

Summary
Chromosome 16p11.2 reciprocal genomic disorder, resulting from recurrent copy-number variants (CNVs), involves intellectual disability, autism spectrum disorder (ASD), and schizophrenia, but the responsible mechanisms are not known. To systemically dissect molecular effects, we performed transcriptome profiling of 350 libraries from six tissues (cortex, cerebellum, striatum, liver, brown fat, and white fat) in mouse models harboring CNVs of the syntenic 7qF3 region, as well as cellular, transcriptional, and single-cell analyses in 54 isogenic neural stem cell, induced neuron, and cerebral organoid models of CRISPR-engineered 16p11.2 CNVs. Transcriptome-wide differentially expressed genes were largely tissue-, cell-type-, and dosage-specific, although more effects were shared between deletion and duplication and across tissue than expected by chance. The broadest effects were observed in the cerebellum (2,163 differentially expressed genes), and the greatest enrichments were associated with synaptic pathways in mouse cerebellum and human induced neurons. Pathway and co-expression analyses identified energy and RNA metabolism as shared processes and enrichment for ASD-associated, loss-of-function constraint, and fragile X messenger ribonucleoprotein target gene sets. Intriguingly, reciprocal 16p11.2 dosage changes resulted in consistent decrements in neurite and electrophysiological features, and single-cell profiling of organoids showed reciprocal alterations to the proportions of excitatory and inhibitory GABAergic neurons. Changes both in neuronal ratios and in gene expression in our organoid analyses point most directly to calretinin GABAergic inhibitory neurons and the excitatory/inhibitory balance as targets of disruption that might contribute to changes in neurodevelopmental and cognitive function in 16p11.2 carriers. Collectively, our data indicate the genomic disorder involves disruption of multiple contributing biological processes and that this disruption has relative impacts that are context specific.
Keywords: genomic disorder, copy-number variation, transcriptome, RNAseq, 16p11.2, CRISPR, cerebral organoid
Graphical abstract
The 16p11.2 genomic disorder involves deletion and duplication of a contiguous set of genes, resulting in neurodevelopmental and other abnormalities. On the basis of comparisons of mouse models and human neuronal cells, the effects of these lesions on genome-wide gene expression and cellular function are highly dependent on cellular context.
Introduction
Reciprocal genomic disorders (RGDs) are syndromes caused by recurrent CNVs generated from non-allelic homologous recombination (NAHR).1 These disorders typically involve altered dosage of multiple genes and are collectively among the greatest contributors to neurodevelopmental disorders (NDDs) and a spectrum of related neuropsychiatric disorders.2, 3, 4, 5, 6 Despite this considerable morbidity, the molecular mechanisms by which these reciprocal rearrangements disrupt development remain largely unknown. Given that NAHR reproducibly alters the dosage of precisely the same sets of genes, the inherent genomic architecture of RGDs has largely prevented assessment of each affected gene’s specific contributions to associated phenotypes. However, the establishment of accessible RGD mouse models, and recent advances in CRISPR-based genome engineering in human induced pluripotent stem cells (hiPSCs), show promise that dissecting specific genetic and molecular underpinnings of RGDs could now be tractable.7
Recurrent deletion (MIM: 611913) and duplication (MIM: 614671) of an ∼743 kb genomic segment of chromosomal locus 16p11.2 underly a relatively common and highly penetrant RGD associated with a spectrum of phenotypes, including autism spectrum disorder (ASD), schizophrenia (SCZ), abnormal head circumference, altered body mass, craniofacial and skeletal anomalies, and predisposition to neuroblastoma.8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 This specific CNV includes a unique ∼593 kb segment, as well as at least one copy equivalent of an ∼150 kb flanking segmental duplicati8on (SD). The unique segment encompasses 27 protein-coding genes, and four protein-coding gene paralogues are located in each SD (Figure 1A). Genes in this region exhibit conserved-order synteny on mouse chromosomal locus 7qF3; three of the genes in the SD are present but not duplicated, and the fourth is absent from the mouse genome. The 16p11.2 genes are involved in a wide variety of cellular functions, including chromatin remodeling (INO80E, HIRIP3 [MIM: 603365]),20,21 ubiquitination (KCTD13 [MIM: 608947]),22 DNA repair (SLX1A [MIM: 615822], SLXB [MIM: 615823]),23 MAP kinase signaling (MAPK3 [MIM: 601795], TAOK2 [MIM: 613199]),24 and neurotransmitter release (DOC2A [MIM: 604567], PRRT2 [MIM: 614386]), among others.25,26 In large-scale exome-sequencing studies of ASD or NDD, no individual genes within 16p11.2 have been implicated as contributors to these disorders on the basis of a significant excess of de novo loss-of-function mutations. Other reports have linked individual genes to these phenotypes; such studies have included one observing an excess of missense variants in MAPK3 among NDD cases,27 a case report where a 118 kb deletion encompassing five genes (MVP [MIM: 605088], CDIPT [MIM: 605893], SEZ6L2 [MIM: 616667], ASPHD1, and KCTD13) segregated with ASD in a three-generation pedigree,28 and most recently publication of a nominal association of coding variants in CORO1A [MIM: 605000] with ASD (false discovery rate [FDR] q < 0.05),29 but none of these results reach stringent statistical thresholds for reproducible association, as well-established ASD and NDD risk loci do. Multiple in vivo studies have also suggested a contribution of reciprocally modulated expression of KCTD13 to the neuroanatomical changes associated with the 16p11.2 RGD, but these findings have not been consistent across studies.22,30, 31, 32, 33 Thus, the precise pathogenic mechanisms associated with reciprocal dosage changes of the 16p11.2 locus and the particular genes that drive them remain to be defined.
Figure 1.

Experimental design and expression profile of RGD genes across samples
(A) Illustration of human 16p11.2 segment and SD and the syntenic 7qF3 region in mouse. Only protein-coding genes are shown, via human Ensembl GRCh37 (version 75) annotation and mouse Ensembl GRCm38 (version 83). Single-guide RNA targeting the SDs to promote a model of NAHR-mediated CNV is indicated by scissors. Mouse models were generated through Cre-loxP-mediated recombination as described.34
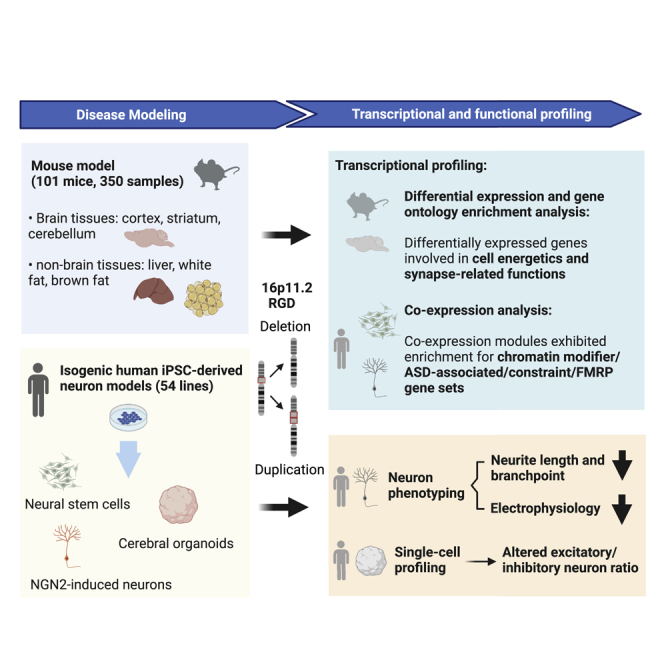
(B) Schematic depiction of the study design and analyses. To systemically dissect molecular functions associated with 16p11.2 RGD, we performed transcriptome analyses of 101 mice with reciprocal CNVs (350 total samples) of the syntenic chromosomal region 7qF3 across cortex, striatum, and cerebellum, as well as three non-brain tissues. Furthermore, we generated NSCs, iNs, and cerebral organoid derivatives of isogenic hiPSCs harboring CRISPR-engineered reciprocal 16p11.2 CNVs and assessed cellular, transcriptional, and single-cell signatures associated with 16p11.2 CNVs.
(C) Expression distribution of 27 protein-coding genes within the 16p11.2 region in WT samples including mouse tissues, NSCs and iNs, and GTEx data. The results revealed 16p11.2 genes with a higher expression level in brain tissue and neurons than in non-brain tissues.
(D) Heatmaps of 16p11.2 region genes' basal expression in mouse tissues (top), hiPSC-derived NSCs and induced neurons (middle), and human tissues (GTEx, transcripts per million) (bottom).
(E) n-fold change (log2) of the protein-coding genes in the CNV and in the flanking regions are shown in coordinate space for deletions in red and duplications in blue across brain tissues (left panel) and human cells (right panel). The light-blue shaded region in the left panel highlights the unique portion of the 16p11.2 CNV region harboring 27 human orthologous protein-coding genes in the mouse 7qF3 segment, whereas pink vertical bars in the right panel highlight the segmental duplication region in the human 16p11.2 segment.
Recent genomic studies of the broad set of genes individually associated with ASD and NDD have suggested a convergence of NDD-associated genes on key functional pathways, including chromatin modification, transcriptional regulation, and synaptic transmission.35,36 Investigations of animal models and tissues have also explored NDD pathogenesis by interrogating protein-protein interactions, quantifying regional and temporal patterns of co-expression,35, 36, 37, 38, 39, 40 and performing in vivo phenotyping during development.41 The resulting data have corroborated the aforementioned processes as contributors but have also revealed neurogenesis-related effects, including altered ratios of cell types in the developing brain. One approach to elucidating pathogenic mechanisms in RGDs is to use global transcriptome analysis of peripheral cells from individuals harboring CNVs.42 However, patient-specific variability in genetic background and cell type-specific expression patterns can complicate identification of regulatory changes most relevant to abnormal neurodevelopment. We previously developed genome-editing methods of modeling NAHR by targeting SDs with CRISPR/Cas9 to produce precise isogenic human cellular models of recurrent RGDs in hiPSCs and derived neuronal lineages.7 Here, we sought to integrate large-scale human and mouse modeling to disentangle the tissue-specific, cell-type-specific, and gene-dosage-specific molecular and transcriptional signatures associated with 16p11.2 RGD. To accomplish this, we examined tissue-specific changes in global gene expression by using mouse models with reciprocal CNVs in 7qF334 and comprehensive analyses of three brain regions (cortex, striatum, and cerebellum) and three non-brain tissues (liver, white fat, and brown fat), along with neural stem cells (NSCs) and neurogenin-2 induced neurons (iNs) derived from isogenic hiPSC lines engineered to model reciprocal 16p11.2 CNV (Figure 1B). We found that these CNVs cause a complex spectrum of distinct and overlapping gene expression changes that reflect both tissue-specific and shared pathway changes. Human cell models carrying 16p11.2 CNVs demonstrate aberrant neuronal phenotypes, including shorter neurites and reduced electrical activity. Finally, single-cell RNA sequencing (scRNAseq) of cerebral organoids harboring 16p11.2 CNVs revealed an altered cell composition with an excitatory/inhibitory neuron imbalance, providing a potential link between 16p11.2 rearrangements and their associated neuropsychiatric phenotypes, including ASD and SCZ.
Methods
CRISPR/Cas9 genome editing and cell-model development
Guide-RNA design, hiPSC culture, and DNA transfection
16p11.2 CRISPR-engineered, isogenic hiPSC lines with deletion or duplication of the 16p11.2 region were generated via the SCORE approach.7 In brief, to design the optimal guide RNA, we first identified all possible 18–25mer guides with Jellyfish and performed a degenerate BLAST search to identify sequences that would uniquely target the 16p11.2 SDs, respectively, with no predicted off-target effects. The gRNA was cloned into pSpCas9(BB)-2A-Puro plasmid with a puromycin resistance marker (pX459, Addgene plasmid 48139) via a BbsI restriction site. Validation of the guide sequence in the gRNA vector was confirmed by Sanger sequencing. Before transfection, the EndoFree Plasmid Maxi Kit was used according to the manufacturer’s (Qiagen’s) instruction for purification of all plasmids.
CRISPR-engineered, isogenic hiPSC lines (KCTD13Het) with deletion of KCTD13 were generated via transfection with CRISPR guide RNA 5′-TAAAAAGGATGGATGTAGGC-3' and 5′-TGCCTGTGTTAGGAGGTATC-3′ with the Amaxa Nucleofector II (Lonza) with Human Stem Cell Nucleofector Kit 1 (Lonza) and program B-016, according to the manufacturer’s instructions. After nucleofection, hiPSCs were cultured in media supplemented with 10 μM Y-27632 dihydrochloride for 24 h prior to selection with puromycin (0.1 μg/mL). After 24 h, surviving hiPSCs were recovered in fresh Essential 8 medium (E8; Gibco, A1517001) for 48 h prior to fluorescence-activated cell sorting (FACS).
The GM08330 hiPSCs43 were used for all studies and maintained in feeder-free culture on plates coated with Matrigel hESC-Qualified Matrix (Corning, 08-774-552) with E8 supplemented with penicillin-streptomycin (Life Technologies, 15140) in a humidified incubator at 37°C with 5% CO2. ReLeSR (STEMCELL Technologies, 05873) was used for routine cell passaging. mFreSR freezing medium (STEMCELL Technologies, 05855) was used for cryopreservation. Y-27632 dihydrochloride (MedChem Express, HY-10583) was added to media at 10 μM for up to 24 h for initial plating and for several subsequent passages.
Single-cell isolation via FACS
To obtain isogenic hiPSC colonies after CRISPR/Cas9 treatment, we isolated single cells via FACS. At 72 h after nucleofection, we dissociated the hiPSCs into a single-cell suspension by using Accutase and resuspended them in DPBS with 10 μM Y-27632 dihydrochloride (Santa Cruz Biotech). All samples were filtered through 5 mL polystyrene tubes with 35-μm-mesh cell strainer caps (BD Falcon 352235) immediately prior to sorting. After the addition of TO-PRO-3 viability dye (Invitrogen), live (TO-PRO-3-) GFP+ hiPSCs were sorted on the BD FACSAria II with a 100 μm nozzle under sterile conditions and plated at one cell per well onto Matrigel-coated 96-well plates. Once multicellular colonies were clearly visible (2–3 days after sorting), they were collected into individual wells of Matrigel-coated 96-well plates by manual picking. Once individual hiPSC colonies were available (∼14 days after sorting), the genomic DNA from those colonies was characterized by copy-number assay.
Copy-number analysis and characterization of cell lines: Quantitative real-time PCR (qRT-PCR), chromosomal microarray analysis, and optical genome mapping
Array-based comparative genomic hybridization (aCGH) was performed on the CytoScan HD array (ThermoFisher) according to the protocol provided by the manufacturer. The assay tests for imbalances (gains or losses) in the genomic DNA sample. This array platform contains ∼2.7 million probes, including 1,953,246 copy-number probes and 743,304 SNP probes, targeting regions throughout the human genome. A genomic imbalance is noted when six or more oligonucleotides show a minimum average log ratio of 0.25 for one-copy gains and −0.50 for one-copy losses; oligonucleotide information is based on the human genome reference build NCBI 37.3 (hg19). It covers >36,000 RefSeq genes and has 1 marker per 880 bases, complete ISCA constitutional coverage (1 marker/384 bases), cancer gene coverage (1 marker/553 bases), X chromosome genes (1 marker/486 bases), and 12,000 OMIM genes (1 marker/659 bases). Genomic imbalances are called with ChAS software (ThermoFisher) when a minimum of 50 consecutive probes is observed for loss and 50 consecutive probes are observed for gain. A 50 Kb size cutoff (i.e., the lower limit of detection) has been established for CNV calls from constitutional specimens at the Jackson Laboratory. This assay does not exclude chromosome anomalies smaller than the assay’s effective resolution. The assay is also not specifically designed to detect mosaicism, uniparental disomy, methylation abnormalities, or other chromosomal rearrangements (including chromosomal translocations, insertions, and inversions). For optical genome mapping, frozen hiPSC pellets were shipped to Bionano Genomics. Isolation of high-molecular-weight DNA, labeling, data assembly, and identification of breakpoint regions were conducted by Bionano Genomics as described.44
Selection and differentiation of TRA-1-60-positive hiPSCs
Lines selected for differentiation underwent magnetic-activated cell sorting (MACS) for expression of the TRA-1-60 cell-surface marker for selection of pluripotent cells. Cells were separated on a MiniMACS Separator (Miltenyi Biotec, 130-090-312) with Anti-TRA-1-60 microbeads (Miltenyi Biotec, 130-100-832) according to the manufacturer’s instructions (∼2 × 106 cells per line). TRA-1-60-positive cells were plated with Y-27632 dihydrochloride (10 μM), expanded, and cryopreserved with mFreSR. Cells within three passages of TRA-1-60 selection were used for differentiation into NSCs with the PSC Neural Induction Medium kit (ThermoFisher, A1647801) according to the manufacturer’s protocol (MAN0008031). For all CRISPR lines, passage 7 stage NSCs were dissociated for RNA-seq.
For differentiation to iNs, TRA-1-60-positive hiPSCs were plated as single cells at 80% confluence on a Matrigel-coated 6-well plate with Y-27632 dihydrochloride. Polybrene (hexadimethrine bromide; Sigma, 107689) was added at 8 mg/mL within 3 h of replating. Cells were incubated with polybrene for 10–15 min prior to the addition of lentivirus. Lentiviral constructs for directed differentiation of hiPSCs into iNs were made as described previously45 and added to polybrene-treated hiPSCs. Cells were incubated with lentivirus for 24 h, followed by a media change with regular E8. At least 48 h after single-cell replating, transduced hiPSCs were cryopreserved and passaged for expansion. Transduced hiPSCs were expanded onto Matrigel-coated T-25 flasks. Once all lines in a batch reached 70%-80% confluence, cells were replated as single cells onto a new T-25 flask with neural maintenance medium (NMM) supplemented with Y-27632 dihydrochloride and 2 μg/mL doxycycline (Clontech, NC0424034) to begin induction of Ngn2 expression and puromycin resistance (day 0), driven by the tetO promoter. The NMM we use in this study is adopted from Shi et al., 2012.46 24 h after re-plating, media were changed to NMM supplemented with 2 mg/L doxycycline (Millipore Sigma, D9891) and 1 μg/mL puromycin (Sigma) so that selection of Ngn2-expressing cells (day 1) could begin. We added fresh NMM with doxycycline and puromycin to cells to continue selection on days 2 and 3. On day 4, cells were detached with Accutase (ThermoFisher, A1110501) and replated onto Poly-L-Ornithine (10 μg/mL; Sigma-Aldrich, P4957)/Laminin (5 μg/mL; Sigma-Aldrich, L2020)-coated plates with NMM supplemented with 2 mg/L doxycycline, 10 mg/L human BDNF (Pro-Spec, CYT-207), and 10 mg/L human NT-3 (PeproTech, 450-03). Prior to replating, cells were counted via a Countess II Automated Cell Counter (Invitrogen, AMQAF1000) with 2.5 × 105 cells plated per well of a 12-well plate. After replating, iNs were not exposed to air, and they required half-media changes every other day. On day 6, fresh NMM with doxycycline, human BDNF, human NT-3, and 2 g/L cytosine β-D-arabinofuranoside (Sigma, C1768-100MG) was added to prevent glial growth. On day 8, a half-media change with fresh NMM with doxycycline, BDNF, and NT-3 was conducted. For subsequent media changes (days 10+), NMM supplemented with only BDNF and NT-3 was added until cells reached day 24 of differentiation, at which time cells were dissociated for RNA-seq.
Generation of cerebral organoids and dissociation for single-cell RNAseq
To generate cortical organoids from hiPSCs, we used TRA-1-60-positive hiPSCs with the protocol described.47,48 In brief, we derived embryoid bodies (EBs) by dissociating hiPSC colonies and plating 9,000 single cells in each well of a 96-well ultra-low attachment plate (Corning 7007). On day 3, half of the media were replaced with embryoid body media without bFGF and Y-27632. On day 7, EBs were moved to 24-well low-attachment plates (Corning 3473). Media were changed every other day. On day 12, EBs were transferred to a droplet of Matrigel in 6-well low-attachment plates (Corning 3471). Media were changed every 3-4 days, and organoids were moved to an orbital shaker placed in the incubator.
Single-cell suspensions of cerebral organoids were prepared with the Worthington Papain Dissociation System (Worthington Biochemical, LK003153) and previously described adjustments.49 In short, cerebral organoids at 6 months were moved to 60 mm dishes, to which a solution of Papain/DNase was added. Using a new, sterile razor blade for each, we minced organoids to form <1 mm pieces and incubated them at 37°C for 30 min on an orbital shaker set to 70 rpm. Each sample was then mixed with a 1 mL pipette and returned for an additional 10 min incubation. Cells were then triturated with a 10 mL pipette and transferred to new conical tubes to allow debris to settle. These suspensions were transferred to new tubes containing protease inhibitor solution, inverted to mix, and then passed through 40 μm cell strainers into new tubes, which were centrifuged at 300 × g for 7 min. The resulting pellets were resuspended in DPBS with 0.04% BSA between 900 and 1,000 cells/μL and with viability ranging from 87-98%, according to the Countess II Automated Cell Counter. Additional wash/resuspension steps were omitted to promote cell viability.
Neurite dynamics measurement
Neurite dynamics measurement was performed on a live-cell imaging system, Incucyte ZOOM system (Sartorius) with the automated IncuCyte NeuroTrack analysis platform. Time-lapse images were acquired under incubated conditions at 37°C and 5% CO2, as described,50 but with some modifications regarding image analyses. In brief, iNs were plated onto transparent 96-well plates at a density of 17,000 cells per well with IncuCyte NucLight Rapid Red Reagent (1:2000) and imaged every hour (nine image locations from each well) over 7 days at a resolution of 0.61 μm/pixel. The number of biologically independent lines per genotype in the experiment were WT n = 3, 16pDel n = 2, 16pDup n = 3, and KCTD13Het n = 2.
Phase-contrast (cells) and red-channel (nuclei) images (1,392 ×1,040 pixels) were segmented with Essen IncuCyte NeuroTrack software (2018A), and time-course data for neurite length, neurite branchpoints, and number of nuclei were exported. The numbers of images analyzed per group were WT n = 170, 16pDel n = 118, 16pDup n = 105, and KCTD13Het n = 87. Further analysis was performed on these metrics with custom software written in Matlab (R2018b). Images with a nuclear count less than 200 were omitted from analysis. We calculated neurite length per nucleus and neurite branchpoints per nucleus by dividing neurite length and neurite branchpoints by the average number of nuclei detected during the first 8–12 h of the imaging period. These metrics were then smoothed via the robust Lowess method with a window of 10 data points (20 h). Cumulative neurite outgrowth and branchpoints were computed from the sum of the first difference of the smoothed metrics. Outliers were removed by mutant group with median + 1.5∗ inter quartile range. Time course plots were generated using Gramm for Matlab (Morel, 2018). One-way ANOVA with Tukey post-hoc comparisons were used for statistical comparisons between mutant groups.
Microelectrode array (MEA) electrophysiology
To track spontaneous activity in neuronal cultures, we plated 60k iNs (day 5) per well of a 48-well CytoView MEA plate (Axion BioSystems). NMM supplemented with only BDNF and NT-3 were added until cells reached day 24 of differentiation as described above. On day 24, iN culture medium switched to BrainPhys Neuronal Medium (STEMCELL Technologies), and then half of the medium was changed every three days. Starting on day 25, we made extracellular recordings on the Axion Maestro Pro machine (Axion BioSystems) to monitor spontaneous activity within the culture. All recordings were performed in BrainPhys and were started 5 min after the MEA plates were placed on the recording chamber for a duration of 15 min. The raw signals were acquired real-time and analyzed offline with Axion’s Integrated Studio Navigator v3.4.1 software (Axion BioSystems). Measurements obtained by the manufacturer-set thresholds included activity, electrode burst, network burst, synchrony, and oscillation metrics. For assessment of the strength of synaptic connections, Synchrony is measured as the area under the normalized cross-correlation, a unitless measure between 0 and 1.51 A value of 1 means spikes are perfectly synchronous, whereas 0 indicates perfectly asynchronous. For assessment of the functional networks, oscillation is a measure of how the spikes from all of the neurons in a well are organized in time as the coefficient of variation in inter-spike intervals for each electrode; this coefficent is averaged across electrodes in the well. High values indicate action potentials are not coordinated across neurons in the network. Spike raster plots were created in Neural Metric Tool v3.2.5 software (Axion BioSystems). The statistical results were created in Axion’s Integrated Metric Plotting Tool v2.4.4 software (Axion BioSystems). The MEA experiment was performed on two independent plates on different dates, named as two different batches. Data were analyzed from the wells with ≥8 active electrodes/well and normalized by corresponding wild-type (WT) mean within each batch. To test the statistical significance between groups (16pDel, 16pDup and KCTD13Het) and WT samples, we employed a multivariate linear model (∼ edit + batch). The recordings from two 48-well MEA plates were included in the analysis on the basis of the time point with the highest average neuron activity shown in the WT. The numbers of biologically independent lines for MEA analysis are WT n = 2, 16pDel n = 2, 16pDup n = 2, and KCTD13Het n = 2. The numbers of replicates per group were WT n = 15, 16pDel n = 24, 16pDup n = 24, and KCTD13Het n = 18 in the analysis reflected in Figure 5.
Figure 5.

RGD neurons displayed aberrant electrophysiological properties
(A) Overview of the study design. iNs were differentiated from hiPSCs as described in the methods section. On day, 5 iNs were replated onto MEA plates with NMM. The neural activities were recorded after the culture medium switched to BrainPhys Neuronal Medium (day 24).
(B–E) Representative temporal raster plots from iN models demonstrating the activity over time for all electrodes in the well. Each plot is 30 s for sufficient spike and burst resolution, and horizontal rows correspond to 16 electrodes. Abbreviations are as follows: wild type (WT), 16p11.2 deletion (16pDel), 16p11.2 duplication (16pDup), and KCTD13 heterozygous deletion (KCTD13Het). Raster plots were generated with Neural Metric Tool v3.2.5 software (Axion Biosystems).
(F) Neuron activity (normalized weighted-mean firing rate). 16pDel and 16pDup neurons displayed significantly lower activity than WT neurons (∗∗p < 0.01), whereas KCTD13Het neurons displayed a level of activity comparable to that of the WT (p = 0.222). Data are presented as means ± SEM, and normalized data points are plotted.
(G) Neuron synchrony (normalized synchrony Index). 16pDel, 16pDup, and KCTD13Het neurons displayed significantly lower synchrony than WT neurons (∗p < 0.05, ∗∗p < 0.01). Data are presented as means ± SEM, and normalized data points are plotted.
(H) Neuron network oscillation (normalized network ISI coefficient of variation). 16pDel and 16pDup neurons displayed significantly lower oscillation than WT neurons (∗∗∗p < 0.001), whereas KCTD13Het neurons displayed a level of activity comparable to that of the WT (p = 0.276). The number of samples per group was n = 15 (WT), n = 24 (16pDel), n = 24 (16pDup), and n = 18 (KCTD13Het). Data are presented as means ± SEM, and normalized data points are plotted.
Transcriptomics
Mouse models for 16p RGD, samples, and underlying datasets
The 16p11.2 mouse models with reciprocal CNVs of the syntenic 7qF3 region were created at the Cold Spring Harbor Laboratory by A. Mills and colleagues, as previously described in Horev et al.,34 and provided by the Jackson Laboratories (stock numbers 013128 and 013129). After receipt, mice were housed in the animal facility of the Massachusetts General Hospital (Boston, MA), provided with constant access to a standard diet of food and water, and maintained on a 12 h light/dark cycle. All procedures were designed to minimize pain and discomfort, under approved IACUC protocols of the Massachusetts General Hospital, in accordance with National Institutes of Health (NIH) guidelines. Analyses were performed with animals of both sexes. Dissection of mouse tissues was performed simultaneously for all mice at 8 weeks of age. The mouse samples were split into five RNA extraction batches (labeled as B1–B5) and three sequencing batches (labeled as DS1–DS3) (Table S1). An overview of the underlying datasets and samples used for these analyses is shown in Figure 1B, and more details can be found in Table S1.
In brief, the following models and samples were analyzed. (1) 16p11.2 CNV mice. 350 RNAseq libraries from 101 mice with 16p11.2 CNV. In the initial 16 mice, we evaluated six tissues (liver, white fat, brown fat, cerebellum, striatum, and cortex), which enabled exploration of 16p11.2 tissue-specific effects; and in the 85 replication mice, we restricted our analysis to brain tissues (cortex, striatum, and cerebellum). (2) 16p11.2 CRISPR hiPSC-derived NSCs (n = 28) and iNs (n = 24). NSC samples were composed of two batches: batch 1 (WT n = 6, 16pDup n = 8) and batch 2 (WT n = 6, 16pDel n = 8).
Strand-specific RNAseq library preparation
All RNA samples were extracted with Trizol reagent according to the manufacturer’s (Invitrogen’s) instruction. RNA sample quality (based on RNA integrity number [RIN]) and quantity were determined on an Agilent 2200 TapeStation, and between 500 and 100 ng of total RNA was used for library preparation. 1 μL of diluted (1:100) External RNA Controls Consortium (ERCC) RNA Spike-In Mix (Thermo Fisher) was added to each sample; mix 1 and mix 2 were alternated for each well in a batch.
350 mouse RNASeq libraries were prepared with a customized version of the strand-specific dUTP method (188 libraries)42,52 and the Stranded mRNA Library Kit (Illumina) (162 libraries), and the same TrueSeq kit was used in the preparation of 52 human cell line RNAseq libraries (28 NSCs, 24 iNs). Both library preparation methods used polyA capture to enrich mRNA, followed by stranded reverse transcription and chemical shearing to make appropriate stranded cDNA inserts for the library. Libraries were finished by the addition of both sample-specific barcodes and adapters for Illumina sequencing and then between 10 and 15 rounds of PCR amplification. We evaluated the final concentration and size distribution of libraries by using 2200 TapeStation and/or qPCR with the Library Quantification Kit (KK4854, Kapa Biosystems). We multiplexed libraries by pooling equimolar amounts of each prior to sequencing. 350 RNASeq libraries were sequenced on multiple lanes of an Illumina HiSeq 2000 or 2500 platform, generating median 38M paired-end reads of 50, 51, and 75 bp. 52 human cell line RNAseq libraries were sequenced on multiple lanes of an Illumina HiSeq 2500 platform, generating median 33.8M paired-end reads of 75 bp.
RNA sequencing quality control
Quality of sequence reads was assessed by fastQC (version 0.10.1). We generated gene-based counts for mouse and human RNAseq libraries by aligning sequence reads to the mouse reference genome, GRCm38 (v83), and the human reference genome, GRCh37 (v75), and relying on Ensembl gene annotations of these reference genomes by using STAR (version 2.4.2a)53 with parameters “--outSAMunmapped Within --outFilterMultimapNmax 1 --outFilterMismatchNoverLmax 0.1 --alignIntronMin 21 --alignIntronMax 0 --alignEndsType Local --quantMode GeneCounts --twopassMode Basic.” We assessed the quality of alignments by using custom scripts in Picard Tools, RNASeQC,54 and SamTools.55 These quality-checking assessments and exploratory analyses identified one outlier RNAseq in cerebellum samples, Dp835, one outlier in human NSC, H7, and five outliers in human iNs (deletions B5, H7, and C3 and duplications B9 and C5). These samples exhibited high duplication rate varying between 55 and 89% and low estimated library sizes between 3.4 and 20.5 M Dp835 was also an outlier in other quality control metrics including exonic rate (0.41), intergenic rate (0.24) and intronic rate (0.35). Further exploratory analyses including clustering and principal-component analysis (PCA) were implemented in R (version 3.4) with DESeq2 (version 1.18.1)56 and custom scripts. Exploratory analyses identified a striatum sample, t1992, as an outlier in batch 1 samples, which also exhibited a high chimeric-pair percentage (6.73%). These outliers were excluded from further analyses, including those of differential expression and co-expression. Our analyses were restricted to mouse genes with 1-to-1 human orthologs, which were obtained from the Mouse Genome Informatics database in July 2020.
Expression analyses of the CNV region
Genomics coordinates of the engineered region, spanning from Slx1b to Sept1, in 16p11.2 deletion and duplication mouse models were obtained from Horev et al.34 Because original coordinates were based on the mm9 mouse reference genome, they were further converted to mm10 coordinates with Liftover (University of California at Santa Cruz) chr7: 126,688,926–127,218,445. The Ensembl GRCm38 (v. 83) annotations showed that this region harbored 35 protein-coding genes: 27 human orthologous protein-coding genes; two human orthologous segmental duplication genes (Slx1b and Bola2), which were not duplicated in the mouse genome; four additional genes (Cd2bp2, Tbc1d10b, Mylpf, and Sept1) that were centromeric to the CNV and beyond the human 16p11.2 CNV segment; and mouse-specific genes Gm42742 and Pagr1b, although the latter was removed in Figure 1A because it was not a separate gene and has been removed in the most recent mouse genome. Breakpoints of human 16p11.2 CNV regions in our CRISPR/Cas9-treated cell lines were determined as Chr16: 29,487,574–30,226,919 on the basis of human reference genome GRCh37 (v. 75) and CRISPR guides. This region harbored 32 protein-coding genes on the basis of the Ensembl GRCh37 (v. 75) gene annotation. In Figures 1A and 1D, we used the new gene name TLCD3B (MIM: 615175)/Tlcd3b for a 16p11.2/7qF3 gene that was named as FAM57B/Fam57b in the reference genomes (GRCh37 v. 75 and GRCm38 v. 83) used in our analysis. An uncharacterized gene, RP11-37C12.3 (ENSG00000258130), was excluded from the region because it was annotated as a pseudogene in the most recent human reference genome. To estimate the expression levels of SD genes BOLA2B, BOLA2 [MIM: 613182], SLX1A, SLX1B, SULT1A3 [MIM: 600641], and SULT1A4 [MIM: 615819] in human cell lines, we generated two reference genomes in which genes at one SD region were masked with bedtools maskdata.57 This approach allowed better estimation of expression levels of unmasked SD genes. To this end, we first realigned the sequence reads to these reference genomes by using STAR with the above-described parameters. Next, we identified the sequence reads that mapped to the SD genes’ exons with no mismatch, and from this we estimated the expression of a particular SD gene by counting the fragments represented by two mated reads that were mapped to the same gene. Using these count data for six SD genes in the original raw-count matrix, we converted raw-count expressions of SD genes to transcripts per million (TPM). Similarly, applying the DESeq2 + SVA pipeline to the same count data matrix, we estimated changes of these six SD genes in comparisons of deletion versus wild type and duplication versus wild type across human cell lines. We assessed the statistical significance of the deviation of observed n-fold changes of SD genes from expected n-fold changes (n = 0.75 for deletion; n = 1.25 for duplication) in each comparison by applying two-tailed one sample t test.
Differential-gene-expression analyses
Differential expression (DE) analyses performed on genes that passed the expression threshold in a given comparison were conducted with R/Bioconductor packages DESeq2 (v.1.18.1).56 To determine genes that passed the expression threshold for a particular comparison, we first calculated count-per-million (cpm) expression values of genes across the samples used in the comparison. cpm expression of ith gene in sample j was defined as 106 × Ci/LSj, where Ci is raw counts of the ith gene and LSj is the library size of jth sample. We used the total number of uniquely mapped reads reported by STAR for a given sample for the library size of that sample. Next, we calculated the cpm expression threshold corresponding to 10 counts for the particular comparison by using the equation 106 × 10/median(LS), where median(LS) is the median value of library sizes of samples used in the comparison. cpm thresholds varied between 0.27 and 0.38 across the comparisons for which we performed DE analysis. All the genes, regardless of their type (e.g., protein coding, antisense), that had expression values in cpm equal or greater than the cpm expression threshold in at least 50% of samples in either condition (e.g., Del vs WT) were further analyzed in the DE analysis. We performed DE analysis for each type of mouse tissue and human cell line. In these analyses, CNV type (deletion or duplication) was compared with CNV-type-matched wild-type samples in mouse non-brain tissues (e.g., Del vs Del WT). When we compared deletion or duplication to their CNV-matched wild types in brain tissues, we also added wild types not specific for a certain CNV type from the third batch (B3) or the second dataset (DS2) to each of their corresponding wild types (e.g., Del vs WT + Del WT). Deletion and duplication samples were compared to the same wild types in human iNs, whereas deletion and duplication samples were compared to their separate WTs in human NSCs. To account for unknown sources of variation in the expression data, we estimated surrogate variables (SVs) for each comparison by using the R/Bioconductor package Surrogate Variable Analysis (SVA version 3.26) by setting ∼ genotype as the full model and ∼1 as the reduced model.58,59 Differentially expressed genes (DEGs) were identified at false discovery rate (FDR) < 0.1 via the Wald test under the design “∼ genotype + SVs,” where FDR was calculated according to the Benjamini Hochberg procedure.60 In these analyses, DESeq2’s independent filtering and cooksCutoff options were turned off. Protein-coding DEGs were used in the further downstream analyses, including enrichment analyses and comparisons of DEGs across different comparisons. Statistical significance of overlap of DEGs between different comparisons was assessed by a one-sided Fisher’s exact test or, equivalently, a hypergeometric test.
Gene co-expression network analyses
Gene co-expression network analysis for each mouse brain tissue and human cell type was performed separately with R package Weighted Correlation Network Analysis (WGCNA version 1.61).61 For this analysis, we used log2-transformed SVA-corrected counts under the signed network option; we set the minimum module size to 50 and merged modules with >75% similarity. We generated SVA-corrected counts for each brain tissue and human cell line by combining deletion, duplication, and wild-type samples after removing the outlier samples that were described above. SVA was applied to the union of analyzed genes in deletion versus wild-type and duplication versus wild-type comparisons relying on the full model ∼ genotype (deletion, duplication, wild-type) and the reduced model ∼1. Genes in the 7qF3 engineered region and the 16p11.2 region were excluded in the further co-expression analyses of mouse and human samples, respectively. We set negative SVA-corrected counts to zero and log2 transformed them after adding 1 to the count matrix. To identify additional outlier samples in co-expression analysis, we adapted the procedure described by Oldham et al.62 as follows. We first computed the average Pearson’s correlation coefficient for each sample by correlating that sample with other samples within each mouse brain tissue and human cell type. From these sample-specific average correlation values, we further computed tissue- or cell-type-specific average correlation values and standard deviations. Within each mouse brain tissue or human cell type, we identified samples with average correlation values at least three standard deviations lower than tissue- or cell-type-specific average correlation values as outlier samples. In doing so, we identified the following samples as outliers in co-expression analyses: D809 for mouse cerebellum, w1514 and w1516 for mouse cortex, w1210 for mouse striatum, and C5 for human NSCs. These outlier samples were excluded from further analyses. Soft power was selected such that scale-free topology fit (R2) > 0.85. Module membership for each was re-evaluated on the basis of the module membership p_value such that genes with p ≥ 0.01 were marked as unassigned (grey module). To identify the modules highly correlated with deletion and duplication in each co-expression analysis, we investigated how expression patterns of module genes represented by module eigengenes correlated with four situations in which deletion, duplication, and wild-type samples were represented by the following contrasts: (1) dosage effect (−1, log2(3/2), 0); (2) genotype effect (“cnv,””cnv,””wt”); (3) deletion versus duplication + wt (“cnv,””wt,””wt”); and (4) duplication vs deletion + wt (“wt,””cnv,””wt”). The statistical significance of the relationship between module eigengenes and the above contrasts was assessed via linear regression models, eigengenei ∼ vectorj, where i and j index the module in a particular mouse brain tissue or human cell type and one of the four situations described above, respectively. Modules with p < 1 × 10−5 and size ≥30 protein coding genes were selected for further analyses. To profile the expression pattern of module genes across the human neurodevelopment, we generated an expression matrix for selected modules by using the PsychENCODE expression data63 and calculated module eigengenes for each module by employing WGCNA. Next, we calculated the mean module eigengene value for each of nine developmental windows per module as described.63 We assessed the statistical significance of module overlaps by employing a one-sided Fisher’s exact test or, equivalently, a hypergeometric test.
Overrepresentation analysis
To assign biological significance to selected protein-coding genes, including DEGs, DEGs shared between two or more comparisons, and module genes identified in co-expression analysis, we used a one-tailed Fisher’s exact test (equivalent to a hypergeometric test) to perform overrepresentation analysis (ORA) of these genes for Gene Ontology (GO) “biological process” terms,64 which we retrieved from the MSigDB (v7.4) database,65 synaptic gene ontologies (SynGO version 1.1),66 and curated gene sets. ORA was performed only for protein-coding genes, where both selected and background gene sets contained only protein-coding genes. For calculations of the enrichment p value for a particular selected gene set, analyzed protein-coding genes from which selected genes were chosen constituted the background gene set. For example, if ORA would be performed for DEGs from a deletion versus wild type comparison in mouse cortex tissue, then the background set was all the protein-coding genes that were analyzed in this analysis—in other words, all the protein-coding genes that passed the expression threshold. Similarly, if ORA would be performed for a particular module identified in co-expression analysis of mouse cortex samples, then the background gene set was all the protein-coding genes analyzed in this co-expression analysis. In the case of ORA of selected genes shared between two or more comparisons, the background gene set was an intersection of background gene sets for each comparison. In these analyses, both human and mouse genes were represented by their symbol identifiers, and one-to-one human orthologs were used for the latter group. Furthermore, only GO terms with at least 10 associated genes in the background set were considered.
Generation of single-cell RNAseq libraries
Dissociated cells were maintained on ice no longer than 30 min prior to being loaded onto the 10× Chromium single-cell controller (10x Genomics, PN-120263). scRNA-seq libraries were prepared with the Chromium Single Cell 3′ Library & Gel Bead Kit v3 (PN-1000075) according to the manufacturer’s instructions (CG000183) with a Chromium Single Cell B Chip (PN-1000073) for a targeted cell recovery of 5,000 cells. Post-library construction quality control and quantification were performed with both a High-Sensitivity D1000 ScreenTape (Agilent, 5067-5582) and qPCR via the Universal KAPA Library Quantification Kit (Roche, KK4824). Final libraries were pooled according to molar concentrations and submitted for sequencing on the Illumina NovaSeq S4 platform; there were an average of eight libraries per lane at 2.5 billion reads per lane.
Single-cell RNAseq analysis
Each scRNA library was aligned against Ensembl human transcriptome reference GRCh38 version 92 via the 10X CellRanger (v. 3.0.2) program with 5,000 expected cells. We then processed each aligned library in Seurat v. 4.0.0.901067 independently by filtering out cells with the highest and lowest 2.5% quantile RNAs. All filtered libraries were merged, followed by an unsupervised clustering via the UMAP method.68 The number of cell clusters were determined with resolution of 0.2. The unsupervised-cluster labels were transferred to annotated neuron cell types heuristically, on the basis of the expression levels of the selected cell type markers. Empirically, clusters 0, 4, 5, and 6 were labeled as excitatory neurons, whereas clusters 1 and 3 were labeled as inhibitory neurons. Cluster 2 was labeled as astroglia cells.
We then independently analyzed excitatory neurons, inhibitory neurons, and astroglia cells by using. unsupervised clustering via the UMAP method to subdivide cell types. The number of cell clusters was determined with resolution of 0.2. Meanwhile, WGCNA v1.70.369 was applied on highly variable genes from excitatory neurons, inhibitory neurons and astroglia cells, respectively, with soft power of 8, 7 and 5. In each cell type, raw modules were merged with a dissimilarity threshold of 0.25. To identify the genotype effect on each module, the average expression of genes associated with a module in a given genotype was compared to that in WT. Wilcoxon Rank Sum and Signed Rank test was implemented to determine the significance. Bonferroni correction was applied to p values from all identified modules in all the three cell types. To reveal whether a module was enriched for a certain group of genes, Fisher’s exact test was implemented.
Creation of schematic diagrams
The graphical abstract and figures of the study design were created with BioRender.com.
Results
The genomic properties of the 16p11.2 RGD locus
As described above, the human 16p11.2 RGD locus involves ∼743 kb of genomic sequence that includes a unique ∼593 kb segment, as well as at least one copy equivalent of an ∼150 kb flanking SD. The unique segment encompasses 27 protein-coding genes, and four gene paralogs are located in each SD (Figure 1A). Across these 31 protein-coding genes in humans, 30 (with the exception of ZG16 [MIM: 617311]) are expressed at detectable levels (≥0.1 TPM) from bulk RNA sequencing (RNAseq) across at least one of 13 brain regions profiled in the Genotype-Tissue Expression (GTEx) project; the highest fraction of genes were expressed in the frontal cortex (30/31, 97%).70 To place the genomic properties of the 16p11.2 RGD, and the genes therein, within the context of 19 NAHR-mediated RGDs profiled in a study by Collins et al.71 in which both deletion and duplication were associated with a spectrum of disease phenotypes, we note that the proximal 16p11.2 CNV ranked fifth for total number of protein-coding genes among all RGD segments but ranked first after normalization by RGD size (i.e., it is the most gene-dense RGD profiled). The 16p11.2 RGD also ranked fourth for normalized density of constrained genes intolerant to loss-of-function (LoF) variation as defined as the bottom (decile or sextile) of the LoF observed over the expected upper-bound fraction (LOEUF) metric in the Genome Aggregation Database (gnomAD).72 When we further scrutinized these 19 NAHR-mediated GD segments for more nuanced metrics of dosage sensitivity (i.e., intolerance to haploinsufficiency [pHaplo] or triplosensitivity [pTriplo]) provided in Collins et al.,71 the 16p11.2 CNV ranked second in terms of the combined number of predicted haploinsufficient and/or triplosensitive genes when data were normalized by GD size. These data collectively suggest that the functional consequences of the reciprocal 16p11.2 CNV are likely due to a number of dosage-sensitive loci and not concentrated for all phenotypes on a single “driver gene” as are those in regions such as 15q11-13 (UBE3A [MIM: 601623] in Angelman syndrome [MIM: 105830]73) or 17q21 (KANSL1 [MIM: 612452] in Koolen-de Vries syndrome [MIM: 610443]74), among many others.
Transcriptional profiling of 16p11.2 RGD in mouse and human models
To identify the transcriptional consequences of the 16p11.2 CNVs in the brain, we used both mouse models of deletion and duplication of the 7qF3 region of synteny conservation and neuronal derivatives of 16p11.2 RGD hiPSC models (Figures 1A and 1B). These mice display 16p11.2-relevant phenotypes because of CNVs that include orthologs of the 27 unique 16p11.2 protein-coding genes and of two genes (SLX1A and BOLA2) that are located in the flanking human segmental duplication and which are not duplicated in the mouse.34 The reciprocal mouse CNVs also include four genes located outside the human segment: Cd2bp2, Tbc1d10b, Mylpf, and Sept1. We performed transcriptional profiling in brain tissue on a collection of 101 mice (12 deletion, 31 duplication, and 58 wild-type [WT] littermates) across cortex, striatum, and cerebellum (302 total libraries from brain tissue) and examined three non-brain tissues (liver, white fat, and brown fat) of relevance to 16p11.2 RGD phenotypes in a subset of 16 mice (four deletion, four duplication, and eight WT littermates; 48 total libraries from non-brain tissue). In summary, we profiled tissue-specific expression patterns from mouse 7qF3 CNVs across 350 RNAseq libraries (Table S1).
For the comparison with human cellular models, we generated isogenic hiPSC lines with 16p11.2 CNVs by using the CRISPR SCORE method7 and genotyped them by quantitative real-time PCR (qRT-PCR) and genome-wide array-based comparative genomic hybridization (aCGH). For a subset of lines, we additionally performed nanopore sequencing and generated direct label and stain (DLS) optical genome maps (Bionano Genomics) (Figure S1A, see methods). We then differentiated the hiPSCs into NSCs (n = 27 lines; 12 WT, seven deletion, and eight duplication), iNs (n = 19 lines; six WT, seven deletion, and six duplication), and cerebral organoids (n = 8) to assess disease-relevant cellular and transcriptomic signatures (Figure 1B). Cellular identities of all lines were verified with cell type-specific marker gene expression in the RNAseq data (Figure S1B).
We first reviewed the local expression patterns of genes within and near the RGD segment by CNV genotype. The expression levels of genes within the CNV interval in WT mice varied widely by tissue. In general, their expression levels and the significance of altered expression caused by the CNV were relatively greater in brain tissues, with some exceptions, such as Qprt, which was predominantly expressed in the liver (Figures 1C and 1D, Table S2). GTEx data show a comparable overall pattern, where 16p11.2 CNV genes are more highly expressed in human brain tissues than in non-brain tissues and where the human NSCs and iNs showed brain-like expression, except for QPRT [MIM: 606248] (Figures 1C and 1D). In both mouse and human experiments, expression changes of most genes within the CNV segment reflected their dosage loss or gain, as expected (Figures 1E and S1C). We found no consistent evidence of dosage compensation from the unaltered allele in the brain regions, as we had observed previously for human lymphoblastoid cells.42 The fact that the SD genes NPIPB12 and NPIPB13 are members of a dispersed set of paralogs (absent in the mouse genome) precludes their individual quantification, whereas the paralog pairs BOLA2/BOLA2B, SLX1A/SLX1B, and SULT1A3/SULT1A4 each constitute four copy equivalents in the WT human lines. The human deletion and duplication cell lines lost and gained one SD copy, respectively, corresponding to expected 0.75- and 1.25-fold changes in expression for these three paralogue pairs. The average n-fold expression change of these SD genes in human cell lines did not deviate significantly from these expectations (iN deletion, n = 0.79 ± 0.07, p = 0.17; iN duplication, n = 1.25 ± 0.04, p = 0.84; NSC duplication, n = 1.18 ± 0.11, p = 0.18), except that SD genes were significantly more downregulated than expected in the NSC deletion samples (n = 0.64 ± 0.10, p = 0.047).
A few genes in mouse tissues and human cell lines did not show altered expression levels consistent with an expected CNV effect. Gdpd3 exhibited highly variable expression across all six mouse tissues, previously reported behavior attributed to genetic background differences of the parental mouse strains.34 Genes with higher-than-expected dysregulation include Kif22 in brown fat, Mylpf in liver, Qprt and Zg16 in white fat (Figure S1C), and TLCD3B in human NSCs (6.6-fold, Figure 1E). Overall, genes in the engineered CNV region were expressed more variably in the non-brain tissues than in the brain tissues. Principal-component analysis based on the expression profile of the 27 CNV genes separated all tissues (Figure S1D).
Genome-wide transcriptional changes in the RGD models
We next asked whether genes outside the 16p11.2 region showed altered expression patterns in our RGD models. In the mouse models, the number of genes differentially expressed (on the basis of FDR <0.1) as a result of 7qF3 deletion or duplication (excluding the CNV genes) varied greatly across tissues (Figure 2A, Table S3). Although most of these effects were tissue specific, there was significantly greater sharing of DEGs than expected by chance across the brain tissues (Figure 2A); such sharing that was evident for both for deletion- and duplication-associated DEGs (Figures S2A and S2B). Conversely, within each brain region there were fewer DEGs shared between deletion and duplication models (Figure S2C). Interestingly, many of these DEGs were similarly upregulated or downregulated in both CNV models (i.e., perturbed non-reciprocally). It is possible that for some genes (e.g., Ccdc101 and Kdm8) near the CNV region, differential expression could be due to position effects observed in brain tissues but not in peripheral tissues. Only one gene, Kctd21, was differentially expressed as aresult of both 7qF3 CNVs in all three brain tissues; deletion was associated with upregulation, and duplication was associated with downregulation (i.e., reciprocal dysregulation). The overall pattern of gene-expression changes observed in brain tissue was not observed in peripheral tissues, where there was little sharing of DEGs across liver tissue, brown fat, and white fat or between these tissues and the brain regions (Figure 2A). With the notable exception of liver tissue, these peripheral tissues, like the brain tissues, lacked significant DEG sharing between deletion and duplication models, (Figure S2C). Overall, our mouse DEG analyses highlight significant shared effects of the CNVs that are greatest in the brain but also reveal many more distinct tissue-specific reciprocal and non-reciprocal impacts.
Figure 2.

Transcriptional profiling and disease specific brain signatures in 16p11.2 transgenic mouse and human neuron models
(A) Overlap among the DEGs observed between mouse brain tissues, peripheral tissues, and human cells.
(B) Overlap among the DEGs observed between NSCs and iNs.
(C) GO enrichment analysis is shown as bar plots for comparisons of mouse brain tissues, human NSCs and iNs. Enrichments at a nominal level and at FDR <0.1 are marked in light blue and blue, respectively.
(D) SynGO enrichment analysis for comparisons of mouse brain tissues, human NSCs, and iNs. Enrichments at nominal level (p < 0.05) and at FDR <0.1 are marked in light blue and blue, respectively.
(E) The pathways enriched for shared mouse brain DEGs.
(F) GO Biological Process terms enriched for DEGs shared between human NSCs and iNs.
In the human cell models, the number of global changes in gene expression varied strikingly between NSCs and iNs (Figures 2A and 2B, Table S3); the former yielded many more significant DEGs due to either 16p11.2 deletion or duplication. Despite the surprising paucity of iN DEGs, there was significant sharing of DEGs between NSCs and iNs (p = 7.55 × 10⁻7), although these effects were less significant than the stronger sharing exhibited across the mouse brain regions (p < 1× 10⁻13) (Figures 2A, S2A and S2B). However, the NSC DEGs showed more significant evidence than the brain regions of sharing between deletion and duplication effects (p = 6.12× 10⁻7), although only 31 of the 81 shared DEGs exhibited reciprocally altered expression (Figures 2B and S2C). Thus, like the adult tissues of the mouse model, the human models of NSC and maturing neuron developmental stages point to a combination of shared and nonshared effects across cell types, in that there is evidence for both reciprocal and non-reciprocal changes in gene expression.
Gene Ontology enrichment analysis of differentially expressed genes
To explore the potential functional ramifications of differential expression in these models, we performed Gene Ontology (GO) “biological process” term enrichment (Figure 2C and Table S4). No GO terms were enriched at an FDR-significant level (FDR <0.1) among mouse cortex DEGs, but the mouse cerebellar and striatal deletion-elicited DEGs both showed significant enrichments for a number of GO terms related to energy metabolism, for example oxidative phosphorylation and electron transport chain. Although the significant terms revealed by deletion-elicited DEGs largely overlapped between these two brain regions, a few were tissue-specific, including proton transmembrane transport in cerebellum and tricarboxylic acid cycle in striatum. In cerebellum, the duplication CNV elicited a much larger number of DEGs, which revealed enrichments for a number of neuronal-development-related terms, including neuron differentiation and neurogenesis (Figure 2C). Therefore, we repeated our enrichment analyses by using the expert-curated SynGO database, which was developed for studying synaptic biology and includes a number of additional GO “biological process” and “cellular components” terms (Figure 2D and Table S5). Notably, known NDD-associated genes are enriched for SynGO terms, suggesting that leveraging this database could provide insights into pathogenic mechanisms. The cerebellum 7qF3 duplication DEGs showed significant enrichments for a series of synapse-related terms, including synapse, synapse organization, process in the synapse, and postsynapse. In contrast, cortex 7qF3 duplication DEGs were enriched for synaptic vesicle priming and extrinsic component of presynaptic membrane (Figure 2D).
Given the significant sharing of DEGs across brain regions, we also performed GO-term enrichment analysis for the 223 unique DEGs shared by at least two brain regions. These showed significant enrichment only for the energy-metabolism-related terms cellular respiration and energy derivation by oxidation of organic compounds (Figure 2E, Table S4), both at lower significance levels than those of the distinct energy-metabolism-related terms enriched among cerebellum and striatum DEGs (Figure 2C, Table S4). These data indicate that many additional DEGs that are not shared across the brain regions contribute to the increased significance of the energy-metabolism-related terms in individual brain regions, suggesting that analysis focused solely on shared effects across tissues does not fully capture the extent of a biological process altered as a result of the CNV.
Consistent with the findings from the mouse brain regions, the iN DEGs associated with duplication showed FDR-significant GO-term enrichment for energy-metabolism-related terms, among many others (Figure 2C, Table S4). The iN duplications also revealed significance for a variety of terms related to translation, mRNA metabolism and protein targeting, and some of the RNA metabolism terms (RNA catabolic process, RNA metabolic process) were shared with NSC duplication-elicited DEGs, although the latter showed fewer GO enrichments overall. Not surprisingly, the SynGO enrichment analyses of DEGs from the iNs and NSCs differed substantially, and significant terms resulted only with the more differentiated iNs, where SynGO: metabolism was prominent, along with translation at both presynapse and postsynapse (Figure 2D, Table S5). Several of the significant terms overlapped with the mouse brain analysis of cerebellar duplication-elicited DEGs. Interestingly, the 81 genes shared between 16p11.2 deletion and duplication NSCs were not significantly enriched for any GO terms, failing to support a common process disrupted by the reciprocal dosage changes in this cell type. By contrast, analysis of the 28 DEGs shared between the NSC and iN DEGs yielded significant enrichment for a series of energy metabolism-related terms (Figure 2F, Table S4) and for terms related to synaptic protein translation (Figure S2D, Table S5) pointing to these processes as disrupted across both cell types.
None of the GO terms from brain-tissue analyses emerged as significant in the peripheral tissues (Figure S3A and S3B). Brown-fat DEGs revealed no significant GO-term enrichment, whereas the deletion- and duplication-elicited DEGs each yielded a distinct set of significant enrichments in liver and white fat. Liver deletion DEGs showed top enrichments for a series of terms related to muscle or actin-based contraction and filament sliding, monocarboxylic acid metabolism, and lipid metabolism, whereas those due to duplication revealed terms such as connective tissue development, chondrocyte differentiation, and apoptotic process. In white fat, the deletion DEGs detected most prominently a series of terms related to organic acid metabolism, whereas the duplication DEGs revealed terms related to inflammation.
Overall, the mouse DEG and enrichment analyses suggest that in the brain, the CNVs produce both shared effects, whose strength varies greatly across the three brain regions, and effects that are largely region specific. In the peripheral tissues, the significant differences are largely tissue and dosage specific. Analyses of the human 16p11.2 models reinforce the view that, at the level of FDR-significant differences, the gene-expression pattern due to the reciprocal CNVs is largely cell-type specific even when it involves the same biological process and that disruption of genes involved in energetics and synapse-related functions is a feature shared across mouse brain and human neuronal cells.
Co-expression analyses of mouse tissues and human cell lines
The finding that similar biological processes are revealed in different tissues by largely distinct sets of CNV-elicited DEGs indicates that an analysis limited to FDR-significant DEGs does not adequately capture the biological impact of the RGD. Consequently, as a complementary route that utilized all of the gene expression data in defining the processes disrupted by the CNVs, we performed weighted gene correlation network analysis (WGCNA).61 In view of the largely tissue- and region-specific pattern of DEGs and the forced differential expression of the genes contained in the CNV region, we applied WGCNA individually to each brain region, peripheral tissue, and human cell type after excluding the 16p11.2 CNV genes. For a given analysis, we combined deletion, duplication, and wild-type datasets, performed WGCNA, and then examined the eigengenes of the resulting modules for fit to (1) a reciprocal CNV effect (CNV dosage), (2) an effect driven solely by deletion (deletion versus duplication + WT) or by duplication (duplication versus deletion + WT), or (3) a similar effect induced by both deletion and duplication (genotype versus WT). Within each co-expression module that showed a significant fit at p < 1 × 10−5, we tested the member genes for enrichment of GO “biological process” terms and SynGO terms as well as enrichment of a variety of neurodevelopment-associated gene sets. The results for mouse brain regions and human cells are shown in Figure 3 and for mouse peripheral tissues are shown in Figure S3C. The latter yielded only one significant co-expression module in the liver and none in brown fat or white fat. The full lists of co-expression modules in the mouse brain and human-cell data are shown in Tables S6A and S6B, respectively. Module eigengenes of all the modules identified by WGCNA for six mouse tissues and human NSCs and iNs are shown in Table S7.
Figure 3.

Weighted gene co-expression network analysis (WGCNA) of mouse brain and human cell transcriptome
modules that are statistically significantly associated with deletion (Del) and duplication (Dup) genotypes at p < 1 × 10−5. The first- and second-row annotations indicate for each column the source tissue or cell line for modules and whether module genes are more highly expressed in prenatal or postnatal stages. The first panel, named “eigengene genotype correlations,” is colored to show the statistical significance of the eigengene correlation for the listed module. The second panel, named “differentially expressed genes,” is colored to show the statistical significance of the overlap between module members and differentially more- or less-expressed genes (FDR < 0.1) from the corresponding mouse tissue or human cell line with deletion (Del) or duplication (Dup) genotype. The third and fourth panels list the number of genes overlapping between the module and literature-curated gene lists (disease gene sets and functional gene sets, respectively) and are colored to show the statistical significance of the overlap. The lists include ASD-associated genes,29 NDD-associated genes,29 genes identified in the DDD,75 rare variants in genes associated with SCZ,76 chromatin modifiers,77 loss-of-function-intolerance-constrained genes (LOEUF <0.35) as reported by the genome aggregation database consortium,72 FMRP targets,78 synaptic genes from SynGO v1.1,66 and Rho GTPase cycle genes.65,79 The heatmap color scale highlights the statistical significance in −log10 scale, and numbers in the heatmap cells are Pearson’s correlation coefficients (the first panel) or the number of genes shared between gene sets and modules (other panels). Numbers within the parentheses next to module names show the number of protein-coding genes in the co-expression modules identified in the mouse tissue, human NSC, and iN modules.
Analysis of the cerebellum (CE modules) and cortex (CO modules) yielded four co-expression modules each (CE1–CE4 and CO1–CO4, respectively) that showed a significant fit to one or more of the models tested, and the striatum (S modules) revealed five significant modules (S1–S5) (Figure 3). For most of these co-expression modules, the eigengenes showed the greatest significance for a continuous reciprocal effect of CNV dosage, albeit with different relative contributions from deletion and duplication, as evidenced by the lesser significance achieved for solely a deletion effect (deletion versus duplication + WT) and solely a duplication effect (duplication versus deletion + WT). Notable exceptions, such as the cerebellar CE4 module and the striatal S5 module, provided greater significance for an effect limited to deletion (only enriched for deletion-upregulated DEGs). In contrast, the cortex CO2 module revealed significance related to duplication (only enriched for duplication-downregulated DEGs). Notably, no module was significant for gene-expression effects driven in the same direction by both deletion and duplication. The cerebellum CE1 module, whose eigengene showed a positive correlation for 16p duplication, displayed the most significant enrichment for ASD,29 NDD,29 genes identified in the Deciphering Developmental Disorder Study (DDD),75 schizophrenia (SCZ) genes,76 chromatin modifiers,77 loss-of-function (LoF)-constrained genes,72 mRNA targets of fragile X messenger ribonucleoprotein (FMRP targets),78 and Rho GTPase cycle genes,65,79 which was the top significantly enriched term from the REACTOME database.79 These disease and functional gene sets have been described previously in relation to 16p11.2 CNV genes and associated neurodevelopmental and psychiatric disorders.2, 3, 4, 5, 6 Other modules, including CE3, CE4, CO1–CO4, and S1–S3, displayed selective enrichments for disease and functional gene sets. The results of co-expression enrichment indicate significant disturbance of the transcriptome caused by 16p11.2 CNVs, but with effects that are variable across brain regions. The distinct impact on various brain regions may indicate a potential link between tissue-specific contributions to a spectrum of phenotypes associated with 16p11.2 RGD.
To gain insights into disease development and progression, we then compared expression patterns of member genes from co-expression modules across human brain developmental time points by using the expression data from the PsychENCODE project63 (Figure S3D). Except for CO2, S4, NSC2, and NSC4, most of the modules exhibited high expression in the prenatal stage, suggesting that the impact of 16p11.2 CNVs is significant in early developmental stages (Figure 3 and Figure S3D). Module CO2, highly expressed in the postnatal stage, displayed significant enrichment for LoF-constrained genes,72 FMRP targets,78 and synaptic genes,66 strongly suggesting that this module contributes mainly to the abnormalities in the cortex (Figure 3).
GO-term enrichment revealed biological processes associated with the various co-expression modules (Figure 3, Tables S8A and S9). Although some mouse brain modules (CO1, CO2, S3, and S5) provided comparatively weak or no support for GO enrichment (FDR > 1 × 10−5), most pointed to multiple terms that had moderate to high support (FDR < 1 × 10−5) and that were most often shared with several other modules across all three brain regions, indicating disruption of some of the same processes across the brain. For example, the top-scoring term in cortex (CO4, translational termination) was also significant in CE4 and S4. The top-scoring terms in cerebellum modules CE2 and CE4 (cellular respiration and cotranslational protein targeting to membrane) were also significant in S4 and CO4, respectively. The top-scoring term in striatum (S4, mitochondrion organization) was also significant in CO4 and CE2. Most of the shared terms across the modules were related to energy metabolism (e.g., cellular respiration; oxidative phosphorylation; mitochondrion organization; aerobic respiration; ATP synthesis-coupled electron transport), to RNA metabolism (e.g., RNA processing; mRNA processing; RNA splicing; mRNA metabolic process; RNA catabolic process), to translation (e.g., peptide metabolic process; translational initiation; translational elongation; translational termination; and peptide biosynthetic process), or to protein targeting (e.g., establishment of protein localization to endoplasmic reticulum; protein targeting). Other GO enrichments that were detected at FDR < 1 × 10−5 in only a single module typically also received weaker support (1 × 10−5 < FDR < 0.1) in some other modules, but some enrichments implicated distinctly region-specific effects (e.g., cerebellum CE1, homophilic cell adhesion via plasma membrane adhesion molecules; striatal S4, proteasomal ubiquitin-dependent protein catabolic process). Interestingly, while pointing to cell adhesion, the CE1 module did not detect any of the energy metabolism, RNA metabolism, translation, or protein-targeting terms, yet it was notably enriched for disease and functional gene sets. These results suggest that the co-expression of these genes in the largest module observed (2,563 genes) (Table S8A) might reflect convergence of components of multiple biological processes and thereby represent disease pathways that are not well captured by individual GO terms defined from normal biological processes.
A similar analysis of the less abundant data from human cell lines yielded six significant modules in NSCs (NSC1–NSC6) and one in iNs (iN1) (Figure 3, Tables S8B and S9), generally of smaller size and lower significance than the modules detected in the mouse brain. In individual instances, these modules favored reciprocal effects (NSC1) or effects primarily due to 16p11.2 deletion (NSC2, NSC4–NSC6) or to duplication (NSC3, iN1). NSC1 and iN1 modules displayed common enrichments for disease and functional gene sets, including NDD, SCZ, and LoF-constrained genes, whereas iN1 yielded a distinct enrichment for synaptic genes. In GO enrichment analysis, only NSC6 yielded a significant functional category at FDR < 1 × 10−5, and most of these were among the RNA metabolism terms noted above for the mouse model. Weaker support (1 × 10−5 < FDR < 0.1) was obtained for other terms, including a variety of terms related to cellular morphogenesis (iN1, NSC4), cell substrate adhesion (NSC1), cell-cell adhesion, and neurogenesis (NSC4) and energy metabolism (NSC6).
Overall, the co-expression analysis yielded both distinct and common enrichments across tissues and cells, suggesting that there are critical genes shared by particular modules. We performed a pairwise comparison of the modules that exhibited enrichment for ASD or NDD gene sets at p < 0.1 to define shared genes and signatures (Figure S3E). Amongst these comparisons, CE1 and NSC1, both containing genes highly expressed in the prenatal stage, displayed the most significant overlap (p < 1 × 10−10) between mouse and human cell line modules: 423 shared genes were significantly enriched for disease gene sets, chromatin modifiers,77 LoF-constrained genes,72 FMRP targets78 and the Rho GTPase cycle.65,79 Interestingly, these two modules were correlated in opposite directions with CNV dosage, suggesting that some genes that impact the system during the early neurodevelopmental stages can respond to perturbation by deletion and duplication in a context-specific manner (Figure S3E).
SynGO analysis of the mouse brain and human-cell WGCNA modules revealed fewer significant terms overall at FDR <0.1 (Table S9). In the mouse brain, these were limited to CE2 and CE4, the latter of which revealed the greatest significance. However, again CE1 differed; its top terms were related to synaptic organization and function (maintenance of alignment of postsynaptic density and presynaptic active zone; postsynaptic spectrin-associated cytoskeleton organization; regulation of presynaptic membrane potential), whereas the most significant enrichments in both CE2 and CE4 related to translation (e.g., SynGO: postsyn_ribosome postsynaptic ribosome, SynGO: presyn_ribosome presynaptic ribosome, and GO: translation at synapse). The NSC and iNs shared enrichment of terms related to synaptic organization (e.g., synapse organization), whereas the top hits in each were anchored component of presynaptic active zone membrane and SynGO: synprocess process in the synapse, respectively.
The neurons with 16p11.2 RGD display aberrant spatiotemporal neurite dynamics
To determine whether the changes in gene expression caused by the 16p11.2 deletion and duplication lesions were reflected in the functional properties of neurons carrying these lesions, we evaluated whether the 16p11.2 RGD alters the morphological and electrophysiological properties of iNs. To assess neurite dynamics, we performed morphological analysis of the 16p11.2 RGD iNs by using the IncuCyte real-time live-cell imaging system (Sartorius) over seven days (Figure 4A) in comparison with WT cells and with cells heterozygous for inactivation of KCTD13, which is a candidate driver gene in the 16p CNV. Figure 4B shows the corresponding neurite outgrowth images with image segmentation. One-way ANOVA indicated significant differences in cumulative neurite length (F = 25.14, p = 4.12 × 10−15, df = 3) and neurite branchpoints (F = 33.14, p = 1.81 × 10−19, df = 3) across WT, 16p11.2 deletion (16pDel), 16p11.2 duplication (16pDup), and KCTD13 heterozygous deletion (KCTD13Het) lines. The iNs with 16p11.2 CNVs showed lower neurite length and reduced numbers of neurite branchpoints (Figures 4C and 4D), whereas the WT and KCTD13Het iNs displayed comparable values (Figures 4C and 4D). Post-hoc comparisons with the Tukey HSD test of the 16pDel (neurite length—mean = 140.30, SE = 7.52; neurite branchpoints— mean = 3.10, SE = 0.25) and 16pDup (neurite length—mean = 139.68, SE = 7.98; neurite branchpoints—mean = 3.79, SE = 0.26) iNs confirmed significantly decreased total neurite length (p = 4.31 × 10−9 and p = 5.37 × 10−9, respectively, Figure 4E) and branchpoints (p = 3.77 × 10−9 and p = 4.85 × 10−9, respectively, Figure 4F) when compared to the WT (neurite length—mean = 203.73, SE = 6.27; neurite branchpoints—mean = 5.77, SE = 0.21). The 16p11.2 CNV iNs were also significantly different from the KCTD13Het iNs (neurite length—mean = 207.24, SE = 8.76; neurite branchpoints—mean = 6.01, SE = 0.29) in terms of both neurite length (p = 4.44 × 10−8 and p = 7.47 × 10−8, respectively, Figure 4E) and neurite branchpoints (p = 3.77 × 10−9 and p = 1.14 × 10−7, respectively, Figure 4F). The 16p11.2 CNV iNs were not significantly different from each other, and there was no significant difference between the WT and KCTD13Het iNs. Taken together, these findings strongly suggest that 16p11.2 CNV results in neurite outgrowth and branching deficits, that some of the genes in the region are involved in these mechanisms, and that deletion of KCTD13 alone does not recapitulate these neuronal deficits.
Figure 4.

16p11.2 RGD neurons revealed altered neurite dynamic features
(A) Experimental design. iNs were differentiated from hiPSCs as described in the methods section. On day 5, iNs were replated onto 96-well plates and imaged over 7 days with the Incucyte ZOOM system (Sartorius).
(B) IncuCyte images of iNs at 0, 4, and 8 days after being plated with overlaid neurite (blue) and nucleus (magenta) segmentation masks (scale bar represents 50 μm).
(C) The results for cumulative neurite length. 16p11.2 deletion (16pDel) and duplication (16pDup) iNs showed significant differences in neurite length compared to that of WT and KCTD13 heterozygous deletion (KCTD13Het) iNs at the p < 0.05 level (one-way ANOVA). The shaded area indicates SEM.
(D) Similar results for cumulative neurite branchpoints. 16pDel and 16pDup iNs showed significant differences in neurite branchpoints compared to those of WT and KCTD13Het at the p < 0.05 level (one-way ANOVA). The shaded area indicates SEM.
(E) Post-hoc comparisons of neurite length via the Tukey HSD test. 16pDel and 16pDup neurons displayed significantly reduced neurite length in comparison to WT neurons (∗∗∗p < 0.001), whereas KCTD13Het neurons displayed neurite length comparable to that of WT neurons (p > 0.05).
(F) Post-hoc comparisons of neurite branchpoints via the Tukey HSD test. Compared to WT neurons, 16pDel and 16pDup neurons displayed significantly fewer neurite branchpoints (∗∗∗p < 0.001), whereas the number of neurite branchpoints displayed by KCTD13Het neurons was comparable to that displayed by the WT group (p > 0.05). The numbers of images per group were n = 170 (WT), n = 118 (16pDel), n = 105 (16pDup), and n = 87 (KCTD13Het).
16p11.2 RGD neurons exhibit altered electrophysiological features
To assess the electrophysiological features of the 16p11.2 RGD neuronal cultures, we measured spontaneous neuronal firing by using multi-electrode arrays (MEAs), a non-invasive platform for simultaneous recording of electric signals from multiple electrodes, to study electrophysiology in vitro (Figure 5A). We differentiated iNs, replated 60k day 5 iNs onto MEA plates, then continued differentiation and recorded their activity directly for a period of time (Figure 5A). Representative temporal raster plots illustrating timestamps of spikes over 1 min of continuous recording; overlaid representative waveforms are shown in Figures 5B–5E, respectively, for WT, 16pDel, 16pDup, and KCTD13Het iNs. One-way ANOVA indicated significant differences in neuronal activity (normalized weighted mean firing rate, F = 4.28, p = 8 × 10−3, df = 3), functional connections between neurons (normalized synchrony, F = 4.69, p = 5 × 10−3, df = 3), and functional networks (normalized oscillation, F = 5.21, p = 3 × 10−3, df = 3) across WT, 16pDel, 16pDup, and KCTD13Het lines. We observed a significant effect of 16p CNVs on neuronal activity (WT—mean = 1, SE = 0.09; 16pDel—mean = 0.65, SE = 0.05, p = 1.02 × 10−3; 16pDup—mean = 0.65, SE = 0.06, p = 3.23 × 10−3) (see methods), whereas WT and KCTD13Het (mean = 0.85, SE = 0.12) were not significantly different (p = 2.23 × 10−1) (Figure 5F). The neurons with 16p11.2 CNVs and KCTD13Het all displayed significantly reduced synchrony (WT—mean = 1, SE = 0.22; 16pDel: mean = 0.52, SE = 0.08, p = 7.97e-3; 16pDup: mean = 0.51, SE = 0.08, p = 8.54 × 10−3; KCTD13Het: mean = 0.43, SE = 0.05, p = 1.13 × 10−2) (Figure 5G). However, only 16pDel and 16pDup neurons exhibited significantly reduced oscillation (deletion—mean = 0.77, SE = 0.04, p = 7.39 × 10−5; duplication—mean = 0.78, SE = 0.04, p = 5.1 × 10−5) (Figure 5H) in comparison to WT (mean = 1, SE = 0.05), whereas KCTD13Het neurons were not significantly different (mean = 0.91, SE = 0.06, p = 2.76 × 10−1). As was the case with the morphometric phenotyping, these data indicate that changes in dosage of 16p11.2 genes affect the electrophysiological properties of iNs.
Altered cell complement in 16p RGD cerebral organoid model
The observation of an impact on the development and function of iNs by both 16p11.2 deletion and duplication, coupled with the fact that a number of the significant WGCNA modules from mouse brain and human NSCs were enriched for members of a module defined in early human neurodevelopment (‘M2’ in Figure 3), prompted us to assess potential neurodevelopmental deficits in cerebral organoids. We differentiated a subset of hiPSC lines with 16p CNVs or KCTD13 heterozygous inactivation into cerebral organoids by using the protocols as described47,48 and performed scRNAseq on 6-month-old organoids (n = 2 WT, 2 16pDel deletion, 2 16pDup duplication, and 2 KCTD13Het) to investigate genotype-specific single-cell signatures (Figure 6A). These data were analyzed by uniform manifold approximation and projection (UMAP), GO enrichment, and co-expression analysis (WGCNA). Expression of canonical marker genes identified excitatory neurons, inhibitory neurons, and astroglia as three major cell classes in the cerebral organoids (Figures 6B and S4A). A fourth cell population that did not pass criteria and was annotated as “unknown” expressed limited oligodendrocyte and microglia-related markers (Figures 6B and S4A). The cerebral organoids carrying 16p11.2 CNVs displayed reciprocally altered ratios of excitatory to inhibitory neurons in comparison to WT (Figure 6C); 16pDel and 16pDup organoids had relatively more inhibitory and excitatory neurons, respectively. The KCTD13Het organoids showed no dramatic changes in cell ratio as compared to WT, again suggesting that the observed functional changes are a result of the combinatorial effects of many genes in the region and that the altered excitatory/inhibitory neuron ratio seen with the 16p11.2 deletion is not driven by KCTD13 alone.
Figure 6.

Altered neurodevelopmental signatures in 16p11.2 cerebral organoids
(A) Experimental design. 16p11.2 organoids were differentiated from hiPSCs, as described in the methods section. The organoids at 6 months were dissociated as single-cell suspensions and further processed by 10× Chromium and sequenced on an Illumina NovaSeq S4 platform.
(B) Clustering of organoid cells in the UMAP space, with cell types assigned.
(C) Proportion of cells in each cell type per genotype (WT, 16pDup, 16pDel, and KCTD13Het).
(D) Modules’ average expression change in each genotype compared to WT. The circle size represents the significance of expression changes in terms of negative-log10-transformed Bonferroni-corrected p values, and the color gradient represents the strength of expression changes in the log-transformed scale. In inhibitory neurons, Inhibitory_ME2 contains all the six HVGs from the 16p11.2 region and is positively correlated with 16p11.2 CNV dosage, as expected.
(E and F) Normalized expression of CALB2 (E) and the eigengene expression of Inhibitory_ME4 (F) across the inhibitory neuron population in the UMAP space.
To dissect the underlying molecular patterns that led to genetic lesions and cell-composition imbalance in these organoids, we investigated gene co-expression modules that are correlated with various 16p genotypes (Table S10). To avoid data sparsity from scRNA, we used only highly variable genes (HVGs) in each cell population. Although two modules from astroglia, “ME3” and “ME10,” showed overall upregulation due to 16p11.2 deletion and to heterozygous KCTD13 deletion, respectively, indicating the potential for non-neuronal effects, the most significant correlation with 16p11.2 gene dosage was found in the “ME4” module of inhibitory neurons. Notably, the inhibitory ME4 module was negatively correlated with 16p11.2 CNV dosage (Figure 6D and Table S10) and showed modest enrichment for NDD-associated genes (Figure S4B), supporting the importance of an effect on inhibitory neurons. Indeed, GO enrichment analysis of ME4 member genes revealed significance for terms relevant to cell morphogenesis, neurogenesis, and neuron differentiation (Figure S4C and Table S11), whereas SynGO enrichment analysis revealed no significant terms. Moreover, a number of the top 10 genes most correlated with the ME4 module eigengene were highly relevant to GABAergic inhibitory neuron function (Figure S4D and Table S12). Among these, GAD2 (MIM: 138275) encodes a GABA-synthesizing enzyme, and DLX2 (MIM: 126255) and DLX5 (MIM: 600028) specify GABA interneuron progenitor transcription factors.80 We further explored which subtype of inhibitory neurons was represented by the inhibitory ME4 module by using six subtype markers, including CALB1 (MIM: 114050), CALB2 (MIM: 114051), NPY (MIM: 162640), PVALB (MIM: 168890), SST (MIM: 182450), and VIP (MIM: 192320). Interestingly, we found that the expression pattern of CALB2 (encoding calretinin) across the inhibitory neuron population (Figure 6E) was most similar to the expression pattern of the inhibitory ME4 module eigengene (Figure 6F), indicating that the majority cell type represented in the ME4 module is the calretinin-positive GABAergic inhibitory neuron. Thus, both neuronal ratios and gene expression changes in the organoid analyses point most directly to GABAergic inhibitory neurons and the associated excitatory/inhibitory balance as a target of disruption that could contribute to neurodevelopmental and cognitive deficits in both 16p11.2 deletion and duplication.
Discussion
The 16p11.2 RGD is associated with a variety of prominent neurodevelopmental and other phenotypes, including features that are shared (e.g., NDD, ASD, seizure), mirrored (e.g., macrocephaly or microcephaly; obesity or low body weight), or distinct (e.g., predisposition to neuroblastoma among deletion subjects, schizophrenia among duplication subjects).81, 82, 83 Each of these features is likely to be driven by haploinsufficiency or triplosensitivity for one or more genes or combinations of genes within the 16p11.2 region. Indeed, it is conceivable that each of the shared or mirrored phenotypes results from the pathway disruptions that ensue from altered expression of the same critical CNV region gene or set of genes in both deletion and duplication individuals. However, although mutational analysis of persons with NDD in the absence of 16p11.2 dosage change have pointed to several different genes in the CNV region as potential contributors, none has been identified as unequivocally causal. Consequently, we sought to gain insight into this RGD through the shared (both reciprocal and non-reciprocal) and distinct transcriptional changes associated with deletion and duplication of the entire region.
Our overall findings in both the mouse and hiPSC model systems indicate that the expression of genes in the CNV segment directly reflects their gene dosage and is typically highest in the brain but that the same dosage change results in significantly altered expression of largely different sets of genes outside the CNV segment in different tissues and cells. There was more sharing of these altered non-CNV genes than expected by chance, particularly across the brain regions, but those genes that were shared represented a small minority of the total DEGs and only a yet smaller subset of these displayed a reciprocal effect (i.e., expression altered in opposite directions by deletion vs. duplication). However, GO enrichment analyses of the significantly altered genes, and particularly co-expression of genes whose expression was correlated with CNV dosage, provided evidence for several commonly disrupted biological processes across the mouse brain regions and human neurons. These results were most prominent for alterations in energy metabolism, mRNA metabolism, translation and protein targeting. These alterations were not observed in the peripheral tissues, which exhibited disruption of distinct biological processes.
Interestingly, although the vast majority of DEGs did not display a significant reciprocal effect of deletion and duplication on their expression, those WGCNA co-expression modules that correlated with CNV dosage were revealing of similar GO terms across the brain and human cell analyses, indicating that this enrichment is driven by more subtle reciprocal alterations of many genes involved in these biological processes. However, some significant co-expression modules, particularly the cerebellar module CE1, suggest the existence of additional interconnected effects on a large set of genes that shows limited GO enrichment but that may be important in contributing to abnormal phenotypes. Expression of the genes in CE1 is positively correlated with CNV dosage—there is an apparently larger effect of deletion than duplication—and boasts strongly significant enrichment for LoF-constrained genes and FMRP target genes, along with enrichment for NDD-associated genes, genes identified in the DDD, chromatin modifiers, and genes from a co-expression module (M2) defined very early in human neurodevelopment. However, unlike the extensive sets of GO enrichments for most other significant mouse brain modules, CE1 is most significant for the term homophilic cell adhesion via plasma membrane adhesion molecules and shows weaker support for other terms related to neuronal development (e.g., neuron differentiation; neuron development; neurogenesis) and adhesion (e.g., cell-cell adhesion via plasma membrane; biological adhesion) (Table S9). This contrast indicates that the large CE1 module and modules CO1, CO2, S3, and S5, which did not show any strong GO enrichments, might each reflect convergence of 16p11.2 dosage-elicited expression changes for subsets of non-CNV genes drawn from multiple different biological processes. Alternatively, they could result from a diversity of 16p11.2 dosage-elicited responses in the different cell types within each region.
Together, the notable similarities and many differences in transcriptomic alterations between brain regions, peripheral tissues, and human neuronal cells indicate that the CNV dosage changes of the same set of genes have impacts, both in terms of biological processes disrupted and in terms of the genes most significantly altered within those processes, that are context-specific. The importance of cellular context is reinforced by differences in the functional impact of the 16p11.2 CNV in our iNs (both deletion and duplication result in reduced numbers of neurites and branchpoints and in reduced electrical activity) compared to dopaminergic neurons (larger soma and hyperexcitability due to deletion and increased neurite numbers due to duplication) derived from the same hiPSCs.84 In the former, neuronal homeostasis appears to reflect the “Goldilocks principle,” in that too little or too much expression of the 16p11.2 genes results in the same cellular phenotype, similar to the observation that some individual genes cause ASD both by haploinsufficiency and by triplosensitivity.85, 86, 87, 88 By contrast, in the latter context and in our organoid analysis, decreased and increased 16p11.2 gene expression have different consequences. The context specificity of the effects of 16p11.2 deletion, along with the potential effects of genetic background, are also reflected in a comparison of our data with those of Roth et al.,89 who sequenced RNA from cortical neural rosettes of 13 16p11.2 deletion individuals and seven controls. In this mixed genetic background, they identified only 93 significant DEGs outside the deletion region, and none of these overlapped with DEGs from our isogenic comparison of 16pDel NSCs or iNs. Neither did the orthologues of their DEGs appear among our mouse-brain-region DEGs, with the exception of two genes in striatum (Ext1 and Lhfpl3). Similarly, they detected no enriched GO terms at FDR < 0.05, and the 25 GO term enrichments that they reported at p < 0.05 did not overlap with those detected by DEGs from our 16pdel NSCs and iNs. Consequently, it is likely that multiple CNV genes and, consequently, disrupted biological processes contribute to the phenotypic features of the 16p11.2 RGD, potentially through a combination of different effects in different contributing cell types. For example, the cortex, cerebellum, and striatum are associated with a variety of functions, including movements, motor behaviors, learning, and cognition functions, and they are also implicated in various neurological diseases.90 Recent data have shown that the cerebellum, known for sensory-motor control, also plays a role in social cognition and emotion as a result of its cortical connections. Consequently, the wide range of symptoms represented on the autism spectrum could result from the disruption of circuits involving all three of these brain regions.91 Our findings suggest that the cortex-striatum-cerebellum network could suffer distinct disruption at each of its nodes, even if through different impacts on similar biological processes, and that these disruptions might all contribute to some degree to the ultimate neurodevelopmental phenotypes. Similarly, any phenotypes initiated in the periphery are extremely likely to be due to different biological processes than those critical to neurological phenotypes.
The biological processes that were most commonly disrupted as a result of 16p11.2/7qF3 CNV in the context of brain or cultured neuronal cells were energy metabolism, mRNA metabolism, translation, and protein targeting. Although a number of the genes in the 16p11.2 CNV segment could participate in or impact on these processes, it is difficult to point to one as individually responsible for any of these disruptions. On the other hand, a number of the genes in the region have been implicated, primarily through model systems, as having potential impact on neurodevelopmental processes. These include, for example, SEZ6L2 (synapse numbers, dendritic morphology, and neuritogenesis),92,93 CORO1A (filopodia formation, required for initial neurite formation),94,95 DOC2A (spontaneous neurotransmission associated with its calcium-dependent translocation),25,96 TLCD3B (altered composition and levels of sphingolipids and glycerolipids associated with cellular membranes, leading to synaptic protein mislocalization),97 TAOK2 (brain size, neural connectivity, and excitatory transmission)98; PRRT2 (neuronal excitability)99; and MAPK3 (dendritic alterations of cortical pyramidal neurons).100 Recently, integration of genetic regulation of gene expression with genome-wide association data from human cohorts pointed to INO80E as the potential driver of schizophrenia due to 16p11.2 duplication and to both SPN (MIM: 182160) and INO80E as contributors to the 16p11.2-deletion-associated increase in body mass index.101 Other model studies have also pointed to interaction between 16p11.2 genes as potentially being responsible for abnormal phenotypes.102,103 Consequently, our transcriptomic studies suggest that the various phenotypes observed in the 16p11.2 RGD most likely result from both individual genes and gene interactions operating within the context of different cell types.
In the context of human glutamatergic iNs, the net functional effects of 16p11.2 deletion and duplication were similar in terms of neuronal morphometry and electrophysiology, yet in the context of cerebral organoids, these lesions produced reciprocal effects with respect to the content of GABAergic inhibitory neurons. Perhaps the importance of cellular context and specificity of phenotype is best exemplified by KCTD13. On the basis of modeling in zebrafish,32 this gene has been implicated previously as the source of the macrocephaly and microcephaly phenotypes associated with 16p11.2 deletion and duplication, respectively. Recently, RhoA and the associated Rho GTPase signaling pathway has been implicated in the link between KCTD13 and 16p11.2 RGD phenotypes.22,33,104 However, in our human iN model system, KCTD13 did not show haploinsufficiency for the neurite outgrowth and branching phenotypes and was minimally different from WT for the electrophysiological measures. Similarly, in cerebral organoids, KCTD13 did not show a disrupted ratio of inhibitory versus excitatory neurons. Consequently, KCTD13 does not appear to be a contributor to these phenotypes in this context, but this does not preclude its contribution to the RGD phenotypes via other cellular or developmental contexts. Notably, in our co-expression analysis, cerebellum module CE1 and striatum module S1 yielded significant enrichment for the Rho GTPase cycle, but module iN1 and the cortex modules CO1–CO4 did not,22,33,104 suggesting that the contribution of KCTD13 dosage could be limited to 16p11.2 RGD phenotypes involving cerebellum and striatum.
In the future, the organoid model approach might offer the best available experimental route to assessing the effects of the 16p11.2 CNV and its constituent genes on human-relevant phenotypes that are generated through interaction of cell types in a developmental context. For example, in a 16p11.2 cohort study, the differences in intracranial volume, gray and white matter volume, cortical thickness, and surface area in CNV carriers affected regions known to exhibit structural abnormalities in ASD and SCZ.82 Moreover, immunohistochemical studies of both ASD and SCZ brains have shown a decrease in the caudate nucleus of the density of calretinin-positive interneurons.105,106 Genome-wide genetic analyses in humans have also supported the long-standing hypothesis that ASD involves disruption of the excitatory-inhibitory balance.36,107,108 Consistent with ths hypothesis, our data from cerebral organoids indicate that dose-dependent differences in the ratio of excitatory to inhibitory neurons are caused by the 16p11.2 CNV and that this system might offer a route to further dissect the pathways underlying this difference. By contrast, it has been reported that CALB2-positive GABAergic inhibitory neurons show greater abundance in adult primates than in rodents.109,110 In addition, there are other subtypes that are numerous in primates but are missing or greatly reduced in mouse cortex.109,111 Therefore, fundamental differences in species context could potentially preclude the discovery of phenotype-associated signatures that are absent in the brains of model organisms.
In summary, we have demonstrated by using isogenic hiPSC-derived neurons and mouse models that transcriptomic, morphological, electrophysiological, and cell-fate signatures of the 16p11.2 CNV are highly context dependent. Although they provide evidence for disruption of a number of critical processes, most notably energy metabolism, mRNA metabolism, translation, and protein targeting, and for the disruption of neuronal development and function, the details of these shared pathway disruptions vary by brain region, tissue, and cell type. The shared pathway disruptions are also accompanied by alterations specific to the brain region, tissue, or cell type. Delineation of the individual contributions of causal genes, gene-gene interactions and dosage change (deletion, duplication or both, either reciprocal or shared) presents a complex problem that will most likely require a multi-faceted approach. However, our work suggests that the human cerebral organoid could be a particularly valuable tool for exploring human neurodevelopmental phenotypes of 16p11.2 RGD, as well as ASD and SCZ more generally.8,17
Data and code availability
The accession number for the RNA sequence data reported in this paper is NCBI GEO: GSE211103. The code used in this study is available upon request.
Acknowledgments
We thank S. Haggarty and S. Sheridan (Center for Genomic Medicine, Massachusetts General Hospital) for generously providing the control hiPSC line. These studies were supported by funding from the Simons Foundation Autism Research Initiative (SFARI 328656 and 573206 [M.E.T.] and SFARI 308955 [J.F.G.]), the Nancy Lurie Marks Family Foundation (J.F.G. and M.E.T.), the U.S. National Institutes of Health (NS093200, HD096326, MH115957, HD104224, MH123155, and GM061354 to J.F.G. and M.E.T.; MH123804 to R.P.), and Autism Speaks (J.F.G.). C.E.D.E. was the recipient of a Rubicon Fellowship from the Netherlands Organisation for Scientific Research (NWO).
Declaration of interests
J.F.G. is a founder and member of the scientific advisory board of Triplet Therapeutics and has been a paid consultant to Biogen, Pfizer, and Wave Biosciences. M.E.T. receives research funding and/or reagents from Illumina, Levo Therapeutics, and Microsoft. E.M. receives research funding and reagents from PTC Therapeutics. The following co-authors are currently employed by for-profit companies or non-profit organizations: P.R. is founder and CEO of Kumuda; K.M. is currently employed by Tornado Bio; T.A. is a founder and CEO of Independent Data Lab and OmicsChart; and A.H. is currently employed by Bionano Genomics.
Published: September 23, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2022.08.012.
Contributor Information
Michael E. Talkowski, Email: mtalkowski@mgh.harvard.edu.
James F. Gusella, Email: gusella@helix.mgh.harvard.edu.
Web resources
BioRender, https://biorender.com/
BLAST, https://blast.ncbi.nlm.nih.gov/Blast.cgi
fastQC, a quality control tool for high throughput sequence data, https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Gene Expression Omnibus (GEO), https://www.ncbi.nlm.nih.gov/geo/
Gene Ontology website, http://geneontology.org/docs/download-ontology/
Genotype-Tissue Expression Project, https://gtexportal.org/home/
Jackson Laboratory, 16p11.2 syntenic deletion mouse strain (B6129S-Del(7Slx1b-Sept1)4Aam/J), https://www.jax.org/strain/013128
Jackson Laboratory, 16p11.2 syntenic duplication mouse strain (B6129S-Del(7Slx1b-Sept1)5Aam/J), https://www.jax.org/strain/013129
Liftover UCSC, https://genome.ucsc.edu/cgi-bin/hgLiftOver
MGI-Mouse Vertebrate Homology, http://www.informatics.jax.org/homology.shtml
MSigDB Molecular Signature Database, http://www.gsea-msigdb.org/gsea/msigdb/index.jsp
NCBI gene data, https://ftp.ncbi.nlm.nih.gov/gene/DATA/
NIMH Data Archive, https://nda.nih.gov/
OMIM, http://www.omim.org/
Picard Tools, https://broadinstitute.github.io/picard/
PsychEncode,https://psychencode.synapse.org/
Synaptic Gene Ontologies, https://www.syngoportal.org/
Supplemental information
References
- 1.Gu W., Zhang F., Lupski J.R. Mechanisms for human genomic rearrangements. Pathogenetics. 2008;1:4. doi: 10.1186/1755-8417-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cooper G.M., Coe B.P., Girirajan S., Rosenfeld J.A., Vu T.H., Baker C., Williams C., Stalker H., Hamid R., Hannig V., et al. A copy number variation morbidity map of developmental delay. Nat. Genet. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geschwind D.H., Flint J. Genetics and genomics of psychiatric disease. Science. 2015;349:1489–1494. doi: 10.1126/science.aaa8954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Girirajan S., Campbell C.D., Eichler E.E. Human copy number variation and complex genetic disease. Annu. Rev. Genet. 2011;45:203–226. doi: 10.1146/annurev-genet-102209-163544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sebat J., Lakshmi B., Malhotra D., Troge J., Lese-Martin C., Walsh T., Yamrom B., Yoon S., Krasnitz A., Kendall J., et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stefansson H., Rujescu D., Cichon S., Pietiläinen O.P.H., Ingason A., Steinberg S., Fossdal R., Sigurdsson E., Sigmundsson T., Buizer-Voskamp J.E., et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tai D.J.C., Ragavendran A., Manavalan P., Stortchevoi A., Seabra C.M., Erdin S., Collins R.L., Blumenthal I., Chen X., Shen Y., et al. Engineering microdeletions and microduplications by targeting segmental duplications with CRISPR. Nat. Neurosci. 2016;19:517–522. doi: 10.1038/nn.4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Battaglia A., Novelli A., Bernardini L., Igliozzi R., Parrini B. Further characterization of the new microdeletion syndrome of 16p11.2-p12.2. Am. J. Med. Genet. 2009;149:1200–1204. doi: 10.1002/ajmg.a.32847. [DOI] [PubMed] [Google Scholar]
- 9.Bedoyan J.K., Kumar R.A., Sudi J., Silverstein F., Ackley T., Iyer R.K., Christian S.L., Martin D.M. Duplication 16p11.2 in a child with infantile seizure disorder. Am. J. Med. Genet. 2010;152A:1567–1574. doi: 10.1002/ajmg.a.33415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shiow L.R., Paris K., Akana M.C., Cyster J.G., Sorensen R.U., Puck J.M. Severe combined immunodeficiency (SCID) and attention deficit hyperactivity disorder (ADHD) associated with a coronin-1A mutation and a chromosome 16p11.2 deletion. Clin. Immunol. 2009;131:24–30. doi: 10.1016/j.clim.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steinberg S., De Jong S., Mattheisen M., Costas J., Demontis D., Jamain S., Pietiläinen O.P.H., Lin K., Papiol S., Huttenlocher J., et al. Common variant at 16p11.2 conferring risk of psychosis. Mol. Psychiatry. 2014;19:108–114. doi: 10.1038/mp.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zufferey F., Sherr E.H., Beckmann N.D., Hanson E., Maillard A.M., Hippolyte L., Macé A., Ferrari C., Kutalik Z., Andrieux J., et al. A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. J. Med. Genet. 2012;49:660–668. doi: 10.1136/jmedgenet-2012-101203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Degenhardt F., Priebe L., Herms S., Mattheisen M., Mühleisen T.W., Meier S., Moebus S., Strohmaier J., Groß M., Breuer R., et al. Association between copy number variants in 16p11.2 and major depressive disorder in a German case-control sample. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012;159B:263–273. doi: 10.1002/ajmg.b.32034. [DOI] [PubMed] [Google Scholar]
- 14.Gimeno-Ferrer F., Albuquerque D., Guzmán Luján C., Marcaida Benito G., Torreira Banzas C., Repáraz-Andrade A., Ballesteros Cogollos V., Aleu Pérez-Gramunt M., Galán Gómez E., Quintela I., Rodríguez-López R. The effect of copy number variations in chromosome 16p on body weight in patients with intellectual disability. J. Hum. Genet. 2019;64:221–231. doi: 10.1038/s10038-018-0545-5. [DOI] [PubMed] [Google Scholar]
- 15.Hempel M., Rivera Brugués N., Wagenstaller J., Lederer G., Weitensteiner A., Seidel H., Meitinger T., Strom T.M. Microdeletion syndrome 16p11.2-p12.2: Clinical and molecular characterization. Am. J. Med. Genet. 2009;149A:2106–2112. doi: 10.1002/ajmg.a.33042. [DOI] [PubMed] [Google Scholar]
- 16.Jacquemont S., Reymond A., Zufferey F., Harewood L., Walters R.G., Kutalik Z., Martinet D., Shen Y., Valsesia A., Beckmann N.D., et al. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature. 2011;478:97–102. doi: 10.1038/nature10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCarthy S.E., Makarov V., Kirov G., Addington A.M., McClellan J., Yoon S., Perkins D.O., Dickel D.E., Kusenda M., Krastoshevsky O., et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat. Genet. 2009;41:1223–1227. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Owen D., Bracher-Smith M., Kendall K.M., Rees E., Einon M., Escott-Price V., Owen M.J., O’Donovan M.C., Kirov G. Effects of pathogenic CNVs on physical traits in participants of the UK Biobank 11 Medical and Health Sciences 1103 Clinical Sciences. BMC Genom. 2018;19:867. doi: 10.1186/s12864-018-5292-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Redaelli S., Maitz S., Crosti F., Sala E., Villa N., Spaccini L., Selicorni A., Rigoldi M., Conconi D., Dalprà L., et al. Refining the phenotype of recurrent rearrangements of chromosome 16. Int. J. Mol. Sci. 2019;20:1095. doi: 10.3390/ijms20051095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding J., Yu C., Sui Y., Wang L., Yang Y., Wang F., Yao H., Xing F., Liu H., Li Y., et al. The chromatin remodeling protein INO80 contributes to the removal of H2A.Z at the p53-binding site of the p21 gene in response to doxorubicin. FEBS J. 2018;285:3270–3285. doi: 10.1111/febs.14615. [DOI] [PubMed] [Google Scholar]
- 21.Lorain S., Quivy J.-P., Monier-Gavelle F., Scamps C., Lécluse Y., Almouzni G., Lipinski M. Core Histones and HIRIP3, a Novel Histone-Binding Protein, Directly Interact with WD Repeat Protein HIRA. Mol. Cell Biol. 1998;18:5546–5556. doi: 10.1128/mcb.18.9.5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Escamilla C.O., Filonova I., Walker A.K., Xuan Z.X., Holehonnur R., Espinosa F., Liu S., Thyme S.B., López-García I.A., Mendoza D.B., et al. Kctd13 deletion reduces synaptic transmission via increased RhoA. Nature. 2017;551:227–231. doi: 10.1038/nature24470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fekairi S., Scaglione S., Chahwan C., Taylor E.R., Tissier A., Coulon S., Dong M.Q., Ruse C., Yates J.R., Russell P., et al. Human SLX4 Is a Holliday Junction Resolvase Subunit that Binds Multiple DNA Repair/Recombination Endonucleases. Cell. 2009;138:78–89. doi: 10.1016/j.cell.2009.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Z., Cobb M.H. Regulation of Stress-responsive Mitogen-activated Protein (MAP) Kinase Pathways by TAO2. J. Biol. Chem. 2001;276:16070–16075. doi: 10.1074/jbc.M100681200. [DOI] [PubMed] [Google Scholar]
- 25.Courtney N.A., Briguglio J.S., Bradberry M.M., Greer C., Chapman E.R. Excitatory and Inhibitory Neurons Utilize Different Ca2+ Sensors and Sources to Regulate Spontaneous Release. Neuron. 2018;98:977–991.e5. doi: 10.1016/j.neuron.2018.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valente P., Castroflorio E., Rossi P., Fadda M., Sterlini B., Cervigni R.I., Prestigio C., Giovedì S., Onofri F., Mura E., et al. PRRT2 Is a Key Component of the Ca2+-Dependent Neurotransmitter Release Machinery. Cell Rep. 2016;15:117–131. doi: 10.1016/j.celrep.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coe B.P., Stessman H.A.F., Sulovari A., Geisheker M.R., Bakken T.E., Lake A.M., Dougherty J.D., Lein E.S., Hormozdiari F., Bernier R.A., Eichler E.E. Neurodevelopmental disease genes implicated by de novo mutation and copy number variation morbidity. Nat. Genet. 2019;51:106–116. doi: 10.1038/s41588-018-0288-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crepel A., Steyaert J., De La Marche W., De Wolf V., Fryns J.P., Noens I., Devriendt K., Peeters H. Narrowing the critical deletion region for autism spectrum disorders on 16p11.2. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011;156:243–245. doi: 10.1002/ajmg.b.31163. [DOI] [PubMed] [Google Scholar]
- 29.Fu J.M., Satterstrom F.K., Peng M., Brand H., Collins R.L., Dong S., Wamsley B., Klei L., Wang L., Hao S.P., et al. Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat. Genet. 2022;18 doi: 10.1038/s41588-022-01104-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arbogast T., Razaz P., Ellegood J., McKinstry S.U., Erdin S., Currall B., Aneichyk T., Lerch J.P., Qiu L.R., Rodriguiz R.M., et al. Kctd13-deficient mice display short-term memory impairment and sex-dependent genetic interactions. Hum. Mol. Genet. 2019;28:1474–1486. doi: 10.1093/hmg/ddy436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blaker-Lee A., Gupta S., McCammon J.M., De Rienzo G., Sive H. Zebrafish homologs of genes within 16p11.2, a genomic region associated with brain disorders, are active during brain development, and include two deletion dosage sensor genes. Dis. Model. Mech. 2012;5:834–851. doi: 10.1242/dmm.009944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Golzio C., Willer J., Talkowski M.E., Oh E.C., Taniguchi Y., Jacquemont S., Reymond A., Sun M., Sawa A., Gusella J.F., et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 2012;485:363–367. doi: 10.1038/nature11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin G.N., Corominas R., Lemmens I., Yang X., Tavernier J., Hill D.E., Vidal M., Sebat J., Iakoucheva L.M. Spatiotemporal 16p11.2 Protein Network Implicates Cortical Late Mid-Fetal Brain Development and KCTD13-Cul3-RhoA Pathway in Psychiatric Diseases. Neuron. 2015;85:742–754. doi: 10.1016/j.neuron.2015.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horev G., Ellegood J., Lerch J.P., Son Y.E.E., Muthuswamy L., Vogel H., Krieger A.M., Buja A., Henkelman R.M., Wigler M., Mills A.A. Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc. Natl. Acad. Sci. USA. 2011;108:17076–17081. doi: 10.1073/pnas.1114042108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Rubeis S., He X., Goldberg A.P., Poultney C.S., Samocha K., Cicek A.E., Kou Y., Liu L., Fromer M., Walker S., et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Satterstrom F.K., Kosmicki J.A., Wang J., Breen M.S., De Rubeis S., An J.Y., Peng M., Collins R., Grove J., Klei L., et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. 2020;180:568–584.e23. doi: 10.1016/j.cell.2019.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grove J., Ripke S., Als T.D., Mattheisen M., Walters R.K., Won H., Pallesen J., Agerbo E., Andreassen O.A., Anney R., et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019;51:431–444. doi: 10.1038/s41588-019-0344-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krishnan A., Zhang R., Yao V., Theesfeld C.L., Wong A.K., Tadych A., Volfovsky N., Packer A., Lash A., Troyanskaya O.G. Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder. Nat. Neurosci. 2016;19:1454–1462. doi: 10.1038/nn.4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parikshak N.N., Luo R., Zhang A., Won H., Lowe J.K., Chandran V., Horvath S., Geschwind D.H. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155:1008–1021. doi: 10.1016/j.cell.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Willsey A.J., Sanders S.J., Li M., Dong S., Tebbenkamp A.T., Muhle R.A., Reilly S.K., Lin L., Fertuzinhos S., Miller J.A., et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155:997–1007. doi: 10.1016/j.cell.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Willsey H.R., Exner C.R.T., Xu Y., Everitt A., Sun N., Wang B., Dea J., Schmunk G., Zaltsman Y., Teerikorpi N., et al. Parallel in vivo analysis of large-effect autism genes implicates cortical neurogenesis and estrogen in risk and resilience. Neuron. 2021;109:1409–1804.e8. doi: 10.1016/j.neuron.2021.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blumenthal I., Ragavendran A., Erdin S., Klei L., Sugathan A., Guide J.R., Manavalan P., Zhou J.Q., Wheeler V.C., Levin J.Z., et al. Transcriptional consequences of 16p11.2 deletion and duplication in mouse cortex and multiplex autism families. Am. J. Hum. Genet. 2014;94:870–883. doi: 10.1016/j.ajhg.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheridan S.D., Theriault K.M., Reis S.A., Zhou F., Madison J.M., Daheron L., Loring J.F., Haggarty S.J. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS One. 2011;6:e26203. doi: 10.1371/journal.pone.0026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kriegova E., Fillerova R., Minarik J., Savara J., Manakova J., Petrackova A., Dihel M., Balcarkova J., Krhovska P., Pika T., et al. Whole-genome optical mapping of bone-marrow myeloma cells reveals association of extramedullary multiple myeloma with chromosome 1 abnormalities. Sci. Rep. 2021;11:14671. doi: 10.1038/s41598-021-93835-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y., Pak C., Han Y., Ahlenius H., Zhang Z., Chanda S., Marro S., Patzke C., Acuna C., Covy J., et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013;78:785–798. doi: 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shi Y., Kirwan P., Livesey F.J. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc. 2012;7:1836–1846. doi: 10.1038/nprot.2012.116. [DOI] [PubMed] [Google Scholar]
- 47.Lancaster M.A., Knoblich J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014;9:2329–2340. doi: 10.1038/nprot.2014.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quadrato G., Nguyen T., Macosko E.Z., Sherwood J.L., Min Yang S., Berger D.R., Maria N., Scholvin J., Goldman M., Kinney J.P., et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature. 2017;545:48–53. doi: 10.1038/nature22047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Velasco S., Kedaigle A.J., Simmons S.K., Nash A., Rocha M., Quadrato G., Paulsen B., Nguyen L., Adiconis X., Regev A., et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature. 2019;570:523–527. doi: 10.1038/s41586-019-1289-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Song J.J., Oh S.M., Kwon O.C., Wulansari N., Lee H.S., Chang M.Y., Lee E., Sun W., Lee S.E., Chang S., et al. Cografting astrocytes improves cell therapeutic outcomes in a Parkinson’s disease model. J. Clin. Invest. 2018;128:463–482. doi: 10.1172/JCI93924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Halliday D.M., Rosenberg J.R., Breeze P., Conway B.A. Neural spike train synchronization indices: definitions, interpretations, and applications. IEEE Trans. Biomed. Eng. 2006;53:1056–1066. doi: 10.1109/TBME.2006.873392. [DOI] [PubMed] [Google Scholar]
- 52.Levin J.Z., Yassour M., Adiconis X., Nusbaum C., Thompson D.A., Friedman N., Gnirke A., Regev A. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat. Methods. 2010;7:709–715. doi: 10.1038/nmeth.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Graubert A., Aguet F., Ravi A., Ardlie K.G., Getz G. RNA-SeQC 2: Efficient RNA-seq quality control and quantification for large cohorts. Bioinformatics. 2021;37:3048–3050. doi: 10.1093/bioinformatics/btab135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leek J.T., Johnson W.E., Parker H.S., Jaffe A.E., Storey J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leek J.T. svaseq: removing batch effects and other unwanted noise from sequencing data. Nucleic Acids Res. 2014;42:e161. doi: 10.1093/nar/gku864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- 61.Langfelder P., Horvath S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinf. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oldham M.C., Konopka G., Iwamoto K., Langfelder P., Kato T., Horvath S., Geschwind D.H. Functional organization of the transcriptome in human brain. Nat. Neurosci. 2008;11:1271–1282. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li M., Santpere G., Imamura Kawasawa Y., Evgrafov O.V., Gulden F.O., Pochareddy S., Sunkin S.M., Li Z., Shin Y., Zhu Y., et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science. 2018;362:eaat7615. doi: 10.1126/science.aat7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koopmans F., van Nierop P., Andres-Alonso M., Byrnes A., Cijsouw T., Coba M.P., Cornelisse L.N., Farrell R.J., Goldschmidt H.L., Howrigan D.P., et al. SynGO: an evidence-based, expert-curated knowledge base for the synapse. Neuron. 2019;103:217–234.e4. doi: 10.1016/j.neuron.2019.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Satija R., Farrell J.A., Gennert D., Schier A.F., Regev A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015;33:495–502. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Becht E., McInnes L., Healy J., Dutertre C.A., Kwok I.W.H., Ng L.G., Ginhoux F., Newell E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2019;37:38–44. doi: 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- 69.Zhang B., Horvath S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005;4:Article17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 70.Lonsdale J., Thomas J., Salvatore M., Phillips R., Lo E., Shad S., Hasz R., Walters G., Garcia F., Young N., et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Collins R.L., Glessner J.T., Porcu E., Lepamets M., Brandon R., Lauricella C., Han L., Morley T., Niestroj L.-M., Ulirsch J., et al. A cross-disorder dosage sensitivity map of the human genome. Cell. 2022;185:3041–3055.e25. doi: 10.1016/j.cell.2022.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141, 456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kishino T., Lalande M., Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 1997;15:70–73. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- 74.Koolen D.A., Kramer J.M., Neveling K., Nillesen W.M., Moore-Barton H.L., Elmslie F.V., Toutain A., Amiel J., Malan V., Tsai A.C.H., et al. Mutations in the chromatin modifier gene KANSL1 cause the 17q21.31 microdeletion syndrome. Nat. Genet. 2012;44:639–641. doi: 10.1038/ng.2262. [DOI] [PubMed] [Google Scholar]
- 75.Kaplanis J., Samocha K.E., Wiel L., Zhang Z., Arvai K.J., Eberhardt R.Y., Gallone G., Lelieveld S.H., Martin H.C., McRae J.F., et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature. 2020;586:757–762. doi: 10.1038/s41586-020-2832-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Singh T., Poterba T., Curtis D., Akil H., Al Eissa M., Barchas J.D., Bass N., Bigdeli T.B., Breen G., Bromet E.J., et al. Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature. 2022;604:509–516. doi: 10.1038/s41586-022-04556-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Iossifov I., O’Roak B.J., Sanders S.J., Ronemus M., Krumm N., Levy D., Stessman H.A., Witherspoon K.T., Vives L., Patterson K.E., et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Darnell J.C., Van Driesche S.J., Zhang C., Hung K.Y.S., Mele A., Fraser C.E., Stone E.F., Chen C., Fak J.J., Chi S.W., et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gillespie M., Jassal B., Stephan R., Milacic M., Rothfels K., Senff-Ribeiro A., Griss J., Sevilla C., Matthews L., Gong C., et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022;50:D687–D692. doi: 10.1093/nar/gkab1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Al-Jaberi N., Lindsay S., Sarma S., Bayatti N., Clowry G.J. The Early Fetal Development of Human Neocortical GABAergic Interneurons. Cereb. Cortex. 2015;25:631–645. doi: 10.1093/cercor/bht254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weiss L.A., Shen Y., Korn J.M., Arking D.E., Miller D.T., Fossdal R., Saemundsen E., Stefansson H., Ferreira M.A.R., Green T., et al. Association between Microdeletion and Microduplication at 16p11.2 and Autism. N. Engl. J. Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 82.Maillard A.M., Ruef A., Pizzagalli F., Migliavacca E., Hippolyte L., Adaszewski S., Dukart J., Ferrari C., Conus P., Männik K., et al. The 16p11.2 locus modulates brain structures common to autism, schizophrenia and obesity. Mol. Psychiatry. 2015;20:140–147. doi: 10.1038/mp.2014.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.D’Angelo D., Lebon S., Chen Q., Martin-Brevet S., Snyder L.G., Hippolyte L., Hanson E., Maillard A.M., Faucett W.A., Macé A., et al. Defining the Effect of the 16p11.2 Duplication on Cognition, Behavior, and Medical Comorbidities. JAMA Psychiatr. 2016;73:20–30. doi: 10.1001/jamapsychiatry.2015.2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sundberg M., Pinson H., Smith R.S., Winden K.D., Venugopal P., Tai D.J.C., Gusella J.F., Talkowski M.E., Walsh C.A., Tegmark M., Sahin M. 16p11.2 deletion is associated with hyperactivation of human iPSC-derived dopaminergic neuron networks and is rescued by RHOA inhibition in vitro. Nat. Commun. 2021;12:2897. doi: 10.1038/s41467-021-23113-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yauy K., Schneider A., Ng B.L., Gaillard J.B., Sati S., Coubes C., Wells C., Tournaire M., Guignard T., Bouret P., et al. Disruption of chromatin organisation causes MEF2C gene overexpression in intellectual disability: a case report. BMC Med. Genomics. 2019;12:116. doi: 10.1186/s12920-019-0558-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Novara F., Rizzo A., Bedini G., Girgenti V., Esposito S., Pantaleoni C., Ciccone R., Sciacca F.L., Achille V., Della Mina E., et al. MEF2C deletions and mutations versus duplications: a clinical comparison. Eur. J. Med. Genet. 2013;56:260–265. doi: 10.1016/j.ejmg.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 87.Doyle D.Z., Lam M.M., Qalieh A., Qalieh Y., Sorel A., Funk O.H., Kwan K.Y. Chromatin remodeler Arid1a regulates subplate neuron identity and wiring of cortical connectivity. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2100686118. e2100686118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bidart M., El Atifi M., Miladi S., Rendu J., Satre V., Ray P.F., Bosson C., Devillard F., Lehalle D., Malan V., et al. Microduplication of the ARID1A gene causes intellectual disability with recognizable syndromic features. Genet. Med. 2017;19:701–710. doi: 10.1038/gim.2016.180. [DOI] [PubMed] [Google Scholar]
- 89.Roth J.G., Muench K.L., Asokan A., Mallett V.M., Gai H., Verma Y., Weber S., Charlton C., Fowler J.L., Loh K.M., et al. 16p11.2 microdeletion imparts transcriptional alterations in human ipsc-derived models of early neural development. Elife. 2020;9:1–34. doi: 10.7554/eLife.58178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Milardi D., Quartarone A., Bramanti A., Anastasi G., Bertino S., Basile G.A., Buonasera P., Pilone G., Celeste G., Rizzo G., et al. The Cortico-Basal Ganglia-Cerebellar Network: Past, Present and Future Perspectives. Front. Syst. Neurosci. 2019;13:61. doi: 10.3389/fnsys.2019.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Crippa A., Del Vecchio G., Busti Ceccarelli S., Nobile M., Arrigoni F., Brambilla P. Cortico-cerebellar connectivity in Autism Spectrum Disorder: What do we know so far? Front. Psychiatry. 2016;7:20. doi: 10.3389/fpsyt.2016.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Qiu W.Q., Luo S., Ma S.A., Saminathan P., Li H., Gunnersen J.M., Gelbard H.A., Hammond J.W. The Sez6 family inhibits complement by facilitating factor I cleavage of C3b and accelerating the decay of C3 convertases. Front. Immunol. 2021;12:607641. doi: 10.3389/fimmu.2021.607641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yaguchi H., Yabe I., Takahashi H., Watanabe M., Nomura T., Kano T., Matsumoto M., Nakayama K.I., Watanabe M., Hatakeyama S. Sez6l2 regulates phosphorylation of ADD and neuritogenesis. Biochem. Biophys. Res. Commun. 2017;494:234–241. doi: 10.1016/j.bbrc.2017.10.047. [DOI] [PubMed] [Google Scholar]
- 94.Alvarez Juliá A., Frasch A.C., Fuchsova B. Neuronal filopodium formation induced by the membrane glycoprotein M6a (Gpm6a) is facilitated by coronin-1a, Rac1, and p21-activated kinase 1 (Pak1) J. Neurochem. 2016;137:46–61. doi: 10.1111/jnc.13552. [DOI] [PubMed] [Google Scholar]
- 95.Dent E.W., Kwiatkowski A.V., Mebane L.M., Philippar U., Barzik M., Rubinson D.A., Gupton S., Van Veen J.E., Furman C., Zhang J., et al. Filopodia are required for cortical neurite initiation. Nat. Cell Biol. 2007;9:1347–1359. doi: 10.1038/ncb1654. [DOI] [PubMed] [Google Scholar]
- 96.Groffen A.J.A., Friedrich R., Brian E.C., Ashery U., Verhage M. DOC2A and DOC2B are sensors for neuronal activity with unique calcium-dependent and kinetic properties. J. Neurochem. 2006;97:818–833. doi: 10.1111/j.1471-4159.2006.03755.x. [DOI] [PubMed] [Google Scholar]
- 97.Tomasello D.L., Kim J.L., Khodour Y., McCammon J.M., Mitalipova M., Jaenisch R., Futerman A.H., Sive H. 16pdel lipid changes in iPSC-derived neurons and function of FAM57B in lipid metabolism and synaptogenesis. iScience. 2022;25:103551. doi: 10.1016/j.isci.2021.103551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Richter M., Murtaza N., Scharrenberg R., White S.H., Johanns O., Walker S., Yuen R.K.C., Schwanke B., Bedürftig B., Henis M., et al. Altered TAOK2 activity causes autism-related neurodevelopmental and cognitive abnormalities through RhoA signaling. Mol. Psychiatry. 2019;24:1329–1350. doi: 10.1038/s41380-018-0025-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fruscione F., Valente P., Sterlini B., Romei A., Baldassari S., Fadda M., Prestigio C., Giansante G., Sartorelli J., Rossi P., et al. PRRT2 controls neuronal excitability by negatively modulating Na+ channel 1.2/1.6 activity. Brain. 2018;141:1000–1016. doi: 10.1093/brain/awy051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Blizinsky K.D., Diaz-Castro B., Forrest M.P., Schürmann B., Bach A.P., Martin-De-saavedra M.D., Wang L., Csernansky J.G., Duan J., Penzes P. Reversal of dendritic phenotypes in 16p11.2 microduplication mouse model neurons by pharmacological targeting of a network hub. Proc. Natl. Acad. Sci. USA. 2016;113:8520–8525. doi: 10.1073/pnas.1607014113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vysotskiy M., Zhong X., Miller-Fleming T.W., Zhou D., Autism Working Group of the Psychiatric Genomics Consortiumˆ. Bipolar Disorder Working Group of the Psychiatric Genomics Consortiumˆ Integration of genetic, transcriptomic, and clinical data provides insight into 16p11.2 and 22q11.2 CNV genes. Genome Med. 2021;13:172. doi: 10.1186/s13073-021-00972-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.McCammon J.M., Blaker-Lee A., Chen X., Sive H. The 16p11.2 homologs fam57ba and doc2a generate certain brain and body phenotypes. Hum. Mol. Genet. 2017;26:3699–3712. doi: 10.1093/hmg/ddx255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Iyer J., Singh M.D., Jensen M., Patel P., Pizzo L., Huber E., Koerselman H., Weiner A.T., Lepanto P., Vadodaria K., et al. Pervasive genetic interactions modulate neurodevelopmental defects of the autism-associated 16p11.2 deletion in Drosophila melanogaster. Nat. Commun. 2018;9:2548. doi: 10.1038/s41467-018-04882-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Martin Lorenzo S., Nalesso V., Chevalier C., Birling M.C., Herault Y. Targeting the RHOA pathway improves learning and memory in adult Kctd13 and 16p11.2 deletion mouse models. Mol. Autism. 2021;12:1. doi: 10.1186/s13229-020-00405-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Adorjan I., Ahmed B., Feher V., Torso M., Krug K., Esiri M., Chance S.A., Szele F.G. Calretinin interneuron density in the caudate nucleus is lower in autism spectrum disorder. Brain. 2017;140:2028–2040. doi: 10.1093/brain/awx131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Adorjan I., Sun B., Feher V., Tyler T., Veres D., Chance S.A., Szele F.G. Evidence for decreased density of calretinin-immunopositive neurons in the caudate nucleus in patients with Schizophrenia. Front. Neuroanat. 2020;14:581685. doi: 10.3389/fnana.2020.581685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sohal V.S., Rubenstein J.L.R. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry. 2019;24:1248–1257. doi: 10.1038/s41380-019-0426-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rubenstein J.L.R., Merzenich M.M. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Boldog E., Bakken T.E., Hodge R.D., Novotny M., Aevermann B.D., Baka J., Bordé S., Close J.L., Diez-Fuertes F., Ding S.L., et al. Transcriptomic and morphophysiological evidence for a specialized human cortical GABAergic cell type. Nat. Neurosci. 2018;21:1185–1195. doi: 10.1038/s41593-018-0205-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wonders C., Anderson S.A. Cortical interneurons and their origins. Neuroscientist. 2005;11:199–205. doi: 10.1177/1073858404270968. [DOI] [PubMed] [Google Scholar]
- 111.Yáñez I.B., Muñoz A., Contreras J., Gonzalez J., Rodriguez-Veiga E., DeFelipe J. Double bouquet cell in the human cerebral cortex and a comparison with other mammals. J. Comp. Neurol. 2005;486:344–360. doi: 10.1002/cne.20533. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA sequence data reported in this paper is NCBI GEO: GSE211103. The code used in this study is available upon request.