

Abstract
Background:
Recent findings demonstrate that single nucleotide variants can cause non-obstructive azoospermia (NOA). In contrast, copy number variants (CNVs) were only analysed in few studies in infertile men. Some have reported a higher prevalence of CNVs in infertile versus fertile men.
Objectives:
This study aimed to elucidate if CNVs are associated with NOA.
Materials and methods:
We performed array-based comparative genomic hybridisation (aCGH) in 37 men with meiotic arrest, 194 men with Sertoli cell-only phenotype, and 21 control men. We filtered our data for deletions affecting genes and prioritised the affected genes according to the literature search. Prevalence of CNVs was compared between all groups. Exome data of 2,030 men were screened to detect further genetic variants in prioritised genes. Modelling was performed for the protein encoded by the novel candidate gene TEKT5 and we stained for TEKT5 in human testicular tissue.
Results:
We determined the cause of infertility in two individuals with homozygous deletions of SYCE1 and in one individual with a heterozygous deletion of SYCE1 combined with a likely pathogenic missense variant on the second allele. We detected heterozygous deletions affecting MLH3, EIF2B2, SLX4, CLPP and TEKT5, in one subject each. CNVs were not detected more frequently in infertile men compared with controls.
Discussion:
While SYCE1 and MLH3 encode known meiosis-specific proteins, much less is known about the proteins encoded by the other identified candidate genes, warranting further analyses. We were able to identify the cause of infertility in one out of the 231 infertile men by aCGH and in two men by using exome sequencing data.
Conclusion:
As aCGH and exome sequencing are both expensive methods, combining both in a clinical routine is not an effective strategy. Instead, using CNV calling from exome data has recently become more precise, potentially making aCGH dispensable.
Keywords: CNV, meiotic arrest, non-obstructive azoospermia, SCO, SYCE1, WES
1 |. INTRODUCTION
Infertility is a common condition that affects up to 10%–15% of all couples. While the clinical causes are equally distributed between men and women,1 around 10% of men in infertile couples exhibit azoospermia. With this condition, a man cannot father a child naturally.
Azoospermic men are offered a testicular biopsy with the aim of testicular sperm extraction (TESE); if sperm are gained, these can then be used for assisted reproductive technology (ART). Yet, TESE is only successful in around 50% of men with non-obstructive azoospermia (NOA).2,3 According to histological analyses, one testicular phenotype that is a common cause of NOA is Sertoli cell-only (SCO) phenotype when tubules contain no germ cells. Another cause of NOA is meiotic arrest (MeiA), if men exhibit incomplete spermatogenesis, interrupted at meiosis, most commonly during prophase I. In the case of complete bilateral SCO or MeiA, no spermatozoa can be obtained by biopsy and TESE, making it impossible for those men to father a child. Thus, deciphering the underlying genetic causes for SCO and MeiA may help better predict the success of TESE and subsequent ART.
Previous studies have primarily identified single nucleotide variations (SNVs) in several genes as causes for NOA. However, in other diseases such as mental retardation, also copy number variations (CNVs) play a relevant role.4 Fittingly, a higher prevalence of CNVs, most significantly deletions, has been reported in infertile men versus fertile men,5 but results were not conclusive concerning recurrently affected genes—most likely due to rather small group sizes and heterogeneous phenotypes.
To address these issues, we performed array-based comparative genomic hybridisation (aCGH) in larger numbers of highly selected men with complete bilateral SCO and MeiA to identify deletions undetectable by conventional karyotyping. For the first time, we synergistically analysed whole exome sequencing (WES) data in the same men.
2 |. MATERIALS AND METHODS
2.1 |. Study population for array-based comparative genomic hybridisation
The study cohort for aCGH was retrospectively selected from men who attended the Centre of Reproductive Medicine and Andrology (CeRA) in Münster. The cohort comprised 231 men with unexplained azoospermia who underwent testicular biopsy and TESE. Of this cohort, 194 men had complete bilateral SCO (N = 37 from Tüttelmann et al.,6 were re-analysed and included), and 37 men had MeiA. All known clinical and genetic causes for azoospermia (e.g., chemotherapy, AZF deletions, chromosomal aberrations) were excluded. As control group, we used the data from men with normal semen parameters according to WHO criteria7 (N = 21, previously published6). A list of all patients, which were derived from the previous study6 can be found in the supporting information. Descriptive statistics of all men included in the aCGH cohort is shown in Table S1.
2.2 |. Ethics approval
The study protocol was approved by the Ethics Committees (Ref. No. 2010–578-f-S) in accordance with the Helsinki Declaration of 1975.
2.3 |. Array-based comparative genomic hybridisation
Genomic DNA was extracted from peripheral blood using standard methods. CGH was conducted using the Human Genome CGH Microarray Kit 400K (Agilent Technologies, Santa Clara, CA, USA). The median probe spacing was 5.3 kb. The infertile men’s DNA was compared to a pooled male control DNA (Human Genomic DNA Male, Promega, Walldorf, Germany) and hybridised against DNA oligonucleotides representing the whole human genome. Labelling and hybridisation were performed using the SureTag DNA Labeling Kit and Oligo aCGH/ChIP-on-chip Hybridisation Kit (Agilent Technologies) according to the manufacturer’s protocol. Double digestion of 1 μg of both infertile men’s and control DNA was carried out with AluI and RsaI. Consecutive labelling of the DNA using Cy5-dUTP for the infertile men and Cy3-dUTP for the controls was done. Purification of all samples by filtration followed. Hybridisation of the pooled infertile men and control DNA with 25 μg of competitor (COT) DNA was conducted for 40 h at 65°C in the hybridisation oven (Agilent Technologies, Santa Clara, CA, USA) followed by washing steps. The arrays were scanned using a microarray scanner (G2565BA, Agilent Technologies, Santa Clara, CA, USA). A computer-based analysis followed, measuring the spot intensities using the ‘Feature Extraction Software’ (version 10.7, Agilent Technologies, Santa Clara, CA, USA) and Genomic Workbench (Version 7.0.4.0) with the following parameters: aberration algorithm ADM-2, threshold 6.0, fuzzy zero, centralisation and moving average window 1 Mb. Signals larger than four adjacent probes were considered to be a genomic CNV.
2.4 |. Filtering for relevant copy number variants detected by array-based comparative genomic hybridisation
The CNVs were categorised into deletions and duplications. As deletions are assumed to be more likely pathogenic, only deletions were taken into account for further analyses. To consider a putative CNV as a deletion, we set as a criterion a negative log value of below −0.6. CNVs with a log value between −0.6 and −2.5 were considered as heterozygous, while those with a log value below −2.5 were considered as homozygous. CNVs with a log value below −2.5 affecting the sex chromosomes were considered as hemizygous. These filtering criteria slightly differed from those applied in our previous study,6 explaining the small deviations in the number of detected CNVs. Deletions also found in a homozygous state in controls were disregarded if the control deletion covered the infertile man’s deletion with breakpoints varying with a maximum of 1,000 base pairs. Additionally deletions found in a heterozygous state in both infertile men and control men were disregarded. Deletions found in a homozygous state in infertile men while being found in a heterozygous state in a control were not excluded. Also, we excluded deletions that did not affect at least part of a coding gene. Genes that were deleted in more than three men were considered to be a common, non-pathogenic polymorphism and were not considered further. The remaining genes affected by deletions were ranked according to their expression in the testis as available from the GTEx Portal.8 Only protein-coding genes with the highest expression in the testis were further taken into account.
For the genes that remained, we performed an extensive database and literature search using Mouse Genome Informatics (MGI) and PubMed. A special focus was placed on genes in which the corresponding complete knockout (KO) or testis-specific KO mice exhibited reduced fertility or spermatogenic impairment. If not previously described for the respective gene, the mode of inheritance of prioritised genes was determined using DOMINO9 and the loss-of-function observed/expected upper bound fraction (o/e ratio) available in gnomAD database. Deletions in prioritised genes were validated by quantitative Polymerase Chain Reaction (qPCR) using two primer pairs spanning the respective deletion. The primer sequences can be found in Table S2.
2.5 |. Statistical analysis
The prevalence of all CNVs as well as deletions/duplications detected by aCGH was compared between men with SCO, MeiA, and the control group. This analysis was also done with regards to the prevalence of CNVs in each group on each chromosome and with regards to the size of detected CNVs in each group. Comparisons between infertile men and fertile controls were carried out using the two-sample t-test in the case of normally distributed data (e.g., number of CNVs). Otherwise the non-parametric Mann–Whitney test was used. P-values less than 0.05 were considered statistically significant. All calculations were performed with GraphPad Prism version 8.3.1 (GraphPad Software, San Diego, CA, USA).
2.6 |. Whole exome sequencing
WES was performed in the Male Reproductive Genomics (MERGE) study that currently comprised 2,030 men with a diverse spectrum of male infertility phenotypes and 41 normozoospermic controls as previously described.10 A majority of 1,777 men were affected by severe spermatogenic failure (severe oligo-, crypto, or azoospermia). Reads were aligned using GRCh37/hg19. Men with previously identified (genetic) causes for infertility and men with testicular malignancies were excluded from the analyses. WES data of the men with potentially relevant deletions (M1369, M663, M921, M226, M1635, M877 and M1281), detected by aCGH, were analysed to validate the aCGH findings. Additionally, WES data of all men from the MERGE study, including those, in which no aCGH has been performed, were analysed for deletions in the five prioritised genes. To this end, CNVs were called using GATK’s GermlineCNVCaller.11 Seventy-two samples were excluded before CNV calling due to poor quality, high variability or capture panels with too few reference samples for normalisation.
Furthermore, the frequency of CNVs in the general population affecting the prioritised genes was determined using gnomAD structural variation database (gnomAD SVs v2.1) and database of genomic variants (DGV).
Additionally, the WES data of our MERGE study were analysed with a focus on rare stop-gain, frameshift and missense variants (minor allele frequency [MAF] < 0.01 in the gnomAD database, gnomAD v2.1.1) with a Combined Annotation Dependent Depletion (CADD) score > 20 in the prioritised genes. In autosomal-recessive genes, only homozygous or likely compound-heterozygous variants were considered as relevant.
In men with heterozygous deletions in one of the prioritised genes with an autosomal-recessive mode of inheritance, we screened the WES data to identify rare variants (MAF < 0.01) on the second allele, indicating compound heterozygosity.
Next, all patients with potentially relevant CNVs and SNVs listed in Tables 1 and 2 were examined for possible other causes of their infertility. To this end, the exome sequencing data of M1369, M663, M921, M226, M1635, M877, M1281, M2681 and M3187 were screened for rare (MAF < 0.01 for autosomal recessive genes and < 0.001 for autosomal dominant genes, according to the gnomAD database), possibly pathogenic variants (stop-gain, frameshift, splice site and missense variants with a CADD score > 20) in 230 genes that were classified with at least a limited level of evidence for being associated with male infertility according to a recent review.12 The list of genes can be found in the supporting information. Additionally, the genes ADAD2, GCNA, MAJIN, MSH4, MSH5, M1AP, RAD21L1, RNF212, SHOC1, STAG3, SYCP2, TERB1, TERB2 and TRIM71, which have recently been described to be associated with NOA (Table S3), were screened. Only variants detected in genes that quantitatively impair spermatogenesis, leading to non-syndromic infertility were considered for further analyses. Variants in recessive genes were only considered if at least two heterozygous variants in this gene or a homozygous variant were identified in one individual. The pathogenicity of identified variants was assessed based on the guidelines provided by the American College of Medical Genetics (ACMG) and the Association of Molecular Pathology (AMP).13
TABLE 1.
Deletions in prioritised genes in azoospermic men
| Men | Testicular phenotype | Gene symbols; prioritised genes in bold | Region | Start | End | Size (kb) | Deletion of prioritised gene | Genotype | o/e ratio of prioritized genea | Inheritance of prioritized geneb |
|---|---|---|---|---|---|---|---|---|---|---|
| M136916 | MeiA | SYCE1, CYP2E1, SCART1 | 10q26.3 | 135281682 | 135378761 | 97 | Complete | Homozygous | 0.63 | Very likely recessive |
| M3187 | MeiA | SYCE1, CYP2E1, SCART1 | 10q26.3 | 135252347 | 135378802 | 127 | Complete | Homozoygous | 0.63 | Very likely recessive |
| M2681 | SCO | SYCE1, CYP2E1, SCART1 | 10q26.3 | 135256762 | 135393215 | 137 | Complete | Heterozygous | 0.63 | Very likely recessive |
| M663 | SCO | SYCE1, CYP2E1, SCART1 | 10q26.3 | 135252327 | 135378761 | 126 | Complete | Heterozygous | 0.63 | Very likely recessive |
| M921 | MeiA | SYCE1, CYP2E1, SCART1 | 10q26.3 | 135252327 | 135378761 | 126 | Complete | Heterozygous | 0.63 | Very likely recessive |
| M226 | SCO | MLH3, EIF2B2 | 14q24.3 | 75470567 | 75490715 | 20 | Partial | Heterozygous | 0.41 | Very likely recessive |
| M1635 | SCO | SLX4, CLUAP1, NLRC3 | 16p13.3 | 3586849 | 3657147 | 70 | Partial | Heterozygous | 0.68 | Very likely recessive |
| M877 | SCO | TEKT5 | 16p13.13 | 10721351 | 10825301 | 103 | Complete | Heterozygous | 1.22 | Very likely recessive |
| M1281 | SCO | CLPP | 19p13.3 | 6340541 | 6369092 | 28 | Complete | Heterozygous | 0.18 | Very likely recessive |
Abbreviations: MeiA, meiotic arrest; o/e, loss-of-function observed/expected upper bound fraction; SCO, Sertoli cell-only phenotype.
According to gnomAD v2.1.1.
According to DOMINO.
TABLE 2.
Genetic data of infertile men carrying possibly relevant single nucleotide variants in the genes SYCE1, TEKT5 and SLX4
| Individual | Genetic variant |
In silico prediction programs |
MAF | o% ratio | Predicted mode of inheritance | ACMG-AMP classification | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Transcript | Inh. pattern | cDNA change | Protein change | CADD | PolyPhen/SIFT/MutationTaster | gnomAD v2.1.1 | gnomAD v2.1.1 | DOMINO | ||
| M2681 | SYCE1 | NM 130784.4 | AR | c.[430C>T];[0] | p.[Arg144Trp];[0] | 282 | P/D/N | 0,00003187 | 0.63 | Very likely recessive | Likely pathogenic |
| M1767 | TEKT5 | NM 144674.1 | AR | c.[263G>A(;)1022C>T] | p.(Arg88His)(;) (Ala341Va I) | 25.6; 26.0 | P/D/D; P/D/D | 0.00002152; 0.0001034 | 1.22 | Very likely recessive | VUSNUS |
| PIT10 | TEKT5 | NM 144674.1 | AR | c.[531G>C(;)938C>G] | p.(G1u177Asp)(;) (Ser313Cys) | 21.2; 25.7 | B/D/D; D/T/D | 0.002315; 0.00007426 | 1.22 | Very likely recessive | VUSNUS |
| M1626 | SLX4 | NM 032444.3 | AR | c.[2359G>A(;)5468G>A] | p.(G1u787Lys)(;) (Arg1823GIn) | 22.8; 22.7 | B/T/N; B/T/N | 0.001277; 0.00001421 | 0.68 | Very likely recessive | VUSNUS |
| M2023 | SLX4 | NM 032444.3 | AR | c.[2359G>A];[2359G>A] | p.[G1u787Lys]; [G1u787Lys] | 22.7 | B/T/N | 0.001277 | 0.68 | Very likely recessive | VUSNUS |
Abbreviations: AR, autosomal recessive; B, benign; D, damaging, deleterious or disease causing; Inh, inheritance; LoF: loss-of-function; MAF, minor allele frequency; NA, not available; ND, not determined; oe, loss-of-function observed/expected upper bound fraction; P, polymorphism; T, tolerated; VUS, variant of unknown significance.
Deletions, which were primarily detected via analysis of the WES data, were confirmed by qPCR and by single nucleotide polymorphism-array (SNP-array), because array-CGH was no longer available. SNP-array was performed as described in the supporting information. Primers for qPCR are shown in Table S2.
2.7 |. Replication study of TEKT5 variants in an independent population
To validate our findings concerning TEKT5, we sequenced the whole coding region of this gene in an independent cohort of 126 azoospermic men (NOA/hypospermatogenesis, MeiA, SCO) enrolled at Magee Research Institute, Department of OB/GYN and Reproductive Sciences, School of Medicine, University of Pittsburgh in Pittsburgh, PA, USA as described previously.14
2.8 |. In silico analysis of TEKT5
The secondary structure of the TEKT5 protein was predicted using the protein prediction server PSIPred and protein structure and function prediction server I-Tasser. The Pfam database was used to search for protein domains in TEKT5. To visualise the three-dimensional (3D) structure, we used the Phyre2 intensive algorithm to generate a homology model of TEKT5. Structural models were designed with PyMOL Molecular Graphics System (Schrödinger). The in silico program HOPE15 was applied to predict functional consequences of identified TEKT5 variants.
2.9 |. Immunohistochemistry of TEKT5 in fetal and adult human and mouse testes
Paraffin sections from human and murine fetal and adult testis tissue were dewaxed using ProTaqs Clear (ProTaqs Clear, Quartett Immunodiagnostika and Biotechnologie, Berlin, Germany) and rehydrated in decreasing concentrations of ethanol. After rinsing with distilled water and tris-buffered saline (TBS), heat-induced antigen retrieval was performed with citrate buffer (pH 6) and a microwave (12 min). After cooling to room temperature (RT), sections were washed in TBS, and endogenous peroxidase activity was blocked with 3% (v/v) hydrogen peroxidase (H2O2) for 15 min at RT. The reaction was stopped by short incubation with ddH2O and followed by washing with TBS. In order to block non-specific binding sites, sections were incubated with 5% (w/v) bovine serum albumin (BSA) in TBS for 30 min at RT. Subsequently, a primary antibody against TEKT5 (rabbit polyclonal anti-TEKT5, PA5–21157 ThermoFisher Scientific, Langenselbold, Germany, 1:100) was applied, and sections were incubated in a humid chamber at 4°C overnight. After three washes in TBS, sections were incubated with the secondary antibody (goat-anti-rabbit-HRP, sc-2004, Santa Cruz Biotechnology, Heidelberg, Germany, 1:100). Isotype-specific immunoglobin (IgG) was used as a technical control (I5006, Sigma–Aldrich, Munich, Germany). Sections were washed in TBS prior to incubation with 3,3′-diaminobenzidine tetrahydrochloride (DAB) as chromogen (D4168, Sigma–Aldrich, Munich, Germany). Precipitation was stopped with distilled water, and cross sections were counterstained with Mayer’s hematoxylin. Next, sections were dehydrated in increasing concentrations of ethanol and then mounted with Merckoglas® mounting medium (Merck Millipore, Darmstadt, Germany).
3 |. RESULTS
3.1 |. Identification of copy number variants in infertile men
Of the 5211 CNVs that were found in the infertile men by aCGH, 2797 were deletions and 2414 duplications. Of these, 971 deletions had a log value −0.6 and were not found in controls. There were 928 deletions, which covered at least one gene. After excluding all genes that were deleted in more than three men, 577 genes remained. Of these, 150 genes had high testis expression (scaled TPM [transcripts per million] 0.4–1); of these 23 were non-protein-coding genes, which were disregarded (Figure 1).
FIGURE 1.
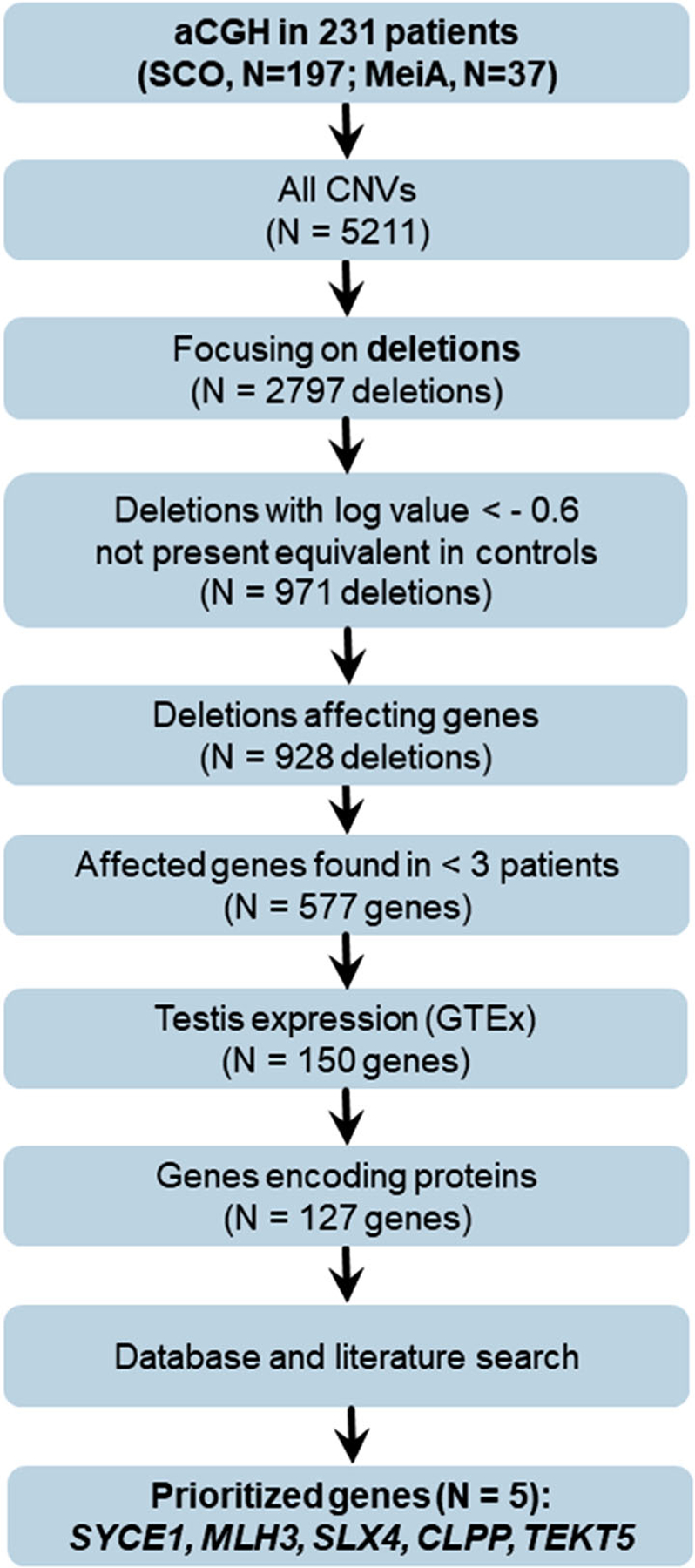
Criteria for prioritizing copy number variants (CNVs) obtained by array-based comparative genomic hybridisation (aCGH) in relevant genes. SCO: Sertoli cell-only phenotype; MeiA: meiotic arrest; GTEx: ‘Genotype-Tissue Expression’ project
After the database and literature search (Table S4), we prioritised the five genes CLPP, MLH3, SLX4, SYCE1 and TEKT5 (Table 1). Overall, we detected one individual with a homozygous deletion and six men with heterozygous deletions in these genes. Clinical data of these men are shown in Table S5. Individual M136916 carried a 97 kb homozygous deletion on chromosome 10, covering the genes SYCE1, SCART1 and CYP2E1 (Figure 2A). M663 and M921 carried heterozygous deletions affecting SYCE1. Additionally, we identified heterozygous deletions affecting large parts of the prioritised genes MLH3 (M226) and SLX4 (M1635) as well as heterozygous deletions of the complete genes TEKT5 (M877) and CLPP (M1281) (Figure 2). In particular, M226 was a carrier of the 20 kb heterozygous deletion on chromosome 14 affecting the genes MLH3 and EIF2B2. M1635 was shown to be a carrier of a 70 kb heterozygous deletion on chromosome 16, leading to a heterozygous loss of a large fraction of SLX4 and CLUAP1 and the complete NLRC3 gene. The deletion was inherited from M1635’s mother as revealed by qPCR analysis in both parents. In M1281, a 29 kb heterozygous deletion on chromosome 19 was detected, exclusively involving the CLPP gene. In M877, aCGH analysis revealed a 104 kb large deletion covering TEKT5. Based on CNV calling from WES data as well as qPCR analysis the deletions in the prioritised genes were confirmed in all seven men (Table 1, Table S6).
FIGURE 2.
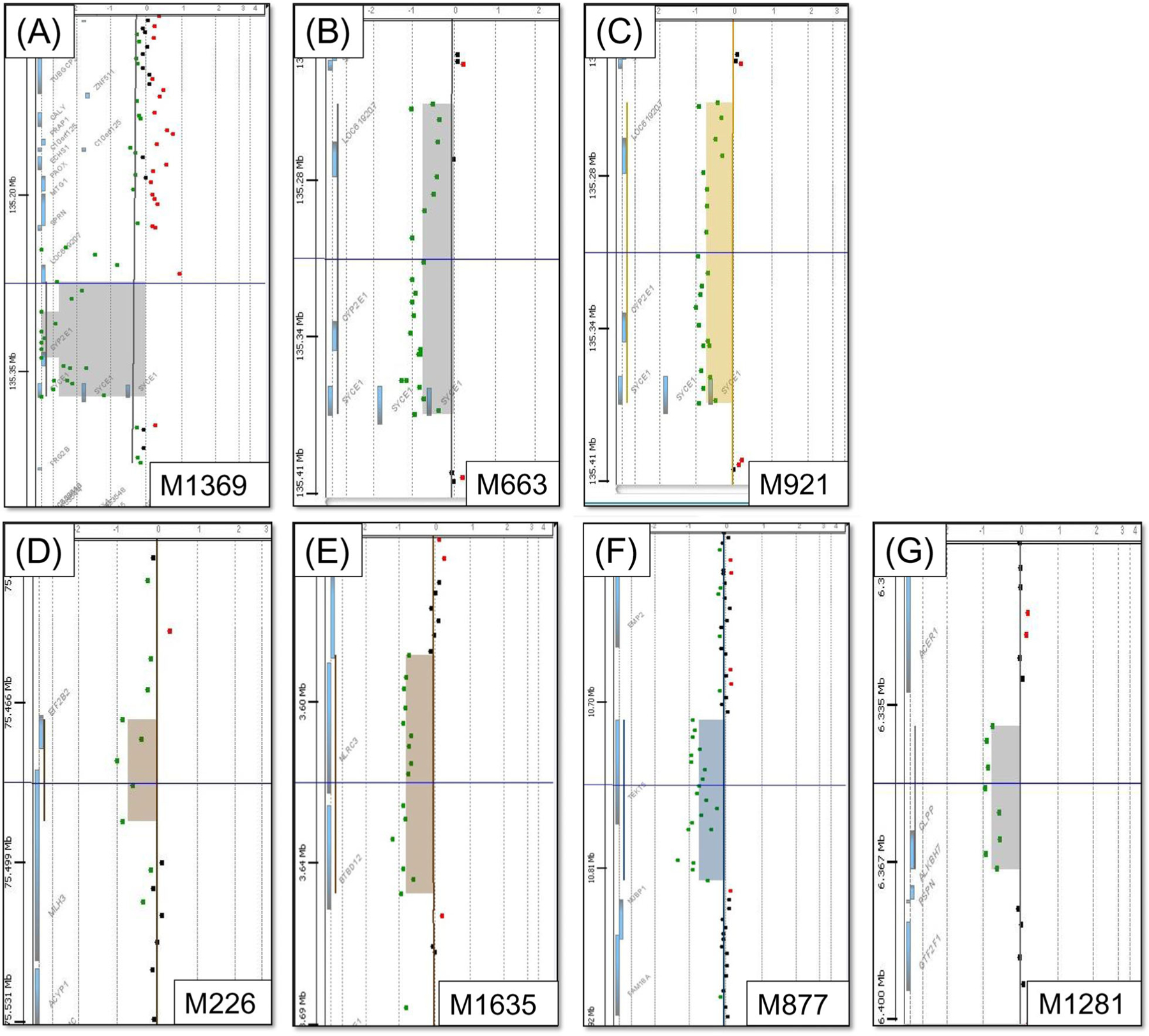
Array-based comparative genomic hybridisation of men with meiotic arrest (MeiA) or Sertoli cell-only phenotype (SCO) exhibiting deletions (indicated in colored areas) in prioritised genes. Dots represent probe position. Red dots represent facultative duplications, green dots represent facultative deletions. Individual M1369 had MeiA and a homozygous deletion of SYCE1, encompassing the genes CYP2E1 and SCART1 (A). M663 had MeiA and was carrier of a heterozygous deletion spanning the genes SYCE1, CYP2E1 and SCART1 (B). The same genes are deleted heterozygously in M921, who had SCO (C). In M226, who also had SCO, we detected a heterozygous deletion encompassing the genes MLH3 and EIF2B2 (D). Individual M1635 had SCO and a heterozygous deletion encompassing the genes SLX4, CLUAP1 and NLRC3 (E). SCO man M877 was found to have a heterozygous deletion of TEKT5 (F). In SCO man M1281 we found a heterozygous deletion of the CLPP gene (G)
When screening WES data for CNVs in the prioritised genes, we detected two additional men with likely causal deletions affecting the genes SYCE1, SCART1 and CYP2E1. M3187 carried a homozygous deletion, while M2681 carried almost the same deletion in a heterozygous state, leading to a heterozygous loss of SYCE1. The deletions in both men were confirmed by qPCR analysis as well as by SNP-array analysis (Figure S1).
In gnomAD structural variation database, no individuals with homozygous deletions affecting the coding regions of SYCE1 and TEKT5 are listed, while no individuals with homo- or heterozygous deletions are listed affecting the coding regions of CLPP, MLH3 and SLX4. In DGV two individuals with deletions affecting SLX4 and MLH3, respectively, 24 individuals with deletions affecting TEKT5 and no individuals with deletions affecting CLPP are listed.
3.2 |. Identification of possibly relevant single nucleotide variations in infertile men
As the infertile men M921, M663, M226, M1635, M1281, M877 and M2681 were carriers of heterozygous deletions, we examined their WES data for rare genetic variants on the second allele of the gene deleted in a heterozygous state. However, in the infertile men M921, M663, M226, M1635 and M1281, no such variants were detected.
In M2681 who is affected by a heterozygous deletion covering i.a. SYCE1, we detected the missense variant c.430C>T p.(Arg144Trp) in SYCE1 resulting in compound-heterozygosity. This missense variant is extremely rare in gnomAD database (MAF = 0.00003541) with no homozygous individuals listed. This variant was classified as likely pathogenic according to the ACMG-AMP criteria (Table S8).
In M877, who was a carrier of the heterozygous deletion in TEKT5, WES revealed the missense variant p.(Val61Ile) (c.181G>A, rs138126929, MAF = 0.00712 in gnomAD) on the second allele of TEKT5, confirming compound heterozygosity.
After filtering the exome sequencing data of the men M1369, M663, M921, M226, M1635, M877, M1281, M1767, M1404, M1626, M1761, M2023 and M2134 for possibly pathogenic variants in the examined genes, one heterozygous missense variant (c.17G>A p.(Gly6Glu)) in PLK4 in M1281, remained as potential alternative cause for infertility.
WES data from the MERGE study including 2,030 men revealed variants, which might potentially affect both alleles, in the genes SLX4 and TEKT5 in four men (genetic data in Table 2, clinical data in Table S7). In the mother of M1767 only the TEKT5 variant c.263G>A p.(Arg88His) was detected (Figure S2), strongly suggesting that the second variant c.1022C>T p.(Ala341Val) was inherited from the father, though his DNA was not available. By sequencing the TEKT5 gene in an independent male infertility cohort from Pittsburgh, PA, USA, we identified one man with MeiA (PIT10) with two heterozygous missense variants (Table 2).
Criteria for ACMG-AMP classification of SNVs listed in Table 2 can be found in Table S8.
3.3 |. Comparison of copy number variants detected by array-based comparative genomic hybridisation between groups
In total, 527 CNVs were detected in 21 men of the control group (24.0 per individual), 795 CNVs in men with MeiA (N = 37, 21.5 per individual) and 4416 CNVs in men with SCO (N = 194, 22.8 per individual). Of these CNVs, 252 were deletions found in the control group (12 per individual), 414 were deletions detected in men with MeiA (11.2 per individual) and 2,383 were deletions detected in men with SCO (12.3 per individual). The prevalence of all CNVs was significantly higher in controls, while no significant differences between the three groups were identified when analysing deletions and duplications separately (Figure S3). To analyse whether the chromosomal distribution of CNVs was different between the groups, the number of CNVs, and deletions/duplications separately, was calculated per chromosome and normalised to 100 men. The chromosomal distribution did not show significantly more sex-chromosomal CNVs in the azoospermic men (Figure S4). Also, the detected CNVs in the infertile men did not cover more base pairs than CNVs from the control group (Figure S5).
3.4 |. Investigation of TEKT5
TEKT5 is, to date, a relatively unknown protein. Therefore, we performed immunohistochemistry (IHC) in fetal and adult testes of mice and humans to analyse the cellular expression of the TEKT5 protein. In E15.5 mice, TEKT5 is highly expressed in fetal germ cells, whereas in the pre-pubertal testes of human samples only very few germ cells were positively stained. In both murine and human adult testicular tissue, TEKT5 can exclusively be observed in the germ cell population, specifically in spermatocytes as well as round and elongated spermatids (Figure 3A–D). In M877 with SCO (Figure 3F), no staining was detected, which further confirms the germ cell-specificity of TEKT5 in human testes.
FIGURE 3.
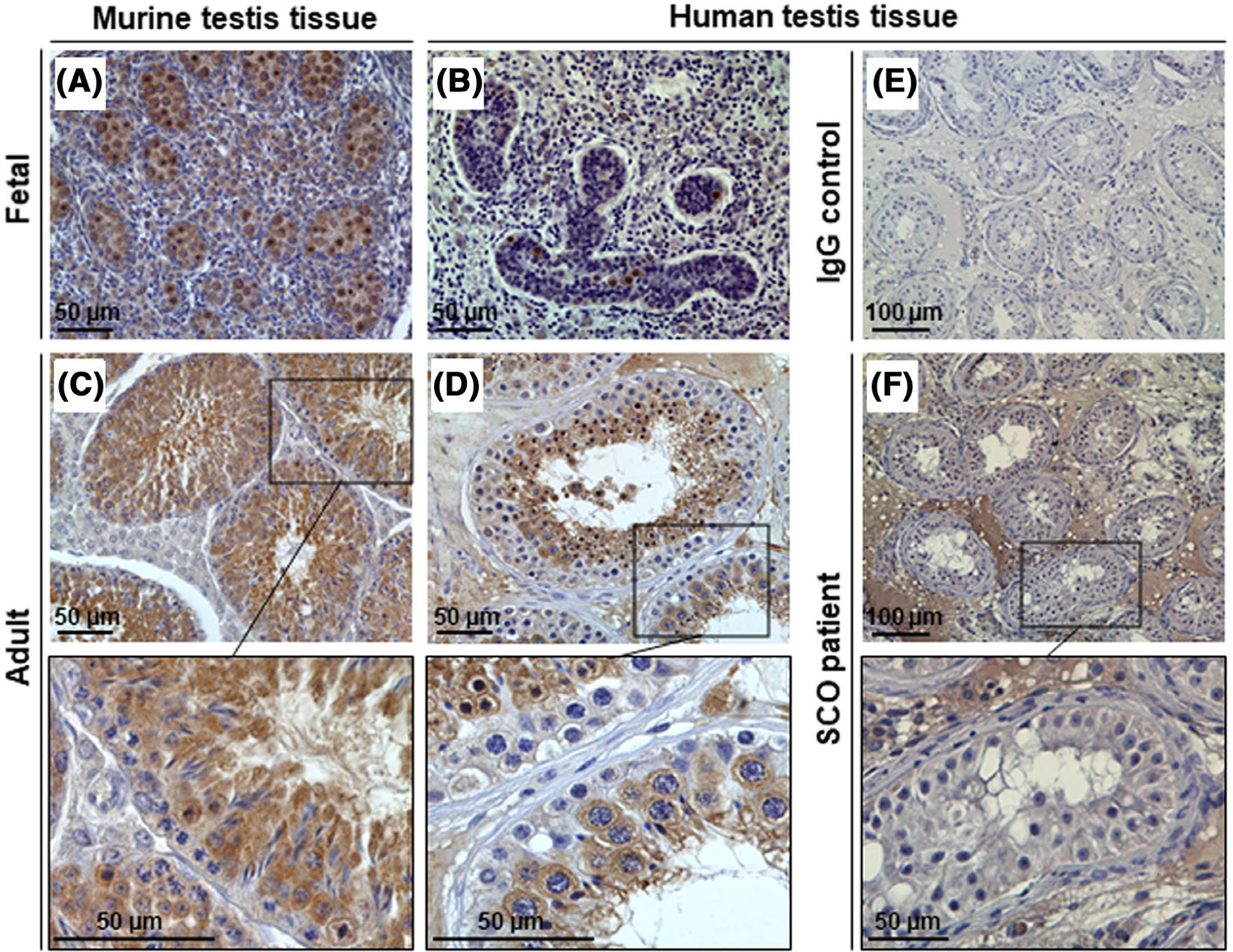
Histological analysis of the TEKT5 protein expression by immunohistochemical staining. (A, B) Testis tissue from fetal mouse and human biopsies was stained with TEKT5 primary antibody (PA5–21157), resulting in a germ cell-specific staining pattern. (C, D) Evaluation of adult murine and human tissue confirmed characteristic protein expression of the germ cell population, mainly including spermatocytes and round and elongated spermatids. (E) Isotype-specific immunoglobin (IgG) was used as a technical control. (F) Man M877, displaying a Sertoli cell-only phenotype (SCO), showed no specific TEKT5 staining since germ cells were absent within the testicular tubules. Scale bars are indicated in each micrograph
While the 3D modelling of the TEKT5 structure does not directly provide evidence for the pathogenicity and the observed phenotype for M877, the larger amino acid (p.(Val61Ile)) could alter the tertiary structure of the protein. It could also affect intermolecular interactions with TEKT5 binding partners (Figure 4).17
FIGURE 4.
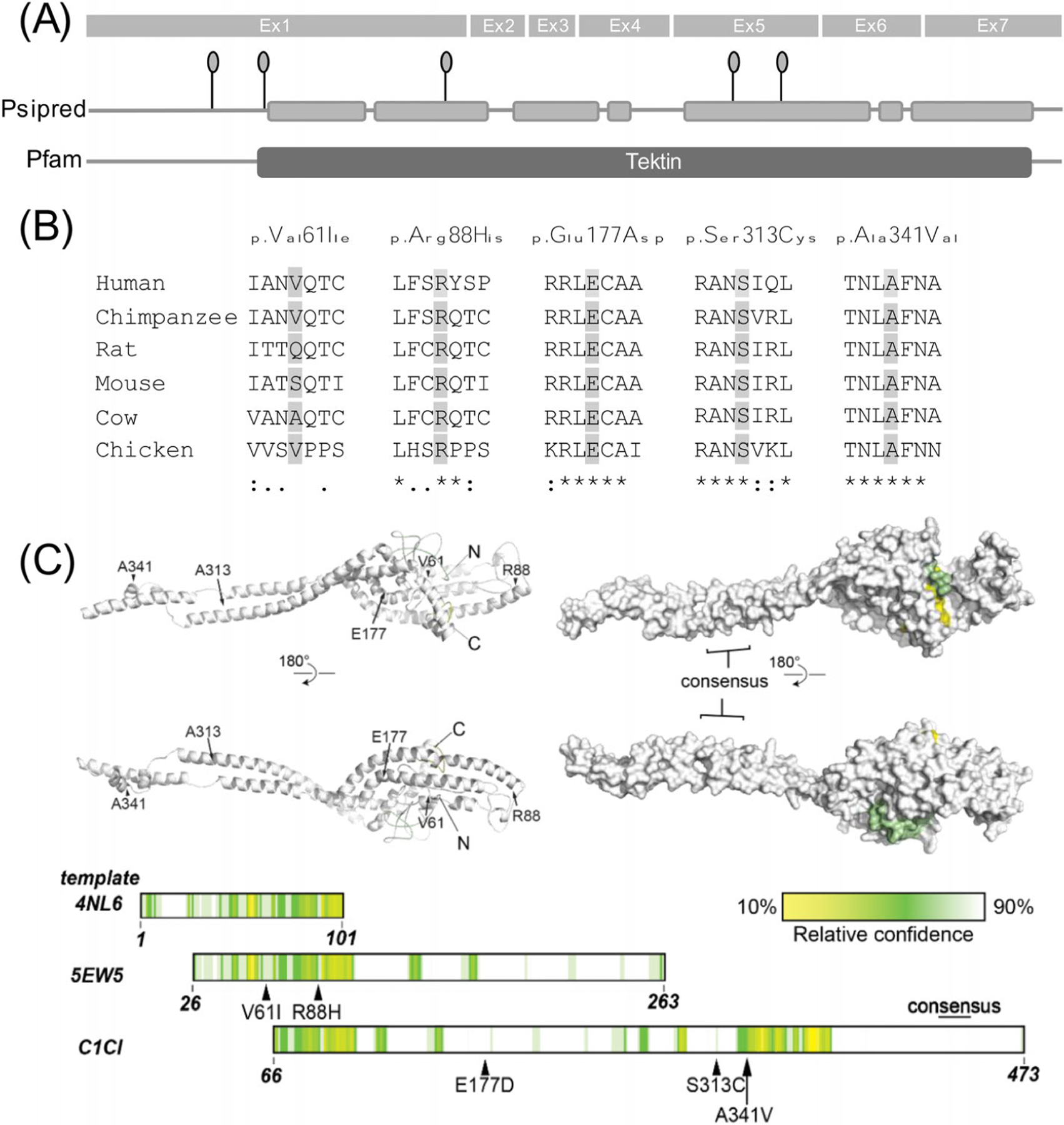
Structure, conservation and modelling of the TEKT5 gene/protein. (A) Exon structure of TEKT5 gene. Linear diagram of secondary structure prediction from PSIPRED of the TEKT5 protein. Grey boxes represent predicted alpha-helices which are linked by predicted coiled structures. Identified rare or novel TEKT5 variants (MAF < 0.01) are depicted as pinheads. Pfam predicts a tektin domain (amino acids 94–476) within the protein. (B) Orthologous alignment of the TEKT5 protein. Conservation of five novel identified amino acid substitutions among orthologous species are highlighted in light grey. (C) Modelling of TEKT5 structure using Phyre2. Left: ribbon representation; right: surface representation. Position of amino acids of interest, and N- and C-termini are indicated. Below: linear organisation of each template used to generate the final model of TEKT5 by Phyre2, coloured by confidence of modelling, as quantified in the bar.
4 |. DISCUSSION
In most cases of NOA-associated male infertility, no underlying genetic cause can be determined. While SNVs, that is, point mutations, are a well-established cause of NOA, CNVs have rarely been examined in infertile men. In this study, we describe the so far largest cohort of well-characterised men with complete bilateral SCO and MeiA and assessed their genetic causes of infertility using primarily aCGH, combined with WES.
Applying aCGH and WES in a cohort of 231 azoospermic men, we identified the cause of MeiA in M1369, who was diagnosed with a homozygous deletion of SYCE1 — a gene previously reported to cause male infertility. Two further men with deletions affecting SYCE1 were identified by CNV calling from WES data. Moreover, as a potential novel candidate gene, we propose the heterozygous deletion in combination with the missense variant in TEKT5 in subject M877 as possible cause for his infertility. M1635 was heterozygous for the deletion of the SLX4 gene with an expected autosomal-recessive mode of inheritance. As we did not detect a second hit in this man, it is unlikely that this deletion alone is responsible for his infertility. The same applies to the gene CLPP; we therefore cannot definitively diagnose a causal effect of the deletion in CLPP in individual M1281 who also carries a heterozygous missense variant in PLK4 that additionally might have an impact. According to gnomAD structural variation database, CNVs in these five genes are very rare or even absent and therefore might be relevant.
SYCE1 encodes synaptonemal complex central element protein 1, which is involved in the formation of the synaptonemal complex during prophase of the first meiotic division.18 Fittingly, male Syce1 KO mice exhibit a lack of post-meiotic stages of germ cells19 and homozygous variants in SYCE1 have been described to cause NOA in men.20–23 According to a very recent gene–disease relationship assessment using clinical guidelines, SYCE1 reaches a strong level of evidence for being associated with male infertility and should therefore be analysed in clinical diagnostics.16 The additional deleted genes in M1369, M2681 and M3187 are SCART1 and CYP2E1. As SCART1 is a non-protein-coding pseudogene and CYP2E1 is exclusively expressed in the liver, we postulate that the homozygous deletion of SYCE1 caused the man’s infertility. The fact that M2681 carries a likely pathogenic missense variant in SYCE1 on the second allele while no rare coding variants in the other two genes were identified in this man, further supports the hypothesis that SYCE1 is indeed the causal gene affected by this deletion.
TEKT5 (tektin 5) is, so far, a poorly studied gene, but it has high expression patterns in both mouse and human testis.24 Moreover, male Tekt5 testis-specific KO mice show a decreased number of spermatids. TEKT5 has been suggested to be involved in human spermatogenesis and spermiogenesis, showing upregulated expression patterns from the late pachytene stage on.24 We found that TEKT5 shows similar cellular localisation in mice and men in both fetal and adult testicular tissue. Thus, we speculate that a lack of or misfolded TEKT5 protein might have consequences for human male fertility. As such, the SCO phenotype observed in man M877 may have been caused by the heterozygous deletion of the entire gene in combination with a missense variant that might impact the tertiary structure of the protein. In particular, this SNV results in a valine-to-isoleucine exchange of the amino acid at position 61, which is moderately conserved (Figure 4). The affected amino acids differ in size, this might lead to an altered tertiary structure. Nonetheless, the in silico algorithms did not predict the amino acid change would have a damaging impact. Thus, only in vitro or in vivo analysis will be able to clarify the functional effect of this variation. In addition, one man (M1767) with maturation arrest at the round spermatid stage was observed to carry two rare missense variants in TEKT5 in our own cohort. According to the prediction program HOPE, the missense variant c.263G>A p.(Arg88His) detected in this patient replaces the conserved, positively charged arginine with the neutral, smaller histidine. The second missense variant in M1767 is c.1022C>T p.(Ala341Val), which introduces the larger amino acid valine instead of alanine at a conserved position. Another man (PIT10) with MeiA and two heterozygous variants was identified in an independent study cohort. The missense variant c.531G>C p.(Glu177Asp) in PIT10 replaces the negatively charged glutamine, which is located in an α-helix, with the smaller, neutral asparagine. The other missense variant c.938C>G p.(Ser313Cys) in PIT10 introduces a more hydrophobic residue at position 313. Still, it remains unclear if the variants in either men are located in trans. The fact that M877 was diagnosed with SCO, while M1767 and PIT10 had maturation or MeiA, does not contradict TEKT5 as joint underlying cause, because such a phenotypic spectrum is also observed in patients with pathogenic variants in other well-established male infertility genes such as TEX14.25 It remains elusive if the detected variants in TEKT5 are the cause of the individuals’ impaired spermatogenesis and functional analyses are required to further interpret the pathogenicity of these missense variants.
MLH3 (MutL-homolog 3) encodes a mismatch repair protein which is involved in prophase of the first meiotic division.26,27Mlh3 KO mice are infertile due to meiotic impairment and a severe depletion of spermatocytes.28 Recently, a homozygous frameshift variant was reported as the cause of NOA in one man;29 therefore, an autosomal-recessive mode of inheritance can be assumed. However, we were not able to detect a likely pathogenic variant on the second MLH3 allele in M226. In M226, the heterozygous deletion further covers the EIF2B2 gene, a gene in which compound-heterozygous variants have been described to cause premature ovarian insufficiency (POI) in women,30 which is in some instances the equivalent to MeiA in men. Although we did not detect an SNV on the second EIF2B2 allele of M226, we cannot exclude that the heterozygous deletion of EIF2B2 was involved in M226’s NOA. Also, an oligogenic mode of inheritance is conceivable, as homozygous Eif2b2 KO mice do not exhibit impaired fertility due to preweaning lethality.31 However, according to STRING database EIF2B2 and MLH3 do not interact with each other, making an oligogenic inheritance pattern rather unlikely.
Biallelic mutations in SLX4 (also known as BTBD12 or FANCP) are associated with Fanconi anaemia due to a defects in DNA repair.32,33 During murine spermatogenesis, SLX4 facilitates primordial germ cell proliferation and is involved in meiotic recombination.34 In humans, SLX4 interacts with FANCM, a gene described to cause NOA.35,36 M1635 was a heterozygous carrier of the SLX4 deletion, but we did not find a second hit on the other allele. The 70 kb deletion found in M1635 also spans the genes CLUAP1 and NLRC3. For both genes we found no link to infertility based on the literature search. Taken together, we cannot establish the definitive genetic diagnosis for man M1635, but the heterozygous deletion of both SLX4 and CLUAP1 might produce a synergistic effect, though no interaction of SLX4 and CLUAP1 has been described; further investigations are necessary to clarify their respective impact.
Biallelic mutations of CLPP are associated with Perrault syndrome, involving ovarian insufficiency in women;37 a homozygous mutation of CLPP has been reported to cause azoospermia in one man.38 This gene’s relevance for human infertility is underlined by the fact that Clpp KO mice of both sexes are infertile.39 The predicted mode of inheritance of CLPP is “very likely autosomal-recessive”.9 We identified a heterozygous deletion of CLPP in M1281, but, due to the lack of a variant on the second allele, we cannot provide sufficient reason for his infertility. M1635 did also not exhibit any other signs of Perrault syndrome such as sensorineural deafness.
Some studies have suggested that the recurringly detected deletion “CNV67” is an X-chromosomal variant leading to infertility in men, while others have contradicted this finding.40,41 Of note, we did not detect any CNVs involving this chromosomal region in our large cohort. This does not exclude an impact of CNV67 on male infertility but the relevance of these CNVs remains controversial.42
Previous studies have reported that infertile men carry overall more CNVs compared to controls, thus hinting at a greater instability of their genomes, which would be the ultimate cause of these men’s infertility.6,43 In contrast to our own previous, smaller and some other similar studies, the prevalence of all CNVs, deletions/duplications separately, and CNVs specifically on the sex chromosomes was not significantly higher in infertile men compared with control men. Thus, we could not support this assumption. In this respect, the small size of the control cohort (21 men) should be pointed as limitation in the current analysis, which does not allow drawing general conclusions. To overcome this issue, large databases like gnomAD SV and DGV are indispensable. However, no phenotypic information is available and gnomAD data were obtained by CNV calling from sequencing data making them difficult to compare to our dataset.
Taken together, we were able to confidently determine the cause of infertility in three individuals (M1369, M2681 and M3187) by aCGH and CNV calling from WES data and therefore provide further evidence for SYCE1 association with NOA. Both methods, aCGH/SNP-array and WES, are expensive, so combining both in clinical diagnostics of male infertility due to spermatogenic failure is not an effective strategy. Instead, using WES read depth data to predict CNVs has recently become more precise, making aCGH/SNP-array more and more dispensable.44 Indeed, the homozygous deletion in SYCE1, which was detected in M1369 by aCGH, would also have been detected solely by WES. Moreover, the deletions affecting SYCE1 in M2681 and M3187 were primarily detected by WES and only secondary confirmed by SNP-array, underlining rapid technological advances.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the infertile men for agreeing to a research evaluation of their data and DNA and the physicians who took care of them at the Centre of Reproductive Medicine and Andrology, Münster. The technical assistance of Mandy Hoffmann, Steffi Burkhart and Laura Hankamp is gratefully acknowledged. Special thanks go to Jana Emich and Linda Ebbert for her support with reasonable filtering, database, and literature search and to Dr. Celeste Brennecka for language editing. The German Research Foundation fundered Clinical Research Unit ‘Male Germ Cells’ (DFG, CRU326 grants to SS, CF, and FT); supported by NIH grants, Grant numbers: P50 HD096723 and R21 HD080755 (to ANY).
Funding information
German Research Foundation (DFG), Grant/Award Number: CRU326; NIH, Grant/Award Numbers: P50 HD096723, R21 HD080755
Footnotes
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
SUPPORTING INFORMATION
Additional supporting information can be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
The published paper includes all datasets generated or analysed during this study. The exceptions are the primary WES data as well as the aCGH data that have not been deposited in a public repository because the individual’s consent did not allow this. Additional information is available from the corresponding author on request.
REFERENCES
- 1.Agarwal A, Mulgund A, Hamada A, Chyatte MR. A unique view on male infertility around the globe. Reprod Biol Endocrinol 2015;13:37. 10.1186/s12958-015-0032-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vloeberghs V, Verheyen G, Haentjens P, Goossens A, Polyzos NP, Tournaye H. How successful is TESE-ICSI in couples with non-obstructive azoospermia? Hum Reprod 2015;30:1790–1796. [DOI] [PubMed] [Google Scholar]
- 3.Corona G, Minhas S, Giwercman A, et al. Sperm recovery and ICSI outcomes in men with non-obstructive azoospermia: A systematic review and meta-analysis. Hum Reprod Update 2019;25:733–757. [DOI] [PubMed] [Google Scholar]
- 4.Guilmatre A, Dubourg C, Mosca A-L, et al. Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch Gen Psychiatry 2009;66:947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krausz C, Riera-Escamilla A. Genetics of male infertility. Nat Rev Urol 2018;15:369–384. [DOI] [PubMed] [Google Scholar]
- 6.Tüttelmann F, Simoni M, Kliesch S, et al. Copy number variants in patients with severe oligozoospermia and sertoli-cell-only syndrome. PLoS One 2011;6:e19426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.World Health Organization (WHO). Examination and Processing of Human Semen 6th ed. WHO; 2021. [Google Scholar]
- 8.The Gtex Consortium. The genotype-tissue expression (GTEx) project. Nat Genet 2014;45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quinodoz M, Royer-Bertrand B, Cisarova K, Di Gioia SA, Superti-Furga A, Rivolta C. DOMINO: using machine learning to predict genes associated with dominant disorders. Am J Hum Genet 2017;101:623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Araujo TF, Friedrich C, Grangeiro CHP, et al. Sequence analysis of 37 candidate genes for male infertility: Challenges in variant assessment and validating genes. Andrology 2020;8:434–441. [DOI] [PubMed] [Google Scholar]
- 11.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Houston BJ, Riera-Escamilla A, Wyrwoll MJ, et al. A systematic review of the validated monogenic causes of human male infertility: 2020 update and a discussion of emerging gene–disease relationships. Hum Reprod Update 2021;28:15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yatsenko AN, Georgiadis AP, Röpke A, et al. X-linked TEX11 mutations, meiotic arrest, and azoospermia in infertile men. N Engl J Med 2015;372:2097–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Venselaar H, te Beek TAH, Kuipers RKP, Hekkelman ML, Vriend G. Protein structure analysis of mutations causing inheritable diseases: An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics 2010;11:548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wyrwoll MJ, Köckerling N, Vockel M, et al. Genetic architecture of azoospermia—Time to advance the standard of care. Eur Urol 2022. [DOI] [PubMed]
- 17.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 2015;10:845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geisinger A, Benavente R. Mutations in genes coding for synaptonemal complex proteins and their impact on human fertility. Cytogenet Genome Res 2017;150:77–85. [DOI] [PubMed] [Google Scholar]
- 19.Schramm S, Fraune J, Naumann R, et al. A novel mouse synaptonemal complex protein is essential for loading of central element proteins, recombination, and fertility. PLoS Genet 2011;7:e1002088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang N, Wen Y, Guo X, et al. A screen for genomic disorders of infertility identifies MAST2 duplications associated with nonobstructive azoospermia in humans. Biol Reprod 2015;93:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maor-Sagie E, Cinnamon Y, Yaacov B, et al. Deleterious mutation in SYCE1 is associated with non-obstructive azoospermia. J Assist Reprod Genet 2015;32:887–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pashaei M, Rahimi Bidgoli MM, Zare-Abdollahi D, et al. The second mutation of SYCE1 gene associated with autosomal recessive nonobstructive azoospermia. J Assist Reprod Genet 2020;37:451–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krausz C, Riera-Escamilla A, Moreno-Mendoza D, et al. Genetic dissection of spermatogenic arrest through exome analysis: Clinical implications for the management of azoospermic men. Genet Med 2020;22:1956–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aoki N, Matsui Y. Comprehensive analysis of mouse cancer/testis antigen functions in cancer cells and roles of TEKT5 in cancer cells and testicular germ cells. Mol Cell Biol 2019;39:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fakhro KA, Elbardisi H, Arafa M, et al. Point-of-care whole-exome sequencing of idiopathic male infertility. Genet Med 2018;20:1365–1373. [DOI] [PubMed] [Google Scholar]
- 26.Santucci-Darmanin S The DNA mismatch-repair MLH3 protein interacts with MSH4 in meiotic cells, supporting a role for this MutL homolog in mammalian meiotic recombination. Hum Mol Genet 2002;11:1697–1706. [DOI] [PubMed] [Google Scholar]
- 27.Wang TF, Kleckner N, Hunter N. Functional specificity of MutL homologs in yeast: Evidence for three Mlh1-based heterocomplexes with distinct roles during meiosis in recombination and mismatch correction. Proc Natl Acad Sci U S A 1999;96:13914–13919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lipkin SM, Moens PB, Wang V, et al. Meiotic arrest and aneuploidy in MLH3-deficient mice. Nat Genet 2002;31:385–390. [DOI] [PubMed] [Google Scholar]
- 29.Chen S, Wang G, Zheng X, et al. Whole-exome sequencing of a large Chinese azoospermia and severe oligospermia cohort identifies novel infertility causative variants and genes. Hum Mol Genet 2020;29:2451–2459. [DOI] [PubMed] [Google Scholar]
- 30.Liu H, Wei X, Sha Y, et al. Whole-exome sequencing in patients with premature ovarian insufficiency: Early detection and early intervention. J Ovarian Res 2020;13:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bult CJ, Blake JA, Smith CL, et al. Mouse genome database (MGD) 2019. Nucleic Acids Res 2019;47:D801–D806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stoepker C, Hain K, Schuster B, et al. SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat Genet 2011;43:138–141. [DOI] [PubMed] [Google Scholar]
- 33.Crossan GP, Van Der Weyden L, Rosado IV, et al. Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat Genet 2011;43:147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holloway JK, Mohan S, Balmus G, et al. Mammalian BTBD12 (SLX4) protects against genomic instability during mammalian spermatogenesis. PLoS Genet 2011;7:e1002094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kasak L, Punab M, Nagirnaja L, et al. Bi-allelic recessive loss-of-function variants in FANCM cause non-obstructive azoospermia. Am J Hum Genet 2018;103:200–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin H, Ma H, Hussain S, et al. A homozygous FANCM frameshift pathogenic variant causes male infertility. Genet Med 2019;21:62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Newman WG, Friedman TB, Conway GS, Demain LA. Perrault Syndrome University of Washington; 1993. [PubMed] [Google Scholar]
- 38.Demain LAM, Urquhart JE, O’Sullivan J, et al. Expanding the genotypic spectrum of Perrault syndrome. Clin Genet 2017;91:302–312. [DOI] [PubMed] [Google Scholar]
- 39.Gispert S, Parganlija D, Klinkenberg M, et al. Loss of mitochondrial peptidase clpp leads to infertility, hearing loss plus growth retardation via accumulation of CLPX, mtDNA and inflammatory factors. Hum Mol Genet 2013;22:4871–4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krausz C, Giachini C, Lo Giacco D, et al. High-resolution X chromosome-specific array-CGH detects new CNVs in infertile males. PLoS One 2012;7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma X, Kuete M, Gu X, Zhou H, Xiong C, Li H. Recurrent deletions of the X chromosome linked CNV64, CNV67, and CNV69 shows geographic differences across China and no association with idiopathic infertility in men. PLoS One 2017;12:e0185084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vockel M, Riera-Escamilla A, Tüttelmann F, Krausz C. The X chromosome and male infertility. Hum Genet 2019;140:203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lopes AM, Aston KI, Thompson E, et al. Human spermatogenic failure purges deleterious mutation load from the autosomes and both sex chromosomes, including the gene DMRT1. PLoS Genet 2013;9:e1003349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marchuk DS, Crooks K, Strande N, et al. Increasing the diagnostic yield of exome sequencing by copy number variant analysis. PLoS One 2018;13:e0209185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published paper includes all datasets generated or analysed during this study. The exceptions are the primary WES data as well as the aCGH data that have not been deposited in a public repository because the individual’s consent did not allow this. Additional information is available from the corresponding author on request.