

Abstract
Heart failure with preserved ejection fraction (HFpEF) is defined by increased left ventricular (LV) stiffness, impaired vascular compliance, and fibrosis. Although systemic inflammation, driven by comorbidities, has been proposed to play a key role, the precise pathogenesis remains elusive. To test the hypothesis that inflammation drives endothelial dysfunction in HFpEF, we used cardiosphere-derived cells (CDCs), which reduce inflammation and fibrosis, improving function, structure, and survival in HFpEF rats. Dahl salt-sensitive rats fed a high-salt diet developed HFpEF, as manifested by diastolic dysfunction, systemic inflammation, and accelerated mortality. Rats were randomly allocated to receive intracoronary infusion of CDCs or vehicle. Two weeks later, inflammation, oxidative stress, and endothelial function were analyzed. Single-cell RNA sequencing of heart tissue was used to assay transcriptomic changes. CDCs improved endothelial-dependent vasodilation while reducing oxidative stress and restoring endothelial nitric oxide synthase (eNOS) expression. RNA sequencing revealed CDC-induced attenuation of pathways underlying endothelial cell leukocyte binding and innate immunity. Exposure of endothelial cells to CDC-secreted extracellular vesicles in vitro reduced VCAM-1 protein expression and attenuated monocyte adhesion and transmigration. Cell therapy with CDCs corrects diastolic dysfunction, reduces oxidative stress, and restores vascular reactivity. These findings lend credence to the hypothesis that inflammatory changes of the vascular endothelium are important, if not central, to HFpEF pathogenesis.
NEW & NOTEWORTHY We tested the concept that inflammation of endothelial cells is a major pathogenic factor in HFpEF. CDCs are heart-derived cell products with verified anti-inflammatory therapeutic properties. Infusion of CDCs reduced oxidative stress, restored eNOS abundance, lowered monocyte levels, and rescued the expression of multiple disease-associated genes, thereby restoring vascular reactivity. The salutary effects of CDCs support the hypothesis that inflammation of endothelial cells is a proximate driver of HFpEF.
Keywords: cell therapy, endothelial dysfunction, HFpEF, inflammation, oxidative stress
INTRODUCTION
Despite similar clinical manifestations, heart failure (HF) with preserved ejection fraction (HFpEF; EF ≥ 50%) differs from HF with reduced EF (HFrEF; EF < 50%) in being a particularly challenging clinical syndrome refractory to routine medical approaches (1–4). With the possible exception of SGLT2 inhibitors (5), no drug or device has been convincingly shown to substantially improve morbidity or mortality in HFpEF, which is ever increasing in prevalence. In exploring this urgent unmet medical need, we found that cardiosphere-derived cells (CDCs) have strong disease-modifying bioactivity in HFpEF. These cardiac stromal/progenitor cells, which are distinct from discredited c-kit-positive cells (6, 7), exhibit multilineage potential and clonogenicity (8, 9) but work primarily through indirect mechanisms (10, 11). At least 75 independent laboratories worldwide have generated CDCs and verified their bioactivity (e.g., see Refs. 12–16). The first experience with cell therapy in HFpEF came from studies of CDCs in Dahl salt-sensitive (DS) rats (17). These animals have genetic insulin resistance and hypercholesterolemia (18) but, when fed normal chow, are otherwise indistinguishable from wild type. In contrast, a high-salt diet precipitates hypertension and HFpEF with accelerated mortality (17, 19, 20). This model exhibits several features typical of human HFpEF (17, 21, 22), with clinically relevant comorbidities (insulin resistance, hyperlipidemia, and hypertension, all common in patients with HFpEF; 2). A single intracoronary dose of rat CDCs, injected after HFpEF, was already evident, reversed key functional abnormalities of HFpEF, decreased fibrosis and inflammation, and attenuated mortality, without reducing blood pressure (17). CDCs also reversed adverse electrical remodeling in HFpEF rat hearts, thereby reducing the propensity for sudden death (22).
The anti-inflammatory, immunomodulatory, and antifibrotic effects of CDCs present a novel opportunity for mechanistic dissection of HFpEF. A widely cited hypothesis for the pathogenesis of HFpEF posits that systemic inflammation (23, 24), secondary to various comorbidities, leads to oxidative stress of the endothelium (24). Reduced bioavailability of nitric oxide (NO) and endothelial nitric oxide synthase (eNOS) uncoupling produce vasoconstrictive endothelial dysfunction (25), which in turn stimulates monocyte infiltration and proinflammatory macrophages, leading to fibrosis (26, 27). Although many of these processes have been well documented in diverse preclinical scenarios, few have been confirmed in well-validated HFpEF models. Here, we sought to investigate the roles of inflammation and endothelial dysfunction in DS rats with phenotypically verified HFpEF. Specifically, we tested whether functional improvements resulting from CDC therapy were associated with changes in oxidative stress, innate immunity, and vascular reactivity, to test the inflammatory/endothelial hypothesis of HFpEF pathogenesis.
MATERIALS AND METHODS
In Vivo Studies
In vivo experimental protocols were performed on 7-wk-old male Dahl salt-sensitive rats (DS; Charles River, Wilmington, MA) that were fed 0.3% NaCl (low salt, No. AIN-76A, D10001i, Research Diets) or 8% NaCl (high salt, No. AIN-76A, D05011703i, Research Diets) diets (21, 28). Only male adult rats were used since female rats are relatively resistant to excess dietary salt (29). At 14 wk of age, high-salt diet animals were randomly allocated to receive placebo [100 μL phosphate-buffered saline (PBS)] or allogeneic rat CDCs (5 × 105 cells in 100 μL PBS) via left ventricular (LV) cavity injection with aortic cross-clamp (17, 22). We previously characterized rat CDCs and demonstrated their antigenic and functional similarity to human CDCs (30, 31). All rats were euthanized at 16 wk of age for tissue collection and downstream analyses. All studies were performed at Cedars-Sinai Medical Center. The study was approved by and performed in accordance with Institutional Animal Care and Use Committee (IACUC) guidelines.
Echocardiography
Rats were anesthetized with isoflurane (5% induction, 2% maintenance) for transthoracic echocardiography (Vevo3100, VisualSonics). Heart images were acquired in the parasternal long-axis view for systolic function (EF) and the apical four-chamber view for diastolic function (E/A and E/e′); E and A waves were measured between the tips of the mitral valve, e′ waves were measured at the septal corner of the mitral annulus. Three separate measurements were acquired, analyzed, and averaged to quantify systolic and diastolic function.
Endothelial Function
Acetylcholine (ACh, No. A6625, Sigma) infusion was performed to assess ACh-mediated dilation (32). For the ACh infusion experiment, basal measurements were recorded before ACh infusion (single bolus, 10 µg/kg, via tail vein). Measurements were acquired at 10- or 20-s intervals over a 5-min period. Peak change in diameter was determined 30 s after ACh infusion. Tail-cuff plethysmography was performed as reported previously (33).
Flow Cytometry
Blood was collected in heparinized tubes. Red blood cells were lysed with ACK buffer (No. A1049201, Invitrogen), and then white blood cells were washed (Perm/Wash, BD Biosciences) and stained with the appropriate fluorescently conjugated antibodies (Cd45: No. 561588; CD11b: No. 562102; Granulocyte: No. 554907). Cells were analyzed with a spectral cell analyzer (SA3800, Sony).
Fibrosis
Midventricular heart tissues were sectioned and stained for Picrosirius red per the manufacturer’s protocol (Abcam No. ab150681). Each slide was scanned with a Leica SCN400 Slide Scanner. Fractional myocardial fibrosis (red pixels over the total pixels) was quantified using ImageJ software (National Institutes of Health, NIH).
Cell Culture
Cardiosphere-derived cells and extracellular vesicles.
Primary human CDCs were isolated from nondiseased primary heart tissue, as described (34). To isolate extracellular vesicles (EVs), CDCs were grown to confluence, washed four times with PBS (to remove residual FBS-derived EVs), and then incubated in serum-free media at 37°C. Fifteen days later, conditioned media was collected, filtered (0.45 μm, MilliporeSigma), and concentrated using ultrafiltration by centrifugation with a 100-kDa molecular mass cutoff (MWCO; MilliporeSigma). EVs were analyzed for particle number and size distribution using nanotracking analysis (Nanosight NS300, Malvern). Conditioned media was advanced into the microfluidics chamber of the NS300 using an automated syringe pump and then particles were visualized (×20 magnification) and captured by video (30 frames/s). Four representative cycles were collected for each sample, and measurements for concentration and size were averaged.
Endothelial cells.
Rat aortic endothelial cells (RAOECs) were purchased (No. R304-05A, Millipore-Sigma) and cultured according to the manufacturer’s recommendations.
Coculture.
Monocytes (THP-1) were purchased (No. TIB-202, ATCC) and cultured according to the manufacturer’s recommendations. Before coculture experiments, THP-1 cells and RAOECs were prestained with CFSE (No. C34554, Thermo Fisher Scientific) and wheat germ agglutinin (WGA-647; No. W32466, Thermo Fisher Scientific).
Molecular Biology
Quantitative RT-PCR.
RNA was isolated and purified from tissue samples using QIAzol lysis reagent and an RNeasy kit (No. 73404, QIAGEN). To assess changes in mRNA expression within cells or EVs, we used Taqman technology (Applied Biosystems, Foster City, CA). Briefly, cDNA was synthesized from mRNA using the High-Capacity cDNA Reverse Transcription Kit (No. 4388950, Applied Biosystems) per the manufacturer’s protocol. The resulting cDNA was standardized across samples and then mixed with master mix and designated primer sets (Thermo Fisher Scientific). The following predesigned TaqMan primer sets were purchased: Vcam1, Icam1, e-selectin, and Hprt.
Western blot analysis.
Tissue was placed in RIPA lysis buffer (supplemented with HALT protease and phosphatase inhibitors, No. 89901 and No. 78442, Thermo Fisher Scientific) and mechanically disrupted (BeadRuptor, OMNI). The resulting suspensions were centrifuged at 14,000 g, and the protein supernatants were collected. Protein concentrations were assessed using a bicinchoninic acid (BCA) assay (No. 23227, Thermo Fisher Scientific) and measured by spectrophotometry (SpectraMax iD3, Molecular Devices). Protein samples were prepared for gel electrophoresis with the NuPAGE system (Invitrogen) per the manufacturer’s protocol. Proteins (10–30 μg) were resolved on 4–20% gradient gels, transferred onto a polyvinylidene fluoride membrane, blocked at room temperature with 5% BSA solution, and incubated with the following primary antibodies: eNOS (sc-136977, 1:500, Santa Cruz Biotechnology), ICAM1 (No. ab171123, 1:500, Abcam), VCAM1 (No. ab134047, 1:500, Abcam), and GAPDH (No. 3683S, 1:3,000, Cell Signaling). All primary antibodies were incubated overnight with blocking solution at 4°C. A secondary antibody (horseradish peroxidase-conjugated, 1:5,000) was incubated for 60 min at room temperature. Bands were detected following activation with ECL substrate (Thermo Fisher Scientific) and visualization using a ChemiDoc MP Imaging System (Bio-Rad).
Dihydroethidium.
Vascular sections were incubated with dihydroethidium (DHE; 10 μM in Tyrode’s solution; No. D11347, Thermo Fisher) for 20 min at room temperature. Slides were washed (Tyrode’s solution), mounted with DAPI (Fluoroshield, Sigma Aldrich), and then imaged by confocal microscopy (Leica). Fluorescence images were captured in at least four areas per each aortic ring, using identical imaging settings across all samples. Changes in mean fluorescence intensity and integrated density were determined using ImageJ software.
Single-Cell RNA Sequencing and Statistical Analyses
Sample preparation.
At endpoint (16 wk of age), DS rats were heparinized (4,000 IU/kg ip) and euthanized, and hearts were collected (35). The ascending aorta was cannulated and retrogradely perfused (10 mL/min) on a Langendorff system with Ca2+-free Tyrode’s solution, consisting of (in mM) 137 NaCl, 5.4 KCl, 0.33 NaH2PO4, 1 MgCl2, 10 HEPES, and 10 d-glucose (pH 7.4) at 37°C. Five minutes later, an enzyme solution (type II collagenase; 1.66 mg/mL, No. LS004176, Worthington Biomedical) and type XIV protease (0.13 mg/mL, No. P5147, Sigma, reconstituted in Ca2+-free Tyrode’s solution) was introduced. After 20 min of digestion, Tyrode’s solution (containing 0.2 mM CaCl2) was perfused through the heart. Five minutes later, the heart was removed, and the LV was excised, minced, resuspended, and filtered (100 μm) to generate a single-cell suspension.
Single-cell library construction.
Libraries were prepared per the Single Cell 3′ v2 Reagent Kits User Guide (10× Genomics, Pleasanton, CA). Cell suspensions from tubes 1 and 2 were loaded on the Chromium Controller instrument (10× Genomics) to generate single-cell Gel Bead-In-EMulsions (GEMs). GEM-RT was performed in a Veriti 96-well thermal cycler (Thermo Fisher Scientific, Waltham, MA). GEMs were then harvested, and the cDNA was amplified and cleaned up with SPRIselect Reagent Kit (Beckman Coulter, Brea, CA). Indexed sequencing libraries were constructed using Chromium Single-Cell 3′ Library Kit for enzymatic fragmentation, end repair, A-tailing, adapter ligation, ligation cleanup, sample indexing, and PCR cleanup. The barcoded sequencing libraries were quantified by quantitative RT-PCR (qPCR) using the KAPA Library Quantification Kit for Illumina platforms (KAPA Biosystems, Wilmington, MA). Sequencing libraries were loaded on a NextSeq500 (Illumina, San Diego, CA) with a custom sequencing setting (26 bp for Read 1 and 98 bp for Read 2) to obtain a sequencing depth of ∼200,000 reads/cell.
Data analysis.
Reference sequences for the sample tags were generated using BD Biosciences multiplexing tools. The demultiplexed raw reads were aligned using STAR (19; v.2.5.1) with default parameters to the sample-tags sequences and a custom rat rn6 transcriptome reference (GENCODE Release v95), containing all protein-coding and long noncoding RNA genes. Expression counts for each gene in all samples were collapsed into unique molecular identifier (UMI) counts using Cell Ranger (v.3.1.0, 10× Genomics). Singlets (droplets with one sample tag), multiplets (droplets with multiple sample tags), and negatives (droplets with no sample tags) were identified based on their distinctive t-distribution stochastic neighbor embedding (tSNE) clusters. Multiplets, negatives, and aberrant cells/debris (>15% of reads mapping to mitochondrial genes or <300 expressed genes) were removed from further analysis.
UMI counts were normalized by multiplying the size factor (calculated based on the median UMI count for all cells) and then dividing the total UMI counts for each cell. Principal component analysis was run on the normalized gene-barcode matrix to reduce the number of feature dimensions and obtain two-dimensional population dynamic projections. The top 50 principal components were selected in R using the package Seurat (v.2.2), and these were used as input for tSNE and uniform manifold approximation and projection (UMAP) plotting (20), nearest neighbor graph construction, and cluster identification. Clusters were identified using a resolution of 0.8 in the Seurat function FindClusters. Subsequently, cluster-specific marker genes were identified with the Seurat function FindAllMarkers with the default settings except “only.pos = TRUE.” Cluster cell types were identified using CellAssign (36) and a curated list of potential marker genes. The endothelial cell cluster was subclustered by creating a subset of those cells and performing the aforementioned processing steps from PCA to marker discovery on this subset. Subcluster identities were then mapped back onto the cell barcodes from the total data set. Also for endothelial cells, a trajectory analysis was conducted using Monocle3 (37).
Statistical analyses.
Differential gene expression between treatments or between clusters was assessed using the Seurat function FindMarkers and a log fold change threshold of 0.1. The statistical test was the default nonparametric Wilcoxon rank-sum test. Raw P values were adjusted for multiple hypothesis testing using the Benjamini–Hochberg method. Genes with an adjusted P < 0.05 were considered exclusively expressed in each single subgroup and used for drawing heat maps using ggplot2 (v.2.2.1) in R (v.3.4.1).
Statistics for Nongenomic Data
Results are expressed as means ± SE. Differences between groups at a single time point were assessed using either two-tailed unpaired Student’s t test or Mann–Whitney test where data failed Kolmogorov–Smirnov test of normality or one-way ANOVA. Differences between groups at multiple time points were assessed using repeated-measures (RM) ANOVA. Post hoc testing was performed by Holm–Sidak’s test to correct for multiple comparisons. All statistical analyses were performed using Prism 9 software (GraphPad), and only differences with P < 0.05 were considered statistically significant.
RESULTS
Rat Model of Heart Failure With Preserved Ejection Fraction
When placed on a high-salt diet, DS rats develop HFpEF with diastolic dysfunction and objective signs of HF (including exercise intolerance, cachexia, and pulmonary congestion; 17, 21, 28). Here, we implemented the DS rat model by randomizing animals to low-salt (LS, as control; 0.3% NaCl) or high-salt (HS; 8% NaCl) diets from 7- to 16-wk of age (Fig. 1A). In contrast to LS rats, HS rats developed hypertension by 16 wk (Fig. 1B), along with diastolic dysfunction [elevated E/e′ and decreased E/A (LS, 1.6 ± 0.05 vs. HS, 1.3 ± 0.03; P < 0.05) ratios] and preserved ejection fraction (Fig. 1, C and D). At endpoint, HS rats exhibited leukocytosis, with relative lymphopenia, granulocytosis, and monocytosis (Fig. 1E). This pattern is one of heightened inflammation (38).
Figure 1.

High-salt diet promotes heart failure with preserved ejection fraction (HFpEF) and systemic inflammation. A: experimental workflow using Dahl salt-sensitive rats fed high-salt (HS; HFpEF induction) and low-salt (LS; control) diets. B: blood pressure (BP) measurements. C: representative pulsed-Doppler mitral inflow and medial annulus velocities. D: quantitative analyses of percent ejection fraction (top) and E/e′ ratio (bottom). Graphs depict means ± SE with n = 4–9/group. Statistical significance was determined using RM-ANOVA followed by Holm–Sidak’s multiple corrections test (B), unpaired Student’s t test (D, top, and E), or Mann–Whitney test (D, bottom). *P < 0.05 and ***P < 0.001. A, late diastolic mitral flow from atrial filling; A′, late diastolic mitral annulus velocity; E, early diastolic filling; E′, mitral annulus early diastolic velocity; WBC, white blood cell.
Consistent with previous findings (39), an HS diet increases systemic inflammation (Fig. 1E). Given that CDCs attenuate inflammation (17), we collected blood from CDC- or vehicle-infused HS DS rats (Fig. 2A) and analyzed myeloid cells by flow cytometry (Fig. 2B). CDC-infused animals exhibited a trend toward reduced circulating granulocytes, but monocytes were unchanged (Fig. 2C). We previously observed functional and structural benefits of CDCs in HFpEF rats along with decreases in circulating inflammatory cytokines (17). These findings are congruent with the observed improvements in diastolic function (Supplemental Table S1; all Supplemental Material is available at https://doi.org/10.6084/m9.figshare.20043350) and the attenuation of fibrosis (Fig. 2, D and E) with CDCs in HFpEF.
Figure 2.

Cardiosphere-derived cells (CDCs) attenuate inflammation and fibrosis in heart failure with preserved ejection fraction (HFpEF). A: experimental workflow of HFpEF animals receiving intracoronary (ic) delivery vehicle or CDCs. B: representative flow plots and gating strategy of peripheral blood immune cells. C: quantitative analysis of granulocytes and monocytes. D: representative Picrosirius red-stained heart sections. E: quantitative analysis of fibrosis. Graphs depict means ± SE with n = 4–6/group. Statistical significance was determined using RM-ANOVA followed by Holm–Sidak’s test (C) or unpaired Student’s t test (D). *P < 0.05, **P < 0.01, and ***P < 0.001. HA, high salt.
Endothelial Dysfunction and the Therapeutic Effect of Rat CDCs in HFpEF
Inflammation and endothelial dysfunction are intertwined mechanistically (40), but little direct evidence supports a central role in HFpEF pathogenesis. We measured endothelial-dependent brachial artery vasodilation by infusing ACh in the three experimental groups (Fig. 3A). At peak flow (30 s following ACh), HFpEF rats that received CDCs had higher blood flow velocities compared with their vehicle counterparts (Fig. 3B), thus exhibiting improved brachial vasodilation (Fig. 3, C and D). These findings reveal that impaired endothelial-dependent vasodilation in HFpEF is improved by CDCs.
Figure 3.

Endothelial dysfunction in heart failure with preserved ejection fraction (HFpEF). A: experimental workflow to assess endothelial function in vivo. B: representative images of brachial artery flow velocity. C: change in brachial artery diameter over time. D: quantitative analysis of change in peak diameter (30 s). E: dihydroethidium (DHE) staining of vascular rings. F: relative difference in DHE fluorescence in E. G: Western blot analysis of endothelial nitric oxide synthase (eNOS) expression in vascular tissue. Graphs depict means ± SE with n = 5–7/group. Statistical significance was determined using one-way ANOVA followed by Holm–Sidak’s test. *P < 0.05. ACh, acetylcholine; CDC, cardiosphere-derived cells; RFU, relative fluorescence units.
Through the synthesis and release of NO by endothelial cells (41), eNOS is an essential factor regulating vascular tone. Under inflammatory stress, as in hypertension or diabetes, eNOS may lose expression or functionally decouple to produce reactive oxygen species (ROS; i.e., superoxide and peroxynitrite) instead of NO, thereby causing vascular stiffness (42). Aortic rings from vehicle-injected HS-fed DS rats exhibited higher levels of dihydroethidium fluorescence than their CDC-treated counterparts (Fig. 3, E and F), indicative of higher ROS levels. Consistent with this finding, total eNOS levels were low in vehicle-infused HFpEF animals but were partially restored with CDCs (Fig. 3G).
Rat CDCs Alter the Phenotype of Endothelial Cells Within the Heart
To probe transcriptomic changes in endothelial cells, rat hearts (n = 3/group; i.e., control, HFpEF, and HFpEF + CDC) were perfused and enzymatically digested for single-cell RNA sequencing (scRNA-seq; Fig. 4A). Unique cell clusters were visualized with tSNE analysis (Fig. 4B). Focusing on the endothelial population demarcated by Cdh5 expression (Fig. 4C), we analyzed genes that were differentially expressed among experimental groups by Ingenuity Pathway Analysis (IPA, QIAGEN) (Fig. 4D). CDCs suppressed a variety of disease-associated terms, including leukocyte movement, cell migration, and leukocyte infiltration in endothelial cells (Fig. 4D).
Figure 4.

Single-cell RNA sequencing (scRNA-seq) of heart failure with preserved ejection fraction (HFpEF) hearts. A: isolated cells from hearts were processed using Chromium 10 × 3′DEG chemistry. B: t-distribution stochastic neighbor embedding (tSNE) plot of identified cells pooled from all groups. C: tSNE plot highlighting scaled and normalized unique molecular identifier (UMI) counts for endothelial gene Cdh5 [vascular endothelial-cadherin (VE-cadherin)]. D: pathway analysis of gene expression profile in endothelial cells. E: pseudotime expression analysis of endothelial cells based on uniform manifold approximation and projection (UMAP) clustering. F: overlay of endothelial cells on pseudotime UMAP plot. Blue, phosphate-buffered saline (PBS); red, cardiosphere-derived cells (CDCs); green, control. G: pie showing the fractional population of cells assigned to each subcluster relative to the total number of endothelial cells.
Trajectory inference or pseudotime ordering is an in silico method of projecting relationships between cells based on changes in global gene expression patterns within individual cells. We hypothesized that nondiseased (control) cells would provide a reference starting point along a continuum of cell changes toward disease; if so, CDCs might revert some of the HFpEF-associated changes toward control levels. To perform pseudotime ordering on endothelial cells, samples were pooled from all three groups, visualized by uniform manifold approximation and projection (UMAP) plots, and analyzed. We positioned time 0 at the center of the control cell cluster and projected cells from HFpEF and HFpEF + CDC hearts relative to that set point (Fig. 4E). In contrast to HFpEF endothelial cells (blue dot, Fig. 4F), which clustered at the distal point of the time plot, HFpEF + CDC endothelial cells (red dot, Fig. 4F) were found in close proximity to control endothelial cells (green dot, Fig. 4F).
Although scRNA-seq trajectory analysis provided an unbiased manner to characterize endothelial cells during HFpEF induction and in response to CDCs, such analysis may obscure an underlying redistribution of endothelial cell subpopulations (43). Analysis of endothelial cell subpopulations identified six subclusters, with subcluster 1 being the most abundant in healthy hearts (Fig. 4G). Conversely, PBS-injected HFpEF animals revealed a marked loss of subcluster 1 along with a gain of subcluster 0 (Fig. 4G), which was associated with multiple diseases by IPA analysis (Supplemental Fig. S1). On the other hand, CDC-infused HFpEF hearts exhibited partial reversion of these subpopulations toward the healthy distribution (Fig. 4G). Further verification of these interpretations comes from pseudotemporal plots of all six endothelial cell subpopulations (Supplemental Fig. S2). These findings, although inferential, are consistent with the conjecture that CDCs reverse the endothelial gene expression profile from HFpEF toward control. Moreover, expression profiling of key proinflammatory genes revealed the rapid anti-inflammatory effects of CDCs on the myocardial endothelium (Fig. 5). Although the focus here is on endothelial cells, CDCs also modulate key pathways in macrophages, fibroblasts, and cardiomyocytes (Supplemental Fig. S3).
Figure 5.
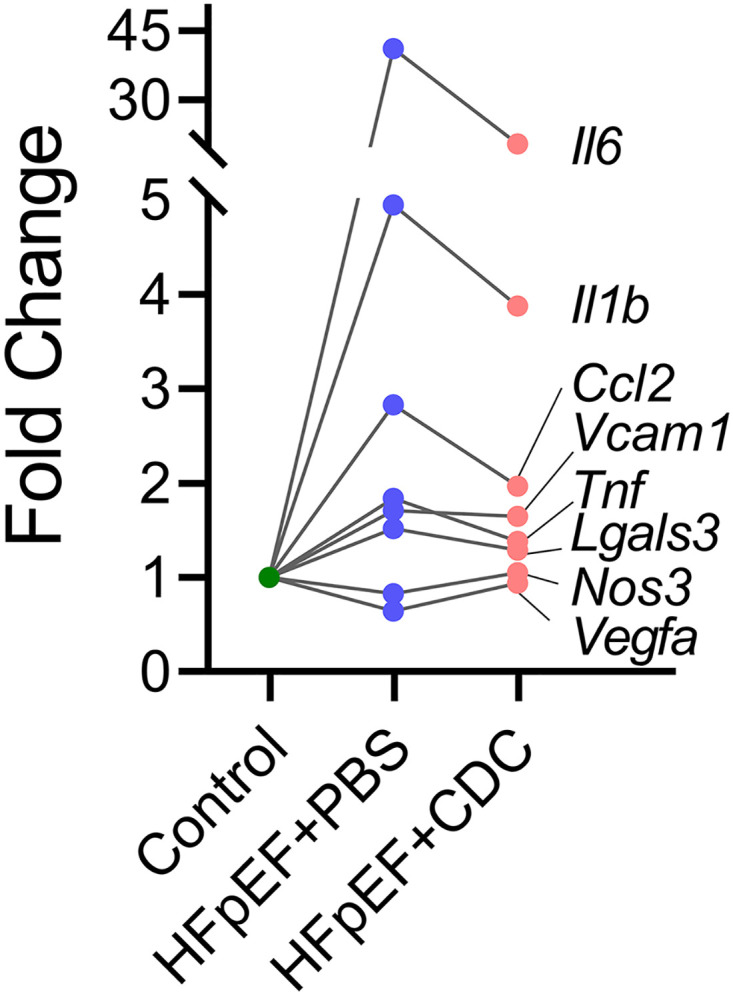
Relative gene expression analysis of proinflammatory genes within endothelial cells. CDC, cardiosphere-derived cell; HFpEF, heart failure with preserved ejection fraction.
Adhesion Molecule Expression in HFpEF
To probe the relevance of scRNA-seq findings, we performed a series of in vitro experiments. As shown in Fig. 6A, aortic rings from HFpEF + CDC rats were more angiogenic than those isolated from HFpEF + vehicle, producing tubes with greater length when placed in Matrigel media. Given the extensive evidence that CDCs act by secreting extracellular vesicles (CDCev), we used an in vitro assay to probe CDCev effects on the cell adhesion. In RAOECs stimulated with TNFα (Fig. 6B), CDCev exposure decreased Vcam1 and Icam1, but not e-selectin, expression (Fig. 6C). At the protein level, VCAM-1 was confirmed to be reduced by CDCev, although ICAM-1 was not much changed (Fig. 6, D and E). To assess whether the reduced adhesion molecule expression reflects changes in adhesion, we added monocytes (THP-1) to the RAOEC monolayer (Fig. 6F). As expected, monocytes bound to the monolayer (Fig. 6G); monocyte attachment and transendothelial cell migration were reduced by CDCev exposure (Fig. 6, G and H). Thus, evidence at multiple levels (isolated endothelial cells, aortic rings, and in vivo) supports the notion that CDCs enhance endothelial function in HFpEF. Taken together, the findings affirm the initial hypothesis that systemic inflammation drives endothelial stress and further rationalize the roles of endothelial cells and monocytes/macrophages in the pathogenesis of HFpEF.
Figure 6.

Endothelial cell adhesion molecule expression and monocyte adhesion. A: representative images of endothelial cell outgrowth from aortic rings grown on Matrigel (left) and endothelial tube length measurements from aortic rings (right). B: schematic of in vitro assays. C: dose-dependent reduction in adhesion molecule gene expression in rat aortic endothelial cells (RAOECs). D: protein expression of VCAM-1 and ICAM-1. E: quantification of immunoblots in D. F: confocal images depicting THP-1 monocyte adhesion to a RAOEC monolayer. G: representative images (left) and quantification of THP-1 monocytes attached to a RAOEC monolayer (right). H: transmigration of THP-1 cells through RAOEC monolayer. Graphs depict means ± SE with n = 5–8/group. Statistical significance was determined using unpaired Student’s t test (A, E, G, and H) or one-way ANOVA followed by Holm–Sidak’s test. *P < 0.05 and ***P < 0.001. CDCev, cardiosphere-derived cell extracellular vesicles; THP-1, monocytes.
DISCUSSION
In 2013, Paulus and Tschöpe (24) presented a novel hypothesis detailing HFpEF pathogenesis. They posited a central role for systemic inflammation, which emerges from underlying comorbidities (e.g., obesity, hypertension, diabetes, and chronic obstructive pulmonary disease), as the unifying thread underlying the protean manifestations of HFpEF. This chronic proinflammatory state was proposed to alter vascular function by promoting endothelial inflammation (ROS production) and reducing NO bioavailability. Concomitantly, endothelial adhesion molecule expression is enhanced, which promotes leukocyte transmigration, secretion of TGF-β, and conversion of fibroblasts to activated myofibroblasts that deposit larger amounts of extracellular matrix. The resulting cardiomyocyte hypertrophy and extracellular matrix accumulation bolsters LV stiffness and impaired relaxation. Meanwhile, functional reserve is limited by impaired vascular responsiveness secondary to endothelial dysfunction.
In models of acute ischemic injury (i.e., myocardial infarction), CDCs and their secreted EVs suppress robust inflammatory responses by modifying macrophage function (11, 44, 45). In the context of Paulus and Tschöpe’s hypothesis, we demonstrate that CDCs confer an anti-inflammatory effect with rapid onset that lasts for at least 2 wk, and, in so doing, modify endothelial function in HFpEF (Fig. 7). CDCs delivered via the intracoronary route migrate into the coronary vasculature where they juxtapose to vascular endothelial cells and release paracrine factors (e.g., EVs). In DS rats, HS diet precipitates hypertension, diastolic dysfunction, and systemic inflammation (17). CDCs suppress endothelial ROS production and reestablish eNOS activity. By doing so, CDCs alleviate oxidative stress and vascular stiffness, resulting in improved endothelial-dependent relaxation. Despite the overall therapeutic benefits, including extended survival, blood pressure was not much changed in CDC-treated HFpEF rats (17). In vitro, CDCev suppress VCAM1 expression, prevent monocyte adhesion, and inhibit diapedesis. Cardiac scRNA-seq findings support the concepts that inflammation is heightened in HFpEF and suppressed by CDCs. Thus, the effects of CDCs not only support the overall Paulus/Tschöpe hypothesis but also provide granular detail as to precisely how the various elements interact to generate and perpetuate the insidious HFpEF phenotype.
Figure 7.

Endothelial dysfunction in heart failure with preserved ejection fraction (HFpEF). Multiple lines of evidence implicate inflammation of endothelial cells as a major pathogenic factor in rats with HFpEF. Inflammation drives reactive oxygen species (ROS) production, reduces endothelial nitric oxide synthase (eNOS) expression, and favors accumulation of monocytes; together, these factors lead to vascular dysfunction. Treatment with cardiosphere-derived cells (CDCs) reduced ROS production, restored eNOS abundance, lowered monocyte levels, and rescued the expression of multiple disease-associated genes, thereby restoring vascular reactivity. SMCs, smooth muscle cells.
Several caveats are worth emphasizing. The DS rat HFpEF model is extremely aggressive, with a >50% mortality within 2 mo of starting high-salt diet (17, 22, 28). Diastolic dysfunction develops within 6–7 wk; inflammation and fibrosis are intense. As we previously described in this model, it is perhaps not surprising that a single dose of CDCs, given when the HFpEF is already established, will have benefits that last to the endpoint 4 wk later (17), given the anti-inflammatory and immunomodulatory nature of these cells (46). Therefore, because of the limitations of the DS model, the therapeutic efficacy of CDCs should be verified in different models of HFpEF and address sex differences (29, 47). Moreover, the prevalence of HFpEF increases with age (47). Young CDCs have the capacity to rejuvenate old Fisher-344 animals (48), which are prone to HFpEF (49, 50), but further investigations are necessary to test whether CDCs alleviate aging-dependent endothelial dysfunction.
In actual clinical practice, however, HFpEF is a chronic, indolent disease. The ongoing Regress-HFpEF trial was designed 6 years ago with single intracoronary dosing of CDCs (NCT02941705), but given what we know now, a more logical regimen going forward may be that used successfully in another chronic form of HF: the cardiomyopathy associated with Duchenne muscular dystrophy (DMD). In the HOPE-2 trial (13), patients with advanced DMD were randomized to receive either placebo or CDCs, intravenously, every 3 mo. After 1 year, the patients who had received CDCs exhibited preserved LV function and structure, unlike the placebo group, which deteriorated significantly. Given the promising results of HOPE-2, multiple intravenous CDC doses delivered sequentially over time may be a rational approach to test in patients with HFpEF. Our laboratory and others have shown that CDCev act as mediators of the CDC therapeutic effect in various forms of cardiomyopathy, modulating numerous disease-associated pathways to favor tissue healing and repair (51–55). Although we and others identified macrophages as functional recipients of CDCev (11, 45), endothelial cells physically interact with the bloodstream and might be first responders to CDCev.
Together, our data emphasize the mechanistic role of inflammation in HFpEF. Further work will be required to elucidate the precise relationships among endothelial cells and classical inflammatory cells (macrophages, neutrophils, and lymphocytes) in HFpEF. Although our findings do not exclude important roles for other cell types, clearly endothelial cells play a major pathogenic role.
SUPPLEMENTAL DATA
Supplemental Table S1 and Supplemental Figs. S1–S3: https://doi.org/10.6084/m9.figshare.20043350.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants R01HL124074 and R01HL133835 (to E.M.) and R01 HL142579 (to G.d.C.) and American Heart Association Postdoctoral Fellowship 836665 (to T.M.).
DISCLOSURES
E.M. holds the Mark S. Siegel Family Foundation Distinguished Chair of the Cedars-Sinai Medical Center. G.d.C. was a paid consultant for Capricor Therapeutics during the study. E.M. owns founder’s stock in Capricor Therapeutics. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
G.d.C. and E.M. conceived and designed research; G.d.C., T.M., X.W., F.H., A.A., N.N., L.L., M.T., and P.K. performed experiments; G.d.C., T.M., X.W., A.R., F.H., A.A., N.N., D.W., Y.W., and P.K. analyzed data; G.d.C., T.M., and E.M. interpreted results of experiments; G.d.C. and T.M. prepared figures; G.d.C., T.M., and E.M. drafted manuscript; G.d.C., T.M., E.C., and E.M. edited and revised manuscript; G.d.C., T.M., X.W., A.R., F.H., A.A., N.N., D.W., Y.W., L.L., M.T., P.K., E.C., and E.M. approved final version of manuscript.
REFERENCES
- 1. Kelly JP, Mentz RJ, Mebazaa A, Voors AA, Butler J, Roessig L, Fiuzat M, Zannad F, Pitt B, O'Connor CM, Lam CSP. Patient selection in heart failure with preserved ejection fraction clinical trials. J Am Coll Cardiol 65: 1668–1682, 2015. doi: 10.1016/j.jacc.2015.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mentz RJ, Kelly JP, von Lueder TG, Voors AA, Lam CSP, Cowie MR, Kjeldsen K, Jankowska EA, Atar D, Butler J, Fiuzat M, Zannad F, Pitt B, O'Connor CM. Noncardiac comorbidities in heart failure with reduced versus preserved ejection fraction. J Am Coll Cardiol 64: 2281–2293, 2014. doi: 10.1016/j.jacc.2014.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Scantlebury DC, Borlaug BA. Why are women more likely than men to develop heart failure with preserved ejection fraction? Curr Opin Cardiol 26: 562–568, 2011. doi: 10.1097/HCO.0b013e32834b7faf. [DOI] [PubMed] [Google Scholar]
- 4. Tschöpe C, Van Linthout S. New insights in (inter)cellular mechanisms by heart failure with preserved ejection fraction. Curr Heart Fail Rep 11: 436–444, 2014. doi: 10.1007/s11897-014-0219-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Anker SD, Butler J, Filippatos G, Ferreira JP, Bocchi E, Böhm M, et al. Empagliflozin in heart failure with a preserved ejection fraction. N Engl J Med 385: 1451–1461, 2021. doi: 10.1056/NEJMoa2107038. [DOI] [PubMed] [Google Scholar]
- 6. van Berlo JH, Kanisicak O, Maillet M, Vagnozzi RJ, Karch J, Lin S-CJ, Middleton RC, Marbán E, Molkentin JD. c-kit+ cells minimally contribute cardiomyocytes to the heart. Nature 509: 337–341, 2014. doi: 10.1038/nature13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cheng K, Ibrahim A, Hensley MT, Shen D, Sun B, Middleton R, Liu W, Smith RR, Marbán E. Relative roles of CD90 and c-kit to the regenerative efficacy of cardiosphere-derived cells in humans and in a mouse model of myocardial infarction. J Am Heart Assoc 3: e001260, 2014. doi: 10.1161/JAHA.114.001260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Davis DR, Zhang Y, Smith RR, Cheng K, Terrovitis J, Malliaras K, Li T-S, White A, Makkar R, Marbán E. Validation of the cardiosphere method to culture cardiac progenitor cells from myocardial tissue. PLoS One 4: e7195, 2009. doi: 10.1371/journal.pone.0007195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith RR, Barile L, Cho HC, Leppo MK, Hare JM, Messina E, Giacomello A, Abraham MR, Marbán E. Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation 115: 896–908, 2007. doi: 10.1161/CIRCULATIONAHA.106.655209. [DOI] [PubMed] [Google Scholar]
- 10. Chimenti I, Smith RR, Li T-S, Gerstenblith G, Messina E, Giacomello A, Marbán E. Relative roles of direct regeneration versus paracrine effects of human cardiosphere-derived cells transplanted into infarcted mice. Circ Res 106: 971–980, 2010. doi: 10.1161/CIRCRESAHA.109.210682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Couto G, Gallet R, Cambier L, Jaghatspanyan E, Makkar N, Dawkins JF, Berman BP, Marbán E. Exosomal microRNA transfer into macrophages mediates cellular postconditioning. Circulation 136: 200–214, 2017. doi: 10.1161/CIRCULATIONAHA.116.024590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aghila Rani KG, Kartha CC. Effects of epidermal growth factor on proliferation and migration of cardiosphere-derived cells expanded from adult human heart. Growth Factors 28: 157–165, 2010. doi: 10.3109/08977190903512628. [DOI] [PubMed] [Google Scholar]
- 13. McDonald CM, Marbán E, Hendrix S, Hogan N, Ruckdeschel Smith R, Eagle M, Finkel RS, Tian C, Janas J, Harmelink MM, Varadhachary AS, Taylor MD, Hor KN, Mayer OH, Henricson EK, Furlong P, Ascheim DD, Rogy S, Williams P, Marbán L; HOPE-2 Study Group. Repeated intravenous cardiosphere-derived cell therapy in late-stage Duchenne muscular dystrophy (HOPE-2): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 399: 1049–1058, 2022. doi: 10.1016/S0140-6736(22)00012-5. [DOI] [PubMed] [Google Scholar]
- 14. Mishra R, Vijayan K, Colletti EJ, Harrington DA, Matthiesen TS, Simpson D, Goh SK, Walker BL, Almeida-Porada G, Wang D, Backer CL, Dudley SC, Wold LE, Kaushal S. Characterization and functionality of cardiac progenitor cells in congenital heart patients. Circulation 123: 364–373, 2011. doi: 10.1161/CIRCULATIONAHA.110.971622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Singh S, Chakravarty T, Chen P, Akhmerov A, Falk J, Friedman O, Zaman T, Ebinger JE, Gheorghiu M, Marbán L, Marbán E, Makkar RR. Allogeneic cardiosphere-derived cells (CAP-1002) in critically ill COVID-19 patients: compassionate-use case series. Basic Res Cardiol 115: 36, 2020. doi: 10.1007/s00395-020-0795-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Takehara N, Tsutsumi Y, Tateishi K, Ogata T, Tanaka H, Ueyama T, Takahashi T, Takamatsu T, Fukushima M, Komeda M, Yamagishi M, Yaku H, Tabata Y, Matsubara H, Oh H. Controlled delivery of basic fibroblast growth factor promotes human cardiosphere-derived cell engraftment to enhance cardiac repair for chronic myocardial infarction. J Am Coll Cardiol 52: 1858–1865, 2008. doi: 10.1016/j.jacc.2008.06.052. [DOI] [PubMed] [Google Scholar]
- 17. Gallet R, de Couto G, Simsolo E, Valle J, Sun B, Liu W, Tseliou E, Zile MR, Marbán E. Cardiosphere-derived cells reverse heart failure with preserved ejection fraction (HFpEF) in rats by decreasing fibrosis and inflammation. JACC Basic Transl Sci 1: 14–28, 2016. doi: 10.1016/j.jacbts.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wendt N, Schulz A, Qadri F, Bolbrinker J, Kossmehl P, Winkler K, Stoll M, Vetter R, Kreutz R. Genetic analysis of salt-sensitive hypertension in Dahl rats reveals a link between cardiac fibrosis and high cholesterol. Cardiovasc Res 81: 618–626, 2009. doi: 10.1093/cvr/cvn263. [DOI] [PubMed] [Google Scholar]
- 19. Qu P, Hamada M, Ikeda S, Hiasa G, Shigematsu Y, Hiwada K. Time-course changes in left ventricular geometry and function during the development of hypertension in Dahl salt-sensitive rats. Hypertens Res 23: 613–623, 2000. doi: 10.1291/hypres.23.613. [DOI] [PubMed] [Google Scholar]
- 20. Rapp JP, Dene H. Development and characteristics of inbred strains of Dahl salt-sensitive and salt-resistant rats. Hypertension 7: 340–349, 1985. [PubMed] [Google Scholar]
- 21. Cho JH, Zhang R, Kilfoil PJ, Gallet R, de Couto G, Bresee C, Goldhaber JI, Marbán E, Cingolani E. Delayed repolarization underlies ventricular arrhythmias in rats with heart failure and preserved ejection fraction. Circulation 136: 2037–2050, 2017. doi: 10.1161/CIRCULATIONAHA.117.028202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cho JH, Kilfoil PJ, Zhang R, Solymani RE, Bresee C, Kang EM, Luther K, Rogers RG, de Couto G, Goldhaber JI, Marbán E, Cingolani E. Reverse electrical remodeling in rats with heart failure and preserved ejection fraction. JCI Insight 3: e121123, 2018. doi: 10.1172/jci.insight.121123. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23. Mesquita T, Lin Y-N, Ibrahim A. Chronic low-grade inflammation in heart failure with preserved ejection fraction. Aging Cell 20: e13453, 2021. doi: 10.1111/acel.13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 62: 263–271, 2013. doi: 10.1016/j.jacc.2013.02.092. [DOI] [PubMed] [Google Scholar]
- 25. Cuijpers I, Simmonds SJ, van Bilsen M, Czarnowska E, González Miqueo A, Heymans S, Kuhn AR, Mulder P, Ratajska A, Jones EAV, Brakenhielm E. Microvascular and lymphatic dysfunction in HFpEF and its associated comorbidities. Basic Res Cardiol 115: 39, 2020. doi: 10.1007/s00395-020-0798-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van Heerebeek L, Franssen CPM, Hamdani N, Verheugt FWA, Somsen GA, Paulus WJ. Molecular and cellular basis for diastolic dysfunction. Curr Heart Fail Rep 9: 293–302, 2012. doi: 10.1007/s11897-012-0109-5. [DOI] [PubMed] [Google Scholar]
- 27. Kakkar R, Lee RT. Intramyocardial fibroblast myocyte communication. Circ Res 106: 47–57, 2010. doi: 10.1161/CIRCRESAHA.109.207456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mesquita T, Zhang R, Cho JH, Zhang R, Lin Y-N, Sanchez L, Goldhaber JI, Yu JK, Liang JA, Liu W, Trayanova NA, Cingolani E. Mechanisms of sinoatrial node dysfunction in heart failure with preserved ejection fraction. Circulation 145: 45–60, 2022. doi: 10.1161/CIRCULATIONAHA.121.054976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Elkholey K, Morris L, Niewiadomska M, Houser J, Ramirez M, Tang M, Humphrey MB, Stavrakis S. Sex differences in the incidence and mode of death in rats with heart failure with preserved ejection fraction. Exp Physiol 106: 673–682, 2021. doi: 10.1113/EP089163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Malliaras K, Li T-S, Luthringer D, Terrovitis J, Cheng K, Chakravarty T, Galang G, Zhang Y, Schoenhoff F, Van Eyk J, Marbán L, Marbán E. Safety and efficacy of allogeneic cell therapy in infarcted rats transplanted with mismatched cardiosphere-derived cells. Circulation 125: 100–112, 2012. doi: 10.1161/CIRCULATIONAHA.111.042598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marbán E, Liao K. On the cellular origin of cardiosphere-derived cells (CDCs). Basic Res Cardiol 117: 12, 2022. doi: 10.1007/s00395-022-00914-x. [DOI] [PubMed] [Google Scholar]
- 32. Machin DR, Leary ME, He Y, Shiu Y-T, Tanaka H, Donato AJ. Ultrasound assessment of flow-mediated dilation of the brachial and superficial femoral arteries in rats. J Vis Exp 54762, 2016. doi: 10.3791/54762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wilde E, Aubdool AA, Thakore P, Baldissera L, Alawi KM, Keeble J, Nandi M, Brain SD. Tail-cuff technique and its influence on central blood pressure in the mouse. J Am Heart Assoc 6: e005204, 2017. doi: 10.1161/JAHA.116.005204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Makkar RR, Kereiakes DJ, Aguirre F, Kowalchuk G, Chakravarty T, Malliaras K, Francis GS, Povsic TJ, Schatz R, Traverse JH, Pogoda JM, Smith RR, Marbán L, Ascheim DD, Ostovaneh MR, Lima JAC, DeMaria A, Marbán E, Henry TD. Intracoronary ALLogeneic heart STem cells to Achieve myocardial Regeneration (ALLSTAR): a randomized, placebo-controlled, double-blinded trial. Eur Heart J 41: 3451–3458, 2020. doi: 10.1093/eurheartj/ehaa541. [DOI] [PubMed] [Google Scholar]
- 35. Kilfoil PJ, Lotteau S, Zhang R, Yue X, Aynaszyan S, Solymani RE, Cingolani E, Marbán E, Goldhaber JI. Distinct features of calcium handling and β-adrenergic sensitivity in heart failure with preserved versus reduced ejection fraction. J Physiol 598: 5091–5108, 2020. doi: 10.1113/JP280425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang AW, O'Flanagan C, Chavez EA, Lim JLP, Ceglia N, McPherson A, Wiens M, Walters P, Chan T, Hewitson B, Lai D, Mottok A, Sarkozy C, Chong L, Aoki T, Wang X, Weng AP, McAlpine JN, Aparicio S, Steidl C, Campbell KR, Shah SP. Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nat Methods 16: 1007–1015, 2019. doi: 10.1038/s41592-019-0529-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32: 381–386, 2014. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Toft P, Lillevang ST, Tønnesen E, Svendsen P, Höhndorf K. Redistribution of lymphocytes following E. coli sepsis. Scand J Immunol 38: 541–545, 1993. doi: 10.1111/j.1365-3083.1993.tb03238.x. [DOI] [PubMed] [Google Scholar]
- 39. Afsar B, Kuwabara M, Ortiz A, Yerlikaya A, Siriopol D, Covic A, Rodriguez-Iturbe B, Johnson RJ, Kanbay M. Salt intake and immunity. Hypertension 72: 19–23, 2018. doi: 10.1161/HYPERTENSIONAHA.118.11128. [DOI] [PubMed] [Google Scholar]
- 40. He H, Xu J, Warren CM, Duan D, Li X, Wu L, Iruela-Arispe ML. Endothelial cells provide an instructive niche for the differentiation and functional polarization of M2-like macrophages. Blood 120: 3152–3162, 2012. doi: 10.1182/blood-2012-04-422758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Förstermann U, Münzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation 113: 1708–1714, 2006. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- 42. Mahmud A, Feely J. Arterial stiffness is related to systemic inflammation in essential hypertension. Hypertension 46: 1118–1122, 2005. doi: 10.1161/01.HYP.0000185463.27209.b0. [DOI] [PubMed] [Google Scholar]
- 43. Tombor LS, John D, Glaser SF, Luxán G, Forte E, Furtado M, Rosenthal N, Baumgarten N, Schulz MH, Wittig J, Rogg E-M, Manavski Y, Fischer A, Muhly-Reinholz M, Klee K, Looso M, Selignow C, Acker T, Bibli S-I, Fleming I, Patrick R, Harvey RP, Abplanalp WT, Dimmeler S. Single cell sequencing reveals endothelial plasticity with transient mesenchymal activation after myocardial infarction. Nat Commun 12: 681, 2021. doi: 10.1038/s41467-021-20905-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. de Couto G, Liu W, Tseliou E, Sun B, Makkar N, Kanazawa H, Arditi M, Marbán E. Macrophages mediate cardioprotective cellular postconditioning in acute myocardial infarction. J Clin Invest 125: 3147–3162, 2015. doi: 10.1172/JCI81321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. de Couto G, Jaghatspanyan E, DeBerge M, Liu W, Luther K, Wang Y, Tang J, Thorp EB, Marbán E. Mechanism of enhanced mertk-dependent macrophage efferocytosis by extracellular vesicles. Arterioscler Thromb Vasc Biol 39: 2082–2096, 2019. doi: 10.1161/ATVBAHA.119.313115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marbán E. A mechanistic roadmap for the clinical application of cardiac cell therapies. Nat Biomed Eng 2: 353–361, 2018. doi: 10.1038/s41551-018-0216-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dunlay SM, Roger VL, Redfield MM. Epidemiology of heart failure with preserved ejection fraction. Nat Rev Cardiol 14: 591–602, 2017. doi: 10.1038/nrcardio.2017.65. [DOI] [PubMed] [Google Scholar]
- 48. Grigorian-Shamagian L, Liu W, Fereydooni S, Middleton RC, Valle J, Cho JH, Marbán E. Cardiac and systemic rejuvenation after cardiosphere-derived cell therapy in senescent rats. Eur Heart J 38: 2957–2967, 2017. doi: 10.1093/eurheartj/ehx454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mesquita TRR, Zhang R, de Couto G, Valle J, Sanchez L, Rogers RG, Holm K, Liu W, Marbán E, Cingolani E. Mechanisms of atrial fibrillation in aged rats with heart failure with preserved ejection fraction. Heart Rhythm 17: 1025–1033, 2020. doi: 10.1016/j.hrthm.2020.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Valero-Muñoz M, Backman W, Sam F. Murine models of heart failure with preserved ejection fraction: a “fishing expedition”. JACC Basic Transl Sci 2: 770–789, 2017. doi: 10.1016/j.jacbts.2017.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ibrahim AG-E, Cheng K, Marbán E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Reports 2: 606–619, 2014. doi: 10.1016/j.stemcr.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gallet R, Dawkins J, Valle J, Simsolo E, de Couto G, Middleton R, Tseliou E, Luthringer D, Kreke M, Smith RR, Marbán L, Ghaleh B, Marbán E. Exosomes secreted by cardiosphere-derived cells reduce scarring, attenuate adverse remodelling, and improve function in acute and chronic porcine myocardial infarction. Eur Heart J 38: 201–211, 2017. doi: 10.1093/eurheartj/ehw240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rogers RG, Fournier M, Sanchez L, Ibrahim AG, Aminzadeh MA, Lewis MI, Marbán E. Disease-modifying bioactivity of intravenous cardiosphere-derived cells and exosomes in mdx mice. JCI Insight 4: 130202, 2019. doi: 10.1172/jci.insight.130202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dawkins JF, Ehdaie A, Rogers R, Soetkamp D, Valle J, Holm K, Sanchez L, Tremmel I, Nawaz A, Shehata M, Wang X, Prakosa A, Yu J, Van Eyk JE, Trayanova N, Marbán E, Cingolani E. Biological substrate modification suppresses ventricular arrhythmias in a porcine model of chronic ischaemic cardiomyopathy. Eur Heart J 43: 2139–2156, 2022. doi: 10.1093/eurheartj/ehac042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lin Y-N, Mesquita T, Sanchez L, Chen Y-H, Liu W, Li C, Rogers R, Wang Y, Li X, Wu D, Zhang R, Ibrahim A, Marbán E, Cingolani E. Extracellular vesicles from immortalized cardiosphere-derived cells attenuate arrhythmogenic cardiomyopathy in desmoglein-2 mutant mice. Eur Heart J 42: 3558–3571, 2021. doi: 10.1093/eurheartj/ehab419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1 and Supplemental Figs. S1–S3: https://doi.org/10.6084/m9.figshare.20043350.