

Keywords: acute kidney injury, exosomes, fibrinolysis, ischemia-reperfusion, microvasculature
Abstract
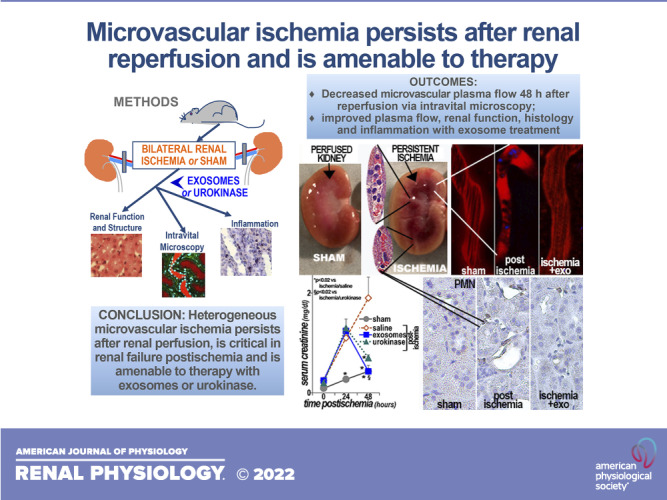
Ischemic acute kidney injury is common, deadly, and accelerates the progression of chronic kidney disease, yet has no specific therapy. After ischemia, reperfusion is patchy with early and persistent impairment in regional renal blood flow and cellular injury. We tested the hypothesis that intrarenal coagulation results in sustained renal ischemia following reperfusion, using a well-characterized model. Markedly decreased, but heterogeneous, microvascular plasma flow with microthrombi was found postischemia by intravital microscopy. Widespread tissue factor expression and fibrin deposition were also apparent. Clotting was accompanied by complement activation and inflammation. Treatment with exosomes derived from renal tubular cells or with the fibrinolytic urokinase, given 24 h postischemia when renal failure was established, significantly improved microvascular flow, coagulation, serum creatinine, and histological evidence of injury. These data support the hypothesis that intrarenal clotting occurs early and the resultant sustained ischemia is a critical determinant of renal failure following ischemia; they demonstrate that the coagulation abnormalities are amenable to therapy and that therapy results in improvement in both function and postischemic inflammation.
NEW & NOTEWORTHY Ischemic renal injury carries very high morbidity and mortality, yet has no specific therapy. We found markedly decreased, heterogeneous microvascular plasma flow, tissue factor induction, fibrin deposition, and microthrombi after renal ischemia-reperfusion using a well-characterized model. Renal exosomes or the fibrinolytic urokinase, administered after renal failure was established, improved microvascular flow, coagulation, renal function, and histology. Data demonstrate that intrarenal clotting results in sustained ischemia amenable to therapy that improves both function and postischemic inflammation.
INTRODUCTION
Ischemia to the kidney and other organs is deadly and expensive (1–12). Patients with acute kidney injury (AKI) have a higher mortality rate (>50%) than those with myocardial infarction (13). In addition to the immeasurable suffering, AKI care has been estimated to be more costly than lung and skin cancer combined (14). Despite significant advances in understanding the mechanisms of tubular cell injury, death, and regeneration, and in supportive care in AKI, there is no specific therapy and mortality has not improved substantially in decades (15). Thus, considerable effort is underway to prevent or reverse AKI, which has emerged as a major health problem (16).
Renal injury triggers conserved protective responses, which presumably evolved to constrain damage and restore homeostasis and normal function. For example, early responses, coagulation and inflammation, can limit fluid loss and clear dead cells and debris (17–19). Subsequent epithelial and mesenchymal adaptations reestablish anatomic integrity and organ function (20). These protective responses have been described, to various degrees, in ischemic (17, 21), chemically induced (22), and infection-related (23) AKI.
Our previous work showed that intravenous renal tubular cell transplants protect rat kidneys injured by different insults, ischemic and nephrotoxic (24–27). We also showed that infusions of exosomes derived from either rat or human renal tubular cells are more effective at protecting postischemic rat kidneys, even when given after 24 h of reperfusion (17, 21). On the other hand, untreated ischemia triggers major chronic renal changes in equally ischemic rats (17, 21). Of note, postischemic renal function improves within 24 h of exosome injection. This rapid effect is not consistent with presumably slower processes, such as cell repair and regeneration. Thus, we hypothesized that the acute effects of exosomes were in part due to prevention of microvascular dysfunction (21).
The renal prothrombotic response was recognized many years ago (19); it is initiated when circulating coagulation factors make contact with renal epithelial tissue factor (TF or F3) (28) in conjunction with enhanced renal fibrinogen synthesis (29). These early steps are potentially harmful, as they activate the coagulation cascade, with platelet aggregation, thrombus formation, and associated complement activation (28). Although some existing data have suggested benefits with prophylactic heparin (30, 31), others have not (32, 33). A human study has suggested that heparin has the potential to prevent but not treat thrombi (34). The renal fibrinolytic response, however, can restore blood flow after clotting occurs (35). Thus, the aim of this study was to test the hypotheses that microvascular ischemia persists after reperfusion, that improving flow with thrombolysis will improve function, and that exosomes, given after ischemia, will improve renal microvascular perfusion. These hypotheses were evaluated in a well-tested model of renal ischemia-reperfusion, and the findings support the concept that postischemic reperfusion is uneven, complicated by unresolved and extensive microthromboses, likely causing zones of cell death and eventual fibrosis (17, 21).
MATERIALS AND METHODS
Human Renal Tubular Cells
Primary human renal tubular cells (21) were obtained from donated normal human kidneys declined for renal transplantation. Our research with human kidney cells from discarded normal kidneys was reviewed and exempted by the Institutional Review Board of Indiana University and approved by the Indiana Donor Network. The kidney cortex was digested with 6 mg/dL collagenase type 4 (Worthington, Lakewood, NJ) at 37°C for 1 h. The renal tubules were separated from damaged cells and debris by centrifugation at 46 rcf for 3 min and subsequently cultured in S1 medium for 3 days under normoxic conditions (37°C, 5% CO2). Each 2 L of S1 medium contains F-12 HAM (10.7 g), DMEM (8.32 g), l-glutamine (0.29 g), HEPES (4.78 g), sodium pyruvate (0.11 g), phenol red (3.2 mL), and 7% sodium bicarbonate solution (82.8 mL) (pH 7.4). S1 medium also contained hepatocyte growth factor (200 ng/mL), epidermal growth factor (400 ng/mL) (R&D Systems, Minneapolis, MN), hydrocortisone (100 µg/mL), insulin (35 µg/mL), transferrin (32 µg/mL), sodium selenite (42 ng/mL) (Sigma, St. Louis MO), 10% FBS, and 1% antibiotic/antimycotic solution. Cultured renal tubular cells were then either used for experiments or frozen at −80°C in Iscove’s modified Dulbecco’s medium with 30% FBS and 10% DMSO.
Cellular Hypoxia and d-Dimer Measurements
Cells were either cultured under standard normoxic conditions (37°C, 5% CO2) or hypoxic conditions (37°C, <1% O2 and 5% CO2) in a hypoxic chamber (SCI-tive Dual, Baker Ruskinn, Sanford, ME). Fibrinolytic activity of human renal tubular cells was estimated from d-dimer production. Cells were cultured in either normoxic medium for 48 h or normoxic medium for 24 h and then switched to hypoxic medium, without or with 1.8e9 exosomes/dish, for an additional 24 h. Cells were also cultured without or with 1 mg of human thrombin and 0.1 mg of purified human fibrinogen (Sigma-Aldrich, St. Louis, MO). d-Dimer levels were measured in the culture media by the clinical laboratory of the Indianapolis Veterans Affairs Hospital using a d-Dimer HS kit and read on an ACL Top 500 analyzer (Instrumentation Laboratory, Bedford, MA).
Exosome Collection
Human renal tubular cells were plated at 30% confluence in 225-cm2 culture flasks with S1 medium that had been previously depleted of exosomes by sequential centrifugation (17, 21). Cells were cultured for 3–4 days under standard normoxic conditions, and their exosomes were collected by sequential centrifugation: 500 rcf for 5 min, recovering the supernatant; 10,000 rcf for 25 min, recovering the supernatant; vacuum filter with 0.2 µm to remove larger extracellular vesicles (apoptotic bodies and microvesicles); and 100,000 rcf for 70 min, recovering the pellet (17, 21). The exosome pellet was suspended in PBS. The number and size of exosomes were determined using a nanoparticle analyzer, ZetaView, from Particle Metrix (Mebane, NC). Exosome measurements were conducted by The Islet and Physiology Core of the Center for Diabetes and Metabolic Diseases at Indiana University. The number of exosomes injected into each rat averaged 2.4e10.
Animal Protocols
Experiments were performed on 8-wk-old male Sprague–Dawley rats from Charles River Laboratories (n = 5–8 rats/group), fed standard chow, and housed under standard conditions (17, 21, 27, 36). Rat experiments were in complete agreement with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, 8th ed.) and Animal Research: Reporting of In Vivo Experiments guidelines and approved by the Institutional Animal Care and Use Committee at Indiana University. Rats were anesthetized with inhaled isoflurane (0.5–1%) and placed on a homeothermic table to maintain a body core temperature of ∼37°C. Adequate anesthesia was ensured before any surgical procedure. Two separate sets of rats were studied. The first set included three groups; in two groups, both renal pedicles were occluded for 50 min with microaneurysm clamps (17, 21). One group was terminated 24 h after occlusion, and the other group was terminated 48 h following occlusion. The third control, or sham group, underwent an identical surgical procedure except that the renal pedicles were not clamped. The second set comprised four additional groups of rats. In three groups, both renal pedicles were occluded for 50 min with microaneurysm clamps. In the fourth group, sham rats underwent an identical surgical procedure except that the renal pedicles were not clamped. One group of ischemic rats received 500 µL of normal saline in the tail vein 24 h after occlusion (untreated ischemic). The second ischemic group received a tail vein injection of exosomes in 500 µL normal saline 24 h after occlusion (exosome treated). The third group of ischemic rats received 10 U/kg of human urokinase (Sigma) dissolved in 500 µL of normal saline 24 h after occlusion (urokinase treated). Rats were then subjected to intravital microscopy, blood samples were collected, and rats were terminated. Maintenance of presurgery body weight (within 5%) and systolic blood pressure ≥100 [measured via tail cuff (IITC amplifier, Woodland Hills, CA)] (37) was confirmed before intravital microscopy was performed. Separate groups of animals were followed for 6 days, and transcripts expressed in the kidneys were evaluated by next-generation sequencing as previously described (17). Rats were randomly assigned to groups using web-based random number generation. Injections and tissue analysis were performed in a blinded manner. Sample size calculation was based on prior outcome (serum creatinine means and SD).
Intravital Imaging
Microvascular flow was quantified by intravital imaging using an Olympus Fluoview 1000 confocal/multiphoton microscope (Olympus, Center Valley, PA) (38–41). Briefly, after ensuring adequate anesthesia, the kidneys were exteriorized and Hoescht (500 µg in 0.5 mL physiological saline, Molecular Probes, Eugene, OR) was injected intravenously to allow visualization of nuclei. Microvascular plasma flow was visualized after the infusion of Texas red or fluorescein-conjugated dextran (150 kDa, Molecular Probes, 800 µg in 0.5 mL of physiological saline), and serial images were acquired approximately every 0.5 s. Plasma flow was quantified as movement of dye-excluding erythrocytes in line scans (with correction for microvascular angle). Quantification was performed on blinded images (38–41).
Histology and Immunohistochemistry
Kidneys were fixed in 3.8% paraformaldehyde and paraffin embedded, and 5-µm sections were obtained for Masson’s trichrome to stain connective tissue and periodic acid-Schiff to image cellular morphology (17, 21). The percentage of tubules in the outer medulla that showed loss of cells, luminal debris, and/or tubular dilation was estimated on coded sections without the knowledge of the experimental group to which the animals belonged. Renal neutrophils were visualized using primary anti-rat neutrophil rabbit antibody (No. ABIN2586050, antibodies-online.com, 1:1,000 dilution) and secondary antibody with horseradish peroxidase tag (No. SK-4805, Vector, Burlingame, CA, 1:2,000 dilution). Neutrophils were counted in blinded renal sections. Damaged tubules and capillary thrombi were counted in blinded periodic acid-Schiff stained sections. Other deparaffinized kidney sections were incubated overnight at 4°C with primary goat anti-complement component 3 (C3) antibody (No. c312-A, Alpha Diagnostics, San Antonio, TX, 1:50 dilution), rabbit anti-tissue factor (No. ab151748, Abcam, Cambridge, MA, 1:100 dilution), and rabbit anti-fibrinogen (No. NBP1-33582, Novus, Centennial, CO, 1:200 dilution). Sections were then washed three times in PBS for 5 min and incubated with Texas red-conjugated donkey anti-rabbit (No. 111-075-045, Jackson ImmunoResearch, 1:200 dilution) for 1 h, followed by triple washing with PBS. All antibodies used were validated for specificity by the suppliers. Images were collected without knowledge of the experimental group using a Leica DMI 3000B fluorescence microscope with secondary antibody controls used for each antigen, and images were analyzed using Metamorph (Universal Imaging, Downingtown, PA).
Renal Gene Expression
Quantitative real-time PCR was used to quantify mRNA from rat kidneys (17, 21). RNA was extracted from the kidneys with the RNeasy Plus mini Kit (Qiagen, Germantown, MD). An AffinityScript cDNA Synthesis Kit (Agilent Technologies, Santa Clara, CA) was used to make cDNA from 1 µg mRNA. RNA was subsequently amplified and quantified via Mx3005P QPCR System (Agilent Technologies) along with PrimeTime Gene Expression Master Mix and PrimeTime qPCR Primer Assays (Integrated DNA Technologies, Coralville, IA) specific to the following rat genes: C3, C1q, and C2 complement components, intercellular adhesion molecule (ICAM), transforming growth factor-β1 (TGF-β1), superoxide dismutase (SOD), catalase (CAT), and heat shock protein 27.
Statistical Analysis
For continuous variables, ANOVA was used to determine if differences among mean values reached statistical significance. Tukey’s test was used to correct for multiple comparisons. Categorical variables were analyzed using Fishers’ exact test. The null hypothesis was rejected at P < 0.05. RNA-sequencing data were mapped and quantified as previously described (17).
RESULTS
Renal Function and Structure Postischemia
Sprague–Dawley rats were subjected to sham surgery or bilateral renal ischemia for 50 min and followed for 24 or 48 h of reperfusion. Serum creatinine, a measure of impaired postischemic renal function, is shown with kidney images in Fig. 1. Serum creatinine increased after 24 h of reperfusion, and it doubled 24 h later. This rapid rise in serum creatinine indicates that glomerular filtration was negligible during the initial 2 days of reperfusion. Gross kidney anatomy, 24- and 48 h postischemia, revealed broadly distributed red radial streaks; histological sections showed thrombotic erythrocyte aggregates in these areas. We also found early and unequivocal evidence of clotting in the form of tubular tissue factor (TF or F3) expression and fibrin deposits in glomerular and peritubular capillaries (Fig. 1). The fibrin plugs (red blood cells and fibrin) indicated compromised renal blood flow (no reflow) during reperfusion (42).
Figure 1.

Postischemic renal failure and coagulation. A: serum creatinine levels (n = 5 male rats) were stable in sham rats but increased rapidly in rats after bilateral renal artery occlusion for 50 min (ischemia, time 0). B: bisected kidneys showed bloody streaks representing microthrombi postischemia. The insets show aggregated red blood cells proximate to intravascular white blood cells in a periodic acid-Schiff-stained section. The red blood cell aggregates were seen in both capillaries and arterioles. Means (±SE) kidney weights are shown on the bottom (n = 3 male rats). C: kidneys of control (sham) rats did not express tissue factor (F3). However, renal epithelial F3 was induced within 24 h following bilateral renal ischemia for 50 min (24 h) and remained upregulated for at least another 24 h (48 h). The graph shows the quantification of F3 induction as the percent area of the F3 immunofluorescence signal in 42 microscopic fields (n = 3 male rats/group). In this and subsequent graphs, the horizontal bars show mean values. Nuclei were counterstained blue with Hoechst (*P < 0.05 vs. sham). Scale bar = 100 µm. D: fibrin was absent from the kidneys of control (sham) rats. However, renal fibrin was broadly deposited in capillaries within 24 h following bilateral renal ischemia for 50 min (24 h) and persisted for at least another day (48 h). The graph shows the quantification of the fibrin immunofluorescence signal in 53 microscopic fields (n = 3 male rats/group). Scale bar = 100 µm. Nuclei were counterstained blue with Hoechst (P < 0.01 vs. sham). E: kidney mRNA fold changes, by RT-PCR, between sham rats (0 h) and rats that sustained bilateral renal ischemia for 50 min and 24 or 48 h (24 and 48 h) reperfusion postischemia are shown. mRNA encoding complement components C3, C1q, and C2, intercellular adhesion molecule 1 (ICAM), and transforming growth factor-β1 (TGF) were elevated 24 and/or 48 h postischemia. In contrast, transcripts for the antioxidants superoxide dismutase-1 (SOD) and catalase (CAT) were suppressed 24 and 48 h postischemia. Stress heat shock protein 27 (HSP) also increased 24 and 48 h postischemia (n = 3 male rats/group). *Significantly different than sham; §significantly different than 24 h (P < 0.05 for both). Statistical analyses were determined using ANOVA.
The rapid inflammatory response postischemia included complement component C3, which increased within 24 h of reperfusion (43), along with complement components C1q and C2. There were also increases in mRNAs encoding ICAM, TGF-β1, and the stress response indicator heat shock protein 27. In contrast, there was prompt suppression of the antioxidant transcripts SOD and CAT (Fig. 1). This pattern of responses, seen in our prior work, can be sustained over several days postischemia (17).
To further examine the role of coagulation in ischemic renal injury, we tested the hypotheses that impaired microvascular flow persists after reperfusion, that enhancing fibrinolysis would improve function, and that one mechanism of protection with exosomes is improved microvascular perfusion. In the untreated (saline vehicle) ischemia group, there was widespread tubular damage and broad microthrombus formation. These changes were averted by treatment with exosomes (2.4e10 exosomes/rat) or urokinase (10 U/kg) 24 h postischemia after renal failure was established (Fig. 2). Significantly less tubular injury and medullary thrombi were found in both treated groups. Serum creatinine levels (48 h postischemia) were in agreement with the morphology as well as our prior studies (17, 21, 24): sham rats, 0.6 ± 0.1 mg/dL (means ± SE); untreated ischemic rats, 1.4 ± 0.1 mg/dL (significantly higher than all groups, P < 0.05); exosome-treated ischemic rats, 0.7 ± 0.03 mg/dL (P < 0.02 vs. untreated ischemia); and urokinase-treated ischemic rats, 0.8 ± 0.14 mg/dL (P < 0.03 vs. untreated ischemia). Using multiphoton intravital microscopy (38–41), we found marked attenuation of capillary plasma flow as well as adherent leukocytes and multiple thrombi in untreated postischemic kidneys. In contrast, capillary thrombi and leukocytes were nearly absent in postischemic rats injected with exosomes or with urokinase. Quantification of erythrocyte velocity was used to evaluate plasma flow and showed heterogeneous and significantly slower capillary flow in postischemic kidneys than in kidneys from sham or treated postischemic rats (Table 1). The distribution of measured capillary red blood cell velocity rates is shown in Fig. 3, and it demonstrated a significant drift to slower, but heterogeneous, rates in ischemic rats compared with the other three groups. Complete obstruction was seen in 10% of capillaries in the ischemia/saline group (P < 0.001 vs. other groups). This may be an underestimate since capillaries in which obstruction occurred outside of the imaged area would not be visualized at all. Furthermore, aggregate data showed that capillary red blood cell velocity in untreated rats 2 days postischemia was depressed by 58% compared with the sham group (Table 1). This was comparable with that seen with heat-inactivated exosomes (399 ± 37 µM/s) (17) but significantly worse than that found after treatment with exosomes or urokinase (Table 1) or with proximal tubule cell transplants (627 ± 37 µM/s) (21). Nevertheless, the renal microvasculature was preserved postischemia (Fig. 4), indicating that restoration of blood flow is possible.
Figure 2.

Histology. Representative periodic acid-Schiff (PAS)-stained kidney sections from four groups are shown. The left images (A) show the medulla and the right images (B) show the cortex in control (sham), untreated ischemic, and ischemic rats treated with exosomes (+EXO) or urokinase (+URO). Tubular damage (example at arrow), cast formation (*), and microthrombi and neutrophils (arrowhead) were seen in postischemic (untreated) kidneys. C: the graphs show the percentage of damaged tubules (top), medullary capillaries with thrombi (middle) (n = 8–150 fields/group), and serum creatinine 48 h following surgery (bottom, n = 5 male rats/group) with mean values as bars. *P < 0.05 vs. ischemia/saline (for creatinine); *P < 0.001 vs. ischemia/saline for damaged tubules and medullary thrombi; §P < 0.001 vs. ischemia/exosomes. Scale bar = 100 µm. Statistical analyses were determined using ANOVA.
Table 1.
Red blood cell velocity after ischemia or sham surgery
| Sham | Ischemia/Saline | Ischemia/Exosomes | Ischemia/Urokinase | |
|---|---|---|---|---|
| Animal weight, g | 248 ± 11 | 227 ± 6 | 254 ± 12 | 240 ± 7 |
| Kidney weight/100 g body wt | 0.63 ± 0.01 | 0.72 ± 0.01 | 0.60 ± 0.02 | 0.65 ± 0.01 |
| Systolic blood pressure, mmHg | 107 ± 1.8 | 105 ± 1.5 | 108 ± 2.4 | 107 ± 1.9 |
| Red blood cell velocity, µM/s | 823.8 ± 36.9 | 350.7 ± 16.6* | 759.9 ± 31.8† | 603.7 ± 107† |
| Obstructed vessels, % | 0 ± 0 | 19 ± 2.6‡ | 0 ± 0§ | 9.5 ± 4.7 |
P < 0.001 vs. sham;
P < 0.001 vs. ischemia/saline via ANOVA;
P < 0.001 vs. sham;
§P < 0.001 vs. ischemia/saline via Fisher’s exact test.
Figure 3.

Microvascular plasma flow following reperfusion. Representative multiphoton intravital images and quantitation of capillary plasma flow 48 h after reperfusion or sham surgery are shown (A). Fluorescein-conjugated dextran (150 kDa) and Hoechst were used to label the vascular space and nuclei, respectively. The top left image shows unobstructed plasma flow following sham surgery. In contrast, areas of no flow (absence of green signal), dye extravasation (arrows), and adherent leukocytes (arrowheads) are seen following untreated ischemia (ISCH; middle top). Plasma flow was improved in the groups treated with exosomes (ISCH/EXO; middle bottom) or urokinase (ISCH/UROKINASE; bottom) after ischemia. Scale bar = 100 µm. The middle images (B) show representative line scans, which illustrate the red blood cell (RBC) trajectories used to calculate plasma flow. The right images (C) show the distribution of capillary flow velocities. In sham kidneys, RBC velocities were normally distributed and ranged between 400 and 2,000 µM/s, with mean peaks at ∼600 and 800 µM/s (n = 96 unconnected vessels). In contrast, renal RBC velocity in untreated ischemic rats (ischemia/saline) was significantly slower, with a peak in the lowest range of 200 µM/s (n = 104). RBC velocities in exosome-treated (ischemia/exosomes, n = 171) or urokinase-treated (ischemia/urokinase, n = 126), ischemic rats showed normal distributions, peaking at ∼600 µM/s. *Significantly different than ischemia/saline; §significantly different than ischemia/exosomes (P <0.01). The inset (D) shows the mean number of intravascular leukocytes per mm2 (with means as horizontal bars). *Significantly lower than ischemia/saline (P <0.05). n = 5 male rats/group. Statistical analyses were determined using Fisher’s exact test (distribution of capillary flow) and ANOVA (leukocyte numbers).
Figure 4.

Preserved microvascular structure postischemia. Representative images and quantification of renal peritubular microvasculature labeled with rhodamine (red)-conjugated Lycopersicon esculentum (tomato) lectin are shown. The microvascular structure is largely preserved 48 h following 50 min of renal ischemia. Quantification of the labeled area (n = 180 microscopic fields, 3 male rats/group) showed no significant differences between sham surgery and postischemia. These data demonstrate that relief of coagulation has the potential to restore perfusion/relieve hypoxia as the microvascular structure to carry blood flow is intact. Scale bar = 100 µm. Statistical analysis was determined using ANOVA.
Coagulation
The renal coagulation pathway is markedly activated in rats with ischemic AKI, as shown in Table 2. These data were from rats subjected to 50 min of bilateral renal ischemia but terminated after 6 days of reperfusion (17). It is noteworthy that renal transcripts revealed a sustained prothrombotic state following reperfusion, including marked increases in tissue factor (F3), fibrinogen isoforms (fgA, fgB, and fgG), and plasminogen activator inhibitor 1. The prothrombotic state was likely enhanced by downregulation of renal tissue factor pathway inhibitor and also by suppression of the renal fibrinolytic pathway, potentially limiting renal clot dissolution. The critical initiator of the thrombotic state in those earlier experiments was likely hypoxia-dependent activation of renal tissue factor (44). It is noteworthy that these prothrombotic responses were prevented by exosome treatment after renal failure was established (17).
Table 2.
Anticoagulants and procoagulants
| Fold Change |
||
|---|---|---|
| Ischemia/Sham | Ischemia/Ischemia + Exosomes | |
| F2 (prothrombin) | 0.52 | 1.75NS |
| F3 (tissue factor) | 1.92 | 0.72 |
| Serpine1 (plasminogen activator inhibitor 1) | 5.9 | 1.89 |
| fgA (fibrinogen A) | 3.5 | 2.36 |
| fgB (fibrinogen B) | 33.3 | 5.3 |
| fgG (fibrinogen G) | 14.3 | 2.2 |
| Tissue factor pathway inhibitor | 0.69 | 1.35 |
| Protc (protein C) | 0.43 | 1.72 |
| Serpinc1 (antithrombin) | 1.0NS | 1.2NS |
| Plaur (urokinase) | 1.75 | 1.44NS |
| CD11 (itgam) | 4.54 | 1.74 |
| Selp (platelet selectin) | 2.22 | 2.45 |
| Sell (leukocyte selectin) | 3.15 | 1.45NS |
| Sele (endothelial selectin) | 1.0NS | 1.34NS |
| Intercellular adhesion molecule-1 | 3.12 | 1.88 |
| Interleukin-1 receptor-1 | 2.3 | 2.28 |
| OLR1 (LOX-1) | 4.76 | 2.85 |
P < 0.05 unless marked as not significant (NS).
In the present work, tissue factor expression was induced in renal tubules of untreated ischemic rats by 48 h after reperfusion compared with sham or postischemic rats treated with single injections of exosomes or urokinase (Figs. 1 and 5). The postischemia prothrombotic state was also evidenced by widespread deposition of fibrin in peritubular capillaries. In contrast, renal fibrin was negligible in sham rats as well as in postischemic rats treated with exosomes or urokinase. In other words, infusions of exosomes or urokinase after 24 h of reperfusion prevented or significantly limited renal clotting (Fig. 5).
Figure 5.

Tissue factor and fibrin. A: immunoreactive tissue factor (F3; red) was undetectable in the kidneys of control (SHAM) rats, but it was widely distributed in renal tubules of untreated ischemic rats (ISCHEMIA/SALINE). *Area amplified in the inset depicting F3 expression in the vicinity of a microthrombus (arrow). In contrast, F3 expression was barely detectable in ischemic rats treated with exosomes (ISCHEMIA/EXOSOMES) or in ischemic rats treated with urokinase (ISCHEMIA/UROKINASE). The graph shows the percent area of the F3 immunofluorescence signal in each group (n = 9–11 fields and 3 male rats/group), with mean values as horizontal bars. Nuclei were counterstained blue with Hoechst (P < 0.01 vs. sham and §P = 0.058 vs. ischemia/exosomes). B: representative images showing immunoreactive fibrin in kidneys harvested 48 h after surgery. Fibrin (red) was undetectable in the kidneys of control (SHAM) rats, but it was broadly deposited in peritubular capillaries of untreated ischemic rats (ISCHEMIA/SALINE). In contrast, fibrin deposition was markedly reduced in the kidneys of ischemic rats infused with exosomes (ISCHEMIA/EXOSOMES) or urokinase (ISCHEMIA/UROKINASE). The graph shows the percent area of the fibrin immunofluorescence signal in all microscopic fields (n = 12–14 microscope fields and 3 male rats/group). Nuclei were counterstained blue with Hoechst (*P < 0.01 vs. ischemia/saline). Scale bars = 100 µm. Statistical analysis was determined using ANOVA.
Fibrinolysis
To further examine this pathway in a system in which more factors could be controlled, cultured primary human renal tubular cells were subjected to hypoxia and fibrin degradation (fibrinolysis) and quantified. The kidney cells had considerable basal fibrinolytic activity, measured as the formation of d-dimers after 48 h of culture under normoxic conditions. However, fibrinolysis was markedly inhibited by sequential hypoxia/reoxygenation. It is also noteworthy that exosomes added during reoxygenation attenuated the suppression of fibrinolysis seen in hypoxia/reoxygenation (Fig. 6).
Figure 6.

Fibrinolytic activity. Human tubular cells were cultured under normoxic (48 h) or hypoxic (<1% O2 for 24 h/reoxygenation for 24 h) conditions in the presence of fibrin. In the hypoxia/exosome group, exosomes were added during the reoxygenation period. Confluent human renal tubular cells cultured without fibrin (cells) and fibrin alone (without cells, to measure nonenzymatic fibrin degradation) were included as controls. *Significantly different than normoxia (P < 0.001); §significantly different than hypoxia (P < 0.001). n = 4 replicates/group. The horizontal bars represent mean values. Statistical analysis was determined using ANOVA.
Inflammation
Renal neutrophil infiltration in conjunction with activation of renal C3 component of complement are recognized as inflammatory responses following ischemia (17, 21) and, along with clotting, are features of AKI (18, 45). Many more neutrophils, as well as activation of C3, were found in untreated ischemia compared with sham surgery kidneys (17, 21). The infusion of exosomes abrogated the expression of both inflammatory parameters, whereas urokinase was less effective (Fig. 7).
Figure 7.

Renal inflammation. A: neutrophils (polymorphonuclear neutrophils, PMN; top images, brown) were very rare after sham (SHAM) surgery but significantly increased after ischemia (ISCHEMIA/SALINE). Exosomes and, to a lesser extent, urokinase given 24 h postischemia resulted in significantly fewer renal PMN. Scale bar = 100 µm. B: similarly, immunoreactive C3 (red, bottom images) was markedly increased postischemia (ISCHEMIA/SALINE) and significantly improved in the ischemia/exosome (EXO) and ischemia/urokinase groups. The results (number of PMN and percent area C3) are quantified in the graphs on the right, with horizontal bars showing mean values. Scale bar = 50 µm. n = 30–40 fields/group for PMN and 11–16 fields/group for C3, 3 or 4 male rats/group. Nuclei were counterstained blue with hematoxylin (top images) or Hoechst (bottom images). hpf, high-power field. For PMN, *P < 0.05 vs. ischemia/saline and §P < 0.01 vs. ischemia/exosomes; for C3, *P < 0.01 vs. ischemia/saline and §P < 0.01 vs. ischemia/exosomes). Statistical analysis was determined using ANOVA.
DISCUSSION
Ischemic renal injury is increasingly recognized as a major health problem (16). In addition to the high morbidity and mortality associated with acute kidney injury, renal ischemia can worsen function in kidney diseases of other etiologies and is nearly universal in transplanted kidneys (46, 47), leading to delayed graft function and contributing to chronic allograft nephropathy. In the present study, we found that renal function was negligible during reperfusion (48 h) following renal artery occlusion. The postischemic kidneys were markedly congested and had broadly distributed streaks of microthrombi. The postischemic kidneys expressed tissue factor (F3) and glomerular and peritubular capillary deposition of fibrin was prominent, yet microvascular structure was preserved. The expression of tissue factor and fibrin, although involving a small portion of total tissue area, represent a substantial portion of the capillary bed, and the differences between ischemia and sham groups were markedly different (P < 0.0001). Based on preparative data and our earlier studies (17, 21), we hypothesized that renal clotting and subsequent ischemic AKI could be prevented by treatment with normal human kidney exosomes. The exosomes were given noninvasively (intravenously) 24 h postischemia, after renal failure was present. We found that exosomes averted renal capillary thrombosis with near correction of depressed renal function. These data support decreasing coagulation as one mechanism of benefit with exosomes, in addition to improvement in other pathophysiological factors (17, 21). Due to their multiple beneficial effects as well as their stability, immunotolerance, biocompatibility, lack of toxicity, ability to cross physical barriers, and inability to replicate, exosomes are an attractive therapeutic and have reached clinical trials (48–52).
We also verified the central role of renal clotting in postischemic AKI by restoring function with infusion of the thrombolytic urokinase. Hence, we hypothesized that renal microvascular clotting is an important mechanism of AKI, an idea further supported by a set of transcripts that illustrated the renal prothrombotic state 6 days postischemia. These unpublished RNA-sequencing data were collected during a related project (17) and are included here because they show a remarkable perturbation of normal coagulation/fibrinolysis balance relevant to our present work. In the older study, several key procoagulant components of thrombosis were activated, including fibrinogen transcripts, and antithrombotic transcripts were inhibited.
Renal protection by exosomes was striking, as they prevented renal injury postischemia, including key prothrombotic and inflammatory responses (17). It is noteworthy that exosomes were given after renal failure was established. Although urokinase mRNA was activated by renal ischemia, it was not stimulated in ischemic rats treated with exosomes. Moreover, fibrinolytic activity was present in renal tubular cells, was suppressed by hypoxia/reoxygenation, and was partly, but not fully, normalized by exosomes. Thus, fibrinolysis may contribute to improvement with exosomes but is not likely the main mechanism of improvement with exosomes. Our present and prior data (17, 21) support multiple beneficial effects of exosomes, including limiting inflammation, consistent with their abundant cargo. We propose that the level of protection was so extensive that stereotypical postischemic responses were not elicited or perhaps not even needed. In other words, data (17, 21) show that exosomes limit early ischemic injury so that coagulation, inflammation, and cell death do not follow. Consequently, capillary blood flow was maintained when exosomes were given within 24 h of the occlusion. We found that exosomes are taken up by renal endothelial cells, although they are predominantly found in tubular epithelial cells (17, 21). This is consistent with the findings of Brodsky et al. (53), who demonstrated better renal function with transplantation of umbilical vein endothelial cells or human embryonic kidney cells expressing endothelial nitric oxide synthase and concluded that cell-cell interactions are critical in recovery postischemia. Our data also indicate that renal damage in postischemic AKI is caused, in part, by the maladaptive prothrombotic response, which, if left unattended, obstructs capillaries during reperfusion. This is consistent with the work of others who found improvement in renal blood flow and function with the anticoagulant activated protein C (54) or its enhancer thrombomodulin (55) in other models of AKI. These ideas presuppose that advancing zones of no reflow and renal hypoxia are dominant features during reperfusion (56). Decreases in total renal blood flow (or volume) after reperfusion have been demonstrated using a variety of techniques (for example, see Refs. 57–59). The present study used intravital multiphoton microscopy to directly visualize and quantify the decreases in renal blood flow following reperfusion. In addition, we demonstrate the heterogeneity of local perfusion after ischemia-reperfusion and the response to potential therapeutic agents, indicating that decreased perfusion is not due to acute capillary loss and is amenable to treatment. We emphasize that no reflow does not necessarily mean complete cessation of blood flow, but the phenomenon can occur with obstruction such that blood velocity is slowed to a degree that compromises nutrients and oxygen delivery given the tight control required to assure homeostasis (60).
The phenomenon of no reflow, first described in the heart, is due to residual capillary occlusions following restoration of arterial flow (61) and is the main cause of continued poor cardiac function during reperfusion (56). Subsequently, it was revealed that progressive hypoxic endothelial dysfunction aggravated the prothrombotic state (56). Renal no reflow has also been documented (42, 62, 63), along with endothelial cell hypoxic injury and loss (42, 53, 64). Ischemic renal endothelial injury likely contributes to thrombus formation (65). Our findings are consistent with those of others showing persistent vascular obstruction postischemia (66), which is heterogeneous (42, 67), including in patients following transplant (62). Alternatively, the areas of obstruction could be the result of erythrocyte trapping, medullary venous obstruction (68), and cell swelling (42, 69), as opposed to primary “no reflow.” Our data, however, are consistent with abnormal coagulation as the thrombotic urokinase improves microvascular flow as well as renal function and structure. Although some investigators (66) have demonstrated improvement in histological evidence of injury with pretreatments to decrease medullary venous obstruction and red blood cell trapping in experimental renal ischemia, others (70) have not.
The delicate balance between prothrombotic and profibrinolytic elements maintains homeostasis and renal perfusion in the steady state. This equilibrium is disrupted by ischemia and becomes prothrombotic and proinflammatory during reperfusion. Prothrombotic alterations escalate and can promote capillary thrombosis and the transition from AKI to CKD (71). Exosomes, given after 24 h of reperfusion, either thwart ischemic stress or attenuate acute postischemic responses and prevent the conversion to CKD (17, 21). Urokinase is also effective after 24 h of reperfusion, presumably by lysing emerging clots and promoting restoration of blood flow at early time points. We acknowledge that anticoagulation may not be beneficial (34, 72), most likely because anticoagulants cannot acutely remove forming clots from the capillary circulation. In summary, these data support the hypothesis that intrarenal clotting occurs early and is a critical determinant of renal failure following ischemia. Although we are not aware of specific biomarkers, these changes parallel findings in humans where fibrin deposition is found in AKI (73, 74) and a prothrombotic profile is found in renal allografts with delayed graft function (75). Thrombin generation has also been found to be an independent risk factor for AKI following cardiac surgery (76). Human data also suggest that nonvitamin K antagonist oral anticoagulants, which inhibit thrombin, are associated with a decreased incidence of AKI compared with warfarin (34). Of the nonvitamin K antagonist oral anticoagulants, the risk was lower with dabigatran, the direct thrombin inhibitor, than with those that inhibit factor Xa (34). Our data demonstrate that the coagulation abnormalities are amenable to therapy with exosomes or urokinase in this model and that therapy results in improvement in function and postischemic inflammation.
Perspectives and Significance
The kidney has tremendous ability to survive a variety of insults and to recover function. Evolutionarily conserved responses to preserve homeostasis often involve a delicate balance as illustrated by coagulation and fibrinolysis. Our data, demonstrating sustained ischemia following apparent restoration of renal blood flow, show that coagulation, which protects from excessive blood loss, can also prolong tissue deprivation from oxygen and essential nutrients. However, the preservation of the capillary network allows the restoration of plasma flow. We also found fibrinolytic activity in renal tubular epithelia. The data are consistent with an integrative physiology view of renal function and dysfunction with endothelial and tubular injury and function closely coupled and interdependent. It also follows that a multipronged approach (with exosomes as a proof of principal) may be needed to effectively treat renal injury.
GRANTS
This work was supported by Veterans’ Affairs Merit Review Awards (to J.H.D. and K.J.K.), National Institutes of Health Grant AI156756, Indiana Clinical and Translational Sciences Institute (funded, in part, by National Institutes of Health Grant UL1TR002529), and Paul Teschan Research awards of Dialysis Clinic (to K.J.K.).
DISCLAIMER
The content does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.H.D. and K.J.K. conceived and designed research; D.X., J.M.D., and K.J.K. performed experiments; J.H.D., D.X., J.M.D., and K.J.K. analyzed data; J.H.D., J.M.D., and K.J.K. interpreted results of experiments; D.X. and K.J.K. prepared figures; J.H.D. and K.J.K. drafted manuscript; K.J.K. edited and revised manuscript; J.H.D., D.X., J.M.D., and K.J.K. approved final version of manuscript.
ACKNOWLEDGMENTS
Imaging was performed at the Indiana Center for Biomedical Imaging and nanotracker analysis at the Islet and Physiology Core of the Center for Diabetes and Metabolic Diseases, both at the Indiana University School of Medicine.
REFERENCES
- 1. Havasi A, Borkan SC. Apoptosis and acute kidney injury. Kidney Int 80: 29–40, 2011. doi: 10.1038/ki.2011.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Agarwal A, Dong Z, Harris R, Murray P, Parikh SM, Rosner MH, Kellum JA, Ronco C; Acute Dialysis Quality Initiative XIII Working Group. Cellular and molecular mechanisms of AKI. J Am Soc Nephrol 27: 1288–1299, 2016. doi: 10.1681/ASN.2015070740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Faubel S, Chawla LS, Chertow GM, Goldstein SL, Jaber BL, Liu KD; Acute Kidney Injury Advisory Group of the American Society of Nephrology. Ongoing clinical trials in AKI. Clin J Am Soc Nephrol 7: 861–873, 2012. doi: 10.2215/CJN.12191111. [DOI] [PubMed] [Google Scholar]
- 4. Kwon O, Ahn K, Zhang B, Lockwood T, Dhamija R, Anderson D, Saqib N. Simultaneous monitoring of multiple urinary cytokines may predict renal and patient outcome in ischemic AKI. Ren Fail 32: 699–708, 2010. doi: 10.3109/0886022X.2010.486496. [DOI] [PubMed] [Google Scholar]
- 5. Lameire N, Vanmassenhove J, Lewington A. Did KDIGO guidelines on acute kidney injury improve patient outcome? Intensive Care Med 43: 921–923, 2017. doi: 10.1007/s00134-017-4740-1. [DOI] [PubMed] [Google Scholar]
- 6. Nath KA. Pathogenesis of acute renal failure. In: Textbook of Nephrology (4th ed.), edited by Massry SG, Glassock RJ. Baltimore, MD: Williams and Wilkins, 2000. [Google Scholar]
- 7. Tang C, Dong Z. Epigenetic regulation in acute kidney injury: new light in a dark area. Kidney Int 88: 665–668, 2015. doi: 10.1038/ki.2015.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stallons LJ, Funk JA, Schnellmann RG. Mitochondrial homeostasis in acute organ failure. Curr Pathobiol Rep 1: 169–177, 2013. doi: 10.1007/s40139-013-0023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. García-Ortuño LE, Bobadilla NA. Integrative view of the mechanisms that induce acute kidney injury and its transition to chronic kidney disease. Rev Invest Clin 70: 261–268, 2018. doi: 10.24875/RIC.18002546. [DOI] [PubMed] [Google Scholar]
- 10. Ishiko S, Goligorsky MS. Ways and means of cellular reconditioning for kidney regeneration. Am J Nephrol 53: 96–107, 2022. doi: 10.1159/000522050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Han SJ, Williams RM, Kim M, Heller DA, D’Agati V, Schmidt-Supprian M, Lee HT. Renal proximal tubular NEMO plays a critical role in ischemic acute kidney injury. JCI Insight 5: e139246, 2020. doi: 10.1172/jci.insight.139246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grange C, Skovronova R, Marabese F, Bussolati B. Stem cell-derived extracellular vesicles and kidney regeneration. Cells 8: 1240, 2019. doi: 10.3390/cells8101240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chawla LS, Amdur RL, Shaw AD, Faselis C, Palant CE, Kimmel PL. Association between AKI and long-term renal and cardiovascular outcomes in United States veterans. Clin J Am Soc Nephrol 9: 448–456, 2014. doi: 10.2215/CJN.02440213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anonymous. Calculating the cost. Health Serv J 121, Suppl 1: 2011. [PubMed] [Google Scholar]
- 15. Wald R, McArthur E, Adhikari NK, Bagshaw SM, Burns KE, Garg AX, Harel Z, Kitchlu A, Mazer CD, Nash DM, Scales DC, Silver SA, Ray JG, Jo F. Changing incidence and outcomes following dialysis-requiring acute kidney injury among critically ill adults: a population-based cohort study. Am J Kidney Dis 65: 870–877, 2015. doi: 10.1053/j.ajkd.2014.10.017. [DOI] [PubMed] [Google Scholar]
- 16. Siew ED, Davenport A. The growth of acute kidney injury: a rising tide or just closer attention to detail? Kidney Int 87: 46–61, 2015. doi: 10.1038/ki.2014.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dominguez JH, Liu Y, Gao H, Dominguez JM 2nd, Xie D, Kelly KJ. Renal tubular cell-derived extracellular vesicles accelerate the recovery of established renal ischemia reperfusion injury. J Am Soc Nephrol 28: 3533–3544, 2017. doi: 10.1681/ASN.2016121278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kelly KJ, Burford JL, Dominguez JH. The post-ischemic inflammatory syndrome: a critical mechanism of progression in diabetic nephropathy. Am J Physiol Renal Physiol 297: F923–F931, 2009. doi: 10.1152/ajprenal.00205.2009. [DOI] [PubMed] [Google Scholar]
- 19. Losonczy G, Hársing L. Simultaneous activation of coagulation and fibrinolysis after severe renal ischemia in rats. Nephron 32: 180–184, 1982. doi: 10.1159/000182840. [DOI] [PubMed] [Google Scholar]
- 20. Suárez-Álvarez B, Liapis H, Anders HJ. Links between coagulation, inflammation, regeneration, and fibrosis in kidney pathology. Lab Invest 96: 378–390, 2016. doi: 10.1038/labinvest.2015.164. [DOI] [PubMed] [Google Scholar]
- 21. Dominguez JM 2nd, Dominguez JH, Xie D, Kelly KJ. Extracellular microvesicles from renal tubules reverse kidney ischemia-reperfusion injury in rats. PLoS One 13: e0202550, 2018. doi: 10.1371/journal.pone.0202550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Darwish MA, Abo-Youssef AM, Khalaf MM, Abo-Saif AA, Saleh IG, Abdelghany TM. Vitamin E mitigates cisplatin-induced nephrotoxicity due to reversal of oxidative/nitrosative stress, suppression of inflammation and reduction of total renal platinum accumulation. J Biochem Mol Toxicol 31: 1–9, 2017. doi: 10.1002/jbt.21833. [DOI] [PubMed] [Google Scholar]
- 23. Melican K, Boekel J, Månsson LE, Sandoval RM, Tanner GA, Källskog O, Palm F, Molitoris BA, Richter-Dahlfors A. Bacterial infection-mediated mucosal signalling induces local renal ischaemia as a defence against sepsis. Cell Microbiol 10: 1987–1998, 2008. doi: 10.1111/j.1462-5822.2008.01182.x. [DOI] [PubMed] [Google Scholar]
- 24. Kelly KJ, Kluve-Beckerman B, Zhang J, Dominguez JH. Intravenous cell therapy for acute renal failure with serum amyloid A protein-reprogrammed cells. Am J Physiol Renal Physiol 299: F453–F464, 2010. doi: 10.1152/ajprenal.00050.2010. [DOI] [PubMed] [Google Scholar]
- 25. Kelly KJ, Zhang J, Han L, Kamocka M, Miller C, Gattone VH 2nd, Dominguez JH. Improved structure and function in autosomal recessive polycystic rat kidneys with renal tubular cell therapy. PLoS One 10: e0131677, 2015. doi: 10.1371/journal.pone.0131677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kelly KJ, Zhang J, Han L, Wang M, Zhang S, Dominguez JH. Intravenous renal cell transplantation with SAA1-positive cells prevents the progression of chronic renal failure in rats with ischemic-diabetic nephropathy. Am J Physiol Renal Physiol 305: F1804–F1812, 2013. doi: 10.1152/ajprenal.00097.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kelly KJ, Zhang J, Wang M, Zhang S, Dominguez JH. Intravenous renal cell transplantation for rats with acute and chronic renal failure. Am J Physiol Renal Physiol 303: F357–F365, 2012. doi: 10.1152/ajprenal.00680.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yan SF, Lu J, Zou YS, Kisiel W, Mackman N, Leitges M, Steinberg S, Pinsky D, Stern D. Protein kinase C-β and oxygen deprivation. A novel Egr-1-dependent pathway for fibrin deposition in hypoxemic vasculature. J Biol Chem 275: 11921–11928, 2000. doi: 10.1074/jbc.275.16.11921. [DOI] [PubMed] [Google Scholar]
- 29. Craciun FL, Ajay AK, Hoffmann D, Saikumar J, Fabian SL, Bijol V, Humphreys BD, Vaidya VS. Pharmacological and genetic depletion of fibrinogen protects from kidney fibrosis. Am J Physiol Renal Physiol 307: F471–F484, 2014. doi: 10.1152/ajprenal.00189.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Druid H, Rammer L. Protective effect on postischemic renal edema by anticoagulation. Nephron 60: 319–323, 1992. doi: 10.1159/000186772. [DOI] [PubMed] [Google Scholar]
- 31. Gedik HS, Korkmaz K, Erdem H, Karakilic E, Lafci G, Ankarali H. Protective effect of heparin in the end organ ischemia/reperfusion injury of the lungs and heart. J Cardiothorac Surg 7: 123, 2012. doi: 10.1186/1749-8090-7-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shin CS, Han JU, Kim JL, Schenarts PJ, Traber LD, Hawkins H, Traber DL. Heparin attenuated neutrophil infiltration but did not affect renal injury induced by ischemia reperfusion. Yonsei Med J 38: 133–141, 1997. doi: 10.3349/ymj.1997.38.3.133. [DOI] [PubMed] [Google Scholar]
- 33. Tillet S, Giraud S, Delpech PO, Thuillier R, Ameteau V, Goujon JM, Renelier B, Macchi L, Hauet T, Mauco G. Kidney graft outcome using an anti-Xa therapeutic strategy in an experimental model of severe ischaemia-reperfusion injury. Br J Surg 102: 132–142, 2015. doi: 10.1002/bjs.9662. [DOI] [PubMed] [Google Scholar]
- 34. Yao X, Tangri N, Gersh BJ, Sangaralingham LR, Shah ND, Nath KA, Noseworthy PA. Renal outcomes in anticoagulated patients with atrial fibrillation. J Am Coll Cardiol 70: 2621–2632, 2017. doi: 10.1016/j.jacc.2017.09.1087. [DOI] [PubMed] [Google Scholar]
- 35. Longstaff C, Kolev K. Basic mechanisms and regulation of fibrinolysis. J Thromb Haemost 13, Suppl 1: S98–S105, 2015. doi: 10.1111/jth.12935. [DOI] [PubMed] [Google Scholar]
- 36. Kelly KJ, Sutton TA, Weathered N, Ray N, Caldwell EJ, Plotkin Z, Dagher PC. Minocycline inhibits apoptosis and inflammation in a rat model of ischemic renal injury. Am J Physiol Renal Physiol 287: F760–F766, 2004. doi: 10.1152/ajprenal.00050.2004. [DOI] [PubMed] [Google Scholar]
- 37. Kelly KJ. Distant effects of experimental renal ischemia/reperfusion injury. J Am Soc Nephrol 14: 1549–1558, 2003. doi: 10.1097/01.asn.0000064946.94590.46. [DOI] [PubMed] [Google Scholar]
- 38. Dunn KW, Sandoval RM, Kelly KJ, Dagher PC, Tanner GA, Atkinson SJ, Bacallao RL, Molitoris BA. Functional studies of the kidney of living animals using multicolor two-photon microscopy. Am J Physiol Cell Physiol 283: C905–C916, 2002. doi: 10.1152/ajpcell.00159.2002. [DOI] [PubMed] [Google Scholar]
- 39. Kelly KJ, Dominguez JH. Treatment of the post-ischaemic inflammatory syndrome of diabetic nephropathy. Nephrol Dial Transplant 25: 3204–3212, 2010. doi: 10.1093/ndt/gfq217. [DOI] [PubMed] [Google Scholar]
- 40. Kelly KJ, Sandoval RM, Dunn KW, Molitoris BA, Dagher PC. A novel method to determine specificity and sensitivity of the TUNEL reaction in the quantitation of apoptosis. Am J Physiol Cell Physiol 284: C1309–C1318, 2003. doi: 10.1152/ajpcell.00353.2002. [DOI] [PubMed] [Google Scholar]
- 41. Temm C, Dominguez JH. Microcirculation: nexus of comorbidities in diabetes. Am J Physiol Renal Physiol 293: F486–F493, 2007. doi: 10.1152/ajprenal.00503.2006. [DOI] [PubMed] [Google Scholar]
- 42. Flores J, DiBona DR, Beck CH, Leaf A. The role of cell swelling in ischemic renal damage and the protective effect of hypertonic solute. J Clin Invest 51: 118–126, 1972. doi: 10.1172/JCI106781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kelly KJ, Liu Y, Zhang J, Dominguez JH. Renal C3 complement component: feed forward to diabetic kidney disease. Am J Nephrol 41: 48–56, 2015. doi: 10.1159/000371426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yan SF, Mackman N, Kisiel W, Stern DM, Pinsky DJ. Hypoxia/Hypoxemia-Induced activation of the procoagulant pathways and the pathogenesis of ischemia-associated thrombosis. Arterioscler Thromb Vasc Biol 19: 2029–2035, 1999. doi: 10.1161/01.atv.19.9.2029. [DOI] [PubMed] [Google Scholar]
- 45. Zuk A, Bonventre JV. Acute kidney injury. Annu Rev Med 67: 293–307, 2016. doi: 10.1146/annurev-med-050214-013407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ojo AO, Wolfe RA, Held PJ, Port FK, Schmouder RL. Delayed graft function: risk factors and implications for renal allograft survival. Transplantation 63: 968–974, 1997. doi: 10.1097/00007890-199704150-00011. [DOI] [PubMed] [Google Scholar]
- 47. Weber M, Dindo D, Demartines N, Ambühl PM, Clavien PA. Kidney transplantation from donors without a heartbeat. N Engl J Med 347: 248–255, 2002. doi: 10.1056/NEJMoa020274. [DOI] [PubMed] [Google Scholar]
- 48. Ha D, Yang N, Nadithe V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: current perspectives and future challenges. Acta Pharm Sin B 6: 287–296, 2016. doi: 10.1016/j.apsb.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vlassov AV, Magdaleno S, Setterquist R, Conrad R. Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim Biophys Acta 1820: 940–948, 2012. doi: 10.1016/j.bbagen.2012.03.017. [DOI] [PubMed] [Google Scholar]
- 50. NV Lakshmi Kavya A, Subramanian S, Ramakrishna S. Therapeutic applications of exosomes in various diseases: a review. Biomater Adv 134: 112579, 2022. doi: 10.1016/j.msec.2021.112579. [DOI] [PubMed] [Google Scholar]
- 51. Silva AKA, Morille M, Piffoux M, Arumugam S, Mauduit P, Larghero J, . et al. Development of extracellular vesicle-based medicinal products: a position paper of the group “Extracellular Vesicle translatiOn to clinicaL perspectiVEs—EVOLVE France”. Adv Drug Deliv Rev 179: 114001, 2021. doi: 10.1016/j.addr.2021.114001. [DOI] [PubMed] [Google Scholar]
- 52. Thomas SC, Kim JW, Pauletti GM, Hassett DJ, Kotagiri N. Exosomes: biological pharmaceutical nanovectors for theranostics. Front Bioeng Biotechnol 9: 808614, 2021. doi: 10.3389/fbioe.2021.808614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brodsky SV, Yamamoto T, Tada T, Kim B, Chen J, Kajiya F, Goligorsky MS. Endothelial dysfunction in ischemic acute renal failure: rescue by transplanted endothelial cells. Am J Physiol Renal Physiol 282: F1140–F1149, 2002. doi: 10.1152/ajprenal.00329.2001. [DOI] [PubMed] [Google Scholar]
- 54. Gupta A, Gerlitz B, Richardson MA, Bull C, Berg DT, Syed S, Galbreath EJ, Swanson BA, Jones BE, Grinnell BW. Distinct functions of activated protein C differentially attenuate acute kidney injury. J Am Soc Nephrol 20: 267–277, 2009. doi: 10.1681/ASN.2008030294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sharfuddin AA, Sandoval RM, Berg DT, McDougal GE, Campos SB, Phillips CL, Jones BE, Gupta A, Grinnell BW, Molitoris BA. Soluble thrombomodulin protects ischemic kidneys. J Am Soc Nephrol 20: 524–534, 2009. doi: 10.1681/ASN.2008060593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bouleti C, Mewton N, Germain S. The no-reflow phenomenon: state of the art. Arch Cardiovasc Dis 108: 661–674, 2015. doi: 10.1016/j.acvd.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 57. Braunagel M, Helck A, Wagner A, Schupp N, Brocker V, Reiser M, Notohamiprodjo M, Meiser B, Habicht A. Dynamic contrast-enhanced computed tomography: a new diagnostic tool to assess renal perfusion after ischemia-reperfusion injury in mice: correlation of perfusion deficit to histopathologic damage. Invest Radiol 51: 316–322, 2016. doi: 10.1097/RLI.0000000000000245. [DOI] [PubMed] [Google Scholar]
- 58. Greite R, Thorenz A, Chen R, Jang MS, Rong S, Brownstein MJ, Tewes S, Wang L, Baniassad B, Kirsch T, Bräsen JH, Lichtinghagen R, Meier M, Haller H, Hueper K, Gueler F. Renal ischemia-reperfusion injury causes hypertension and renal perfusion impairment in the CD1 mice which promotes progressive renal fibrosis. Am J Physiol Renal Physiol 314: F881–F892, 2018. doi: 10.1152/ajprenal.00519.2016. [DOI] [PubMed] [Google Scholar]
- 59. Martín-Solé O, Rodó J, García-Aparicio L, Blanch J, Cusí V, Albert A. Effects of platelet-rich plasma (PRP) on a model of renal ischemia-reperfusion in rats. PLoS One 11: e0160703, 2016. doi: 10.1371/journal.pone.0160703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. O'Connor PM. Renal oxygen delivery: matching delivery to metabolic demand. Clin Exp Pharmacol Physiol 33: 961–967, 2006. doi: 10.1111/j.1440-1681.2006.04475.x. [DOI] [PubMed] [Google Scholar]
- 61. Kloner RA, Ganote CE, Jennings RB. The “no-reflow” phenomenon after temporary coronary occlusion in the dog. J Clin Invest 54: 1496–1508, 1974. doi: 10.1172/JCI107898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schmitz V, Schaser KD, Olschewski P, Neuhaus P, Puhl G. In vivo visualization of early microcirculatory changes following ischemia/reperfusion injury in human kidney transplantation. Eur Surg Res 40: 19–25, 2008. doi: 10.1159/000107683. [DOI] [PubMed] [Google Scholar]
- 63. Summers WK, Jamison RL. The no reflow phenomenon in renal ischemia. Lab Invest 25: 635–643, 1971. [PubMed] [Google Scholar]
- 64. Bábíčková J, Klinkhammer BM, Buhl EM, Djudjaj S, Hoss M, Heymann F, Tacke F, Floege J, Becker JU, Boor P. Regardless of etiology, progressive renal disease causes ultrastructural and functional alterations of peritubular capillaries. Kidney Int 91: 70–85, 2017. doi: 10.1016/j.kint.2016.07.038. [DOI] [PubMed] [Google Scholar]
- 65. Kokura S, Yoshida N, Yoshikawa T. Anoxia/reoxygenation-induced leukocyte-endothelial cell interactions. Free Radic Biol Med 33: 427–432, 2002. doi: 10.1016/s0891-5849(02)00852-3. [DOI] [PubMed] [Google Scholar]
- 66. Mason J, Welsch J, Torhorst J. The contribution of vascular obstruction to the functional defect that follows renal ischemia. Kidney Int 31: 65–71, 1987. doi: 10.1038/ki.1987.10. [DOI] [PubMed] [Google Scholar]
- 67. Mason J, Torhorst J, Welsch J. Role of the medullary perfusion defect in the pathogenesis of ischemic renal failure. Kidney Int 26: 283–293, 1984. doi: 10.1038/ki.1984.171. [DOI] [PubMed] [Google Scholar]
- 68. McLarnon SR, Wilson K, Patel B, Sun J, Sartain CL, Mejias CD, Musall JB, Sullivan JC, Wei Q, Chen JK, Hyndman KA, Marshall B, Yang H, Fogo AB, O'Connor PM. Lipopolysaccharide pretreatment prevents medullary vascular congestion following renal ischemia by limiting early reperfusion of the medullary circulation. J Am Soc Nephrol 33: 769–785, 2022. doi: 10.1681/ASN.2021081089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mason J, Joeris B, Welsch J, Kriz W. Vascular congestion in ischemic renal failure: the role of cell swelling. Miner Electrolyte Metab 15: 114–124, 1989. [PubMed] [Google Scholar]
- 70. Andersson G, Jennische E. Lack of casual relationship between medullary blood congestion and tubular necrosis in postischaemic kidney damage. Acta Physiol Scand 130: 429–432, 1987. doi: 10.1111/j.1748-1716.1987.tb08158.x. [DOI] [PubMed] [Google Scholar]
- 71. Mörtberg J, Blombäck M, Wallén Å, He S, Jacobson SH, Spaak J. Increased fibrin formation and impaired fibrinolytic capacity in severe chronic kidney disease. Blood Coagul Fibrinolysis 27: 401–407, 2016. doi: 10.1097/MBC.0000000000000462. [DOI] [PubMed] [Google Scholar]
- 72. Tuuminen R, Jouppila A, Salvail D, Laurent CE, Benoit MC, Syrjälä S, Helin H, Lemström K, Lassila R. Dual antiplatelet and anticoagulant APAC prevents experimental ischemia-reperfusion-induced acute kidney injury. Clin Exp Nephrol 21: 436–445, 2017. doi: 10.1007/s10157-016-1308-2. [DOI] [PubMed] [Google Scholar]
- 73. Kincaid-Smith P. Coagulation and renal disease. Kidney Int 2: 183–190, 1972. doi: 10.1038/ki.1972.93. [DOI] [PubMed] [Google Scholar]
- 74. Koffler D, Paronetto F. Fibrinogen deposition in acute renal failure. Am J Pathol 49: 383–395, 1966. [PMC free article] [PubMed] [Google Scholar]
- 75. Faulk WP, Gargiulo P, McIntyre JA, Bang NU. Hemostasis and fibrinolysis in renal transplantation. Semin Thromb Hemost 15: 88–98, 1989. doi: 10.1055/s-2007-1002691. [DOI] [PubMed] [Google Scholar]
- 76. Scrascia G, Rotunno C, Simone S, Montemurno E, Amorese L, De Palo M, Castellano G, Pertosa GB, Gesualdo L, Paparella D. . Acute kidney injury in high-risk cardiac surgery patients: roles of inflammation and coagulation. J Cardiovasc Med (Hagerstown) 18: 359–365, 2017. doi: 10.2459/JCM.0000000000000343. [DOI] [PubMed] [Google Scholar]