

Abstract
Arthropods harbor a largely undocumented diversity of RNA viruses. Some arthropods, like mosquitoes, can transmit viruses to vertebrates but are themselves parasitized by other arthropod species, such as mites. Very little is known about the viruses of these ectoparasites and how they move through the host–parasite relationship. To address this, we determined the virome of both mosquitoes and the mites that feed on them. The mosquito Aedes communis is an abundant and widely distributed species in Sweden, in northern Europe. These dipterans are commonly parasitized by water mite larvae (Trombidiformes: Mideopsidae) that are hypothesized to impose negative selection pressures on the mosquito by reducing fitness. In turn, viruses are dual-host agents in the mosquito–mite interaction. We determined the RNA virus diversity of mite-free and mite-detached mosquitoes, as well as their parasitic mites, using meta-transcriptomic sequencing. Our results revealed an extensive RNA virus diversity in both mites and mosquitoes, including thirty-seven putative novel RNA viruses that cover a wide taxonomic range. Notably, a high proportion of viruses (20/37) were shared between mites and mosquitoes, while a limited number of viruses were present in a single host. Comparisons of virus composition and abundance suggest potential virus transfer between mosquitoes and mites during their symbiotic interaction. These findings shed light on virome diversity and ecology in the context of arthropod host–parasite–virus relationships.
Keywords: meta-transcriptomics, mosquito-borne viruses, arthropod-borne viruses, virus evolution, virome
Introduction
Arthropods can interact in various ways to establish symbiotic relationships in nature (Kaplan and Eubanks 2005; Peng et al. 2013; Werblow et al. 2015). Among these, parasitic associations have profound effects on host populations and community ecology (Vasquez et al. 2020). This symbiotic strategy allows a parasitic arthropod to exploit the resources of an arthropod host to survive and reproduce. In freshwater ecosystems, parasitic associations can be observed between water mites and other arthropods such as crustaceans and insects (Pozojević et al. 2019; Vasquez et al. 2020). However, such biotic associations are not only confined to hosts and parasites. Relatedly, viruses are ubiquitous actors capable of permeating through arthropod symbiotic systems and interacting with either the parasite and/or the ‘base host’, resulting in a dynamic tripartite setting (i.e. host–parasite–virus system) (Di Prisco et al. 2016; Parratt and Laine 2016).
Parasitic mites can act as vectors or activators of viral diseases. For instance, RNA viruses, such as Kashmir bee virus (KBV), sacbrood virus (SBV), and deformed wing virus (DWV), are often detected in honeybee colonies infested with Varroa mites (Shen et al. 2005; Dainat et al. 2009). Importantly, there are major gaps in our current knowledge of the diversity and biology of viruses associated with natural mite populations that parasitize mosquitoes and their vectorial capacity. Indeed, most research on viruses infecting mites are related to pathogens of mammals and plants (Poinar and Poinar 1998; Valiente Moro, Chauve, and Zenner 2005; Yu and Tesh 2014). In the same way, the relationship between parasitism (e.g. multiparasitism) and virus ecology at the mosquito–mite–virus interface remains to be determined (Auld, Searle, and Duffy 2017).
Water mite larvae (Acari: Parasitengona: Hydrachnid) are obligate ectoparasites of culicid mosquitoes (Werblow et al. 2015). Although the exact nature of the host–parasite relationship between mosquitoes and mites is uncertain, water mites exhibit predatory and parasitic behaviors on larval and adult stage mosquitoes, respectively (Werblow et al. 2015; Atwa, Bilgrami, and Al-Saggaf 2017; Vasquez et al. 2020). During the biotic interaction, water mite larvae often attach to pre-imaginal stages or adult mosquitoes that provide the larvae with nutrients and transport to complete their life cycle (Werblow et al. 2015). Once the larval stage is complete, water mites detach from the mosquito for post-larval (nymphal stages) and adult development, feeding on insect larvae, including mosquito eggs and larvae present in aquatic habitats (Atwa, Bilgrami, and Al-Saggaf 2017; Vasquez et al. 2020). Conversely, parasitism of mosquitoes by mites is usually associated with adverse effects on mosquito fitness (i.e. reduced reproductive ability and survival) (Dos Santos et al. 2016). Among these, mite infestation might impact flight, sexual maturity, and egg production in mosquitoes.
The snow-pool mosquito species, Aedes communis (De Geer 1776), is a monocyclic species with a Holarctic distribution, occurring in Eurasia and North America (Becker et al. 2010). It is commonly found not only in forested areas such as coniferous and temperate forests but also on the tundra (Medvedev, Aibulatov, and Panyukova 2011). Aedes communis females commonly blood feed during twilight on a variety of vertebrates, including humans, rabbits, birds, rodents, and cattle. In Sweden, A. communis is abundant and widespread in spring and early summer (Lundström et al. 2013). The virome of A. communis is largely unknown but has been shown to include insect-specific viruses (ISVs) from the families Phasmaviridae, Rhabdoviridae, and Solemoviridae of RNA viruses (Öhlund et al. 2019). Sporadic detections of different arboviruses have also been reported from this species (Campbell et al. 1991; Lvov et al. 2015), although it is not considered a vector species for any arbovirus (Campbell et al. 1991).
The use of metagenomic sequencing to characterize virus diversity has revolutionized our understanding of the evolutionary history, ecology, and distribution of RNA viruses in nature (Shi, Zhang, and Holmes 2018; Zhang, Shi, and Holmes 2018), transforming our ability to detect viruses in terms of scalability, speed, and accuracy. In particular, these studies have revealed an enormous number and diversity of viruses in invertebrates, including both ISVs and arboviruses, some of which fall into highly divergent lineages or RNA virus families (Li et al. 2015a; Liu, Chen, and Bonning 2015; Shi et al. 2016a). Herein, we used meta-transcriptomics to reveal the virome diversity of A. communis and their parasitic mites and investigated whether mosquito–mite interactions can facilitate virus transfer among them. For this purpose, we compared the diversity and abundance of RNA viruses in mosquitoes parasitized by mites, mite-free mosquitoes, and parasitic mites to assess the viral community composition in the mosquito–mite interaction.
Methods
Sample collection
Aedes communis mosquitoes were collected within a mosquito control program (https://mygg.se/) across the river Dalälven floodplains in central Sweden (60.2888° N, 16.8970° E) between weeks 25 and 35 in 2014, 2019, and 2020, using the Centers for Disease Control and Prevention miniature light traps (CDC-traps) baited with dry ice. Morphological identification of mosquitoes was conducted using a stereomicroscope and the key provided by Becker et al. (2010) on a chilled table. The mosquitoes collected were examined under a stereoscopic microscope for the presence of mites (Mideopsis sp.). Detected mites were removed and, together with the mosquitoes, were separated into groups of mites (K), mite-free mosquitoes (M), and mite-detached mosquitoes (MK) (infection load = 1–20 mites per mosquito) (Supplementary Table S1). The collected specimens were kept at −80°C until molecular processing.
Sample processing and sequencing
Samples were processed in three groups corresponding to M (n = 80), MK (n = 80), and K (n = 160). In total, twenty-four sequencing libraries were prepared, eight libraries for each group. Samples were homogenized in pools of ten mosquitoes or twenty mites, using ZR BashingBead 0.1 mm (Zymo Research, Irvine, CA) for 180 s using a TissueLyzer II (Qiagen). Total RNA was extracted from the homogenates using the ZymoBIOMICS DNA/RNA Miniprep Kit (Zymo Research, Irvine, California, USA) according to the manufacturer’s instructions. Ribosomal RNA (rRNA)-depleted RNA was extracted from each sample using the Ribo-Zero Gold kit (Illumina). Whole-transcriptome libraries were constructed using DNA nanoball technology (paired-end sequencing) on a DNBseq platform. Library preparation and sequencing were performed by the Beijing Genomics Institute, Hong Kong. For taxonomic identification of the most likely genus of mites detached from the mosquitoes, we compared our contigs against a custom database, including Cox-1 amino acid sequences from mites (Trombidiformes; taxid: 83136). We also assessed the variation in Cox-1 gene abundance across libraries using different reference sequences of Mideopsis sp. (Supplementary Fig. S2).
Sequence data processing
Sequence read quality assessment was performed with FastQC v0.11.8 (Andrews 2010) and summarized using the MultiQC tool (Ewels et al. 2016). Ribosomal reads of Archaea, Bacteria, and Eukarya were filtered from the meta-transcriptomic data with the SortMeRNA v2.1b software (Kopylova, Noé, and Touzet 2012). Reads were assembled into contigs using the metagenomic assembler MEGAHIT v1.2.9 with default settings (Li et al. 2015b). A meta-transcriptome assembly evaluation was conducted using QUAST v4.3 (Gurevich et al. 2013). To reduce false positives in the detection of viruses due to index-hopping, putative viruses were considered as present in a library if the total read count was 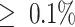 of the highest count for that virus across the libraries with at least two reads per sample. Taxonomic profiling of metagenomic data was conducted using CCMetagen v.1.2.4 (Marcelino et al. 2020) (summary data available at doi: 10.6084/m9.figshare.20499726).
of the highest count for that virus across the libraries with at least two reads per sample. Taxonomic profiling of metagenomic data was conducted using CCMetagen v.1.2.4 (Marcelino et al. 2020) (summary data available at doi: 10.6084/m9.figshare.20499726).
The sequencing reads and viral sequences identified in this study have been deposited in the SRA (Bioproject: PRJNA838788; Biosamples: SAMN28502431–SAMN28502454; SRA accession codes: SRR19268734–SRR19268757) and GenBank (ON860444–ON860480, OP555115-OP555127) databases, respectively.
Virus abundance and host association inference
Abundance was quantified as the number of reads per million mapped reads (RPM). Reads were mapped to the viral assemblies and the Cox-1 gene as host marker (JX040509.1 and MN362385.1) using the BBMap tool v.37.98 (sourceforge.net/projects/bbmap/). Contig assemblies were compared against the National Center for Biotechnology Information (NCBI) nucleotide (NCBI-nt) and non-redundant protein database (NCBI-nr) using DIAMOND v.2.0.9 with e-value cutoffs ≥1E−10 and ≥1E−4, respectively. To infer likely virus–host associations, we considered available data on (i) the virus prevalence within and between arthropod groups, (ii) abundance estimates, (iii) the closest hits in the BLAST/nr search, and (iv) phylogenetic relationships. To establish a likely host association, at least three of the four criteria had to be compatible.
Taxonomic assignment and protein annotation
Taxonomic information was collected from the NCBI Entrez taxonomy database using the NCBI-taxonomist tool v1.2.1 (https://pypi.org/project/ncbi-taxonomist/) (Buchmann and Holmes 2020). Open reading frame (ORF) detection and sequence translation were performed on contigs >1,000 nt with the program getORF v.6.6.0 (-minsize 600 -find 0), EMBOSS (Rice, Longden, and Bleasby 2000). Classification of proteins and domain detection on predicted ORFs were performed using the InterProScan v5.51-85.0 software (Jones et al. 2014) with default search parameters, and the HMMER v3.3 program (hmmscan search) against the Pfam and PROSITE databases (Finn, Clements, and Eddy 2011). To identify and annotate highly divergent viruses that were missed in the DIAMOND BLASTX search or that had similarities to taxonomically unassigned viruses, orphan contigs and unclassified viruses were run through the RdRp-scan resource with e-value 1E−6 in the hmmscan search (Charon et al. 2022). The completeness and quality of viral sequences were assessed by visual inspection and execution of the CheckV pipeline (Nayfach et al. 2020) and Prodigal v.2.6.3 (Hyatt et al. 2012).
Statistical analyses
To determine whether virus abundance levels differed significantly among the K, M, and MK groups, we assessed the normality of the data corresponding to RPM values (raw and log10 transformed) by visual inspection and using the Shapiro–Wilk test. Since the data did not follow a normal distribution, comparisons were made using the Kruskal–Wallis chi-squared test and Pairwise Wilcoxon Rank Sum Test. All analyses were performed using the packages rstatix (Kassambara 2021) and ggpubr (Kassambara 2020) in R (R Core Team 2021) (available at https://www.R-project.org/).
Phylogenetic analysis
Viral protein sequences for the RNA-dependent RNA polymerase (RdRp) identified in this study were aligned to a set of representative sequences publicly available at NCBI/GenBank according to the virus family, using Clustal Omega v.1.2.4 with default settings. The reference Quenyavirus sequences were obtained from Obbard et al. (2020). We assigned provisional names to novel viruses based on geographic locations from where they were collected. Selection of the best-fit model of sequence evolution and phylogenetic relationships within the virus families were assessed using the maximum likelihood (ML) method available in IQ-TREE v1.6.12 (-m TEST -alrt 1000 -bb 1000 -nt 4 -bnni) (Nguyen et al. 2015; Hoang et al. 2018). Nodal support was estimated with SH-aLRT and the ultrafast bootstrap (UFBoot). A total of 1,000 replicates were run for both approaches, and we used the option bnni to avoid overestimating branch supports with UFBoot. Tree visualization was conducted using the R software packages ggtree and ggplot2 (Wickham 2016; Yu et al. 2017).
Results
Extenstive RNA virome diversity in A. communis and their parasitic mites
A total of 160 mosquitoes and their parasitic mites were collected and pooled into twenty-four separate libraries, representing three different groups, to characterize the virome of each host and assess the virus prevalence across mites and in both mite-free and mite-detached mosquitoes. Overall, we generated between 48.5 and 74 million pair reads per library, of which ∼76 per cent corresponded to rRNA content. Meta-transcriptomic reads were de novo assembled into partial viral genomes from which we identified thirty-seven novel RNA viruses based on the presence of a viral RdRp: these represented eighteen families and fifteen orders of positive-stranded RNA (n = 8), negative-stranded RNA (n = 18), and double stranded RNA (n = 6) (Table 1). Three viruses were only classified to the level of phylum or class. One additional virus was taxonomically unclassified. The newly discovered viruses shared between 25.2 and 80.7 per cent amino acid sequence similarity to the RdRp of the closest viral hit in the NCBI-nr (Table 1).
Table 1.
List of putative viruses discovered in this study and present in mite/mosquito hosts. Each viral sequence was compared with the NCBI non-redundant (nr) database using DIAMOND BLASTX. Hosts are represented with letters corresponding to mites (K), mite-free mosquitoes (M), and mite-detached mosquitoes (MK).
| Virus name | Contig name | Length | Provisional classification | Best hit in the NCBI/nr | Similarity | E-value | Host |
|---|---|---|---|---|---|---|---|
| Smedsang bunya-like virus | k119_16122 | 2,250 | Bunyavirales | BBQ05095.1 RNA-dependent RNA polymerase (Culex pseudovishnui bunya-like virus) | 34.5 | 1.34E−100 | K, M, and MK |
| Avesta bunya-like virus | k119_3430 | 1,925 | Bunyavirales | QNS17451.1 RNA-dependent RNA polymerase, partial (Serbia bunya-like virus 1) | 57.4 | 2.84E−249 | K, M, and MK |
| Heby virus | k119_4879 | 1,754 | Bunyavirales | QGA70945.1 RNA-dependent RNA polymerase (Salari virus) | 69 | 8.28E−286 | M and MK |
| Buska virus | k119_17401 | 7,663 | Bunyavirales | AJG39275.1 RNA-dependent RNA polymerase (Zhee mosquito virus) | 39.4 | 0 | K, M, and MK |
| Gaddsjo leishbunyavirus | k119_11873 | 2,442 |
Bunyavirales
/Leishbuviridae |
ANJ59510.1 putative RNA-dependent RNA polymerase (Leptomonas moramango leishbunyavirus) | 80.7 | 0 | M and MK |
| Sater virus | k119_979 | 13,942 |
Bunyavirales
/Nairo-like |
YP_009300680.1 RNA-dependent RNA polymerase (Shayang spider virus 1) | 25.4 | 1.42E−278 | K and MK |
| Fallet virus | k119_5606 | 7,027 |
Jingchuvirales
/Chuviridae |
API61887.1 RNA-directed RNA polymerase (Chuvirus Mos8Chu0) | 63 | 0 | K, M, and MK |
| Hede virus | k119_2521 | 1,378 |
Amarillovirales
/Flaviviridae |
YP_009179222.1 polyprotein (Xinzhou spider virus 2) | 31 | 2.07E−53 | K |
| Broddbo narna-like virus | k119_1307 | 3,122 |
Wolframvirales
/Narnaviridae |
APG77272.1 RNA-dependent RNA polymerase, partial (Wenling narna-like virus 6) | 35.9 | 6.23E−179 | K |
| Hytton narna-like virus | k119_17837 | 3,140 |
Wolframvirales/ Narnaviridae |
AGW51768.2 putative RNA-dependent RNA polymerase-like protein (Ochlerotatus-associated narna-like virus 2) | 73.4 | 0 | K, M, and MK |
| Hedemora virus | k119_6373 | 16,014 |
Mononegavirales/ Rhabdo-like |
YP_009304476.1 RNA-dependent RNA polymerase (Tacheng tick virus 7) | 29.2 | 5.70E−211 | K |
| Sonnboviken virus | k119_1814 | 1,016 |
Mononegavirales/ Lispi-like |
QMP82230.1 RNA-dependent RNA polymerase, partial (Megalopteran arli-related virus OKIAV106) | 25.2 | 2.02E−13 | K |
| Fors virus | k119_3330 | 3,921 |
Mononegavirales/ Lispi-like |
YP_009342285.1 RNA-dependent RNA polymerase (Wuchang romanomermis nematode virus 2) | 33.5 | 7.53E−171 | K |
| Osterbannback virus | k119_16137 | 13,129 | Mononegavirales | QRW42735.1 RNA-dependent RNA polymerase (Gordis virus) | 37.6 | 0 | K, M, and MK |
| Bro virus | k119_22347 | 6,397 |
Mononegavirales/ Xinmoviridae |
BBQ04817.1 RNA-dependent RNA polymerase (Culex tritaeniorhynchus anphevirus) | 41.2 | 0 | M |
| Malby virus | k119_10539 | 6,320 |
Mononegavirales/ Xinmoviridae |
BBQ04817.1 RNA-dependent RNA polymerase (Culex tritaeniorhynchus anphevirus) | 40 | 0 | K, M, and MK |
| Pelarsalen rhabdo-like virus | k119_4181 | 6,711 |
Mononegavirales/ Rhabdo-like |
QHA33680.1 RdRp (Atrato Rhabdo-like virus 3) | 48.6 | 0 | K, M, and MK |
| Tierp virus | k119_3941 | 2,428 |
Articulavirales/ Orthomyxoviridae |
QRW42655.1 polymerase PB1 (Usinis virus) | 57.6 | 4.35E−311 | K and MK |
| Husby virus | k119_19965 | 2,448 |
Articulavirales/ Orthomyxoviridae |
QGA70921.1 RNA-dependent RNA polymerase (Wuhan mosquito virus 4) | 55.9 | 3.01E−305 | K, M, and MK |
| Kagbo partiti-like virus | k119_14506 | 1,722 |
Durnavirales/ Partitiviridae |
APG78217.1 RdRp (Hubei partiti-like virus 22) | 60 | 2.19E−248 | MK |
| Ormpussen virus | k119_12042 | 1,630 |
Durnavirales/ Partitiviridae |
AWY11085.1 orf1 (galbut virus) | 36 | 8.71E−90 | K, M, and MK |
| Hebron partiti-like virus | k119_1779 | 1,699 |
Durnavirales/ Partitiviridae |
APG78260.1 RdRp (Hubei partiti-like virus 19) | 50.6 | 7.23E−194 | K, M, and MK |
| Hundmyran chaq-like virus | k119_19664 | 1,476 |
Durnavirales/ Partitiviridae |
AKH40308.1 orf1 (Chaq virus*) | 52.9 | 3.8E−103 | K, M, and MK |
| Nor picorna-like virus | k119_7745 | 9,260 |
Picornavirales/ Iflaviridae |
AWC26954.1 polyprotein (Culex picorna-like virus 1) | 53.9 | 0 | M |
| Dalkarlsbo virus | k119_10044 | 2,238 | Quenyaviridae | QIQ61196.1 putative RNA-dependent RNA polymerase (Nete virus) | 39.7 | 5.40E−158 | K, M, and MK |
| Morgongava virus | k119_3685 | 5,653 |
Muvirales/ Qinviridae |
QGA70948.1 RNA-dependent RNA polymerase (Vittskovle virus) | 33.9 | 3.85E−226 | K, M, and MK |
| Hallarsbo virus | k119_7784 | 4,121 |
Muvirales/ Qinviridae |
QLJ83493.1 RNA-dependent RNA polymerase (Fitzroy Crossing qinvirus 1) | 33 | 1.09E−222 | K, M, and MK |
| Berg reo-like virus | k119_12531 | 4,277 |
Reovirales/ Sedoreoviridae |
QHA33824.1 putative RdRp (Atrato reo-like virus) | 63.8 | 0 | M |
| Koversta virus | k119_2089 | 3,950 |
Ghabrivirales/ Totiviridae |
QHA33712.1 RdRp (Embera virus) | 54.2 | 0 | M |
| Disbo virus | k119_15173 | 2,401 |
Ghabrivirales/ Totiviridae |
YP_009552795.1 RNA-dependent RNA polymerase (Diatom colony-associated dsRNA virus 11) | 34.2 | 2.53E−131 | M |
| Karbo virus | k119_13033 | 1,727 |
Tymovirales/ Tymoviridae |
YP_009551972.1 polyprotein (Alfalfa virus F) | 44.5 | 5.20E−139 | M and MK |
| Ginka virga-like virus | k119_5852 | 2,312 |
Martellivirales/ Virgaviridae |
QHA33742.1 polyprotein (Atrato Virga-like virus 3) | 57.1 | 4.76E−285 | M and MK |
| Baggbo virus | k119_4924 | 4,407 | Unclassified Sobeliviralesa |
QIS87998.1 RNA-dependent RNA polymerase (Khabarov virus) | 42.7 | 1.87E−75 | K |
| Sala virus | k119_14050 | 5,220 | Unclassified Ellioviricetesa |
AGW51765.1 RNA-dependent RNA polymerase-like protein (uncultured virus) | 39.3 | 0 | K, M, and MK |
| Nedre virus | k119_4180 | 7,799 | Unclassified Ellioviricetesa |
AGW51765.1 RNA-dependent RNA polymerase-like protein (uncultured virus) | 39.2 | 0 | K and MK |
| Kvarnon virus | k119_3338 | 1,763 | Ortervirales/Metaviridae | QPF16710.1 putative RNA-dependent RNA polymerase (Aedes aegypti to virus 2) | 51.3 | 7.60E−194 | K, M, and MK |
| Fullsta virus | k119_9880 | 2,263 | Unclassified Pisuviricotaa |
QFR59041.1 putative RNA-dependent RNA polymerase, partial (Hanyang virus) | 41.5 | 2.57E−157 | K, M, and MK |
Chaq virus is often considered either a satellite virus or a segment or galbut virus.
Previously unclassified viruses annotated using RdRp-scan [1].
Notably, the mite-specific viruses were highly divergent. BLASTX similarity searches revealed that the majority of the virus contigs were related to arthropod-associated viruses (29/37), although we identified three viruses to be associated with those previously identified in nematodes (Wuchang romanomermis nematode virus 2; similarity = 33.5 per cent), protozoans (Leptomonas moramango leishbunyavirus; similarity = 80.7 per cent), and algae (diatom colony-associated dsRNA virus 11; similarity = 34.2 per cent). Likewise, we identified three viruses in the families Narnaviridae and Tymoviridae that are most often associated with fungi or plants (Table 1).
Based on the phylogenetic analysis, we identified several putative novel viruses in mosquitoes and mites that shared close relationships to known RNA viruses within the families Chuviridae, Flaviviridae, Metaviridae, Narnaviridae, Orthomyxoviridae, Partitiviridae, Iflaviridae, Qinviridae, Quenyaviridae, Sedoreoviridae, Totiviridae, and Tymoviridae (Figs 1–5). Of particular note was a novel pestivirus, tentatively named Hede virus, that exhibited ∼31 per cent amino acid sequence similarity to Xinzhou spider virus 2 previously discovered in spiders (Araneae), and the novel Kvarnon virus that shared ∼51 per cent similarity to the errantivirus Aedes aegypti To virus 2 (Metaviridae) (Table 1 and Figs 3B and 4C) (Shi et al. 2016b). Similarly, we identified viruses related to members of the Leishbunyaviridae and Nairoviridae within the order Bunyavirales, as well as members of the Rhadboviridae, Lispiviridae, and Xinmoviridae in the order Mononegavirales of single-strand negative-sense RNA viruses (Fig. 1). A small number of the novel viruses identified here was grouped with unclassified RNA virus sequences in the Bunyavirales and Mononegavirales. Due to the limited similarity shared between the novel and known viruses, we only recovered partial genome/replicase sequences encoding conserved domains, such as the RdRp and MTase, as well as segments encoding uncharacterized proteins (Supplementary Table S3 and Fig. 5).
Figure 1.
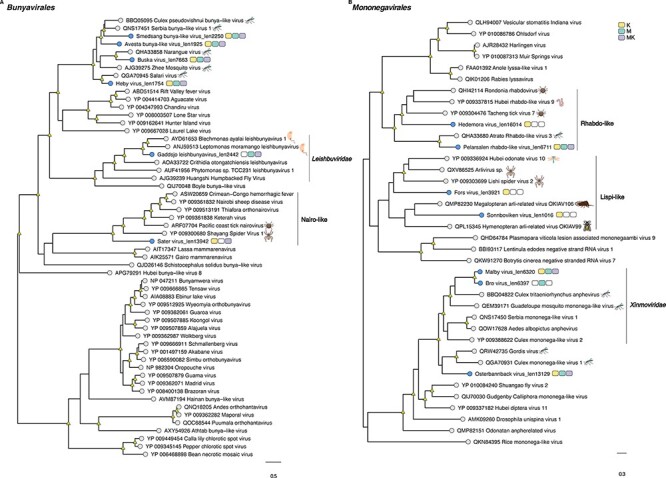
Phylogenetic relationships among the viruses found in this study and reference sequences within the RNA virus orders (A) Bunyavirales and (B) Mononegavirales. Phylogenetic trees were estimated using the Q.pfam + F + I + Γ4 substitution model. Novel viruses are indicated by blue tip points, and hosts are represented with three-pack bars corresponding to mite (K; yellow), mite-free mosquito (M; green), and mite-detached mosquito samples (MK; purple). Trees are based on the amino acid sequences of the putative RdRp. Nodal support values ≥80 per cent SH-aLRT and ≥95 per cent UFboot are denoted with yellow triangles at nodes. Scale bars indicate the number of amino acid substitutions per site and the trees are mid-point rooted for clarity only. Host species information (animal icons) is shown for the closest relatives of the novel viruses.
Figure 3.
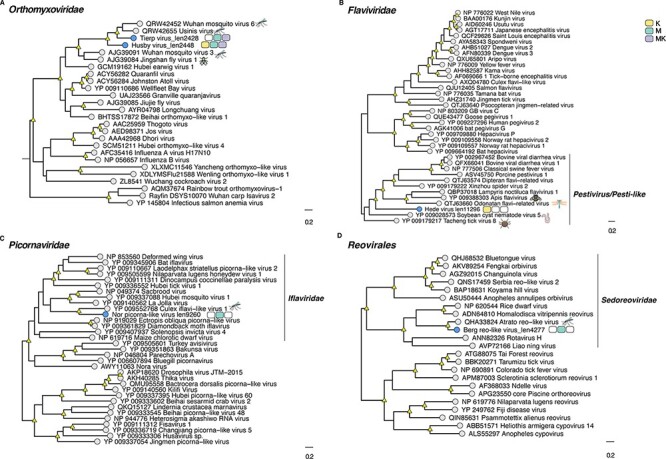
Phylogenetic relationships among the viruses found in this study and reference sequences within the RNA virus families (A) Flaviviridae, (B) Orthomyxoviridae, (C) Picornavirales, and (D) Sedoreoviridae. Phylogenetic trees were estimated using the VT + F + I + G4 (Flaviviridae, Picornavirales, and Sedoreoviridae) and Q.pfam + F + I+ Γ4 (Orthomyxoviridae) substitution models. Novel viruses are indicated by blue tip points, and hosts are represented with three-pack bars corresponding to mite (K; yellow), mite-free mosquito (M; green), and mite-detached mosquito samples (MK; purple). Trees are based on the amino acid sequences of the putative RdRp. Nodal support values ≥80 per cent SH-aLRT and ≥95 per cent UFboot are denoted with yellow triangles at nodes. Scale bars indicate the number of amino acid substitutions per site and the trees are mid-point rooted for clarity only. Host species information (animal icons) is shown for the closest relatives of the novel viruses.
Figure 4.
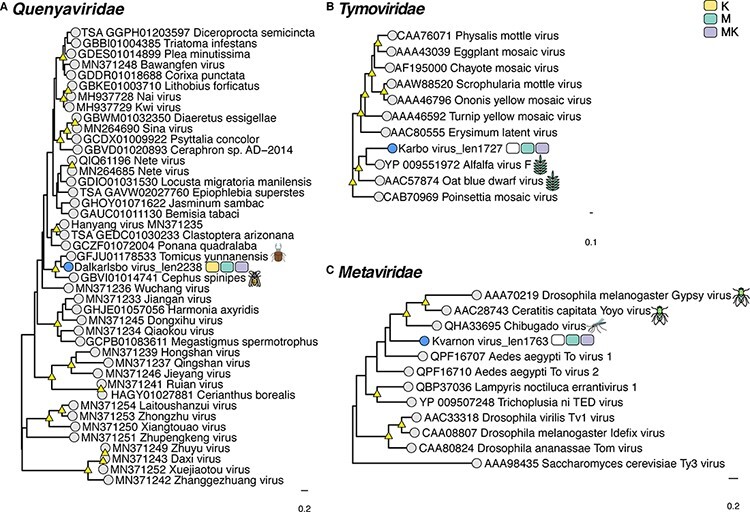
Phylogenetic relationships among the viruses found in this study and reference sequences within the RNA virus families (A) Quenyaviridae (B) Tymoviridae, and (C) Metaviridae. Phylogenetic trees were estimated using the rtREV + F + I+ Γ4 substitution model. Novel viruses are indicated by blue tip points and hosts are represented with three-pack bars corresponding to mite (K; yellow), mite-free mosquito (M; green), and mite-detached mosquito samples (MK; purple). Trees are based on the amino acid sequences of the putative RdRp or RT. Nodal support values ≥80 per cent SH-aLRT and ≥95 per cent UFboot are denoted with yellow triangles at nodes. Scale bars indicate the number of amino acid substitutions per site and the trees are mid-point rooted for clarity only. Host species information (animal icons) is shown for the closest relatives of the novel viruses.
Figure 5.
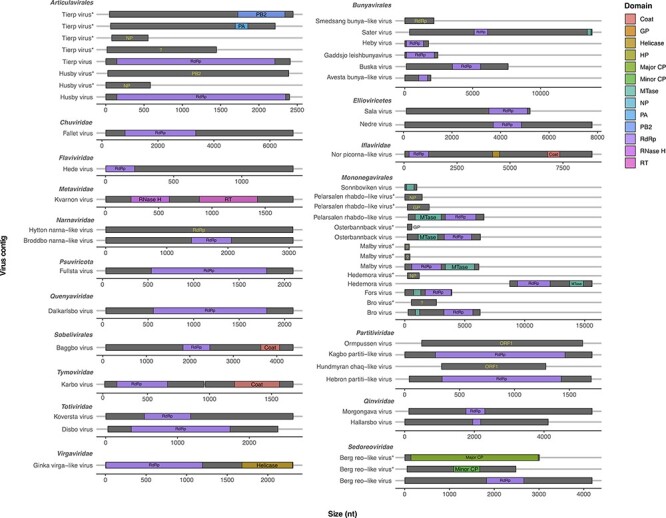
Schematic representation of protein domains found in the viral sequences identified in this study. Putative novel viruses are grouped by virus family. Diagrams represent predicted ORFs (gray), while domains are displayed as colored boxes (see legend). ORFs lacking conserved domains are annotated (text labels) based on the closest hit in the BLASTX search. The question marks (?) represent ORFs encoding hypothetical/unknown proteins. The ORFs size is shown as number of nucleotides, and each ORF is located along the contig according to the predicted coordinates. Multiple segments of a virus are indicated by asterisks (*). GP: glycoprotein; HP: hypothetical protein; CP: core protein; MTase: metyltransferase; NP: nucleoprotein; PA: polymerase acidic protein; PB2: polymerase basic protein 2; RdRp: RNA-dependent RNA polymerase; RNase H: ribonuclease H; RT: reverse transcriptase.
Although the newly discovered Baggbo virus, Fullsta virus, Sala virus, and Nedre virus shared limited similarity with unclassified viruses (similarity = 39.2–42.7 per cent) (Table1), we provided a broad taxonomic assignment for these viruses within the Sobelivirales, Ellioviricetes, and Pisuviricota (Supplementary Table S2). Also of note was that the newly identified viruses for which the taxonomic status could be assigned fell into distinct clades within several families, helping to fill the gaps in the phylogenies of these groups. In other cases, the putative viruses identified here are grouped together as sister taxa to each other. For example, the Hallarsbo virus fell as a sister taxon to Morgongava virus (Qinviviridae) as part of a clade of mosquito-associated viruses (Fig. 2D), as did Malby virus and Bro virus (Xinmoviridae) (Fig. 1B). Finally, some of the newly identified viral sequences occupied basal phylogenetic positions, such as Heby virus (Bunyavirales), which was a sister to a clade comprising the newly discovered viruses Smedsang bunya-like virus, Avesta bunya-like virus, Buska virus, and their closest known relatives in mosquitoes (Fig. 1A). Similarly, Sater virus (a Nairo-like virus) shared common ancestry with other tick nairoviruses (Fig. 1A).
Figure 2.
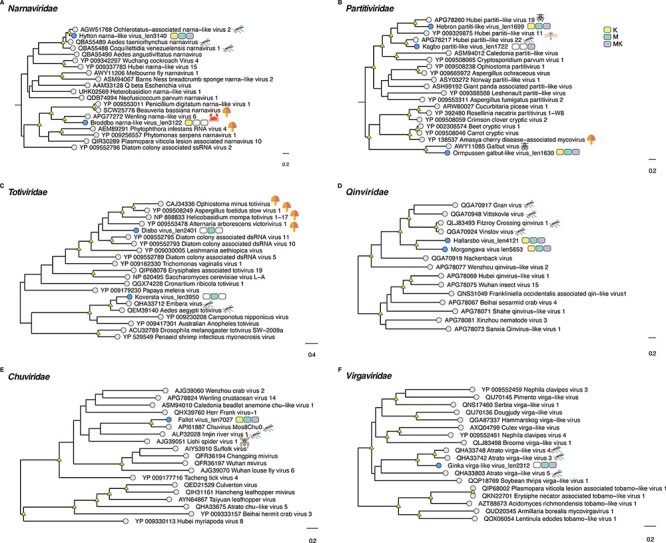
Phylogenetic relationships among the viruses found in this study and reference sequences within the RNA virus families (A) Narnaviridae, (B) Partitiviridae, (C) Totiviridae, (D) Qinviridae, (E) Chuviridae, and (F) Virgaviridae. Phylogenetic trees were estimated using the VT + F + I + G4 (Narnaviridae, Partitiviridae, Virgaviridae) and Q.pfam + F + I+ Γ4 (Totiviridae, Qinviridae, Chuviridae) substitution models. Novel viruses are indicated by blue tip points and hosts are represented with three-pack bars corresponding to mite (K; yellow), mite-free mosquito (M; green), and mite-detached mosquito samples (MK; purple). Trees are based on the amino acid sequences of the putative RdRp. Nodal support values ≥80 per cent SH-aLRT and ≥95 per cent UFboot are denoted with yellow triangles at nodes. Scale bars indicate the number of amino acid substitutions per site and the trees are mid-point rooted for clarity only. Host species information (animal icons) is shown for the closest relatives of the novel viruses.
Composition and distribution of RNA viruses reveal host connectivity
To assess the differences in the virome composition between groups, we determined virus prevalence across all the sequencing libraries generated here. This revealed a similar number of viruses present in M (n = 29), MK (n = 25), and K (n = 26) (Fig. 6). Notably, six viruses were specific to the mite (K) libraries, eighteen viruses were shared between mosquitoes (M/MK) and mites (K), whereas five viruses were exclusively present in the mite-free (M) libraries (Table 1, Fig. 6). The shared viruses were classified within the Chuviridae, Narnaviridae, Orthomyxoviridae, Partitiviridae, and Quenyaviridae, as well as those assigned to the orders Bunyavirales and Mononegavirales (Table 1, Fig. 6). Cross-reference between some MK and K pools was concordant with these results (Supplementary Fig. S1). Within the Partitiviridae, Ormpussen galbut-like virus and Hundmyran chaq-like virus exhibited limited sequence similarity to galbut (similarity = 36 per cent) and Chaq virus (similarity = ∼ 53 per cent), respectively (Table 1). Remarkably, these viruses also co-occurred in most libraries (7/10) (Fig. 7, Supplementary Fig. S1). In general, we found at least four viruses per library, with the exception of the mite-free mosquito library M3, which harboured seventeen novel viruses (Fig. 7).
Figure 6.
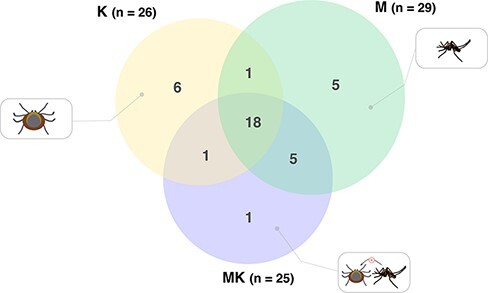
Comparison of the number of viruses shared between mites (K), mite-free mosquitoes (M), and mite-detached mosquito libraries (MK). The total number of viruses is indicated for each group. The size of the circle is proportional to the number of viruses identified in each host group. Color coding is the same as in Figs 1–4.
Figure 7.
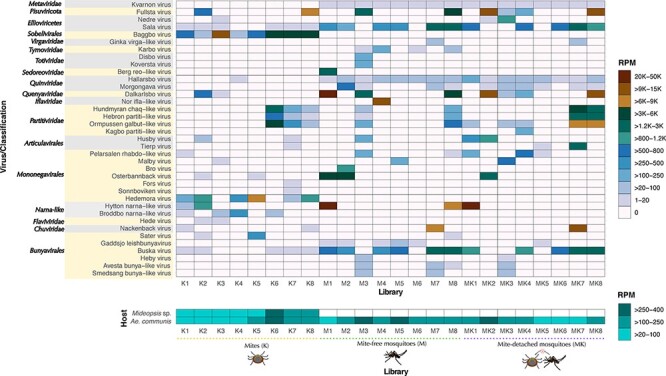
Overview of virus abundance quantified as reads per million (RPM). The newly discovered viruses are indicated on the y-axis. Abundance levels are color-coded as a heat map as specified in the legend. The queried libraries corresponding to mite (K), mite-free mosquito (M), and mite-detached mosquito (MK) are shown on the x-axis. Host abundance levels based on the Cox-1 gene marker are represented in the bottom panel.
Although we expected that all viruses present in MK libraries (i.e. mosquitoes previously infested with mites) would also be found in either M or K libraries, Kagbo partiti-like virus was only recovered from a single MK library (Figs 6 and 7). In contrast, we observed common viruses among host-specific libraries, such as Hallarsbo virus (Qinviridae) and Kvarnon virus (Metaviridae) in mosquitoes and Baggbo virus (Sobelivirales) and Hedemora virus (Mononegavirales) in mites (Table 1, Fig. 7). Only the putative Buska virus (Bunyavirales) was broadly distributed among most of the libraries (17/24).
Virus abundance levels suggest host associations and virome connectivity between mosquitoes and mites
We next assessed virus abundance for each newly discovered virus compared to host gene markers (Fig. 7). Accordingly, virus abundance varied from 1 to 47,863 RPM (Fig. 7). In contrast, the abundance of the reference mitochondrial gene marker Cox-1 was more stable across host-specific libraries, ranging between 40 and 307 RPM for Mideopsis sp. and 43–348 RPM for A. communis. Importantly, reads from the mosquito host were detected in all the libraries (Fig. 7). Comparisons of virus abundances between groups K, M, and MK revealed no significant differences (KW = 2.68 P-value = 0.2617). We considered viruses with values >1,000 RPM (>0.1 per cent of ribosomal-depleted RNA) to be highly abundant. For example, Hytton narna-like virus (Narnaviridae, RPM = 20–39,637) exhibited the highest abundance and was present across mite and mosquito libraries (Fig. 7). Nearly all viruses exclusively detected in mites showed very high abundance levels, such as Baggbo virus (Sobelivirales, RPM = 84–10,251), which was highly prevalent among K libraries (Figs 5 and 7). We were also able to identify some viruses that were abundant in mosquito libraries but were still present in mites at negligible levels, including Buska virus (Bunyavirales, RPM = 1–1,885), Osterbannback virus (Partitiviridae, RPM = 1–3771), and Fallet virus (Chuviridae, RPM = 6–11,594), supporting the idea that these viruses are likely more associated with mosquitoes (Fig. 7). Likewise, although both Kvarnon virus (Metaviridae, RPM = 6–19) and Hallarsbo virus (Quinviridae, RPM = 2–93) were at low abundance, they were stably expressed and highly prevalent in M libraries while scarce in K libraries. It is also notable that viruses restricted to single libraries, such as Kagbo partiti-like virus (Partitiviridae, RPM = 125) and Disbo virus (Totiviridae, RPM = 135), were at low abundance (Fig. 7). Overall, these results revealed differences in virus composition and abundance that might help demonstrate virus–host associations as well as connectivity (i.e. potential virus transfer) through the host–parasite system.
Discussion
Host–parasite relationships between mosquitoes and mites have impact on both arthropod and ecosystem ecology. However, aspects such as host and parasite virome diversity and composition have largely been neglected within the mite–mosquito interaction. Here, we provide an overview of the diversity of RNA viruses in these arthropods, comparing their virome profile to investigate possible transfer events between both hosts. In line with previous studies on arthropod viromes (Li et al. 2015a; Obbard et al. 2020; Pettersson et al. 2017; Shi et al. 2016a), we observed a high abundance of many diverse viruses, suggesting that many more arthropod viruses remain to be discovered (Harvey and Holmes 2022; Junglen and Drosten 2013; Shi et al. 2016a).
Although mites are known to be vectors of some pathogens of medical and veterinary importance for vertebrate hosts (Gubler 1988; Hubálek, Rudolf, and Nowotny 2014; Mullen and O’Connor 2019; Weaver and Reisen 2010; Yu and Tesh 2014), these arthropods can also act as vectors of viral agents to other arthropods. For example, Varroa mites (Varroa destructor) appear to mediate transmission of KBV, SBV, and DWV to honey bees Apis mellifera (Shen et al. 2005). As a consequence, the interaction between parasites and their hosts is also likely to lead to the transfer of viruses. We investigated the virome of mosquitoes and their infesting mites to reveal mosquito–mite interactions. A key result was that a number of viruses were commonly present in both mosquitoes and mite samples (Fig. 6), indicating that the transfer of viruses is likely to occur when parasitic mites feed on dipteran hosts (Dolja and Koonin 2018). Further research is needed to assess whether these viruses are able to infect, replicate, and spread in both arthropod hosts, as opposed to being of dietary origin or infecting components of the microbiome (Supplementary Fig. S3) (Obbard 2018). In the latter case, the presence of partitiviruses, such as Ormpussen galbut-like virus and Hebron partiti-like virus, both widely distributed among mite and mosquito samples, suggest the presence of common fungal and/or protozoan microbiota (Figs 2B and 7). Nonetheless, we cannot definitively exclude possible contamination with microorganisms present on the surface of arthropods or derived from sample processing.
The repertoire of putative viruses identified in this study spanned different viral families previously reported in mites and mosquitoes (Table 1, Figs 1–4) (Chang et al. 2021; Junglen and Drosten 2013; Li et al. 2015a; Shi et al. 2016a). In particular, prior to this work, there were a relatively limited number of viruses recorded in mites that parasitize other arthropods, representing the Chuviridae, Dicistroviridae, Iflaviridae, and Rhabdoviridae families (Shen et al. 2005; Dietzgen et al. 2014; Niu et al. 2019). Our results expand this virus diversity to include viruses within the orders Sobelivirales and Mononegavirales and the family Flaviviridae (Figs 1B, 3B, and 7). The occurrence of flaviviruses has been previously recorded in acarid ectoparasites parasitizing natural bird populations (Santillán et al. 2015; Kovalev and Yakimenko 2021). In contrast, the highly divergent Hede virus (likely partial RdRp) found in mites was most closely related to arthropod-specific long genome flaviviruses (Table 1, Figs 3B and 5) (Paraskevopoulou et al. 2021), with related viruses documented in ticks (Tokarz et al. 2014; Xu et al. 2022). Therefore, the presence of highly divergent viruses in mites suggests a hidden diversity in Acari.
A key outcome of this study was the presence of viruses restricted to either mosquito or mite libraries, which we hypothesized to correspond to host-specific viruses or those associated with the host microbiota. For example, the presence of the Baggbo virus at abundant levels across several K samples (Table 1, Fig. 7) suggests that these arthropods might serve as natural carriers of this virus. In contrast, Kvarnon virus, which was found at low abundance in the majority of the M and MK libraries, yet not the K libraries, is presumed to derive from symbionts in A. communis mosquitoes (Supplementary Fig. S3). However, due to the small sample size, caution is needed in the formulation of definitive virus–host associations.
Insect-specific viruses (ISVs) in mosquitoes represent the Bunyaviridae, Sedoreoviridae, Iflaviridae, Mononegavirales, and Flaviviridae (Roundy et al. 2017). Bro virus (Xinmoviridae), Nor ifla-like virus (Iflaviridae), and Berg reo-like virus (Sedoreoviridae) described here may also constitute ISVs as they were found in high abundance and grouped with other viruses reported in mosquitoes (Figs 1–5) (Pettersson et al. 2019). In contrast, the novel totiviruses are more likely to have a fungal or protozoan origin (Fauver et al. 2016; Wu et al. 2020; Coatsworth et al. 2021) (Supplementary Fig. S3). Finally, among the set of newly discovered viruses, the broad distribution of the Buska virus across most libraries (Fig. 7) is consistent with that reported for its closest match—Zhee mosquito virus in Coquillettidia richardii and Aedes spp. (Öhlund et al. 2019). Together with the substantial variation in the abundance of Buska virus between mite and mosquito libraries, we hypothesized that this bunyavirus might infect and replicate well in A. communis mosquitoes.
Given the occurrence of Kagbo partiti-like virus in a single MK library and the lack of detection in K and M samples, its true host association is difficult to assign (Figs 2B and 7). Despite our thorough examination of the samples, it is possible that small remnants of mouthparts contaminated with fungi or protozoa were still present in the M/MK libraries, which may explain the sporadic occurrence of the novel Kagbo partiti-like virus. Conversely, the presence of contaminant viral sequences might offer an alternative explanation (Porter et al. 2021). It has previously been shown that contaminant viruses can not only be derived from multiple sources, including specimen surface contamination, reagents, controls, and cell culture, but can also be introduced at any step in sample preparation and sequencing (Batson et al. 2021; Cobbin et al. 2021; Porter et al. 2021). Interestingly, the closest relative to Kagbo partiti-like virus (Fig. 2B) has been reported in Culex modestus, Culex vishnui, Culex tritaeniorhynchus, Culex quinquefasciatus, Culex pipiens, and Culex torrentium and A. aegypti from different geographic globally (Faizah et al. 2020; Öhlund et al. 2019; Pettersson et al. 2019; Shi et al. 2019; Wang et al. 2021).
Previous work on arthropod viral transcriptomes strongly suggests that galbut and chaq virus are associated with a satellite–helper virus system or are part of the same segmented RNA virus (Batson et al. 2021; Cross et al. 2020; Shi et al. 2018; Webster et al. 2015), although key aspects of the system are still poorly understood. In this context, the co-occurrence of the novel partitiviruses Ormpussen galbut-like virus and Hundmyran chaq-like virus, distantly related to galbut virus- and chaq virus-like sequences, respectively, which further supports the notion of an existing relationship between these viruses, extending this association to more distant viral relatives. It is important to note that we detected a similar pattern of co-occurrence for Hebron partiti-like virus (Fig. 7). This observation agrees with previous studies reporting the presence of multiple partitiviruses in samples (Webster et al. 2015; Faizah et al. 2020), although, based on the available data, we were unable to determine whether Hebron partiti-like virus is specifically associated with Ormpussen galbut-like virus and Hundmyran chaq-like sequences.
It is also important to consider the co-occurrence of multiple virus taxa within libraries regardless of the arthropod host (Fig. 7). Accordingly, our analysis revealed a heterogeneous diversity (i.e. composition and abundance) of RNA viruses within libraries. These differences might reflect underlying interactions at the host–parasite–virus interface. Indeed, viral infection can shape host–parasite relationships by impacting ectoparasite virulence and imposing differential selective pressures on the hosts in question (Di Prisco et al. 2016; Parratt and Laine 2016). However, the interactions between viruses carried by the parasite host and the base host have been poorly explored (Díaz-Muñoz 2019), and viruses can also interact with host microbiota (Jagdale and Joshi 2018; Hahn et al. 2020; Altinli, Schnettler, and Sicard 2021). Understanding the implications of such symbiotic relationships in arthropods is therefore of importance (Altinli, Schnettler, and Sicard 2021).
Significance, limitations, and future directions
Our findings provide preliminary baseline evidence for understanding the structure of the RNA virome in mosquitoes and their parasitic mites. A holistic understanding will require research addressing open questions on such major topics as host associations and competence, as well as the effect of virus infection on host biology. This study extends the current diversity of RNA viruses in arthropods and provides high-resolution insights into the RNA viral metagenome in the context of host–parasite interactions. Future research efforts should be addressed to determine the impact of these viruses on host–parasite relationships as well as the ecological and evolutionary implications for this and other tripartite systems
Supplementary Material
Acknowledgements
This work was supported by Stiftelsen P. E. Lindahls stipendiefond, natural sciences (Grant No. LN2018-0059), and the European Union’s Horizon 2020 research and innovation programme under Grant No. 874735 (VEO). J.H.O.P. is funded by the Swedish research council FORMAS (Grant No: 2015-710) and Vetenskapsrådet (Grant No: 2020-02593). E.C.H. is funded by an ARC Australian Laureate Fellowship (FL170100022). The authors are thankful to Pernilla Wahlqvist for mosquito collection and identification.
Contributor Information
Ayda Susana Ortiz-Baez, Sydney Institute for Infectious Diseases, School of Medical Sciences, The University of Sydney, Sydney, NSW 2006, Australia.
Edward C Holmes, Sydney Institute for Infectious Diseases, School of Medical Sciences, The University of Sydney, Sydney, NSW 2006, Australia.
Justine Charon, Sydney Institute for Infectious Diseases, School of Medical Sciences, The University of Sydney, Sydney, NSW 2006, Australia.
John H-O Pettersson, Sydney Institute for Infectious Diseases, School of Medical Sciences, The University of Sydney, Sydney, NSW 2006, Australia; Clinical Microbiology and Hospital Hygiene, Uppsala University Hospital, Dag Hammarskjölds väg 38, Uppsala SE-751 85, Sweden; Zoonosis Science Center, Department of Medical Biochemistry and Microbiology, University of Uppsala, Husargatan 3, C8:3, Uppsala SE-751 23, Sweden.
Jenny C Hesson, Zoonosis Science Center, Department of Medical Biochemistry and Microbiology, University of Uppsala, Husargatan 3, C8:3, Uppsala SE-751 23, Sweden; Biologisk Myggkontroll, Nedre Dalälven Utvecklings AB, Vårdsätravägen 5, Uppsala SE 75646, Sweden.
Supplementary data
Supplementary data are available at Virus Evolution online.
Conflict of interest:
None declared.
References
- Altinli M., Schnettler E., and Sicard M. (2021) ‘Symbiotic Interactions between Mosquitoes and Mosquito Viruses’, Frontiers in Cellular and Infection Microbiology, 11: 694020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. (2010) ‘FastQC: A Quality Control Tool for High Throughput Sequence Data.’ <https://www.bioinformatics.babraham.ac.uk/projects/fastqc/> accessed 08 2021.
- Atwa A. A., Bilgrami A. L., and Al-Saggaf A. I. M. (2017) ‘Host–Parasite Interaction and Impact of Mite Infection on Mosquito Population’, Revista Brasileira de Entomologia, 61: 101–6.doi: 10.1016/j.rbe.2017.03.005 [DOI] [Google Scholar]
- Auld S. K. J. R., Searle C. L., and Duffy M. A. (2017) ‘Parasite Transmission in a Natural Multihost–Multiparasite Community’, Philosophical Transactions of the Royal Society B: Biological Sciences, 372: 1–10.doi: 10.1098/rstb.2016.0097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batson J. et al. (2021) ‘Single Mosquito Metatranscriptomics Identifies Vectors, Emerging Pathogens and Reservoirs in One Assay’, eLife, e68353.doi: 10.7554/eLife.68353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker N. et al. (2010) Mosquitoes and Their Control, 2nd edn. Berlin: Springer Science & Business Media.doi: 10.1007/978-3-540-92874-4 [DOI] [Google Scholar]
- Buchmann J. P., and Holmes E. C. (2020) ‘Collecting and Managing Taxonomic Data with NCBI-taxonomist’, Bioinformatics, 36: 5548–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell G. L. et al. (1991) ‘Isolation of Jamestown Canyon Virus from Boreal Aedes Mosquitoes from the Sierra Nevada of California’, American Journal of Tropical Medicine and Hygiene, 44: 244–9.doi: 10.4269/ajtmh.1991.44.244 [DOI] [PubMed] [Google Scholar]
- Chang T. et al. (2021) ‘Arthropods and the Evolution of RNA Viruses’, bioRxiv.doi: 10.1101/2021.05.30.446314 [DOI] [Google Scholar]
- Charon J. et al. (2022) ‘RdRp-Scan: A Bioinformatic Resource to Identify and Annotate Divergent RNA Viruses in Metagenomic Sequence Data’, Virus Evolution, 8: veac082.doi: 10.1093/VE/VEAC082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coatsworth H. et al. (2021) ‘Intrinsic Variation in the Vertically Transmitted Insect-specific Core Virome of Aedes aegypti’, bioRxiv, 31: 2545–61. [DOI] [PubMed] [Google Scholar]
- Cobbin J. C. et al. (2021) ‘Current Challenges to Virus Discovery by Meta-transcriptomics’, Current Opinion in Virology, 51: 48–55. [DOI] [PubMed] [Google Scholar]
- Cross S. T. et al. (2020) ‘Partitiviruses Infecting Drosophila melanogaster and Aedes aegypti Exhibit Efficient Biparental Vertical Transmission’, Journal of Virology, 94: e01070–20.doi: 10.1128/jvi.01070-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dainat B. et al. (2009) ‘The Ectoparasitic Mite Tropilaelaps mercedesae (Acari, Laelapidae) as a Vector of Honeybee Viruses’, Insectes Sociaux, 56: 40–3.doi: 10.1007/s00040-008-1030-5 [DOI] [Google Scholar]
- De Geer C. (1776) ‘Mémoires pour servir à l’histoire des Insectes’, Tome 6. P., Hesselburg, Stockholm. [Google Scholar]
- Di Prisco G. et al. (2016) ‘A Mutualistic Symbiosis between A Parasitic Mite and A Pathogenic Virus Undermines Honey Bee Immunity and Health’, Proceedings of the National Academy of Sciences of the United States of America, 113: 3203–8.doi: 10.1073/PNAS.1523515113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-Muñoz S. L. (2019) ‘Uncovering Virus-virus Interactions by Unifying Approaches and Harnessing High-throughput Tools’, mSystems, 4: e00121–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietzgen R. G. et al. (2014) ‘Dichorhavirus: A Proposed New Genus for Brevipalpus Mite-transmitted, Nuclear, Bacilliform, Bipartite, Negative-strand RNA Plant Viruses’, Archives of Virology, 159: 607–19.doi: 10.1007/S00705-013-1834-0 [DOI] [PubMed] [Google Scholar]
- Dolja V. V., and Koonin E. V. (2018) ‘Metagenomics Reshapes the Concepts of RNA Virus Evolution by Revealing Extensive Horizontal Virus Transfer’, Virus Research, 244: 36–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Santos E. B. et al. (2016) ‘Mites (Acari: Trombidiformes) Parasitizing Mosquitoes (Diptera: Culicidae) in an Atlantic Forest Area in Southern Brazil with a New Mite Genus Country Record’, Experimental and Applied Acarology, 69: 323–33.doi: 10.1007/s10493-016-0045-2 [DOI] [PubMed] [Google Scholar]
- Ewels P. et al. (2016) ‘MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report’, Bioinformatics, 32: 3047–8.doi: 10.1093/bioinformatics/btw354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faizah A. N. et al. (2020) ‘Deciphering the Virome of Culex vishnui Subgroup Mosquitoes, the Major Vectors of Japanese Encephalitis, in Japan’, Viruses, 12: 1–26.doi: 10.3390/v12030264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauver J. R. et al. (2016) ‘West African Anopheles gambiae Mosquitoes Harbor a Taxonomically Diverse Virome Including New Insect-specific Flaviviruses, Mononegaviruses, and Totiviruses’, Virology, 498: 288–99. [DOI] [PubMed] [Google Scholar]
- Finn R. D., Clements J., and Eddy S. R. (2011) ‘HMMER Web Server: Interactive Sequence Similarity Searching’, Nucleic Acids Research, 39: W29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubler D. J. (1988) ‘The Arboviruses: Epidemiology and Ecology’. In: Monath TP (ed.) Dengue, Vol. II: pp. 223–60. CRC Press: Boca Raton, FL. [Google Scholar]
- Gurevich A. et al. (2013) ‘QUAST: Quality Assessment Tool for Genome Assemblies’, Bioinformatics, 29: 1072–5.doi: 10.1093/bioinformatics/btt086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn M. A. et al. (2020) ‘Characterization of Viruses in a Tapeworm: Phylogenetic Position, Vertical Transmission, and Transmission to the Parasitized Host’, The ISME Journal, 14: 1755–67.doi: 10.1038/S41396-020-0642-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey E., and Holmes E. C. (2022) ‘Diversity and Evolution of the Animal Virome’, Nature Reviews Microbiology, 20: 321–34.doi: 10.1038/s41579-021-00665-x [DOI] [PubMed] [Google Scholar]
- Hoang D. T. et al. (2018) ‘UFBoot2: Improving the Ultrafast Bootstrap Approximation’, Molecular Biology and Evolution, 35: 518–22.doi: 10.1093/molbev/msx281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubálek Z., Rudolf I., and Nowotny N. (2014) ‘Arboviruses Pathogenic for Domestic and Wild Animals’, Advances in Virus Research, 89: 201–75.doi: 10.1016/B978-0-12-800172-1.00005-7 [DOI] [PubMed] [Google Scholar]
- Hyatt D. et al. (2012) ‘Gene and Translation Initiation Site Prediction in Metagenomic Sequences’, Bioinformatics, 28: 2223–30. [DOI] [PubMed] [Google Scholar]
- Jagdale S. S., and Joshi R. S. (2018) ‘Enemies with Benefits: Mutualistic Interactions of Viruses with Lower Eukaryotes’, Archives of Virology, 163: 821–30. [DOI] [PubMed] [Google Scholar]
- Jones P. et al. (2014) ‘InterProScan 5: Genome-scale Protein Function Classification’, Bioinformatics (Oxford, England), 30: 1236–40.doi: 10.1093/bioinformatics/btu031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junglen S., and Drosten C. (2013) ‘Virus Discovery and Recent Insights into Virus Diversity in Arthropods’, Current Opinion in Microbiology, 16: 507–13.doi: 10.1016/J.MIB.2013.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan I., and Eubanks M. D. (2005) ‘Aphids Alter the Community-wide Impact of Fire Ants’, Ecology, 86: 1640–9.doi: 10.1890/04-0016 [DOI] [Google Scholar]
- Kassambara A. (2020). ‘Ggpubr: “Ggplot2” Based Publication Ready Plots’. <https://cran.r-project.org/package=ggpubr> accessed 04 2022.
- Kassambara A. (2021). ‘Rstatix: Pipe-friendly Framework for Basic Statistical Tests’. <https://rpkgs.datanovia.com/rstatix/> accessed 04 2022.
- Kopylova E., Noé L., and Touzet H. (2012) ‘SortMeRNA: Fast and Accurate Filtering of Ribosomal RNAs in Metatranscriptomic Data’, Bioinformatics, 28: 3211–7. [DOI] [PubMed] [Google Scholar]
- Kovalev S. Y., and Yakimenko V. V. (2021) ‘Kama Virus (KAMV) Is an Atypical Representative of the Seabird Tick-borne Flaviviruses’, Virus Genes, 57: 395–9. [DOI] [PubMed] [Google Scholar]
- Li C.-X. et al. (2015a) ‘Unprecedented Genomic Diversity of RNA Viruses in Arthropods Reveals the Ancestry of Negative-sense RNA Viruses’, eLife, 4: 1–26.doi: 10.7554/eLife.05378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D. et al. (2015b) ‘MEGAHIT: An Ultra-fast Single-node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph’, Bioinformatics, 31: 1674–6.doi: 10.1093/bioinformatics/btv033 [DOI] [PubMed] [Google Scholar]
- Liu S., Chen Y., and Bonning B. C. (2015) ‘RNA Virus Discovery in Insects’, Current Opinion in Insect Science, 8: 54–61.doi: 10.1016/J.COIS.2014.12.005 [DOI] [PubMed] [Google Scholar]
- Lundström J. et al. (2013) ‘The Geographic Distribution of Mosquito Species in Sweden’, Journal of the European Mosquito Control Association, 31: 21–35. [Google Scholar]
- Lvov D. K. et al. (2015) Zoonotic Viruses of Northern Eurasia: Taxonomy and Ecology, 1st edn. Oxford: Academic Press. [Google Scholar]
- Marcelino V. R. et al. (2020) ‘CCMetagen: Comprehensive and Accurate Identification of Eukaryotes and Prokaryotes in Metagenomic Data’, Genome Biology, 21: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev S. G., Aibulatov S. V., and Panyukova E. V. (2011) ‘Ecological Features and Distribution of the Mosquito Aedes communis (De Geer, 1776) in the Northwestern European Russia’, Entomological Review, 91: 1108–21.doi: 10.1134/S0013873811090041 [DOI] [PubMed] [Google Scholar]
- Mullen G. R., and O’Connor B. M. (2019) ‘Mites (Acari)’, Medical and Veterinary Entomology, 533–602.doi: 10.1016/B978-0-12-814043-7.00026-1 [DOI] [Google Scholar]
- Nayfach S. et al. (2020) ‘CheckV Assesses the Quality and Completeness of Metagenome-assembled Viral Genomes’, Nature Biotechnology, 39: 578–85.doi: 10.1038/s41587-020-00774-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L.-T. et al. (2015) ‘IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-likelihood Phylogenies’, Molecular Biology and Evolution, 32: 268–74.doi: 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu J. et al. (2019) ‘Three Novel RNA Viruses in the Spider Mite Tetranychus urticae and Their Possible Interactions with the Host RNA Interference Response’, Journal of Invertebrate Pathology, 166: 107228.doi: 10.1016/J.JIP.2019.107228 [DOI] [PubMed] [Google Scholar]
- Obbard D. J. (2018) ‘Expansion of the Metazoan Virosphere: Progress, Pitfalls, and Prospects’, Current Opinion in Virology, 31: 17–23.doi: 10.1016/J.COVIRO.2018.08.008 [DOI] [PubMed] [Google Scholar]
- Obbard D. J. et al. (2020) ‘A New Lineage of Segmented RNA Viruses Infecting Animals’, Virus Evolution, 6: vez061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Öhlund P. et al. (2019) ‘Viromics Reveal a Number of Novel RNA Viruses in Swedish Mosquitoes’, Viruses, 11: 1–18.doi: 10.3390/v11111027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paraskevopoulou S. et al. (2021) ‘Viromics of Extant Insect Orders Unveil the Evolution of the Flavi-like Superfamily’, Virus Evolution, 7: veab030.doi: 10.1093/ve/veab030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parratt S. R., and Laine A. L. (2016) ‘The Role of Hyperparasitism in Microbial Pathogen Ecology and Evolution’, The ISME Journal, 10: 1815–22.doi: 10.1038/ismej.2015.247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng P. et al. (2013) ‘A Color-mediated Mutualism between Two Arthropod Predators’, Current Biology, 23: 172–6.doi: 10.1016/J.CUB.2012.11.057 [DOI] [PubMed] [Google Scholar]
- Pettersson J. H. O. et al. (2017) ‘Characterizing the Virome of Ixodes ricinus Ticks from Northern Europe’, Scientific Reports, 7: 1–7.doi: 10.1038/s41598-017-11439-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson J. H. O. et al. (2019) ‘Meta-transcriptomic Comparison of the RNA Viromes of the Mosquito Vectors Culex pipiens and Culex torrentium in Northern Europe’, Viruses, 11: 1–20.doi: 10.3390/V11111033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poinar G., and Poinar R. (1998) ‘Parasites and Pathogens of Mites’, Annual Review of Entomology, 43: 449–69.doi: 10.1146/annurev.ento.43.1.449 [DOI] [PubMed] [Google Scholar]
- Porter A. F. et al. (2021) ‘Metagenomic Identification of Viral Sequences in Laboratory Reagents’, Viruses, 13: 1–13.doi: 10.3390/v13112122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozojević I. et al. (2019) ‘Is the Spatial Distribution of Lentic Water Mite Assemblages (Acari: Hydrachnidia) Governed by Prey Availability?’, Experimental and Applied Acarology, 77: 487–510. [DOI] [PubMed] [Google Scholar]
- R Core Team (2021) ‘R: A Language and Environment for Statistical Computing’. Vienna, Austria. [Google Scholar]
- Rice P., Longden L., and Bleasby A. (2000) ‘EMBOSS: The European Molecular Biology Open Software Suite’, Trends in Genetics: TIG, 16: 276–7.doi: 10.1016/S0168-9525(00)02024-2 [DOI] [PubMed] [Google Scholar]
- Roundy C. M. et al. (2017) ‘Insect-specific Viruses: A Historical Overview and Recent Developments’, Advances in Virus Research, 98: 119–46.doi: 10.1016/BS.AIVIR.2016.10.001 [DOI] [PubMed] [Google Scholar]
- Santillán M. et al. (2015) ‘New Hosts for the Mite Ornithonyssus bursa in Argentina’, Medical and Veterinary Entomology, 29: 439–43.doi: 10.1111/MVE.12129 [DOI] [PubMed] [Google Scholar]
- Shen M. et al. (2005) ‘The Role of Varroa Mites in Infections of Kashmir Bee Virus (KBV) and Deformed Wing Virus (DWV) in Honey Bees’, Virology, 342: 141–9. [DOI] [PubMed] [Google Scholar]
- Shi C. et al. (2019) ‘Stable Distinct Core Eukaryotic Viromes in Different Mosquito Species from Guadeloupe, Using Single Mosquito Viral Metagenomics’, Microbiome, 7: 1–20.doi: 10.1186/s40168-019-0734-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M. et al. (2016a) ‘Redefining the Invertebrate RNA Virosphere’, Nature, 540: 539–43.doi: 10.1038/nature20167 [DOI] [PubMed] [Google Scholar]
- Shi M. et al. (2016b) ‘Divergent Viruses Discovered in Arthropods and Vertebrates Revise the Evolutionary History of the Flaviviridae and Related Viruses’, Journal of Virology, 90: 659–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2018) ‘No Detectable Effect of Wolbachia wMel on the Prevalence and Abundance of the RNA Virome of Drosophila melanogaster’, Proceedings of the Royal Society B: Biological Sciences, 285: 20181165.doi: 10.1098/rspb.2018.1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M., Zhang Y. Z., and Holmes E. C. (2018) ‘Meta-transcriptomics and the Evolutionary Biology of RNA Viruses’, Virus Research, 243: 83–90.doi: 10.1016/j.virusres.2017.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokarz R. et al. (2014) ‘Virome Analysis of Amblyomma americanum, Dermacentor variabilis, and Ixodes scapularis Ticks Reveals Novel Highly Divergent Vertebrate and Invertebrate Viruses’, Journal of Virology, 88: 11480–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiente Moro C., Chauve C., and Zenner L. (2005) ‘Vectorial Role of Some Dermanyssoid Mites (Acari, Mesostigmata, Dermanyssoidea)’, Parasite, 12: 99–109.doi: 10.1051/parasite/2005122099 [DOI] [PubMed] [Google Scholar]
- Vasquez A. A. et al. (2020) ‘The Biodiversity of Water Mites that Prey on and Parasitize Mosquitoes’, Diversity, 12: 1–26.doi: 10.3390/D12060226 [DOI] [Google Scholar]
- Wang L. et al. (2021) ‘Establishment of Culex modestus in Belgium and a Glance into the Virome of Belgian Mosquito Species’, mSphere, 6: e01229–20.doi: 10.1128/msphere.01229-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver S. C., and Reisen W. K. (2010) ‘Present and Future Arboviral Threats’, Antiviral Research, 85: 328–45.doi: 10.1016/j.antiviral.2009.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster C. L. et al. (2015) ‘The Discovery, Distribution, and Evolution of Viruses Associated with Drosophila melanogaster’, PLoS Biology, 13: e1002210.doi: 10.1371/journal.pbio.1002210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werblow A. et al. (2015) ‘Hyperparasitism of Mosquitoes by Water Mite Larvae’, Parasitology Research, 114: 2757–65.doi: 10.1007/s00436-015-4482-3 [DOI] [PubMed] [Google Scholar]
- Wickham H. (2016) Ggplot2: Elegant Graphics for Data Analysis. New York: Springer. [Google Scholar]
- Wu H. et al. (2020) ‘Abundant and Diverse RNA Viruses in Insects Revealed by RNA-Seq Analysis: Ecological and Evolutionary Implications’, mSystems, 5: e00039–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z. et al. (2022) ‘Virome of Bat-infesting Arthropods: Highly Divergent Viruses in Different Vectors’, Journal of Virology, 96: e01464–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G. et al. (2017) ‘Ggtree: An R Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data’, Methods in Ecology and Evolution, 8: 28–36.doi: 10.1111/2041-210X.12628 [DOI] [Google Scholar]
- Yu X.-J., and Tesh R. B. (2014) ‘The Role of Mites in the Transmission and Maintenance of Hantaan Virus (Hantavirus: Bunyaviridae)’, Journal of Infectious Diseases, 210: 1693–9.doi: 10.1093/infdis/jiu336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.-Z., Shi M., and Holmes E. C. (2018) ‘Using Metagenomics to Characterize an Expanding Virosphere’, Cell, 172: 1168–72.doi: 10.1016/j.cell.2018.02.043 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.