

Abstract
Ameloblastoma (AB) is an odontogenic tumor that arises from ameloblast-lineage cells. Although relatively uncommon and rarely metastatic, AB tumors are locally invasive and destructive to the jawbone and surrounding structures. Standard-of-care surgical resection often leads to disfigurement, and many tumors will locally recur, necessitating increasingly challenging surgeries. Recent genomic studies of AB have uncovered oncogenic driver mutations, including in the mitogen-activated protein kinase (MAPK) and Hedgehog signaling pathways. Medical therapies targeting those drivers would be a highly desirable alternative or addition to surgery; however, a paucity of existing AB cell lines has stymied clinical translation. To bridge this gap, here we report the establishment of 6 new AB cell lines—generated by “conditional reprogramming”—and their genomic characterization that reveals driver mutations in FGFR2, KRAS, NRAS, BRAF, PIK3CA, and SMO. Furthermore, in proof-of-principle studies, we use the new cell lines to investigate AB oncogene dependency and drug sensitivity. Among our findings, AB cells with KRAS or NRAS mutation (MAPK pathway) are exquisitely sensitive to MEK inhibition, which propels ameloblast differentiation. AB cells with activating SMO-L412F mutation (Hedgehog pathway) are insensitive to vismodegib; however, a distinct small-molecule SMO inhibitor, BMS-833923, significantly reduces both downstream Hedgehog signaling and tumor cell viability. The novel cell line resource enables preclinical studies and promises to speed the translation of new molecularly targeted therapies for the management of ameloblastoma and related odontogenic neoplasms.
Keywords: odontogenic tumors, biological models, tumor cell line, oncogene addiction, smoothened receptor, drug evaluation
Introduction
Ameloblastoma (AB) is an odontogenic tumor thought to originate from remnant epithelium of the enamel organ (Effiom et al. 2018). While uncommon, AB is nevertheless impactful because the tumors are locally invasive and highly destructive of critical surrounding structures such as the orbit and skull (McClary et al. 2016). Conventional AB treatment involves radical surgical resection of the tumor and infiltrated structures, which is often disfiguring. Moreover, 10% to 20% of resected tumors recur locally, where subsequent surgeries are more challenging (McClary et al. 2016). New molecular medicines that target the underlying oncogenic driver mutations would be a highly desirable addition or alternative to conventional surgery.
Toward that goal, we and others have applied next-generation DNA sequencing to define the AB driver mutations (Brown et al. 2014; Sweeney et al. 2014). To date, most identified mutations occur in either the mitogen-activated protein kinase (MAPK) pathway or Hedgehog pathway. The MAPK pathway relays growth signals from receptor tyrosine kinases (RTKs) through RAS proteins (KRAS, HRAS, NRAS) to a cytoplasmic cascade of kinases (RAF → MEK → ERK), ultimately inducing genes involved, for example, in cell proliferation (Braicu et al. 2019). The most common AB driver mutation is BRAF-V600E (valine to glutamic acid at amino acid position 600) (Kurppa et al. 2014), which constitutively activates the BRAF kinase (Davies et al. 2002). Other AB drivers in the MAPK pathway include activating mutations in fibroblast growth factor receptor 2 (FGFR2) and the RAS family members KRAS, HRAS, and NRAS. Less commonly, activating mutations have been reported in phosphatidylinositol-4,5-bisphosphate 3-kinase (PIK3CA), which also transduces growth signals from RTKs.
The other major affected pathway in AB is the Hedgehog pathway, which plays important roles in development, including tooth development (Seppala et al. 2017). In the Hedgehog pathway, Hedgehog ligand binds patched 1 (PTCH1), which relieves inhibition of the transmembrane signaling protein smoothened (SMO) (Pak and Segal 2016). SMO signaling activates GLI transcription factors, leading to induction of genes involved in cell differentiation or proliferation. Some AB tumors harbor activating mutations of SMO, most commonly SMO-L412F (and less commonly SMO-W535L) (Brown et al. 2014; Sweeney et al. 2014). SMO mutations occur in AB tumors of the maxilla (upper jawbone), while BRAF mutations are in tumors of the mandible (lower jawbone) (Sweeney et al. 2014).
The discovery of BRAF-V600E mutations in AB immediately suggested a potential utility of BRAF-V600E inhibitors like vemurafenib and dabrafenib, already approved by the Food and Drug Administration (FDA) for melanoma. Supporting this potential, we and others found the AB cell line AM1 (Harada et al. 1998) carries BRAF-V600E and is sensitive to vemurafenib (Brown et al. 2014; Sweeney et al. 2014). Subsequent pilot clinical studies demonstrated efficacy of BRAF-V600E inhibition in shrinking AB tumors (Kaye et al. 2015; Tan et al. 2016).
Instrumental to the translation of BRAF-V600E inhibitors in AB was the availability of the AM1 cell line. Established cell lines provide critical models to evaluate oncogene dependency, as well as drug sensitivity and resistance. A current major roadblock in translating recently discovered AB driver mutations to new molecularly targeted therapies is the paucity of existing AB cell lines. Besides AM1 cells, only 1 other human AB cell line has been reported (Kibe et al. 2013). The primary motivation of our study has been to bridge this gap by creating a new AB cell line resource.
Materials and Methods
Generating AB Cell Lines
Fresh AB tissue was obtained from patients undergoing surgical tumor resection at Stanford Hospital, with institutional review board approval and patient informed consent. Cell lines were generated by conditional reprogramming (CR) (Liu et al. 2017), followed by transduction of TERT. Briefly, AB tissue was minced by scalpel, then incubated in 1× collagenase/hyaluronidase with 1 U dispase (StemCell) at 37°C for 3 h. Tissue was then triturated, washed in 1× phosphate-buffered saline (PBS), and incubated in 1× trypsin (StemCell) at 37°C for 10 min. Cells were next washed, passed through a 40-µM filter, resuspended in complete CR media, and plated in a T25 flask. Complete CR media comprised 10 µM Y-27632 (ROCK inhibitor) in a mix of 25% complete F medium and 75% complete F medium conditioned for 72 h on irradiated murine 3T3-J2 fibroblasts. Complete F medium comprised a Dulbecco’s modified Eagle’s medium (DMEM)/F12 mix augmented with hydrocortisone (25 ng/mL), epidermal growth factor (0.125 ng/mL), insulin (5 µg/mL), cholera toxin (0.1 nM), amphotericin B (250 ng/mL), and gentamicin (10 µg/mL). Early passage (passages 2–4) AB cells were then transduced with TERT by spin infection using pBABE-puro-hTERT (Addgene), followed by selection in 1 µg/mL puromycin. All AB cell lines were STR-fingerprinted (ATCC service) (Appendix Table 1) and are available from the corresponding author. Additional cell lines served as controls, including AM1 (provided by H. Harada, Iwate Medical University), cultured in keratinocyte serum-free medium (KSFM) medium, and CAMA-1 (ATCC), cultured in Eagle’s minimum essential medium (EMEM) medium.
Targeted Deep Sequencing to Identify Driver Mutations
Genomic DNA was isolated from early passage AB cell lines using DNeasy kits (Qiagen). Targeted deep sequencing was done using custom-designed Nimblegen SeqCap EZ Choice Library baits to capture all coding exons of 253 known cancer genes (Appendix Table 2). Nimblegen target capture and DNA-seq library construction were done per the manufacturer’s protocol (SeqCap EZ HyperCap Workflow). Sequencing (150 bp × 2) was done on an Illumina HiSeqX-Ten (Novogene) to an average 19 million mapped reads per sample. Raw DNA-seq data are available from dbGaP (phs002753.v1.p1). To identify candidate driver mutations, Illumina sequence reads were aligned to the hg19 RefSeq genome using BWA (Li and Durbin 2009). Single-nucleotide variants (SNVs) were then identified using Octopus (Cooke et al. 2021). Without matched normal DNA to exclude personal germline SNVs, we focused on known cancer somatic variants reported in the Sanger COSMIC database (Forbes et al. 2010). Candidate driver mutations were validated by polymerase chain reaction (PCR) amplification of genomic DNA followed by Sanger sequencing (Quintara Biosciences); PCR primers are in Appendix Table 3. To confirm driver mutations in the parent AB tumor, genomic DNA was isolated from the matching formalin-fixed, paraffin-embedded (FFPE) tumor block using QIAamp DNA FFPE Tissue Kits (Qiagen). Driver mutations were then PCR amplified and sequenced as above.
Cell Assays
Arsenic trioxide was purchased from Sigma and all other drug stocks from Selleck Chemicals. AB cells were plated at 30,000 to 40,000 cells per 6-well plate in duplicate, and then drug was added the following morning (t = 0) and replenished every 24 h. Cell viability at 72 h was assayed by flow cytometry counts (BD Accuri C6 Plus) gated on propidium iodide–negative single cells from a defined volume. Dose–response (inhibition) curves were plotted using GraphPad Prism (GraphPad Software). Cell apoptosis was assayed at 72 h using Dead Cell Apoptosis Kits (Thermo Fisher) and flow cytometry. Western blot, quantitative reverse transcriptase PCR (qRT-PCR) and RNA interference (RNAi) knockdown methods are detailed in the Appendix Methods. To ensure reproducibility, all the above experiments were replicated at least once.
Transcriptome Profiling
Total RNA was isolated using Qiagen RNeasy kits. RNA-seq libraries were constructed using TruSeq RNA Library Prep Kit v2 (Illumina) and sequenced (50 bp × 1) on an Illumina HiSeq4000 to an average 34 million mapped reads. Raw RNA-seq data are available from dbGaP (phs002753.v1.p1). Transcripts were mapped to the hg19 RefSeq genome and quantified as fragments per kilobase of exon per million mapped reads (FPKM) using TopHat/Cufflinks (Trapnell et al. 2012). Gene set enrichment was done by gene set enrichment analysis (Subramanian et al. 2007).
Hedgehog Pathway Luciferase Reporter Cell Lines
Hedgehog pathway luciferase reporter cell lines were created from SHH-Light2 (Taipale et al. 2000), an NIH-3T3 derivative carrying inducible Gli-Luc (firefly luciferase downstream of a Gli promoter) and constitutive Renilla luciferase (normalization control). Details are in the Appendix Methods. Briefly, we first inactivated endogenous murine Smo by CRISPR/Cas9 (Bauer et al. 2015). A single clone with confirmed biallelic mouse Smo inactivation (SHH-Light2-ΔSmo) was then stably transduced with C-terminal Myc-tagged human SMO wild-type, SMO-L412F, or SMO-W535L (Sweeney et al. 2014). Luciferase assays were done using the Dual-Luciferase Reporter Assay System (Promega).
Results
New Ameloblastoma Cell Lines with Defined Driver Mutations in the MAPK and Hedgehog Pathways
The paucity of existing AB cell lines for basic and preclinical studies motivated us to establish additional cell lines reflecting the spectrum of recently discovered AB driver mutations. To generate AB cell lines, we chose the method of conditional reprogramming (CR) (Liu et al. 2012, 2017) for its reported ease and simplicity. In the CR method, diverse epithelial cell types have been successfully immortalized—without viral oncogenes—simply by culturing the cells in medium conditioned from irradiated 3T3-J2 fibroblasts, with addition of the Rho kinase (ROCK) inhibitor Y-27632.
We obtained fresh AB tissue that was surplus from surgical resections done at Stanford Hospital. Tumor tissue was disaggregated to single cells, which were then plated in complete CR media and serially passaged. In all, we attempted generating CR cell lines from 6 AB cases (Appendix Table 4). However, in contrast to published reports (Liu et al. 2012), the AB cell cultures in CR medium could not be grown beyond 8 to 12 passages, at which point they appeared to undergo cellular senescence. We could successfully bypass this block by transducing early passage AB cells with human telomerase (HTERT). The resultant 6 AB cell lines (named AB1T through AB6T, Appendix Table 4) could be cultured beyond 30 passages with no diminished growth rate. The morphology of the AB cell lines was epithelial-like, where denser cultures exhibited cell clusters and cords reminiscent of the parent tumors (Fig. 1A, B and Appendix Fig. 1). By qRT-PCR, all 6 AB cell lines expressed the ameloblast-associated genes amelotin (AMTN) and odontogenic ameloblast associated (ODAM), as well as E-cadherin (CDH1), at comparable or higher levels than control AM1 ameloblastoma cells (Appendix Fig. 2).
Figure 1.
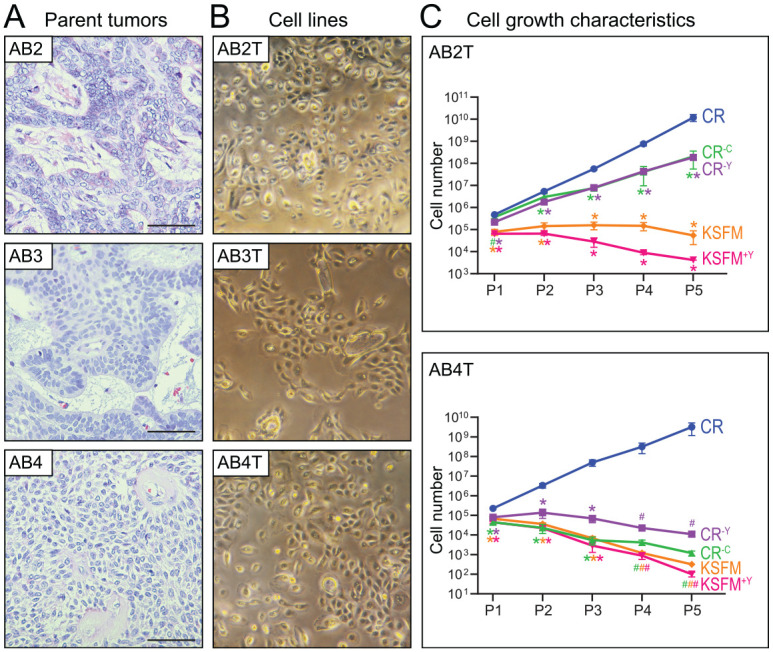
Morphology and growth characteristics of newly established ameloblastoma (AB) cell lines. (A) Histology of AB parent tumors from which the cell lines were derived. Hematoxylin and eosin stains shown for representative parent tumors AB2, AB3, and AB4; bar is 100 µM. (B) Morphology of corresponding TERT-transduced AB cell lines (AB2T, AB3T, and AB4T) cultured in conditional reprogramming (CR) medium. Photographed using an inverted light microscope at 10× magnification. (C) Growth rate of AB2T and AB4T cells cultured in different media formulations. CR–C and CR–Y are deficient in the conditioned component or the ROCK inhibitor (Y27632), respectively. Equal cell numbers were plated in duplicate and viable cell numbers counted (by flow cytometry) over 5 serial passages. Error bars represent 1 SD. #P < 0.10, *P < 0.05 (2-sided Student’s t test versus CR medium). Note the lack of significance in AB4T P4 and P5 is attributable to the small sample size (duplicates) and to the relatively larger SDs in CR P4 and P5 data points (note the log scale). KSFM, keratinocyte serum-free medium.
Given the transduction of HTERT, we sought to evaluate the continued necessity of specialized CR culture medium. We attempted to grow 2 of the cell lines, AB2T and AB4T, in CR medium lacking either the conditioning on irradiated fibroblasts or else the ROCK inhibitor (Y-27632). In both instances, cell growth was either substantially diminished (AB2T) or unsupported (AB4T) (Fig. 1C). Similarly, the AB cell lines did not proliferate in complete keratinocyte serum-free medium (KSFM), a medium reported to support growth of AM1 and AM3 cells (Kibe et al. 2013) (Fig. 1C).
We next determined whether the 6 newly established AB cell lines harbored any of the driver mutations recently reported by us and others (Brown et al. 2014; Sweeney et al. 2014), including activating mutations in the MAPK and Hedgehog pathways. Using genomic DNA from early passage AB cell lines, we carried out targeted deep Illumina sequencing of ~250 cancer genes (Fig. 2 and Appendix Table 2). In total, 5 of the 6 AB cell lines were found to harbor known AB driver mutations, including AB1T (NRAS-Q61R), AB2T (KRAS-G12R and SMO-L412F), AB3T (FGFR2-C383R, SMO-L412F, and PIK3CA-N345K), AB4T (BRAF-V600E), and AB6T (NRAS-Q61K) (Fig. 2 and Appendix Table 4). All the above mutations were verified by Sanger sequencing and confirmed to be present in the parent AB tumors from which the cell line was derived (Fig. 2).
Figure 2.

Defining the driver mutations in new ameloblastoma (AB) cell lines. Each panel pair depicts AB cell line DNA-seq reads indicative of driver mutation (left: Integrative Genomics Viewer screenshot), along with Sanger sequencing validation of mutation in the parent tumor (right: sequence chromatogram). (A) NRAS-Q61R mutation in AB1T cells. (B) NRAS-Q61K mutation in AB6T cells. (C) SMO-L412F mutation in AB2T cells. (D) KRAS-G12R mutation in AB2T cells. (E) SMO-L412F mutation in AB3T cells. (F) FGFR2-C383R mutation in AB3T cells. (G) PIK3CA-N345K mutation in AB3T cells. (H) BRAF-V600E mutation in AB4T cells.
AB Cell Lines with MAPK Pathway Mutations Are Sensitive to Pathway Inhibition
Having newly generated AB cell lines with defined driver mutations, we next sought to apply them as preclinical models for drug sensitivity testing, focusing first on the MAPK pathway. AB4T cells, which harbor a BRAF-V600E mutation, were sensitive to the BRAF-V600E inhibitor vemurafenib, although somewhat less so than AM1 ameloblastoma cells (which also carry BRAF-V600E) (Fig. 3A and Appendix Table 5). Control CAMA-1 (breast cancer) cells that carry no activating MAPK pathway mutation (Sanger COSMIC database) were insensitive to vemurafenib.
Figure 3.

Targeting mitogen-activated protein kinase (MAPK) pathway mutations in ameloblastoma (AB) cells. (A) BRAF inhibition with vemurafenib in AB cell line with BRAF-V600E mutation (AB4T). Dose–response (inhibition) curves depict cell viability (assayed in triplicate by flow cytometry) over a 4-log range of drug concentration. Error bars represent 1 SD, and IC50 values are reported in Appendix Table 5. (B) MEK inhibition with GDC-0623 in AB cell lines with KRAS-G12R (AB2T), NRAS-Q61R (AB1T), and NRAS-Q61K (AB6T), plus control CAMA-1 cells. Error bars represent 1 SD, and IC50 values are reported in Appendix Table 5. (C) Verification of on-target MEK inhibition (with GDC-0623), by reduced phospho-ERK levels on Western blot. (D) MEK inhibition in AB2T cells (KRAS-G12R) drives morphologic changes; 1 µM GDC-0623 at 72 h versus vehicle control. (E) MEK inhibition by GDC-0623 in AB2T cells (KRAS-G12R) drives transcriptome changes enriched for tooth development genes (gene set enrichment analysis). Core enrichment genes—those in the leading edge of the running enrichment score—are listed in Appendix Table 6.
The 3 AB cell lines harboring RAS mutations—AB1T (NRAS-Q61R), AB2T (KRAS-G12R), and AB6T (NRAS-Q61K)—were each sensitive to GDC-0623, an allosteric MEK inhibitor (Hatzivassiliou et al. 2013), at low nanomolar concentration (Fig. 3B). Control CAMA-1 cells were insensitive (Fig. 3B), as previously reported (Barretina et al. 2012). Reduced phospho-ERK levels by Western blot confirmed MEK inhibition (Fig. 3C).
MEK inhibition of RAS-mutant AB cells led to growth arrest and large flat cells characteristic of cell differentiation and/or senescence (Fig. 3D). Transcriptome analysis of GDC-0623–treated AB2T cells (KRAS-G12R), by gene set enrichment analysis, revealed enriched expression of annotated “tooth development” genes (Pemberton et al. 2007) (Fig. 3E and Appendix Table 6), consistent with cell differentiation. Known ameloblast-associated genes AMTN and ODAM were overexpressed 162-fold and 15-fold, respectively (Appendix Table 7).
AB Cell Lines with SMO-412F Mutation Are Vulnerable to Hedgehog Pathway Inhibition
We next evaluated the AB cell lines as preclinical models for Hedgehog pathway inhibition. Two of the cell lines, AB2T and AB3T, both derived from maxillary tumors, carry the SMO-L412F mutation that constitutively activates the Hedgehog pathway (Sweeney et al. 2014). We first evaluated small interfering RNAs (siRNA) targeting SMO. In AB2T cells, 4 different SMO siRNAs, as well as a pool of all 4 (siSMO pool), markedly reduced both SMO messenger RNA (mRNA) levels and AB cell proliferation, compared to a nontargeting siRNA pool (siNTC) (Fig. 4A). SMO knockdown also reduced cell proliferation in AB3T cells (Fig. 4B), and in both AB2T and AB3T cells, the reduced cell numbers were attributable at least in part to increased apoptosis (Fig. 4C). On-target Hedgehog pathway inhibition by siSMO was verified by the reduced expression of Hedgehog pathway target gene GLI1 (Fig. 4D).
Figure 4.

Hedgehog pathway dependency in ameloblastoma (AB) cells with SMO mutation. (A) SMO knockdown by multiple different small interfering RNAs (siRNAs) reduces viable cell counts in AB2T cells (SMO-L412F). Four different siRNAs are each compared to a nontargeting control (NTC) siRNA pool, assaying SMO messenger RNA levels (left) and viable cell numbers (right, by flow cytometry). Mean and 1 SD shown. *P < 0.05, **P < 0.01 (2-sided Student’s t test). (B) SMO knockdown (by siSMO pool) reduces viable cell counts in 2 different cell lines with SMO-L412F mutation. Time course done in triplicate shown for AB2T cells (left) and AB3T cells (right). Mean and 1 SD shown. *P < 0.05, **P < 0.01 (2-sided Student’s t test; for each time point, in comparison to siNTC pool). (C) SMO knockdown (by siSMO pool) drives cell apoptosis in AB cell lines with SMO-L412F mutation. Apoptosis assayed in triplicate by annexin staining (flow cytometry). Mean and 1 SD shown. *P < 0.05 (2-sided Student’s t test). (D) SMO knockdown (by siSMO pool) diminishes downstream Hedgehog pathway signaling, assayed in triplicate by Hedgehog pathway target GLI1 transcript levels (by quantitative reverse transcriptase polymerase chain reaction [qRT-PCR], normalized to GAPDH). Mean and 1 SD shown. **P < 0.01 (2-sided Student’s t test). (E) Downstream Hedgehog pathway inhibitor arsenic trioxide (ATO) reduces cell viability (assayed in triplicate by flow cytometry) of AB2T cells (SMO-L412F) at submicromolar concentrations. IC50 value in Appendix Table 5. (F) Verification of on-target Hedgehog pathway inhibition (with ATO), by reduced GLI1 transcript levels (qRT-PCR). RNA was isolated after 48 h of drug treatment. Assay done with technical quadruplicates; error bars indicate RQmax/RQmin.
AB2T cells were also sensitive to the Hedgehog pathway inhibitor arsenic trioxide (ATO) (Fig. 4E), thought to act downstream of SMO (Beauchamp and Uren 2012). ATO inhibited AB2T cell proliferation at comparable concentrations (IC50 ~100 nM) to its inhibition of Hedgehog pathway (assayed by GLI1 transcript levels; Fig. 4F), consistent with but not proving an on-target Hedgehog pathway effect.
In recent years, several small-molecule inhibitors of SMO have been reported (Pietrobono and Stecca 2018). All were primarily developed to inhibit wild-type SMO, whereas their activity against activating SMO mutations (including the SMO-L412F mutation common in AB) has remained largely unknown. To address that question, we first evaluated 6 different SMO inhibitors—vismodegib, glasdegib, PF-5274857, sonidegib, taladegib, and BMS-833923—for their ability to inhibit Hedgehog signaling by SMO-L412F and SMO-W535L, using a heterologous NIH3T3-based Gli-Luciferase reporter assay (detailed in Materials and Methods and Appendix Fig. 3). While all 6 SMO antagonists inhibited Hedgehog pathway signaling from wild-type SMO, only 3 (sonidegib, taladegib, and BMS-833923) significantly inhibited SMO-L412F (Fig. 5A). BMS-833923 exhibited the most potent SMO-L412F inhibition, comparable to ATO, and also showed activity against SMO-W535L (Fig. 5A). Indeed, in a dose–response assay in the Gli-Luciferase reporter cells (Fig. 5B), BMS-833923 showed comparable potency against SMO-L412F as did FDA-approved vismodegib against wild-type SMO (compare dark blue and pink curves in Fig. 5B).
Figure 5.

Targeting SMO mutations in ameloblastoma (AB) cells. (A) Efficacy small-molecule SMO inhibitors (each at 1 µM, except for arsenic trioxide [ATO] at 8 µM) versus SMO wild-type, SMO-L412F, and SMO-W535L. Hedgehog pathway inhibition assayed using a Gli-Luciferase reporter in NIH-3T3 cells with murine Smo replaced by human wild-type or mutant SMO. Assayed in triplicate, mean and 1 SD shown. *P < 0.01, **P < 0.01 (2-sided Student’s t test). (B) SMO inhibition with BMS-833923 in NIH-3T3 Gli-Luciferase reporter cells harboring SMO wild-type, SMO-L412F, and SMO-W535L. Dose–response (inhibition) curves depict Hedgehog pathway activation (assayed in triplicate by dual luciferase) over a 4-log range of drug concentration. Error bars represent 1 SD, and IC50 values are reported in Appendix Table 5. (C) SMO inhibition comparing BMS-833923 versus vismodegib in AB2T (SMO-L412F) and AB3T (SMO-L412F) cells. Dose–response curves depict cell viability assayed in triplicate by flow cytometry. Error bars represent 1 SD, and IC50 values are reported in Appendix Table 5. (D) Verification of on-target Hedgehog pathway inhibition (with 1 µM BMS-83923 versus 1 µM vismodegib or DMSO control) in AB2T cells (by reduced GLI1 transcript levels). RNA was isolated after 48 h of drug treatment. Quantitative reverse transcriptase polymerase chain reaction was done with technical quadruplicates. Error bars indicate RQmax/RQmin.
We next evaluated BMS-833923, the most potent SMO-L412F inhibitor in the Gli-Luciferase reporter assay, on AB2T and AB3T cells that harbor SMO-L412F. While both AB cell lines were relatively resistant to vismodegib, they were sensitive to BMS-833923 (Fig. 5C). Consistent with the dose–response (inhibition) findings, BMS-833923 but not vismodegib reduced transcript levels of the Hedgehog pathway target gene GLI1 in AB2T cells (Fig. 5D). In control experiments, AB cells (AB4T and AB6T) without SMO-L412F were, as expected, less sensitive to BMS-833923, while vismodegib even mildly enhanced cell proliferation at higher doses (Appendix Fig. 4).
Discussion
We report the generation of 6 new human AB cell lines, at once quadrupling the previous number available. Our success is likely attributable, at least in part, to our use of conditional reprogramming. The CR method is robust and simple—relying on conditioned medium (now even commercially available, e.g., StemCell Technologies) plus ROCK inhibitor, rather than viral oncoproteins. Indeed, we succeeded in establishing all 6 of 6 AB cell lines attempted. However, unlike the original CR protocol (Liu et al. 2012) and our recent experience generating a single canine AB cell line (Saffari et al. 2019), establishing human AB cell lines required the addition of hTERT, ostensibly to overcome replicative senescence. In the CR approach, the CR medium (conditioned on irradiated murine 3T3-J2 fibroblasts) is thought to contribute soluble factors that induce telomerase to bypass replicative senescence (Liu et al. 2012). It is therefore notable that both CR medium and hTERT are required to maintain the human AB cell lines, pointing to a possible additional function of the conditioned CR medium.
The 6 new AB cell lines together cover the full spectrum of reported AB driver mutations, including activating mutations in BRAF, KRAS, NRAS, FGFR2, SMO, and PIK3CA. Previously, only a single AB cell line (AM1) (Harada et al. 1998) represented solely the BRAF mutation. Notably, the AB2T and AB3T cell lines, to our knowledge, represent the first established human tumor cell lines (of many thousands across dozens of cancer types) to carry activating mutations of SMO. These cell lines will thus newly enable studies of SMO mutation in human ameloblastoma and with direct relevance to other SMO mutation-driven human cancers, including meningioma (Clark et al. 2013) and vismodegib-resistant basal cell carcinoma (Atwood et al. 2015).
While 5 of the 6 AB cell lines each carry 1 or more AB driver mutations, 1 line (AB5T) harbors no known AB driver. Our targeted Illumina sequencing covered 253 known cancer genes, including all reported AB driver genes. Defining the oncogenic driver mutation(s) in AB5 cells will require whole-exome or whole-genome sequencing, which may also uncover additional driver mutations in the other AB cell lines.
Leveraging the new AB cell lines, we carried out several proof-of-principle studies on AB oncogene dependency and drug sensitivity. Within the MAPK signaling pathway, we confirmed AB4T (BRAF-V600E) sensitivity to the BRAF-V600E inhibitor vemurafenib. In recent pilot studies, BRAF-V600E inhibitors (used off-label) have shown clinical efficacy with AB tumor regression (Kaye et al. 2015; Tan et al. 2016). We also demonstrated sensitivity of AB1T (NRAS-Q61R), AB2T (KRAS-G12R), and AB6T (NRAS-Q61K) cells to the downstream MEK inhibitor GDC-0623, which both inhibits MEK and blocks feedback-mediated RAF/MEK activation. In particular, GDC-0623 appears to drive AB cell differentiation, marked by the expression of tooth development genes. Thus, GDC-0623, with a phase I clinical trial completed for metastatic solid tumors (Cheng and Tian 2017), may find utility in the treatment of RAS mutation-driven ameloblastoma.
Taking advantage of the 2 AB cell lines (AB2T and AB3T) with SMO-L412F mutation, we also carried out oncogene dependency and drug sensitivity studies of the Hedgehog pathway. Both cell lines are dependent on SMO activation, evidenced by SMO knockdown driving cell death. AB cells with SMO-L412F are also sensitive to ATO, which inhibits the Hedgehog pathway downstream of SMO (Beauchamp and Uren 2012). Thus, ATO may find clinical utility in AB patients with SMO mutations. However, ATO inhibits other cell signaling pathways and carries some unwanted toxicity (Wang et al. 2020). Thus, targeted inhibition of mutant SMO itself may be desirable.
Toward that end, we also leveraged the AB cell lines with SMO-L412F mutation for preclinical investigations of small-molecule SMO inhibitors. Of 6 SMO inhibitors surveyed, BMS-833923 showed particular promise in inhibiting SMO-L412F (and SMO-W535L) activity by Gli-Luciferase reporter and in growth inhibition of AB2T and AB3T (SMO-L412F) cells. Phase I clinical trials of BMS-833923 were recently completed for basal cell nevus syndrome, multiple myeloma, gastrointestinal cancer, and small cell lung cancer (Pietrobono and Stecca 2018). The future development of compounds selective for mutant SMO-L412F (over wild-type SMO), analogous to the BRAF-V600E selective inhibitors, may also prove advantageous.
In summary, we have generated 6 new human AB cell lines that together cover the complete spectrum of AB driver mutations. As our findings demonstrate, these cell lines will prove valuable in investigating AB oncogenic pathways and for preclinical studies of new pharmacologic treatments. Those studies should accelerate the incorporation of precision medicines for treating and managing patients with ameloblastoma.
Author Contributions
J. Nguyen, J.R. Pollack, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; P.S. Saffari, A.S. Pollack, contributed to data acquisition, analysis, and interpretation, critically revised the manuscript; S. Vennam, X. Gong, contributed to data analysis and interpretation, critically revised the manuscript; R.B. West, contributed to conception, design, data acquisition, and interpretation, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
Supplemental material, sj-docx-1-jdr-10.1177_00220345221100773 for New Ameloblastoma Cell Lines Enable Preclinical Study of Targeted Therapies by J. Nguyen, P.S. Saffari, A.S. Pollack, S. Vennam, X. Gong, R.B. West and J.R. Pollack in Journal of Dental Research
Acknowledgments
We thank the Stanford Tissue Procurement Facility for assistance with AB tissue acquisition, the Stanford Genome Sequencing Service Center for Illumina sequencing, and the Stanford Shared FACS Facility for operator-assisted cell sorting.
Footnotes
A supplemental appendix to this article is available online.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the NIDCR R01 DE026502 (J.R. Pollack and R.B. West).
References
- Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M, Ally MS, Kim J, Yao C, Chang AL, et al. 2015. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 27(3):342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. 2012. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 483(7391):603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer DE, Canver MC, Orkin SH. 2015. Generation of genomic deletions in mammalian cell lines via CRISPR/Cas9. J Vis Exp. (95):e52118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchamp EM, Uren A. 2012. A new era for an ancient drug: arsenic trioxide and Hedgehog signaling. Vitam Horm. 88:333–354. [DOI] [PubMed] [Google Scholar]
- Braicu C, Buse M, Busuioc C, Drula R, Gulei D, Raduly L, Rusu A, Irimie A, Atanasov AG, Slaby O, et al. 2019. A comprehensive review on MAPK: a promising therapeutic target in cancer. Cancers. 11(10):1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown NA, Rolland D, McHugh JB, Weigelin HC, Zhao L, Lim MS, Elenitoba-Johnson KS, Betz BL. 2014. Activating FGFR2-RAS-BRAF mutations in ameloblastoma. Clin Cancer Res. 20(21):5517–5526. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Tian H. 2017. Current development status of MEK inhibitors. Molecules. 22(10):1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark VE, Erson-Omay EZ, Serin A, Yin J, Cotney J, Ozduman K, Avsar T, Li J, Murray PB, Henegariu O, et al. 2013. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science. 339(6123):1077–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke DP, Wedge DC, Lunter G. 2021. A unified haplotype-based method for accurate and comprehensive variant calling. Nat Biotechnol. 39(7):885–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. 2002. Mutations of the BRAF gene in human cancer. Nature. 417(6892):949–954. [DOI] [PubMed] [Google Scholar]
- Effiom OA, Ogundana OM, Akinshipo AO, Akintoye SO. 2018. Ameloblastoma: current etiopathological concepts and management. Oral Dis. 24(3):307–316. [DOI] [PubMed] [Google Scholar]
- Forbes SA, Tang G, Bindal N, Bamford S, Dawson E, Cole C, Kok CY, Jia M, Ewing R, Menzies A, et al. 2010. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 38(Database issue):D652–D657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada H, Mitsuyasu T, Nakamura N, Higuchi Y, Toyoshima K, Taniguchi A, Yasumoto S. 1998. Establishment of ameloblastoma cell line, AM-1. J Oral Pathol Med. 27(5):207–212. [DOI] [PubMed] [Google Scholar]
- Hatzivassiliou G, Haling JR, Chen H, Song K, Price S, Heald R, Hewitt JF, Zak M, Peck A, Orr C, et al. 2013. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature. 501(7466):232–236. [DOI] [PubMed] [Google Scholar]
- Kaye FJ, Ivey AM, Drane WE, Mendenhall WM, Allan RW. 2015. Clinical and radiographic response with combined BRAF-targeted therapy in stage 4 ameloblastoma. J Natl Cancer Inst. 107(1):378. [DOI] [PubMed] [Google Scholar]
- Kibe T, Fuchigami T, Kishida M, Iijima M, Ishihata K, Hijioka H, Miyawaki A, Semba I, Nakamura N, Kiyono T, et al. 2013. A novel ameloblastoma cell line (AM-3) secretes MMP-9 in response to Wnt-3a and induces osteoclastogenesis. Oral Surg Oral Med Oral Pathol Oral Radiol. 115(6):780–788. [DOI] [PubMed] [Google Scholar]
- Kurppa KJ, Caton J, Morgan PR, Ristimaki A, Ruhin B, Kellokoski J, Elenius K, Heikinheimo K. 2014. High frequency of BRAF V600E mutations in ameloblastoma. J Pathol. 232(5):492–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Krawczyk E, Suprynowicz FA, Palechor-Ceron N, Yuan H, Dakic A, Simic V, Zheng YL, Sripadhan P, Chen C, et al. 2017. Conditionalreprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat Protoc. 12(2):439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B, Timofeeva OA, Nealon C, Dakic A, Simic V, et al. 2012. Rock inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol. 180(2):599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClary AC, West RB, Pollack JR, Fischbein NJ, Holsinger CF, Sunwoo J, Colevas AD, Sirjani D. 2016. Ameloblastoma: a clinical review and trends in management. Eur Arch Otorhinolaryngol. 273(7):1649–1661. [DOI] [PubMed] [Google Scholar]
- Pak E, Segal RA. 2016. Hedgehog signal transduction: key players, oncogenic drivers, and cancer therapy. Dev Cell. 38(4):333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemberton TJ, Li FY, Oka S, Mendoza-Fandino GA, Hsu YH, Bringas P, Jr, Chai Y, Snead ML, Mehrian-Shai R, Patel PI. 2007. Identification of novel genes expressed during mouse tooth development by microarray gene expression analysis. Dev Dyn. 236(8):2245–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrobono S, Stecca B. 2018. Targeting the oncoprotein smoothened by small molecules: focus on novel acylguanidine derivatives as potent smoothened inhibitors. Cells. 7(12):272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffari PS, Vapniarsky N, Pollack AS, Gong X, Vennam S, Pollack AJ, Verstraete FJM, West RB, Arzi B, Pollack JR. 2019. Most canine ameloblastomas harbor HRAS mutations, providing a novel large-animal model of RAS-driven cancer. Oncogenesis. 8(2):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seppala M, Fraser GJ, Birjandi AA, Xavier GM, Cobourne MT. 2017. Sonic Hedgehog signaling and development of the dentition. J Dev Biol. 5(2):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP. 2007. GSEA-P: a desktop application for gene set enrichment analysis. Bioinformatics. 23(23):3251–3253. [DOI] [PubMed] [Google Scholar]
- Sweeney RT, McClary AC, Myers BR, Biscocho J, Neahring L, Kwei KA, Qu K, Gong X, Ng T, Jones CD, et al. 2014. Identification of recurrent SMO and BRAF mutations in ameloblastomas. Nat Genet. 46(7):722–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, Scott MP, Beachy PA. 2000. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 406(6799):1005–1009. [DOI] [PubMed] [Google Scholar]
- Tan S, Pollack JR, Kaplan MJ, Colevas AD, West RB. 2016. BRAF inhibitor treatment of primary BRAF-mutant ameloblastoma with pathologic assessment of response. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 122(1):e5–e7. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with Tophat and Cufflinks. Nat Protoc. 7(3):562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QQ, Jiang Y, Naranmandura H. 2020. Therapeutic strategy of arsenic trioxide in the fight against cancers and other diseases. Metallomics. 12(3):326–336. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-docx-1-jdr-10.1177_00220345221100773 for New Ameloblastoma Cell Lines Enable Preclinical Study of Targeted Therapies by J. Nguyen, P.S. Saffari, A.S. Pollack, S. Vennam, X. Gong, R.B. West and J.R. Pollack in Journal of Dental Research