

Abstract
Neural tube closure (NTC) is crucial for proper development of the brain and spinal cord and requires precise morphogenesis from a sheet of cells to an intact three-dimensional structure. NTC is dependent on successful regulation of hundreds of genes, a myriad of signaling pathways, concentration gradients, and is influenced by epigenetic and environmental cues. Failure of NTC is termed a neural tube defect (NTD) and is a leading class of congenital defects in the United States and worldwide. Though NTDs are all defined as incomplete closure of the neural tube, the pathogenesis of an NTD determines the type, severity, positioning, and accompanying phenotypes. In this review we survey pathogenesis of NTDs relating to disruption of cellular processes arising from genetic mutations, altered epigenetic regulation, and environmental influences by micronutrients and maternal condition.
1. INTRODUCTION
Neural tube defects
NTDs are devastating congenital defects which affect approximately 300,000 babies each year, 88,000 of which will be deadly, and the remaining cases often will experience significant impacts to the length and quality of life (Zaganjor et al., 2016). These statistics are most likely on the low side, as less than half of the 194 World Health Organization member states report data on NTDs. The outcome of an NTD depends heavily on where the neural folds fail to close. Failure to close in the developing brain causes exencephaly, which ultimately results in loss of brain tissue and anencephaly, which is lethal (Fig1 A,B)(Wilde et al., 2014). Failure to close more caudally leads to spina bifida or myelomeningocele (Fig 1 C,D), which has much better outcomes but the lifetime cost of care for spina bifida can be up to $700,000 (Grosse et al., 2016). Spina bifida occulta conversely is a mild defect caused by a gap in bone(s) of the spine and the neural tissue is not exposed
Figure 1: NTD phenotypes.
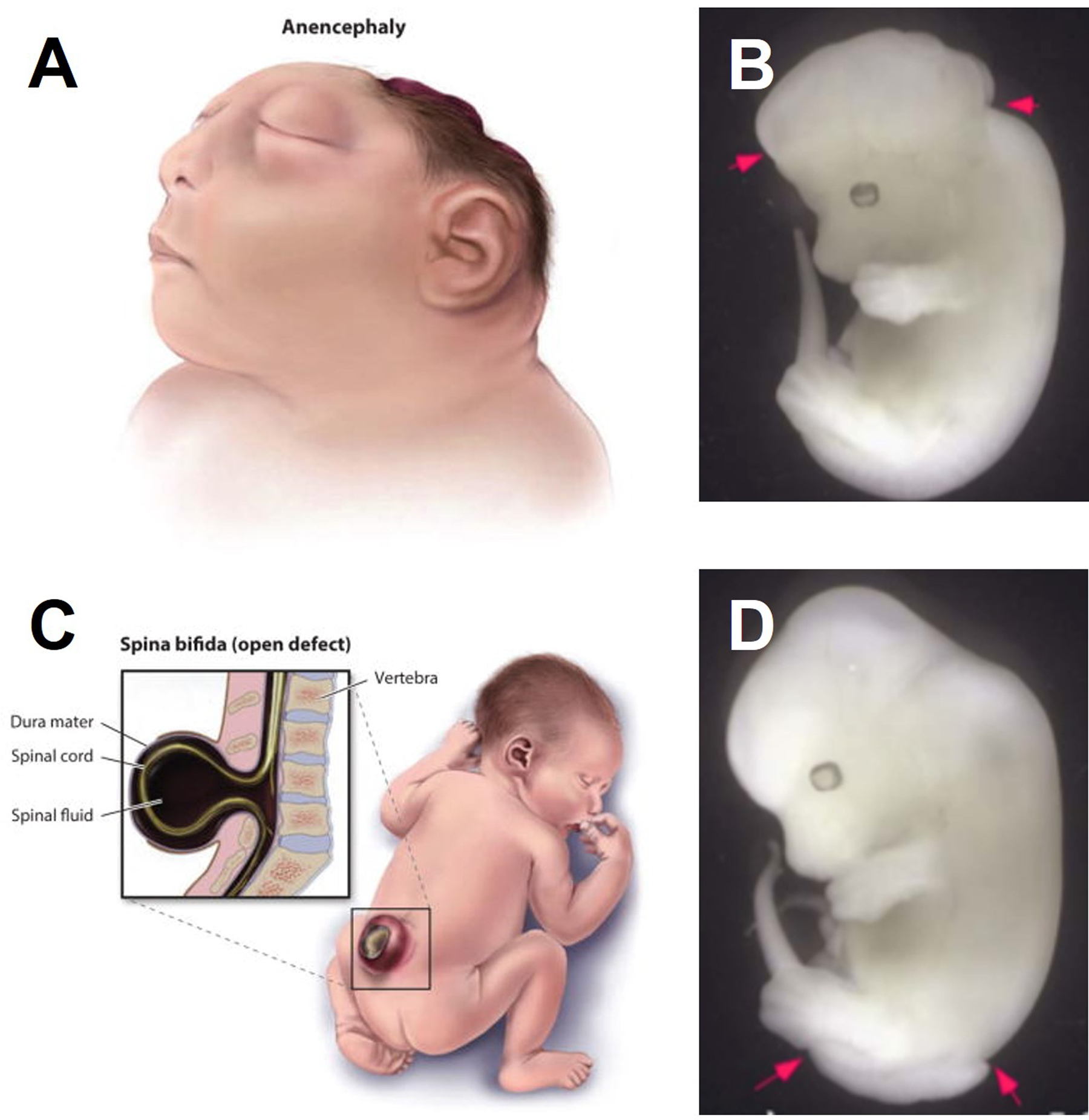
A) Human anencephaly B) Mouse exencephaly C) Human spina bifida D) Mouse spina bifida
Fig 1A,C from Wilde, 2014
Neural tube closure
This review emphasizes specific cellular processes and tissues that mediate the major steps of NTC, which are briefly covered here. Formation of the neural tube begins in humans at approximately day 18 and is completed by the fourth week of pregnancy (O’Rahilly & Müller, 1994). The process of NTC is similar in mice but starts at approximately embryonic day 8 and is completed by day 10 (Gray & Ross, 2011). Formation of the neural tube which will become the brain and most of the spinal cord is completed by a process called primary NTC, while neural tube formation below the sacral region is completed by secondary neural tube formation. The process of primary NTC begins with a flat sheet of epithelial cells overlying the notochord and mesoderm (Fig. 2A). Neural induction specifies the neuroepithelium (NE, green) from the non-neural ectoderm (NNE or surface ectoderm; blue). The first major morphogenetic event is cell intercalation which drives NE cells toward the midline resulting in medial-lateral narrowing and rostral-caudal lengthening of the NE. Morphogenesis continues through the elevation of the neural folds (Fig. 2B,C) which is driven by proliferation within the mesoderm and NE, and polarized actomyosin contractions which create hinge points at the medial and dorsolateral positions to shift the rising neural folds from a convex to concave orientation (Fig. 2C). As the neural folds approach each other, the NE and NNE cells separate, cell protrusions extend across the gap and each cell layer adheres with its partner tissue on the opposing fold (called fusion) to form a closed neural tube covered by NNE. Arising at the border between the NE and NNE are pre-migratory neural crest cells, which begin to delaminate during this last step of NTC and migrate to become neural crest cells. This process of NT closure does not occur simultaneously along the rostral-caudal axis. In humans, closure initiates at two discrete sites: one initiation site lies at the forebrain and zippers backwards towards the hindbrain, the other initiation point occurs at the hindbrain/ cervical boundary and zippers both rostrally towards the midbrain and caudally towards the sacral region where secondary neural tube formation will occur (O’Rahilly & Müller, 2002). Mice utilize a third closure point which lies between these two, and zippers both rostrally and caudally (Nikolopoulou et al., 2017). The following sections will analyze individual cellular processes which can fail and lead to an NTD. The overwhelming majority of examples are contextualized within the process of primary neural tube closure, as most NTDs result from defects in primary NTC. Secondary neural tube formation, which occurs in the most caudal region, is not accomplished by large morphogenetic changes, and instead relies on the condensation of tail-bud mesenchyme and subsequent migration and epithelialization to form a lumen (Catala, 2021).
Figure 2: Process of NTC.
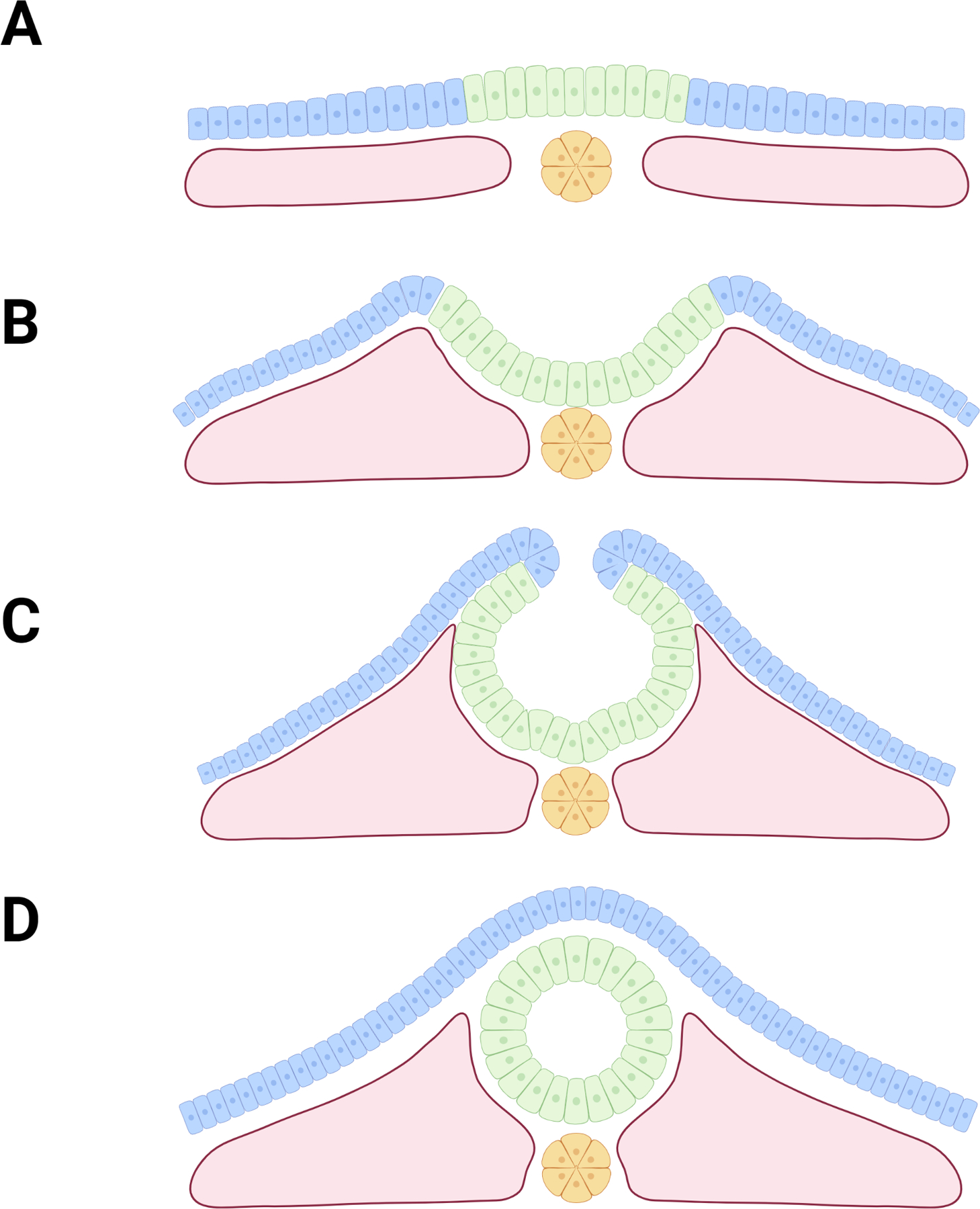
A) NTC begins with a flat sheet of epithelial cells (NE cells shown in green, NNE cells shown in blue) overlaying the notochord (yellow) and mesoderm (pink). B) Convergent extension and proliferation drive the bulging of the mesoderm and formation of neural folds. C) Actomyosin contractions form medial and dorsolateral hinge points as neural folds bend and approach one another. D) Neural folds adhere to partner tissue and fuse, forming intact neural tube covered by NNE
2. CELLULAR PROCESSES UNDERLYING NTDS
NTC is a highly orchestrated process which requires coordination between a myriad of signaling pathways and cellular behaviors. NTC is driven by a multitude of genes, which presents many points of failure leading to NTDs. When attempting to understand the causes of neural tube defects, one can start by surveying the known genetic perturbations which can manifest as NTDs. Animal models that show NTD phenotypes have been crucial for elucidating mechanism of NTC and gene action. Indeed, over 300 genes in mice play critical roles in NTC and over 80 genes in human are associated with NTDs to date (Kakebeen & Niswander, 2021; Wilde et al., 2014). However, most of the animal studies have investigated the effect of single gene, and often homozygous mutations, whereas the risk in humans of presenting with an NTD is multi-factorial and often polygenic, rather than simple mendelian inheritance due to single gene mutations (Lee & Gleeson, 2020; Neumann et al., 1994; Zohn, 2012). In other words, human NTDs are thought to be due to interactions between multiple gene variants and genetic modifiers. Nonetheless, we can learn much about the broad cellular processes required for NTC as highlighted by individual gene examples. Although there is not a strict correspondence between the genes associated with NTDs in mouse with those discovered from human NTD cases to date, there is excellent correspondence when considered in the context of the biological processes affected, such as outlined in the rest of this review. As we consider the genetic contributions towards NTDs, we will be drawing upon animal models to contextualize the cellular processes involved in NTC, each with their own tolerances to perturbations and failure points which may result in NTDs.
2.1. Proliferation
The process of NTC involves extensive morphogenetic changes to the embryo, accommodated in part by spatially and temporally directed increases in cell division. Too little proliferation can result in not enough material to form an intact neural tube, whereas too much proliferation can change the tissue geometry and inhibit closure. Proliferation defects often manifest during primary NTC, excluding Xenopus where proliferation is not an important component of early embryogenesis (Nikolopoulou et al., 2017). In chick and mouse models and human NTC, all embryonic cells are rapidly proliferating at this stage. Focusing on the tissues that contribute to neural tube formation (neuroepithelium, non-neural ectoderm, mesoderm), proliferation within these tissues needs to keep pace with one another for successful NTC.
Perturbation of proliferation within the NE cells of the closing neural tube is represented by many NTD models (Harris & Juriloff, 2010). This includes defects in canonical WNT/β-catenin signaling, which plays a key role in maintaining proliferative balance within the neural plate. Mouse lines which harbor Pax3 mutations, a downstream effector of WNT/β-catenin, display both exencephaly and spina bifida - highlighting its importance spanning the rostro-caudal axis (Epstein et al., 1991). Pax3 mutant mice display decreased proliferation and premature neuronal differentiation of dorsal NE cells - suggesting that WNT and PAX3 work to maintain the NE in a proliferative state (Fig. 3A) (Sudiwala et al., 2019). Loss of proliferation capacity in Pax3 mutant mice prevents the neural folds from meeting, disrupting zippering of the mid and hindbrain neural folds (Fleming & Copp, 2000). Furthermore, the spatial restriction of PAX3 within the dorsal NE highlights the importance of regulated proliferation, even within similarly fated cells. This restriction of PAX3 dorsally is accomplished in part by the CDX family of proteins which interact with posteriorizing WNT signals and act as inducers for Pax3 transcription, the loss of which results in a truncated anteroposterior axis, fused somites, and craniorachischisis (Ferras et al., 2012; Ramakrishnan et al., 2021; Savory et al., 2011). CDX2, which localizes with PAX3 dorsally, seems to be particularly important as it also regulates PTK7 which interacts with multiple WNT receptors to fine-tune canonical and non-canonical WNT signaling (Berger et al., 2017; Hayes et al., 2013). The loss of Ptk7 mimics phenotypes found in Cdx1/2 conditional knockouts and results in 100% penetrance of craniorachischisis (Lu et al., 2004; Savory et al., 2011; M. Wang et al., 2015). Pax3 restriction to the dorsal NE is also controlled by ventral SHH signaling, which attenuates WNT signaling through inhibition of ZIC2 (Sanchez-Ferras et al., 2014).
Figure 3: Disruption of NE proliferation.
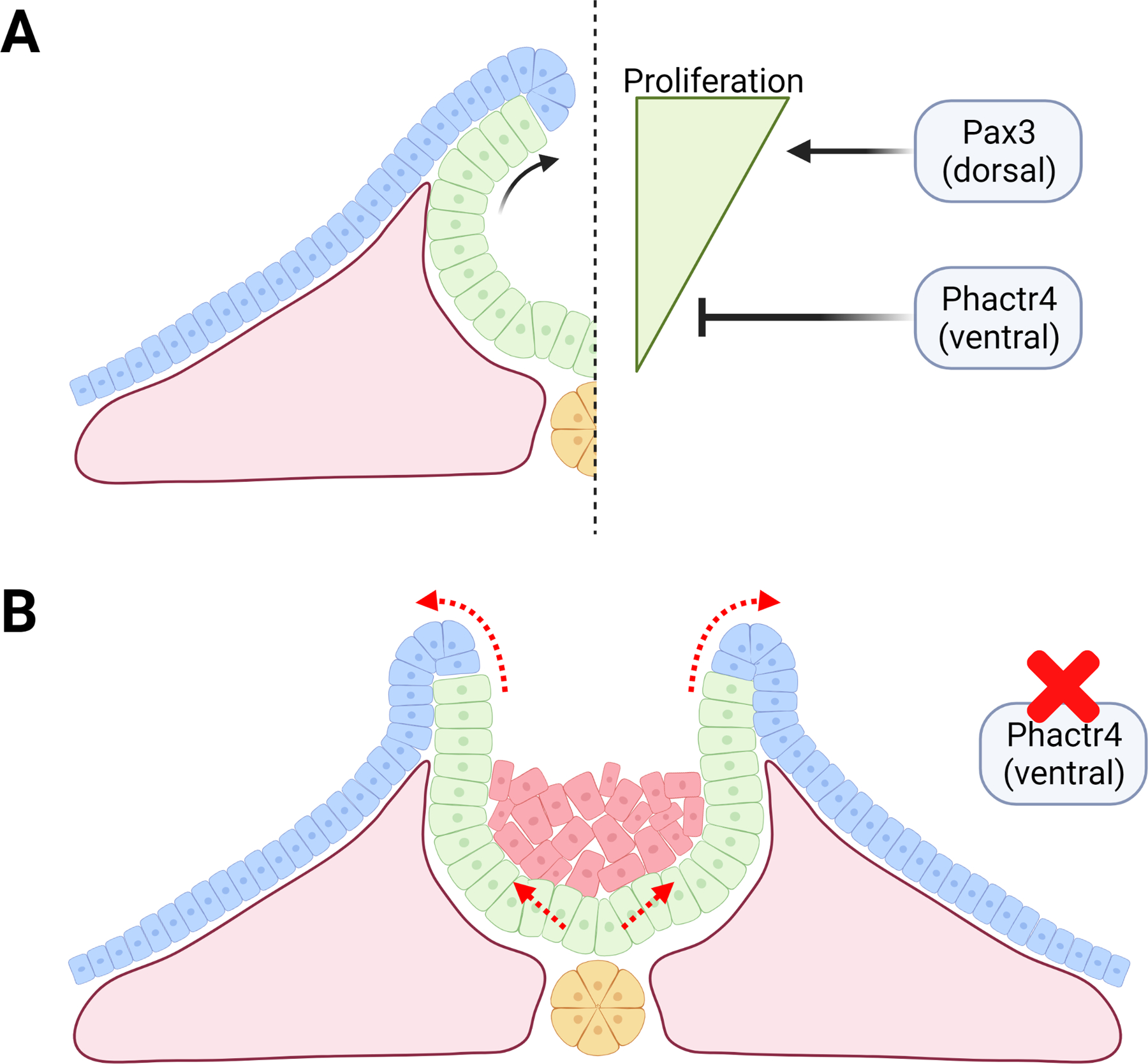
A) Illustration of proper balance of dorsal/ventral proliferation. Pax3 acts to increase proliferation dorsally, while Phactr4 acts to inhibit proliferation ventrally. B) Mice lacking Phactr4 display abnormal ventral NE proliferation (unexpected cells shown in red). Buildup of cells ventrally changes the geometry, which does not allow proper neural fold bending and inhibits closure.
There is a gradient of proliferation along the dorsal-ventral axis of the NE at the time of NTC with higher proliferation on the dorsal side through WNT/PAX3 and lower rate of proliferation of the ventral side (Fig. 3A). PHACTR4 normally serves to restrict proliferation in the ventral NE at the time of NTC. Phactr4 mutation in mice results in faster cell cycle in NE cells leading to too many cells, especially in the ventral neural tube, and exencephaly (Fig. 3B) (Kim et al., 2007). Similarly, mice harboring mutations of the Nap1/2 genes experience increased mitotic events in neuronal precursors with exencephaly and spina bifida correlated with an overabundance of NE cells (Rogner et al., 2000).
Disrupted proliferation within the NE is also represented by NTD animal models whose mutations affect proliferation more indirectly by causing cells to prematurely differentiate and therein reducing their proliferative capacity. Several of these models fall within the Notch pathway, which works to ensure that NE specified cells retain proliferative capacity until NTC is complete (Hatakeyama & Kageyama, 2006). Notch itself is a marker of neuronal differentiation, however components of the Notch pathway act to inhibit Notch signaling and retain NE cells in a proliferative state (Engler et al., 2018). When components of the Notch pathway (Hes1, Hes3, Rbpj) are perturbed, Notch expression is increased, NE cells prematurely differentiate, resulting in either spina bifida or exencephaly (Harris & Juriloff, 2007; Ishibashi et al., 1995). Premature differentiation has also been proposed to cause NTDs through mechanical stiffening of the cells, which might disrupt dorsolateral hinge point bending, interfere with neural crest cell release, or inhibit adhesion between neural folds (Copp et al., 2003). An argument can be made that all three may be simultaneously possible. ZIC2 plays roles in both the roof plate cells and in regulating dorsolateral hinge point bending which is necessary for fold adhesion (Ybot-Gonzalez et al., 2007). Interestingly Zic2 is regulated along with Notch by the transcription factor FOXD5, with feedback from Notch signaling regulating Foxd5 expression itself (B. Yan et al., 2009). In disrupted Notch signaling mouse models, NTDs might occur from either premature neuronal differentiation alone, or alongside FOXD5 feedback associated with ZIC2 perturbation. This aligns with observations in Zic2 mutant mice and zebrafish, which display lack of dorsolateral hinge point bending (Nyholm et al., 2009; Ybot-Gonzalez et al., 2007).
Looking beyond NE progenitor cells, NTD models associated with disrupted proliferation in other tissues are fewer in number. Within the mesoderm for example, cell migration seems to play a larger role, yet proper proliferative capacity is necessary to expand the mesoderm population and to form the mesenchyme bulges which will help elevate the neural folds (Fig. 4A) (Nikolopoulou et al., 2017). Twist1 and Cart1 knockout lines demonstrate reduced proliferative capacity within the mesenchyme, interfering with the formation of the biconvex neural folds and resulting in exencephaly (Fig. 4B) (Z. F. Chen & Behringer, 1995; Zhao et al., 1996, p. 1). It is noteworthy though that caudal NTC appears to be less sensitive to disruption of the mesoderm, as demonstrated by physical depletion of the paraxial mesoderm still allowing subsequent NTC (van Straaten et al., 1993).
Figure 4: Disruption of mesoderm proliferation.
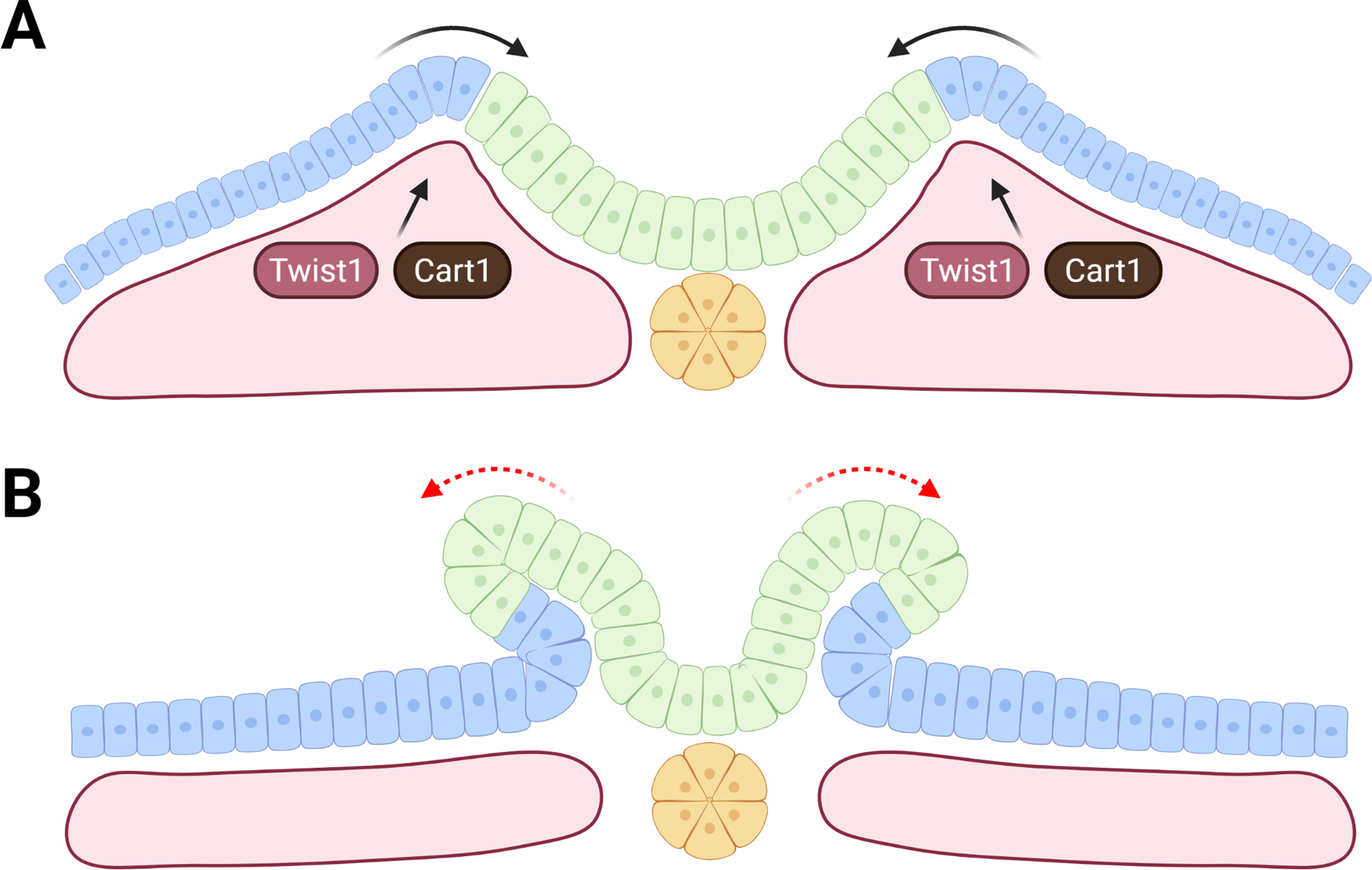
A) Illustration of proper mesoderm proliferation and migration, highlighting Twist1 and Cart1 as necessary for bulging of mesoderm. B) Loss of Twist1 or Cart1 causes NE proliferation to outpace mesoderm growth, preventing the neural folds to adopt a concave orientation.
Proliferative defects in non-neural ectoderm cells (NNE) are even more rare, with cell structural changes and adhesion playing a larger role. Mutations which lengthen the cell cycle (Gadd45a, TrP53) can reduce overall cell numbers in both the NE and NNE leading to an NTD, albeit with relatively low penetrance (Harris & Juriloff, 2007). These mutations have also been proposed to reduce genomic stability, which could lead to cell death and reduced cell numbers (Hollander et al., 1999). In more caudal regions, reduction of Grhl3 is associated with an imbalance of proliferation between the NE and caudal hindgut resulting in spina bifida (Gustavsson et al., 2007). These phenotypes associated with Grhl3 mutation may actually be localized to uncommitted stem-like cells along the neural plate border as this phenotype can be rescued when paired with a deficiency of Dkk1/Kremen1 – canonical WNT signaling antagonists which promote NE specification within uncommitted neural plate border cells (Kimura-Yoshida et al., 2015).
2.2. Migration
Cell migration plays a key role in the processes of: convergent-extension which shapes the neural plate, mesoderm migration to help elevate the neural folds, clearance of neural crest cells, and lumen formation in secondary neural tube formation.
During early stages of primary NTC, the first key cell migration step involves intercalation of cells toward the midline, which narrows and lengthens the neural plate along the medial-lateral axis and the rostral-caudal axis, respectively (Fig. 5A,B). Animal NTD models in which these convergent-extension movements are disrupted display a wide neural plate and ultimately a large gap between the neural folds such that the folds cannot meet in the midline (Fig. 5C)(Harris & Juriloff, 2010). This cell migration is heavily dependent on planar cell polarity (PCP) signaling and the response seems to be tissue dependent (Shindo & Wallingford, 2014). In the mesenchyme, migration is accomplished by shortening of cell-cell junctions and cellular protrusions which exert traction to produce a crawling motion, with the orientation of the protrusions dependent on the distance from the midline mesoderm and notochord (Butler & Wallingford, 2018; Ezin et al., 2006). The positioning of the protrusions which drive intercalation is thought to be regulated by the non-canonical WNT-Frizzled PCP pathway, with extension of the protrusions regulated downstream of WNT in both a Dishevelled-dependent and independent manner (Keller, 2002). Mutations which disrupt the Dishevelled pathway in Xenopus affect convergent-extension and are marked by more but less stable and randomly oriented protrusions (Wallingford & Harland, 2002). In the neural plate, cell migration is also thought to be controlled by PCP signaling, but instead seems to be mainly accomplished by shortening of cell-cell junctions (Nishimura et al., 2012). This is accomplished by pulsed actomyosin contractions that are restricted to or enriched at mediolaterally oriented cell-cell junctions (Butler & Wallingford, 2018). These actomyosin contractions will be discussed further in the cell structure section but are dependent upon polarization within the cell. NTD mice deficient for PCP regulators PTK7 and Vangl2 display varying degrees of polarity loss, and NE cells in both cases fail to apically constrict (M. Williams et al., 2014), leading to the most severe form of NTD called craniorachischisis in which the neural folds fail to meet in the midline along most of the rostral-caudal axis. Similarly in humans, mutations in the non-canonical WNT-Frizzled PCP pathway are associated with NTDs, including craniorachischisis (Kibar et al., 2001).
Figure 5: Disruption of cell migration & intercalation.
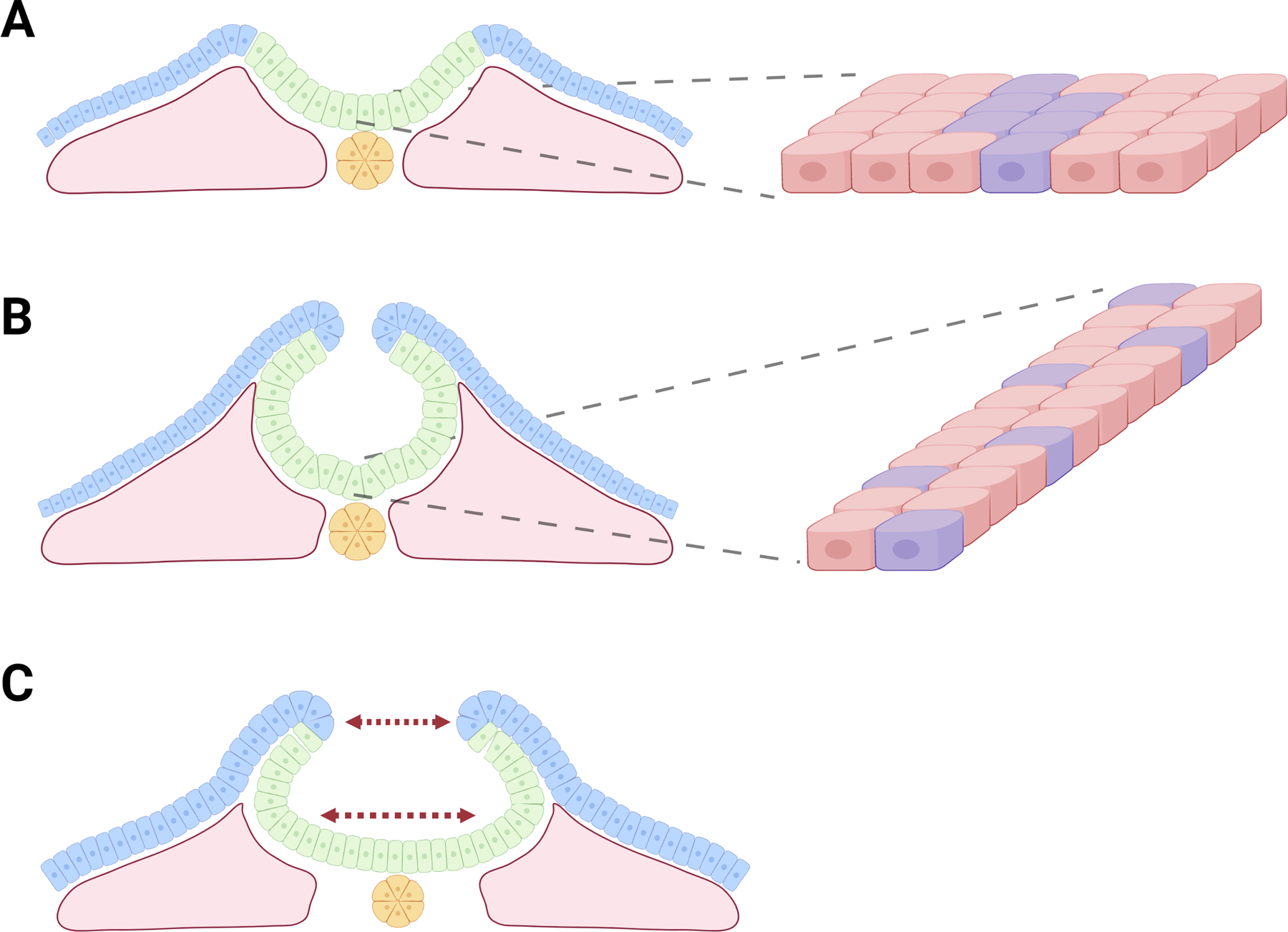
A) Illustration of NE cell sheet with selected cells marked in purple. B) Convergent extensions movements drive the narrowing and lengthening of the cell sheet due to cell migration and intercalation. C) Disruptions to cell migration & intercalation result in a wide neural plate which prevents contact of neural folds.
Completion of primary NTC requires that neural plate border cells, also referred to as pre-migratory neural crest cells – delaminate from the intact neural tube and NNE. Failure of neural crest cells to migrate can lead to NTDs, which might stem from inhibition of neural fold bending (Copp et al., 2003). The process of neural crest formation and migration requires neural plate border cells to undergo epithelial to mesenchymal transition (EMT). EMT is highlighted by loss of adherens junctions (specifically E-cadherin together with Catenins and actin rings), the loss of cell polarity, separation from surrounding epithelial tissue (delamination), and migration through extracellular matrix to adjacent tissues (Acloque et al., 2009). In mice, neural crest cells which overexpress the gap junction protein Connexin 43 fail to migrate which leads to exencephaly (Ewart et al., 1997). Furthermore, sequestration of chondroitin sulphate proteoglycans (a family of molecules important in regulating adhesion) in rats prevents the disruption of fibronectin-containing ECM necessary for neural crest migration, which delays dorsolateral bending of the neural folds (Morriss-Kay & Tuckett, 1989). As neural crest cells can be observed detaching from the neural folds before NTC is complete, it has been suggested that the failure of neural plate border cells to differentiate and thus migrate may inhibit the transition from biconvex to biconcave morphology of the neural plate (Copp et al., 2003).
In secondary neural tube formation, evidence also points towards the importance of cell migration and intercalation. Mesodermal cells undergo a mesenchymal to epithelial transition (MET) to form an epithelial tube that then fuses with the neural tube formed by primary NTC. When the mesodermal cells are prevented from undergoing epithelialization, the cells fail to participate in neural tube formation, and instead accumulate in the neural canal (Nakaya et al., 2004) Chick embryos in which SMAD3 is disrupted, a receptor involved in Transforming Growth Factor β (TGF-β) signaling, display disrupted secondary neural tube formation marked by multiple small lumen (Gonzalez-Gobartt et al., 2021). SMAD3 seems unique among other SMAD proteins in directing cell migration during secondary neural tube formation (Gonzalez-Gobartt et al., 2021). TGF-β signaling has been previously implicated in chick brain medial hinge point (MHP) formation (Nikolopoulou et al., 2017). Together these data have intriguing implications for possible cell migration & intercalation in the formation of the MHP in chick and mouse, as SMAD2 & SMAD3 are highly active at the MHP while other SMAD proteins involved in BMP signaling are down regulated (Amarnath & Agarwala, 2017). Furthermore, a role for cell migration in MHP formation may be obfuscated by the ability of dorsolateral hinge point bending to overcome the loss of MHP in mice (Ybot-Gonzalez et al., 2002).
2.3. Cell death
With NTC being a highly proliferative process, the subsequent death of some of these newly formed cells may seem counter-intuitive or even counter-productive. Indeed, the involvement of apoptosis in NTC is still a subject of debate, with evidence that it may be dispensable in the context of mammalian NTC (Massa et al., 2009). Still, cell death is observed and may be necessary within the neural folds (Cecconi et al., 1998; Urase et al., 2003). Loss of apoptosis in mouse models has been observed to accompany disrupted neural fold bending and delayed NTC in the mid- and hind-brain (Yamaguchi et al., 2011). Regardless of the necessity of apoptosis for NTC, there is evidence that abnormally increased apoptosis can underlie NTDs in animal models, and this can result from genetic insult, nutritional deficiencies, or pharmacological insults (Kakebeen & Niswander, 2021).
Arguments for the necessity of apoptosis in NTC are supported by a myriad of mice NTD models (mutations in Mapk8/Mapk9, Apaf1, Casp3, Casp9, and Cytc) which result in decreased apoptosis and lead to midbrain exencephaly. Most of these mutant also show forebrain protrusion or cauliflower-like overgrowth not observed in other mouse NTD models (Harris & Juriloff, 2007). In Xenopus, the pro-apoptotic function of Barhl2 has been demonstrated to be crucial for NTC, and is thought to normally limit the number of Chordin- and Shh-expressing cells in the prospective notochord and floorplate (Juraver-Geslin & Durand, 2015).
On the other hand, increased apoptosis can also increase the risk for NTDs. Analysis of human NTD autopsies found that hallmarks of anencephaly cases include higher amounts of apoptotic cells, higher expression of p53, and lower expression of Pax3 (L. Wang, Lin, Yi, et al., 2017). In human spina bifida autopsies, Pax3 is diminished, yet p53 expression and the number of apoptotic cells resemble controls, suggesting that rostral NTC may be more sensitive than caudal NTC to increases in apoptosis (L. Wang, Lin, Yi, et al., 2017). Within animal NTD models, increased apoptosis is represented in Bcl10 and MEKK4 mutant mice (Harris & Juriloff, 2007). MEKK4 is specifically found in NE cells, and while MEKK4 deficient cells demonstrate massively elevated apoptosis and highly penetrant NTDs, proliferation of NE cells seems to be largely unaffected (Chi et al., 2005). Alteration in the levels of micronutrients can lead to increased cell apoptosis and NTDs. For example, treating mouse embryos at the time of NTC with TPEN, a zinc chelator to model zinc deficiency, leads to stabilization of p53, increased apoptosis and failed NTC. The TPEN-induced NTD can be rescued by pharmacological inhibition of p53 or overexpression of a E3 ubiquitin ligase which targets p53 for degradation (H. Li, Zhang, & Niswander, 2018). These experiments not only demonstrated a mechanism for zinc-deficiency associated NTD risk, but also provide evidence that increased apoptosis can drive NTDs as opposed to simply accompanying them. Perhaps in a similar fashion, an excess of the drug all-trans-retinoic acid (RA) has also been demonstrated to induce caudal NTDs in mice and rats, which is accompanied by an increase in apoptosis (Kakebeen & Niswander, 2021; Wei et al., 2012). Notably, as discussed later, increased apoptosis is thought to be a driver of diabetes associated NTDs(Gao & Gao, 2007).
2.4. Structural Changes
NTC relies upon proper proliferation, cell death, and migration as previously discussed, but these cellular processes in turn are often dependent on cell structural changes. The directionality of cell migration, oriented cell division, actomyosin constriction, and cell shape is highly dependent on the cell polarity (Nikolopoulou et al., 2017). It is not surprising then that a large number of NTD animal models are rooted in perturbed planar cell polarity (PCP) signaling (Harris & Juriloff, 2007; Juriloff & Harris, 2012).
In the context of the mesenchyme, we have already discussed disruptions to the Disheveled pathway which randomize cellular protrusions and prevent cell crawling (Wallingford & Harland, 2002). Mutations to Ptk7 and Vangl2 similarly disrupt convergent extension in both the mesenchyme and NE by disrupting cell-cell junction shortening required for cell migration (M. Williams et al., 2014). The proper cell-cell junction regulation in these mutants is dependent on restriction of actomyosin contractions to the mediolateral junctions. This restriction appears to be driven by the polarity of the cell imparted by asymmetric enrichment of PCP proteins Vangl2 and Prickle2 (Butler & Wallingford, 2018). Knockdown of Prickle2 in Xenopus, which is spatiotemporally coordinated at shrinking junctions, disrupts convergent extension (Butler & Wallingford, 2018). Though Prickle2 is not associated with NTDs in human, it is noteworthy that the gene family member Prickle1 has multiple documented mutations in human patients which are thought to pre-dispose to NTD risk (Bosoi et al., 2011). Though Vangl2 and Prickle2 may contribute to the actomyosin contractions, the directionality of this enrichment is thought to be influenced by the Fat/Dsch/Fjx1 pathway (Badouel et al., 2015). Fat1–4 and Dsch1/2 are atypical cadherins which are phosphorylated by Fjx1 to affect their binding affinity which is proposed to contextualize local cell behaviors to a larger axial orientation (Mangione & Martín-Blanco, 2018). The exact interaction between the Fat/Dsch/Fjx1 pathway and PCP signaling is still in question, however Fat1−/− mice display forebrain NTDs and Fat4−/− mice exhibit caudal defects and a broad spinal cord (Ciani et al., 2003; Nikolopoulou et al., 2017; Saburi et al., 2008).
In addition to the need for cell-cell junction shortening due to PCP signaling in the neural plate, NE cells contain unique points for cell structural regulation and failure. The formation of the dorsal-lateral-hinge point (DLHP) is an intersection of several such pathways (Fig. 6A). Bending of the neural plate in more rostral regions is dependent on actomyosin contractions, however, this regulation differs from NE cell-cell junction shortening (Haigo et al., 2003). The myosin contraction is carried out by the small GTPase RhoA (Arnold et al., 2018). Though there is no RhoA NTD model, Gef-H1 and RhoGap which act upstream in the neural folds both display severe exencephaly phenotypes accompanied by significant basal actin accumulation (Brouns et al., 2000; Itoh et al., 2014). Similarly, disruption of ROCK, LIMK, and Cofilin which act downstream of RhoA to regulate actin polymerization and turnover result in abnormal actin accumulation, failure of neuroepithelial folding, and NTDs (Fig. 6B) (Escuin et al., 2015). Interestingly Blebbistatin, a myosin inhibitor which blocks actomyosin cross-linking, rescues NTDs accompanied by abnormal actin accumulation. Conversely, the drug Jasplakinolide, which blocks actin depolymerization, significantly delays NTC (Cramer, 1999; Escuin et al., 2015). With the large influence that actin function and turnover has over DLHP bending, it is perhaps not a surprise that disrupted localization of ROCK would yield similar phenotypes to the previously discussed perturbations of its downstream signaling components. Indeed, disruption of Shroom3, which has been proposed to localize ROCK to apical cell junctions, results in failed neuroepithelial bending and severe exencephaly (Nishimura & Takeichi, 2008). A separate NTD model arising from failed neuroepithelial bending involves the gene encoding the protein Noggin. Noggin normally works to inhibit BMP signaling at more caudal regions of the NE where its own expression is no longer repressed by Sonic Hedgehog signaling (Ybot-Gonzalez et al., 2007). Noggin knockout mice display spina bifida marked by increased BMP signaling activity and failure of neuroepithelial bending at the DLHP (Stottmann et al., 2006).
Figure 6: Disruption of actomyosin contraction.
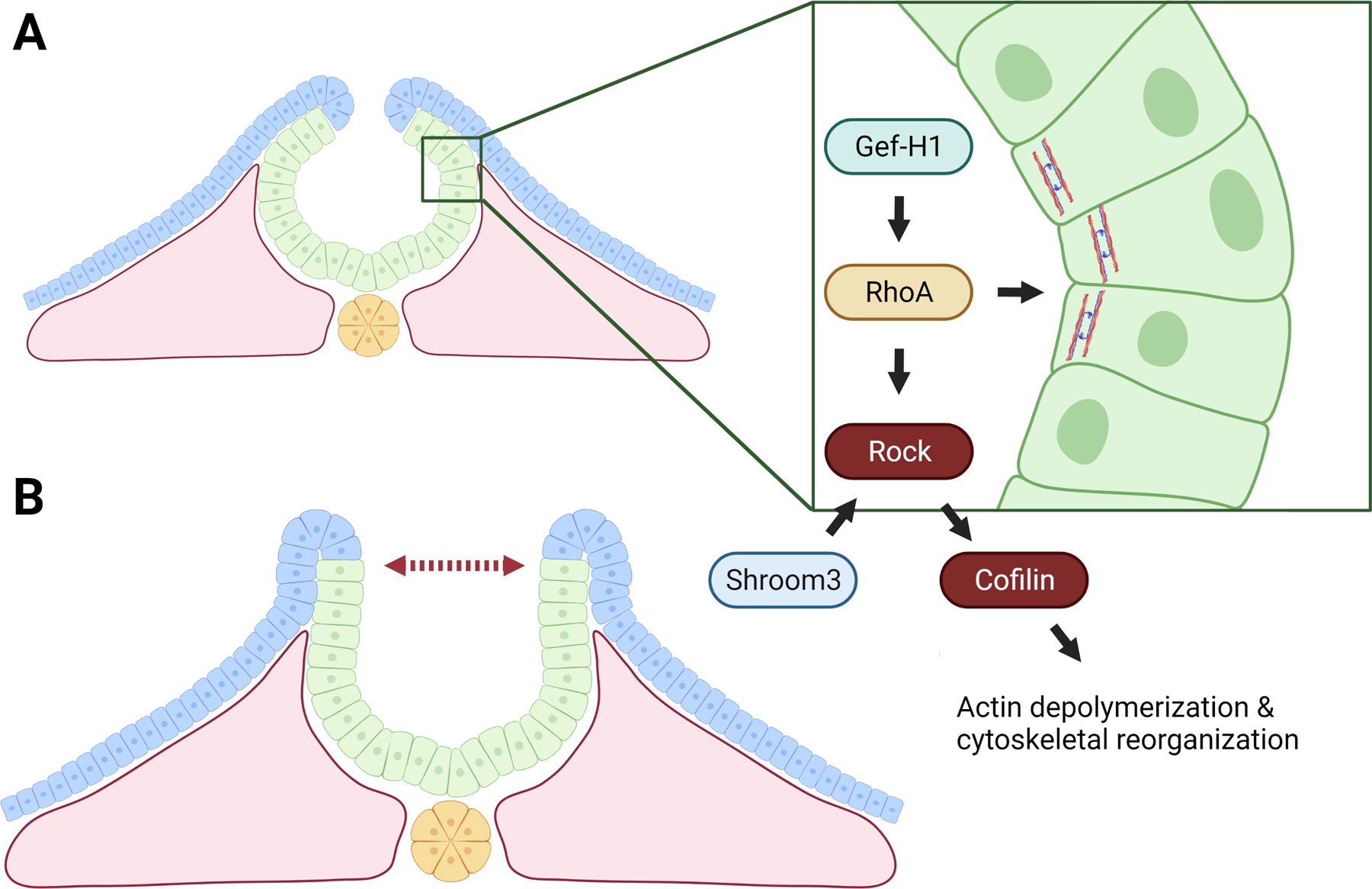
A) Illustration of components necessary for proper localization, contraction, and turnover of actomyosin. B) Disruption of actomyosin contraction inhibits formation of hinge points and neural fold bending.
The NNE is unique in that NTD animal models that affect this tissue are represented almost exclusively by disruptions to cellular structure (Rolo et al., 2016). This is most clearly evident by the role that the NNE and its surface proteins play in neural fold fusion (Copp et al., 2003). As the neural folds undergo fusion or zippering along the rostral-caudal axis, the NNE cells exhibit protrusive behavior and this differs along the axis. Two sets of protrusions identified as important for NTC are long filopodia and ruffle-like lamellipodia, with filopodia thought to be important to cranial and upper spinal cord NTC, and lamellipodia more crucial to later most caudal closure (Rolo et al., 2016). The GRHL3 transcription factor is important in controlling some of these NNE cell structural elements. Knockout of Grhl3 displays loss of sheet-like lamellipodia thought to be crucial for caudal neural fold fusion (Jaffe & Niswander, 2021). Loss of Rac1, more directly linked to lamellipodia production, mimics Grhl3 loss phenotypes and suggests GRHL3 as an upstream regulator of Rac1 (Jaffe & Niswander, 2021; Rolo et al., 2016). This preferential role in the regulation of lamellipodia might be reflected in the infrequency of Grhl3 mutants to produce cranial NTDs, whereas spina bifida is a completely penetrant phenotype (Nikolopoulou et al., 2017). The family member Grhl2 is expressed uniformly within the NNE and upon loss demonstrates both cranial and spinal NTDs (Brouns et al., 2011; Rifat et al., 2010). GRHL2 regulates epithelial morphogenesis through its downstream targets Claudin4, Rab25, and E-cadherin. Although none of these downstream targets are represented by NTD models, this could be due to their critical roles during earlier stages of embryogenesis (Harris & Juriloff, 2010; Pyrgaki et al., 2011; Senga et al., 2012). Notably though, antisense oligonucleotide knockdown of E-cadherin leads to cranial NTDs, pointing towards a role in GRHL2 maintaining epithelial cohesion through tight junctions (B. Chen & Hales, 1995), and as seen by loss of NNE integrity in mouse Grhl2 mutants (Ray & Niswander, 2016). It has been proposed the presence of N-cadherin at junctions may be sufficient to maintain neural identity (Punovuori et al., 2019), and a similar role for GRHL2 and E-cadherin in maintaining epithelial identity is supported by the observation that Grhl2 loss is accompanied by mesenchymal markers in the NNE (Ray & Niswander, 2016).
3. EPIGENETICS OF NTDS
We previously defined individual genetic mutations which can produce an NTD, most often when gene function is largely ablated in animal models. We also discussed how a variety of genes regulate critical cellular processes. In humans it is thought that NTD penetrance results from a combination of genetic and environmental perturbations, which can be tolerated to a certain level, above which an NTD becomes a possibility. As epigenetic regulators act globally on the genome, it might be expected that mutations in epigenetic regulators could globally alter gene expression. Reflecting on the complexity of the cellular processes involved in NTC, it may seem that even small noise introduced by epigenetic disruption may lead to NTDs; however, variability in gene expression seems to be tolerated and even a necessary aspect to maintain plasticity required in NTC (Pujadas & Feinberg, 2012). This partly points towards the extent of epigenetic disruption needed to induce an NTD, but perhaps also emphasizes the multi-factorial nature of NTDs and the interplay between epigenetic, genetic, and environmental factors. Of epigenetic regulators that act globally to modify gene expression, disruptions to DNA methylation, histone modifications, and chromatin structure are enriched in animal models of NTD (Kakebeen & Niswander, 2021; Wilde et al., 2014). The context that individual NTD epigenetic models provide will undoubtedly improve as researchers begin to cross-reference global epigenetic changes against the expanding knowledge created by the encyclopedia of coding elements project (ENCODE) which includes spatial and temporal profiles of regulatory elements within mouse development (J. E. Moore et al., 2020).
3.1. DNA methylation
The majority of the genome (70%−90%) harbors enzymatically added methyl groups at CpG dinucleotides which are established and maintained by DNA methyltransferases (DNMT)(Jones & Baylin, 2002). DNA methylation is essential for silencing retroviral elements, regulating tissue-specific gene expression, genomic imprinting, and X chromosome inactivation (L. D. Moore et al., 2013). CpG islands are defined as >500bp stretches with >55% GC content, and an observed/expected CpG ratio of 0.65 (Bird et al., 1985; Takai & Jones, 2002) Approximately 70% of gene promoters lie within CpG islands, and methylation in these regions is thought to contribute towards tissue specific transcriptional regulation through exclusion (Miranda & Jones, 2007) or attraction (Blattler & Farnham, 2013) of transcription factors and accessibility of DNA (L. D. Moore et al., 2013; Saxonov et al., 2006).
Disruption of methylation globally through depletion of the DNA methylation maintenance enzyme DNMT1 is detrimental to embryogenesis, however early embryonic death and ubiquity of function impedes the study of DNMT1 in the context of NTDs (E. Li et al., 1992; Wilde et al., 2014). Instances where disruption of global methylators result in NTDs are represented by mice lacking de novo methyltransferases DNMT3B and DNMT3L; however, mice lacking the other major de novo methyltransferase DNMT3A, die early in embryogenesis (Hata et al., 2002; Okano et al., 1999). Although these de novo methyltransferases can methylate DNA in a non-specific manner, they are also thought to methylate in a site-specific manner through protein complexes which contain tissue specific transcription factors (Hervouet et al., 2018; Yagi et al., 2020). These site-directed methylation behaviors have led to some insight to their role in NTC. It has been demonstrated that DNMT3A is required for methylation and proper transcription of Polycomb group target developmental genes, and has been proposed to act as switch mediating neural crest fate transition (Hu & Rosenfeld, 2012; Yagi et al., 2020). Knockdown of DNMT3A during NTC upregulates neural genes such as Sox2 and down regulates neural crest specifier genes (FoxD3, Sox10, and Snail2), broadening the dorsal neural tube and leading to expansion of NE cells over neural crest cells (Hu & Rosenfeld, 2012). The NTD phenotype observed following loss of methylation of Sox2 and neural crest genes points to the possible involvement of site directed methylation in other NTD animal models. Further evidence for specific methylation influencing NTDs can be found in human NTD cases where increased methylation within the Pax3 gene body has been observed (Lin et al., 2019; L. Wang, Lin, Zhang, et al., 2017). This is mirrored in mice treated with a benzoapyrene, which demonstrate hypermethylation of Pax3 and NTDs (Lin et al., 2019).
3.2. Histone modifications
The ability of the nucleus to contain such large amounts of DNA is made possible by its organization and compaction into chromosomes. This organization is rooted in the wrapping of DNA around nucleosomes, composed of four homodimers of core histone proteins H2A, H2B, H3, and H4 (Rosenfeld et al., 2009). Post-translational modifications to the N-terminal tails of these histones are thought to regulate the interaction between DNA and nucleosomes, and subsequently the packaging and accessibility of the DNA to transcription factors (Kouzarides, 2007). There are eight different post-translational modifications that can affect more than 60 sites, with multiple modifications possible at each site (Kouzarides, 2007). This leads to a nearly endless combination of histone modifications. Moreover, the influence these marks exert over transcriptional regulation is also thought to be affected by neighboring proteins with their own combination of modifications. Overall, the influence of one or multiple histone states on transcription has been deemed the “histone code” (Jenuwein & Allis, 2001). Similarly to DNA methylation, histone modifiers act on a global scale, however the different marks and myriad of enzymes which produce local histone states lends itself to more direct interrogation of the roles of histone modifiers on NTC.
The most widely studied histone modifications are acetylation and methylation, a trend which is reflected in current NTD mice models (Harris & Juriloff, 2007). Histone acetylation is thought to decrease the affinity of DNA to nucleosomes, increasing the accessibility of the DNA and is associated often with increased transcription (Kouzarides, 2007). Mice harboring mutations in histone acetyltransferases (HATs) GCN5 and CBP/p300, which deposit acetyl marks, display exencephaly; and even mice heterozygous for CBP or p300 mutations display exencephaly (Bu et al., 2007; Partanen et al., 1999). These HAT mutations are thought to act through disruption of metabolic pathways such as glucose metabolism needed for NTC, which has interesting implications for pregnant people with diabetes who have a higher risk of NTD births (Wilde et al., 2014). Histone de-acetyltransferases (HDACs) serve the opposite role and loss of HDACs Sirt1 and HDAC4 leads to dysregulation of P53 and Runx2 respectively and are associated with exencephaly in mouse mutants (H.-L. Cheng et al., 2003; Vega et al., 2004). Treatment with HDAC inhibitor trichostatin A or anticonvulsant valproic acid are thought to produce NTDs through a similar pathway, as they display disruption of HDACs and hyperacetylation of histones (Göttlicher et al., 2001; Murko et al., 2013). Beyond specific HDACs, HDACs in general have been implicated in the regulation of differentiation, and have been demonstrated to be necessary to inhibit BMP2/4 signaling in the developing mouse forebrain (Shakèd et al., 2008; Wilde et al., 2014). Though there is much less evidence for histone methylation specifically affecting NTC, the loss of histone demethyltransferase FBXL10 in mice leads to early NE cell death and exencephaly (Fukuda et al., 2011).
4. ENVIRONMENTAL INFLUENCES
The impact that environmental factors exert on NTD risk is apparent when studying global statistics. NTD rates vary significantly by geographic location, and can be stratified even further by income status (Zaganjor et al., 2016). Many of these disparities can be tied to nutritional status as discussed below. Also the relatively high frequency of NTDs has allowed researchers and epidemiologists to make connections to a variety of maternal conditions and teratogenic agents.
4.1. Folic acid
The micronutrient most closely associated with NTD incidence is folate, found widely in the diet, especially in leafy green vegetables, and synthetically as Folic Acid (FA). This relationship was first recognized by a midwife named Catherina Schrader in 18th century Holland. Schrader’s meticulous record keeping showed the correlation between poor crop years and higher incidence of NTDs (Michie, 1991). In 1965, a paper detailing a case-control study marked the recognition of an inverse relationship between deficient folate metabolism and increased NTD incidence (Hibbard & Smithells, 1965). Further clinical trials in the 1980’s and 1990’s confirmed the protective nature of FA in reducing NTD cases. Now, FA is widely used to fortify grains to avoid maternal folate deficiency (“Prevention of Neural Tube Defects,” 1991). Starting in 1998 the United States enriched cereal grains with 140μg FA/100g, which has prevented an estimated 1,300 cased of NTDs annually (J. Williams et al., 2015). Eighty countries around the world also fortify their grains with FA. According to the Food Fortification Initiative, countries with higher FA supplementation have lower NTD incidence. However, this association, according to an epidemiological study done in 2020, may be confounded by socio-economic class (Qu et al., 2021). Still, women who had a previous NTD affected pregnancy are less likely to have a second NTD affected pregnancy after supplementing FA in the diet. Additionally, a randomized control trial in China showed that when pregnant women regularly took periconceptional FA, there was an 85% reduction in NTD risk (Berry et al., 1999). Thus, many epidemiological studies have established the efficacy of FA fortification on NTD prevention, although debate continues over the details of supplementation and public implementation.
Despite the focus on folate status, maternal folate deficiency itself is not enough to produce a NTD in mice, and instead requires a predisposing mutation in a folate-sensitive gene (Greene et al., 2009). Similarly, in humans it is believed that NTDs are multi-factorial in which genetic variants (inherited or de novo) along with environmental factors act in combination to affect NTD risk (Wilde et al., 2014). This has led to investigations to identify NTD gene mutations that are responsive to folate levels. Studies in mice have used FA deficient and FA supplemented regimes to begin to uncover the interplay between genetic and environmental factors. First, we will discuss genes within the folate one-carbon metabolism (OCM) pathway and then highlight that many of the FA responsive genes have no apparent roles in FA metabolism or other processes related to this pathway, indicating the broader impact of the OCM pathway on nucleotides, proteins, and lipids during NTC.
Folate is processed in the cell within the OCM pathway. Folic acid must be reduced to produce the body’s biologically active form, tetrahydrofolate (THF). Further reduction and the addition of a methyl group yields 5Me-THF. 5Me-THF can then be used as methyl carrier for transfer of one-carbon units to many molecules within the cell (Imbard et al., 2013). This pathway is essential for the biosynthesis of purines and thymidylate, remethylation of homocysteine to methionine, and from there making S-adenosylmethionine which is the universal methyl donor in the cell (Suh et al., 2001). It is thought that these key metabolites may be at suboptimal levels at critical closure time points in folate deficient mothers.
As highlighted earlier, the coordination of proliferation is critical for NTC. The folate pathway is necessary for production of the building blocks for DNA synthesis, and hence proliferation has been a focus of study in FA pathway mutants. Mutations that affect folate uptake and metabolism are a prime example of the correlation between folate levels and NTD incidence. Deletion of the Folate Receptor 1 (Folr1, Folbp1) in mouse causes NTD and the embryos are severely growth retarded (Piedrahita et al., 1999). The evidence in humans is less clear but there are reports of autoantibodies against folate receptors associated with NTD risk (Cabrera et al., 2008; Rothenberg et al., 2004). MTHFD1L is an enzyme in the mitochondria responsible for catalyzing the last step in the flow of one-carbon units (formate) from the mitochondria to the cytosol. Deletion of this gene in mice results in severe NTDs, including craniorachischisis, exencephaly, and a “wavy” neural tube (Momb et al., 2013). Mthfd1L mutant mice show reduced cellular proliferation in the brain after completion of NTC (Shin et al., 2019). Gene variants in the mitochondrial OCM pathway are associated with increased NTD risk in humans including MTHFD1L, AMT, and GLDC, and this increased NTD risk is borne out in studies in mice (Narisawa et al., 2012; Parle-McDermott et al., 2009). Within the cytosolic folate pathway, loss of serine hydroxymethyltransferase SHMT1, which utilizes serine to form one-carbon units, causes NTDs in mice, but only on a folate deficient diet, through decreased flux through the thymidylate biosynthesis pathway (Beaudin et al., 2011). It has been reported that SHMT1 C1420T parental alleles increase NTD risk (K Rebekah et al., 2017). Variants in MTHFD1, a tri-functional enzyme which catalyzes three steps in the folate cycle, are also associated with increased NTD risk (De Marco et al., 2002; Parle-McDermott et al., 2009; Zheng et al., 2015) and disease alleles show altered flux through thymidylate biosynthesis pathway (Field et al., 2015). These data link the importance of the folate OCM pathway in the regulation of proliferation and NTC.
Another proposed mechanism by which FA may affect the penetrance of NTDs is modulation of the epigenome through OCM metabolites, via the methionine cycle and methylation of substrates including DNA, RNA and proteins. In animal models, methionine cycle inhibitors or mutations in DNA methyltransferase proteins can cause NTDs (Dunlevy et al., 2006; Okano et al., 1999). The MTHFR enzyme catalyzes the synthesis of 5Me-MTHF, which is the methyl donor for homocysteine to regenerate methionine through the methionine synthase (MTR) and methionine synthase reductase (MTRR) enzymes. MTHFR has been studied for association with NTDs in human cases. In a case-control study within Italian families, it was determined that the families and mothers who have experienced an NTD are enriched in a MTHFR polymorphism A1298C (De Marco et al., 2002). Similarly, MTHFR polymorphism C677T is associated with a greater risk for NTDs and has been found to result in low plasma folate levels and higher levels of homocysteine, as well as diminished DNA methylation, which could be improved by increased FA status (Friso et al., 2002; van der Put et al., 1995). MTR and MTRR variants are also associated with NTD (Cai et al., 2019; Ouyang et al., 2013) and altered homocysteine levels (Gaughan et al., 2001). These data indicate the importance of the methionine cycle in NTC. However, the interrelationship between the three OCM cycles – folate cycle, methionine cycle, and mitochondrial glycine cleavage cycle has been demonstrated; where, a mutation in one arm of the OCM pathway can alter the flux of one-carbon units in the other cycles. Thus, it may be an oversimplification to view the effect of a mutation as limited to an individual OCM cycle but instead to consider how it may affect the partitioning of one-carbon units between these three cycles that together are critical for NTC (Leung et al., 2017).
Epigenetic changes are also possible as FA supplementation over multiple generations exacerbates FA sensitive mutations (Marean et al., 2011). It has been demonstrated that there is a trans-generational effect of a mutation in the methionine synthase reductase gene. Mtrr deficiency in either maternal grandparent of offspring can result in developmental defects of the grandprogeny and global DNA hypomethylation (Padmanabhan et al., 2013). These data suggest that modulations in folate metabolism can be passed down epigenetically for many generations.
Notably, many of the FA responsive genes identified in mice have no apparent roles in FA metabolism or other processes related to this pathway. For example, PAX3 is necessary for successful NTC by regulating cell proliferation and suppressing neuronal differentiation. Although levels of individual folate cycle species are not altered in Pax3 mutants (Sudiwala et al., 2019), supplementation with FA prevents NTD, whereas FA deficiency increases NTD incidence (Burren et al., 2008; Wlodarczyk et al., 2006). FA supplementation helps to drive cell cycle progression which is proposed to prevent NTD in Pax3 mutants (Sudiwala et al., 2019). In most cases, however, how FA levels act to prevent or exacerbate NTD incidence in animal models of FA sensitive gene mutations is yet unclear. Additional processes impacted by FA levels but not directly associated with the OCM pathway have been uncovered. There is a correlation between ubiquitination of Histone 2A (H2A) under low FA conditions, and this can lead to modulation of the expression of NTC associated genes (Pei et al., 2019). Additionally, the formation of cilia can be modulated by FA supplementation (our lab unpublished data), and folate is thought to promote ciliogenesis by regulating the methylation of Septin2, an important ciliogenesis gene (Toriyama et al., 2017). The underlying mechanisms for how FA modulates NTC in the case of folate sensitive genes is likely a balance of all of the processes outlined above as well as yet to be discovered processes.
4.2. Non-FA micronutrients
In cases where FA does not protect against NTDs, other micronutrients have been examined. The quest for other factors that may prevent NTD has been driven in part by epidemiological studies and by an understanding of the biology underlying NTC and gene function in mouse models which do not respond to FA. Here we summarize evidence for an impact on NTD incidence by inositol, iron, zinc, and retinoic acid. Inositol is important in cell transduction and is an important component of phosphoinositol lipids. Inositol deficiency can increase NTD risk and inositol supplementation can decrease NTD incidence in curly tail mouse mutants, which are FA-resistant, and in diabetic rodent models (Reece et al., 1993). Inositol supplementation was used as a preventative treatment for recurrent NTD in humans, with no detriment to the mother or the fetus (Cavalli et al., 2011). This led to a small clinical trial (PONTI: Prevention of Neural Tube Defects by Inositol) which showed inositol supplementation is safe and showed promising results relative to NTD risk (Greene et al., 2016). These results have led to speculation that combined inositol and FA treatment will have a more optimal effect among women who have had a NTD affected pregnancy.
Iron deficiency is the most common micronutrient deficiency among pregnant women, and the World Health Organization estimates that 40% of pregnant women are iron deficient or anemic. Despite the importance of iron in numerous cellular reactions and oxygen transport, its effect on NTC has been little studied. Epidemiological studies have differed as to whether there is a correlation between iron status and NTD risk (Felkner et al., 2005; Molloy et al., 2014). In mouse, mutation of the Ferroportin1 (Fpn1) gene, which transports iron from the visceral endoderm to the embryo, causes a high NTD rate, as does culture of wildtype mouse embryos in an iron chelator (Mao et al., 2010). Fpn1 mutants do not respond to FA but iron supplementation can reduce NTD incidence (Stokes et al., 2017). However, iron levels may also influence folate metabolism (Herbig & Stover, 2002). Indeed, in mouse it was found that iron supplementation can cause folate deficiency even in wildtype dams and embryos (Stokes et al., 2017). In a study of a mouse mutant for the Lrp2 gene, which mediates endocytosis of the FA receptor as well as uptake of transferrin bound iron, high FA supplementation was able to prevent NTDs, while iron supplementation alone had no effect, or in combination iron worked against the positive effects of FA supplementation (Sabatino et al., 2017). This highlights the interrelationship between iron and folate metabolism and lends caution to an oversimplification of the effect of an individual nutrient without a broader look at other possible confounding effects, both genetic and environmental.
Zinc is another vital micronutrient and almost 3,000 proteins interact with zinc. Epidemiological studies indicate that zinc deficiency is associated with elevated NTD risk whereas zinc supplementation has been correlated with decreased risk as well as increased risk, perhaps due to intracellular versus peripheral levels of zinc depending on uptake (H. Li, Zhang, & Niswander, 2018). Knockout in the mouse of three zinc transporters (ZIP1, and double knockout of ZIP1 and ZIP3) on a zinc deficient diet leads to a 2–3 fold increase of risk for NTDs (Dufner-Beattie et al., 2006). This implies that zinc transporters are essential for the distribution and retention of zinc, and this is important for NTC. Zinc deficiency has been linked to p53 ubiquitylation, through the zinc binding protein, MDM2, which leads to p53 stabilization and abnormal apoptosis, resulting in NTDs (H. Li, Zhang, & Niswander, 2018). Conversely, experimentally induced zinc accumulation by exposure to zinc oxide nanoparticles cause NTDs in chickens and mice, likely due to abnormal apoptosis. This apoptosis is thought to result from endoplasmic reticulum related stress induced by intracellular calcium concentrations (Y. Yan et al., 2021). These studies indicate imbalance in the level of zinc may contribute to NTDs and that further studies of maternal and fetal zinc status in NTD cases are warranted.
Retinoic acid (RA) is another micronutrient implicated in NTDs. RA is a vitamin A derivative and a proper spatial gradient of RA is required for development of the hindbrain and spinal cord (Araya García, 2017). During normal development, RA suppresses BMP, another key factor in neural tube patterning. Cyp26a1 is an enzyme responsible for maintaining precise concentrations of RA, and in Cyp26a1 knockout mice, NTC fails (Abu-Abed et al., 2001). FBXO30 ubiquitin ligase can promote ubiquitin-mediated degradation of Retinoic acid receptor gamma and hence can positively regulate BMP signaling. A case-control study in humans revealed that human NTD cases are enriched for mutations in vitamin A related processing genes (H. Li, Zhang, Chen, et al., 2018). In NTD cases with high levels of retinol, BMP target genes are downregulated and FBXO30 is dysregulated (X. Cheng et al., 2019). Together the data indicate that when the RA pathway is unbalanced due to deficiency or high dosage, the patterning of the neural tube cannot proceed as normal. Overall, micronutrients play a key and dynamic role during NTC, and the roles of these nutrients and how they influence each other requires further investigation in order to understand the causes of NTDs and the most effective types of therapies, depending on the genetic makeup of individuals.
4.2. Diabetes
Shifting to a consideration of maternal factors associated with NTD risk, maternal diabetes and obesity are significant risk factors for NTDs (Zabihi & Loeken, 2010). The embryo does not develop pancreatic function until week 7 of pregnancy, and thus relies on the metabolism of the mother during neural tube formation (Lupo et al., 2012). It is proposed that altered metabolism in the mother can detrimentally affect NTC. This is supported by correlation between SNPs in maternal glucose metabolism genes (FTO, LEP, TCF7l2, LEPR, GLUT1, Glut2, and HK1) and increased NTD risk(Loeken, 2020; Lupo et al., 2012; Wilde et al., 2014). The exact mechanism is still under debate, however animal models point towards decreased proliferation and increased apoptosis within the NE (Gao & Gao, 2007). Chemically induced diabetes in mice with streptozotocin has implicated several pathways including: increased oxidative stress and transcription of Jnk1/2 which can trigger pro-apoptotic pathways (X. Li et al., 2012), upregulation of miR-200b and subsequent repression of its targets leading to ER stress (Gu et al., 2016), and activation of the Foxo3a - Caspase8 pathway which upregulates apoptosis (Yang et al., 2013). Further evidence for the latter pathway is demonstrated through the reduction of NTDs in streptozotocin mice via knockout of Foxo3a (Xu et al., 2021). Evidence from another diabetic mouse model, the nonobese diabetic mouse strain which exhibits high rates of NTDs, points towards defects in cell migration when exposed to high amounts of glucose both in vitro, and in-vivo (Herion et al., 2019). This suggests convergent extension as another diabetic sensitive NTC process. The complexity in parsing diabetes associated contributions towards NTD risk is driven home by the observation that more than a third of genes involved in NTC have their expression altered in maternal diabetes models (Salbaum & Kappen, 2010).
Conclusion
Large-scale sequencing of NTD samples by researchers across the globe will be key in moving beyond testing of genes identified in animal studies to a more comprehensive characterization of human risk alleles and to uncover the multigenic nature of NTDs in humans. Efforts to bring together cohort samples and sequencing data, such as the Spina Bifida Sequencing Consortium (https://sbseqconsortium.org), will greatly increase our ability to pinpoint candidate genes for future evaluation of NTD risk. Current sequencing studies include samples from NTD cases conceived post-FA fortification as well as cases prior to FA fortification or from countries that do not yet include FA fortification. This holds the possibility of identifying NTD risk alleles that differ in these populations, which may reflect FA sensitivity. It is speculated that the burden of de novo mutations and/or the genetic landscape may differ pre- and post-FA fortification, perhaps representing a shift in the threshold of severity or number of gene variants in NTD cases after decades of FA supplementation (Lee & Gleeson, 2020). Most sequencing efforts are focused on identifying germline mutations, but an additional challenge arises from the idea that somatic mutations at the NTD lesion site may contribute to NTD. Recent sequencing studies of the lesion site have discovered tissue mosaicism with somatic mutations in proteins of the Mediator complex as well as PCP genes(Tian et al., 2021). Moreover, experimental studies in mouse showed that as few as 16% of cells carrying a Vangl2 mutation can non-autonomously prevent apical constriction in neighbouring cells to prevent NTC(Galea et al., 2021). Also, increased signatures of DNA mismatch repair deficiency have been associated with low folate status(H. Li et al., 2020). In the future, it is possible that single-cell sequencing may reveal the extent to which localized perturbations, rather than germline mutations, contribute to NTD. The overall goal will be to provide better genetic counselling for genetic risk factors and more personalized consideration of nutrient supplements for better outcomes of all births.
Funding Information
NIH NINDS R01-NS110887
NIH NICHD P01-HD104436
Footnotes
Conflict of Interest The Authors declare that there is no conflict of interest.
Contributor Information
David M. Engelhardt, University of Colorado at Boulder.
Cara A. Martyr, University of Colorado at Boulder.
Lee Niswander, University of Colorado at Boulder.
References
- Abu-Abed S, Dollé P, Metzger D, Beckett B, Chambon P, & Petkovich M (2001). The retinoic acid-metabolizing enzyme, CYP26A1, is essential for normal hindbrain patterning, vertebral identity, and development of posterior structures. Genes & Development, 15, 226–240. 10.1101/gad.855001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, & Nieto MA (2009). Epithelial-mesenchymal transitions: The importance of changing cell state in development and disease. The Journal of Clinical Investigation, 119(6), 1438–1449. 10.1172/JCI38019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amarnath S, & Agarwala S (2017). Cell-cycle-dependent TGFβ-BMP antagonism regulates neural tube closure by modulating tight junctions. Journal of Cell Science, 130(1), 119–131. 10.1242/jcs.179192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya García CA (2017). Formation of Neural Tube. In Reference Module in Biomedical Sciences. Elsevier. 10.1016/B978-0-12-801238-3.11055-4 [DOI] [Google Scholar]
- Arnold C, Demirel E, Feldner A, Genové G, Zhang H, Sticht C, Wieland T, Hecker M, Heximer S, & Korff T (2018). Hypertension-evoked RhoA activity in vascular smooth muscle cells requires RGS5. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology, 32(4), 2021–2035. 10.1096/fj.201700384RR [DOI] [PubMed] [Google Scholar]
- Badouel C, Zander MA, Liscio N, Bagherie-Lachidan M, Sopko R, Coyaud E, Raught B, Miller FD, & McNeill H (2015). Fat1 interacts with Fat4 to regulate neural tube closure, neural progenitor proliferation and apical constriction during mouse brain development. Development, 142(16), 2781–2791. 10.1242/dev.123539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudin AE, Abarinov EV, Noden DM, Perry CA, Chu S, Stabler SP, Allen RH, & Stover PJ (2011). Shmt1 and de novo thymidylate biosynthesis underlie folate-responsive neural tube defects in mice. The American Journal of Clinical Nutrition, 93(4), 789–798. 10.3945/ajcn.110.002766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger H, Wodarz A, & Borchers A (2017). PTK7 Faces the Wnt in Development and Disease. Frontiers in Cell and Developmental Biology, 5. https://www.frontiersin.org/article/10.3389/fcell.2017.00031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry RJ, Li Z, Erickson JD, Li S, Moore CA, Wang H, Mulinare J, Zhao P, Wong LY, Gindler J, Hong SX, & Correa A (1999). Prevention of neural-tube defects with folic acid in China. China-U.S. Collaborative Project for Neural Tube Defect Prevention. The New England Journal of Medicine, 341(20), 1485–1490. 10.1056/NEJM199911113412001 [DOI] [PubMed] [Google Scholar]
- Bird A, Taggart M, Frommer M, Miller OJ, & Macleod D (1985). A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell, 40(1), 91–99. 10.1016/0092-8674(85)90312-5 [DOI] [PubMed] [Google Scholar]
- Blattler A, & Farnham PJ (2013). Cross-talk between Site-specific Transcription Factors and DNA Methylation States*. Journal of Biological Chemistry, 288(48), 34287–34294. 10.1074/jbc.R113.512517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosoi CM, Capra V, Allache R, Trinh VQ-H, De Marco P, Merello E, Drapeau P, Bassuk AG, & Kibar Z (2011). Identification and characterization of novel rare mutations in the planar cell polarity gene PRICKLE1 in human neural tube defects. Human Mutation, 32(12), 1371–1375. 10.1002/humu.21589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouns MR, De Castro SCP, Terwindt-Rouwenhorst EA, Massa V, Hekking JW, Hirst CS, Savery D, Munts C, Partridge D, Lamers W, Köhler E, van Straaten HW, Copp AJ, & Greene NDE (2011). Over-expression of Grhl2 causes spina bifida in the Axial defects mutant mouse. Human Molecular Genetics, 20(8), 1536–1546. 10.1093/hmg/ddr031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouns MR, Matheson SF, Hu KQ, Delalle I, Caviness VS, Silver J, Bronson RT, & Settleman J (2000). The adhesion signaling molecule p190 RhoGAP is required for morphogenetic processes in neural development. Development (Cambridge, England), 127(22), 4891–4903. [DOI] [PubMed] [Google Scholar]
- Bu P, Evrard YA, Lozano G, & Dent SYR (2007). Loss of Gcn5 Acetyltransferase Activity Leads to Neural Tube Closure Defects and Exencephaly in Mouse Embryos. Molecular and Cellular Biology, 27(9), 3405–3416. 10.1128/MCB.00066-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burren KA, Savery D, Massa V, Kok RM, Scott JM, Blom HJ, Copp AJ, & Greene NDE (2008). Gene–environment interactions in the causation of neural tube defects: Folate deficiency increases susceptibility conferred by loss of Pax3 function. Human Molecular Genetics, 17(23), 3675–3685. 10.1093/hmg/ddn262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MT, & Wallingford JB (2018). Spatial and temporal analysis of PCP protein dynamics during neural tube closure. ELife, 7, e36456. 10.7554/eLife.36456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera RM, Shaw GM, Ballard JL, Carmichael SL, Yang W, Lammer EJ, & Finnell RH (2008). Autoantibodies to folate receptor during pregnancy and neural tube defect risk. Journal of Reproductive Immunology, 79(1), 85–92. 10.1016/j.jri.2008.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai C-Q, Fang Y-L, Shu J-B, Zhao L-S, Zhang R-P, Cao L-R, Wang Y-Z, Zhi X-F, Cui H-L, Shi O-Y, & Liu W (2019). Association of neural tube defects with maternal alterations and genetic polymorphisms in one-carbon metabolic pathway. Italian Journal of Pediatrics, 45(1), 37. 10.1186/s13052-019-0630-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catala M (2021). Overview of Secondary Neurulation. Journal of Korean Neurosurgical Society, 64(3), 346–358. 10.3340/jkns.2020.0362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli P, Tonni G, Grosso E, & Poggiani C (2011). Effects of inositol supplementation in a cohort of mothers at risk of producing an NTD pregnancy. Birth Defects Research. Part A, Clinical and Molecular Teratology, 91(11), 962–965. 10.1002/bdra.22853 [DOI] [PubMed] [Google Scholar]
- Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, & Gruss P (1998). Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell, 94(6), 727–737. 10.1016/s0092-8674(00)81732-8 [DOI] [PubMed] [Google Scholar]
- Chen B, & Hales BF (1995). Antisense Oligonucleotide Down-Regulation of E-Cadherin in the Yolk Sac and Cranial Neural Tube Malformations1. Biology of Reproduction, 53(5), 1229–1238. 10.1095/biolreprod53.5.1229 [DOI] [PubMed] [Google Scholar]
- Chen ZF, & Behringer RR (1995). Twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes & Development, 9(6), 686–699. 10.1101/gad.9.6.686 [DOI] [PubMed] [Google Scholar]
- Cheng H-L, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, & Chua KF (2003). Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proceedings of the National Academy of Sciences, 100(19), 10794–10799. 10.1073/pnas.1934713100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Pei P, Yu J, Zhang Q, Li D, Xie X, Wu J, Wang S, & Zhang T (2019). F-box protein FBXO30 mediates retinoic acid receptor γ ubiquitination and regulates BMP signaling in neural tube defects. Cell Death & Disease, 10(8), 1–17. 10.1038/s41419-019-1783-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi H, Sarkisian MR, Rakic P, & Flavell RA (2005). Loss of mitogen-activated protein kinase kinase kinase 4 (MEKK4) results in enhanced apoptosis and defective neural tube development. Proceedings of the National Academy of Sciences, 102(10), 3846–3851. 10.1073/pnas.0500026102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciani L, Patel A, Allen ND, & ffrench-Constant C (2003). Mice Lacking the Giant Protocadherin mFAT1 Exhibit Renal Slit Junction Abnormalities and a Partially Penetrant Cyclopia and Anophthalmia Phenotype. Molecular and Cellular Biology. 10.1128/MCB.23.10.3575-3582.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp AJ, Greene NDE, & Murdoch JN (2003). The genetic basis of mammalian neurulation. Nature Reviews Genetics, 4(10), 784–793. 10.1038/nrg1181 [DOI] [PubMed] [Google Scholar]
- Cramer LP (1999). Role of actin-filament disassembly in lamellipodium protrusion in motile cells revealed using the drug jasplakinolide. Current Biology, 9(19), 1095–1105. 10.1016/S0960-9822(99)80478-3 [DOI] [PubMed] [Google Scholar]
- De Marco P, Calevo MG, Moroni A, Arata L, Merello E, Finnell RH, Zhu H, Andreussi L, Cama A, & Capra V (2002). Study of MTHFR and MS polymorphisms as risk factors for NTD in the Italian population. Journal of Human Genetics, 47(6), 319–324. 10.1007/s100380200043 [DOI] [PubMed] [Google Scholar]
- Dufner-Beattie J, Huang Z, Geiser J, Xu W, & Andrews G (2006). Mouse ZIP1 and ZIP3 genes together are essential for adaptation to dietary zinc deficiency during pregnancy. Genesis (New York, N.Y. : 2000), 44, 239–251. 10.1002/dvg.20211 [DOI] [PubMed] [Google Scholar]
- Dunlevy LPE, Burren KA, Chitty LS, Copp AJ, & Greene NDE (2006). Excess methionine suppresses the methylation cycle and inhibits neural tube closure in mouse embryos. FEBS Letters, 580(11), 2803–2807. 10.1016/j.febslet.2006.04.020 [DOI] [PubMed] [Google Scholar]
- Engler A, Zhang R, & Taylor V (2018). Notch and Neurogenesis. In Borggrefe T & Giaimo BD (Eds.), Molecular Mechanisms of Notch Signaling (pp. 223–234). Springer International Publishing. 10.1007/978-3-319-89512-3_11 [DOI] [Google Scholar]
- Epstein DJ, Vekemans M, & Gros P (1991). Splotch (Sp2H), a mutation affecting development of the mouse neural tube, shows a deletion within the paired homeodomain of Pax-3. Cell, 67(4), 767–774. 10.1016/0092-8674(91)90071-6 [DOI] [PubMed] [Google Scholar]
- Escuin S, Vernay B, Savery D, Gurniak CB, Witke W, Greene NDE, & Copp AJ (2015). Rho-kinase-dependent actin turnover and actomyosin disassembly are necessary for mouse spinal neural tube closure. Journal of Cell Science, 128(14), 2468–2481. 10.1242/jcs.164574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewart JL, Cohen MF, Meyer RA, Huang GY, Wessels A, Gourdie RG, Chin AJ, Park SM, Lazatin BO, Villabon S, & Lo CW (1997). Heart and neural tube defects in transgenic mice overexpressing the Cx43 gap junction gene. Development (Cambridge, England), 124(7), 1281–1292. [DOI] [PubMed] [Google Scholar]
- Ezin AM, Skoglund P, & Keller R (2006). The presumptive floor plate (notoplate) induces behaviors associated with convergent extension in medial but not lateral neural plate cells of Xenopus. Developmental Biology, 300(2), 670–686. 10.1016/j.ydbio.2006.09.004 [DOI] [PubMed] [Google Scholar]
- Felkner MM, Suarez L, Brender J, Scaife B, & Hendricks K (2005). Iron status indicators in women with prior neural tube defect-affected pregnancies. Maternal and Child Health Journal, 9(4), 421–428. 10.1007/s10995-005-0017-3 [DOI] [PubMed] [Google Scholar]
- Ferras OS-, Coutaud B, Djavanbakht Samani T, Tremblay I, Souchkova O, & Pilon N (2012). Caudal-related Homeobox (Cdx) Protein-dependent Integration of Canonical Wnt Signaling on Paired-box 3 (Pax3) Neural Crest Enhancer. The Journal of Biological Chemistry, 287(20), 16623–16635. 10.1074/jbc.M112.356394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field MS, Kamynina E, Watkins D, Rosenblatt DS, & Stover PJ (2015). Human mutations in methylenetetrahydrofolate dehydrogenase 1 impair nuclear de novo thymidylate biosynthesis. Proceedings of the National Academy of Sciences, 112(2), 400–405. 10.1073/pnas.1414555112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming A, & Copp AJ (2000). A genetic risk factor for mouse neural tube defects: Defining the embryonic basis. Human Molecular Genetics, 9(4), 575–581. 10.1093/hmg/9.4.575 [DOI] [PubMed] [Google Scholar]
- Friso S, Choi S-W, Girelli D, Mason JB, Dolnikowski GG, Bagley PJ, Olivieri O, Jacques PF, Rosenberg IH, Corrocher R, & Selhub J (2002). A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proceedings of the National Academy of Sciences of the United States of America, 99(8), 5606–5611. 10.1073/pnas.062066299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda T, Tokunaga A, Sakamoto R, & Yoshida N (2011). Fbxl10/Kdm2b deficiency accelerates neural progenitor cell death and leads to exencephaly. Molecular and Cellular Neurosciences, 46(3), 614–624. 10.1016/j.mcn.2011.01.001 [DOI] [PubMed] [Google Scholar]
- Galea GL, Maniou E, Edwards TJ, Marshall AR, Ampartzidis I, Greene NDE, & Copp AJ (2021). Cell non-autonomy amplifies disruption of neurulation by mosaic Vangl2 deletion in mice. Nature Communications, 12(1), 1159. 10.1038/s41467-021-21372-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q, & Gao Y-M (2007). Hyperglycemic condition disturbs the proliferation and cell death of neural progenitors in mouse embryonic spinal cord. International Journal of Developmental Neuroscience: The Official Journal of the International Society for Developmental Neuroscience, 25(6), 349–357. 10.1016/j.ijdevneu.2007.08.002 [DOI] [PubMed] [Google Scholar]
- Gaughan DJ, Kluijtmans LA, Barbaux S, McMaster D, Young IS, Yarnell JW, Evans A, & Whitehead AS (2001). The methionine synthase reductase (MTRR) A66G polymorphism is a novel genetic determinant of plasma homocysteine concentrations. Atherosclerosis, 157(2), 451–456. 10.1016/s0021-9150(00)00739-5 [DOI] [PubMed] [Google Scholar]
- Gonzalez-Gobartt E, Blanco-Ameijeiras J, Usieto S, Allio G, Benazeraf B, & Martí E (2021). Cell intercalation driven by SMAD3 underlies secondary neural tube formation. Developmental Cell, 56(8), 1147–1163.e6. 10.1016/j.devcel.2021.03.023 [DOI] [PubMed] [Google Scholar]
- Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, & Heinzel T (2001). Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. The EMBO Journal, 20(24), 6969–6978. 10.1093/emboj/20.24.6969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J, & Ross ME (2011). Neural tube closure in mouse whole embryo culture. Journal of Visualized Experiments: JoVE, 56, 3132. 10.3791/3132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene NDE, Leung K-Y, Gay V, Burren K, Mills K, Chitty LS, & Copp AJ (2016). Inositol for the prevention of neural tube defects: A pilot randomised controlled trial. The British Journal of Nutrition, 115(6), 974–983. 10.1017/S0007114515005322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene NDE, Stanier P, & Copp AJ (2009). Genetics of human neural tube defects. Human Molecular Genetics, 18(R2), R113–R129. 10.1093/hmg/ddp347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse SD, Berry RJ, Mick Tilford J, Kucik JE, & Waitzman NJ (2016). Retrospective Assessment of Cost Savings From Prevention: Folic Acid Fortification and Spina Bifida in the U.S. American Journal of Preventive Medicine, 50(5 Suppl 1), S74–S80. 10.1016/j.amepre.2015.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Yu J, Dong D, Zhou Q, Wang J-Y, Fang S, & Yang P (2016). High Glucose-Repressed CITED2 Expression Through miR-200b Triggers the Unfolded Protein Response and Endoplasmic Reticulum Stress. Diabetes, 65(1), 149–163. 10.2337/db15-0108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson P, Greene NDE, Lad D, Pauws E, de Castro SCP, Stanier P, & Copp AJ (2007). Increased expression of Grainyhead-like-3 rescues spina bifida in a folate-resistant mouse model. Human Molecular Genetics, 16(21), 2640–2646. 10.1093/hmg/ddm221 [DOI] [PubMed] [Google Scholar]
- Haigo SL, Hildebrand JD, Harland RM, & Wallingford JB (2003). Shroom Induces Apical Constriction and Is Required for Hingepoint Formation during Neural Tube Closure. Current Biology, 13(24), 2125–2137. 10.1016/j.cub.2003.11.054 [DOI] [PubMed] [Google Scholar]
- Harris MJ, & Juriloff DM (2007). Mouse mutants with neural tube closure defects and their role in understanding human neural tube defects. Birth Defects Research Part A: Clinical and Molecular Teratology, 79(3), 187–210. 10.1002/bdra.20333 [DOI] [PubMed] [Google Scholar]
- Harris MJ, & Juriloff DM (2010). An update to the list of mouse mutants with neural tube closure defects and advances toward a complete genetic perspective of neural tube closure. Birth Defects Research. Part A, Clinical and Molecular Teratology, 88(8), 653–669. 10.1002/bdra.20676 [DOI] [PubMed] [Google Scholar]
- Hata K, Okano M, Lei H, & Li E (2002). Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development, 129(8), 1983–1993. 10.1242/dev.129.8.1983 [DOI] [PubMed] [Google Scholar]
- Hatakeyama J, & Kageyama R (2006). Notch1 expression is spatiotemporally correlated with neurogenesis and negatively regulated by Notch1-independent Hes genes in the developing nervous system. Cerebral Cortex (New York, N.Y.: 1991), 16 Suppl 1, i132–137. 10.1093/cercor/bhj166 [DOI] [PubMed] [Google Scholar]
- Hayes M, Naito M, Daulat A, Angers S, & Ciruna B (2013). Ptk7 promotes non-canonical Wnt/PCP-mediated morphogenesis and inhibits Wnt/β-catenin-dependent cell fate decisions during vertebrate development. Development (Cambridge, England), 140(8), 1807–1818. 10.1242/dev.090183 [DOI] [PubMed] [Google Scholar]
- Herbig AK, & Stover PJ (2002). Regulation of Folate Metabolism by Iron. In Massaro EJ & Rogers JM (Eds.), Folate and Human Development (pp. 241–262). Humana Press. 10.1007/978-1-59259-164-0_12 [DOI] [Google Scholar]
- Herion NJ, Kruger C, Staszkiewicz J, Kappen C, & Salbaum JM (2019). Embryonic cell migratory capacity is impaired upon exposure to glucose in vivo and in vitro. Birth Defects Research, 111(14), 999–1012. 10.1002/bdr2.1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervouet E, Peixoto P, Delage-Mourroux R, Boyer-Guittaut M, & Cartron P-F (2018). Specific or not specific recruitment of DNMTs for DNA methylation, an epigenetic dilemma. Clinical Epigenetics, 10(1), 17. 10.1186/s13148-018-0450-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbard E, & Smithells RW (1965). FOLIC ACID METABOLISM AND HUMAN EMBRYOPATHY. The Lancet, 285(7398), 1254. 10.1016/S0140-6736(65)91895-7 [DOI] [Google Scholar]
- Hollander MC, Sheikh MS, Bulavin DV, Lundgren K, Augeri-Henmueller L, Shehee R, Molinaro TA, Kim KE, Tolosa E, Ashwell JD, Rosenberg MP, Zhan Q, Fernández-Salguero PM, Morgan WF, Deng CX, & Fornace AJ (1999). Genomic instability in Gadd45a-deficient mice. Nature Genetics, 23(2), 176–184. 10.1038/13802 [DOI] [PubMed] [Google Scholar]
- Hu Q, & Rosenfeld MG (2012). Epigenetic regulation of human embryonic stem cells. Frontiers in Genetics, 3, 238. 10.3389/fgene.2012.00238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbard A, Benoist J-F, & Blom HJ (2013). Neural Tube Defects, Folic Acid and Methylation. International Journal of Environmental Research and Public Health, 10(9), 4352–4389. 10.3390/ijerph10094352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi M, Ang SL, Shiota K, Nakanishi S, Kageyama R, & Guillemot F (1995). Targeted disruption of mammalian hairy and Enhancer of split homolog-1 (HES-1) leads to up-regulation of neural helix-loop-helix factors, premature neurogenesis, and severe neural tube defects. Genes & Development, 9(24), 3136–3148. 10.1101/gad.9.24.3136 [DOI] [PubMed] [Google Scholar]
- Itoh K, Ossipova O, & Sokol SY (2014). GEF-H1 functions in apical constriction and cell intercalations and is essential for vertebrate neural tube closure. Journal of Cell Science, 127(11), 2542–2553. 10.1242/jcs.146811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe E, & Niswander L (2021). Loss of Grhl3 is correlated with altered cellular protrusions in the non-neural ectoderm during neural tube closure. Developmental Dynamics: An Official Publication of the American Association of Anatomists, 250(5), 732–744. 10.1002/dvdy.292 [DOI] [PubMed] [Google Scholar]
- Jenuwein T, & Allis CD (2001). Translating the Histone Code. Science. 10.1126/science.1063127 [DOI] [PubMed] [Google Scholar]
- Jones PA, & Baylin SB (2002). The fundamental role of epigenetic events in cancer. Nature Reviews Genetics, 3(6), 415–428. 10.1038/nrg816 [DOI] [PubMed] [Google Scholar]
- Juraver-Geslin HA, & Durand BC (2015). Early development of the neural plate: New roles for apoptosis and for one of its main effectors caspase-3. Genesis (New York, N.Y.: 2000), 53(2), 203–224. 10.1002/dvg.22844 [DOI] [PubMed] [Google Scholar]
- Juriloff DM, & Harris MJ (2012). A consideration of the evidence that genetic defects in planar cell polarity contribute to the etiology of human neural tube defects. Birth Defects Research. Part A, Clinical and Molecular Teratology, 94(10), 824–840. 10.1002/bdra.23079 [DOI] [PubMed] [Google Scholar]
- K Rebekah P, Tella S, Buragadda S, Tiruvatturu MK, & Akka J (2017). Interaction between Maternal and Paternal SHMT1 C1420T Predisposes to Neural Tube Defects in the Fetus: Evidence from Case-Control and Family-Based Triad Approaches. Birth Defects Research, 109(13), 1020–1029. 10.1002/bdr2.23623 [DOI] [PubMed] [Google Scholar]
- Kakebeen AD, & Niswander L (2021). Micronutrient imbalance and common phenotypes in neural tube defects. Genesis, 59(11), e23455. 10.1002/dvg.23455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller R (2002). Shaping the Vertebrate Body Plan by Polarized Embryonic Cell Movements. Science. 10.1126/science.1079478 [DOI] [PubMed] [Google Scholar]
- Kibar Z, Vogan KJ, Groulx N, Justice MJ, Underhill DA, & Gros P (2001). Ltap, a mammalian homolog of Drosophila Strabismus/Van Gogh, is altered in the mouse neural tube mutant Loop-tail. Nature Genetics, 28(3), 251–255. 10.1038/90081 [DOI] [PubMed] [Google Scholar]
- Kim T-H, Goodman J, Anderson KV, & Niswander L (2007). Phactr4 Regulates Neural Tube and Optic Fissure Closure by Controlling PP1-, Rb-, and E2F1-Regulated Cell-Cycle Progression. Developmental Cell, 13(1), 87–102. 10.1016/j.devcel.2007.04.018 [DOI] [PubMed] [Google Scholar]
- Kimura-Yoshida C, Mochida K, Ellwanger K, Niehrs C, & Matsuo I (2015). Fate Specification of Neural Plate Border by Canonical Wnt Signaling and Grhl3 is Crucial for Neural Tube Closure. EBioMedicine, 2(6), 513–527. 10.1016/j.ebiom.2015.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T (2007). Chromatin Modifications and Their Function. Cell, 128(4), 693–705. 10.1016/j.cell.2007.02.005 [DOI] [PubMed] [Google Scholar]
- Lee S, & Gleeson JG (2020). Closing in on Mechanisms of Open Neural Tube Defects. Trends in Neurosciences, 43(7), 519–532. 10.1016/j.tins.2020.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung K-Y, Pai YJ, Chen Q, Santos C, Calvani E, Sudiwala S, Savery D, Ralser M, Gross SS, Copp AJ, & Greene NDE (2017). Partitioning of One-Carbon Units in Folate and Methionine Metabolism Is Essential for Neural Tube Closure. Cell Reports, 21(7), 1795–1808. 10.1016/j.celrep.2017.10.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Bestor TH, & Jaenisch R (1992). Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell, 69(6), 915–926. 10.1016/0092-8674(92)90611-F [DOI] [PubMed] [Google Scholar]
- Li H, Wang X, Zhao H, Wang F, Bao Y, Guo J, Chang S, Wu L, Cheng H, Chen S, Zou J, Cui X, Niswander L, Finnell RH, Wang H, & Zhang T (2020). Low folate concentration impacts mismatch repair deficiency in neural tube defects. Epigenomics, 12(1), 5–18. 10.2217/epi-2019-0279 [DOI] [PubMed] [Google Scholar]
- Li H, Zhang J, Chen S, Wang F, Zhang T, & Niswander L (2018). Genetic contribution of retinoid-related genes to neural tube defects. Human Mutation, 39(4), 550–562. 10.1002/humu.23397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Zhang J, & Niswander L (2018). Zinc deficiency causes neural tube defects through attenuation of p53 ubiquitylation. Development, 145(24), dev169797. 10.1242/dev.169797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Weng H, Xu C, Reece EA, & Yang P (2012). Oxidative Stress–Induced JNK1/2 Activation Triggers Proapoptotic Signaling and Apoptosis That Leads to Diabetic Embryopathy. Diabetes, 61(8), 2084–2092. 10.2337/db11-1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Ren A, Wang L, Santos C, Huang Y, Jin L, Li Z, & Greene NDE (2019). Aberrant methylation of Pax3 gene and neural tube defects in association with exposure to polycyclic aromatic hydrocarbons. Clinical Epigenetics, 11(1), 13. 10.1186/s13148-019-0611-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeken MR (2020). Mechanisms of Congenital Malformations in Pregnancies with Pre-existing Diabetes. Current Diabetes Reports, 20(10), 54. 10.1007/s11892-020-01338-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Borchers AGM, Jolicoeur C, Rayburn H, Baker JC, & Tessier-Lavigne M (2004). PTK7/CCK-4 is a novel regulator of planar cell polarity in vertebrates. Nature, 430(6995), 93–98. 10.1038/nature02677 [DOI] [PubMed] [Google Scholar]
- Lupo PJ, Canfield MA, Chapa C, Lu W, Agopian AJ, Mitchell LE, Shaw GM, Waller DK, Olshan AF, Finnell RH, & Zhu H (2012). Diabetes and obesity-related genes and the risk of neural tube defects in the national birth defects prevention study. American Journal of Epidemiology, 176(12), 1101–1109. 10.1093/aje/kws190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangione F, & Martín-Blanco E (2018). The Dachsous/Fat/Four-Jointed Pathway Directs the Uniform Axial Orientation of Epithelial Cells in the Drosophila Abdomen. Cell Reports, 25(10), 2836–2850.e4. 10.1016/j.celrep.2018.11.036 [DOI] [PubMed] [Google Scholar]
- Mao J, McKean DM, Warrier S, Corbin JG, Niswander L, & Zohn IE (2010). The iron exporter ferroportin 1 is essential for development of the mouse embryo, forebrain patterning and neural tube closure. Development (Cambridge, England), 137(18), 3079–3088. 10.1242/dev.048744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marean A, Graf A, Zhang Y, & Niswander L (2011). Folic acid supplementation can adversely affect murine neural tube closure and embryonic survival. Human Molecular Genetics, 20(18), 3678–3683. 10.1093/hmg/ddr289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa V, Savery D, Ybot-Gonzalez P, Ferraro E, Rongvaux A, Cecconi F, Flavell R, Greene NDE, & Copp AJ (2009). Apoptosis is not required for mammalian neural tube closure. Proceedings of the National Academy of Sciences, 106(20), 8233–8238. 10.1073/pnas.0900333106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michie CA (1991). Neural tube defects in 18th century. The Lancet, 337(8739), 504. 10.1016/0140-6736(91)93453-G [DOI] [PubMed] [Google Scholar]
- Miranda TB, & Jones PA (2007). DNA methylation: The nuts and bolts of repression. Journal of Cellular Physiology, 213(2), 384–390. 10.1002/jcp.21224 [DOI] [PubMed] [Google Scholar]
- Molloy AM, Einri CN, Jain D, Laird E, Fan R, Wang Y, Scott JM, Shane B, Brody LC, Kirke PN, & Mills JL (2014). IS LOW IRON STATUS A RISK FACTOR FOR NEURAL TUBE DEFECTS? Birth Defects Research. Part A, Clinical and Molecular Teratology, 100(2), 100–106. 10.1002/bdra.23223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momb J, Lewandowski JP, Bryant JD, Fitch R, Surman DR, Vokes SA, & Appling DR (2013). Deletion of Mthfd1l causes embryonic lethality and neural tube and craniofacial defects in mice. Proceedings of the National Academy of Sciences of the United States of America, 110(2), 549–554. 10.1073/pnas.1211199110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, Adrian J, Kawli T, Davis CA, Dobin A, Kaul R, Halow J, Van Nostrand EL, Freese P, Gorkin DU, Shen Y, He Y, Mackiewicz M, Pauli-Behn F, Williams BA, … Weng Z (2020). Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature, 583(7818), 699–710. 10.1038/s41586-020-2493-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore LD, Le T, & Fan G (2013). DNA Methylation and Its Basic Function. Neuropsychopharmacology, 38(1), 23–38. 10.1038/npp.2012.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morriss-Kay G, & Tuckett F (1989). Immunohistochemical localisation of chondroitin sulphate proteoglycans and the effects of chondroitinase ABC in 9- to 11-day rat embryos. Development (Cambridge, England), 106(4), 787–798. [DOI] [PubMed] [Google Scholar]
- Murko C, Lagger S, Steiner M, Seiser C, Schoefer C, & Pusch O (2013). Histone deacetylase inhibitor Trichostatin A induces neural tube defects and promotes neural crest specification in the chicken neural tube. Differentiation, 85(1), 55–66. 10.1016/j.diff.2012.12.001 [DOI] [PubMed] [Google Scholar]
- Nakaya Y, Kuroda S, Katagiri YT, Kaibuchi K, & Takahashi Y (2004). Mesenchymal-epithelial transition during somitic segmentation is regulated by differential roles of Cdc42 and Rac1. Developmental Cell, 7(3), 425–438. 10.1016/j.devcel.2004.08.003 [DOI] [PubMed] [Google Scholar]
- Narisawa A, Komatsuzaki S, Kikuchi A, Niihori T, Aoki Y, Fujiwara K, Tanemura M, Hata A, Suzuki Y, Relton CL, Grinham J, Leung K-Y, Partridge D, Robinson A, Stone V, Gustavsson P, Stanier P, Copp AJ, Greene NDE, … Kure S (2012). Mutations in genes encoding the glycine cleavage system predispose to neural tube defects in mice and humans. Human Molecular Genetics, 21(7), 1496–1503. 10.1093/hmg/ddr585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann PE, Frankel WN, Letts VA, Coffin JM, Copp AJ, & Bernfield M (1994). Multifactorial inheritance of neural tube defects: Localization of the major gene and recognition of modifiers in ct mutant mice. Nature Genetics, 6(4), 357–362. 10.1038/ng0494-357 [DOI] [PubMed] [Google Scholar]
- Nikolopoulou E, Galea GL, Rolo A, Greene NDE, & Copp AJ (2017). Neural tube closure: Cellular, molecular and biomechanical mechanisms. Development (Cambridge, England), 144(4), 552–566. 10.1242/dev.145904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T, Honda H, & Takeichi M (2012). Planar cell polarity links axes of spatial dynamics in neural-tube closure. Cell, 149(5), 1084–1097. 10.1016/j.cell.2012.04.021 [DOI] [PubMed] [Google Scholar]
- Nishimura T, & Takeichi M (2008). Shroom3-mediated recruitment of Rho kinases to the apical cell junctions regulates epithelial and neuroepithelial planar remodeling. Development, 135(8), 1493–1502. 10.1242/dev.019646 [DOI] [PubMed] [Google Scholar]
- Nyholm MK, Abdelilah-Seyfried S, & Grinblat Y (2009). A novel genetic mechanism regulates dorsolateral hinge-point formation during zebrafish cranial neurulation. Journal of Cell Science, 122(Pt 12), 2137–2148. 10.1242/jcs.043471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, & Li E (1999). DNA Methyltransferases Dnmt3a and Dnmt3b Are Essential for De Novo Methylation and Mammalian Development. Cell, 99(3), 247–257. 10.1016/S0092-8674(00)81656-6 [DOI] [PubMed] [Google Scholar]
- O’Rahilly R, & Müller F (1994). Neurulation in the normal human embryo. Ciba Foundation Symposium, 181, 70–82; discussion 82–89. 10.1002/9780470514559.ch5 [DOI] [PubMed] [Google Scholar]
- O’Rahilly R, & Müller F (2002). The two sites of fusion of the neural folds and the two neuropores in the human embryo. Teratology, 65(4), 162–170. 10.1002/tera.10007 [DOI] [PubMed] [Google Scholar]
- Ouyang S, Li Y, Liu Z, Chang H, & Wu J (2013). Association between MTR A2756G and MTRR A66G polymorphisms and maternal risk for neural tube defects: A meta-analysis. Gene, 515(2), 308–312. 10.1016/j.gene.2012.11.070 [DOI] [PubMed] [Google Scholar]
- Padmanabhan N, Jia D, Geary-Joo C, Wu X, Ferguson-Smith AC, Fung E, Bieda MC, Snyder FF, Gravel RA, Cross JC, & Watson ED (2013). Mutation in folate metabolism causes epigenetic instability and transgenerational effects on development. Cell, 155(1), 81–93. 10.1016/j.cell.2013.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parle-McDermott A, Pangilinan F, O’Brien KK, Mills JL, Magee AM, Troendle J, Sutton M, Scott JM, Kirke PN, Molloy AM, & Brody LC (2009). A Common Variant in MTHFD1L is Associated with Neural Tube Defects and mRNA Splicing Efficiency. Human Mutation, 30(12), 1650–1656. 10.1002/humu.21109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partanen A, Motoyama J, & Hui CC (1999). Developmentally regulated expression of the transcriptional cofactors/histone acetyltransferases CBP and p300 during mouse embryogenesis. The International Journal of Developmental Biology, 43(6), 487–494. [PubMed] [Google Scholar]
- Pei P, cheng X, Yu J, Shen J, Li X, Wu J, Wang S, & Zhang T (2019). Folate deficiency induced H2A ubiquitination to lead to downregulated expression of genes involved in neural tube defects. Epigenetics & Chromatin, 12(1), 69. 10.1186/s13072-019-0312-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piedrahita JA, Oetama B, Bennett GD, van Waes J, Kamen BA, Richardson J, Lacey SW, Anderson RG, & Finnell RH (1999). Mice lacking the folic acid-binding protein Folbp1 are defective in early embryonic development. Nature Genetics, 23(2), 228–232. 10.1038/13861 [DOI] [PubMed] [Google Scholar]
- Prevention of neural tube defects: Results of the Medical Research Council Vitamin Study. MRC Vitamin Study Research Group. (1991). Lancet (London, England), 338(8760), 131–137. [PubMed] [Google Scholar]
- Pujadas E, & Feinberg AP (2012). Regulated Noise in the Epigenetic Landscape of Development and Disease. Cell, 148(6), 1123–1131. 10.1016/j.cell.2012.02.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punovuori K, Migueles RP, Malaguti M, Blin G, Macleod KG, Carragher NO, Pieters T, van Roy F, Stemmler MP, & Lowell S (2019). N-cadherin stabilises neural identity by dampening anti-neural signals. Development (Cambridge, England), 146(21), dev183269. 10.1242/dev.183269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyrgaki C, Liu A, & Niswander L (2011). Grainyhead-like 2 regulates neural tube closure and adhesion molecule expression during neural fold fusion. Developmental Biology, 353(1), 38–49. 10.1016/j.ydbio.2011.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Lin S, Bloom MS, Wang X, Ye B, Nie Z, Ou Y, Mai J, Wu Y, Gao X, Xiao X, Tan H, Liu X, Chen J, & Zhuang J (2021). Maternal folic acid supplementation mediates the associations between maternal socioeconomic status and congenital heart diseases in offspring. Preventive Medicine, 143, 106319. 10.1016/j.ypmed.2020.106319 [DOI] [PubMed] [Google Scholar]
- Ramakrishnan A-B, Chen L, Burby PE, & Cadigan KM (2021). Wnt target enhancer regulation by a CDX/TCF transcription factor collective and a novel DNA motif. Nucleic Acids Research, 49(15), 8625–8641. 10.1093/nar/gkab657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray HJ, & Niswander LA (2016). Grainyhead-like 2 downstream targets act to suppress epithelial-to-mesenchymal transition during neural tube closure. Development, 143(7), 1192–1204. 10.1242/dev.129825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece EA, Homko CJ, Wu YK, & Wiznitzer A (1993). Metabolic fuel mixtures and diabetic embryopathy. Clinics in Perinatology, 20(3), 517–532. [PubMed] [Google Scholar]
- Rifat Y, Parekh V, Wilanowski T, Hislop NR, Auden A, Ting SB, Cunningham JM, & Jane SM (2010). Regional neural tube closure defined by the Grainy head-like transcription factors. Developmental Biology, 345(2), 237–245. 10.1016/j.ydbio.2010.07.017 [DOI] [PubMed] [Google Scholar]
- Rogner UC, Spyropoulos DD, Le Novère N, Changeux J-P, & Avner P (2000). Control of neurulation by the nucleosome assembly protein-1–like 2. Nature Genetics, 25(4), 431–435. 10.1038/78124 [DOI] [PubMed] [Google Scholar]
- Rolo A, Savery D, Escuin S, de Castro SC, Armer HEJ, Munro PMG, Molè MA, Greene NDE, & Copp AJ (2016). Regulation of cell protrusions by small GTPases during fusion of the neural folds. ELife, 5, e13273. 10.7554/eLife.13273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JA, Wang Z, Schones DE, Zhao K, DeSalle R, & Zhang MQ (2009). Determination of enriched histone modifications in non-genic portions of the human genome. BMC Genomics, 10, 143. 10.1186/1471-2164-10-143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg SP, da Costa MP, Sequeira JM, Cracco J, Roberts JL, Weedon J, & Quadros EV (2004). Autoantibodies against folate receptors in women with a pregnancy complicated by a neural-tube defect. The New England Journal of Medicine, 350(2), 134–142. 10.1056/NEJMoa031145 [DOI] [PubMed] [Google Scholar]
- Sabatino JA, Stokes BA, & Zohn IE (2017). Prevention of neural tube defects in Lrp2 mutant mouse embryos by folic acid supplementation. Birth Defects Research, 109(1), 16–26. 10.1002/bdra.23589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saburi S, Hester I, Fischer E, Pontoglio M, Eremina V, Gessler M, Quaggin SE, Harrison R, Mount R, & McNeill H (2008). Loss of Fat4 disrupts PCP signaling and oriented cell division and leads to cystic kidney disease. Nature Genetics, 40(8), 1010–1015. 10.1038/ng.179 [DOI] [PubMed] [Google Scholar]
- Salbaum JM, & Kappen C (2010). Neural tube defect genes and maternal diabetes during pregnancy. Birth Defects Research. Part A, Clinical and Molecular Teratology, 88(8), 601–611. 10.1002/bdra.20680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Ferras O, Bernas G, Laberge-Perrault E, & Pilon N (2014). Induction and dorsal restriction of Paired-box 3 (Pax3) gene expression in the caudal neuroectoderm is mediated by integration of multiple pathways on a short neural crest enhancer. Biochimica Et Biophysica Acta, 1839(7), 546–558. 10.1016/j.bbagrm.2014.04.023 [DOI] [PubMed] [Google Scholar]
- Savory JGA, Mansfield M, Rijli FM, & Lohnes D (2011). Cdx mediates neural tube closure through transcriptional regulation of the planar cell polarity gene Ptk7. Development (Cambridge, England), 138(7), 1361–1370. 10.1242/dev.056622 [DOI] [PubMed] [Google Scholar]
- Saxonov S, Berg P, & Brutlag DL (2006). A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proceedings of the National Academy of Sciences of the United States of America, 103(5), 1412–1417. 10.1073/pnas.0510310103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senga K, Mostov KE, Mitaka T, Miyajima A, & Tanimizu N (2012). Grainyhead-like 2 regulates epithelial morphogenesis by establishing functional tight junctions through the organization of a molecular network among claudin3, claudin4, and Rab25. Molecular Biology of the Cell, 23(15), 2845–2855. 10.1091/mbc.E12-02-0097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakèd M, Weissmüller K, Svoboda H, Hortschansky P, Nishino N, Wölfl S, & Tucker KL (2008). Histone Deacetylases Control Neurogenesis in Embryonic Brain by Inhibition of BMP2/4 Signaling. PLOS ONE, 3(7), e2668. 10.1371/journal.pone.0002668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M, Vaughn A, Momb J, & Appling DR (2019). Deletion of neural tube defect-associated gene Mthfd1l causes reduced cranial mesenchyme density. Birth Defects Research, 111(19), 1520–1534. 10.1002/bdr2.1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo A, & Wallingford JB (2014). PCP and septins compartmentalize cortical actomyosin to direct collective cell movement. Science (New York, N.Y.), 343(6171), 649–652. 10.1126/science.1243126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes BA, Sabatino JA, & Zohn IE (2017). High levels of iron supplementation prevents neural tube defects in the Fpn1ffe mouse model. Birth Defects Research, 109(2), 81–91. 10.1002/bdra.23542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stottmann RW, Berrong M, Matta K, Choi M, & Klingensmith J (2006). The BMP antagonist Noggin promotes cranial and spinal neurulation by distinct mechanisms. Developmental Biology, 295(2), 647–663. 10.1016/j.ydbio.2006.03.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudiwala S, Palmer A, Massa V, Burns AJ, Dunlevy LPE, de Castro SCP, Savery D, Leung K-Y, Copp AJ, & Greene NDE (2019). Cellular mechanisms underlying Pax3-related neural tube defects and their prevention by folic acid. Disease Models & Mechanisms, 12(11), dmm042234. 10.1242/dmm.042234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh JR, Herbig AK, & Stover PJ (2001). New Perspectives on Folate Catabolism. Annual Review of Nutrition, 21(1), 255–282. 10.1146/annurev.nutr.21.1.255 [DOI] [PubMed] [Google Scholar]
- Takai D, & Jones PA (2002). Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proceedings of the National Academy of Sciences, 99(6), 3740–3745. 10.1073/pnas.052410099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian T, Cao X, Chen Y, Jin L, Li Z, Han X, Lin Y, Wlodarczyk BJ, Finnell RH, Yuan Z, Wang L, Ren A, & Lei Y (2021). Somatic and de novo Germline Variants of MEDs in Human Neural Tube Defects. Frontiers in Cell and Developmental Biology, 9. https://www.frontiersin.org/article/10.3389/fcell.2021.641831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toriyama M, Toriyama M, Wallingford JB, & Finnell RH (2017). Folate-dependent methylation of septins governs ciliogenesis during neural tube closure. The FASEB Journal, 31(8), 3622–3635. 10.1096/fj.201700092R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urase K, Kouroku Y, Fujita E, & Momoi T (2003). Region of caspase-3 activation and programmed cell death in the early development of the mouse forebrain. Brain Research. Developmental Brain Research, 145(2), 241–248. 10.1016/j.devbrainres.2003.07.002 [DOI] [PubMed] [Google Scholar]
- van der Put NM, Steegers-Theunissen RP, Frosst P, Trijbels FJ, Eskes TK, van den Heuvel LP, Mariman EC, den Heyer M, Rozen R, & Blom HJ (1995). Mutated methylenetetrahydrofolate reductase as a risk factor for spina bifida. Lancet (London, England), 346(8982), 1070–1071. 10.1016/s0140-6736(95)91743-8 [DOI] [PubMed] [Google Scholar]
- van Straaten HW, Hekking JW, Consten C, & Copp AJ (1993). Intrinsic and extrinsic factors in the mechanism of neurulation: Effect of curvature of the body axis on closure of the posterior neuropore. Development (Cambridge, England), 117(3), 1163–1172. [DOI] [PubMed] [Google Scholar]
- Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E, McAnally J, Pomajzl C, Shelton JM, Richardson JA, Karsenty G, & Olson EN (2004). Histone Deacetylase 4 Controls Chondrocyte Hypertrophy during Skeletogenesis. Cell, 119(4), 555–566. 10.1016/j.cell.2004.10.024 [DOI] [PubMed] [Google Scholar]
- Wallingford JB, & Harland RM (2002). Neural tube closure requires Dishevelled-dependent convergent extension of the midline. Development, 129(24), 5815–5825. 10.1242/dev.00123 [DOI] [PubMed] [Google Scholar]
- Wang L, Lin S, Yi D, Huang Y, Wang C, Jin L, Liu J, Zhang Y, & Ren A (2017). Apoptosis, Expression of PAX3 and P53, and Caspase Signal in Fetuses with Neural Tube Defects. Birth Defects Research, 109(19), 1596–1604. 10.1002/bdr2.1094 [DOI] [PubMed] [Google Scholar]
- Wang L, Lin S, Zhang J, Tian T, Jin L, & Ren A (2017). Fetal DNA hypermethylation in tight junction pathway is associated with neural tube defects: A genome-wide DNA methylation analysis. Epigenetics, 12(2), 157–165. 10.1080/15592294.2016.1277298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, De Marco P, Merello E, Drapeau P, Capra V, & Kibar Z (2015). Role of the planar cell polarity gene Protein tyrosine kinase 7 in neural tube defects in humans. Birth Defects Research Part A: Clinical and Molecular Teratology, 103(12), 1021–1027. 10.1002/bdra.23422 [DOI] [PubMed] [Google Scholar]
- Wei X, Li H, Miao J, Zhou F, Liu B, Wu D, Li S, Wang L, Fan Y, Wang W, & Yuan Z (2012). Disturbed apoptosis and cell proliferation in developing neuroepithelium of lumbo-sacral neural tubes in retinoic acid-induced spina bifida aperta in rat. International Journal of Developmental Neuroscience, 30(5), 375–381. 10.1016/j.ijdevneu.2012.03.340 [DOI] [PubMed] [Google Scholar]
- Wilde JJ, Petersen JR, & Niswander L (2014). Genetic, Epigenetic, and Environmental Contributions to Neural Tube Closure. Annual Review of Genetics, 48(1), 583–611. 10.1146/annurev-genet-120213-092208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J, Mai CT, Mulinare J, Isenburg J, Flood TJ, Ethen M, Frohnert B, Kirby RS, & Centers for Disease Control and Prevention. (2015). Updated estimates of neural tube defects prevented by mandatory folic Acid fortification—United States, 1995–2011. MMWR. Morbidity and Mortality Weekly Report, 64(1), 1–5. [PMC free article] [PubMed] [Google Scholar]
- Williams M, Yen W, Lu X, & Sutherland A (2014). Distinct Apical and Basolateral Mechanisms Drive Planar Cell Polarity-Dependent Convergent Extension of the Mouse Neural Plate. Developmental Cell, 29(1), 34–46. 10.1016/j.devcel.2014.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarczyk BJ, Tang LS, Triplett A, Aleman F, & Finnell RH (2006). Spontaneous neural tube defects in splotch mice supplemented with selected micronutrients. Toxicology and Applied Pharmacology, 213(1), 55–63. 10.1016/j.taap.2005.09.008 [DOI] [PubMed] [Google Scholar]
- Xu C, Shen W-B, Reece EA, Hasuwa H, Harman C, Kaushal S, & Yang P (2021). Maternal diabetes induces senescence and neural tube defects sensitive to the senomorphic rapamycin. Science Advances. 10.1126/sciadv.abf5089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi M, Kabata M, Tanaka A, Ukai T, Ohta S, Nakabayashi K, Shimizu M, Hata K, Meissner A, Yamamoto T, & Yamada Y (2020). Identification of distinct loci for de novo DNA methylation by DNMT3A and DNMT3B during mammalian development. Nature Communications, 11(1), 3199. 10.1038/s41467-020-16989-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Shinotsuka N, Nonomura K, Takemoto K, Kuida K, Yosida H, & Miura M (2011). Live imaging of apoptosis in a novel transgenic mouse highlights its role in neural tube closure. Journal of Cell Biology, 195(6), 1047–1060. 10.1083/jcb.201104057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan B, Neilson KM, & Moody SA (2009). Notch signaling downstream of foxD5 promotes neural ectodermal transcription factors that inhibit neural differentiation. Developmental Dynamics : An Official Publication of the American Association of Anatomists, 238(6), 1358–1365. 10.1002/dvdy.21885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Wang G, Luo X, Zhang P, Peng S, Cheng X, Wang M, & Yang X (2021). Endoplasmic reticulum stress-related calcium imbalance plays an important role on Zinc oxide nanoparticles-induced failure of neural tube closure during embryogenesis. Environment International, 152, 106495. 10.1016/j.envint.2021.106495 [DOI] [PubMed] [Google Scholar]
- Yang P, Li X, Xu C, Eckert RL, Reece EA, Zielke HR, & Wang F (2013). Maternal Hyperglycemia Activates an ASK1–FoxO3a–Caspase 8 Pathway That Leads to Embryonic Neural Tube Defects. Science Signaling. 10.1126/scisignal.2004020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ybot-Gonzalez P, Cogram P, Gerrelli D, & Copp AJ (2002). Sonic hedgehog and the molecular regulation of mouse neural tube closure. Development (Cambridge, England), 129(10), 2507–2517. [DOI] [PubMed] [Google Scholar]
- Ybot-Gonzalez P, Gaston-Massuet C, Girdler G, Klingensmith J, Arkell R, Greene NDE, & Copp AJ (2007). Neural plate morphogenesis during mouse neurulation is regulated by antagonism of Bmp signalling. Development (Cambridge, England), 134(17), 3203–3211. 10.1242/dev.008177 [DOI] [PubMed] [Google Scholar]
- Zabihi S, & Loeken MR (2010). Understanding diabetic teratogenesis: Where are we now and where are we going? Birth Defects Research. Part A, Clinical and Molecular Teratology, 88(10), 779–790. 10.1002/bdra.20704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaganjor I, Sekkarie A, Tsang BL, Williams J, Razzaghi H, Mulinare J, Sniezek JE, Cannon MJ, & Rosenthal J (2016). Describing the Prevalence of Neural Tube Defects Worldwide: A Systematic Literature Review. PLOS ONE, 11(4), e0151586. 10.1371/journal.pone.0151586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Behringer RR, & de Crombrugghe B (1996). Prenatal folic acid treatment suppresses acrania and meroanencephaly in mice mutant for the Cart1 homeobox gene. Nature Genetics, 13(3), 275–283. 10.1038/ng0796-275 [DOI] [PubMed] [Google Scholar]
- Zheng J, Lu X, Liu H, Zhao P, Li K, & Li L (2015). MTHFD1 polymorphism as maternal risk for neural tube defects: A meta-analysis. Neurological Sciences: Official Journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology, 36(4), 607–616. 10.1007/s10072-014-2035-7 [DOI] [PubMed] [Google Scholar]
- Zohn IE (2012). Mouse as a model for multifactorial inheritance of neural tube defects. Birth Defects Research. Part C, Embryo Today: Reviews, 96(2), 193–205. 10.1002/bdrc.21011 [DOI] [PubMed] [Google Scholar]