

Abstract
Lipocalin 2 (LCN2) is highly expressed in several infectious and inflammatory disorders. However, the expression level and underlying mechanism of LCN2 in inflammatory bowel disease (IBD) are poorly understood. The current study used murine IBD models and LPS-activated macrophages to elucidate the role of LCN2 in IBD pathogenesis. The levels of LCN2 protein and concentration were confirmed to be much higher in the colons of colitis-induced mice compared with healthy mice using immunohistochemistry, western blotting and ELISA assay. In vitro, the level of LCN2 in RAW264.7 macrophages increased significantly following LPS stimulation and diminished markedly upon using NF-κB-specific inhibitors. Assembly of the NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome was inhibited when LCN2 expression was knocked down, as evidenced by decreased NLRP3, ASC-1 and caspase-1 activation. Furthermore, secretion and maturation of IL-1β was attenuated when LCN2 was silenced in LPS-stimulated macrophages. Together, these results suggested that LCN2 directly upregulated the NLRP3 inflammasome complex via NF-κB activation in response to stimulating macrophages with LPS, and that it acted as a pro-inflammatory regulator in macrophage activation modulated by NF-κB activation. Overall, LCN2 may serve as a promising target for the prevention and treatment of IBD.
Keywords: lipocalin 2, lipopolysaccharide, NF-ĸB, NOD- LRR- and pyrin domain-containing protein 3, IL-1β, inflammasome, inflammation, inflammatory bowel disease
Introduction
Inflammatory bowel disease (IBD), including mainly Crohn's disease (CD) and ulcerative colitis (UC), is an immune-mediated chronic and relapsing inflammatory disorder resulting from dysregulation of the mucosal immune response in the gastrointestinal track (1). The etiologies of IBD remain elusive as they involve complex interactions between genetic, environmental, and immunoregulatory factors (2). Furthermore, patients with long-standing UC are prone to colorectal cancer (CRC). Although colitis-associated CRC accounts for only 1–2% of all CRC cases, IBD with colon involvement is among the top three risk factors for CRC (3). Therefore, researchers are interested in identifying target genes for IBD to reduce the overall risk of colitis-associated CRC.
Macrophages have a central role in intestinal homeostasis, being strategically positioned in the subepithelial lamina propria to clear microbes that breach the epithelial barrier. Activated macrophages prolifically secrete inflammatory cytokines in the gut, and the imbalance of cytokines is an important part of the pathogenesis of IBD (4). Lipopolysaccharide (LPS), a vital biological ingredient of the Gram-negative bacterial cell wall, is well known to trigger inflammation due to its ability to activate macrophages (5). One major target of LPS is toll-like receptor 4, which induces the activation of nuclear factor-kappa B (NF-κB) (6). NF-κB activation promotes the phosphorylation of IκB-α, which results in translocation of the NF-κB subunit into the nucleus. Finally, the subunit binds to the promoter regions of target genes and induces the transcription of pro-inflammatory cytokines, including tumor necrosis factor-alpha (TNF-α), interleukin-1β (IL-1β), and IL-6 (7).
Lipocalins (LCNs) are a family of proteins with various functions, including regulation of immune responses, cell growth, metabolism, iron transport, and prostaglandin synthesis (8). LCN2, also known as oncogene 24p3 neutrophil gelatinase-associated lipocalin, is a 25-kDa secreted glycoprotein that was initially purified from neutrophil granules (9). LCN2 can bind hydrophobic molecules, including retinoids, fatty acids, and various steroids (10). The LCN2 promoter region contains the binding sites of several transcription factors, including NF-κB, STAT1/3, CREB, and C/EBP-β, indicating a potential role of LCN2 in regulating inflammation and metabolism (11–13).
Emerging evidence has shown that LCN2 is highly associated with inflammatory conditions, and that it plays a role in modulating macrophage activation in the adipose tissue, lungs, liver, and the brain (14–18). Moreover, the upregulation of LCN2 in IBD patients with UC has recently been reported, suggesting that LCN2 can be a sensitive biomarker for intestinal inflammation (19,20). In a previous study, we suggested that the pro-inflammatory cytokine IL-6 induces LCN2 promoter activity via NF-κB/STAT3 activation, and that increased LCN2 may serve as an indicator of CRC development in a chronic inflammation setting (21). Considering that LCN2 is associated with intestinal inflammation, IBD as well as the CRC supportive microenvironment may rely, at least in part, on the regulation of LCN2 in activated macrophages. However, the underlying mechanism of LCN2 in activated macrophages as a target for IBD has not been established. A new perspective of LCN2 under inflammatory conditions would provide greater insight into its role in the tumor microenvironment. The aim of this study was to investigate the role and molecular mechanism of LCN2 in regulating inflammation, especially inflammasome assembly, using the murine macrophage cell line RAW264.7.
Materials and methods
Chemicals and reagents
LPS and parthenolide were purchased from Sigma-Aldrich (St. Louis, MO). Bay 11-7085 was obtained from Calbiochem (St. Louis, MO). LPS was dissolved in 1× phosphate-buffered saline (PBS) to a concentration of 1 mg/ml and stored at −20°C. Parthenolide and BAY 11-7082 were dissolved in dimethylsulfoxide (Sigma-Aldrich) and stored at −20°C in the dark. Dextran sulfate sodium (DSS) was purchased from MP Biochemicals (Irvine, CA, USA).
IBD murine model
Forty specific-pathogen-free, female, six-week-old BALB/c mice were purchased from Orient Bio, Inc. (Seongnam, Korea). Mice were given ad libitum access to water and standard rodent food until they reached the desired weight (18–20 g). They were maintained on a 12/12 hour light/dark cycle under specific-pathogen-free conditions. All procedures were reviewed and approved by Jeonbuk National University Animal Care and Use Committee (Approval no: CBNU 2018-001). Following the guideline of Institutional Animal Care and Use Committee (IACUC), over 20% of body weight loss of animal model in oncology studies is defined as severe suffering for humane endpoint decisions, although other signs were not detected. Death of mice was verified through cessation of breathing and the loss of heartbeats.
Mice were randomly assigned to either the control (n=10) or the IBD group (n=12) after they were weighed. The drinking water of the IBD group was laced with 3% DSS for 7 days. Following the guideline of EU, over 20% of body weight loss of animal model in oncology studies is defined as severe suffering for humane endpoint decisions, although other signs were not detected. Following sacrifice of animals from both groups by cervical dislocation, their body weights were measured. All colons were removed from the cecum to the anus and their lengths were measured. The colons were cut open longitudinally along the main axis and washed with 1X PBS (pH 7.4). After gross examination, half of each groups' specimens were fixed in 10% neutral-buffered formalin for histological and immunohistochemical analyses. The remaining colons were used for enzyme-linked immunosorbent assay (ELISA) and western blot analyses.
Histological staining and immunohistochemistry
Paraffin-embedded samples were cut into 5-µm sections and stained with hematoxylin and eosin for light microscopic examination. The sections were immunostained with anti-LCN2/NGAL antibody (Abcam, Cambridge, UK, catalog number: ab216462, dilution: 5 µg/ml), visualized by appropriate biotin-conjugated secondary antibodies, followed by immunoperoxidase detection with HRP/DAB (ABC) Detection IHC Kit (Abcam, catalog number: ab64261). The colon sections were captured using a Leica DM750 (Wetzlar, Germany) photomicroscope.
LCN2 ELISA
Mouse LCN2 ELISA Kits (R&D Systems) were used to measure LCN2 levels in mouse colon tissue lysates, per the manufacturer's instructions. All samples were assayed in duplicate and compared with a standard curve to quantitate the expression level.
Cell culture
The murine macrophage cell line RAW264.7 was purchased from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in Dulbecco's modified Eagle's medium (Gibco®, Carlsbad, CA, USA), supplemented with 10% fetal bovine serum (Gibco®), 100 units penicillin (Invitrogen, Carlsbad, CA, USA), and 100 units streptomycin (Invitrogen, Carlsbad, CA, USA), in a humidified 5% CO2 environment at 37°C.
RNA isolation and reverse transcription-PCR (RT-PCR)
Total RNA was isolated from cultured cells using TRIzol reagent (Invitrogen, Eugene, OR) and cDNA was synthesized using SuperScript II Reverse Transcriptase (Invitrogen), according to the manufacturer's protocol. The expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. The following primer sequences were used: mouse LCN2, 5′-GGACCAGGGCTGTCGCTACT-3′ (forward) and 5′-GGTGGCCACTTGCACATTGT-3′ (reverse), GAPDH, 5′-ACCACAGTCCATGCCATCAC-3′ (forward) 5′-TCCACCACCCTGTTGCTGTA-3′ (reverse), generating a 120- and 452-bp product, respectively. After initial denaturation at 95°C for 1 min, PCR was performed for various cycles (30 sec at 94°C, 1 min at annealing temperature, and 2 min at 72°C) using Taq polymerase. Reaction products (10 µl) were separated on 2% agarose gels and stained with RedSafe™ (Intron, Daejeon, Korea). DNA band intensity was analyzed by densitometry using an NαBI imager (Neogene Science, Suwon, Korea).
Small interfering RNA (siRNA) for inhibiting specific gene expression
siRNA, used for targeted silencing of the mouse LCN2 (NCBI Ref Seq NM_008491) gene, was purchased from Santa Cruz Biotechnology (catalog number: sc-60044, CA, USA). Specific siRNAs and scrambled siRNA (for negative controls, catalog number: AM4637, Invitrogen, TX, USA) were transfected into RAW264.7 cells using TransIT-X2® transfection reagent (Mirus Bio, WI, USA). Briefly, Opti-MEM I Reduced-Serum Medium, transfection reagent and 30 nM of siRNA were gently mixed and incubated at room temperature for 30 min for complexes to form. The complexes were added drop-wise to the cell culture plates and the cells were incubated in 37°C incubator for 72 h. Then the cells were used further experiments.
Protein extraction
Cells were harvested and resolved in a lysis buffer (20 mM Tris-HCl (pH 7.5), 137 mM NaCl, 10% glycerol (v/v), 1% Triton X-100 (v/v), 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, and protease inhibitor cocktail). After centrifugation at 16,000 × g for 15 min, the supernatants were used as cytoplasmic extracts. To extract the nuclear fraction, cells were resuspended in 150 µl of buffer A [10 mM HEPES (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 0.4% Nonidet P-40 (v/v), and protease inhibitor cocktail], incubated for 20 min on ice, and centrifuged at 2,300 × g for 5 min. The resulting pellets were resolved in 100 µl of buffer C [20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (pH 7.9), 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM ethylenediamine tetraacetic acid, 0.5 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, and protease inhibitor cocktail], incubated for 30 min on ice, and centrifuged at 16,000 × g for 15 min. The resulting supernatants were used as nuclear extracts.
For whole cell extracts, the cells were resolved by RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, and protease inhibitors) and centrifuged at 13,200 rpm at 4°C for 30 min. The resulting supernatants were used as whole cell extracts.
Western blotting
After extraction, the protein concentration was measured using a Bradford Reagent (Sigma-Aldrich). Conditioned media (100 µl) was collected and concentrated using a speed vacuum concentrator (Thermo Scientific™, MA, USA). Fifty microgram of cell protein and concentrated media were loaded onto 10~13.5% of sodium dodecyl sulfate-polyacrylamide gels. After transferring and blocking, each polyvinylidene difluoride membrane (PVDF, 0.2 µm, Bio-rad, USA) was probed with various antibodies [anti-LCN2 (R&D systems, catalog number: AF1757, 1:2,000) anti-p-IκB-α (Santa Cruz Biotechnology, catalog number: SC-8404, 1:1,000), anti-p65 (Santa Cruz Biotechnology, catalog number: SC-8008, 1:1,000), anti-NLRP3 (Adipoge, catalog number; AG-20B-0014, 1:2,000), anti-ASC (GeneTex, catalog number: GTX105780, 1:2,000), anti-caspase-1 p20 (Adepogen, catalog number: AG-20B-0042, 1:2,000), anti-IL-1β (R&D systems, catalog number: AF-401-NA, 1:2,000), anti-GAPDH (GeneTex, catalog number: GTX100118, 1:2,000) and anti-Lamin B (Santa Cruz Biotechnology, catalog number: SC-6216, 1:1,000)]. Antibody-antigen binding was detected using enhanced ECL prime (GE Healthcare, NJ, USA), captured by FUSION FX Image Analyzer (VILBER Lourmat, France) and analyzed by Evolution-Capt software (VILBER Lourmat).
Inflammatory cytokine detection
RAW264.7 cells were stimulated with LPS and then transfected with mouse LCN2 siRNA or scrambled siRNA. After the indicated time, concentrations of the inflammatory cytokines TNF-α (catalog number: 88-7324), IL-18 (catalog number: BMS618-3), and IL-1β (catalog number 88-7013) in the conditioned media were determined using corresponding commercial ELISA kits (Invitrogen).
Statistical analyses
The data are presented as the mean ± standard error of at least three independent experiments performed in duplicate. Microsoft Excel 5.0, and Graphpad Prism 5.0 was used to perform two-tailed t-tests for in vivo experiments, and analysis of variance (ANOVA) for in vitro ELISA assay. The differences between groups were compared by the analysis of variance, followed by post hoc Tukey's test to correct for multiple comparisons. A P-value <0.05 was considered statistically significant.
Results
LCN2 levels are highly elevated in DSS-induced colitis models
To assess whether the expression of LCN2 in the colon differs between the control and the IBD groups, we analyzed their pathological symptoms and colonic LCN2 levels. The severity of colonic inflammation is strongly correlated with body weight loss. Compared with the control group, mice with DSS-induced colitis exhibited significant weight loss by ~15.05±3.49% (Fig. 1A left graph). Next, the entire colons were collected from these mice to examine the gross morphological damage. The colonic mucosal lesions in the colitis group were obvious, with a wide range of hyperemia, edema, and bleeding, compared with those in the control group (Fig. 1A). The colons in the colitis group were considerably shorter than those in the control group (Fig. 1A right graph). Furthermore, histological evaluation showed that colonic tissues from the colitis group were characterized by epithelial cell destruction, crypt deformation, ulceration, and inflammatory cells infiltrating the lamina propria and submucosa (Fig. 1B).
Figure 1.

Expression of LCN2 is specifically induced in the colon tissue of DSS-induced colitis mice. (A) Macroscopic appearance of inflammation in the colon (upper panel). Body weight and colon length were statistically analyzed (lower panel). (B) Images of hematoxylin and eosin staining (magnification, ×10) of colonic mucosal tissue sections from mice were captured using microscopy. (C) Total tissue extracts were prepared after animal sacrifice and analyzed for LCN2 and actin antibody by western blotting. Actin was used as a loading control. (D) After homogenizing colonic tissues, level of LCN2 in tissues was detected using ELISA. (E) IHC images of LCN2 expression in the colonic tissues from control and colitis mice show positively bound cells in brown (magnification, ×20). **P<0.01, ***P<0.001. LCN2, lipocalin 2; DSS, dextran sulfate sodium; IHC, immunohistochemical.
Next, we examined the LCN2 protein levels in colon tissues using western blotting (Fig. 1C). LCN2 levels were higher in the colon tissues from the colitis group than in those from the control group (Fig. 1C). The ELISA results also indicated that LCN2 levels in mice with colitis (10.35±1.821 pg/µg of protein) were 12-fold higher than in healthy mice (0.699±0.673 pg/µg of protein; Fig. 1D). Finally, immunohistochemistry also revealed more LCN2-positive cells in the colitis group than in the control group, in line with the western blotting and ELISA results (Fig. 1E).
LCN2 is specifically stimulated by LPS in murine macrophages
To investigate whether macrophages are one of the main cellular targets for LCN2 regulation in IBD, we analyzed the gene and protein expression of LCN2 in RAW264.7 macrophages after stimulating them with LPS. LPS strongly induced LCN2 mRNA and protein levels in a dose-dependent manner in RAW264.7 cells (Fig. 2A). Moreover, stimulation for 24 h with the lowest concentration of LPS led to increased LCN2 release in the conditioned media of RAW264.7 cells.
Figure 2.

LPS specifically promotes the expression and secretion of LCN2 in macrophages. (A and B) After stimulation with varying (A) concentrations or (B) periods of LPS for the indicated time periods, RAW264.7 cells were isolated, and the conditioned media was collected. Western blotting and RT-PCR were performed. The top images are from the RT-PCR, and the lower images are western blots. GAPDH was used as a loading control. LPS, lipopolysaccharide; LCN2, lipocalin 2; RT-PCR, reverse transcription-PCR.
To confirm the time-dependency of this effect, RAW264.7 cells were stimulated with LPS for the indicated time periods and the expression and secretion of LCN2 were measured (Fig. 2B). We found that LPS induced the intracellular expression and secretion of LCN2 in a time-dependent manner. These results indicate that the inflammatory mediator LPS strongly induces LCN2 gene transcription, translation, and secretion in macrophages.
NF-ĸB activation is required for LPS-induced LCN2 expression
Exposing macrophages to LPS activates the transcription factor NF-κB, which regulates the transcription of various inflammation-responsive genes (22–24). After determining that 50 ng/ml of LPS maximally induced LCN2 in macrophages, we first assessed the degradation of IĸB-α and the nuclear translocation of NF-ĸB in RAW264.7 cells in response to LPS stimulation for 1–24 h. LPS-induced LCN2 decreased the expression of IĸB-α and p65 in the cytosolic fraction between 1~6 h (Fig. 3A). In contrast, the expression of p65 in the nuclear fraction was the greatest at the 3 h time point and decreased slightly thereafter.
Figure 3.

LPS-induced LCN2 is dependent on NF-κB activation in macrophages. (A) After stimulation with 50 ng/ml LPS for the indicated time, cytosolic and nuclear extracts of RAW264.7 cells were prepared and used to determine activation of the NF-ĸB pathway. Actin and lamin B were used as loading controls. (B) Cells were pretreated with PT and BAY 11-7082 (NF-κB-specific inhibitor), and the cells were stimulated with LPS for 24 h. Then, the levels of LCN2 mRNA and protein were detected using RT-PCR (upper panel) and western blot analysis (lower panel). GAPDH was used as a loading control. LPS, lipopolysaccharide; LCN2, lipocalin 2; PT, parthenolide; RT-PCR, reverse transcription-PCR; IκB-α, NF-kappa-B inhibitor α.
Next, we examined whether the expression of LCN2 depended upon NF-κB activation. Pretreatment with the NF-ĸB-specific inhibitors BAY 11-7082 and parthenolide suppressed the expression of LCN2 in LPS-stimulated RAW264.7 cells (Fig. 3B), suggesting that NF-ĸB activation is required for LCN2 expression in LPS-stimulated macrophages.
LCN2 regulates activation of the NLRP3 inflammasome in LPS-stimulated murine macrophages
The well-characterized NLRP3 (NOD-, LRR-, and pyrin domain-containing protein 3) inflammasome is a multiprotein complex that orchestrates an inflammatory response, which contributes to IBD through increased epithelial permeability and a detrimental immune response against invading commensal bacteria in the gut (25). NF-κB is a central mediator of the priming signal that activates the NLRP3 inflammasome (26). Thus, we investigated whether the NLRP3 inflammasome was influenced by LCN2 in LPS-stimulated RAW264.7 cells (Fig. 4). After scrambled siRNA transfection and LPS stimulation, the protein levels of NLRP3, ASC, and cleaved Caspase 1 were strikingly augmented. Interestingly, the expression of these inflammasome-related proteins was dramatically attenuated when siRNA targeting LCN2 was used instead of the scrambled siRNA, suggesting that LCN2 regulates the activation of the NLRP3 inflammasome in LPS-stimulated macrophages.
Figure 4.
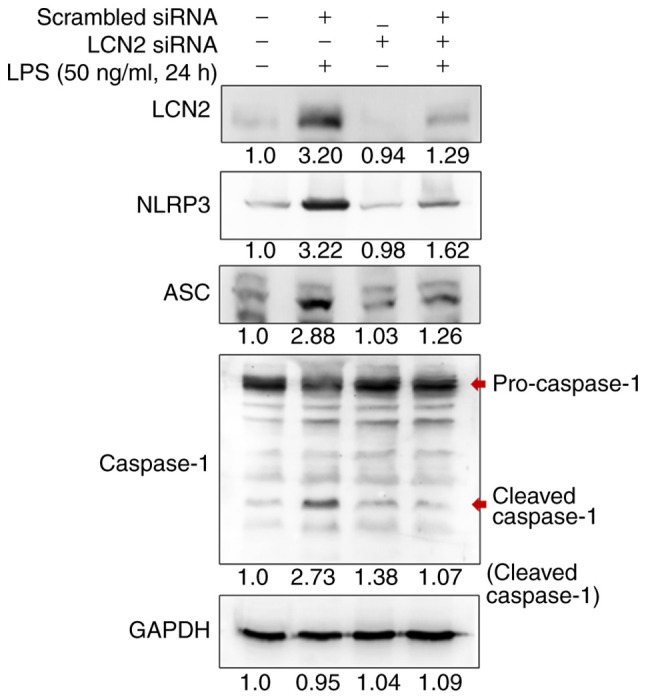
LCN2 is required for NLRP3 inflammasome activation in macrophages. After being transfected with scrambled or LCN2 siRNA, LPS-stimulated cells were isolated for western blotting. The protein levels of NLRP3, ASC and cleaved Caspase 1 were detected as indicators of the status of inflammasome activation. GAPDH was used as a loading control. LCN2, lipocalin 2; NLRP3, NOD-, LRR-, and pyrin domain-containing protein 3; siRNA, small interfering RNA; LPS, lipopolysaccharide; ASC, apoptosis-associated speck-like protein containing a CARD.
LCN2 is responsible for the production of IL-1β in LPS-stimulated murine macrophages
The activated NLRP3 inflammasome serves as a platform for the activation of caspase-1, which induces maturation of the inflammatory cytokines IL-1β and IL-18 (27). Thus, we first measured the concentrations of these two cytokines in media from RAW264.7 cell cultures stimulated with LPS for 6, 24, and 48 h. After 48 h of LPS stimulation, the concentration of IL-1β was markedly increased in RAW264.7 cells, which was prominently suppressed by LCN2 silencing (Fig. 5A). However, LCN2 silencing did not affect the LPS-induced IL-18 levels.
Figure 5.

LPS-induced LCN2 leads to production and secretion of the pro-inflammatory cytokine IL-1β in macrophages. (A) After being transfected with scrambled or LCN2 siRNA, RAW264.7 cells were stimulated by LPS for the indicated time periods. Then, IL-1β, TNF-α and IL-18 were quantified in conditioned media by ELISA. (B) After being transfected with scrambled or LCN2 siRNA, LPS-stimulated cells were isolated for western blotting to detect the levels of mature IL-1β. GAPDH was used as a loading control. ***P<0.001. LPS, lipopolysaccharide; LCN2, lipocalin 2; siRNA, small interfering RNA; scr, scrambled siRNA; siLCN2, LCN2 siRNA.
We confirmed the above observation by measuring the intracellular IL-1β protein levels by western blotting. LPS dramatically induced the matured form of the IL-1β protein, which was diminished when LCN2 was silenced in RAW264.7 cells (Fig. 5B). These findings indicate that LCN2 is involved in the maturation and secretion of the pro-inflammatory cytokine IL-1β in LPS-stimulated macrophages.
Discussion
The pathogenesis of IBD is unknown, but the uncontrolled immune response of genetically predisposed individuals to environmental factors is considered to be the cause (28). In the present study, we demonstrated that LCN2 in activated macrophages is a powerful mediator of the NLRP3 inflammasome via NF-ĸB signaling and is eventually responsible for secretion of the pro-inflammatory cytokine IL-1β. Based on our results, we propose a new molecular mechanism explaining the pro-inflammatory role of LCN2 in activated macrophages (Fig. 6), thereby, increasing our understanding of IBD.
Figure 6.

Schematic representation of the proposed mechanisms of LCN2 in macrophages activated by LPS. LPS, lipopolysaccharide; LCN2, lipocalin 2; TLR, Toll-like receptor; ASC, apoptosis-associated speck-like protein containing a CARD; NLRP3, NOD-, LRR-, and pyrin domain-containing protein 3; IκB-α, NF-kappa-B inhibitor α.
Interestingly, LCN2 in IBD may serve as a biomarker to distinguish active from inactive inflammation. In 2011, Oikonomou et al suggested that serum LCN2 levels are particularly elevated in patients with active IBD, correlating with established markers of inflammation and disease activity (29). Moreover, Østvik et al showed that LCN2 staining of colonic biopsies was significantly increased in cases of active UC and CD compared with inactive UC and CD (30). Our in vivo experiments also demonstrate that LCN2 levels were augmented in colonic tissues from DSS-induced animal models. These findings suggest that LCN2 is involved in the pathophysiology of IBD, and is associated with activity and severity of the disease.
When the intestine is invaded by pathogens that can cross the damaged epithelial cell barrier, the intrinsic defense cells in the epithelium, especially macrophages, produce and secrete pro-inflammatory cytokines (31–33). LPS, a component of the microbial pathogens, can directly activate macrophages and trigger inflammation (34). Hence, attenuating the LPS-induced inflammatory response in macrophages is considered an attractive therapeutic strategy for numerous acute and chronic inflammatory diseases. Emerging evidence has shown that LCN2 plays a role in regulating LPS-stimulated macrophages. Guo et al and Du et al showed that LCN2 was an essential factor in the inflammatory response of macrophages to LPS (35,36). Likewise, our results (Fig. 2) confirm that LPS strongly induces LCN2 expression in macrophages.
The transcription factor NF-ĸB, induced in macrophages by LPS, controls the expression of numerous target genes, such as inducible nitric oxide synthase, cyclooxygenase-2, TNF-α, IL-1β, IL-18, and IL-6, all of which play important roles in immune and inflammatory responses (37). Activated NF-κB has been detected in lamina propria macrophages, epithelial cells from biopsy specimens, and cultured cells from IBD patients (38). Therefore, NF-κB is a potential target for IBD therapy, and nonsteroidal anti-inflammatory drugs, such as 5-amino salicylic acid, inhibit the NF-ĸB signaling pathway (39). Although substantial evidence exists for the involvement of NF-κB in IBD, further investigation is needed to elucidate the underlying mechanisms and the precise role of NF-κB-related molecules in IBD pathogenesis. In the present study, we suggest that LPS-induced upregulation of LCN2 strongly depends upon NF-κB activation, and LCN2 mediates the NF-κB-induced NLRP3 inflammasome complex.
LCN2 is widely known to play various biological roles; however, its specific role in different inflammatory conditions is inconsistent. For example, LCN2 was reported to play a pro-inflammatory role in autoimmune encephalomyelitis models of neuroinflammation (40). Moreover, it was shown to exert both pro-inflammatory and iron-sequestering effects along the respiratory mucosa in response to bacterial enterobactin (41). In contrast, Du et al used LCN2 knockout mice to show that it alleviates LPS-induced gut inflammation (36). Guo Guo et al also reported that LCN2 plays an anti-inflammatory role in macrophage polarization, potentially by modulating the activation of the NF-κB-STAT3 loop (35). Our results indicate that LPS-induced LCN2 in macrophages drives severe inflammation by enhancing inflammasome assembly and IL-1β secretion. Through our present study, we demonstrate the critical role of LCN2 in IBD pathogenesis.
The NLRP3 inflammasome, a cytosolic multiprotein complex, is well known to be involved in the development and progression of numerous inflammatory disorders (42). Once activated by stimuli such as LPS, NLRP3 recruits the adaptor protein ASC to promote recruitment and activation of caspase-1, resulting in the production and maturation of inflammatory cytokines (43). NF-κB is a central mediator of the priming signal of NLRP3 inflammasome activation and induces the transcriptional expression of NLRP3 (44). The function of LCN2 in NF-κB-mediated activation of the NLRP3 inflammasome was investigated in experimental models of heart failure in 2017. Song E. and colleagues reported that LCN2 activates the NLRP3 inflammasome by inducing the release of the high-mobility group box 1 protein in primary cardiac fibroblasts from neonatal rats (45). Consistent with these findings, we found that LCN2 regulates LPS-induced NLRP3 inflammasome assembly in macrophages, which contributes to IBD pathogenesis.
The NLRP3 inflammasome is critical for the processing and maturation of IL-1β and is rapidly emerging as a crucial regulator of intestinal homeostasis (46,47). Activated NF-κB transcriptionally induces the expression of the 35-kDa pro-IL-1β, but the maturation and secretion of 17-kDa IL-1β requires the activation of the NLRP3 inflammasome (48,49). Elevated levels of IL-1β were found in colonic biopsies and isolated myeloid cells, correlating with the severity of IBD (50). In the present study, we observed that LCN2 silencing strongly repressed IL-1β maturation and secretion in LPS-stimulated macrophages, indicating that LCN2 regulates IL-1β production and maturation via NF-κB/NLRP3 inflammasome signaling in macrophages.
In addition, IL-1β, produced mainly by the activated NLRP3 inflammasome, can promote tumorigenesis in cancer cells (51). Several publications have demonstrated that IL-1β-specific upregulation of LCN2 is controlled by the activation of NF-κB in cancer cells. Cowland et al reported that stimulating lung cancer cells with IL-1β induced NF-κB to bind to the LCN2 promoter and upregulate its expression (52). In 2006, they also reported that specific induction of LCN2 by IL-1β in lung adenocarcinoma cells critically depends on activation of the NF-κB cofactor IκB-ζ (53). Collectively, this evidence suggests that upregulation of LCN2 in macrophages results in the production and secretion of IL-1β to the outer membrane of macrophages, where it subsequently induces NF-κB-mediated LCN2 expression in cancer cells, leading to cancer development and carcinogenesis. Therefore, the positive feedback between LCN2 and IL-1β most likely contributes to the pathogenesis of inflammation-driven cancer.
IL-18, an IL-1 family member, has pleiotropic functions and is also chronically elevated in several inflammatory diseases (54). Like IL-1β, the production and release of IL-18 also depends on inflammasome activation. Interestingly, IL-1β is only induced in response to inflammatory stimuli, but IL-18 is constitutively expressed. In other words, a cellular repository of IL-18 already exists even before an inflammatory stimulus, and is ready to be activated and released by the inflammasome (55,56). However, how different inflammatory signals regulate the expression of IL-18 and IL-1β remains poorly studied. In 2017, Zhu et al reported that type I interferon (IFN) signaling is required for the induction of IL-18, but not IL-1β, which points to a critical and differential role for type I IFN in regulating IL-18 signaling (57). A correlation between LCN2 and IL-18 has not directly been demonstrated; however, Østvik et al showed that LCN2 in intestinal epithelial cells is not affected by IFN-γ stimulation (30). In the present study, we observed that LCN2 modulated the levels of IL-1β but did not affect LPS-induced IL-18. Collectively, LCN2 is mostly irrelevant to IFN-dependent IL-18 in macrophages. However, further study is necessary to understand the interactions between LCN2 and IFN-dependent IL-18 in macrophages and IBD pathogenesis.
In conclusion, our results show that LCN2 directly upregulates the NLRP3 inflammasome complex by NF-κB activation in response to stimulation of macrophages by LPS, consequently leading to the production and maturation of IL-1β. We have presented novel findings on the role and mechanism of LCN2 in IBD pathophysiology. Moreover, this study clearly highlights the role of LCN2 as a potential target for the NLRP3 inflammasome in the field of IBD therapy.
Acknowledgements
Not applicable.
Funding Statement
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT; grant no. 2020R1C1C1012108) and the Fund of the Biomedical Research Institute of Jeonbuk National University Hospital (grant no. CUH2021-0041) in Korea.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors' contributions
SWK participated in the design of the study. SLK and MWS carried out the in vitro and in vivo experiments. SWK and SLK confirm the authenticity of all the raw data. SLK wrote the draft and performed the statistical analysis. SWK edited the draft and supervised all experimental procedures. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
All procedures of animal experiments were reviewed and approved by Jeonbuk National University Animal Care and Use Committee (approval no. CBNU 2018-001). Animal experiments were performed in strict compliance with European guidelines and regulations on protection of animals used for scientific purposes (EC Directive 2010/63/EU).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sartor RB. Mechanisms of disease: Pathogenesis of Crohn's disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol. 2006;3:390–407. doi: 10.1038/ncpgasthep0528. [DOI] [PubMed] [Google Scholar]
- 3.Lennard-Jones JE, Morson BC, Ritchie JK, Williams CB. Cancer surveillance in ulcerative colitis. Experience over 15 years. Lancet. 1983;2:149–152. doi: 10.1016/S0140-6736(83)90129-0. [DOI] [PubMed] [Google Scholar]
- 4.Sanchez-Munoz F, Dominguez-Lopez A, Yamamoto-Furusho JK. Role of cytokines in inflammatory bowel disease. World J Gastroenterol. 2008;14:4280–4288. doi: 10.3748/wjg.14.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gioannini TL, Weiss JP. Regulation of interactions of Gram-negative bacterial endotoxins with mammalian cells. Immunol Res. 2007;39:249–260. doi: 10.1007/s12026-007-0069-0. [DOI] [PubMed] [Google Scholar]
- 6.Krappmann D, Wegener E, Sunami Y, Esen M, Thiel A, Mordmuller B, Scheidereit C. The IkappaB kinase complex and NF-kappaB act as master regulators of lipopolysaccharide-induced gene expression and control subordinate activation of AP-1. Mol Cell Biol. 2004;24:6488–6500. doi: 10.1128/MCB.24.14.6488-6500.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kueanjinda P, Roytrakul S, Palaga T. A novel role of numb as a regulator of pro-inflammatory cytokine production in macrophages in response to toll-like receptor 4 (vol 5, 12784, 2015) Sci Rep. 2015;5:12784. doi: 10.1038/srep12784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tong ZM, Wu XL, Ovcharenko D, Zhu JX, Chen CS, Kehrer JP. Neutrophil gelatinase-associated lipocalin as a survival factor. Biochem J. 2005;391:441–448. doi: 10.1042/BJ20051020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kjeldsen L, Johnsen AH, Sengelov H, Borregaard N. Isolation and primary structure of ngal, a novel protein associated with human neutrophil gelatinase. J Biol Chem. 1993;268:10425–10432. doi: 10.1016/S0021-9258(18)82217-7. [DOI] [PubMed] [Google Scholar]
- 10.Akerstrom B, Flower DR, Salier JP. Lipocalins: Unity in diversity. Biochim Biophys Acta. 2000;1482:1–8. doi: 10.1016/S0167-4838(00)00137-0. [DOI] [PubMed] [Google Scholar]
- 11.Shen F, Hu Z, Goswami J, Gaffen SL. Identification of common transcriptional regulatory elements in interleukin-17 target genes. J Biol Chem. 2006;281:24138–24148. doi: 10.1074/jbc.M604597200. [DOI] [PubMed] [Google Scholar]
- 12.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat Rev Immunol. 2011;11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- 13.Zhao P, Stephens JM. STAT1, NF-κB and ERKs play a role in the induction of lipocalin-2 expression in adipocytes. Mol Metab. 2013;2:161–170. doi: 10.1016/j.molmet.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo H, Jin D, Chen X. Lipocalin 2 is a regulator of macrophage polarization and NF-κB/STAT3 pathway activation. Mol Endocrinol. 2014;28:1616–1628. doi: 10.1210/me.2014-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warszawska JM, Gawish R, Sharif O, Sigel S, Doninger B, Lakovits K, Mesteri I, Nairz M, Boon L, Spiel A, et al. Lipocalin 2 deactivates macrophages and worsens pneumococcal pneumonia outcomes. J Clin Invest. 2013;123:3363–3372. doi: 10.1172/JCI67911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borkham-Kamphorst E, de Leur EV, Zimmermann HW, Karlmark KR, Tihaa L, Haas U, Tacke F, Berger T, Mak TW, Weiskirchen R. Protective effects of lipocalin-2 (LCN2) in acute liver injury suggest a novel function in liver homeostasis. Biochim Biophys Acta. 2013;1832:660–673. doi: 10.1016/j.bbadis.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 17.Srinivasan G, Aitken JD, Zhang BY, Carvalho FA, Chassaing B, Shashidharamurthy R, Borregaard N, Jones DP, Gewirtz AT, Vijay-Kumar M. Lipocalin 2 deficiency dysregulates iron homeostasis and exacerbates endotoxin-induced sepsis. J Immunol. 2012;189:1911–1919. doi: 10.4049/jimmunol.1200892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang YY, Foncea R, Deis JA, Guo H, Bernlohr DA, Chen XL. Lipocalin 2 expression and secretion is highly regulated by metabolic stress, cytokines, and nutrients in adipocytes. Plos One. 2014;9:e96997. doi: 10.1371/journal.pone.0096997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chassaing B, Srinivasan G, Delgado MA, Young AN, Gewirtz AT, Vijay-Kumar M. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. Plos One. 2012;7:e44328. doi: 10.1371/journal.pone.0044328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nielsen OH, Gionchetti P, Ainsworth M, Vainer B, Campieri M, Borregaard N, Kjeldsen L. Rectal dialysate and fecal concentrations of neutrophil gelatinase-associated lipocalin, interleukin-8, and tumor necrosis factor-alpha in ulcerative colitis. Am J Gastroenterol. 1999;94:2923–2928. doi: 10.1016/S0002-9270(99)00495-5. [DOI] [PubMed] [Google Scholar]
- 21.Kim SL, Shin MW, Seo SY, Kim SW. Lipocalin 2 potentially contributes to tumorigenesis from colitis via IL-6/STAT3/NF-κB signaling pathway. Biosci Rep. 2022;42:BSR20212418. doi: 10.1042/BSR20212418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Dea E, Hoffmann A. NF-κB signaling. Wiley Interdiscip Rev Syst Biol Med. 2009;1:107–115. doi: 10.1002/wsbm.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharif O, Bolshakov VN, Raines S, Newham P, Perkins ND. Transcriptional profiling of the LPS induced NF-kappaB response in macrophages. BMC Immunol. 2007;8:1. doi: 10.1186/1471-2172-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allen IC, TeKippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB, Herfarth HH, Jobin C, Ting JPY. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med. 2010;207:1045–1056. doi: 10.1084/jem.20100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023. doi: 10.1038/sigtrans.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19:477–489. doi: 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han X, Ding S, Jiang H, Liu G. Roles of macrophages in the development and treatment of gut inflammation. Front Cell Dev Biol. 2021;9:625423. doi: 10.3389/fcell.2021.625423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oikonomou KA, Kapsoritakis AN, Theodoridou C, Karangelis D, Germenis A, Stefanidis I, Potamianos SP. Neutrophil gelatinase-associated lipocalin (NGAL) in inflammatory bowel disease: Association with pathophysiology of inflammation, established markers, and disease activity. J Gastroenterol. 2012;47:519–530. doi: 10.1007/s00535-011-0516-5. [DOI] [PubMed] [Google Scholar]
- 30.Ostvik AE, Granlund AV, Torp SH, Flatberg A, Beisvag V, Waldum HL, Flo TH, Espevik T, Damås JK, Sandvik AK. Expression of toll-like receptor-3 is enhanced in active inflammatory bowel disease and mediates the excessive release of lipocalin 2. Clin Exp Immunol. 2013;173:502–511. doi: 10.1111/cei.12136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baumgart DC, Carding SR. Inflammatory bowel disease: Cause and immunobiology. Lancet. 2007;369:1627–1640. doi: 10.1016/S0140-6736(07)60750-8. [DOI] [PubMed] [Google Scholar]
- 32.Geremia A, Biancheri P, Allan P, Corazza GR, Di Sabatino A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun Rev. 2014;13:3–10. doi: 10.1016/j.autrev.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 33.Geremia A, Arancibia-Carcamo CV. Innate lymphoid cells in intestinal inflammation. Front Immunol. 2017;8:1296. doi: 10.3389/fimmu.2017.01296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.West MA, Seatter SC, Bellingham J, Clair L. Mechanisms of reprogrammed macrophage endotoxin signal transduction after lipopolysaccharide pretreatment. Surgery. 1995;118:220–228. doi: 10.1016/S0039-6060(05)80327-7. [DOI] [PubMed] [Google Scholar]
- 35.Guo H, Jin D, Chen X. Lipocalin 2 is a regulator of macrophage polarization and NF-κB/STAT3 pathway activation. Mol Endocrinol. 2014;28:1616–1628. doi: 10.1210/me.2014-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Du H, Liang L, Li J, Xiong Q, Yu X, Yu H. Lipocalin-2 Alleviates LPS-induced inflammation through alteration of macrophage properties. J Inflamm Res. 2021;14:4189–4203. doi: 10.2147/JIR.S328916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 38.Rogler G, Brand K, Vogl D, Page S, Hofmeister R, Andus T, Knuechel R, Baeuerle PA, Schölmerich J, Gross V. Nuclear factor kappaB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology. 1998;115:357–369. doi: 10.1016/S0016-5085(98)70202-1. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Z, Liu N, Chen X, Zhang F, Kong T, Tang X, Yang Q, Chen W, Xiong X, Chen X. UCHL1 regulates inflammation via MAPK and NF-κB pathways in LPS-activated macrophages. Cell Biol Int. 2021;45:2107–2117. doi: 10.1002/cbin.11662. [DOI] [PubMed] [Google Scholar]
- 40.Nam Y, Kim JH, Seo M, Kim JH, Jin M, Jeon S, Seo JW, Lee WH, Bing SJ, Jee Y, et al. Lipocalin-2 protein deficiency ameliorates experimental autoimmune encephalomyelitis: The pathogenic role of lipocalin-2 in the central nervous system and peripheral lymphoid tissues. J Biol Chem. 2014;289:16773–16789. doi: 10.1074/jbc.M113.542282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bachman MA, Miller VL, Weiser JN. Mucosal lipocalin 2 has pro-inflammatory and iron-sequestering effects in response to bacterial enterobactin. PLoS Pathog. 2009;5:e1000622. doi: 10.1371/journal.ppat.1000622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz AJ, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248–255. doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abderrazak A, Syrovets T, Couchie D, El Hadri K, Friguet B, Simmet T, Rouis M. NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015;4:296–307. doi: 10.1016/j.redox.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sutterwala FS, Haasken S, Cassel SL. Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci. 2014;1319:82–95. doi: 10.1111/nyas.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song E, Jahng JW, Chong LP, Sung HK, Han M, Luo C, Wu D, Boo S, Hinz B, Cooper MA, et al. Lipocalin-2 induces NLRP3 inflammasome activation via HMGB1 induced TLR4 signaling in heart tissue of mice under pressure overload challenge. Am J Transl Res. 2017;9:2723–2735. [PMC free article] [PubMed] [Google Scholar]
- 46.Zaki MH, Lamkanfi M, Kanneganti TD. The Nlrp3 inflammasome: Contributions to intestinal homeostasis. Trends Immunol. 2011;32:171–179. doi: 10.1016/j.it.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haneklaus M, Gerlic M, O'Neill LA, Masters SL. MiR-223: Infection, inflammation and cancer. J Intern Med. 2013;274:215–226. doi: 10.1111/joim.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mariathasan S, Monack DM. Inflammasome adaptors and sensors: Intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- 49.Martinon F, Burns K, Tschopp J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426. doi: 10.1016/S1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 50.Reinecker HC, Steffen M, Witthoeft T, Pflueger I, Schreiber S, MacDermott RP, Raedler A. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn's disease. Clin Exp Immunol. 1993;94:174–181. doi: 10.1111/j.1365-2249.1993.tb05997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klampfer L. Cytokines, inflammation and colon cancer. Curr Cancer Drug Targets. 2011;11:451–464. doi: 10.2174/156800911795538066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cowland JB, Sorensen OE, Sehested M, Borregaard N. Neutrophil gelatinase-associated lipocalin is up-regulated in human epithelial cells by IL-1 beta, but not by TNF-alpha. J Immunol. 2003;171:6630–6639. doi: 10.4049/jimmunol.171.12.6630. [DOI] [PubMed] [Google Scholar]
- 53.Cowland JB, Muta T, Borregaard N. IL-1beta-specific up-regulation of neutrophil gelatinase-associated lipocalin is controlled by IkappaB-zeta. J Immunol. 2006;176:5559–5566. doi: 10.4049/jimmunol.176.9.5559. [DOI] [PubMed] [Google Scholar]
- 54.Canna SW, Girard C, Malle L, de Jesus A, Romberg N, Kelsen J, Surrey LF, Russo P, Sleight A, Schiffrin E, et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J Allergy Clin Immunol. 2017;139:1698–16701. doi: 10.1016/j.jaci.2016.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: Back to the future. Immunity. 2013;39:1003–1018. doi: 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sims JE, Smith DE. The IL-1 family: Regulators of immunity. Nat Rev Immunol. 2010;10:89–102. doi: 10.1038/nri2691. [DOI] [PubMed] [Google Scholar]
- 57.Zhu Q, Kanneganti TD. Cutting edge: Distinct regulatory mechanisms control proinflammatory cytokines IL-18 and IL-1β. J Immunol. 2017;198:4210–4215. doi: 10.4049/jimmunol.1700352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.