

Abstract
Many extracellular matrix (ECM) associated proteins that influence ECM properties have Thrombospondin type 1 repeats (TSRs) which are modified with O-linked fucose. The O-fucose is added in the endoplasmic reticulum to folded TSRs by the enzyme Protein O-fucosyltransferase-2 (POFUT2) and is proposed to promote efficient trafficking of substrates. The importance of this modification for function of TSR-proteins is underscored by the early embryonic lethality of mouse embryos lacking Pofut2. To overcome early lethality and investigate the impact of the Pofut2 knockout on the secretion of POFUT2 substrates and on extracellular matrix properties in vivo, we deleted Pofut2 in the developing limb mesenchyme using Prrx1-Cre recombinase. Loss of Pofut2 in the limb mesenchyme caused significant shortening of the limbs, long bones and tendons and stiff joint resembling the musculoskeletal dysplasias in human and in mice with mutations in ADAMTS or ADAMTSL proteins. Limb shortening was evident at embryonic day 14.5 where loss of O-fucosylation led to an accumulation of fibrillin 2 (FBN2), decreased BMP and IHH signaling, and increased TGF-β signaling. Consistent with these changes we saw a decrease in the size of the hypertrophic zone with lower levels of Collagen-X. Unexpectedly, we observed minimal effects of the Pofut2 knockout on secretion of two POFUT2 substrates, CCN2 or ADAMTS17, in the developing bone. In contrast, CCN2 and two other POFUT2 substrates important for bone development, ADAMTS6 and 10, showed a decrease in secretion from POFUT2-null HEK293T cells in vitro. These combined results suggest that the impact of the Pofut2 mutation is cell-type specific. In addition, these observations raise the possibility that the O-fucose modification on TSRs extends beyond promoting efficient trafficking of POFUT2 substrates and has the potential to influence their function in the extracellular environment.
Keywords: Thrombospondin type I repeats, Fucosylation, Fibrillins, TGF-β, Collagen
Introduction
Proteins containing Thrombospondin Type 1 Repeat (TSR) motifs (~60 a.a.) play critical roles in cellular processes such as cell-cell attachment, extracellular matrix (ECM) remodeling, cell proliferation and apoptosis [1]. Most TSR-containing proteins are modified with an O-linked Glucoseβ1–3fucose (glucose-fucose) disaccharide. This modification is predicted to promote their efficient folding and secretion [2]. The disaccharide is added in the endoplasmic reticulum to properly folded TSRs by the sequential action of Protein O-fucosyltransferase 2 (POFUT2) and β3glucosyltransferase (B3GLCT). POFUT2 recognizes the TSR fold and adds O-linked fucose directly to the serine or threonine in the consensus sequence, C1-X-X-(S/T)-C2 (Group 1 TSRs) and C2-X-X-(S/T)-C3 (Group 2 TSRs), located within a loop of the native TSR fold [3–5]. POFUT2 mediated O-fucosylation of TSRs is highly specific, and the related O-fucosyltransferase POFUT1, which recognizes and modifies epidermal growth factor (EGF) repeats, cannot modify TSRs and POFUT2 does not modify EGFs [4–6] . Once POFUT2 O-fucosylates the TSR, B3GLCT then extends the fucose with glucose to form the O-linked glucose-fucose disaccharide [7,8].
Based on the presence of the POFUT2 consensus located within the TSR loop, forty-nine proteins are predicted to be modified by POFUT2 [9]. Among these substrates, nearly half belong to the ADAMTS (ADisintegrin And Metalloproteinase with Thrombo-Spondin motifs) and ADAMTS-like (ADAMTSL) family of proteins [9,10]. ADAMTS proteins are known to modulate turnover of ECM components [11]. More recently, ADAMTS and ADAMTSL proteins were implicated in fibrillin microfibril assembly and stability [12]. Other substrates for POFUT2 include thrombospondins (THBS), the CCN family (named for cysteine-rich protein 61 (Cyr61), connective tissue growth factor (CTGF), nephroblastoma overexpressed gene (NOV)), adhesion G protein-coupled receptor B (ADGRB) or brain-specific angiogenesis inhibitor (BAI) family, and other secreted or membrane bound glycoproteins [13–18]. Overall, these proteins are critical for normal development of organs including the eye, vasculature, lymphatic system, heart, bones, skeletal structures, kidney, lungs, dermis, and urogenital system [9,19,20].
In secretion assays using either POFUT2 siRNA knockdown or CRISPR-Cas9 knockout in HEK293T cell lines, preventing O-fucosylation blocks secretion of POFUT2 substrates including ADAMTS9, ADAMTS13, ADAMTS17, ADAMTS20, ADAMTSL1, ADAMTSL2, TSP1, and Sco-spondin [19,21–24]. These results suggest that the O-fucose modification is essential for trafficking of POFUT2 substrates to the cell surface or extracellular environment. Consistent with this prediction, mouse Pofut2 mutants are embryonic lethal and display gastrulation defects similar to Adamts9 mutants [25,26]. In contrast, CCN1 secretion was reduced but not blocked in human fibrosarcoma cell line HT1080 following siRNA knockdown of POFUT2 or mutation of the POFUT2 modified threonine in CCN1 [27]. These observations raise the possibility that there are cell-based differences in sensitivity to loss of POFUT2. Moreover, given the recent demonstration that the O-fucose on TSR3 of the POFUT2 substrate ADGRB1 (BAI1) interacts with its ligand, loss of the disaccharide could also impact function of POFUT2 substrates by altering their interactions in the extracellular environment [28].
To begin understanding the physiological role of O-fucosylation for POFUT2 substrate secretion and function, we deleted Pofut2 in the developing limb mesenchyme using Prrx1-Cre recombinase mediated conditional deletion. Chondrocytes derived from the limb mesenchyme provide an excellent tissue for assessing the in vivo consequences of blocking O-fucosylation of TSR on secretion of substrates, ECM remodeling, and cell signaling since they are well adapted for secretion and ECM remodeling and express multiple TSR-containing POFUT2 substrates. In this in vivo model system, we evaluated the effects of the Pofut2 mutation on matrix composition, signaling, and secretion of two POFUT2 substrates, ADAMTS17 and CCN2 (also called CTGF), during skeletal development. The results of these studies provide evidence that developing cartilage has the capacity to secrete unfucosylated POFUT2 substrates and raises the possibility that altered matrix characteristics and cell-signaling observed in Pofut2 mutant limb mesenchyme result from altered function of secreted substrate(s) lacking O-fucose.
Results
Long bones and digits were shortened in Prrx1-Pofut2 animals
Loss of Pofut2 in mice causes early embryonic lethality due to gastrulation defects likely resulting from defects in ADAMTS9 secretion and/or function [25]. For this reason, to better understand how loss of POFUT2 impacts substrate function and extracellular matrix properties, we employed conditional knockout of Pofut2 in the developing limb mesenchyme using Prrx1-cre [29]. Overall, Prrx1-cre mediated deletion of Pofut2 in the limb mesenchyme resulted in mice with significant shortening of the limbs and stiff joints (Fig. 1A-D). In addition, the Achilles tendon in Prrx1-Pofut2 mutants was significantly shorter than in control mice (Fig. S1). Comparison of Alcian blue (cartilage) and Alizarin red (bone) stained skeletons of control (Pofut2-Flox/+; Prrx1-Cre/+) and Prrx1-Pofut2 conditional mutants (Pofut2-Flox/Δ;Prrx1-Cre+) revealed shorter bones and digits without gross malformations in mutants when compared to controls (Fig. 1A-D). Close examination of the disarticulated tibia/fibulas revealed a fully penetrant defect in the postnatal fusion of proximal tibia/fibula in Prrx1-Pofut2 conditional mutant limbs (Fig. 1B, Fig. S2).
Figure 1. Pofut2 is essential for normal limb bone development.

(A) Comparison of skeletal preparations and (B) individual appendicular bones from 43 day old female littermates, control (Pofut2-Flox/+;Prrx1-Cre/+) (left) and Prrx1-Pofut2 mutant (Pofut2-Flox/Δ;Prrx1-Cre/+) (right). Arrows in panel B indicate the region of fusion between distal tibia and fibula in control (left) and failure to fuse in Prrx1-Pofut2 conditional mutants. Panel B abbreviations: fr, front; F/+ for Pofut2-Flox/+;Prrx1-Cre/+, and F/Δ for Pofut2-Flox/Δ;Prrx1-Cre/+. (C-D) Comparison of bone measurements from disarticulated skeletal prep bones from left forelimb (C) and hind limb (D) isolated from 4 animals per genotype. Panels C-D abbreviations: Sc, scapula; Hu, humerus; Ra, Radius; Mc, Metacarpal; Ph, phalanges; Fe, femur; Ti, tibia; Mt, metatarsal. (E-L’) Comparison of representative histological sections from control and Prrx1-Pofut2 mutant limbs through the middle of P22 knee joint (E-E’, I-I’) or P24 proximal tibial growth plate (F-H’, J-L’). Sections were stained with hematoxylin and eosin (H&E) (E-F’, I-J’), Masson’s trichrome (MTC) (G, K), picrosirius red (PSR) (H, L) and picrosirius red under polarized light (H’, L’). Black brackets indicate regions of chondrocyte development in the growth plate, including resting zone (RZ), proliferating zone (PZ), prehypertrophic zone (PHZ) and hypertrophic zone (HZ). Rectangles in F and J represent regions magnified in F’ and J’. Growth plate regions in F’ and J’ are separated by dashed lines. Red arrows indicate region of contact between femur and tibia (I-I’). Abbreviations (E-L): Fe, Femur; Ti, Tibia; M, meniscal cartilage; AC, articular cartilage. Data from control (red dots) and Prrx1-Pofut2 mutants (black dots) were evaluated for statistical significance using Wilcoxon rank test (C and D). *p≤0.05. Scale bars in panel A (5 mm), panel B (1 mm), panels E and I (500 μm), panels E’, F, G, H, H’, I’, J, K, L,-L’ (200 μm) and panels F’ and J’ (100 μm).
Histological comparison of joints from postnatal (P) 22–24 day-old females showed close apposition of femur and tibia articular cartilage and reduced meniscal cartilage in Prrx1-Pofut2 mutants (Fig. 1I-I’) compared to control littermates (Fig. 1E-E’). It is unclear whether the positioning of the bones or alternatively shortening of tendon/ligaments (such as noted for the Achilles tendon (Fig. S1)) contribute to the stiff joints. Comparison of proximal tibial growth plates identified well organized growth plates in Prrx1-Pofut2 conditional mutants and controls (Fig. 1F-H’ and J-L’). However, compared to controls (Fig. 1F-H’), Pofut2-Prrx1 mutant growth plates (Fig. 1J-L’) showed a slight increase in overall height of the growth plate (controls 521±43 μm versus mutant 543±21 μm, p=0.0356) with a decrease in the hypertrophic zone (control 213±17 μm vs mutant 191±21 μm, p=0.0005). MicroCT comparison of tibia identified reduced trabecular spacing and increased trabecular thickness in Prrx1-Pofut2 mutants compared to controls and confirmed the close apposition of femur and proximal tibia (Fig. S2 and S3). In addition, Prrx1-Pofut2 mutant tibia cortical bone had reduced periosteal (the total volume of the bone) and endosteal volume (the region inside the cortical bone) with smaller minimum diameter compared to controls (Fig. S3).
Embryonic day 14.5 tibia in Prrx1-Pofut2 mutants have greatly reduced hypertrophic zone
Prrx1-Pofut2 mutants were distinguishable at birth by their shorter limbs, suggesting that the mutation had a significant impact on POFUT2 substrate secretion or function during primary endochondral ossification. The E14.5 Prrx1-Pofut2 mutant limbs were considerably shorter and also appear to have altered blood vessel formation compared to controls (Fig. 2A-B, S4). Hematoxylin and eosin staining of sectioned limb buds revealed that the hypertrophic zone of Prrx1-Pofut2 mutant tibia was shorter than control littermates (Fig. 2C-D). To identify which specific tissue could be affected by loss of Pofut2, we evaluated Pofut2 expression at embryonic day (E) 14.5 in the developing limb bud (Fig. 2E). Pofut2 was expressed throughout the developing limb bud, with transcripts visible in the proliferative (Fig. 2F), hypertrophic and perichondrial (Fig. 2G) regions in tibia. At this time, Prrx1-cre efficiently converted the Rosa26mTmG reporter from red to green in tissues derived from the limb mesenchyme, including the developing bone (Fig. 2H-J). Since Pofut2 was not deleted in the soft tissues of the developing limb bud and the limb vascularization originates from lumber intersegmental arteries and is remodeled in consort with the specific stage of limb development [30], abnormal blood vessel formation in Prrx1-Pofut2 mutants could reflect a delay in limb development or a secondary effects due to loss of POFUT2 in the limb mesenchyme. The critical role of POFUT2 substrates like ADAMTS/L in skeletal development [31,32] along with our observations of greatly reduced hypertrophic zone in the Prrx1-Pofut2 mutants suggested that loss of O-fucose from POFUT2 substrates impacted chondrocyte development in the mutants.
Figure 2. Loss of POFUT2 in developing limb mesenchyme caused significant shortening of limbs at E14.5.
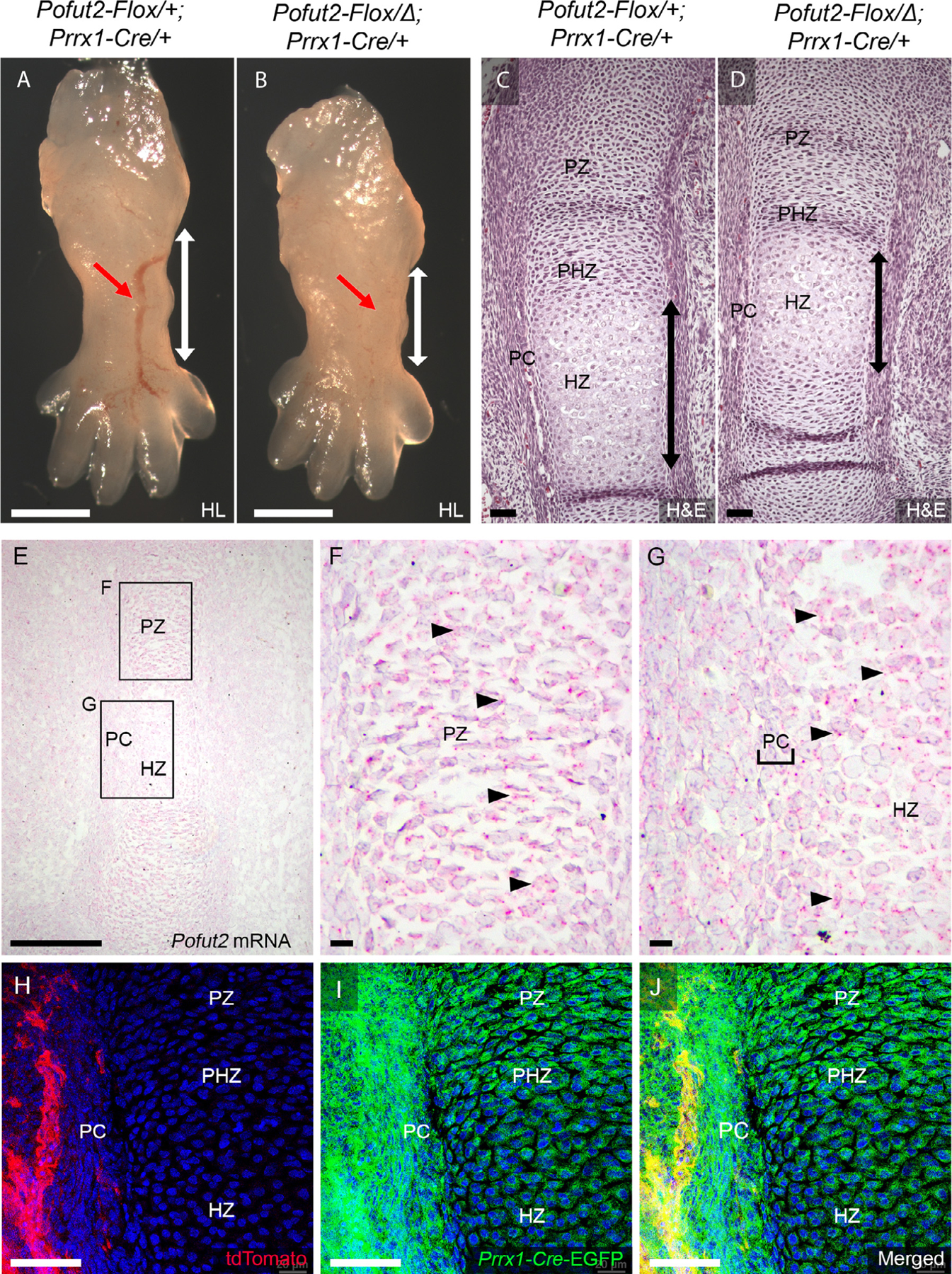
(A-D) Representative images comparing wholemount (A,B) and Hematoxylin and Eosin stained sections (C,D) from E14.5 hind limb (HL) of control (Pofut2-Flox/+;Prrx1-Cre/+) (A,C) and Prrx1-Pofut2 mutant (Pofut2-Flox/Δ;Prrx1-Cre) (B, D). Red arrows in A and B indicate developing blood vessels. White vertical arrows in A and B indicate tibia region. Black vertical arrows in C and D indicate the extent of the hypertrophic zone. (E-G) Analysis of Pofut2 mRNA expression at E14.5 using RNAscope ®. Pofut2 mRNA appears as red dots (arrowheads) overlying cells counterstained with hematoxylin (purple). Rectangles in panel E indicate tibia regions magnified in panels F and G. Pofut2 transcripts were present throughout the proliferating zone (PZ) (F) and in the perichondrium (PC) (indicated by brackets) and hypertrophic zone (HZ) cells (G). (H-J) Assessment of Cre-mediated recombination in E14.5 embryo limb buds isolated from ROSA26mTmG reporter female mated to Prrx1-Cre/+ male mice. Limb section showing unrecombined tissues (tdTomato-red) outside the developing tibia (H), recombined reporter in developing tibia (EGFP-green) (I), and merged channels (J). Scale Bars: in panels A-B (1 mm), panels C-D (200 μm), panel E (500 μm), panels F-G (100 μm), H-J (50 μm).
Fibrillin-2 accumulated in Pofut2 null limb
During chondrocyte differentiation into hypertrophic chondrocytes, several POFUT2 substrates, including ADAMTS6, 10, 17, and ADAMTSL2, modulate proteolysis and assembly of fibrillins into microfibril networks [23,32–34]. Earlier studies suggest that as development proceeds, Fbn1 expression becomes dominant and expression of Fbn2 substantially declines in mice [35]. Coinciding with this, as chondrocytes begin to differentiate into hypertrophic chondrocytes, fibrillin-1 (FBN1) expression increases and fibrillin-2 (FBN2) decreases [35–37]. To determine whether microfibril assembly was impacted by loss of O-fucosylation, we compared the localization of FBN2 and FBN1 in Prrx1-Pofut2 mutants and controls (Fig. 3, S5 and S6). In control specimens, FBN2 was detected in the developing tibia (Fig. 3A-B’) and regions of soft tissue (Fig. 3A). The FBN2 microfibrils were discontinuous in the developing chondrocytes throughout the tibia (Fig. 3A-B’). By comparison, increased FBN1 staining was detected throughout the limb bud of controls (Fig. 3E-F’, J). In contrast, we detected strong continuous FBN2 staining in Prrx1-Pofut2 mutants around the chondrocytes and perichondrium with levels increased relative to controls (Fig. 3C-D’, I). Moreover, FBN1 levels were decreased in hypertrophic and perichondrium regions of Prrx1-Pofut2 mutants compared to controls (Fig. 3G-H’, J). Similar abnormalities in microfibril assembly and limb shortening were observed in Adamts6, Adamts10, Adamts17 and Adamtsl2 mouse mutants [33,38,39].
Figure 3. Elevated FBN2 and reduced FBN1 in E14.5 Pofut2 mutant developing tibia.
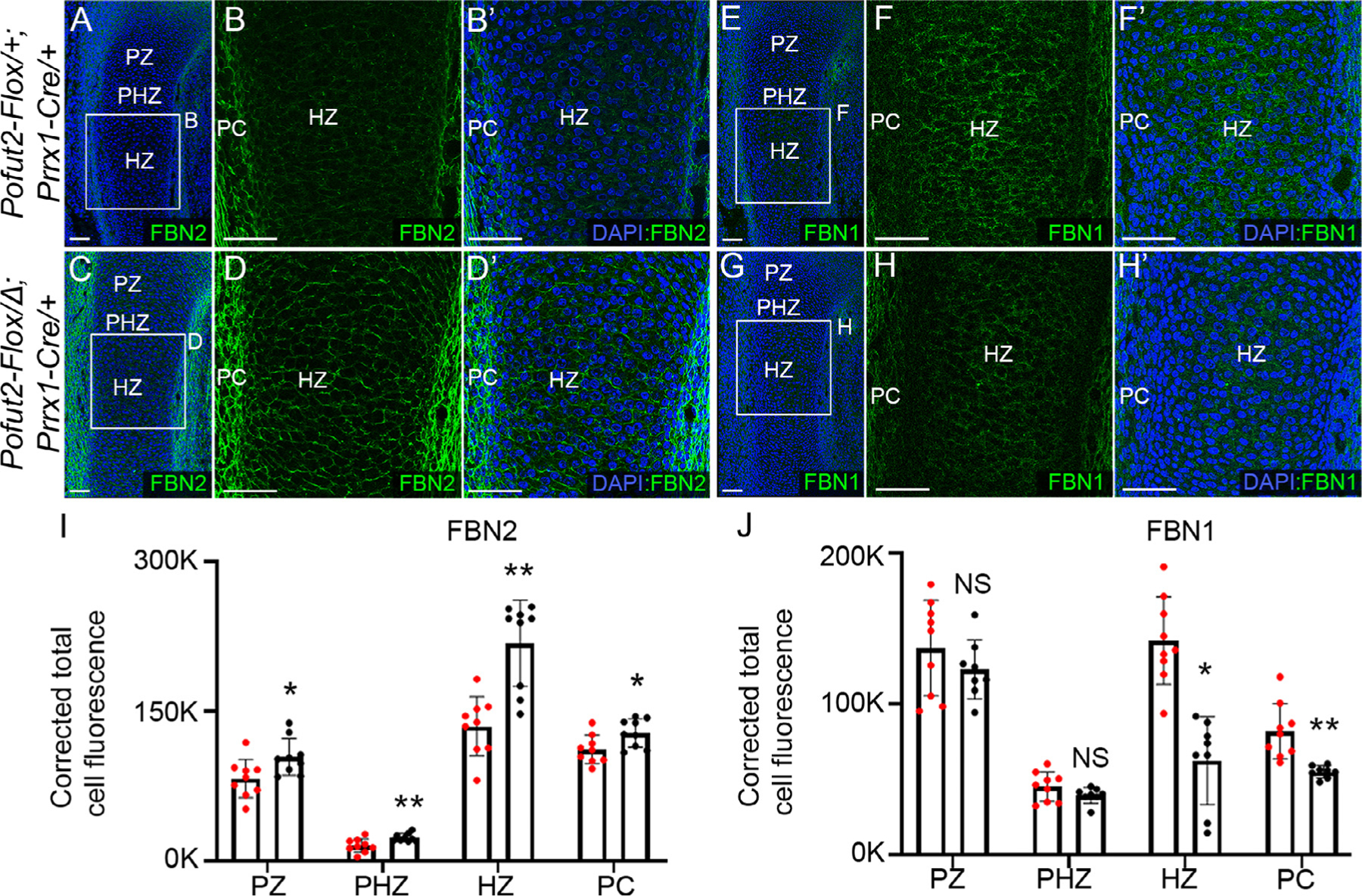
(A-H’) Representative maximum projection images of fibrillin2 (FBN2) (A-D’) and fibrillin1 (FBN1) (E-H’) localization in E14.5 hind limbs. (A and C) Lower magnification images of control (Pofut2-Flox/+;Prrx1-Cre/+) (A) and Prrx1-Pofut2 mutant (Pofut2-Flox/Δ;Prrx1-Cre) (C) hind limbs immunostained for FBN2 (green) and counterstained with DAPI (blue). Rectangles indicate locations of higher magnification images shown in panels B-B’ and D-D’. (E and G) Lower magnification images of control (E) and Prrx1-Pofut2 mutant (G) hind limbs immunostained for FBN1 (green) and counter stained with DAPI (blue). Rectangles indicate location of higher magnification images shown in panels F-F’ and H-H’. (I-J) Quantification of FBN2 (I) and FBN1 (J) immunofluorescence signals in the specified regions of tibia sections (see Figs. S5 and S6). Abbreviations: PZ, proliferative zone, PHZ, prehypertrophic zone; PC, perichondrium; and HZ, hypertrophic zone. Analyses were performed using a single hind limb isolated from three embryos per genotype (n=3) with 2–3 sections per limb (see Figs. S5 and S6). Data from control (red dots) and Prrx1-Pofut2 mutants (black dots) were evaluated for statistical significance using unpaired, two-tailed t-test: *p≤0.05, **p≤0.01 and NS; not significant (I and J). Scale bars: panels A–H’ 50 μm.
Loss of Pofut2 upregulated TGF-β and downregulated BMP signaling
In mouse ADAMTS/L mutants, changes in FBN composition altered TGF-β and BMP signaling and contributed to shortened limbs [33,36,38–40]. To determine whether loss of O-fucosylation on POFUT2 substrates similarly affected TGF-β and BMP signaling, we compared the level of pSMAD2 and pSMAD1/5/9 in the Prrx1-Pofut2 mutants and their control littermates (Fig. 4, S7 and S8). Elevated levels of pSMAD2 were detected in Prrx1-Pofut2 mutant proliferative and hypertrophic zones compared to controls (Fig. 4A-H). In contrast, Prrx1-Pofut2 mutants showed reduced overall levels of pSMAD1/5/9 with reduced nuclear localization compared to controls (Fig. 4I-P).
Figure 4. Elevated TGF-β and reduced BMP signaling in Pofut2 mutant tibia.
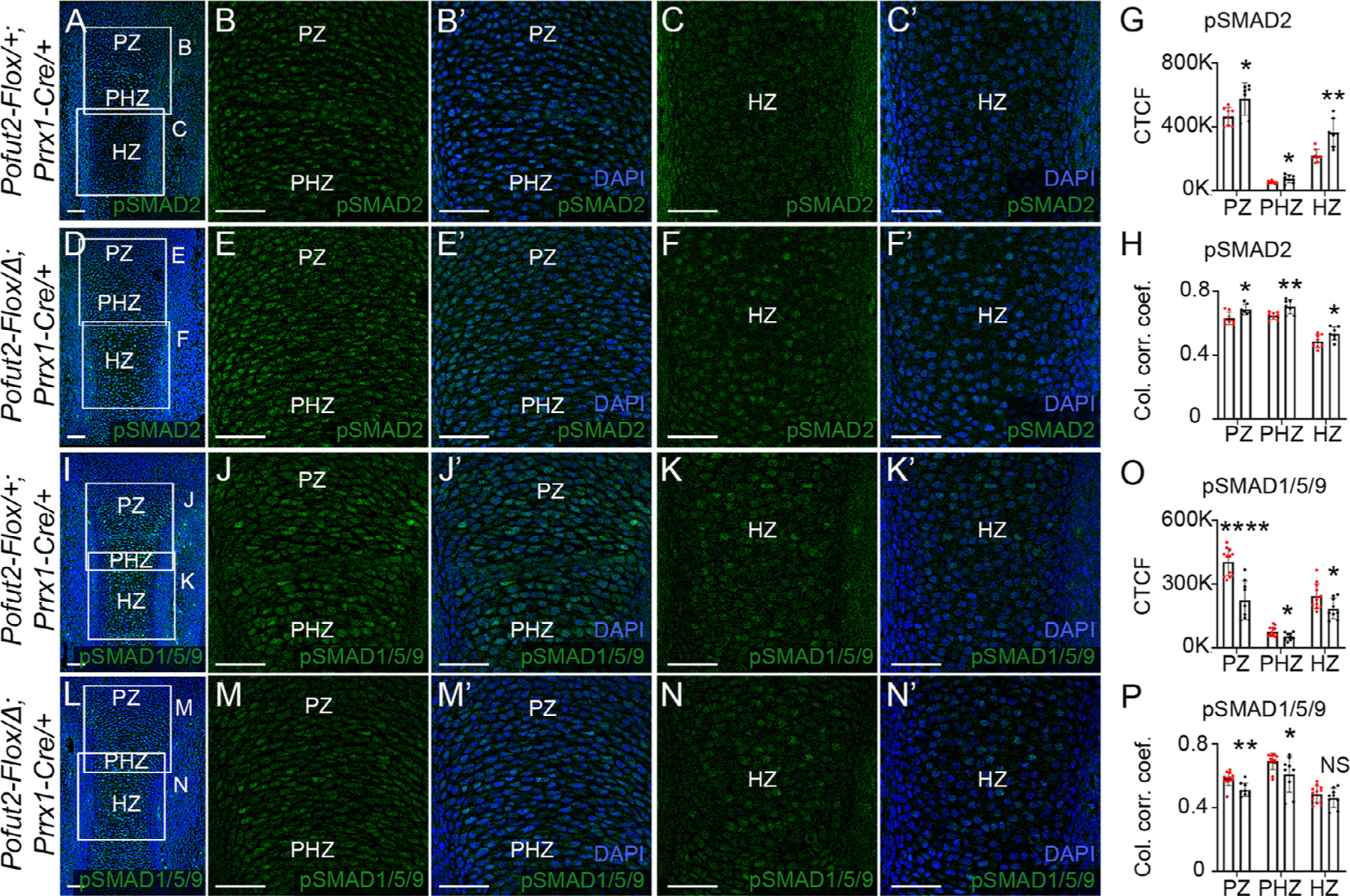
(A-F’) Representative maximum projection images of pSMAD2 (green) immunolocalization and DAPI (blue) in developing tibia from E14.5 control (Pofut2-Flox/+;Prrx1-Cre/+) (A-C’) and Prrx1-Pofut2 mutant (Pofut2-Flox/Δ;Prrx1-Cre) (D-F’) limbs. (A and D) Boxed regions in low magnification images indicate positions of higher magnification images showing proliferating (PZ) and prehypertrophic (PHZ) (B-B’, E-E’) and hypertrophopic (HZ) (C-C’, F-F’) zones. (G-H) Quantification of pSMAD2 immunofluorescence signals (G) and pSMAD2-DAPI nuclear colocalization by estimation of the colocalization correlation (corr.) coefficient (H) in the PZ, PHZ and HZ (see Figs. S7). (I-N’) Representative maximum projection images of pSMAD1/5/9 (green) immunolocalization and DAPI (blue) in the developing tibia from E14.5 control (I-K’) and Prrx1-Pofut2 mutant (L-N’) limbs. (I and L) Lower magnification images with boxed regions indicating position of higher magnification images showing the proliferative (PZ) and prehypertrophic (PHZ) (J-J’, M-M’) and hypertrophopic (HZ) (K-K’, N-N’) zones. (O-P) Quantification of pSMAD1/5/9 immunofluorescence signals (O) and pSMAD1/5/9-DAPI nuclear colocalization by estimation of the colocalization correlation (corr.) coefficient (P) in the PZ, PHZ and HZ regions. Analyses were performed using a single hind limb isolated from minimum of three embryos per genotype (n=3) with 2–4 sections per limb (Fig. S8). White squares indicate magnified regions of tibia sections. Data from control (red dots) and Prrx1-Pofut2 mutants (black dots) were evaluated for statistical significance using unpaired, two-tailed t-test: *p≤0.05, **p≤0.01, ****p≤0.0001, and NS; not significant (G, H, O and P). Scale bars: 50 μm.
Loss of Pofut2 reduced the size of hypertrophic zone during primary endochondral ossification
Mutations in Adamts genes can also impact ECM proteins such as collagens that are important for maintaining the growth plate cytoarchitecture, chondrocyte differentiation and digit development [11,32,33,41,42]. We assessed the impact of the Prrx1-Pofut2 mutation on Collagen-II (COL-II), Collagen-X (COL-X), and EDU incorporation in E14.5 limb buds (Fig. 5, S9, S10). In controls, COL-II was highly expressed throughout the region of proliferating chondrocytes (region of strong EDU labeling), and present at low levels in the differentiating hypertrophic chondrocytes (Fig. 5A and A’). In contrast, COL-X was abundant in the non-proliferating hypertrophic zone (Fig. 5B and B’).
Figure 5. Loss of Pofut2 increased COL-II and reduced COL-X in the developing tibia.

(A-A’ and C-C’) Maximum projection images of COL-II localization (green), DAPI (blue) counterstaining, and EDU labeling (pink) in E14.5 developing tibia from control (Pofut2-Flox/+;Prrx1-Cre/+) (A-A’) and Prrx1-Pofut2 mutant (Pofut2-Flox/Δ;Prrx1-Cre) (C-C’) limbs. (A and C) Low magnification images with white rectangles indicating regions corresponding to higher magnification images in A’ and C’. (B-B’ and D-D’) Maximum projection images of COL-X (green) immunolocalization and DAPI (blue) counter-staining from control (B-B’) and Prrx1-Pofut2 mutant (D-D’) limbs. (B and D) Low magnification images with white rectangles indicating regions corresponding to higher magnification images in B’ and D’. (E-G) Quantification of percent EDU positive nuclei (E), corrected total cell fluorescence for COL-II (F) and COL-X (G) immunostaining in proliferating zone (PZ), prehypertrophic zone (PHZ) and hypertrophic zone (HZ) cells. Analyses were completed for a single hind limb isolated from minimum of three animals per genotype (n=3) and three sections per limb analyzed (see Figs. S9, S10). Data from control (red dots) and Prrx1-Pofut2 mutants (black dots) were evaluated for statistical significance using unpaired, two-tailed t-test: *p≤0.05, **p≤0.01, and NS; not significant (E, F and G). Scale bars: panels A–D’ 50 μm.
In Prrx1-Pofut2 mutants, COL-II, COL-X, and EDU labeling was similarly distributed to controls (Fig. 5C-D’). However, we detected an increase in the number of EDU positive cells in the proliferating and prehypertrophic regions of Prrx1-Pofut2 mutants compared to controls (Fig. 5 A, A’, C, C’, E, F, and S9). In addition, the level of COL-II, trended higher, in mutants with statistically significant elevated levels in the prehypertrophic regions (Fig. 5F). Consistent with the decreased size of the hypertrophic zone, the region of tibia expressing COL-X and levels of COL-X within the region were reduced in Prrx1-Pofut2 mutants (Fig. 5D, D’, G, S10) when compared to controls (Fig. 5B, B’, G and S10). Levels of apoptosis were similar in control and mutant limbs, based on cleaved caspase 3 immunohistochemistry (Fig. S11). Taken together, increased COL-II and reduced COL-X with increased proliferation and unaltered apoptosis suggested that there was a delay in the transition of prehypertrophic chondrocytes to hypertrophic chondrocytes.
Prrx1-Pofut2 limbs have reduced hedgehog signaling and elevated RUNX2
Changes in COL-II and COL-X distribution and delayed differentiation of hypertrophic chondrocytes were also described in Indian hedgehog (Ihh) mutants, raising the possibility that loss of O-fucosylation disrupted IHH signaling [43]. To determine whether the Prrx1-Pofut2 mutation impacts IHH signaling or a transcriptional regulator of Ihh, Runt-related transcription factor 2 (RUNX2) [44,45], we evaluated Ptch1 mRNA (Fig. 6), a downstream target of IHH signaling, by RNAscope® and the localization of RUNX2 protein using immunohistochemistry in E14.5 limb buds. Although the Ptch1 mRNA localization was detected throughout control and the Prrx1-Pofut2 mutant limbs (Fig. 6A, B, S12), the transcript levels were decreased in the proliferative, prehypertrophic and hypertrophic zone of the Prrx1-Pofut2 mutants compared to their littermates (Fig. 6A-C). Nuclear localized RUNX2 was detected throughout the developing bone in both the control and Prrx1-Pofut2 mutants (Fig. 6D,G and S13A and B). However, nuclear RUNX2 levels were increased in the proliferative and hypertrophic zone of Prrx1-Pofut2 mutants (Fig. 6G-K) compared to controls (Fig. 6D-F’, J, K). The elevated levels of RUNX2 and reduced IHH signaling in Prrx1-Pofut2 mutants compared to controls (Fig. 6) reflect a delay in chondrocyte maturation rather than a shift to osteoblastic cell fate (Fig. S14).
Figure 6. Loss of Pofut2 reduced hedgehog signaling and upregulated RUNX2 in the developing E14.5 tibia.
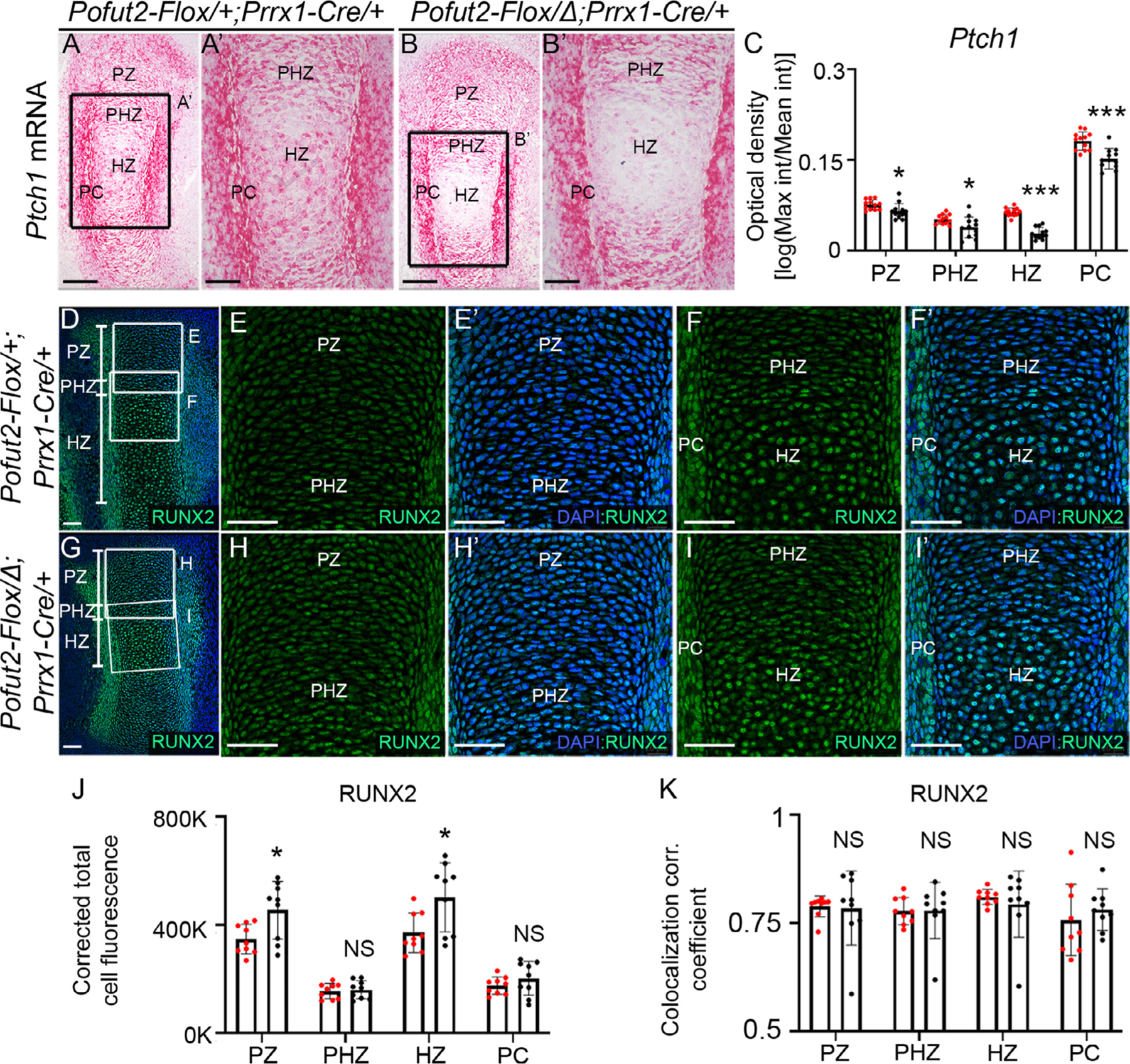
(A-C) Analysis of Ptch1 mRNA expression (a downstream target of hedgehog signaling) using RNAscope ® assay in control (Pofut2-Flox/+;Prrx1-Cre/+) (A-A’) and Prrx1-Pofut2 mutants (Pofut2-Flox/Δ;Prrx1-Cre) (B-B’). Ptch1 mRNA appears as red dots or patches. Rectangles in A and B indicate magnified regions of tibia sections in A’ and B’. (A-B’) Ptch1 is expressed in proliferating zone (PZ), prehypertrophic zone (PHZ), hypertrophic zone (HZ) and perichondrium (PC) cells. Expression patterns were observed in a single hind limb isolated from four embryos per genotype (n=4) and 3 sections per limb. (C) Optical density in the PZ, PHZ, HZ and PC regions was quantified as a log measure ratio of maximum and mean intensity (int) (see Fig. S12). (D-I’) Maximum projection images of RUNX2 immunostaining in control (D-F’) and Prrx1-Pofut2 mutants (G-I’). (D and G) Lower magnification image with rectangles indicating location of higher magnification images of the PZ shown in E-E’ and H-H’ and PHZ and HZ shown in F-F’ and I-I’. (J) Quantification of RUNX2 immunofluorescent signals using corrected total cell fluorescence. (K) Determination of RUNX2-DAPI colocalization by estimation of the colocalization correlation (corr.) coefficient in the PZ, PHZ, HZ and PC (see Fig. S13A and B). Analyses were performed using a single hind limb isolated from three embryos (n=3) per genotype and three sections per limb. Data from control (red dots) and Prrx1-Pofut2 mutants (black dots) were evaluated for statistical significance using unpaired, two-tailed t-test: *p≤0.05, ***p≤0.0001 and NS; not significant (C and D). Scale bars in panels A and B 200 μm, panels A’ and B’ and panels D-I’ 50 μm.
ADAMTS6, ADAMTS10 and CCN2 are modified with O-fucose glycans and secretion was significantly reduced in POFUT2-null HEK293T cells
Mutations in Adamts6, Adamts10, Adamts17, Adamtsl2 and Ccn2 cause significant limb shortening [15,33,38,39,46,47]. Secretion of all substrates tested to date including, ADAMTS17 and ADAMTSL2 is significantly reduced in POFUT2-null HEK293T cells [19,21–25]. This raised the possibility that impaired secretion of one or more POFUT2 substrates caused limb shortening in Prrx1-Pofut2 mutants. To evaluate whether ADAMTS6, ADAMTS10 and CCN2 were modified by POFUT2, we performed mass spectral glycoproteomic analysis of full-length ADAMTS6, ADAMTS10 and CCN2 expressed in HEK293T cells. ADAMTS6 and 10 have 5 TSRs with 3 predicted to be modified, and CCN2 has 1 TSR predicted to be modified (Fig. 7). Mass spectral analysis demonstrated that TSR1, 2 and 3 of ADAMTS6 were fully modified with the O-fucose disaccharide (Fig. 7A, Fig. S15A, C, D). TSR1 of ADAMTS6 was also modified with a C-mannose (Fig. S15B). TSR1 of ADAMTS10 was modified by both O-fucose disaccharide and a C-mannose (Fig. 7B, Fig. S15E), but TSR4 was not modified. TSR4 contains a proline immediately prior to the predicted modification site (C1TPSC2), suggesting a proline in this position may block modification (Fig. 7B, Fig. S15F). We could not detect the peptide with the consensus sequence from TSR3 of ADAMTS10. The single TSR of CCN2 was mainly modified with O-fucose monosaccharide and a small amount of disaccharide (Fig. 7C, Fig. S15G). Thus, ADAMTS6, ADAMTS10, and CCN2 were all modified with O-fucose glycans.
Figure 7. Loss of POFUT2 significantly reduced secretion of ADAMTS6, ADAMTS10 and CCN2 from HEK293T cells.

Cartoons in A-C illustrate the domain structure of ADAMTS6 (A), ADAMTS10 (B) and CCN2 (C). TSRs are depicted by ovals, and red ovals indicate TSRs that have a consensus sequence for POFUT2 mediated O-fucosylation. Mass spectral analysis of peptides containing the consensus sequence from ADAMTS6, ADAMTS10, and CCN2 are shown in Fig. S15, and glycan modifications identified by these analyses are indicated on the TSR domains. Solid red triangle indicates fucose, solid blue circle indicates glucose, and solid green circle (below TSR) indicates mannose. The white “X” mark in the ADAMTS10 TSR4 indicates that there was no O-fucose modification on this TSR even though it contains the POFUT2 consensus sequence. The “?” over TSR3 of ADAMTS10 indicates that we were unable to identify the appropriate peptide in our analysis. The empty circle above CCN2 TSR1 indicates that less 10% of O-linked fucose (solid red triangle) on this TSR was extended with glucose. Normalized results from cell-based assays used to measure the effects of the POFUT2 mutation on the secretion of substrates (ADAMTS6, ADAMTS10, and CCN2) are shown below each domain map. Substrates were tagged with Myc, and GFP was used as a transfection control. HEK293 are wild-type cells, P2 KO are POFUT2-null HEK293T cells, and P2 KO + P2 are POFUT2-null cells co-transfected with wild-type POFUT2. Western blots used for quantitation of protein levels in the medium and cell lysate are shown in Fig. S16. Three biological triplicates were performed, and data were evaluated for statistical significance using unpaired, one-tailed t-test. *P≤0.05, **P≤0.01, n.s. not significant. Abbreviations: SP, signal peptide; Pro, propeptide; Cat, catalytic domain; Dis, disintegrin domain; Cys, cys-rich domain; Spa, spacer domain; PLAC, protease and lacunin domain; IGFBP, insulin-like growth factor binding protein; VWC, von willebrand factor type C domain; GHB, glycoprotein hormone beta chain homologues.
To test if loss of O-fucose modification affected secretion of ADAMTS6, ADAMTS10 or CCN2 in vitro, we evaluated secretion of ADAMTS6, ADAMTS10 and CCN2 expressed in HEK293T wild-type and POFUT2-null cells (Fig. 7). We chose in vitro secretion assays due to lack of validated antibodies against ADAMTS6 and ADAMTS10 for immunohistochemistry. Tagged versions of full length ADAMTS6, ADAMTS10 and CCN2 were secreted efficiently from the wild-type HEK293T cells (Fig. 7, Fig. S16). In contrast, loss of POFUT2 resulted in significant decrease in the secretion of all three (Fig. 7, Fig. S16). Co-transfection of POFUT2-null cells with a plasmid encoding wild-type POFUT2 rescued the secretion defect (Fig.7, Fig. S16). These data suggest that ADAMTS6, ADAMTS10 and CCN2 require O-fucose modification for efficient secretion from HEK293T cells.
In vivo loss of Pofut2 minimally impacted ADAMTS17 and CCN2 secretion
To determine whether POFUT2 was essential for secretion of POFUT2 substrates in vivo, we focused on two substrates with validated antibodies, CCN2 (with a single O-fucosylated TSR) and ADAMTS17 (with 4 of 5 TSRs O-fucosylated [23]) (Fig. 8, S17 and S18). In control E14.5 limbs, strong extracellular localization of CCN2 was detected in the proliferative and prehypertrophic zone with reduced signal detected in the hypertrophic zone (Fig. 8A-D’ and S17) and was similar to previously described CCN2 localization patterns [47]. ADAMTS17 was strongly expressed in the perichondrium with lower levels in the proliferative and hypertrophic zone (Fig. 8K-N’ and S18), a pattern consistent with the previously described mRNA localization [47]. In controls, both CCN2 and ADAMTS17 staining was distinct from the perinuclear localization of the ER chaperone BiP (Fig. 8C, D’, M, N’ and S19), providing evidence that the CCN2 and ADAMTS17 signals represented secreted substrate. In contrast to results from cell-based secretion assays, extracellular CCN2 and ADAMTS17 was detected in Prrx1-Pofut2 mutant limbs (Fig. 8E-H’, O-R’). In Prrx1-Pofut2 mutants, CCN2 levels were slightly reduced in the proliferative region but comparable in other regions (Figs. 8I, S17), and ADAMTS17 levels were comparable to controls throughout the developing bone (Figs. 8S, S18). As in controls, BiP localization in Prrx1-Pofut2 mutants was distinct from CCN2 and ADAMTS17 signals (Fig. 8G-H’ and Q-R’)) with no difference in the levels of BiP (Fig. 8J and T). These combined results suggest that loss of POFUT2 had minimal effect on the secretion of CCN2 and ADAMTS17 in vivo and did not elicit the unfolded protein response (Fig. S20). This outcome suggests that loss of secretion of POFUT2 substrates in the absence of O-fucosylation is cell-type specific.
Figure 8. Loss of Pofut2 minimally impacted secretion of CCN2 and ADAMTS17 in developing tibia.

(A-H’) Maximum projection images of E14.5 hind limb sections from control (Pofut2-Flox/+;Prrx1-Cre/+) (A-D’, K-N’) and Prrx1-Pofut2 mutant (Pofut2-Flox/Δ;Prrx1-Cre/+) (E-H’, O-R’) embryos immunostained with CCN2 (red) and BiP (green) and counterstained with DAPI (blue) (A-H’) or immunostained with ADAMTS17 and BiP and counterstained with DAPI (K-R’). (A and E) Lower magnification images showing all channels with white boxes indicating regions of higher magnification showing CCN2 (red) (B and F), BiP (green) (C and G), and DAPI (blue) (B-C). (D-D’ and H-H’) Merged channels with digitally enlarged panels (D’ and H’). (I-J) Quantification of CCN2 (I) and BiP (J) immunofluorescence signals in defined regions of proliferating zone (PZ), prehypertrophic zone (PHZ) and hypertrophic zone (HZ) cells (see Fig. S17). (K-R’) Lower magnification images showing ADAMTS17 (red), BiP (green), and DAPI (blue) with white boxes indicating regions of higher magnification showing ADAMTS17 (L,P), BiP (M,Q), and DAPI (M-N, Q-R). (N-N’ and R-R’) Merged channels with digitally enlarged panels (N’ and R’). (S-T) Quantification of ADAMTS17 (S) and BiP (T) immunofluorescence signals in defined regions PZ, PHZ, HZ and perichondrium (PC) cells (see Fig. S18). Analyses were performed using a single hind limb isolated from minimum of three embryos per genotype (n=3) and three sections per limb. Data from control (red dots) and Prrx1-Pofut2 mutants (black dots) were evaluated for statistical significance using unpaired, two-tailed t-test: *p≤0.05, **p≤0.01 and NS; not significant (E, F, K and L). Scale bars: 50 μm.
Discussion
Here we used the Prrx1-Pofut2 knockout to investigate the consequences of blocking POFUT2-mediated O-fucosylation in the developing tibia. Loss of Pofut2 in the limb mesenchyme caused significant shortening of the limb bones and tendons and abnormalities in the knee joint. We demonstrated that the amount of FBN2 versus FBN1 was altered and altered cell signaling during primary ossification likely contribute to shortened limbs in Prrx1-Pofut2 mutants (Fig. 9). Results from these studies also uncovered unexpected cell-type and/or substrate specific sensitivities to loss of O-fucosylation.
Figure 9. O-linked fucose modification of POFUT2 substrates are essential for ECM remodeling and signaling regulation during primary endochondral ossification.
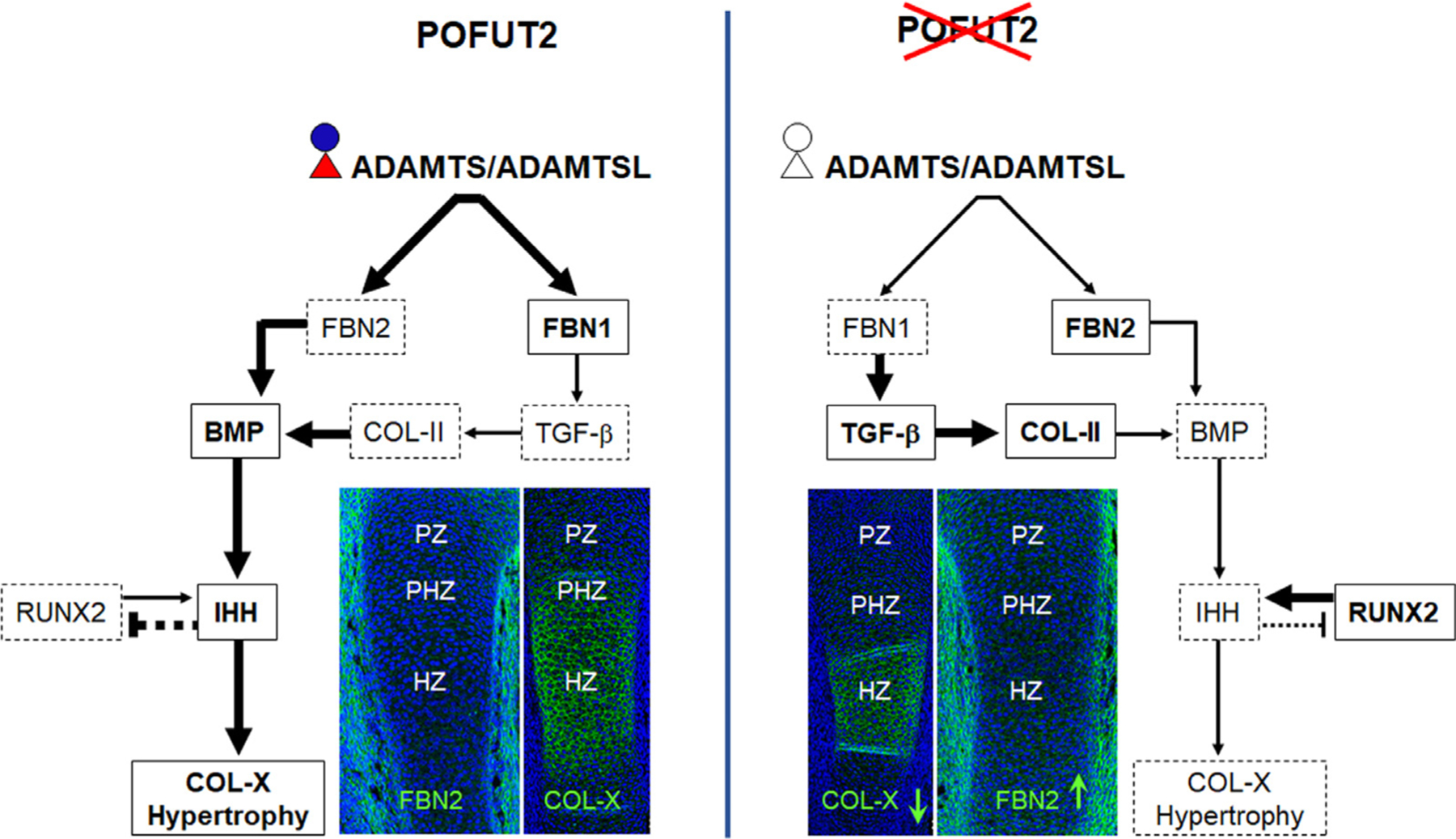
(Left) In control, O-fucosylated ADAMTS/L proteins enhance FBN2 proteolysis and FBN1 biogenesis [23, 33, 34, 46, 50]. Reduced FBN2 and elevated FBN1 decreases the bioavailability TGF-β . In response to reduced TGF-β signaling COL-II synthesis decreases in the proliferating zone. Reduced FBN2 increases the bioavailability of BMPs [36, 52]. Ultimately, BMP signaling promotes IHH signaling essential for chondrocytes differentiation and COL-X synthesis and reduces RUNX2 [43, 44, 57, 60, 84]. (Right) In Prrx1-Pofut2 mutants, impaired O-fucosylation and reduced secretion or altered function of ADAMTS/L proteins elevated FBN2 and reduced FBN1 assembly. Thus, reduced FBN1 could contribute to increased TGF-β, and increased FBN2 to decreased BMP signaling. Increased TGF-β signaling could contribute to elevated COL-II. Whereas, reduced BMP signaling could contribute to elevated RUNX2 and decreased IHH which could lead to decreased chondrocyte hypertrophy and COL-X synthesis. Red triangle and blue circle indicate fucose and glucose, respectively, and black triangle and circle indicate fucose and glucose are absent. Bold fonts in solid rectangles and light fonts in dotted rectangles indicate increased and decreased protein levels, respectively. Thick and thin arrows/or inhibition sign indicate strong and weak signaling, respectively.
The skeletal phenotypes in Prrx1-Pofut2 mutants likely resulted from blocking O-fucosylation on one or more POFUT2 substrates including: ADAMTS6, ADAMTS10, ADAMTS17, ADAMTSL2, and CCN2. Mutations in genes encoding these proteins cause limb shortening [15,33,38,39,48]. Adamts10 mutants showed growth plate organization similar to that observed in Prrx1-Pofut2 mutants, with severely shortened and malformed bones observed when both Adamts6 and Adamts10 are lost [33]. Moreover, short stature, stiff joint and delayed bone age are also observed in Geleophysic dysplasia and Weill-Marchesani syndrome, caused by mutations in ADAMTSL2, ADAMTS17, or ADAMTS10 [32].
During primary endochondral ossification, the action of ADAMTS/TSL proteins promotes turnover of FBN2 and assembly of microfibrils comprised primarily of FBN1 [33,39] (Fig. 9). Since FBN1 and FBN2 lack TSRs and are not modified by POFUT2, altered Fibrillin levels likely stem from the indirect effects of the mutation on POFUT2 substrates that regulate Fibrillin levels. Results from cell-based assays suggest that ADAMTS6 and ADAMTS10 mediate the FBN2 turnover and FBN1 microfibril network assembly through physical interactions with FBN1 and proteolytic cleavage of FBN2 [33,34,46]. Adamts10 mutants showed increased levels FBN2 and poorly organized FBN1 in the ciliary zonules [34,48], suggesting that reduced function of ADAMTS10 contributes to altered FBN patterns in Prrx1-Pofut2 mutants. In human retinal epithelial cells, ADAMTS10 negatively regulates ADAMTS6 expression and loss of ADAMTS10 elevated levels of ADAMTS6 transcripts, which likely could compensate for reduced levels of the related ADAMTS10 [49]. In contrast, because POFUT2 modulates protein function post-translationally, we expect that loss of POFUT2 would simultaneously impact the level of functional ADAMTS6 & ADAMTS10 protein (as well as other TSR proteins). Other POFUT2 substrates that could modulate FBN1 function include ADAMTS17 and ADAMTSL2, which are proposed to regulate assembly of FBN1 microfibrils by physically interacting with FBNs [23,50,51]. Thus, the elevated levels of FBN2 and reduced levels of FBN1 observed in the Prrx1-Pofut2 mutant limbs are consistent with impaired function of one or more of these POFUT2 substrates (Fig. 9).
Changes in the composition of the FBN microfibril network in developing chondrocytes correlates with decreased TGF-β and increased BMP signaling required for chondrocyte hypertrophy (Fig. 9). FBN1 limits the bioavailability of TGF-β through interaction with latent TGF-β binding proteins (LTBP) [52], and reduced FBN1, LTBP1 and LTBP3 enhanced TGF-β signaling in Adamtsl2 mutants [39] (Fig. 9). Conversely, FBN2 limits the bioavailability of BMPs [40,53], and increased levels of FBN2 correlate with decreased BMP signaling in Adamts6 and Adamts17 mutants [33,38] (Fig. 9).
The elevated TGF-β and reduced BMP signaling in Prrx1-Pofut2 developing tibia are consistent with altered FBN microfibril network composition that would result from reduced function of ADAMTS/TSL (Fig. 9). In the proliferating zone, TGF-β signaling promotes COL-II synthesis and negatively regulates BMP signaling [53–55] (Fig. 9). The elevated levels of COL-II in Prrx1-Pofut2 mutants raises the possibility that reduced function of one or more POFUT2 substrates could affect COL-II proteolysis, either directly or indirectly. In mice, elimination of the collagenase cleavage site in COL-II (Bailey mutants), resulted in elevated levels of Col-II, expanded the growth plate, and abnormal angiogenic invasion of the growth plate in postnatal bone [56]. This raises the possibility that defects in growth plate angiogenesis later in development could also contribute to the shortened limb phenotypes in Prrx1-Pofut2 mutants.
Increased BMP signaling in the prehypertrophic zone activates IHH signaling in the hypertrophic zone [57,58]. High IHH signaling and low RUNX2 stimulates COL-X expression and chondrocyte maturation and differentiation [43,57,59,60] (Fig. 9). In the Prrx1-Pofut2 mutants, decreased BMP signaling likely contributed to elevated RUNX2 and lower levels of Ptch1 mRNA/IHH signaling resulting in lower levels of COL-X and decreased region of cartilage hypertrophy (Fig. 9). Notably, Ihh and Runx2 mutant mice had severely shortened limbs [43,61]. Whereas Col-X mutant mice had shortened limbs with compressed growth plates and atypical distribution of matrix in the trabecular regions and often results in metaphyseal chondrodysplasia and short stature in human [62–64].
Altered function of secreted unfucosylated CCN2 could also potentially impact bone growth through modulation of TGF-β, BMP, WNT, and/or VEGF signaling [65–67]. Surface plasmon resonance and solid phase binding assay demonstrate that CCN2 strongly associates with BMP-2, TGF-β1 and TGF-β3 [66]. In cell-based assays, CTGF antagonized BMP and enhanced TGFβ signaling [66], and injection of Xenopus blastomeres with ctgf mRNA generates developmental abnormalities consistent with anti-BMP activity [67]. Although the effects of the mouse Ccn2 knockout on BMP and TGF-β signaling in bone development was not directly measured, loss of Ccn2 did not significantly increase Ihh mRNA [15], as would be expected from elevated BMP signaling. For these reasons, altered BMP and TGF-β signaling in Prrx1-Pofut2 mutants at E14.5 is more likely to result from altered fibrillin composition than from altered CCN2 function. Altered CCN2 function in Prrx1-Pofut2 mutant limbs could impact bone growth at later developmental stages since the TSR domain in CCN2 is proposed to mediate its interactions with LRP6 (Wnt signaling) and VEGF signaling [65], both of which are required for osteoblast differentiation and angiogenesis at later stage of embryonic limb development [15,68]. In the Ccn2 mutants, changes in the matrix proteins MMP9, link protein, and aggrecan significantly reduce VEGF signaling and PECAM expression providing evidence that shortened limbs in the Ccn2 knockout result from defects in growth plate angiogenesis at later stage of limb development [15].
O-fucosylation could impact extracellular function of POFUT2 substrates
Combined, results from cell-based secretion assays, in vitro folding/refolding assays, and structural analysis of O-fucosylated TSRs provide overwhelming evidence that O-fucosylation is required for efficient secretion of POFUT2 substrates from HEK293T cells and suggest that O-fucosylation stabilizes the native fold of TSRs and accelerates the overall rate of protein folding in vitro (Fig. 7 and [2,19,21–24, 27]). Based on these observations, we expected that the major effect of the Prrx1-Pofut2 mutation on limb growth would be caused by decreased stability/secretion of substrates and potentially by activation of the unfolded protein response due to accumulated misfolded substrate. However, here we provided evidence that blocking O-fucosylation did not activate the unfolded protein response in developing limb and that CCN2 and ADAMTS17 were secreted at levels close to or comparable to controls. Although this contrasts with results from cell-based secretion assays of POFUT2 substrates (Fig 7 and [19,21–23]), it suggests that distinct cell types are differentially affected by loss of POFUT2-mediated O-fucosylation of TSRs. This difference in sensitivity to loss of O-fucosylation could result from differences in the array of substrates expressed in the cell or from differences in the trafficking capacity of cells (i.e. HEK293T versus developing chondrocytes). Similar cell-specific secretion defects were reported for loss of POFUT1-mediated O-fucosylation of NOTCH1 epidermal growth factor (EGF) repeats, where O-fucosylation was essential for efficient trafficking in HEK293T cells but not required for trafficking in mouse embryonic stem cells [69,70].
The secretion of unfucosylated CCN2 and ADAMTS17 in Prrx1-Pofut2 mutant developing tibia also raises the possibility that the O-fucose modification could be important for the extracellular function of POFUT2 substrates including both intramolecular interactions as well as intermolecular interactions with ligands or ECM components. This prediction is consistent with the recent demonstration that an O-fucose modification on TSR3 of Brain-specific angiogenesis inhibitor 1 (BAI1) (also known as adhesion-GPCRs) is part of the ligand binding site and is essential for high affinity interactions with its ligand RTN4 (also known as NoGo) required for dendritic arborization, axonal elongation and synapse formation [28]. In the same way, O-linked fucose glycans on Notch EGFs are directly involved with ligand interactions [71,72]. Combined, the results from these studies suggest that shortened limb phenotypes in Prrx1-Pofut2 mutants likely resulted from the effects of the mutation on multiple POFUT2 substrates and underscore the potential that blocking O-fucosylation will have cell-type specific effects on secretion. Future studies will be important for addressing the possibility that O-fucosylation of POFUT2 substrate TSRs influences their functional interactions.
Experimental Procedures
Ethics statement
All animal research was carried out in accordance with relevant national and international regulations and protocols. The Office of Laboratory Animal Welfare (OLAW), assurance number D-16–00006, at Stony Brook University has been approved by the National Institutes of Health (A3011–01). The animal studies were approved by the Stony Brook University Institutional Animal Care and Use Committee (IACUC), which followed all of the guidelines outlined in: Public Health Service Policy on Humane Care and Use of Laboratory Animals, distributed by the National Institutes of Health’s Office of Laboratory Animal Welfare; Animal Welfare Act and Animal Welfare Regulations, distributed by the United States Department of Agriculture; and Public Health Service Policy on Humane Care and Use of Laboratory Animals, distributed by the Office of Laboratory Animal Welfare, NIH; Animal Welfare Act and Animal Welfare Regulations distributed by United States Department of Agriculture and Guide for the Care and Use of Laboratory Animals distributed by the National Research Council. Stony Brook University animal facilities are accredited by Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC International).
Mice and genotyping
The Pofut2-Floxed allele was previously described [25]. To generate conditional Pofut2 knockout in the limb mesenchyme, we crossed Pofut2-Lox/+;Prrx1-Cre homozygous males and Pofut2-Flox/Flox females that generated Pofut2-Flox/+;Prrx1-Cre/+ (control) and Pofut2-Flox/Δ; Prrx1-Cre/+ (Prrx1-Pofut2 knockout) for the study. Details of mouse crosses to generate the homozygous male Pofut2-Lox/+;Prrx1-Cre and Pofut2-Flox/Flox females are described in the supplementary method. To identify limb tissues expressing Cre-recombinase, we crossed hemizygotes Prrx1-Cre transgenic mice with Rosa26mTmG (MGI: 3716464) reporter mice [73]. All mice were housed and maintained under controlled temperature and light (22°C, 12 h light and 12 h dark) with access to sufficient food and water. Tissue lysis conditions, primers and conditions for the genotyping is presented in Supplemental Table S1 and S2.
Skeletal preparations and imaging
Skeletal preparations from 3- to 8-week-old mice were prepared as described previously [74]. Briefly, the mice were eviscerated, and soft tissues were removed. The carcass was then fixed in 95% ethanol overnight and permeabilized with acetone at room temperature for 24 hours. Then the skeletons were stained with Alcian blue [0.03%] (Cat. no. A3157, Sigma) [0.03% w/v Alcian blue, 80% ethanol and 20% acetic acid] for 24 hours. The skeletons were then washed with several changes of 70% ethanol and transferred to alizarin red [0.0025%] (Cat. no. A5533, Sigma) [0.0025% w/v Alizarin red in 1% KOH] for 24 hours. Skeletons were then cleared sequentially 1% KOH, 0.5% KOH in 50% glycerol and finally cleared and stored in 100% glycerol. Skeletons and disarticulated bones were photographed in glycerol using a Zeiss Discovery V8 microscope, AxioCam MRc camera and AxioVisionLE program (Zeiss). Whole skeleton images were reconstructed from two or three images using Adobe Photoshop photomerge feature. For bone measurement, skeletons were disarticulated, and individual limb bones were photographed and measured using ImageJ (http://imagej.net/). Data were evaluated for significance using Wilcoxon rank test.
MicroCT
Left hind limbs from 3-week-old and 22–24 days old Pofut2 control (Pofut2-Flox/+;Prrx1-Cre/+) (n=6) and Pofut2 knockout (Pofut2-Flox/Δ;Prrx1-Cre/+) (n=6) mice were fixed in 70% ethanol after removal of surrounding soft tissues. Micro CT images from whole tibias, mid diaphysis (5% of total tibia length) and proximal tibia metaphysis (5% of total tibia length) were obtained using microCT (Scanco μ40). The cortical region measurements at mid-diaphysis included cortical thickness, volume, minimum and maximum diameters, bone mineral density, and polar moment of inertia. Trabecular measurements included endosteal bone volume and fraction, connectivity, trabecular spacing, and trabecular bone density [75,76]. These measurements were compared using Wilcoxon rank test. The detailed procedure for bone parameters measurements is in a supplementary file.
Plasmids for analysis of ADAMTS6, ADAMTS10, and CCN2
Mammalian expression plasmids for full-length mouse ADAMTS6 (pSecTag2B-ADAMTS6) [77] and human ADAMTS10 (pcDNA3.1-ADAMTS10) [78] were generously supplied by Dr. Suneel S. Apte (Cleveland Clinic). A full-length mouse Ccn2 cDNA plasmid (RefSeq: NM_010217.2) was purchased from Sino Biological Inc. (Catalog number: MG51217-U). To generate a mammalian expression construct, the full-length ORF was amplified by PCR using CloneAmp HiFi PCR Premix (TaKaRa Bio Inc.) with primers 5’- TTTAAACTTAAGCTTGCCAC-CATGCTCGCCTCC −3’ and 5’- CCCTCTAGACTC-GAGCGCCATGTCTCCGTACAT −3’, which include Hind III or Xho I restriction enzyme sites for subcloning, respectively. The fragment was sub-cloned into pcDNA4/TO/Myc-His A (Invitrogen) using T4 polynucleotide ligase (NEB). All three of these plasmids encode C-terminal Myc-His6 tags.
Mass Spectral Analysis of ADAMTS6, ADAMTS10, and CCN2 O-fucosylation
For mass spectral analysis of ADAMTS6, ADAMTS10, and CCN2, the corresponding plasmids (pSecTag2B-ADAMTS6, pcDNA3.1-ADAMTS10 and pcDNA4-CCN2) were transiently transfected separately into wild type HEK293T cells, media was harvested 3 days post-transfection, and each target protein was purified over a nickel-nitrilotriacetic acid (Ni-NTA) column (VWR) as previously described [22]. Purified proteins were reduced and alkylated as described [22]. ADAMTS6 and ADAMTS10 were digested with Trypsin and CCN2 with Chymotrypsin. All digestions were performed as previously described [22] and analyzed on a Q-Exactive mass spectrometer equipped with an Easy nanoLC 1000 and analyzed also as previously described [22]. Peak lists and .raw data files were generated using Xcalibur software set to its default settings. Raw data files were analyzed using Proteome Discoverer 2.1.0.81 (Thermo Fisher) and were searched against protein-specific databases (mouse ADAMTS6 database, accession number D3Z1A5; human ADAMTS10 database, accession number Q9H324; mouse CCN2 database, accession number P29268). Byonic software version 2.10.5 (Protein Metrics) was used as a module inside Proteome Discoverer for identifying peptides with glycan modifications. EIC for all peptides were generated using Xcalibur Qual Browser 4.0.27.19 (Thermo Fisher).
Cell-based secretion assays using wild-type and POFUT2-null HEK293T cells
Wild-type and POFUT2-null HEK293T cells were transiently transfected with plasmids encoding for ADAMTS6, ADAMTS10 or CCN2. For ADAMTS6, 2.4 μg of pSecTag2c-ADAMTS6 plasmid was transfected with 0.1 μg of pEGFP-N1 [79] and 0.5 μg of pSecTag2C empty vector in wild type cells or 0.5 μg of pcDNA4-POFUT2 [25] in POFUT2-null cells using polyethylimine (PEI) as a transfection reagent [80]. For CCN2, 1.8 μg of pcDNA4-CCN2 plasmid was transfected with 0.1 μg of pEGFP-N1 and 0.5 μg of pcDNA4 empty vector in wild type cells or 0.5 μg of pcDNA4-POFUT2 in POFUT2-null cells using (PEI) as a transfection reagent. For ADAMTS10, 2.4 μg of pcDNA3.1-ADAMTS10 plasmid was transfected with 0.1 μg of pEGFP-N1 and 0.2 μg of pcDNA3.1 empty vector in wild type cells or 0.5 μg of pcDNA4-POFUT2 in POFUT2-null cells using Lipofectamine 2000 (Thermo Fisher) as a transfection reagent. Both conditioned media and lysates were harvested and immunoblotted as previously described [21] but Alexa Fluor IRDye® 800CW goat antimouse (LI-COR, Cat. No. 926–32210) and Alexa Fluor IRDye® 680RD goat antirabbit (LI-COR, Cat.No. 926–68071) was used to detect each POFUT2 substrate and GFP respectively. The membrane was imaged and quantified using the Odyssey CLx system and analyzed with Image Studio Lite software (LI-COR Biosciences), and ratios of anti-Myc to anti-GFP were calculated.
Histology and immunohistochemistry
Bones isolated from postnatal mice were fixed in 4% paraformaldehyde, decalcified in EDTA, and processed for paraffin embedding. Embryonic limbs were either fixed in 4% paraformaldehyde or 5% acetic acid in ethanol and processed for paraffin embedding (refer to Supplemental Table S3). Briefly, paraformaldehyde fixed tissues were dehydrated by serially immersing into 30%, 50%, 70%, 80%, 90%, 95% and 100% ethanol and then xylene. Acetic acid fixed tissues were directly transferred to 100% ethanol and then xylene. The tissues were then infiltrated with and embedded in paraffin and processed for sectioning. The embedded tissues were sectioned at 5 μm thickness using a microtome. Limb sections were routinely stained with hematoxylin and eosin (H&E) staining as previously described [21]. Immunofluorescence on tibial paraffin sections were performed as previously described [21]. The sections were incubated with primary antibodies followed by secondary antibodies (Supplementary Table S3). Slides were mounted with DAPI Fluoromount-G® (SouthernBiotech, Cat. no. 0100–20) and cover slipped.
Growth plate size quantification
Hematoxylin and eosin-stained eight mid sections (covering 300 μm thickness) from the middle of proximal tibia growth plates of each animal were photographed at 10 x using a Nikon Optiphot microscope, AxioCam MRc camera and AxioVisionLE program (Zeiss). Sections were then analyzed using ImageJ (http://imagej.net/). Briefly, five parallel lines running from resting to hypertrophic zones were drawn perpendicular to the growth plate and the lengths of each was measured using ImageJ, and average was taken. This gave the height of the growth plate of a section. The height of the hypertrophic regions was measured in similar way. Total 21 to 22 sections from n=3 animals of each genotype were measured and plotted in a graph. Statistical analysis was performed in GraphPad prism software (Version 8) to evaluate significance using unpaired, two-tailed t-test.
EdU incorporation
Cell proliferation was evaluated by measuring incorporation of EdU (5-ethynyl-2’-deoxyuridine). Timed pregnant females were injected intraperitoneally with 200 mg of EdU (Cat no. A10044, Thermo-Scientific) per 10 g (i. e. 20 mg/Kg) of body weight three hours before euthanization. Hindlimbs of embryos were dissected, fixed overnight either in 4% paraformaldehyde or 5 % acetic acid in ethanol. EdU in tibial sections was detected using the Click-iT™ EdU Alexa Fluor™ 594 Imaging Kit (Cat. no.C10339, ThermoScientific) according to manufacturers’ instructions. After the reaction, the sections were mounted with DAPI Fluoromount-G® (SouthernBiotech, Cat. no. 0100–20) and cover slipped. The number of EDU incorporated cells were analyzed in ImageJ (http://imagej.net/) using ‘particle measurement’ functions for both the EDU and DAPI stained cells separately. The EDU incorporation was presented as % EDU over total DAPI stained cells in the region of interests from total of 14 sections from n=4 animals of each genotype groups. Statistical analysis was performed in GraphPad prism software (Version 8) to evaluate significance using unpaired, two-tailed t-test with p<0.05 considered being significant.
RNA extraction and real-time quantitative RT-PCR (qRT-PCR)
RNA extraction and qRT-PCR was performed as described previously [21,81]. Briefly, total RNA was extracted from E14.5 control and Prrx1-Pofut2 mutants limbs using RNeasy® Mini Kit (Qiagen, ID 74104), total of 2 μg RNA from each sample was reverse transcribed using SuperScript™ VILO™ cDNA Synthesis Kit (Invitrogen, Cat. no. 11754050) and qRT-PCR was carried out using PowerTrack™ SYBR Green Master Mix (Thermo Fisher Scientific, Cat. no. A46109) on StepOnePlus™ Real-Time PCR System or with iQ™ SYBR® Green (Bio-Rad, Cat. no. 1708880) on a RealPlex2 MasterCycler (Eppendorf). The results from control (n = 3–6; 3 replicates per sample) and mutant (n = 3–6; 3 replicates per sample) animals for each gene were normalized to those with Gapdh and/or Rpl4 and expressed as fold change ± standard deviations. The mean expression levels from control and mutants were compared using Student’s t-test. The primers of genes used were listed in Supplemental Table S4.
RNAScope assay
Mouse hind limb at embryonic day 14.5 (E14.5) were fixed overnight in 4% PFA and processed for paraffin sectioning (5 μm). RNAScope® assay was carried out as previously described [21] and according to Advanced Cell Diagnostics’ instructions using Pofut2 (465281) and Ptch1 (402811) probes. ACD HybEZ™ II Oven (Advanced Cell Diagnostics, Inc., Hayward, CA) was used for the probe hybridization. Probe binding was visualized by using RNAscope® 2.5 HD Detection Kit (Red) using Mayer’s hematoxylin as a counterstain. The bacterial gene encoding dihydrodipicolinate reductase (DapB; 310043) and Mus musculus gene encoding peptidylprolyl isomerase B (Ppib; 313911) were used as negative and positive controls (Fig. S21), respectively.
Histological sections were photographed using a Nikon Optiphot microscope, AxioCam MRc camera and AxioVisionLE program (Zeiss). Fluorescent images were taken using Leica SP5 or SP8 confocal (Leica, Germany). ImageJ (http://imagej.net/) was used to measure the fluorescence of the images as described previously [21,82]. RNAScope assay was analyzed in ImageJ by calculating the optical density of the region of interests using formula Optical Density = log(Max intensity/Mean intensity) described previously [83] with slight modification. Briefly, optical density was calculated after converting the RGB image to luminance. To eliminate background, optical density from negative control was subtracted from each observation. Optical density was measured in sections obtained from at least four (n=4) animals from each genotype group with a minimum of three sections per animal. Fluorescence was measured in sections obtained from at least three animals (n = 3) from each genotype group with a minimum of three sections per animal. Statistical analysis was performed in GraphPad prism software (Version 8) to evaluate significance using unpaired, two-tailed t-test.
Supplementary Material
Acknowledgement
We would like to thank Professor Dr. Robert P. Mecham (Washington University School of Medicine, St. Louis, MO) for providing anti-fibrillin2 antibody, Professor Dr. Lynn Sakai (Oregon Health and Science University, Portland, OR) for providing anti-fibrillin1 antibody, and Dr. Suneel S. Apte (Cleveland Clinic, Cleveland, OH) for providing the ADAMTS6 and ADAMTS10 expression plasmids. We would like to thank Sulan Xu for assistance with animal breeding, genotyping and histology.
Funding
Supported by grants from the National Institutes of Health (HD090156 to R.S.H. and B.C.H. and P41GM103390 to K.W.M.)
Abbreviations:
- ADAMTS
A Disintegrin And Metalloproteinase with ThromboSpondin motifs
- ADAMTSL
ADAMTS-Like
- ADGRB
Adhesion G protein-coupled receptor B
- BAI
Brain-specific angiogenesis inhibitor
- BMP
Bone morphogenetic protein
- B3GLCT
β3-glucosyltransferase
- CCN
Cellular communication network factor-2
- CTGF
Connective tissue growth factor
- ECM
Extracellular matrix
- EDTA
Ethylenediaminetetraacetic acid
- EGF
Epidermal growth factor-like
- FBN2
Fibrillin 2
- FBN1
Fibrillin 1
- Gapdh
Glyceraldehyde 3-phosphate dehydrogenase
- IHH
Indian hedgehog
- LTBP
Latent transforming growth factor beta binding protein
- MMP
Matrix metalloproteinase
- NOV
Nephroblastoma overexpressed gene
- POFUT2
Protein O-fucosyltransferase-2
- Rpl4
Ribosomal protein L4
- RTN4
Reticulon 4
- RUNX2
Runt-related transcription factor 2
- SMAD
Sma and Mad related protein
- TGF-β
Transforming growth factor-beta
- TSP
Thrombospondin
- TSR
Thrombospondin type 1 repeat
- VEGF
Vascular endothelial growth factor
Footnotes
Supplementary materials
Supplementary material associated with this article can be found in the online version at doi:10.1016/j.matbio.2022.02.002.
References
- [1].Adams JC, Tucker RP, The Thrombospondin Type 1 Repeat (TSR) superfamily: diverse proteins with relatedr-Roles in neuronal development, Dev. Dyn 218 (2000) 280–299. [DOI] [PubMed] [Google Scholar]
- [2].Holdener BC, Haltiwanger RS, Protein O-fucosylation: structure and function, Curr. Opin. Struct. Biol 56 (2019) 78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Valero-Gonzalez J, Leonhard-Melief C, Lira-Navarrete E, Jimenez-Oses G, Hernandez-Ruiz C, Pallares MC, Yruela I, Vasudevan D, Lostao A, Corzana F, Takeuchi H, Haltiwanger RS, Hurtado-Guerrero R, A proactive role of water molecules in acceptor recognition by protein O-fucosyltransferase 2, Nat. Chem. Biol 12 (4) (2016) 240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Luo Y, Nita-Lazar A, Haltiwanger RS, Two distinct pathways for O-fucosylation of epidermal growth factor-like or thrombospondin type 1 repeats, J. Biol. Chem 281 (14) (2006) 9385–9392. [DOI] [PubMed] [Google Scholar]
- [5].Luo Y, Koles K, Vorndam W, Haltiwanger RS, Panin VM, Protein O-fucosyltransferase 2 adds O-fucose to thrombospondin type 1 repeats, J. Biol. Chem 281 (14) (2006) 9393–9399. [DOI] [PubMed] [Google Scholar]
- [6].Li Z, Han K, Pak JE, Satkunarajah M, Zhou D, Rini JM, Recognition of EGF-like domains by the Notch-modifying O-fucosyltransferase POFUT1, Nat. Chem. Biol 13 (7) (2017) 757–763. [DOI] [PubMed] [Google Scholar]
- [7].Sato T, Sato M, Kiyohara K, Sogabe M, Shikanai T, Kikuchi N, Togayachi A, Ishida H, Ito H, Kameyama A, Gotoh M, Narimatsu H, Molecular cloning and characterization of a novel human beta1,3-glucosyltransferase, which is localized at the endoplasmic reticulum and glucosylates O-linked fucosylglycan on thrombospondin type 1 repeat domain, Glycobiology 16 (12) (2006) 1194–1206. [DOI] [PubMed] [Google Scholar]
- [8].Kozma K, Keusch JJ, Hegemann B, Luther KB, Klein D, Hess D, Haltiwanger RS, Hofsteenge J, Identification and characterization of abeta1,3-glucosyltransferase that synthesizes the Glc-beta1,3-Fuc disaccharide on thrombospondin type 1 repeats, J. Biol. Chem 281 (48) (2006) 36742–36751. [DOI] [PubMed] [Google Scholar]
- [9].Schneider M, Al-Shareffi E, Haltiwanger RS, Biological functions of fucose in mammals, Glycobiology 27 (7) (2017) 601–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Leonhard-Melief C, Haltiwanger RS, O-Fucosylation of Thrombospondin Type 1 Repeats, Glycobiology (2010) 401–416. [DOI] [PubMed] [Google Scholar]
- [11].Kelwick R, Desanlis I, Wheeler GN, Edwards DR, The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family, Genome Biol 16 (2015) 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hubmacher D, Apte SS, ADAMTS proteins as modulators of microfibril formation and function, Matrix Biol 47 (2015) 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Moon SY, Shin SA, Oh YS, Park HH, Lee CS, Understanding the role of the BAI subfamily of adhesion G Protein-Coupled Receptors (GPCRs) in pathological and physiological conditions, Genes (Basel) 9 (12) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Amend SR, Uluckan O, Hurchla M, Leib D, Novack DV, Silva M, Frazier W, Weilbaecher KN, Thrombospondin-1 regulates bone homeostasis through effects on bone matrix integrity and nitric oxide signaling in osteoclasts, J. Bone Miner. Res 30 (1) (2015) 106–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ivkovic S, Yoon BS, Popoff SN, Safadi FF, Libuda DE, Stephenson RC, Daluiski A, Lyons KM, Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development, Development 130 (12) (2003) 2779–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tan K, Duquette M, Liu JH, Dong Y, Zhang R, Joachimiak A, Lawler J, Wang JH, Crystal structure of the TSP-1 type 1 repeats: a novel layered fold and its biological implication, J. Cell Biol 159 (2) (2002) 373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mo FE, Muntean AG, Chen CC, Stolz DB, Watkins SC, Lau LF, CYR61 (CCN1) is essential for placental development and vascular integrity, Mol. Cell. Biol 22 (24) (2002) 8709–8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hofsteenge J, Huwiler KG, Macek B, Hess D, Lawler J, Mosher DF, Peter-Katalinic J, C-mannosylation and O-fucosylation of the thrombospondin type 1 module, J. Biol. Chem 276 (9) (2001) 6485–6498. [DOI] [PubMed] [Google Scholar]
- [19].Holdener BC, Percival CJ, Grady RC, Cameron DC, Berardinelli SJ, Zhang A, Neupane S, Takeuchi M, Jimenez-Vega JC, Uddin SMZ, Komatsu DE, Honkanen R, Dubail J, Apte SS, Sato T, Narimatsu H, McClain SA, Haltiwanger RS, ADAMTS9 and ADAMTS20 are differentially affected by loss of B3GLCT in mouse model of Peters plus syndrome, Hum. Mol. Genet 28 (24) (2019) 4053–4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Alford AI, Stephan C, Kozloff KM, Hankenson KD, Compound deletion of thrombospondin-1 and −2 results in a skeletal phenotype not predicted by the single gene knockouts, Bone 153 (2021) 116156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Neupane S, Goto J, Berardinelli SJ, Ito A, Haltiwanger RS, Holdener BC, Hydrocephalus in mouse B3glct mutants is likely caused by defects in multiple B3GLCT substrates in ependymal cells and subcommissural organ, Glycobiology (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang A, Berardinelli SJ, Leonhard-Melief C, Vasudevan D, Liu TW, Taibi A, Giannone S, Apte SS, Holdener BC, Haltiwanger RS, O-Fucosylation of ADAMTSL2 is required for secretion and is impacted by geleophysic dysplasia-causing mutations, J. Biol. Chem 295 (46) (2020) 15742–15753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hubmacher D, Schneider M, Berardinelli SJ, Takeuchi H, Willard B, Reinhardt DP, Haltiwanger RS, Apte SS, Unusual life cycle and impact on microfibril assembly of ADAMTS17, a secreted metalloprotease mutated in genetic eye disease, Sci. Rep 7 (2017) 41871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Vasudevan D, Takeuchi H, Johar SS, Majerus E, Haltiwanger RS, Peters plus syndrome mutations disrupt a noncanonical ER quality-control mechanism, Curr. Biol 25 (3) (2015) 286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Benz BA, Nandadasa S, Takeuchi M, Grady RC, Takeuchi H, LoPilato RK, Kakuda S, Somerville RPT, Apte SS, Haltiwanger RS, Holdener BC, Genetic and biochemical evidence that gastrulation defects in Pofut2 mutants result from defects in ADAMTS9 secretion, Dev. Biol 416 (1) (2016) 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Du J, Takeuchi H, Leonhard-Melief C, Shroyer KR, Dlugosz M, Haltiwanger RS, Holdener BC, O-fucosylation of thrombospondin type 1 repeats restricts epithelial to mesenchymal transition (EMT) and maintains epiblast pluripotency during mouse gastrulation, Dev. Biol 346 (1) (2010) 25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Niwa Y, Suzuki T, Dohmae N, Simizu S, O-Fucosylation of CCN1 is required for its secretion, FEBS Lett 589 (21) (2015) 3287–3293. [DOI] [PubMed] [Google Scholar]
- [28].Wang J, Miao Y, Wicklein R, Sun Z, Wang J, Jude KM, Fernandes RA, Merrill SA, Wernig M, Garcia KC, Su TC €dhof, RTN4/NoGo-receptor binding to BAI adhesion-GPCRs regulates neuronal development, Cell (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ, Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer, Genesis 33 (2) (2002) 77–80. [DOI] [PubMed] [Google Scholar]
- [30].DeSesso JM, Vascular ontogeny within selected thoracoabdominal organs and the limbs, Reprod. Toxicol 70 (2017) 3–20. [DOI] [PubMed] [Google Scholar]
- [31].Stanley S, Balic Z, Hubmacher D, Acromelic dysplasias: how rare musculoskeletal disorders reveal biological functions of extracellular matrix proteins, Ann. N.Y. Acad. Sci 1490 (1) (2021) 57–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Satz-Jacobowitz B, Hubmacher D, The quest for substrates and binding partners: A critical barrier for understanding the role of ADAMTS proteases in musculoskeletal development and disease, Dev. Dyn 250 (1) (2021) 8–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mead TJ, Martin DR, Wang LW, Cain SA, Gulec C, Cahill E, Mauch J, Reinhardt DP, Lo CW, Baldock C, Apte SS, Proteolysis of fibrillin-2 microfibrils is essential for normal skeletal development, bioRxiv preprint (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang LW, Kutz WE, Mead TJ, Beene LC, Singh S, Jenkins MW, Reinhardt DP, Apte SS, Adamts10 inactivation in mice leads to persistence of ocular microfibrils subsequent to reduced fibrillin-2 cleavage, Matrix Biol 77 (2019) 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhang H, Hu W, Ramirez F, DevelopmentaleExpression of fibrillin genes suggests heterogeneity of extracellular microfibrils, J. Cell Biol 129 (1995) 1165–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Thomson J, Singh S, Eckersley A, Cain SA, Sherratt MJ, Baldock C, Fibrillin microfibrils and elastic fibre proteins_ Functional interactions and extracellular regulation of growth factors Seminars in, Cell & Developmental Biology 89 (2019) 109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhang H, Apfelroth SD, Hu W, Davis EC, Sanguineti C, Bonadio J, Mecham RP, Ramirez F, Structure and expression of fibrillin-2, a novel microfibrillar component preferentially located in elastic matrices, J. Cell Biol 124 (5) (1994) 855–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Oichi T, Taniguchi Y, Soma K, Oshima Y, Yano F, Mori Y, Chijimatsu R, Kim-Kaneyama JR, Tanaka S, Saito T, Adamts17 is involved in skeletogenesis through modulation of BMP-Smad1/5/8 pathway, Cell. Mol. Life Sci 76 (23) (2019) 4795–4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Delhon L, Mahaut C, Goudin N, Gaudas E, Piquand K, Le Goff W, Cormier-Daire V, Le Goff C, Impairment of chondrogenesis and microfibrillar network in Adamtsl2 deficiency, FASEB J 33 (2) (2019) 2707–2718. [DOI] [PubMed] [Google Scholar]
- [40].Arteaga-Solis E, Gayraud B, Lee SY, Shum L, Sakai L, Ramirez F, Regulation of limb patterning by extracellular microfibrils, J. Cell Biol 154 (2) (2001) 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dubail J, Apte SS, Insights on ADAMTS proteases and ADAMTS-like proteins from mammalian genetics, Matrix Biol (44–46) (2015) 24–37. [DOI] [PubMed] [Google Scholar]
- [42].Myllyharju J, Extracellular matrix and developing growth plate, Curr. Osteoporos. Rep 12 (4) (2014) 439–445. [DOI] [PubMed] [Google Scholar]
- [43].St-Jacques B, Hammerschmidt M, McMahon AP, Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation, Genes Dev 13 (1999) 2072–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yoshida CA, Yamamoto H, Fujita T, Furuichi T, Ito K, Inoue K, Yamana K, Zanma A, Takada K, Ito Y, Komori T, Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog, Genes Dev 18 (8) (2004) 952–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G, Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation, Cell 89 (1997) 747–754. [DOI] [PubMed] [Google Scholar]
- [46].Kutz WE, Wang LW, Bader HL, Majors AK, Iwata K, Traboulsi EI, Sakai LY, Keene DR, Apte SS, ADAMTS10 protein interacts with fibrillin-1 and promotes its deposition in extracellular matrix of cultured fibroblasts, J. Biol. Chem 286 (19) (2011) 17156–17167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kawaki H, Kubota S, Suzuki A, Lazar N, Yamada T, Matsumura T, Ohgawara T, Maeda T, Perbal B, Lyons KM, Takigawa M, Cooperative regulation of chondrocyte differentiation by CCN2 and CCN3 shown by a comprehensive analysis of the CCN family proteins in cartilage, J. Bone Miner. Res 23 (11) (2008) 1751–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Mularczyk EJ, Singh M, Godwin ARF, Galli F, Humphreys N, Adamson AD, Mironov A, Cain SA, Sengle G, Boot-Handford RP, Cossu G, Kielty CM, Baldock C, ADAMTS10-mediated tissue disruption in Weill-Marchesani syndrome, Hum. Mol. Genet 27 (21) (2018) 3675–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cain SA, Mularczyk EJ, Singh M, Massam-Wu T, Kielty CM, ADAMTS-10 and −6 differentially regulate cell-cell junctions and focal adhesions, Sci. Rep 6 (2016) 35956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sengle G, Tsutsui K, Keene DR, Tufa SF, Carlson EJ, Charbonneau NL, Ono RN, Sasaki T, Wirtz MK, Samples JR, Fessler LI, Fessler JH, Sekiguchi K, Hayflick SJ, Sakai LY, Microenvironmental regulation by fibrillin-1, PLos Genet 8 (1) (2012) e1002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Le Goff C, Mahaut C, Wang LW, Allali S, Abhyankar A, Jensen S, Zylberberg L, Collod-Beroud G, Bonnet D, Alanay Y, Brady AF, Cordier MP, Devriendt K, Genevieve D, Kiper PO, Kitoh H, Krakow D, Lynch SA, Le Merrer M, Megarbane A, Mortier G, Odent S, Polak M, Rohrbach M, Sillence D, Stolte-Dijkstra I, Superti-Furga A, Rimoin DL, Topouchian V, Unger S, Zabel B, Bole-Feysot C, Nitschke P, Handford P, Casanova JL, Boileau C, Apte SS, Munnich A, Cormier-Daire V, Mutations in the TGFbeta binding-protein-like domain 5 of FBN1 are responsible for acromicric and geleophysic dysplasias, Am. J. Hum. Genetic 89 (1) (2011) 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Chaudhry SS, Cain SA, Morgan A, Dallas SL, Shuttleworth CA, Kielty CM, Fibrillin-1 regulates the bioavailability of TGFbeta1, J. Cell Biol 176 (3) (2007) 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Sedlmeier G, Sleeman JP, Extracellular regulation of BMP signaling: welcome to the matrix, Biochem. Soc. Trans 45 (1) (2017) 173–181. [DOI] [PubMed] [Google Scholar]
- [54].Zhu Y, Tao H, Jin C, Liu Y, Lu X, Hu X, Wang X, Transforming growth factor-beta1 induces type II collagen and aggrecan expression via activation of extracellular signal-regulated kinase 1/2 and Smad2/3 signaling pathways, Molecul. Med. Rep 12 (4) (2015) 5573–5579. [DOI] [PubMed] [Google Scholar]
- [55].Wang W, Rigueur D, Lyons KM, TGFbeta signaling in cartilage development and maintenance, Birth Defects Res 102 (1) (2014) 37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gauci SJ, Golub SB, Tatarczuch L, Lee E, Chan D, Walsh NC, Little CB, Stanton H, Lokmic Z, Sims NA, Mackie EJ, Fosang AJ, Disrupted type II collagenolysis impairs angiogenesis, delays endochondral ossification and initiates aberrant ossification in mouse limbs, Matrix Biol 83 (2019) 77–96. [DOI] [PubMed] [Google Scholar]
- [57].Minina E, Wenzel HM, Kreschel C, Karp S, Gaffield W, McMahon AP, Vortkamp A, BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation, Development 128 (2001) 4523–4534. [DOI] [PubMed] [Google Scholar]
- [58].Grimsrud CD, Romano PR, D’Souza M, Puzas JE, Schwarz EM, Reynolds PR, Roiser RN, O’Keefe RJ, BMP signaling stimulates chondrocyte maturation and the expression of Indian hedgehog, J. Orthop. Res 19 (1) (2001) 18–25. [DOI] [PubMed] [Google Scholar]
- [59].Komori T, Molecular mechanism of Runx2-dependent bone development, Mol. Cells 43 (2) (2020) 168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Amano K, Densmore M, Nishimura R, Lanske B, Indian hedgehog signaling regulates transcription and expression of collagen type X via Runx2/Smads interactions, J. Biol. Chem 289 (36) (2014) 24898–24910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T, Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts, Cell 89 (1997) 755–764. [DOI] [PubMed] [Google Scholar]
- [62].Warman ML, Abbott M, Apte SS, Hefferon T, Mcintosh I, Cohn DH, Hecht TH, Olsen BR, Francomano CA, A type X collagen mutation causes Schmid metaphyseal chondrodysplasia, Nat. Genet 5 (1993) 79–82. [DOI] [PubMed] [Google Scholar]
- [63].Jacenko O, LuValle PA, Olsen BR, Spondylometaphyseal dysplasia in mice carrying a dominant negative mutation in a matrix protein specific for cartilage-to-bone transition, Nature 365 (1993) 56–61. [DOI] [PubMed] [Google Scholar]
- [64].Gress CJ, Jacenko O, Growth plate compressions and altered hematopoiesis in collagen X null mice, J. Cell Biol 149 (4) (2000) 983–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Arnott JA, Lambi AG, Mundy CM, Hendesi H, Pixley RA, Owen TA, Safadi FF, Popoff SN, The role of Connective tissue growth factor (CTGF/CCN2) in skeletogenesis, Crit. Rev.™ in Eukaryotic Gene Express 21 (1) (2011) 43–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Maeda A, Nishida T, Aoyama E, Kubota S, Lyons KM, Kuboki T, Takigawa M, CCN family 2/connective tissue growth factor modulates BMP signalling as a signal conductor, which action regulates the proliferation and differentiation of chondrocytes, J. Biochem 145 (2) (2009) 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Abreu JG, Ketpura NI, Reversade B, De Robertis EM, Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-β, Nat. Cell Biol 4 (8) (2002) 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Joeng KS, Schumacher CA, Zylstra-Diegel CR, Long F, Williams BO, Lrp5 and Lrp6 redundantly control skeletal development in the mouse embryo, Dev. Biol 359 (2) (2011) 222–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Takeuchi H, Yu H, Hao H, Takeuchi M, Ito A, Li H, Haltiwanger RS, O-Glycosylation modulates the stability of epidermal growth factor-like repeats and thereby regulates Notch trafficking, J. Biol. Chem 292 (38) (2017) 15964–15973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Stahl M, Uemura K, Ge C, Shi S, Tashima Y, Stanley P, Roles of Pofut1 and O-fucose in mammalian Notch signaling, J. Biol. Chem 283 (20) (2008) 13638–13651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Luca VC, Kim BC, Ge C, Kakuda S, Wu D, Roein-Peikar M, Haltiwanger RS, Zhu C, Ha T, Garcia KC, Notch-Jagged complex structure implicates a catch bond in tuning ligand sensitivity, Science 355 (6331) (2017) 1320–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Luca VC, Jude KM, Pierce NW, Nachury MV, Fischer S, Garcia KC, Structural basis for Notch1 engagement of Delta-like 4, Science 347 (6224) (2015) 847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L, A global double-fluorescent Cre reporter mouse, Genesis 45 (9) (2007) 593–605. [DOI] [PubMed] [Google Scholar]
- [74].Rigueur D, Lyons KM, Whole-mount skeletal staining, Methods Mol. Biol 1130 (2014) 113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Campbell GM, Sophocleous A, Quantitative analysis of bone and soft tissue by micro-computed tomography: applications to ex vivo and in vivo studies, BoneKEy Rep 3 (2014) 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R, Guidelines for assessment of bone microstructure in rodents using micro-computed tomography, J. Bone Miner. Res 25 (7) (2010) 1468–1486. [DOI] [PubMed] [Google Scholar]
- [77].Prins BP, Mead TJ, Brody JA, Sveinbjornsson G, Ntalla I, Bihlmeyer NA, van den Berg M, Bork-Jensen J, Cappellani S, Van Duijvenboden S, Klena NT, Gabriel GC, Liu X, Gulec C, Grarup N, Haessler J, Hall LM, Iorio A, Isaacs A, Li-Gao R, Lin H, Liu CT, Lyytikainen LP, Marten J, Mei H, Muller-Nurasyid M, Orini M, Padmanabhan S, Radmanesh F, Ramirez J, Robino A, Schwartz M, van Setten J, Smith AV, Verweij N, Warren HR, Weiss S, Alonso A, Arnar DO, Bots ML, de Boer RA, Dominiczak AF, Eijgelsheim M, Ellinor PT, Guo X, Felix SB, Harris TB, Hayward C, Heckbert SR, Huang PL, Jukema JW, Kahonen M, Kors JA, Lambiase PD, Launer LJ, Li M, Linneberg A, Nelson CP, Pedersen O, Perez M, Peters A, Polasek O, Psaty BM, Raitakari OT, Rice KM, Rotter JI, Sinner MF, Soliman EZ, Spector TD, Strauch K, Thorsteinsdottir U, Tinker A, Trompet S, Uitterlinden A, Vaartjes I, van der Meer P, Volker U, Volzke H, Waldenberger M, Wilson JG, Xie Z, Asselbergs FW, Dorr M, van Duijn CM, Gasparini P, Gudbjartsson DF, Gudnason V, Hansen T, Kaab S, Kanters JK, Kooperberg C, Lehtimaki T, Lin HJ, Lubitz SA, Mook-Kanamori DO, Conti FJ, Newton-Cheh CH, Rosand J, Rudan I, Samani NJ, Sinagra G, Smith BH, Holm H, Stricker BH, Ulivi S, Sotoodehnia N, Apte SS, van der Harst P, Stefansson K, Munroe PB, Arking DE, Lo CW, Jamshidi Y, Exome-chip meta-analysis identifies novel loci associated with cardiac conduction, including ADAMTS6, Genome Biol 19 (1) (2018) 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Somerville RP, Jungers KA, Apte SS, Discovery and characterization of a novel, widely expressed metalloprotease, ADAMTS10, and its proteolytic activation, J. Biol. Chem 279 (49) (2004) 51208–51217. [DOI] [PubMed] [Google Scholar]
- [79].Schneider M, Kumar V, Nordstrom LU, Feng L, Takeuchi H, Hao H, Luca VC, Garcia KC, Stanley P, Wu P, Haltiwanger RS, Inhibition of Delta-induced Notch signaling using fucose analogs, Nat. Chem. Biol 14 (1) (2018) 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Thomas M, Ge Q, Lu JJ, Chen J, Klibanov AM, Cross-linked small polyethylenimines: while still nontoxic, deliver DNA efficiently to mammalian cells in vitro and in vivo, Pharm. Res 22 (3) (2005) 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Alison VN, Mitche d.R., Kelley WM, Transcript analysis of stem cells, Functional Glycomics 2010, pp. 73–91. [Google Scholar]
- [82].Gavet O, Pines J, Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis, Dev. Cell 18 (4) (2010) 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Fuhrich DG, Lessey BA, Savaris RF, Comparison of HSCORE assessment of endometrial b3 Integrin subunit expression with digital HSCORE using computerized image analysis (ImageJ), Anal. Quantitat. Cytopathol. Histopathol 35 (4) (2013) 210–216. [PMC free article] [PubMed] [Google Scholar]
- [84].Grimsrud CD, Romano PR, D’Souza M, Puzas JE, Schwarz EM, Reynolds PR, Roiser RN, O’Keefe R,J, BMP signaling stimulates chondrocyte maturation and the expression of Indian hedgehog, J. Orthop. Res 19 (2001) 18–25. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.