a resting tension of ∼8 mN for 10 min. We then determined optimal length and measured isometric twitch, tetanic, and fatigue contraction parameters using a 2-Channel Dual-Mode muscle lever system (1200A: Isolated Muscle System—Mouse; Aurora Scientific). We designed and carried out all muscle function testing using 610A Dynamic Muscle Control software (DMC LabBook v6.000, Aurora Scientific). We used high throughput analysis to collect the data from all tests using 611 A Dynamic Muscle Analysis software (DMA v.5.501, Aurora Scientific).
To determine optimal length, we delivered single-twitch pulses (5 V), each separated by 30 s, and adjusted the length of the suspended muscle until a maximal twitch was evoked. We defined the muscle length that evoked maximal twitch force as the muscle optimal length (Lo) and used Lo to estimate muscle cross-sectional area (CSA) for normalization of muscle raw force (Po) to specific force (sPo) (49).
Approximately 4–5 min after the final twitch, we tested maximal tetanic force by applying a train of pulses (150 Hz) for either 200 ms (EDL) or 400 ms (SOL). We defined maximal tetanic force as the highest force elicited after three stimulations, each separated by 5 min. Following the final 5 min rest, we applied one of four, 5-min fatigue protocols, according to duty cycle (tetanus duration/cycle duration; Supplemental Table S1; see https://doi.org/10.6084/m9.figshare.18726560.v1), for both the EDL (None = 0%, LOW = 1%, MOD = 2%, HIGH = 4%) and SOL (None = 0%, LOW = 3%, MOD = 7%, HIGH = 13%). We chose these duty cycles as they extended the range of isometric muscle contraction “workloads” of previous investigations in AMPD1-deficient muscle (23). We also increased the duty cycle length in SOL to promote similar fatigue profiles between the EDL and more fatigue-resistant SOL muscles. Immediately following the final contraction of the fatigue protocols, we blotted the muscles dry and rapidly froze them using liquid nitrogen-cooled steel clamps. We measured muscle mass to the nearest 0.01 mg, keeping the samples frozen, and stored them at −80°C until processing for nucleotide (ultra-performance liquid chromatography, UPLC) and protein (immunoblotting) analyses.
Ultra-Performance Liquid Chromatography
We measured adenine nucleotides (ATP, ADP, and AMP) and degradation products (IMP, inosine, hypoxanthine, xanthine) in the skeletal muscles of half of the animals (n = 24 males, 12 females), as previously established (43, 50, 51). We homogenized EDL and SOL muscles in ice-cold 0.5 N perchloric acid (PCA) with added EDTA (5 mM) using glass-on-glass pestles and tubes (Duall 20, Kontes Glass Company). We then centrifuged homogenates at 20,000 g for 5 min at 4°C and neutralized the supernatant with ice-cold 1 N KOH. The samples were put on ice for 5 min and then centrifuged at 20,000 g for 5 min at 4°C to remove any remaining precipitated proteins and the potassium-perchlorate salts. We measured adenine nucleotides and degradation products using a Waters UPLC H-Class Biosystem (Waters Corp.) with an Acquity UPLC HSS T3 1.8 µm, 2.1 mm × 150 mm column (Waters Corp.). We quantified adenine nucleotides and degradation products by absorbance after calibration using a standard curve from known concentrations of each nucleotide/degradation product, precisely as we have done previously (43, 50, 51).
Protein Immunoblotting
For immunoblotting, we homogenized EDL and SOL muscles from the remaining animals (n = 24 males, 12 females), using a Bio-Gen PRO200 Homogenizer (Pro Scientific), to a 60-fold dilution in ice-cold radio-immunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) with added protease (cOmplete Mini, EDTA-free, Roche Diagnostics) and phosphatase (PhosSTOP, Roche Diagnostics) inhibitors. Following homogenization, samples were rotated end-over-end for 1 h at 4°C and centrifuged at 13,000 g for 30 min at 4°C. We removed the soluble protein-containing supernatant and measured total protein content using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). We combined a small amount of each sample (∼4 µL) to make a universal loading control that was used for normalization of protein quantification between blots. Samples were diluted to a protein concentration of 1 µg/µL in Laemmli’s Buffer (62.5 mM Tris-HCl, 2.5% SDS, 10% glycerol, 0.005% bromophenol blue, 100 mM DTT) and heated for 5 min at 95°C.
Proteins were separated by SDS-PAGE using the Mini-PROTEAN Tetra System (Bio-Rad) with stain-free acrylamide gels (TGX Stain-Free FastCast Acrylamide Kit 7.5%, Bio-Rad). We loaded 10 µg of total protein into each well and then ran the gels at 120 V for either 1 h (AMPD1, AMPK antibodies), 1 h 30 min (SERCA1 and SERCA2) or 1 h 45 min (AMPD3). We then transferred proteins to a polyvinylidene difluoride (PVDF) membrane (Immun-Blot LF PVDF, Bio-Rad) via electrophoretic transfer at 80 V for 1 h. We confirmed equal loading and transfer of proteins after photoactivation of gels and subsequent stain-free blot imaging (ChemiDoc Imaging System, Bio-Rad). Following transfer, we blocked the membranes with 5% bovine serum albumin (BSA) in Tris-buffered saline plus 0.1% Tween-20 (TBS-T; 10 mM Trizma base, 0.15 M NaCl, 0.1% Tween-20, pH 7.8). We applied the following primary antibodies in TBS-T with 5% BSA, at the indicated dilutions, overnight at 4°C: AMPD1 (PA5-23173 at 1:1,000, Invitrogen RRID: AB_2540699), AMPD3 (PA5-76912 at 1:2,000, Invitrogen RRID: AB_2720639), AMPKα (D5A2-5831 at 1:2,000, Cell Signaling Technology RRID: AB_10622186), phospho-AMPKα (Thr172-2531 at 1:2,000, Cell Signaling Technology RRID: AB_330330), phospho-AMPK Substrate Motif (LXRXX(pS/pT)-5759 at 1:2,000, Cell Signaling Technology RRID: AB_10949320), SERCA1 (ab129104 at 1:2,000, abcam RRID: AB_11143243), and SERCA2 (ab150435 at 1:5,000, abcam RRID: AB_2910256). After removing the primary antibodies, we rinsed the membranes with TBS-T, washed the membrane 3 × 5 min in TBS-T, and applied an anti-Rabbit secondary antibody (Anti-rabbit IgG, HRP-linked-7074 at 1:5000, Cell Signaling Technology RRID: AB_2099233) in TBS-T with 5% BSA for 1 h at room temperature. Finally, we rinsed off the secondary antibody with TBS-T, washed 3 × 5 min in TBS-T, and applied a chemiluminescent HRP substrate (Immobilon Western, EMD Millipore) for visualization and quantification of our proteins of interest using the ChemiDoc Imaging System (Bio-Rad) and Image Lab software (v6.0.1, Bio-Rad). All densitometries across lanes were corrected for total protein loaded in each well (measured by stain-free photoactivation) and subsequently normalized to our universal loading control for internal consistency between blots.
Statistics
We used a mixed-model analysis of variance (ANOVA) to determine the effects of sex (male vs. female), plasmid (RNAi vs. Con), and duty cycle (None vs. LOW vs. MOD vs. HIGH). Normality of data was confirmed using the D’Agostino-Pearson normality tests and heteroscedasticity was tested using Spearman’s rank tests. We did not observe any three-way interactions (sex × plasmid × duty cycle) following our mixed-model ANOVA. We therefore turned our attention to exploring plasmid × duty cycle interactions. We carried out post hoc tests only in the event of a significant interaction, using paired t tests with Bonferroni corrections for multiple comparisons. We used GraphPad Prism 9 statistical software (v.9.2.0) to perform all statistical analyses. We set our α a priori at 0.05 and have reported all data as means ± SD.
RESULTS
Muscle Characteristics and AMPD1 Knockdown
The male C57BL/6J mice had greater body mass, EDL mass, and SOL mass than females (P < 0.0001; Supplemental Fig. S1, A–C; see https://doi.org/10.6084/m9.figshare.18726551.v1). However, the differences in muscle mass were explained by the differences in body size, as we did not observe any effect of sex on muscle mass after normalizing to body mass (Supplemental Fig. S1, D and E). In addition, we did not observe any differences in muscle mass between the contralateral muscles that had been treated with either the control plasmid or AMPD1 knockdown (RNAi) plasmid (Supplemental Fig. S1, B–E).
Electroporation of the RNAi encoding plasmid into EDL and SOL muscles reduced AMPD1 protein expression to approximately the same degree in both muscles: −35% in the EDL and −31% in the SOL (Fig. 1, A and B; P < 0.0001). Basal AMPD1 levels (control muscles) were 85% lower in SOL compared with EDL (P < 0.0001; Fig. 1C). Importantly, our knockdown of AMPD1 did not affect expression of the other skeletal muscle AMPD isoform 3, (AMPD3; Fig. 1, D and E) in either muscle. AMPD3 isoform expression was 45-fold higher in control SOL, compared with control EDL (P < 0.0001; Fig. 1F).
Figure 1.

AMP deaminase 1 (AMPD1) knockdown and AMPD isoform expression in EDL and SOL muscle. Ten days after electroporation of the AMPD1-specific miR inhibitory plasmid (RNAi), AMPD1 protein content was reduced in both EDL (A) and SOL (B) muscle. AMPD1 was more abundant in EDL (C) compared with SOL muscle. AMPD1 knockdown did not change AMPD3 content in EDL (D) or SOL (E) muscle. In SOL muscle, AMPD3 protein (F) was more abundant than in EDL muscle. Representative blots and total protein loads imaged after photoactivation for EDL (G) and SOL (H) muscles are provided. *P < 0.05 main effect of treatment (RNAi plasmid) by ANOVA. Connecting lines were drawn to indicate muscles taken from the same animal. n = 24 males and 12 females.
Skeletal Muscle Contraction Kinetics
To test whether loss of AMPD1 would impact muscle contraction characteristics, EDL and SOL muscles were isolated and electrically stimulated ex vivo. AMPD1 knockdown in EDL did not affect twitch force (Fig. 2, A and B), time-to-peak tension (TPT; Fig. 2C), or half-relaxation time (½ RT; Fig. 2D). AMPD1 knockdown also had no impact on maximal isometric force (Fig. 2, E and F). However, during maximal tetanic contractions, AMPD1 knockdown delayed both TPT (19.0 ± 0.02 vs. 19.2 ± 0.02 s; P < 0.0001; Fig. 2G) and ½ RT (17.6 ± 0.62 vs. 18.87 ± 1.20 s; P < 0.0001; Fig. 2H). There were main sex effects, with female mice displaying lower absolute force (P < 0.0001), slower TPT (p < 0.0001), and slower ½ RT (P = 0.0122). However, there were no sex × plasmid interactions, suggesting similar treatment effects between sexes. Thus, despite having no impact on force-generating capacity, AMPD1 knockdown impaired EDL contraction and relaxation kinetics during a single maximal isometric contraction.
Figure 2.

Twitch and isometric contraction characteristics in EDL muscle following AMP deaminase 1 (AMPD1) knockdown. Loss of AMPD1 (inhibitory microRNA, RNAi) had no effect on maximal twitch force (A and B), time-to-peak tension (TPT; C), or half-relaxation time (1/2 RT, D). While RNAi did not affect maximal isometric force (E and F), it did increase both TPT (G) and 1/2 RT (H). #P < 0.05 main effect of sex; *P < 0.05 main effect of treatment (RNAi plasmid) by ANOVA. Connecting lines were drawn to indicate muscles taken from the same animal. n = 48 males and 24 females.
In SOL muscle, AMPD1 knockdown did not affect maximal twitch force, twitch TPT, or ½ RT (Supplemental Fig. S2; see https://doi.org/10.6084/m9.figshare.18726548.v1). Similarly, AMPD1 knockdown in SOL did not alter maximal tetanic force, tetanic TPT, or tetanic ½ RT (Supplemental Fig. S2). Similar to what was observed in EDL, female mice also displayed lower SOL absolute force (P < 0.0001), and slower twitch kinetics (P < 0.0001). Therefore, our knockdown of AMPD1 did not negatively impact the force-generating capacity or contraction/relaxation kinetics of SOL muscle.
Skeletal Muscle Function during Fatiguing Contractions
To examine the effect of AMPD1 knockdown on muscle contractile function during the progression of fatigue, repeated muscle contractions were elicited for 5 min at various duty cycles (Supplemental Table S1). EDL force production was reduced in a dose-dependent manner following LOW (−25%, Fig. 3A), MOD (−55%; Fig. 3B), and HIGH (−70%; Fig. 3C) intensities. AMPD1 knockdown did not affect force production during LOW (Fig. 3A), MOD (Fig. 3B), or HIGH (Fig. 3C) intensities. However, there were significant time × plasmid (RNAi vs. Control) interactions for TPT and ½ RT, indicating slower kinetics, throughout all three fatigue protocols (Fig. 3, D–F; P < 0.0001). These differences were similar for muscle from both male and female mice, as there was not an interactive effect of sex on force production, TPT, or ½ RT.
Figure 3.

Effect of AMP deaminase 1 (AMDP1) knockdown on EDL contractile characteristics during fatiguing contractions. Inhibitory microRNA (RNAi) knockdown of AMPD1 had no effect on force loss during the 5-min protocols of LOW (A), MOD (B), or HIGH fatigue (C). RNAi slowed time-to-peak tension (TPT) during LOW (D), MOD (E), and HIGH (F). Furthermore, RNAi impaired half-relaxation time (½ RT) during LOW (G), MOD (H), and HIGH (I). *P < 0.05 main effect of treatment (RNAi) by ANOVA. Data are represented as means ± SD, n = 10–12 males and 5 or 6 females per group.
In SOL muscle, the 5-min fatiguing contractions were carried out at higher duty cycles than those used for EDL muscle, to produce similar fatigue profiles (Supplemental Table S1). SOL force production was reduced in a dose-dependent manner following LOW (−24%, Supplemental Fig. S3A; see https://doi.org/10.6084/m9.figshare.18726554.v1), MOD (−52%; Supplemental Fig. S3B), and HIGH (−68%; Supplemental Fig. S3C) intensities. AMPD1 knockdown in SOL muscle did not have any effect on force production, TPT, or ½ RT during fatiguing contractions. Our findings suggest that while AMPD1 knockdown does not affect maximal force generating capacity in either EDL or SOL muscle, it does impair contraction/relaxation kinetics in EDL muscle.
[AMP]/p-AMPK-Responses to Fatiguing Contractions
The fatigue protocols used for this study resulted in dose-dependent shifts of the cellular energetic state in both EDL and SOL. These shifts were similar for both males and females (no effect of sex). Therefore, we pooled the data and presented individual values for males and females together. In EDL, there was a significant main effect of duty cycle on all adenine nucleotides (Fig. 4, A–C; all P < 0.05) and degradation products (Table 1; P < 0.05). As duty cycle increased in duration, [ATP] decreased, whereas [AMP] and all other degradation products concomitantly increased. Following fatiguing contractions, AMPD1 knockdown in EDL did not affect total [ATP] or [ADP] (Fig. 4, A and B). However, knockdown of AMPD1 did exaggerate the AMP response to LOW (+125%, P = 0.001) fatiguing contractions, with an increasing trend following MOD (+27%, P = 0.07), but not HIGH (Fig. 4C), contractions. As expected, AMPD1 knockdown reduced cellular [IMP] and [inosine] under baseline and all fatigue conditions (main effect P = 0.0006; Table 1). The exaggerated AMP responses observed with AMPD1 knockdown were accompanied by greater [AMP]/[ATP] ratios with LOW (+93%, P = 0.03) and MOD (+50%, P = 0.003; Fig. 4D) fatigue. AMPD1 knockdown concomitantly exaggerated AMPK phosphorylation after LOW (+18%, P = 0.02) and MOD (+40%, P = 0.02; Fig. 4E) fatigue, as well as, AMPK substrate phosphorylation after LOW (+14%, P = 0.02) and MOD (+17%, P = 0.002; Fig. 4F) fatigue. Taken together, these data demonstrate that AMPD1 helps regulate the AMP and p-AMPK responses to contractions in a workload-dependent manner.
Figure 4.

Impact of AMP deaminase 1 (AMPD1) knockdown (inhibitory microRNA, RNAi) on the [AMP]/AMPK responses to fatiguing contractions in EDL muscle. RNAi had no effect on cellular [ATP] (A) or [ADP] (B) at all contraction duty cycles. RNAi increased [AMP] (C) and the [AMP]/[ATP] (D) ratios following LOW to MOD fatigue, but not HIGH fatigue. In addition, RNAi exaggerated AMPK phosphorylation (E) and downstream AMPK substrate phosphorylation (F) after LOW to MOD fatigue. G: representative trace recording of a typical elution from a contracted muscle. H: representative blots and total protein image following photoactivation. *P < 0.05 duty cycle × plasmid (RNAi) interaction by ANOVA with subsequent post hoc analyses using paired t test with Bonferroni corrections. Connecting lines represent contralateral muscles (control vs. RNAi) taken from the same animal. Data are represented as means ± SD, n = 6 males and 3 females per group.
Table 1.
Degradation products in EDL muscle following fatiguing contractions
| Fatigue Group |
|||||
|---|---|---|---|---|---|
| Cx, µmol/g | Treatment | None | LOW | MOD | HIGH |
| IMP*^ | Control | 0.087 ± 0.081 | 0.183 ± 0.092 | 0.862 ± 0.487 | 2.555 ± 0.783 |
| RNAi | 0.062 ± 0.045 | 0.134 ± 0.114 | 0.601 ± 0.405 | 1.923 ± 0.888 | |
| Inosine*^ | Control | - | 0.005 ± 0.004 | 0.026 ± 0.021 | 0.063 ± 0.030 |
| RNAi | - | 0.004 ± 0.004 | 0.030 ± 0.023 | 0.047 ± 0.027 | |
| Hypoxanthine* | Control | - | - | 0.007 ± 0.005 | 0.020 ± 0.008 |
| RNAi | - | - | 0.009 ± 0.007 | 0.019 ± 0.008 | |
| Xanthine* | Control | - | - | - | 0.005 ± 0.003 |
| RNAi | - | - | - | 0.006 ± 0.005 | |
IMP, inosine monophosphate; iRNA, inhibitory microRNA. For each group, control and RNAi muscles were taken from the same animal. Data are represented as means ± SD, n = 6 males and 3 females per group.
P < 0.05 main effect of duty cycle; ^P < 0.05 main effect of treatment (RNAi); (-) not detectable.
In SOL muscle, the fatiguing protocols also altered the cellular energetic state in a dose-dependent manner. With increasing duty cycle, [ATP] fell and [AMP] rose drastically along (Supplemental Fig. S4; see https://doi.org/10.6084/m9.figshare.18726557.v1) with all other degradation products (Supplemental Table S2; see https://doi.org/10.6084/m9.figshare.18726563.v1). However, AMPD1 knockdown did not affect nucleotide or p-AMPK responses to fatiguing contractions in SOL muscle (Supplemental Fig. S4 and Supplemental Table S2).
SERCA Protein Levels following AMPD1 Knockdown
To explore whether changes in SERCA protein could explain the slower relaxation times observed with AMPD1 knockdown, we quantified changes in SERCA isoform protein content from a random subset of our prepared homogenates. With AMPD1 knockdown, SERCA1 protein content increased in EDL (+100%, P = 0.002; Fig. 5A), but not SOL. In addition, SERCA2 protein content increased in EDL (+243%, P = 0.003; Fig. 5B), with no changes observed in SOL. As the slower ½ RTs were accompanied by higher SERCA protein content, the impaired relaxation kinetics in EDL muscle cannot be explained by changes in SERCA protein content, as higher SERCA density would be expected to speed up overall relaxation kinetics (39).
Figure 5.

Impact of AMP deaminase 1 (AMPD1) knockdown (inhibitory microRNA, RNAi) on SERCA isoform protein content. Following AMPD1 knockdown, EDL muscle displayed higher protein content for both SERCA1 (A), and SERCA2 (B) protein content. C: representative blots and total protein image following photoactivation for EDL muscle. *P < 0.05 for muscle × plasmid interaction using ANOVA increased protein expression following RNAi. Connecting lines represent contralateral muscles (control vs. RNAi) taken from the same animal. n = 8 males and 4 females per group.
DISCUSSION
Herein, we show that partial loss of AMPD1 in skeletal muscle impacts both muscle function and cellular energetic signaling in a muscle-dependent manner. Specifically, we show that AMPD1 knockdown in fast-twitch/glycolytic EDL impairs both contraction and relaxation kinetics during a single isometric contraction and throughout repeated, fatiguing contractions. Furthermore, loss of AMPD1 in EDL muscle results in an accumulation of AMP and exaggerates the p-AMPK responses to contractions at low-to-moderate workloads. These changes were specific to the more fast-twitch/glycolytic EDL muscle and were not observed in the more slow-twitch/oxidative SOL muscle.
RNAi-induced reductions in contraction and relaxation kinetics were only evident during tetanic contractions, not during twitch contractions. The reason for this discrepancy is unclear, though there are several possible explanations. First, it is possible that changes in the series and/or parallel elastic components, which undergo greater strain during the higher forces of tetanic contractions, might reveal differences in kinetics that would not otherwise be apparent at low twitch forces (52). Second, Troponin T of fast-twitch muscle has been found to interact with AMPD, and it has been suggested that this interaction may change calcium sensitivity and regulate muscle function (53). Therefore, loss of AMPD may modulate muscle Ca2+ sensitivity, but this has not been shown in the context of individual contractions (53). Third, the differential observations between twitch and tetanic contraction may be directly tied to differences in the energetic demand. Tetanic contractions have a 10- to 30-fold higher ATP cost compared with a single muscle twitch (54, 55). Furthermore, energetic buffering, a role that AMPD1 serves, affects muscle contraction even at the onset of contractions (56). In light of these possibilities, additional studies are required to determine the precise mechanism whereby AMPD1 knockdown affects tetanic, but not twitch contractions.
The sarco(endo) plasmic reticulum Ca2+ ATPases (SERCAs) of skeletal muscle regulate Ca2+ homeostasis and relaxation kinetics (57–59). There are two primary SERCA isoforms in muscle (SERCA1 across muscle fiber types and SERCA2 predominately in cardiac and slow-twitch fibers), with greater overall density of SERCA leading to enhanced Ca2+ handling and faster relaxation kinetics (39, 59). Therefore, we tested whether AMPD1 knockdown induced changes in SERCA content and/or isoform type. Interestingly, both SERCA1 and SERCA2 protein expression increased in EDL muscle. This is not the first time that impaired relaxation kinetics/Ca2+ handling has been observed in skeletal muscle together with increased SERCA content. Braun et al. (60) observed drastic impairments of Ca2+ handling and increases in both SERCA1 and SERCA2 protein content in SOL muscles from mice following 1 mo of spaceflight. Although increases in SERCA content cannot explain the slower relaxation kinetics in EDL muscle in the current study, these adaptations may represent an insufficient compensation aimed at preventing the decline in muscle function.
Another possible mechanism to explain the effect of AMPD1 knockdown on contractile kinetics could be through changes in myosin heavy chain (MHC), which is a major determinant of contractile speed. Indeed, increased AMPK signaling has been associated with fiber-type shifting from fast IIb myosin heavy chain (MHC) to slower IIa or I MHC (61, 62). Unfortunately, we were unable to directly probe for alterations in MHC composition as we did not use a high-salt buffer (≥300 mM NaCl) for our tissue preparations. Due to the α-helical coiled assembly of myosin dimers, myofibrillar proteins are insoluble in standard buffers (150 mM NaCl) and require specific high-salt buffers to fully solubilize the myofibrils (63). However, our findings suggest that if a shift in MHC composition were present, it is likely small since no changes in twitch contractile kinetics were evident, which should also occur with changes in MHC.
Interestingly, we observed no impairments of contractile function in AMPD1-deficient SOL muscle, despite similar relative reductions in AMPD1 content (∼31%–35%). This may be explained by the differences in AMPD isoform composition between EDL and SOL muscle. SOL muscle demonstrated much lower AMPD1 content (−85%) but drastically greater AMPD3 content (+4,500%). Although this is the first time that the isoform composition between the more glycolytic/fast-twitch EDL and oxidative/slow-twitch SOL has been quantified, it aligns with more general observations that AMPD isoform composition differs between fiber types, with greater AMPD1 protein content and overall AMPD enzyme activity in fast-twitch muscle (15, 23–25, 64). Therefore, it is likely that EDL muscle is inherently more reliant on AMPD1 for energetic buffering than SOL muscle.
In response to our different fatigue protocols, ATP decreased and all purine degradation products increased, clearly demonstrating that our selected contraction parameters for fatigue were sufficient to differentially alter the muscle cellular energetic state. Interestingly, AMPD1 knockdown in EDL resulted in an exaggeration of AMP and p-AMPK responses to contractions, at LOW and MOD, but not HIGH. Although our three different EDL fatigue protocols were at lower relative workloads (duty cycles of 1%, 2%, and 4%) than the 10% duty cycle used by Plaideau et al. (23), direct comparisons between studies is not possible due to the large discrepancy in bath temperatures (20°C vs. 37°C in our study). Such a large difference would affect not only enzyme activities but also functional characteristics of the muscles (65). In part, our lack of an observable effect of AMPD1 knockdown following HIGH workloads may also be confounded by the excessive fatigue (>50% force loss) that these muscles experienced, as such levels of fatigue have been shown to negatively impact fiber recruitment and energy use (66). Furthermore, since changes in energetics during intense contractions may only be transient (67), it is also likely that the observation of measurable changes at this intensity is limited by time constraints during the brief period of muscle collection.
In this study, we included analyses of sex-related differences for all of our variables, which have not yet been included in the skeletal muscle AMPD1 literature. Although female mice displayed lower muscle mass and absolute force production, these differences were accounted for by body mass and muscle cross-sectional area, respectively. This is consistent with sex-related muscle mass and absolute force differences found in the literature (68, 69). In addition, the female mice in this study displayed lower contraction and relaxation rates, consistent with previous investigations exploring sex-based differences in skeletal muscle contraction kinetics (69, 70). Importantly, however, AMPD1 knockdown did not affect any of our measures in a sex-dependent manner. Thus, AMPD1 knockdown impairs muscle contractile function and regulates the adenine nucleotide responses to contractions independent of biological sex.
Our observations of muscle kinetic and energetic differences between the EDL and SOL may help explain the variable published findings regarding AMPD1 deficiency in humans. Using RNAi, we reduced AMPD1 protein content by ∼35% in both muscles, which reflects favorably on the 50% loss of AMPD1 enzyme in individuals who are heterozygous deficient (∼12%–14% of general population) (12, 14, 16). Since the reduction of AMPD1 altered the contractile kinetics and [AMP]/p-AMPK responses in the glycolytic/fast-twitch EDL but had no effect on SOL, the consequences of AMPD1 deficiency might not be apparent in humans with a high percentage of slow-twitch fibers, or in populations with greater mixed-fiber compositions (i.e., untrained or general population) (71–73). Given that elite athletes display optimal skeletal muscle fiber-typing specific to their specialties, it also becomes increasingly important to consider study populations (74–77). This may help explain why AMPD1 mutant allele frequency is lower in power-based sports with athletes displaying greater fast-twitch muscle composition and why homozygous, or complete, AMPD1-deficient individuals are essentially precluded from elite participation in such sports (21, 22). Furthermore, studies that fail to account for individual muscle fiber-type predominance may contribute to the variability of the results in the literature.
In this study, we are the first to characterize the impact of AMPD1 deficiency on skeletal muscle function and energetic signaling after successful knockdown in murine muscle via RNAi, minimizing any life-long effects or possible compensatory adaptations of complete genetic knockout. Specifically, we demonstrate that loss of AMPD1 negatively impacts skeletal muscle function and exaggerates the [AMP]/p-AMPK responses to repeated contractions at low-to-moderate intensities. These effects did not require a complete loss of AMPD1 and were specific to the more glycolytic/fast-twitch EDL muscle. While we confirm previous observations that AMPD1 deficiency does not alter maximal force production, it does lead to impaired contraction and relaxation kinetics. Thus, we demonstrate that the metabolic enzyme AMPD1 plays a role in maintaining skeletal muscle contractile function and regulating the energetic responses associated with repeated contractions in a muscle-specific manner, independent of biological sex.
SUPPLEMENTAL DATA
Supplemental Table S1: https://doi.org/10.6084/m9.figshare.18726560.v1.
Supplemental Table S2: https://doi.org/10.6084/m9.figshare.18726563.v1.
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.18726551.v1.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.18726548.v1.
Supplemental Fig. S3: https://doi.org/10.6084/m9.figshare.18726554.v1.
Supplemental Fig. S4: https://doi.org/10.6084/m9.figshare.18726557.v1.
GRANTS
This project was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award No. R01 AR070200 (to J. J. Brault) and the American Physiological Society Postdoctoral Fellowship Award (to P. S. Hafen).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
P.S.H. and J.J.B. conceived and designed research; P.S.H., A.S.L., C.M., S.G.M., and J.J.B. performed experiments; P.S.H. and A.S.L. analyzed data; P.S.H., A.S.L., and J.J.B. interpreted results of experiments; P.S.H. prepared figures; P.S.H. drafted manuscript; P.S.H., A.S.L., C.M., S.G.M., and J.J.B. edited and revised manuscript; P.S.H., A.S.L., C.M., S.G.M., and J.J.B. approved final version of manuscript.
ACKNOWLEDGMENTS
The graphical abstract was created with BioRender.com and published with permission.
REFERENCES
- 1. Hochachka PW, Matheson GO. Regulating ATP turnover rates over broad dynamic work ranges in skeletal muscles. J Appl Physiol (1985) 73: 1697–1703, 1992. doi: 10.1152/jappl.1992.73.5.1697. [DOI] [PubMed] [Google Scholar]
- 2. Fishbein WN, Armbrustmacher VW, Griffin JL. Myoadenylate deaminase deficiency: a new disease of muscle. Science 200: 545–548, 1978. doi: 10.1126/science.644316. [DOI] [PubMed] [Google Scholar]
- 3. Sabina RL, Swain JL, Olanow CW, Bradley WG, Fishbein WN, DiMauro S, Holmes EW. Myoadenylate daminase deficiency: functional and metabolic abnormalities associated with disruption of the purine nucleotide cycle. J Clin Invest 73: 720–730, 1984. doi: 10.1172/JCI111265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kelemen J, Rice DR, Bradley WG, Munsat TL, DiMauro S, Hogan EL. Familial myoadenylate deaminase deficiency and exertional myalgia. Neurology 32: 857–863, 1982. doi: 10.1212/wnl.32.8.857. [DOI] [PubMed] [Google Scholar]
- 5. Sabina RL, Swain JL, Patten BM, Ashizawa T, O'Brien WE, Holmes EW. Disruption of the purine nucleotide cycle. J Clin Invest 66: 1419–1423, 1980. doi: 10.1172/JCI109995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rico-Sanz J, Rankinen T, Joanisse DR, Leon AS, Skinner JS, Wilmore JH, Rao DC, Bouchard C. Associations between cardiorespiratory responses to exercise and the C34T AMPD1 gene polymorphism in the HERITAGE Family study. Physiol Genomics 14: 161–166, 2003. doi: 10.1152/physiolgenomics.00165.2002. [DOI] [PubMed] [Google Scholar]
- 7. Tarnopolsky MA, Parise G, Gibala MJ, Graham TE, Rush JWE. Myoadenylate deaminase deficiency does not affect muscle anaplerosis during exhaustive exercise in humans. J Physiol 533: 881–889, 2001. doi: 10.1111/j.1469-7793.2001.t01-1-00881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sinkeler SP, Binkhorst RA, Joosten EM, Wevers RA, Coerwinkei MM, Oei TL. AMP deaminase deficiency: study of the human skeletal muscle purine metabolism during ischaemic isometric exercise. Clin Sci (Lond) 72: 475–482, 1987. doi: 10.1042/cs0720475. [DOI] [PubMed] [Google Scholar]
- 9. Verzijl HT, van Engelen BG, Luyten JA, Steenbergen GC, van den Heuvel LP, ter Laak HJ, Padberg GW, Wevers RA. Genetic characteristics of myoadenylate deaminase deficiency. Ann Neurol 44: 140–143, 1998. doi: 10.1002/ana.410440124. [DOI] [PubMed] [Google Scholar]
- 10. Pérez M, Martin MA, Cañete S, Rubio JC, Fernández-Moreira D, San Juan AF, Gómez-Gallego F, Santiago C, Arenas J, Lucia A. Does the C34T mutation in AMPD1 alter exercise capacity in the elderly? Int J Sports Med 27: 429–435, 2006. doi: 10.1055/s-2005-865786. [DOI] [PubMed] [Google Scholar]
- 11. Hanisch F, Joshi P, Zierz S. AMP deaminase deficiency in skeletal muscle is unlikely to be of clinical relevance. J Neurol 255: 318–322, 2008. doi: 10.1007/s00415-008-0530-6. [DOI] [PubMed] [Google Scholar]
- 12. Morisaki T, Gross M, Morisaki H, Pongratz D, Zöllner N, Holmes EW. Molecular basis of AMP deaminase deficiency in skeletal muscle. Proc Natl Acad Sci USA 89: 6457–6461, 1992. doi: 10.1073/pnas.89.14.6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Norman B, Glenmark B, Jansson E. Muscle AMP deaminase deficiency in 2% of a healthy population. Muscle Nerve 18: 239–241, 1995. doi: 10.1002/mus.880180216. [DOI] [PubMed] [Google Scholar]
- 14. Gross M. Clinical heterogeneity and molecular mechanisms in inborn muscle AMP deaminase deficiency. J Inherit Metab Dis 20: 186–192, 1997. doi: 10.1023/a:1005352605421. [DOI] [PubMed] [Google Scholar]
- 15. Norman B, Mahnke-Zizelman DK, Vallis A, Sabina RL. Genetic and other determinants of AMP deaminase activity in healthy adult skeletal muscle. J Appl Physiol (1985) 85: 1273–1278, 1998. doi: 10.1152/jappl.1998.85.4.1273. [DOI] [PubMed] [Google Scholar]
- 16. Gross M, Rötzer E, Kölle P, Mortier W, Reichmann H, Goebel HH, Lochmüller H, Pongratz D, Mahnke-Zizelman DK, Sabina RL. A G468-T AMPD1 mutant allele contributes to the high incidence of myoadenylate deaminase deficiency in the Caucasian population. Neuromuscul Disord 12: 558–565, 2002. doi: 10.1016/S0960-8966(02)00008-1. [DOI] [PubMed] [Google Scholar]
- 17. Rubio JC, Martín MA, Rabadán M, Gómez-Gallego F, San Juan AF, Alonso JM, Chicharro JL, Pérez M, Arenas J, Lucia A. Frequency of the C34T mutation of the AMPD1 gene in world-class endurance athletes: does this mutation impair performance? J Appl Physiol (1985) 98: 2108–2112, 2005. doi: 10.1152/japplphysiol.01371.2004. [DOI] [PubMed] [Google Scholar]
- 18. Lucia A, Martin MA, Esteve-Lanao J, Juan S, Rubio AF, Oliván JC, Arenas J. C34t J. mutation of the AMPD1 gene in an elite white runner. Br J Sports Med 40: e7, 2006. doi: 10.1136/bjsm.2005.019208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ginevičienė V, Jakaitienė A, Pranculis A, Milašius K, Tubelis L, Utkus A. AMPD1 rs17602729 is associated with physical performance of sprint and power in elite Lithuanian athletes. BMC Genet 15: 58, 2014. doi: 10.1186/1471-2156-15-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fischer H, Esbjörnsson M, Sabina RL, Strömberg A, Peyrard-Janvid M, Norman B. AMP deaminase deficiency is associated with lower spring cycling performance in healthy subjects. J Appl Physiol (1985) 103: 315–322, 2007. doi: 10.1152/japplphysiol.00185.2007. [DOI] [PubMed] [Google Scholar]
- 21. Atanasov P, Djaravoa T, Kalinski M, Petrov L, Kaneva R, Mugandani S, Watson G, Jemni M. ACTN3 and AMPD1 polymorphism and genotype combinations in Bulgarian athletes performing Wingate test. J Sports Sci 3: 1–10, 2015. doi: 10.17265/2332-7839/2015.01.001. [DOI] [Google Scholar]
- 22. Cieszczyk P, Ostanek M, Leońska-Duniec A, Sawczuk M, Maciejewska A, Eider J, Ficek K, Sygit K, Kotarska K. Distribution of the AMPD1 C34T polymorphism in Polish power-oriented athletes. J Sports Sci 30: 31–35, 2012. doi: 10.1080/02640414.2011.623710. [DOI] [PubMed] [Google Scholar]
- 23. Plaideau C, Lai Y-C, Kviklyte S, Zanou N, Löfgren L, Andersén H, Vertommen D, Gailly P, Hue L, Bohlooly-Y M, Hallén S, Rider MH. Effects of pharmacological AMP deaminase inhibition and ampd1 deletion on nucleotide levels and ampk activation in contracting skeletal muscle. Chem Biol 21: 1497–1510, 2014. doi: 10.1016/j.chembiol.2014.09.013. [DOI] [PubMed] [Google Scholar]
- 24. Mahnke-Zizelman DK, D'Cunha J, Wojnar JM, Brogley MA, Sabina RL. Regulation of rat AMP deaminase 3 (isoform C) by development and skeletal muscle fibre type. Biochem J 326: 521–529, 1997. doi: 10.1042/bj3260521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van Kuppevelt TH, Veerkamp JH, Fishbein WN, Ogasawara N, Sabina RL. Immunolocalization of AMP-deaminase isozymes in human skeletal muscle and cultured muscle cells: concentration of isoform M at the neuromuscular junction. J Histochem Cytochem 42: 861–868, 1994. doi: 10.1177/42.7.8014469. [DOI] [PubMed] [Google Scholar]
- 26. Cheng J, Morisaki H, Toyama K, Sugimoto N, Shintani T, Tandelilin A, Hirase T, Holmes EW, Morisaki T. AMPD1: a novel therapeutic target for reversing insulin resistance. BMC Endocr Disord 14: 96, 2014. doi: 10.1186/1472-6823-14-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tandelilin AA, Hirase T, Hudoyo AW, Cheng J, Toyama K, Morisaki H, Morisaki T. AMPD1 regulates mTORC1-p70 S6 kinase axis in the control of insulin sensitivity in skeletal muscle. BMC Endocr Disord 15: 11, 2015. doi: 10.1186/s12902-015-0010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cheng J, Morisaki H, Sugimoto N, Dohi A, Shintani T, Kimura E, Toyama K, Ikawa M, Okabe M, Higuchi I, Matsuo S, Kawai Y, Hisatome I, Sugama T, Holmes EW, Morisaki T. Effect of isolated AMP deaminase deficiency on skeletal muscle function. Mol Genet Metab Rep 1: 51–59, 2014. doi: 10.1016/j.ymgmr.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dudley GA, Abraham WM, Terjung RL. Influence of exercise intensity and duration on biochemical adaptations in skeletal muscle. J Appl Physiol Respir Environ Exerc Physiol 53: 844–850, 1982. doi: 10.1152/jappl.1982.53.4.844. [DOI] [PubMed] [Google Scholar]
- 30. Chen Z-P, Stephens TJ, Murthy S, Canny BJ, Hargreaves M, Witters LA, Kemp BE, McConell GK. Effect of exercise intensity on skeletal muscle AMPK signaling in humans. Diabetes 52: 2205–2212, 2003. doi: 10.2337/diabetes.52.9.2205. [DOI] [PubMed] [Google Scholar]
- 31. Tang K, Wagner PD, Breen EC. TNF-α-mediated reduction in PGC-1α may impair skeletal muscle function after cigarette smoke exposure. J Cell Physiol 222: 320–327, 2010. doi: 10.1002/jcp.21955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Augusto V, Padovani CR, Campos GER. Skeletal muscle fiber types in C57BL6J mice. Braz J Morphol Sci 21: 89–94, 2004. http://www.jms.periodikos.com.br/journal/jms/article/587cb4537f8c9d0d058b45ea. [Google Scholar]
- 33. Mahnke-Zizelman DK, Sabina RL. Localization of N-terminal sequences in human AMP deaminase isoforms that influence contractile protein binding. Biochem Biophys Res Commun 285: 489–495, 2001. doi: 10.1006/bbrc.2001.5180. [DOI] [PubMed] [Google Scholar]
- 34. Mattii L, Bianchi F, Falleni A, Frascarelli S, Masini M, Alì G, Chiellini G, Sabbatini ARM. Ultrastructural localization of histidine-rich glycoprotein in skeletal muscle fibers: colocalization with AMP deaminase. J Histochem Cytochem 68: 139–148, 2020. doi: 10.1369/0022155419897573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mattii L, Rossi L, Ippolito C, Alì G, Martini D, Raggi A, Sabbatini ARM. Immunohistochemical localization of histidine-rich glycoprotein in human skeletal muscle: preferential distribution of the protein at the sarcomeric I-band. Histochem Cell Biol 148: 651–657, 2017. doi: 10.1007/s00418-017-1594-0. [DOI] [PubMed] [Google Scholar]
- 36. Rayment I, Holden HM, Whittaker M, Yohn CB, Lorenz M, Holmes KC, Milligan RA. Structure of the actin-myosin complex and its implications for muscle contraction. Science 261: 58–65, 1993. doi: 10.1126/science.8316858. [DOI] [PubMed] [Google Scholar]
- 37. Greene LE, Eisenberg E. The binding of heavy meromyosin to f-actin. J Biol Chem 255: 549–554, 1980. [PubMed] [Google Scholar]
- 38. Zhu Y, Nosek TM. Intracellular milieu changes associated with hypoxia impair sarcoplasmic reticulum Ca2+ transport in cardiac muscle. Am J Physiol Heart Circ Physiol 261: H620–H626, 1991. doi: 10.1152/ajpheart.1991.261.3.H620. [DOI] [PubMed] [Google Scholar]
- 39. Lytton J, Westlin M, Burk SE, Shull GE, MacLennan DH. Functional comparisons between isoforms of the sarcoplasmic or endoplasmic reticulum family of calcium pumps. J Biol Chem 267: 14483–14489, 1992. [PubMed] [Google Scholar]
- 40. Green HJ. Cation pumps in skeletal muscle: potential role in muscle fatigue. Acta Physiol Scand 162: 201–213, 1998. doi: 10.1046/j.1365-201X.1998.0300f.x. [DOI] [PubMed] [Google Scholar]
- 41. Essén-Gustavsson B, Gottlieb-Vedi M, Lindholm A. Muscle adenine nucleotide degradation during submaximal treadmill exercise to fatigue. Equine Exerc Physiol 31: 298–302, 1999. doi: 10.1111/j.2042-3306.1999.tb05238.x. [DOI] [PubMed] [Google Scholar]
- 42. Sahlin K, Broberg S. Adenine nucleotide depletion in human muscle during exercise: causality and significance of AMP deamination. Int J Sports Med 11: S62–S67, 1990. doi: 10.1055/s-2007-1024856. [DOI] [PubMed] [Google Scholar]
- 43. Miller SG, Hafen PS, Law AS, Springer CB, Logsdon DL, O'Connell TM, Witczak CA, Brault JJ. AMP deamination is sufficient to replicate an atrophy-like metabolic phenotype in skeletal muscle. Metabolism 123: 154864, 2021. doi: 10.1016/j.metabol.2021.154864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brault JJ, Jespersen JG, Goldberg AL. Peroxisome proliferator-activated receptor gamma coactivator 1a or 1B overexpression inhibits muscle protein degradation, induction of ubiquitin ligases, and disuse atrophy. J Biol Chem 285: 19460–19471, 2010. doi: 10.1074/jbc.M110.113092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ferey JL, Brault JJ, Smith CA, Witczak CA. Constitutive activation of CaMKKα signaling is sufficient but not necessary for mTORC1 activation and growth in mouse skeletal muscle. Am J Physiol Endocrinol Physiol 307: E686–E694, 2014. doi: 10.1152/ajpendo.00322.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hughes DC, Hardee JP, Waddell DS, Goodman CA. CORP: Gene delivery into murine skeletal muscle using in vivo electroporation. J Appl Physiol (1985) 133: 41–59, 2022. doi: 10.1152/japplphysiol.00088.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kober F, Iltis I, Cozzone PJ, Bernard M. Myocardial blood flow mapping in mice using high-resolution spin labeling magnetic resonance imaging: influence of ketamine/xylazine and isoflurane anesthesia. Magn Reson Med 53: 601–606, 2005. doi: 10.1002/mrm.20373. [DOI] [PubMed] [Google Scholar]
- 48. Mechelinck M, Kupp C, Kruger JC, Habigt MA, Helmedag MJ, Tolba RH, Rossaint R, Hein M. Oxygen inhalation improves postoperative survival in ketamine-xylazine anaesthetised rats: an observational study. PLoS One 14: e0226430, 2019. doi: 10.1371/journal.pone.0226430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brooks SV, Faulkner JA. Contractile properties of skeletal muscles from young, adult and aged mice. J Physiol 404: 71–82, 1988. doi: 10.1113/jphysiol.1988.sp017279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brault JJ, Pizzimenti NM, Dentel JN, Wiseman RW. Selective inhibition of ATPase activity during contraction alters the activation of p38 MAP kinase isoforms in skeletal muscle. J Cell Biochem 114: 1445–1455, 2013. doi: 10.1002/jcb.24486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Law AS, Hafen PS, Brault JJ. Liquid chromatography method for simultaneous quantification of ATP and its degradation products compatible with both UV-Vis and mass spectrometry. J Chromatography B Analyt Technol Biomed Life Sci 1206: 123351, 2022. doi: 10.1016/j.jchromb.2022.123351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Herzog W. The problem with skeletal muscle series elasticity. BMC Biomed Eng 1: 28, 2019. doi: 10.1186/s42490-019-0031-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ronca F, Raggi A. Role of troponin T and AMP deaminase in the modulation of skeletal muscle contraction. Rend Fis Acc Lincei 28: 143–158, 2017. doi: 10.1007/s12210-016-0586-7. [DOI] [Google Scholar]
- 54. Spriet LL. ATP utilization and provision in fast-twitch skeletal muscle during tetanic contractions. Am J Physiol Endocrinol Physiol 257: E595–E605, 1989. doi: 10.1152/ajpendo.1989.257.4.E595. [DOI] [PubMed] [Google Scholar]
- 55. Foley JM, Meyer RA. Energy cost of twitch and tetanic contractions of rat muscle estimated in situ by gated 31P NMR. NMR Biomed 6: 32–38, 1993. doi: 10.1002/nbm.1940060106. [DOI] [PubMed] [Google Scholar]
- 56. Roman BB, Meyer RA, Wiseman RW. Phosphocreatine kinetics at the onset of contractions in skeletal muscle of MM creatine kinase knockout mice. Am J Physiol Cell Physiol 283: C1776–C1783, 2002. doi: 10.1152/ajpcell.00210.2002. [DOI] [PubMed] [Google Scholar]
- 57. Smith IC, Bombardier E, Vigna C, Tupling AR. ATP consumption by sarcoplasmic reticulum Ca(2)(+) pumps accounts for 40–50% of resting metabolic rate in mouse fast and slow twitch skeletal muscle. PLoS One 8: e68924, 2013. doi: 10.1371/journal.pone.0068924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Barclay CJ, Woledge RC, Curtin NA. Energy turnover for Ca2+ cycling in skeletal muscle. J Muscle Res Cell Motil 28: 259–274, 2007. doi: 10.1007/s10974-007-9116-7. [DOI] [PubMed] [Google Scholar]
- 59. Tupling AR. The sarcoplasmic reticulum in muscle fatigue and disease: role of the sarco(endo)plasmic reticulum Ca2+-ATPase. Can J Appl Physiol 29: 308–329, 2004. doi: 10.1139/h04-021. [DOI] [PubMed] [Google Scholar]
- 60. Braun JL, Geromella MS, Hamstra SI, Messner HN, Fajardo VA. Characterizing SERCA function in murine skeletal muscles after 35–37 days of spaceflight. Int J Mol Sci 22: 11764, 2021. doi: 10.3390/ijms222111764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. RöCkl KSC, Hirshman MF, Brandauer J, Fujii N, Witters LA, Goodyear LJ. Skeletal muscle adaptation to exercise training: AMP-activated protein kinase mediates muscle fiber type shift. Diabetes 56: 2062–2069, 2007. doi: 10.2337/db07-0255. [DOI] [PubMed] [Google Scholar]
- 62. Mounier R, Théret M, Lantier L, Foretz M, Viollet B. Expanding roles for AMPK in skeletal muscle plasticity. Trends Endocrinol Metab 26: 275–286, 2015. doi: 10.1016/j.tem.2015.02.009. [DOI] [PubMed] [Google Scholar]
- 63. Cosper PF, Leinwand LA. Myosin heavy chain is not selectively decreased in murine cancer cachexia. Int J Cancer 130: 2722–2727, 2012. doi: 10.1002/ijc.26298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dudley GA, Staron RS, Murray TF, Hagerman FC, Luginbuhl A. Muscle fiber composition and blood ammonia levels after intense exercise in humans. J Appl Physiol Respir Environ Exerc Physiol 54: 582–586, 1983. doi: 10.1152/jappl.1983.54.2.582. [DOI] [PubMed] [Google Scholar]
- 65. Ferrara PJ, Verkerke ARP, Brault JJ, Funai K. Hypothermia decreases O2 cost for ex vivo contraction in mouse skeletal muscle. Med Sci Sports Exerc 50: 2015–2023, 2018. doi: 10.1249/MSS.0000000000001673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Barclay CJ, Arnold PD, Gibbs CL. Fatigue and heat production in repeated contractions of mouse skeletal muscle. J Physiol 488: 741–752, 1995. doi: 10.1113/jphysiol.1995.sp021005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hancock CR, Janssen E, Terjung RL. Skeletal muscle contractile performance and ADP accumulation in adenylate kinse-deficient mice. Am J Physiol Cell Physiol 288: C1287–C1297, 2005. doi: 10.1152/ajpcell.00567.2004. [DOI] [PubMed] [Google Scholar]
- 68. Griffin GE, Goldspink G. The increase in skeletal muscle mass in male and female mice. Anat Rec 177: 465–469, 1973. doi: 10.1002/ar.1091770311. [DOI] [PubMed] [Google Scholar]
- 69. Glenmark B, Nilsson M, Gao H, Gustafsson J-A, Dahlman-Wright K, Westerblad H. Difference in skeletal muscle function in males vs. females: role of estrogen receptor-B. Am J Physiol Endocrinol Physiol 287: E1125–E1131, 2004. doi: 10.1152/ajpendo.00098.2004. [DOI] [PubMed] [Google Scholar]
- 70. Haizlip KM, Harrison BC, Leinwand LA. Sex-based differences in skeletal muscle kinetics and fiber-type composition. Physiology (Bethesda) 30: 30–39, 2015. doi: 10.1152/physiol.00024.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Simoneau JA, Lortie G, Boulay MR, Marcotte M, Thibault MC, Bouchard C. Human skeletal muscle fiber type alteration with high-intensity intermittent training. Eur J Appl Physiol Occup Physiol 54: 250–253, 1985. doi: 10.1007/BF00426141. [DOI] [PubMed] [Google Scholar]
- 72. Froese EA, Houston ME. Torque-velocity characteristics and muscle fiber type in human vastus lateralis. J Appl Physiol (1985) 59: 309–314, 1985. doi: 10.1152/jappl.1985.59.2.309. [DOI] [PubMed] [Google Scholar]
- 73. Staron RS, Hagerman FC, Hikida RS, Murray TF, Hostler DP, Crill MT, Ragg KE, Toma K. Fiber type composition of the vastus lateralis muscle of young men and women. J Histochem Cytochem 48: 623–629, 2000. doi: 10.1177/002215540004800506. [DOI] [PubMed] [Google Scholar]
- 74. Trappe S, Luden N, Minchev K, Raue U, Jemiolo B, Trappe TA. Skeletal muscle signature of a champion sprint runner. J Appl Physiol (1985) 118: 1460–1466, 2015. doi: 10.1152/japplphysiol.00037.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Andersen JL, Klitgaard H, Saltin B. Myosin heavy chain isoforms in single fibres from m. vastus lateralis of sprinters: influence of training. Acta Physiol Scand 151: 135–142, 1994. doi: 10.1111/j.1748-1716.1994.tb09730.x. [DOI] [PubMed] [Google Scholar]
- 76. Plotkin DL, Roberts MD, Haun CT, Schoenfeld BJ. Muscle fiber type transitions with exercise training: shifting perspectives. Sports 9: 127, 2021. doi: 10.3390/sports9090127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. D'Antona G, Lanfranconi F, Pellegrino MA, Brocca L, Adami R, Rossi R, Moro G, Miotti D, Canepari M, Bottinelli R. Skeletal muscle hypertrophy and structure and function of skeletal muscle fibres in male body builders. J Physiol 570: 611–627, 2006. doi: 10.1113/jphysiol.2005.101642. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1: https://doi.org/10.6084/m9.figshare.18726560.v1.
Supplemental Table S2: https://doi.org/10.6084/m9.figshare.18726563.v1.
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.18726551.v1.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.18726548.v1.
Supplemental Fig. S3: https://doi.org/10.6084/m9.figshare.18726554.v1.
Supplemental Fig. S4: https://doi.org/10.6084/m9.figshare.18726557.v1.