

Abstract

In recent years, many manganese-based homogeneous catalytic precursors have been developed as powerful alternatives in organic synthesis. Among these, the hydrofunctionalizations of unsaturated C–C bonds correspond to outstanding ways to afford compounds with more versatile functional groups, which are commonly used as building blocks in the production of fine chemicals and feedstock for the industrial field. Herein, we present an account of the Mn-catalyzed homogeneous hydrofunctionalizations of alkenes and alkynes with the main objective of finding catalytic and mechanistic tendencies that could serve as a platform for the works to come.
1. Introduction
Catalytic systems are of paramount importance in the pursuit of economically and atomically efficient processes. Despite the high activities and selectivities obtained from using homogeneous catalysts based on late transition metals (e.g., Pd, Pt, Ru, Ir, and Rh), the change to catalytic systems based on first-row transition metals is highly desirable.1−6
In a particular way, owing to its low toxicity and relatively high abundance in the earth’s crust, manganese (the third most abundant transition metal) has been developed as a promising alternative in catalysis.7 Within the homogeneous area, many reviews have been disclosed about molecularly defined Mn-based catalytic precursors and their reactivity.8−11 Notoriously, excellent results have been reported for C–H activations with further functionalizations12,13 and (transfer) hydrogenation reactions.14−17
Despite the presence of alkenes and alkynes in compounds with direct application, they are commonly used as feedstock for the construction of more complex molecular structures. In particular, the hydrofunctionalizations (addition of H–E; E = H, SiR3, BR2, Ar, NR2, OH, etc.) of alkynes and alkenes allow their conversion into a variety of value-added products that can be used as building blocks or final products with industrial and academic purposes.18−22 Regarding the Mn-catalyzed hydrofunctionalizations of these substrates, a growing tendency for applications could be observed, where the reports of only 3 years of this decade have almost equalized or even surpassed the whole number of reports made in the past decade (Figure 1). It is worth noting that in comparison to the more polarized unsaturated moieties (such as carbonyls, nitriles, and imines), the unsaturated C–C bonds are less prone to react and usually correspond to more challenging tasks, including issues related to the regio- and stereoselectivity of the processes. Nevertheless, a variety of manganese complexes and salts have served to elude these drawbacks, leading to excellent (and even exclusive) results. As it will be presented in more detail, there exists a relationship between the catalyst design and the strategy to be used, including SET and HAT reactions and metal–ligand cooperative pathways among others. Therefore, we consider it valuable to highlight not only the structural and electronic properties of the reported catalysts but also the way in which the interaction of the metal center with the mentioned functional groups has been demonstrated or proposed.
Figure 1.
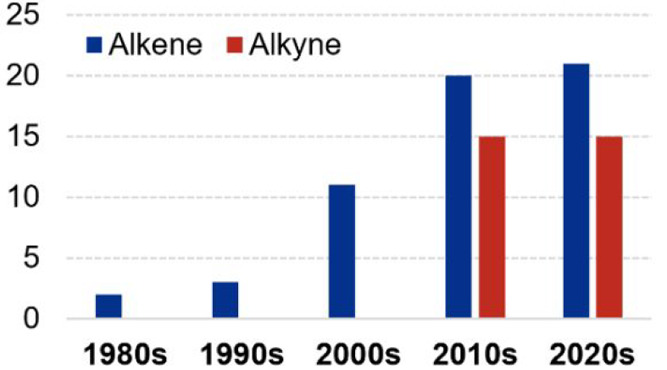
Number of reports per decade on the homogeneous Mn-catalyzed hydrofunctionalizations of alkenes and alkynes.
As was previously mentioned, some reviews have been reported concerning the uses of manganese-based compounds in homogeneous catalysis;8−17 however, most of them are focused on either one specific type of process (e.g., (transfer) hydrogenations or C–H activations) or the reactivity of some specific Mn complexes (for instance, with pincer-type ligands) but not on the state of the Mn catalytic transformations of specific functionalities. Herein, we present an exhaustive collection of the reports about the different homogeneous Mn-catalyzed hydrofunctionalizations of alkenes and alkynes, emphasizing the mechanistic and catalytic descriptions and maintaining a critical approach to the strategies, limitations, and opportunity areas of each type of reactivity reported so far. Thus, we expect that this work can serve to understand the key aspects of the different processes and find tendencies that could lead to potential enhancements toward more robust, versatile, and active catalytic systems. Therefore, this review will include original and complete works as well as isolated reports disclosed to date with direct Mn-catalyzed processes and cocatalyzed methodologies, expecting to serve as a scaffold for further contributions in this growing area.
2. Hydrofunctionalization of Unsaturated C–C Bonds
2.1. Hydrogenation
Hydrogenation is a valuable technique widely used in organic synthesis. Outstanding results in this field have been obtained with homogeneous Mn-based catalytic systems, employing different methodologies and hydrogen sources.23 Albeit the hydrogenation of carbonyl derivates has been more explored, here we present the relevant results disclosed for the reduction of unsaturated C–C bonds with manganese catalysts.
2.1.1. Hydrogenation of Alkenes
Although not Mn catalyzed but mediated, Halpern and Orchin reported the use of [Mn(CO)5H] for the reduction of alkenes in 1977 and 1982, respectively.24 NMR studies by Halpern using a CIDNP technique elucidated a hydrogen-atom transfer (HAT) mechanism operating for the hydrogenation of α-methylstyrene (Scheme 1). Despite these examples not being catalytic, their contributions stated a future common pathway for some Mn-based catalysts.
Scheme 1. HAT Mechanism Proposed for the Mn-Mediated Hydrogenation of α-Methylstyrene with [Mn(CO)5H].

The same type of reactivity was reported by Magnus using Mn-1 (Figure 2) as a catalytic precursor for the selective hydrogenation of α,β-unsaturated ketones in the absence of oxygen.25 Apparently, this system could form in situ a manganese hydride after reacting with phenylsilane at room temperature. Then, this makes an irreversible hydride insertion into the substrate, which acts as a Michael acceptor. Finally, the alcoholysis of the resulting enol and its tautomerization yield the final product. Other alcohols and silanes were not suitable for this transformation, while terminal and more constrained olefins remained unhydrogenated.
Figure 2.

Mn-based catalytic precursors for the selective hydrogenation of alkenes.
In 2014, Shenvi disclosed the use of tert-butyl hydroperoxide (TBHP) for the notoriously improved reactivity of Mn-1 toward the hydrogenation of alkenes (Figure 2).26 They reported a HAT process, maintaining a high stereoselectivity (toward the thermodynamic product) but with higher chemoselectivity as well (no reduction of other normally reactive functional groups). Despite [Co(dpm)2] (dpm = dipivaloylmethanato) also exhibiting catalytic activity, generally higher yields were obtained with [Mn(dpm)3]. Subsequently, the reactivity of this system was improved by changing phenylsilane (PhSiH3) for isopropoxy(phenyl)silane (Ph(iPrO)SiH2) as the reductive agent (Figure 2).27 Shenvi and co-workers found that using this activated silane, the same or even higher yields could be achieved with decreased catalytic loads as well as a greater tolerance toward other polar and non-polar solvents (preferably hexanes). Also, under these conditions, good conversions were obtained with the less active but commercially available [Mn(acac)3] (acac = acetylacetonate). Notably, this catalytic system also triggered the hydrosilylation and hydration of alkenes (vide infra).
On the other hand, the first molecularly defined Mn-based catalyst for the hydrogenation of alkenes was presented by Kirchner and co-workers in 2019.28 In the absence of any additive, they reduced a collection of terminal and disubstituted alkenes employing direct H2 and Mn-2 (Figure 2) as a catalytic precursor; tri- and tetrasubstituted alkenes remained unhydrogenated. They observed that this process is favored with a relatively sterically demanding and electron-rich alkyl diphosphine. The activation of the precatalyst is proposed to occur via a carbonyl migratory insertion into the alkyl moiety with further hydrogenolysis, yielding n-butanal and a coordinatively unsaturated Mn(I)–hydride as the catalytically active species. An experiment in the presence of PMe3, along with DFT calculations, displayed an inner-sphere mechanism for this system, which is summarized in Scheme 2.
Scheme 2. Mechanistic Proposal for the Alkene Hydrogenation with Mn-2.

Another well-defined Mn-based catalytic precursor was described later in 2020 by Khusnutdinova et al. using hydrogen and Mn-3 (Figure 2) in the presence of tBuOK.29 In general, aryl alkenes with electron-withdrawing substituents were less reactive, while internal dialkyl alkenes did not react. Intriguingly, 1-pentadecyne was efficiently over-reduced to pentadecane, but phenylacetylene remained almost inert under these reaction conditions. Analogously to other pincer-based Mn(I)-catalyzed hydrogenation processes,30 this manganese(I) complex with a P,N-donor ligand is proposed to operate via a dearomatization–aromatization pathway initiated by the deprotonation of the methylene arm. This catalytically active species was proposed to scission H2 in a heterolytic manner through a metal–ligand cooperative mechanism followed by an outer-sphere hydrogen addition to the substrate (Scheme 3).
Scheme 3. Mechanistic Proposal for the Alkene Hydrogenation with Mn-3.

A very recent investigation on Mn-catalyzed alkene reduction was reported by Topf’s group in 2021.31 Inspired by the system reported by Sortais in 2017 for the transfer hydrogenation of aldehydes and ketones with isopropanol as the hydrogen source,32 they adapted the use of the system Mn-4 (Figure 2) for the direct hydrogenation of ketones and α,β-unsaturated carbonylic compounds. Interestingly, this system only worked when it was formed in situ; the molecularly defined Mn(I)–picolylamine complex displayed poor activity as a catalytic precursor for these hydrogenations. Nevertheless, this system stands out for the absence of an inert atmosphere and the use of only commercially available components. Although there is not an explicit mechanistic proposal, it is likely that it involves a metal–ligand cooperative (MLC) pathway with an amide-assisted H2 activation.
As was presented, two different strategies have been documented for the catalytic alkene reduction with Mn-based precursors. The first of these works relied on a radical pathway involving HAT reactions. However, none of them included an explicit mechanistic proposal. On the basis of the nature of the catalytic precursors and other types of radical hydrofunctionalizations (vide infra), it is likely that a Mn–H species is formed in the reaction mixture to further react with the alkene, via a HAT step, for the formation of a carbon-centered radical intermediate. Finally, an H-atom abstraction from either solvent or reagents would yield the reduced product. Nonetheless, owing to their remarkable applications in the derivatization of complex substrates, a detailed mechanistic study of these processes is still desirable for a better comprehension of them that serves as an outset for further contributions.
In contrast, the latest works in this field encompass a ligand-tuned strategy for the C–C bond reduction directly with H2. Under these conditions, common organometallic steps are proposed to afford the reduced products, which, at the same time, include two different types of hydrogen activation. For Kirchner’s catalyst, non-classical metallic hydrides are proposed to be involved while the other two works are suggested to entail an MLC pathway. In this vein, inner-sphere and outer-sphere processes were proposed, respectively. For the sake of more sustainable hydrogenation, both decreased H2 pressures and renewable hydrogen sources show important opportunity areas for these ligand-tuned strategies.
2.1.2. Hydrogenation of Alkynes
Unlike the alkene reduction, Mn-catalyzed alkyne semihydrogenations generate two types of isomeric products: E- and Z-alkenes. For that reason, stereoselectivity (the preferable formation of one isomer) along with chemoselectivity (no over-reductions) are common goals for this type of reaction. To date, seven original contributions have been disclosed for this type of reactivity, and they are shown in Figure 3.
Figure 3.

Mn-based catalytic precursors for the semihydrogenation of alkynes.
The first contribution in this field was made by Driess and co-workers in 2018.33 They reported the use of Mn-5 (Figure 3), a Mn(II)–silylene complex, as a precatalyst for the efficient transfer semihydrogenation of internal alkynes to E-olefins with ammonia–borane as the hydrogen source. Since no conversion occurs under H2 atmosphere, it is proposed that the reaction undergoes a direct transfer hydrogenation mechanism rather than a dehydrogenation–hydrogenation pathway. The use of other silylene-based ligands or Mn(0) and Mn(I) precursors led to decreased activities or stereoselectivities. They proposed an in-situ formation of a Mn(I)–hydride as the catalytically active species for which a respective vibration signal was observed at 1900 cm–1.
Close to that date, the opposite stereoselective process was disclosed also with a Mn(II)-based catalytic precursor and using ammonia–borane (along with methanol) as the hydrogen source.34 Azofra and El-Sepelgy achieved the semihydrogenation of a collection of internal alkynes in the presence of Mn-6 (Figure 3), a PNP–Mn(II) complex. Lower conversions were obtained by employing other pincer ligands and solvents (except for ethanol and THF). Similar to the report by Driess, this system did not work under an H2 atmosphere, suggesting a direct transfer hydrogenation pathway. Once again, the in-situ formation of a Mn(I)–hydride is proposed as the catalytically active species, highlighting the concomitant use of ammonia–borane as both the hydrogen source and the catalyst activator.
A simplified and general mechanistic proposal for these two Mn(II)-based catalytic systems is shown in Scheme 4. Initially but not completely sustained, the in-situ reduction of the Mn(II) complex toward a coordinatively unsaturated Mn(I)–hydride affords the catalytically active species, which is capable of interacting with the substrate and continuing with an inner-sphere pathway. While isomerization of the initially formed Z-alkene is considered for the E-alkene formation in both processes, this is clearly more favored with the silylene ligand. Moreover, since there is no other significant difference between the two catalytic systems than the ligand σ-basicity, this comparison suggests that the alkene isomerization is promoted with a more electron-rich catalyst.
Scheme 4. Mechanistic Proposal for the Alkyne Semihydrogenation with Mn-5 and Mn-6.

Subsequently, three similar catalytic precursors were reported in 2020 for the semihydrogenation of alkynes to Z-alkenes. First, Junge and Beller studied the catalytic activity of a collection of Mn(I)–pincer (PNP, NNN, and PNN) complexes toward the direct hydrogenation of alkynes, obtaining better results when using Mn-7 (Figure 3).35 With further experiments, they demonstrated that the Z → E alkene isomerization does not occur at the reaction conditions and that higher temperatures are necessary to observe traces of the over-reduced product. Likewise, evidence of a MLC mechanism was given by changing the NH moiety of the pincer backbone for an N-methylated group, whose complex presented no conversion at the optimized reaction conditions. In addition, the MLC pathway was also supported by DFT calculations, and the good conversion attained by the ex profeso Mn(I)–hydride derived from Mn-7.
The other two reports were from the Rueping group. The first of them corresponded to a reduction under a H2 atmosphere performed by Mn-8 (Figure 3),36 while the second was a transfer hydrogenation process employing Mn-9 (Figure 3) as the precatalyst and methanol as the hydrogen source.37 For the former, the process was favored only in the presence of a strong base (viz. tBuOK) and non-polar solvents, while lower hydrogen pressures helped to minimize the competitive over-reduction process. From the substrate scope, better results were found when electron-withdrawing substituents were present. In the mechanistic proposal, evidence of an outer-sphere MLC pathway was presented with the inert behavior of the N-methylated congener Mn(I)-based complex and the unchanged 31P NMR spectra of the activated species in the presence of equimolar amounts of the model substrate (discarding the formation of a Mn–alkyne complex).
Regarding the other report by Rueping et al., they tested different Mn(I)-based complexes with non-innocent ligands for the transfer hydrogenation of alkynes to Z-alkenes. Excellent results were obtained with Mn-9 (Figure 3) and methanol as the hydrogen source; ethanol and isopropanol were also efficient transfer agents. While excellent conversions were attained when an aryl phosphine moiety was present at the ligand, only a trace amount of the product was observed with its change for an alkyl congener (namely, isopropyl). In addition to other catalytic systems, terminal alkynes proved to be inactive in the hydrogenation reaction. Albeit not in high amounts, over-hydrogenation to alkanes was reported for all of the tested substrates. In addition, the absence of isomerization was demonstrated by independent experiments with Z- and E-stilbenes. On the basis of deuterium-labeled experiments and the lack of activity observed using the N-methylated complex analogous to Mn-9, an outer-sphere pathway was proposed with a MLC mechanism for both the dehydrogenation of the alcohol and the reduction of the alkyne.
In summary, the last three original reports were demonstrated to operate through a MLC mechanism involving an outer-sphere interaction of the alkyne with the catalyst for its Z-selective reduction (Scheme 5). Although there is no evidence for the nature of this hydrogen transfer process (given that it can happen in a concerted manner or in steps), clearly the interaction occurs on only one side of the substrate, resulting in the favored cis isomer formation. Furthermore, isomerization was shown to not happen under these conditions.
Scheme 5. Mechanistic Proposal for the Alkyne Semihydrogenation with Mn-7, Mn-8, and Mn-9.

The latest reports on this topic were documented by Kirchner et al. and our group the same year using Mn(I)–diphosphine complexes as precatalysts. With respect to Kirchner’s work, they reported the semihydrogenation of both terminal and internal alkynes with Mn-2 (Figure 3) as a catalytic precursor, employing direct H2 or being generated in situ by the alcoholysis of KBH4 with methanol.38 It is noteworthy that terminal alkynes react under this methodology, which were reluctant to be reduced in previous catalytic systems. From the substrate scope, over-hydrogenation was detected in the presence of strong electron-withdrawing groups, which was favored when direct H2 was employed. For the mechanistic proposal, isomerization was displayed from the kinetic profile of the reaction and an inner-sphere mechanism was demonstrated with the reaction inhibition in the presence of PPh3, which competes with the substrate for coordination to the metal center. Furthermore, deuterium-labeling experiments revealed that the acidic proton of MeOH is almost exclusively incorporated into the substrate.
On the other hand, our group recently reported the transfer semihydrogenation of internal alkynes to E-alkenes with Mn-10 (Figure 3) as a catalytic precursor and isopropanol as the hydrogen source in the presence of a base.39 By testing different Mn(I)–diphosphine complexes, we observed that a σ-donor alkyl phosphine favored the transfer hydrogenation process. Likewise, from these, the steric influence of the ligand was demonstrated by the better results obtained with isopropyl substituents in comparison to the cyclohexyl. In particular, the Mn(I)–hydride analogous to Mn-10 was shown to be poorly active, although its in-situ formation was observed by 1H NMR of the reaction mixture, which is indicative of an off-cycle resting-state species. This observation also illustrates that not only is a Mn(I)–hydride necessary for the reduction but also a free coordination site is necessary as well for the interaction of the metal center and the substrate. The reaction still showed excellent conversions in the presence of Hg(0) and TEMPO but was inhibited in the presence of PPh3, which denoted a homogeneous system, a non-radical pathway, and an inner-sphere mechanism, respectively. Regarding the mechanistic proposal, reversible alkene isomerization was demonstrated by independent experiments, and its equilibrium is suggested to control the final stereoselectivity of the process. Furthermore, 1H NMR monitoring of the model reaction featured at least one asymmetrical hydride species, which was proposed to perform the catalytic turnover. It is noteworthy that we also observed the Mn-catalyzed hydration of α-keto alkynes toward 1,3-dicarbonylic compounds and derivates (vide infra).
These last two reports use structurally similar catalytic precursors (Figure 3, Mn-2 and Mn-10) for the alkyne semihydrogenation to E-alkenes, however, through different strategies. Making the comparison even more drastic, Kirchner et al. ruled out a transfer hydrogenation process for their system since no acetone (nor byproducts) was observed from the assay with isopropanol. This could be related to the absence of a base, which we identified to be important to promote the β-hydride elimination from the previously formed Mn(I)–alkoxide species or to the activation pathway observed for their precatalyst, which consists of the migratory insertion of the propyl ligand to form an acyl intermediate with the ability to activate different E–H bonds (E = C, B, Si, and H), yielding n-butanal and the respective E-based moiety coordinated to the manganese center. General and summarized mechanisms proposed for both catalytic systems are displayed in Scheme 6.
Scheme 6. Mechanistic Proposals for the Alkyne Semihydrogenation with Mn-2 (A) and Mn-10 (B).

Given that all of the catalytic systems already presented have been performed by well-defined manganese complexes, a few tendencies and intriguing things could be observed from the comparison between each other. First, all of the groups propose a Mn(I)-based complex as the catalytically active species. Although Mn(II)-based precursors were reported by two different groups, both posed their in-situ reduction to Mn(I)–hydride species.
In the early reports, this type of hydrogenation was achieved in the presence of pincer ligands. From these, a pyridyl moiety in the ligand backbone allowed the reaction with Mn(II) precursors since only poor conversions were attained with a NH group instead. Despite this, the latter type of pincer ligands resulted to be the “go-to” option for the Mn(I) centers, allowing the reduction to proceed through a MLC mechanism. This observation denotes the relevance of the oxidation state of the metal center and how it should be considered for the catalyst design as well.
Interestingly, in the work by Junge and Beller, ethyl- and cyclohexylphosphine moieties in ligands provided excellent conversions, while the yield was moderated with the isopropyl congener (precursors analogous to Mn-7, Figure 3). Although there is no evident explanation for this (since the steric and electronic properties are expected to be between the other mentioned substituents), this highlights how each process could be benefited from the ligand fine tuning.
Finally, albeit it could be obvious, it is worth mentioning that for the catalysts with inner-sphere mechanisms, the ability to create a free coordination site is essential. For the Mn(II)-based precatalysts, this was achieved with electron-rich and relatively bulky ligands, which result in tetracoordinate hydride species after their in-situ reduction. On the other hand, Kirchner took advantage of a migratory insertion process to yield a coordinatively unsaturated Mn(I)–acyl intermediate, whereas in our work, it was propitiated by the favored β-hydride elimination after isopropoxide coordination with acetone as a labile ligand.
2.2. Hydrosilylation
The catalytic hydrosilylation reactions are very profitable processes due to the remarkable added value of the products, which are of academic and industrial interest. In particular, addition of silicon compounds to unsaturated C–C bonds leads to organosilicon products, which are commonly employed as coupling agents, cross-linkers, and polymers (such as oils, rubbers, and resins).40 Here, we present the reports on the Mn-catalyzed hydrosilylation of alkenes and alkynes, which take advantage of the dual character of the silanes as hydride donors and radical promoters.
2.2.1. Hydrosilylation of Alkenes
The first report on this topic was published by Faltynek and Pratt in 1983.41 They described the thermal and photochemical activation of Mn-11 (Figure 4) for the hydrosilylation of 1-pentene with heptamethylcyclotetrasiloxane (HMCTS). Notoriously, evidence was given for a radical mechanism under thermal activation (starting with the Mn–Si homolysis), while a coordination pathway was proposed under photochemical conditions. From these, the light-promoted methodology presented higher regio- and chemoselectivity, forming only the linear hydrosilylated product.
Figure 4.

Mn-based catalytic precursors for the hydrosilylation of alkenes.
Later in that decade, a kinetic study about Mn-catalyzed hydrosilylation was presented by Hilal et al.42 They reported the reaction between tertiary silanes and 1-hexene with the commercially available Mn-12 (Figure 4) as a catalytic precursor. Their results suggested a highly selective process, albeit slower than a cobalt analogue. Despite a well-defined order for each reagent involved being determined (along with other tendencies), the mechanism for this process was not clear.
In the report by Shenvi for the HAT-initiated hydrogenation of alkenes (vide supra), they observed the anti-Markovnikov hydrosilylation of a terminal alkene with phenylsilane in the absence of TBHP (Figure 4).27 This reactivity was not detected with isopropoxy(phenyl)silane, which appeared to be a more powerful and versatile reductive agent than phenylsilane. Despite this result being presented as a side reaction in the alkene reduction (suppressed in the presence of TBHP), it highlights the HAT process as a useful initiation methodology for hydrosilylation reactions.
In 2017, Thomas et al. reported the activation of a series of catalytic precursors based on first-row transition metals with sodium tert-butoxide (tBuONa) toward the hydroboration and hydrosilylation of alkenes.43 Among these, they presented the hydrosilylation of 1-octene with phenylsilane, performed by Mn-13 (Figure 4), obtaining modest conversions. The authors focused on the activation methodology and proposed that it occurred via a former interaction between the base and the silane (or borane), resulting in an increase in its reductive capacity. This adduct would later react with the precatalyst for its in-situ reduction.
Subsequently, the same group disclosed a more detailed work about the hydrosilylation and hydroboration (vide infra) of alkenes, employing Mn-14 (Figure 4) and tBuONa as an activator, obtaining excellent regioselectivity toward the linear products.44 From the optimization of the catalytic system, it was observed that excellent yields were also obtained with MeONa and tBuOK, while organometallic reagents usually employed as reductive agents (e.g., EtMgBr, NaBHEt3, and LiAlH4) did not lead to any catalytic activity. On the other hand, the steric effect was an important feature since better results were obtained with bulkier substituents at the ortho position of the N-aryl moiety in the ligand. For the mechanism, a homogeneous and non-radical pathway was demonstrated with tests in the presence of DCT (dibenzo[a,e]-cyclooctatetraene) and TEMPO, respectively. In addition, they reported almost complete inhibition of reactivity in the presence of hydride traps (added after activation time), such as HCl (aq) and the trityl cation, which suggests a Mn–H intermediate as being responsible for the observed catalysis. Furthermore, deuterium-labeling experiments revealed a reversible hydrometalation step. Nevertheless, despite all of the additional experiments and observations, there is not a formal mechanistic proposal for this catalytic system.
Later that year, Trovitch’s group reported the use of the dimeric species Mn-15 (Figure 4) as the catalytic precursor for the hydrosilylation of alkenes.45 Interestingly, the regioselectivity of the process turned out to be substrate dependent, where aliphatic alkenes preferably underwent the anti-Markovnikov hydrosilylation, while styrenes mainly afforded the branched products. This found an explanation in the results of stoichiometric experiments, which revealed that the stereoselectivity was dictated by the alkene insertion into the Mn–H bond. From independent experiments, it was proved that Mn-15 did not react directly with the silane but with the alkene. Furthermore, the monomeric alkyl complex derived from the reaction between the precatalyst and styrene was also active for catalysis and regenerates Mn-15 in the presence of the silane. With all of these results, the authors reported the mechanism displayed in Scheme 7 (the alkene’s substituents were omitted for clarity, but as explained before, the regioselectivity of the hydrometalation is dependent on the substrate nature). Unfortunately, other silanes, such as Ph2SiH2, HSiPh3, and HSi(EtO)3, were unsuitable for this system.
Scheme 7. Mechanistic Proposal for the Alkene Hydrosilylation with Mn-15.

Wang reported the highly chemo- and regioselective anti-Markovnikov alkene hydrosilylation using the readily available Mn-16 (Figure 4).46 Notably, good yields were also obtained under UV irradiation and with aliphatic or aromatic silanes, while [Mn2(CO)10] (Mn-12) was also active for hydrosilylation under light irradiation (but it was barely explored and produced the reduced alkene as a byproduct). For the mechanistic proposal, deuterium-labeling experiments with styrene revealed that the hydrogen atom from the silane was transferred to the benzylic position without further scrambling. Moreover, independent structurally similar Mn–Si and Mn–H complexes were tested, but catalytic activity was observed only for the latter. The Mn–H was also demonstrated to participate in C–H bond formation. Finally, evidence about the radical nature of the process was presented with the lack of reactivity in the presence of TEMPO and the ring-opening detection when using vinyl cyclopropane as a substrate. Considering all of this information, a radical HAT pathway was proposed to be operating for this catalytic system. Simplified mechanisms for the alkene hydrosilylation with Mn-16 and Mn-12 (where L = CO) are displayed in Scheme 8.
Scheme 8. Radical Mechanisms Proposed for the Alkene Hydrosilylation with Mn-16 (A) and Mn-12 (B).

An outstanding work was disclosed by Xie and co-workers in 2021 regarding the divergent silylation of alkenes with [Mn2(CO)10] as a catalyst precursor and through a ligand-tuned metalloradical reactivity strategy.47 This manganese feedstock was tested in the presence of a broad variety of potential ligands for the catalytic hydrosilylation and dehydrogenative silylation of alkenes, with Mn-17 (Figure 4) being the best alternative for the former type of reactivity. It is noteworthy that a variety of complex and light alkenes was successfully hydrosilylated with different silanes under aerobic conditions, observing good regioselectivity toward the linear products. In addition, independent (deuterium-labeling) experiments and DFT calculations revealed that the system involved a radical pathway with the Si–H cleavage being the rate-determining step. The mechanistic proposal for this catalytic system is displayed in Scheme 8B.
Continuing with the use of commercially available manganese precursors, a communication was recently disclosed by Thieuleux’s group about the hydrosilylation of terminal alkenes with Mn-12 (Figure 4) under UV irradiation.48 Excellent yields toward the linear products were obtained at room temperature and in the absence of additives with the industrially relevant MDHM (1,1,1,3,5,5,5-heptamethyltrisiloxane). The authors reported that the process was inhibited in the presence of a radical scavenger (TEMPO) but not with a Hg drop test, which is indicative of a radical pathway and a homogeneous system. Unfortunately, inner and gem-alkenes remained unhydrosilylated, while considerably lower or null activity was observed with other silanes. Due to the nature of the catalytic process, it is likely that a mechanism similar to that presented in Scheme 8B is occurring (with L = CO). Although not too detailed, it is worth noting that this type of reactivity agrees with that reported by Wang et al. but changing the activation strategy.46
As presented, most of these hydrosilylation examples rely on the use of radical strategies. Usually in-situ formation of a Mn-centered radical is proposed, which then reacts with the silane in a HAT process to yield a Si-centered radical intermediate able to react with the alkene. At this point, the resulting C-centered radical intermediate might abstract an H atom from the reaction mixture (e.g., from another molecule of silane or the Mn–H species itself) to finally afford the hydrosilylated product.
In contrast, the catalytic systems with coordination mechanisms started with Mn(II)-based catalytic precursors. Despite one precatalyst being proposed to be reduced in situ to a Mn(I) species whereas the other was observed to remain as a Mn(II) complex, both might undergo similar pathways: a migratory insertion between the Mn–H intermediate and the alkene followed by a σ-bond metathesis with the silane to form the hydrosilylated product and regenerate the catalytically active species.
On the other hand, in terms of the regioselectivity attained, the anti-Markovnikov reaction was notoriously more favored for terminal alkenes, which is usually desirable in organic synthesis. In addition, in view of the justified tendency toward the development of greener catalytic processes, the use of available precursors (in some cases, even in the absence of ligands) is attractive and highlights the potential of Mn-based protocols.
2.2.2. Hydrosilylation of Alkynes
Vinyl silanes can be attained from hydrosilylation of alkynes and have proven to be versatile building blocks for organic synthesis.47 Although these products could also be obtained by dehydrogenative silylation of alkenes, these examples will not be detailed for the purpose of this review. Moreover, albeit less explored than its alkene counterpart, the manganese-catalyzed hydrosilylations of alkynes stand out for the use of readily available manganese sources.
The first report about the alkyne hydrosilylation with manganese was made by Wang’s group in 2018.49 Remarkably, they presented a regioselective and stereodivergent system based on commercially available Mn-16 and Mn-12 (Figure 5) for the formation of a vast collection of E- and Z-vinylsilanes, respectively, with a variety of silanes. For the first system, it was observed that the catalytic activity of Mn-16 can be tuned (even inhibited) in the presence of different ligands with triphenylarsine (AsPh3) being the best promoter, also with great chemo- and stereoselectivities. Interestingly, without an apparent reason, HSiEt3 was the only silane to present a higher affinity for the Z-product. Independent catalytic assays were made with Mn–H and Mn–Si complexes as potential intermediates, but comparable results were attained only with the Mn–Si species. These observations along with a further computational study presented by Li et al.50 serve as evidence for the mechanism presented in Scheme 9A as the plausible pathway for this transformation.
Figure 5.

Mn-based catalytic precursors for the hydrosilylation of alkynes.
Scheme 9. Mechanistic Proposals for the Alkyne Hydrosilylation with Mn-16 (A) and Mn-12 (B).

On the other hand, they observed that [Mn2(CO)10] (Mn-12) favored the formation of hydrosilylated Z-products and hypothesized that this could be occurring through a radical pathway due to the low Mn–Mn bond energy. To their delight, they observed an improved activity and stereoselectivity in the presence of a radical promoter, with dilauroyl peroxide (LPO) being the best alternative. From the optimization, it was also observed that the stereoselectivity of this process was enhanced by bulkier silanes. Finally, for the mechanistic proposal (Scheme 9B), a radical pathway was sustained by the good catalytic activity of the system under UV irradiation (which was already demonstrated to promote Mn–Mn bond homolysis) and its inhibition in the presence of a radical scavenger (viz. TEMPO). It is noteworthy that these examples highlight the potential of manganese in catalysis, with mechanisms proceeding via either classic organometallic reactions or metal-centered radicals.
Continuing with the Z-selective alkyne hydrosilylation with Mn-12 as precatalyst, Zhang, Zhang, and co-workers disclosed a visible-light-promoted protocol.51 Under mild conditions and blue LED irradiation, a wide variety of terminal, internal, and complex alkynes were successfully hydrosilylated with high regio-, chemo-, and stereoselectivities; under similar conditions, the alkyne hydrogermylation was also achieved (vide infra). This process was observed to be favored only in non-polar solvents, and considerably lower yields were attained under thermal activation. In comparison to the earlier mentioned protocol, where good stereoselectivities were only attained with bulky silanes, this catalytic system allows the incorporation of a wide variety of silanes with good to excellent yields. Evidence for a HAT mechanism was given by deuterium-labeling and kinetic isotope effect (KIE) experiments. Furthermore, the silane dimerization was observed in the absence of substrate, and the reaction was suppressed in the presence of a metallic hydride trap (namely, trityl cation) and radical scavengers (such as TEMPO, galvinoxyl, and hydroquinone), indicating the presence of a Mn–H intermediate and a radical pathway, respectively. This mechanistic proposal is similar to that displayed in Scheme 9B.
2.3. Hydroboration
The organoboronates, which can be obtained from the catalytic hydroboration of unsaturated C–C bonds, are highly valuable building blocks for the synthesis of fine chemicals. In particular, they are remarkably used as cross-coupling partners. Furthermore, the catalytic version of this reaction allows the addition of otherwise unreactive boranes and/or affords a regioselectivity unattainable by classical reactions.52 Below, we present the contributions made so far regarding the Mn-catalyzed hydroboration of alkenes and alkynes.
2.3.1. Hydroboration of Alkenes
The first report of any Mn-catalyzed alkene hydroboration was made by Zhang, Zheng, and co-workers in 2016.53 They disclosed a well-defined Mn(II) complex for the selective hydroboration of alkenes, ketones, and aldehydes (Figure 6, Mn-18). Analogous complexes with electronic-modified ligands as well as with chloride instead of alkyl ligands led to considerably lower conversions, highlighting the importance of Mn-18 in the process. Under mild conditions, a variety of styrenes was successfully hydroborated with pinacolborane to their respective Markovnikov products, while the opposite stereoselectivity was preferred with terminal aliphatic alkenes, and null conversion was obtained with geminal alkenes. A radical pathway was discarded by the lack of any ring-opening products in the assay with a substrate containing a cyclopropyl moiety, but no more information was given for a feasible mechanism.
Figure 6.

Mn-based catalytic precursors for the hydroboration of alkenes.
In 2018, Thomas explored the hydrosilylation and hydroboration of alkenes with Mn-14 (Figure 6) as the precatalyst and tBuONa as a promoter.44 For the latter type of reactivity, they reported the addition of pinacolborane in a series of terminal alkenes with excellent regioselectivity toward the linear products. Similar to what was observed in the hydrosilylation process (vide supra), the activation is proposed through an initial formation of a boronate complex with increased reductive character which reduces the Mn(II) precursor. Nevertheless, there is not much more information for a mechanistic proposal.
Also in 2018, Turculet et al. carried out an experimental and computational study of structurally analogous first-row complexes (bearing Mn2+, Fe2+, Co2+, or Ni2+) as precatalysts for the isomerization–hydroboration of structural-isomeric alkenes.54 The complex Mn-19 (Figure 6) was found to be considerably less active and chemoselective than its Fe and Co counterparts for the isomerization and hydroboration of octenes with pinacolborane. By stoichiometric experiments and DFT calculations, it was proposed that this system could be operating via a former metallic hydride formation through a σ-bond metathesis between the precatalyst and the borane. Further olefin insertion (with isomerization, if it is the case) and a final σ-bond metathesis between the Mn–alkyl species and the borane will finally yield the hydroborated product (Scheme 10). The poor yields obtained with Mn-19 were proposed to be related to the electronic properties of the metal center, in particular, the lack of a viable spin crossover mechanism for the Mn–H intermediate.
Scheme 10. Simplified Mechanistic Proposal for the Alkene Hydroboration with Mn-19.

Recently, Karton, de Ruiter, and co-workers disclosed the use of a well-defined Mn(II) complex as a precatalyst for the hydroboration of terminal alkenes with anti-Markovnikov regioselectivity (Figure 6, Mn-20).55 It is noteworthy that they also observed that the Co(II) congener also promoted the hydroboration process but with the prior isomerization of internal alkenes to the terminal isomer; by computational studies, it was proposed that this metal-based divergence is related to the more favored olefin migratory insertion into the metal hydride for a cobalt catalyst. Similar to what was reported by Thomas,44 the formation of the catalytically active species is proposed to occur through the in-situ precursor reduction by the alkoxide-assisted enhancement of the reductive character of the borane. In addition, DFT calculations for the mechanistic proposal suggested that the hemilability of the imino moiety was crucial for the advance of the reaction, as was the non-innocent behavior of the ligand. Finally, a σ-bond metathesis is likely to happen to yield the linear product and regenerate the catalyst. The mechanistic proposal for this system is presented in Scheme 11.
Scheme 11. Mechanistic Proposal for the Alkene Hydroboration with Mn-20.

The last report on this particular topic was recently made by Kirchner’s group.56 They used Mn-2 (Figure 6) for the anti-Markovnikov hydroboration of a collection of terminal alkenes along with the trans-1,2-diboration of terminal alkynes without the presence of any additive. For the alkene transformation, the homogeneity of the system as well as an inner-sphere pathway were demonstrated by a Hg drop test and an assay in the presence of PMe3, respectively. Furthermore, the complex [Mn(H)(dippe)(CO)2(κ1-HBPin)] (with two μ-hydrides bonding both Mn and B atoms) was isolated from the reaction between Mn-2 and pinacolborane, which was observed to also be formed in situ during NMR monitoring of the model reaction. This complex exhibited the same results for hydroboration, so it is proposed to be a resting state species capable of being activated under the reaction conditions. In addition, based on the DFT calculations, it was proposed that the coordinatively unsaturated Mn–BPin complex is the catalytically active species, but no further details were given for a plausible mechanism.
By comparison of the structures of the Mn–based precursors displayed in Figure 6, it could be noted that most of them contain Mn(II)–metal centers; nonetheless, the strategies followed for each system are different. For instance, a radical pathway is proposed for Turculet’s catalyst, while hydride migratory insertions were suggested for the systems of Zhang/Zheng, Thomas, and Karton/de Ruiter. Moreover, Karton and de Ruiter explained their precatalyst activation with the assistance of the non-innocent ligand, which cannot be ruled out to occur in Thomas’ catalyst as well. This apparently important feature of the ligand might unveil some potential further contributions in this area where the borane serves as both the catalyst activator and the reagent in the presence of a base.
In terms of the substrate and product descriptions, the development of protocols that lead to the Markovnikov products and the transformation of geminal alkenes is still desirable; these products might also be important in the pursuit of fine-chemicals synthesis. From these catalytic systems, only that by Zhang/Zheng reported major regioselectivity toward the branched product on the use of styrenes as substrates.
2.3.2. Hydroboration of Alkynes
There is only one report for the manganese-catalyzed alkyne hydroboration. This was made by Rueping and co-workers, bearing Mn-21 and Mn-22 (Figure 7) as catalytic precursors for the syn addition of pinacolborane into a broad collection of alkynes; the former showed better activity for symmetrical internal alkynes, whereas the latter showed better activity for protected propargylic alcohols and amines.57 From the optimization of the process, they observed that non-polar and aprotic solvents were only suitable as well as NaHBEt3 as an activator. Interestingly, both the alkoxide-promoted activation method reported by Thomas et al.44 and a precatalyst already reported for the alkyne semihydrogenation (Figure 3, Mn-6)34 were not applicable for this catalytic system. The homogeneity of the system was demonstrated by a Hg drop test, but there is no information about a mechanistic proposal. Nevertheless, due to the structure of the Mn(II)-based precursor, it is likely that it goes through a Mn–hydride formation via a direct Mn in-situ reduction or a ligand-assisted mechanism owing to the non-innocent character of that type of pincer ligand (vide supra).
Figure 7.

Mn-based catalytic precursor for the hydroboration of alkynes.
2.4. Hydroarylation
The addition of aryl moieties to unsaturated C–C bonds is a powerful process for the formation of C–C bonds with high atom efficiency. Since the late 1980s, C(sp2)–H bond activation with Mn complexes for the further addition of alkenes and alkynes has been disclosed.12,13,58 Although stoichiometric, they stated important set points for the works to come, such as the requirement of a directing group (a functional group that coordinates to the metal center and set the C–H bond to activate) and the proposal of heptacyclic manganese intermediates. The next examples correspond to significant improvements in this type of reactivity, encompassing its transformation into catalytic processes and the broadening of its versatility and selectivity.
Thus far, Mn-catalyzed hydroarylation of alkenes and alkynes has been reported with three different simple Mn precursors but mainly varying either the C–C unsaturated substrate or the aryl coupling partner. For the sake of the current review, these reports will be organized in a more comprehensive manner. The discussion will be centered on the differences and similarities between them.
Of note, other types of reactivities (beyond hydroarylations) have also been documented between C–H activated substrates and unsaturated C–C compounds, but they will not be included here. Nevertheless, they are already encompassed in other reviews.12,13,58
2.4.1. Hydroarylation of Alkenes
The first alkene hydroarylation was reported by Chen, Wang, and co-workers in 2014.59 They used Mn-16 (Figure 8) as a catalytic precursor for the addition of arylpyridines to α,β-unsaturated carbonyls in the presence of dicyclohexylamine. This system presented a high dependence on the solvent and the added base, obtaining considerably better yields with Et2O and the amine just mentioned. Except for [Mn2(CO)10], other transition metal-based precursors presented a lower or null activity. From the substrate scope, the presence of the carbonyl moiety was observed to be crucial in the alkene structure since no conversion was observed in the test with styrene. To demonstrate the viability of a C–H activation step in the catalytic system, the Mn–aryl complex was successfully synthesized and isolated from the reaction mixture in the absence of alkene; this Mn species was not detected in the absence of base, demonstrating its main role as the precatalyst activator. Moreover, the five-membered manganacycle was proved to catalyze the hydroarylation reaction, suggesting it as an intermediate in this process. Additional deuterium-labeled experiments indicated a reversible C–H activation step, while DFT calculations justified the necessity of a carbonyl moiety in the substrate (related to a more stabilized substrate coordination through a Mn–O interaction) and pointed out the olefin insertion step as the most energetically demanding. In Scheme 12 is displayed the simplified mechanism proposed for this catalytic system.
Figure 8.
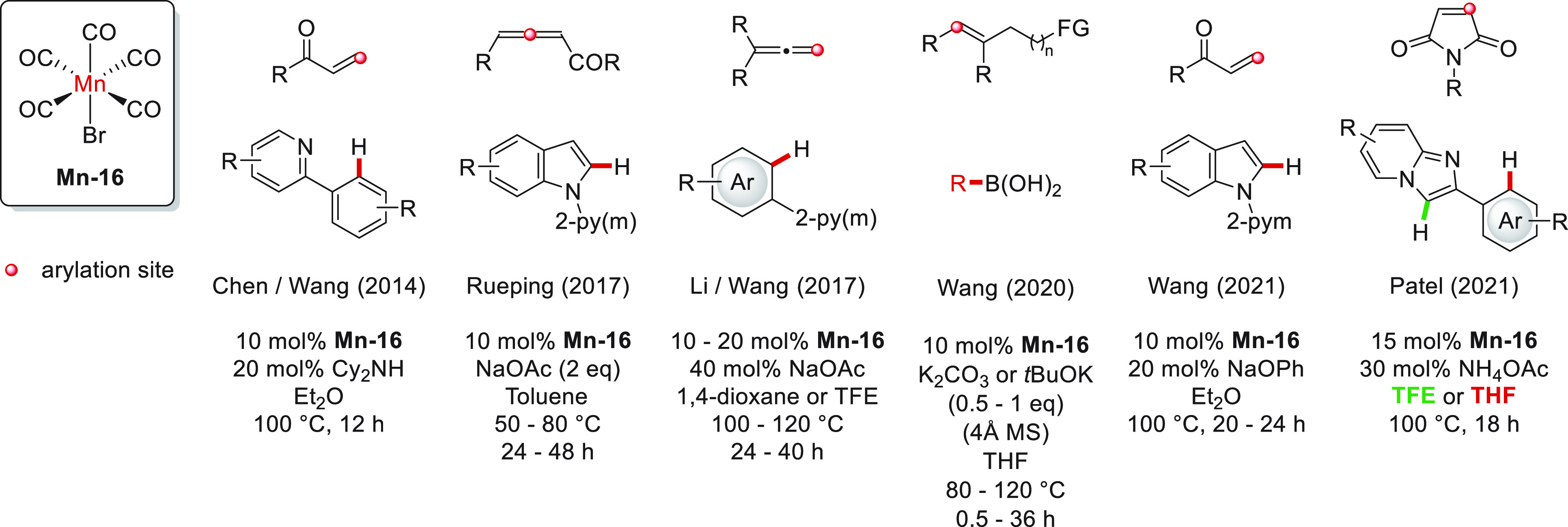
Mn-catalyzed hydroarylation of alkenes with Mn-16 as a catalytic precursor.
Scheme 12. Mechanistic Proposal for the Hydroarylation of α,β-Unsaturated Carbonyls with Mn-16.

Using the same Mn precursor, Rueping et al. reported the regio- and stereoselective hydroarylation of allenes with protected indoles under a slightly basic media and mild reaction conditions (Figure 8).60 For this system, the authors observed a stark increase in the yield upon NaOAc addition and that the reaction was efficient (although in slightly lower yields) at room temperature and under aerobic conditions. Noteworthy, when trisubstituted allenes are used, pyrroloindolones are obtained. For the mechanistic proposal of the hydroarylation process, the five-membered manganacycle (derived from a base-assisted C–H activation) was proved to be catalytically active in the presence of HOAc but not with NaOAc. This observation highlights the synergic behavior of the additive for the reaction, first assisting the C–H activation for the formation of the catalytically active species and then promoting the protodemetalation reaction between the proposed seven-membered manganacycle and the conjugate acid of the initially added base. A plausible mechanism for this system is presented in Scheme 13.
Scheme 13. Mechanistic Proposal for the Hydroarylation of Allenes (simplified as alkenes) with Mn-16.

One more allene hydroarylation with Mn-16 as precatalyst was presented by the groups of Li and Wang in 2017.61 Here, they reported the addition of a wide variety of aryl compounds into terminal allenes in the presence of lower loads of base (Figure 8). In the reaction optimization, they observed a relatively broad tolerance for other solvents but, more important, a better catalytic activity with Mn-16 than [Mn2(CO)10]. Unfortunately, the assays with unsymmetrical allenes presented relatively low stereoselectivities, whereas non-selective alkene isomerization was observed in the products from monosubstituted allenes, and tri- and tetrasubstituted allenes remained unconverted. Further experiments revealed (1) a low KIE value for the C–H activation, indicating that C–H bond cleavage is not the rate-determining step, (2) a reversible C–H activation step, (3) the five-membered manganacycle resulting from the C–H activation step is a catalytic intermediate, and (4) electron-rich indoles react faster. The author’s mechanistic proposal is summarized in Scheme 13.
Not using the same strategy (through a C–H activation) but with the same Mn precatalyst (Mn-16), the Wang group disclosed the hydroarylation of several non-activated alkenes with a variety of aryl boronic acids (Figure 8).62 By this protocol, a series of δ- and γ-arylated amides, ketones, pyridines, and amines could be afforded. This system was highly sensitive for the base employed but even more interestingly to the base counterion as well (in general, K was significantly better than Li, Na, and Cs). With further experiments, the authors proposed the formation of a manganacycle intermediate with the alkene’s functional group interacting with the metal center, which presumably is highly sensitive to water. Then, the regioselectivity of the process was suggested to be related to the easier formation of intermediates with five- and six-membered rings after arylation. On the basis of these observations, the nature of the catalyst, and the information provided for analogous systems reported by Xie et al. (see below), a simplified mechanistic proposal was presented (Scheme 14).
Scheme 14. Mechanistic Proposal for the Hydroarylation of Alkenes with Arylboronic Acids and Mn-16 or Mn-23 as a Catalytic Precursor.

Another Mn-catalyzed hydroarylation of α,β-unsaturated carbonyls was presented in 2021 by Wang’s group, albeit this time with indoles (Figure 8).63 Although this type of reactivity was already attained under their previous system,59 a remarked improvement was achieved under these new reaction conditions. From the optimization of the reaction, it was observed that other Re- and Mn-based precursors (bearing 0 and II oxidation states) led to null or negligible conversions, as do solvents other than Et2O. Moreover, a remarkably better yield was obtained in the presence of NaOPh, since only poor conversions were provided by other bases’ assistance. In stark contrast to other C–H activation methodologies (for instance, see the alkyne hydroarylation with Mn-16), the added base did not assist the initial C–H activation for the formation of the five-membered manganacycle intermediate; however, it was proposed to participate as the conjugate acid (phenol, from the reaction with the in-situ-formed HBr) in the protodemetalation step. Further (deuterium-labeling) experiments revealed that the reaction was faster with electron-rich indoles and the reversibility of the C–H activation step. Except for the base detail, the mechanistic proposal is like that depicted in Scheme 12.
An outstanding report was made also in 2021 for the alkene hydroarylation with Mn-16 as a catalytic precursor. Patel and co-workers reported the base-assisted and solvent-dependent site-selective alkylation of imidazopyridines with a collection of maleimides (Figure 8).64 In particular, with TFE as a solvent, the C–H bond from the C-3 position in the imidazopyridines was activated via an electrophilic distal metalation (supported, among other evidence, by the null reactivity observed with imidazopyridines bearing strong electron-withdrawing substituents), whereas the use of THF led to the well-documented C–H activation of the ortho position at the aryl group by a coordination assistance of the imidazopyridine (the five-membered manganacycle was later detected by ESI-MS). Different solvents and additives produced lower yields and site selectivities. Unfortunately, other alkenes remained unconverted at optimized conditions, while 2-phenylimidazoles yielded the N-alkylated (N–C bond formation from the imidazole and the alkene moieties, respectively) product in both solvents. Catalytic assays in the presence of a radical scavenger discarded a radical pathway for both systems, while experiments with D2O revealed the reversibility of the metalation steps and confirmed the site selectivity observed under different reaction conditions (favored deuterium incorporation at the respective activated site). Moreover, the obtained KIE values for both C–H bond cleavages suggested that they correspond to the rate-determining steps for their catalytic cycles. The simplified mechanism proposed for this novel site-divergent hydroarylation system is displayed in Scheme 15.
Scheme 15. Mechanistic Proposal for the Site-Divergent Hydroarylation of Alkenes with Imidazopyridines and Mn-16 as a Precatalyst.

Although [Mn2(CO)10] (Mn-12) has also shown activity as a precatalyst for some of the already mentioned alkene hydroarylation systems, the yields observed were inferior to those with Mn-16. Nevertheless, in 2017, Gong, Song, and co-workers presented the use of Mn-12 for the hydroarylation of maleimides with different indoles (Figure 9).65 These conditions also tolerated other alkene and aryl compounds but with modest yields in some cases. Of note, poor or null yields were afforded by employing other manganese compounds (with I and II oxidation states) and other transition metal complexes (including noble metals) as catalytic precursors, highlighting the unique activity of Mn-12 for this system. Furthermore, an additive is not necessary for this system, but the addition of dicyclohexylamine (20 mol %) enhanced the conversion of some specific substrates. Afterward, the C–H activation step was demonstrated to be reversible, but its KIE value revealed that this is not related to the rate-determining step. In addition, the five-membered manganacycle, proposed as an intermediate, was independently tested as a catalytic precursor, providing excellent yields. Finally, in Scheme 16 is presented the mechanistic proposal for this system, including (although not well explained) the in-situ oxidation of Mn(0) to Mn(I) with the assistance of the maleimide, which is in excess with respect to the aryl compound and is suggested to be acting as a base (this proposal is based on the five-membered manganacycle formation at optimized conditions, detected only in the presence of the alkene).
Figure 9.

Mn-catalyzed hydroarylation of alkenes with Mn-12 as a catalytic precursor.
Scheme 16. Mechanistic Proposal for the Hydroarylation of Alkenes with Mn-12 as a Catalytic Precursor.

Xie et al. explored the hydroarylation of different types of alkenes with aryl boronic acids and a dimeric Mn(I)-based catalytic precursor (Figure 10). The first of these reports was made in 2020 and reported the β-arylation of a variety of α,β-unsaturated amides in a highly chemo- and regioselective manner.66 Of note, this system also allowed the alkene hydroalkenylation with alkenyl boronic acids (vide infra). An initial comparison of the catalytic activity with [Mn(CO)5Br] (Mn-16) was made for some substrates, observing significantly better yields with Mn-23 as a catalytic precursor and demonstrating to be a more reactive species for this system. The presence of ethanol resulted in a yield enhancement; therefore, it was proposed that it might be operating as a proton source for the protodemetalation step, which was later confirmed by deuterium-labeling experiments. A tentative mechanistic proposal was made for this catalytic system, analogous to what is presented in Scheme 14 but (clearly) involving a different alkene substrate with a N coordination of the amide moiety in the intermediate after the migratory insertion step.
Figure 10.

Mn-catalyzed hydroarylation of alkenes with arylboronic acids and Mn-23 as a catalytic precursor.
The same year an analogous alkene hydroarylation system was disclosed but for the γ-arylation of β,γ-unsaturated amides (Figure 10).67 This protocol stands out for the selective derivatization of densely functionalized internal alkenes, which constantly are challenging substrates. From the optimization of the reaction, the authors observed decreased (even null) yields at lower water concentrations with different organic solvents and using precatalysts based on Pd and Ni; once again, Mn-23 proved to be more active than Mn-16. According to the authors, the observed regioselectivity originated from an interaction of the amide moiety with the metal center, which maintained intact other (terminal) alkenes present in some substrates. This Mn–amide interaction was analyzed by DFT calculations, indicating that the O coordination from the carbonyl is favored over the N coordination. In addition, experiments with D2O revealed that the water present in the reaction media is involved in the protodemetalation step. All of these observations are similar to those previously explained for the analogous Mn-16-based system presented by Wang (Figure 8). Therefore, the mechanistic proposal agrees with that displayed in Scheme 14 but with an easier way to generate the catalytically active species.
The latest of Xie’s reports was presented very recently, encompassing the anti-Markovnikov hydroarylation and hydroalkenylation of both terminal and internal enamides under mild conditions and in surprisingly short reaction times (Figure 10).68 For this process, the catalytic activity was practically independent of the water concentration but highly sensitive to the base and solvent employed. Moreover, they also observed a good tolerance for lower temperatures but poor or null conversions with other catalytic precursors (such as Mn-16 and Ni- and Rh-based complexes), thus highlighting (once again) the remarkable activity of Mn-23 for these processes. Notably, this system was demonstrated to be tolerable to complex substrates, which makes it a powerful alternative for late-stage transformations. A catalytic assay with D2O confirmed the water role as a protic source for the protodemetalation step, but it also revealed a reversible C–H activation in the N-protecting group (the N atom was protected with a Bz motif and derivates; the C–H activation was observed at the ortho position of the phenyl moiety). Although the mechanism still not clear, the authors proposed that it might be analogous to that displayed in Scheme 14, albeit with the enamide instead of the unsaturated amide as the substrate.
As was explained, there are (mainly) two different strategies for the Mn-catalyzed alkene hydroarylation, but both methodologies involved a directing group. In certain cases, there is a prone C–H activation step with mainly 2-pyridine and 2-pyrimidine motifs acting as directing groups, while in those involving aryl boronic acids as transmetalation agents, the directing group was present in the alkene reagent (bearing carbonyl or N-containing functional groups). More interesting comparisons between these two protocols are explained in detail at the end of the alkyne hydroarylation section (see below).
It is noteworthy that the solvent-switched protocol presented by Patel et al.64 corresponds to a unique type of metalation, where the Mn–C bond formation reassembles an electrophilic aromatic substitution pathway, assisted by a resonance effect from the heteroatom at the aryl moiety. Hence, this may correspond to a novel access to C–C bond formations at different aryl positions and/or new further Mn-catalyzed methodologies for the incorporation of whole new collections of aryl motifs.
The major catalytic activity noticed for Mn-23, in comparison to that for Mn-16, for the cross-coupling methodologies was sustained to be related to a more facile generation of a coordinatively unsaturated 16-e– Mn species, which can interact with either of the reagents. Even so, Mn-16 has the benefit of being a readily available substance, which might be a significant characteristic under certain circumstances (like industrial applications). Nevertheless, in some cases, the difference in yields was so critical to still consider this as a significant advantage.
2.4.2. Hydroarylation of Alkynes
In 2013, Chen, Wang, and co-workers reported the first example of the catalytic hydroarylation of terminal alkynes using a commercially available base and manganese precursor (Figure 11).69 This process was highly chemo-, regio-, and stereoselective toward the anti-Markovnikov E-configured hydroarylated product with arylpyridines as the added moiety. The presence of a base improved the conversion by promotion of the C–H activation, enhancing the formation of the catalytically active species; relatively bulky amines were beneficial for the reaction. Interestingly, [Mn2(CO)10] worked as well but not other non-carbonyl Mn species. The same suppressed reactivity was obtained with other transition metal-based carbonyl complexes. These results highlight not only the Mn relevance for this process but also the oxidation state and the coordination environment suitable for this reaction. At optimized conditions, a wide scope of substrates was analyzed, including several variations in the alkyne, aryl, and pyridine motifs. By independent experiments (including deuterium labeling) and DFT calculations, they proposed the mechanism displayed in Scheme 17. In general, they demonstrated that the C–H activation of the arylpyridine was promoted by the base, whose product was isolated and had the same catalytic activity. Furthermore, they proposed the formation of a seven-membered manganacycle as the key step for the chemo-, regio-, and stereoselectivities with a ligand-to-ligand H transfer pathway for the final product formation.
Figure 11.

Mn-catalyzed hydroarylation of alkynes with Mn-16 as a catalytic precursor.
Scheme 17. Simplified Mechanistic Proposal for the Alkyne Hydroarylation of Alkynes with Mn-16 as a Catalytic Precursor.

Using the same catalytic precursor but changing the reaction conditions, in 2015 Lei and Li et al. observed a dichotomy in reactivity between alkynes and indoles (Figure 11).70 On one side, the alkenylation with both terminal and internal alkynes in a highly regio- and stereoselective manner was achieved in the presence of DIPEA and benzoic acid. On the other hand, the dehydrogenative indole cyclization with two arylacetylenes toward carbazoles was attained with DIPEA as the only additive (this reactivity will not be further discussed). For the hydroarylation process, similarly to what was observed in the work just mentioned, the use of other transition metal carbonyl complexes and Mn compounds in other oxidation states led to lower or null conversions; the same occurred with other bases. At the optimized conditions, either alkyne or indole variations were tolerated. For the mechanistic insights, the manganacycle derived from the base-assisted indole’s C–H activation was demonstrated to be an intermediate of the catalytic cycle, while the acid was suggested to be an H transmitter agent. Further deuterium-labeling experiments revealed the C–H activation as the rate-determining step. The mechanism proposed for this catalytic system is consistent with that displayed in Scheme 17.
In 2017, Ackermann’s group reported a continuous flow methodology for the salient hydroarylation of functionalized alkynes without the usual concomitant β-bond cleavage observed in substrates with good leaving groups; this chemoselective hydrofunctionalization allows a variety of late-stage modifications of the products (Figure 11).71 While base additives (which worked for the previous catalytic systems presented here) did not yield the desired product, its change for Brønsted acids (viz. carboxylic acids) appeared to promote the hydroarylation reaction with the suppression of the β-O elimination process. This catalytic system turned out to be applicable to a variety of aryl moieties and propargyl alcohol derivates within very short reaction times (1–20 min) under flow conditions or longer times (16 h) under batch regime. For this system, they found that it was favored with electron-rich aryl compounds along with the catalytic intermediate role of the manganacycle derived from the C–H activation. In addition, computational studies suggest the protodemetalation to proceed via an intramolecular proton transfer; nevertheless, there is no explicit mechanistic proposal.
Another base-assisted hydroarylation of internal alkynes with indoles and Mn-16 as a catalytic precursor along with the Re-catalyzed congener was presented in 2018 by Rueping’s group (Figure 11).72 Although having long reaction times (70 h), this protocol stands out for being carried out in the presence of readily available and relatively cheap reagents. Due to the nature of this catalytic system, it likely operates via the already described manganacycle formation favored by the presence of a base.
The hydroarylation of arylalkynes with amidines was disclosed in 2019 by Wang and co-workers using Mn-16 as a precatalyst and promoted by a Grignard reagent (Figure 11).73 The authors observed that other Mn precursors (in different oxidation states) along with other bases and solvents were not suitable for this system. By competing experiments, they also observed that the process was favored with electron-rich amidines and alkynes with more drastic differences being observed in the latter. Furthermore, they also demonstrated the reversibility of the C–H activation with deuterium-labeling experiments, while the KIE observed (1.76) suggested that C–H activation is not necessarily the rate-determining step. Finally, based on the presented and previous results, the plausible mechanism proposed is like that presented in Scheme 17.
In 2020, Glorius and co-workers described the use of Mn-16 for the highly regio-, chemo-, and stereoselective hydroarylation of a variety of 1,3-diynes employing a plethora of arenes and heteroarenes (Figure 11).74 Of note, when using diynes bearing protected amines or hydroxy moieties, they obtained the one-pot synthesis of pyrroles or furans, respectively. The base was indispensable to obtain efficient yields, while the Mn relevance for this process was demonstrated by the inactive behavior of other transition metal precatalysts (based on Rh, Co, and Ru) already reported for C–H activation and used in reactions with 1,3-diynes. Moreover, for the sake of a mechanistic proposal, the KIE obtained from deuterium-labeling experiments revealed that C–H activation was not the rate-determining step, while competition experiments showed that electron-rich indoles reacted faster. With all of this information and based on the nature of the catalyst, the mechanism proposed by the authors involves the base-assisted Mn-catalyzed C–H activation and the formation of a seven-membered manganacycle (Scheme 17).
In 2021, Xu and Cao et al. presented a detailed work about the hydroarylation of both terminal and internal alkynes with 2-pyridones and Mn-16 as the precatalyst (Figure 11).75 From the optimization of the reaction, the authors observed an important influence of the solvent in the catalytic activity, being better with a non-polar solvent. A similar contrasting difference in activity was observed from the test of different bases, but there is no apparent tendency in reactivity; good results were attained in the presence of KOAc, K2CO3, and NEt3, being better with the former. Of note, the addition of acids as well as the use of different Mn precursors (namely, MnCl2 and Mn2(CO)10) led to decreased or null conversions. Interestingly, the protocols presented by Li, Lei, and co-workers70 and Ackermann et al.71 (Figure 11, second and third reports) were inefficient for the hydroarylation with 2-pyridones, highlighting the reaction conditions that the authors presented. Furthermore, a variety of N-protecting groups (such as Me, Ph, Ac, Boc, Piv, and Ts) and free 2-pyridone were tested but did not react, demonstrating that the 2-pyridine moiety works not only as a protecting group but as a directing group as well. For the mechanistic proposal, the authors observed that the five-membered manganacycle derived from the C–H activation was also active at catalytic loads, which suggests this Mn complex as an intermediate of the catalytic cycle. Moreover, the H/D scrambling observed from deuterium-labeled experiments indicated a reversible C–H activation step, while the KIE values revealed that the C–H activation might be the rate-determining step in the reaction with terminal alkynes but not with the internal alkynes. Finally, the mechanistic proposal was consistent with what is depicted in Scheme 17.
Giving even more added value to the alkyne hydroarylation reaction and envisioning its application in more complex systems, recently, Ackermann and co-workers presented the outstanding hydroarylation of a plethora of propiolates,76 ethynyl–BODIPY’s,77 and ethynyl–carboranes78 with indoles containing different structurally complex biomolecules, such as peptides, natural products, and sugars (Figure 11). All of these protocols were free of peptide racemization and highly tolerant to a variety of functional groups. Furthermore, among the advantages presented by these methodologies, access to peptide-based macrocycles and the non-noble transition metal-catalyzed fluorescent labeling of structurally complex peptides stand out.
For the protocol involving carboranes, the presence of basic or acidic additives led to efficient conversions, being better only with the latter. On the other hand, the indispensability of Mn-16 was proven by the non-activity of either Mn(0) or heavier transition metal-based precursors. Moreover, the reversible C–H activation step was suggested by the H/D scrambling observed using deuterated acetic acid, while a substrate competition experiment revealed a more favored process for electron-rich aryl partners. The mechanistic proposal agrees with the already proposed pathways for analogous alkyne hydroarylations with indoles (Scheme 17).
The most recent report using Mn-16 as a precatalyst for the hydroarylation of alkynes was made by Hajra and co-workers.79 Among the most noteworthy contributions of this work are the incorporation of a new type of aryl compound, 2-arylindazoles, along with their excellent performance in aqueous medium (Figure 11). As was also observed in other catalytic systems of this nature, considerably lower yields were obtained with different Mn-based precursors, solvents (except for 1,4-dioxane), and bases. Finally, deuterium incorporation at the ortho position in the presence of D2O revealed the reversibility of the C–H activation step, whereas its KIE value discarded it as the rate-determining step. Considering these results and analogous systems also based on Mn-16, the mechanistic proposal is similar to that described in Scheme 17.
Regarding the mechanistic proposal, Lynam and Fairlamb et al. provided some crucial insights for the Mn-16-catalyzed hydroarylation through initial C–H activation.80 In general, they were able to characterize the previously speculated short-lived seven-membered manganacycle formed by the alkyne insertion into the previously formed Mn–C bond. Furthermore, for the relatively high catalytic loads (10 mol %) commonly required for the optimal function of these systems, they established a couple of potential deactivation pathways, highlighting the formation of manganese clusters or side reactions derived from the manganacycle intermediate. In addition, they also studied the additive role in the C–H activation and the protodemetalation steps and the photoinduced CO dissociation from the five-membered manganacycle as a possible new methodology for the catalyst’s activation.
With all of the information already presented regarding the reaction conditions and the reagents involved, a few marked constants could be perceived. First, the presence of additives (either basic or acidic) allowed the use of Mn-16 in catalytic amounts (although an improvement of even lower catalytic loads is highly desirable) via the additive-assisted C–H activation and/or protodemetalation. In most of the presented reports, deuterium-labeled experiments revealed H/D scrambling at the C–H-activated bond, which is indicative of a reversible C–H activation step. In addition, the KIE results revealed the C–H activation reaction as the rate-determining step for the process with terminal alkynes; for internal alkynes, this was not observed to be occurring. Subsequently, the seven-membered intermediate formed from the migratory insertion of the alkyne into the already formed Mn–C bond is proposed to be a key step for the high regio- and stereoselectivity observed for these hydroarylation methodologies.
Finally, for the substrates involved in the reaction, broad robustness was observed for both the alkynes and the aryl compounds (albeit internal alkynes usually had lower yields). Although a plethora of functionalized aryl moieties were successfully added into the alkynes, the presence of a directing group was crucial with pyridine and pyrimidine being the most suitable. Other potential directing or protecting groups were inefficient for these protocols. Of note, for the indole case, the N protection with pyridine or pyrimidine was demonstrated to be reversible, highlighting the added value of the products as building blocks for further transformations. In relation to pyridine and pyrimidine as directing groups, their sharp contrasts in reactivity could be attributed not only to the N bonding with the Mn center but also to the electronic properties of these moieties and their interactions with the aryl fragments.
As previously mentioned, Mn-12 also had catalytic activity for some hydroarylation systems, but it was significantly lower than that with Mn-16 in most cases. Nevertheless, two papers were found bearing this Mn species as the precatalyst (Figure 12).
Figure 12.

Mn-catalyzed hydroarylation of alkynes with Mn-12 as a catalytic precursor.
The first of them was made by Wang’s group in 2016.81 They reported the highly selective hydroarylation of alkynes with aromatic nitriles (masked as imidates) in the presence of catalytic amounts of sodium pivalate (Figure 12). Of note, the authors visualized the nitriles’ masking step as a way to avoid side reactions and change the linearity of the nitrile moiety to afford suitable directing groups for the C–H activation process. From the optimization of this process, the authors observed null activity with other potential precatalysts, such as Mn(OAc)2 and Re2(CO)10. In addition, the yields were increased in the presence of bases, obtaining better performance with weaker bases, presumably, due to imidate stability.
The masked substrate’s conversion was favored with the ethanol-added product rather than methanol and isopropanol congeners. Further experiments showed that the C–H activation step is reversible and in competition with the dealcoholization of the aryl substrate; the latter was found to occur faster in the absence of alkyne. In addition, from experiments with terminal alkynes, low deuterium incorporation at the olefinic positions was observed and a high KIE value was determined for the C–H activation step, which suggests that it is the rate-determining step. The proposed mechanism for this system is presented in Scheme 18, which is like that proposed for Mn-16 (vide supra) but without a well-defined precursor’s activation involving an in-situ Mn oxidation.
Scheme 18. Mechanistic Proposal for the Alkyne Hydroarylation with Mn-12 as a Catalytic Precursor.

On the other hand, in 2019, Prabhu and co-workers disclosed the use of Mn-12 as a precatalyst for the hydroarylation of 4-hydroxy-2-alkynoates (propargyl alcohol derivates) with, mainly, protected indoles (Figure 12), including a concomitant intramolecular cyclization to yield arylated lactones.82 Although initial experiments were carried out with [Mn(CO)5Br] (Mn-16), better conversions and regioselectivities were afforded with Mn-12. In addition, they also observed that this process is only favored with basic additives since the addition of an acid (namely, AcOH) almost suppressed the reaction. Furthermore, the authors also noticed that the regioselectivity of the process could be enhanced by fine tuning the C-4 substituents in the alkynoate substrate and that their ethyl esters are more reactive than the methyl congeners. For the sake of a mechanistic proposal, relevant information was attained with further (deuterium-labeling) experiments. For instance: (1) the C–H activation step is reversible and does not correspond to the rate-determining step; (2) at optimized conditions, the in-situ formation of a five-membered manganacycle from the reaction between the indole and the Mn precursor was detected; (3) the H atom of the C-2 indole position ended at the olefinic position. In general, these observations are like those just mentioned for the other Mn-12-based alkyne hydroarylation system; thus, the plausible mechanism is also alike (Scheme 18). Of note, the authors suggested that the regiocontrol observed for this process is related to the steric hindrance of the C-4 substituents in the alkynoate substrate and that the concomitant intramolecular lactonization occurs after the alkyne hydroarylation process.
The last type of methodology used for the alkyne hydroarylation was presented by Xie’s group and is in stark contrast to the previously mentioned examples, which involved a C–H activation step. Here, they used Mn-23 for the hydroarylation of a variety of propargyl alcohol derivates with a plethora of commercially available aromatic boronic acids (Figure 13). These protocols encompass a transmetalation step and a dimeric Mn(I) species as a precatalyst, which apparently is a more reactive Mn precursor than its [Mn(CO)5Br] (Mn-16) congener.
Figure 13.

Mn-catalyzed hydroarylation of alkynes with Mn-23 as a catalytic precursor.
The first of these reports was made in 2018 and dealt with the hydroarylation of unsymmetrical 1,3-diyne alcohols in a regio-, stereo-, and chemoselective manner.83 For this catalytic system, they found a high yield dependence of solvents and additives; in general, better results were attained with non-polar reaction media and relatively weak bases (a marked cation influence was also observed for the base). Of note, worse or negligible conversions were observed using other transition metal-based precursors or [Mn(CO)5Br] (Mn-16), highlighting the relevance of Mn-23 for this catalytic system.
At the optimized conditions, there is not a significant effect of the electronic and steric properties of the alkynes and arylboronic acids on the hydroarylation regioselectivity. Among the key observations of the system are (1) the requirement of a hydroxy group and either aryl or alkynyl substituents in the substrates, (2) the proton at the olefinic position should come from the reaction media, (3) the verification of the transmetalation step viability at reaction conditions, and (4) the stereoselectivity is likely to be defined from the weak interaction between the hydroxy group in the alkyne and the aryl group in the Mn–aryl complex formed after the transmetalation step.
A computational study of this catalytic system was made in 2021 by Yuan, Bi, and co-workers.84 They compared the energetic profiles of the reaction with both Mn-16 and Mn-23 as catalytic precursors and found the main difference between them is in the precatalyst transformation toward the catalytically active species. The energy required for the activation of the mononuclear complex is higher than its dimeric congener with dissociation likely being the rate-determining step of its process and justifying the lower activity observed in the presence of this precursor. On the other hand, the calculated energy required for Mn-23 activation was lower; thus, the diyne migratory insertion step was further identified as the (selectivity- and) rate-determining step. Furthermore, four main steps were proposed for this catalytic cycle: (1) transmetalation, (2) alkyne insertion, (3) protodemetalation, and (4) active catalyst regeneration. Finally, the predominant β-arylation of this methodology was observed to be related to a both kinetically and thermodynamically favored reaction over the other possible insertion modes involving a deformation of the diyne section and the (previously proposed) hydroxy–aryl π interaction. Considering all of this information, a simplified mechanism for this system is presented in Scheme 19; the water is proposed to be formed in situ from the boronic acid.
Scheme 19. Mechanistic Proposal for the 1,3-Diyne Hydroarylation with Arylboronic Acids and Mn-23 as a Catalytic Precursor.

On the other hand, with the same catalytic precursor (Mn-23), in the presence of water, and under an air atmosphere, a considerably broad collection of (complex) propargyl alcohol derivates was hydroarylated with a wide variety of (complex) boronic acids.85 Nevertheless, in comparison to the β-regioselectivity arylation observed in the previous protocol, the γ-arylation was predominant under these new reaction conditions. This change in regioselectivity was proposed to be controlled by both a steric effect (since tertiary alcohols are preferred) and the influence of the inherent hydroxy group in the alkyne. This reaction was observed to proceed in short reaction times and with high regio-, stereo-, and chemoselectivities. Once again, the use of [Mn(CO)5Br] (Mn-16) as the precatalyst led to lower conversions, while insignificant yields were afforded with precursors based on other transition metals. It is worth mentioning that this protocol was demonstrated to be applicable at late-stage synthetic routes, tolerating a variety of functional groups and complex structures.
Regarding the mechanism, it has been recently studied by Yuan, Bi, Xie, and co-workers.86 In this work they found that the activation step proceeds via a bimetallic mechanism, yielding a monomeric catalytically active species, which subsequently undergoes an alkyne migratory insertion, a protodemetalation, and catalyst regeneration, completing the catalytic turnover. From these, precatalyst activation was identified as the rate-determining step, whereas alkyne migratory insertion was proposed to be mainly responsible for the regioselectivity, which is highly dependent on the steric properties of the species involved. For the analyzed tertiary propargyl alcohol derivate, γ-arylation was calculated to be the most kinetically and thermodynamically type of insertion, agreeing with the regioselectivity observed. A simplified version of the mechanism proposed for this catalytic system is displayed in Scheme 20.
Scheme 20. Mechanistic Proposal for the Alkyne Hydroarylation with Arylboronic Acids and Mn-23 as a Catalytic Precursor.

In general, the Mn-catalyzed alkyne hydroarylation has been achieved by two strategies: (1) addition of alkyne into activated C–H bonds and (2) cross-coupling between alkynes and arylboronic acids. Both have been demonstrated to be operable under batch conditions (where the prior formation of Mn-based complexes is not necessary) and highly useful in the construction of C–C bonds even for the late-stage modifications of complex molecules. In addition, broad robustness and tolerance toward air and moisture were also attained in the latest examples. All of these characteristics highlight the potential of these methodologies in the big-scale production of compounds with both academic and industrial interest.
Regarding the hydroarylation processes with Mn-16, it can be observed that the addition of acids led to better conversions in certain cases; nevertheless, there are more examples where the basic additives were more suitable. Despite there not being a clear explanation for this, this observation highlights the potential of the technique after a simple fine tuning of the reaction conditions.
When these two methodologies are compared, the one involving arylboronic acids has the advantage of using commercially available substrates. However, even more importantly, they also can create the C–C bonds at more sites of the aryl motifs; this is not restricted to the confined position determined by a directing group. In contrast, the C–H activation methodology has been more developed, achieving even the incorporation of very complex segments, such as different biomolecules, which shows its highly valuable use at late-stage synthesis routes (nonetheless, there is not an apparent limitation for this to be applicable with organoboron compounds).
Among the opportunity areas of these processes are (1) the decrease in catalytic loads, (2) the expansion of the cross-coupling strategy toward other alkynes, and (3) the pursuit of C–H activation at new sites of the aryl compounds (for instance, see the work presented by Patel et al. for the alkene hydroarylation64).
2.5. Hydration
The hydration of alkenes and alkynes is a powerful technique that allows their transformation into alcohols and enols (which rapidly tautomerize to ketones and aldehydes), respectively. Compared to the unsaturated hydrocarbon reagents, these products are synthetically more versatile due to a more polarized motif that makes them relatively more reactive.87
As will be presented, the Mn-catalyzed hydration strategies are different for alkenes and alkynes. Whereas the former can be visualized as oxidation processes, the latter correspond to a direct reaction with water.
2.5.1. Hydration of Alkenes
In Figure 14, all of the examples found for the homogeneous Mn-catalyzed hydration of alkenes are depicted. Although these processes could be considered hydration reactions (since there is a final incorporation of −H and −OH moieties at the olefinic positions), they do not operate through direct addition of a water molecule but via oxidation in the presence of O2.
Figure 14.

Mn-based catalytic precursors for the hydration of alkenes.
The first report of any homogeneous Mn-catalyzed alkene hydration was made in 1990 by Mukaiyama and co-workers.88 In the presence of catalytic amounts of Mn-1 (originally, it was reported to be the [Mn(dpm)2] complex, but this was corrected later), with phenylsilane, and under an oxygen atmosphere, they achieved the hydration of α,β-unsaturated esters with the hydroxy group at the α position after a final treatment with a Na2S2O3 solution (Figure 14). The authors noted a high dependence on the solvent employed (obtaining only good results with primary and secondary alcohols) and the substituents at the β position of the substrate. For the latter, the authors observed increased regioselectivity toward the β-hydroxy products with the major substitution at this same position, which agrees with the proposed radical mechanism. Although no further details are given for this system, it served as the basis for the research to come.
Ten years later, under relatively similar conditions, Magnus and co-workers reported the transformation of α,β-unsaturated ketones to α-hydroxy ketones after a final quenching with P(OEt)3 (Figure 14).89 Remarkably, this group demonstrated that the [Mn(dpm)2] complex previously reported for Mukaiyama’s system was actually the Mn-1 complex. According to the authors, addition of the phosphite was in order to reduce a peroxide intermediate and yield the final product. Moreover, deuterium-labeling experiments in the absence of oxygen indicated an irreversible hydride addition step, while alkene reduction was identified as a side reaction in the absence of oxygen. Once again, the derivatization of β,β-disubstituted substrates was difficult, even remaining inert in the absence of O2.
More mechanistic insights were obtained from specific stoichiometric experiments, demonstrating the in-situ formation of the intermediate [HMn(dpm)2] (from the mixture of Mn-1, phenylsilane, and isopropanol), which immediately reacts with oxygen in a reversible manner. Finally, a plausible mechanism for this catalytic system was reported (Scheme 21A), but it remains unclear, despite some important observations and corrections being made by other contributions. This is discussed at the end of this section.
Scheme 21. Mechanistic Proposals for the Mn-Catalyzed Hydration of Alkenes: (A) Radical Pathway; (B) Non-radical Pathway.

A little later, the same group reported the hydration of α,β-unsaturated nitriles with Mn-1 as a precatalyst and under similar reaction conditions (Figure 14).90 In stark contrast to the transformation of α,β-unsaturated ketones, only modest conversions were obtained and, in some cases, with low regioselectivity as well. In particular, the mentioned decrease in regioselectivity was observed when there was more than one substituent at the β position of the substrate, while the significantly lower yields noticed for the more active substrates (e.g., olefins difunctionalized with strong electron-withdrawing groups at geminal positions) were related to a considerably favored reduction side reaction even in the presence of O2. Finally, owing to the nature of this catalytic system, it is likely that it operates through a mechanism analogous to that presented in Scheme 21 (vide infra).
In 2004, the Yamada group disclosed the use of Mn-1 for the stereoselective hydration of α,β-unsaturated amides toward (R)-α-hydroxyamides (Figure 14).91 As the authors demonstrated, the stereoselectivity of the reaction derives from the use of substrates containing chiral substituents at the amides’ nitrogen atom; apparently, major stereoselectivities were afforded with bulkier chiral fragments due to the hampered oxygen approximation with a radical intermediate. Although there is not any explicit mechanistic proposal, the authors mentioned that the route depicted in Scheme 21A is likely to happen.
In particular, the applicability of this Mn-1 and iPrOH/O2 system was demonstrated in 2010 by Cassayre and co-workers for a variety of complex substrates.92 Although not all of them proceeded in an efficient manner, some of them did exhibit a high level of regio- and stereoselectivity. Therefore, these observations highlight the potential of this Mn-catalyzed hydration methodology, even for late-stage synthetic steps.
As mentioned in the alkene hydrogenation section (vide supra), in 2016 Shenvi and co-workers reported a very active silane-based reductant system that served as a HAT promoter.27 Among the reactions initiated by this HAT process, they briefly explored the hydration of a terminal aliphatic alkene, obtaining its respective anti-Markovnikov product. They monitored the reaction just mentioned with catalytic amounts of Mn-24 and Mn-1 (Figure 14), varying the solvent and silane. From these experiments, a slightly better and faster conversion was attained with Mn-24 in THF and with isopropoxy(phenyl)silane (Ph(iPrO)SiH2) in comparison to Mn-1 under the same conditions. Of note, albeit for another substrate, Mukaiyama previously reported a negligible catalytic activity of Mn-1 in THF and modest conversions with an Mn–acac complex as the precursor,88 which could point to the silane reagent being an improved promoter for the alkene hydration (similar to what was observed for the hydrogenation reaction).
In 2019, the groups of Donnelly and Rizzacasa disclosed an enhanced hydration of α,β-unsaturated esters toward α-hydroxy esters (compared to the previously mentioned work by Mukaiyama et al.)88 by the use of a Mn(III) complex with a Schiff base ligand (Mn-25, Figure 14).93 Notably, the authors reported that the more reactive silane previously used by Shenvi (Ph(iPrO)SiH2)27 did not present a significant improvement for the reaction; therefore, the reaction conditions remained similar to Mukaiyama’s report. Furthermore, they also found that the treatment with P(OEt)3 was not necessary, as previously reported by Magnus,89 suggesting that the hydroperoxides are not formed in significant amounts or that they could be reduced in situ by the silane in excess. After optimization of the reaction and with this new catalytic precursor, the alkene hydrogenation was suppressed (or present only in low amounts, such as 6%) as a side reaction for most of the tested substrates, providing a much more chemoselective process in comparison to the results with Mn-1. It is worth noting that the β-hydroxy products are preferred when the substrate has a phenyl moiety or two geminal substituents at this same position, which might suggest that a radical pathway is involved; nevertheless, an ionic mechanism could not be ruled out with the given evidence. Unfortunately, a test in the presence of radical scavengers has not been reported, which might shed light over this issue. Thus, both mechanistic proposals are presented in Scheme 21.
As it has been explained, the Mn-catalyzed alkene hydration reports rely on the silane/oxygen mixture to achieve this transformation. Although most of these methodologies are informed for α,β-unsaturated carbonyl compounds (and derivates) with major regioselectivity toward the α-hydroxy products, it was demonstrated that this reactivity could also be extended to terminal alkenes. Therefore, a broader substrate scope along with the development of other potentially applicable techniques would be very beneficial. On the other hand, it is noticeable that a more detailed description of the mechanism involved is highly desirable since the different sorts of selectivities could be improved by its understanding.
2.5.2. Hydration of Alkynes
The alkyne hydration corresponds to a simple yet valuable technique to obtain carbonyl compounds with high atomic efficiency. Ketones and aldehydes (derived from enol intermediates) are the common products of these processes. Nevertheless, albeit the notably Mn-based catalytic activity already observed in the derivatization of alkynes, its participation in the catalytic alkyne hydration remains elusive. In fact, this has only been reported as isolated entries and for specific substrates (Figure 15).
Figure 15.

Mn-based catalytic precursors for the hydration of alkynes.
In 2013, Naka and co-workers reported the Markovnikov catalytic hydration of 1-decyne using a mixture of MnCl2 and a porphyrin ligand (Na4H2TPPS) (Mn-26, Figure 15).94 However, they obtained better results in the presence of a Co(III) metallic center, and they optimized it for that system. Considering that the characteristics observed for the Co-based catalyst can be deemed for its Mn congener as well, there are a few interesting points that are worth mentioning: (1) the presence of hydrophilic motifs in the porphyrin ligand not only allowed the presence of a unique phase during the reaction, but it also facilitated the catalyst recovery; (2) actually, a former hydroalkoxylation is proposed with further hydrolysis of the methyl vinyl ether and/or dimethyl acetal intermediate; (3) better results were observed with electron-deficient porphyrins.
On the other hand, we recently disclosed the use of Mn-10 as a catalytic precursor for the hydration of α-keto alkynes toward 1,3-dicarbonylic compounds and derivates (Figure 15) as a special case in the catalytic transfer semihydrogenation of internal alkynes (vide supra).39 For this type of reactivity, we made some selected experiments and demonstrated the main role of the Mn-based catalyst. Interestingly, from the assay with 4-phenyl-3-butyn-2-one for 4 h at optimized conditions, we obtained a mixture of the respective semihydrogenation and hydration products (8% and 21%, respectively), but after increasing the reaction time to 24 h, the 1,3-dicarbonylic compound was detected as the sole product in quantitative yield. These results made us speculate that the alkene derived from the alkyne semihydrogenation might be an intermediate in the hydration process and could be reacting as a Michael acceptor (enhanced by the Mn–O interaction between the catalyst and the carbonyl motif in the substrate) for the water nucleophilic attack (either coming from the solvents’ trace amounts or generated in situ from the base-promoted acetone self-condensation, whose products were identified by GC-MS). This intermediate could be tautomerized toward the β-hydroxy carbonyl compound and further dehydrogenated (like the iPrOH) to finally yield the observed 1,3-dicarbonylic product. This plausible route is depicted in Scheme 22, where the (de)hydrogenation reactions are proposed to occur as presented in Scheme 6B.
Scheme 22. Plausible Route for the Mn-Catalyzed Hydration of α-Keto Alkynes with Mn-10.

2.6. Hydroamination
The hydroamination of unsaturated C–C bonds is a powerful tool for C–N bond formation, providing value-added products with both academic and industrial applications.95 Although some interesting works have been disclosed with precatalysts based on first-row transition metals, to the best of our knowledge, the Mn-catalyzed hydroamination of alkenes and alkynes remains scarce and only a few (but salient) examples of this have been recently documented for olefin substrates (Figure 16).
Figure 16.

Mn-based catalytic precursors for the hydroamination of alkenes.
The first of these reports was made in 2021 by Qu, Zhang, and co-workers, disclosing the (anti-Markovnikov) hydroamination of (complex) alkenes via the N–F activation of N-fluorosulfonamides under blue light irradiation in the presence of trimethoxysilane and using Mn-12 as a catalytic precursor (Figure 16).96 Although it will not be discussed in this paper, it is worth mentioning that this methodology also afforded the two- and three-component carboamination of alkenes. From the optimization of the hydroamination process, they observed that distinct silanes (except from HSi(OEt)3 and HSi(TMS)3) and other potential precatalysts based on different transition metals led to considerably lower yields. Mechanistically, the authors proved that this process operates via a Mn-assisted amidyl radical formation with the silane participating as both a hydrogen-atom donor and a fluorine-atom acceptor.
These decisive observations led to the mechanistic proposal depicted in Scheme 23 (which is also supported by DFT calculations), where path b is likely to predominate in the early reaction stage, when the substrate’s concentration is higher than the in-situ-generated [Mn(CO)5F] intermediate.
Scheme 23. Mechanistic Proposal for the Hydroamination of Alkenes with Fluorosulfonamides and Mn-12 as a Catalytic Precursor.

The rest of the hydroamination reports proceed via similar pathways, involving allylic alcohols as substrates. In general, these methodologies encompass a hydrogen-borrowing strategy with a tandem reaction composed of a dehydrogenation (yielding an α,β-unsaturated carbonyl compound as intermediate), a Michael addition, and a final hydrogenation (Scheme 24). As will be explained, a metal–ligand cooperative pathway is proposed for every catalytic system with a crucial role of the NH moiety in the ligand.
Scheme 24. Mechanistic Proposal for the Hydroamination of Allylic Alcohols with Mn-27, Mn-28, or Mn-29 Catalytic Precursors.

The first Mn-based catalytic hydroamination of allylic alcohols was documented in 2021 by Junge, Beller, and co-workers.97 In this report, they evaluated the catalytic activity of a collection of Mn(I)-based pincer complexes, obtaining better results when using Mn-27 (Figure 16) as the catalytic precursor for the synthesis of γ-amino alcohols. Almost every Mn(I) complex with PNP-pincer ligands was catalytically active, while NNN-pincer scaffolds provided null conversions. Besides, the process was promoted by ligands with the NH moiety in the backbone (null conversions were attained with the N-methylated congener) and phosphine segments with an electron-rich character and relatively low steric hindrance.
From the optimization of the reaction conditions, the authors observed an important influence of the base and solvent employed, where the best performance was obtained in non-polar and non-oxygen-containing solvents. Although no more mechanistic information was given, a metal–ligand cooperative pathway could be inferred by the inactivity observed for the N-methylated precursor in a similar way to what was presented for some congeners used in outer-sphere hydrogenations (for instance, see alkyne semihydrogenation).
Around the same time, the Maji group employed the same hydroamination strategy but with Mn-28 (Figure 16) as the catalytic precursor.98 This catalytic system stands out owing to the use of a phosphine-free ligand with an active hemilability of a sulfur donor flexible side arm and the notoriously effective transformation of a wide scope of alkenes and amines, including some complex substrates. Once again, the high performance of the reaction was maintained only with non-polar hydrocarbon-based solvents (such as toluene and cyclohexane). On the other hand, they demonstrated the bifunctionality of the ligand in the metal complex, where the amine’s proton is necessary for the reaction progress and the hemilability of the side arm is enhanced by a soft donor atom such as sulfur. Moreover, further experiments revealed that (1) the reaction is hampered by the presence of strong coordinative molecules, (2) the alcohol dehydrogenation might be the rate-determining step, and (3) the β-amino carbonyl compound can be considered as an intermediate species. Finally, in view of this information and the nature of the process, a catalytic cycle like that displayed in Scheme 24 was proposed.
Lastly, Chen, Fan, and co-workers reported the use of a chiral peraza-N6-macrocycle and [Mn(CO)5Br] for the in-situ formation of a complex capable of catalyzing the asymmetric hydroamination of allylic alcohols with excellent regio- and stereoselectivities (Figure 16, Mn-29).99 Interestingly, this system was highly influenced by the added base and the structure of the macrocycles; for the latter, the main changes were observed in the catalytic activity but not in the stereoselectivity. In stark contrast, the use of open-chain ligands (not macrocyclic) led to null conversions.
For the mechanism, the authors envisioned a hydrogen-borrowing cascade route (analogous to what is depicted in Scheme 24), which is consistent with some species identified by HRMS of the reaction mixture (such as the β-amino ketone and the α,β-unsaturated ketone). In addition, the single-crystal structure revealed that the Mn center is linked to the two amino groups closest to the cyclohexyl segment, leading to a certain cavity in the macrocycle that influences (along with proposed non-covalent interactions) the ketone orientation during the concreted hydrogenation step, which ends up promoting the stereoselectivity of the reaction.
2.7. Hydroalkenylation
The homogeneous Mn-catalyzed hydroalkenylation was only found to be documented for alkenes. As previously mentioned, this was presented along with the hydroarylation of alkenes with Mn-23 as the catalytic precursor. Therefore, this is also proposed to follow a cross-coupling mechanism with alkenylboronic acids as the transmetalation agents.
As previously mentioned, in 2020, Xie and co-workers reported both the hydroarylation and the hydroalkenylation (Figure 17) of a series of α,β-unsaturated amides with the aryl or alkenyl incorporation at the β position.66 As the authors mentioned, the major challenge for hydroalkenylation is the protodemetalation as a side reaction, but they were able to avoid it by a simple change of the additive with respect to the hydroarylation process, replacing CsF with K2CO3. Of note, other commonly used simple Mn precursors, such as [Mn2(CO)10] (Mn-12) and [Mn(CO)5Br] (Mn-16), also presented good results for this type of reactivity but not as great as Mn-23. In stark contrast, other transition metal complexes (based on Ni, Pd, and Rh) led to negligible conversions, highlighting the unique and outstanding behavior of Mn for this process. From the substrate scope, the authors observed that the amide motif has a crucial role in the process since α,β-unsaturated carboxylic acids and nitriles give only poor conversions. Moreover, both internal and terminal (aromatic or aliphatic) alkenyl boronic acids were successfully incorporated under the optimized conditions. Finally, the mechanistic insights were identical with those observed for the hydroarylation process (vide supra), resulting in a mechanistic proposal analogous to that displayed in Scheme 14.
Figure 17.

Mn-based catalytic precursors for the hydroalkenylation of alkenes.
One more Mn-catalyzed alkene hydroalkenylation system was reported in 2020 by Xie’s group but for the γ-alkenylation of β,γ-unsaturated amides (Figure 17).67 The generalities of this catalytic system are like those already explained for its hydroarylation counterpart, where the most important achievement is the incorporation of alkenyl segments into densely functionalized internal alkenes. The plausible mechanism for this transformation is depicted in Scheme 14.
The latest report of this type on reactivity was made very recently by the same group.68 Under mild conditions and short reaction times, Xie and co-workers achieved the hydroalkenylation of both terminal and internal enamides employing a variety of alkenyl boronic acids (Figure 17). Since the reaction conditions are identical with the hydroarylation process, its generalities and mechanistic insights are similar as well. Therefore, the mechanistic proposal is like that presented in Scheme 14, albeit with an enamide instead of the unsaturated substrate.
2.8. Hydrohydrazination
Hydrazines are compounds that can be used as feedstock for the synthesis of heterocycles comprising N–N bonds and molecules with biological activity.100 Among the synthetic routes to produce hydrazines is the hydrohydrazination of alkenes with azo compounds, which was briefly documented to be Mn catalyzed (Figure 18).
Figure 18.

Mn-based catalytic precursors for the hydrohydrazination of alkenes.
In 2004, Carreira and co-workers reported the use of Mn-1 for the hydrohydrazination of alkenes with azodicarboxylates and phenylsilane (Figure 18).101 The same results were obtained with [Mn(dpm)2] as the catalytic precursor, but Mn-1 was chosen for being less air sensitive. In addition, they observed full conversions at shorter reaction times (minutes) and room temperature but also lower regioselectivities; therefore, lower temperatures were preferred. Furthermore, the authors showed that another cheaper and more stable silane (viz. PMHS) was also efficient under similar conditions, which highlights its potential for large-scale synthesis. An explicit mechanistic proposal is missing, although the silane is likely to be acting as a reductant agent.
One year later, Yamada and co-workers reported the use of the same catalytic precursor (Mn-1) for the regio- and stereoselective hydrohydrazination of α,β-unsaturated carboxylic compounds toward (S)-α-hydrazinated carboxylates.102 The stereoselectivity of this process is achieved by the presence of chiral substituents linked to the carbonyl moiety but was more effective with the simultaneous use of bulkier azo compounds.
Although not many mechanistic insights were presented for these catalytic systems, due to the nature of the precatalyst and the presence of a silane, it is likely that they operate through radical pathways (for instance, see the hydrogenation, hydrosilylation, and hydration of alkenes with Mn-1).
2.9. Hydrophosphorylation
The hydrophosphorylation of C–C unsaturated groups corresponds to a versatile and atomically efficient route toward organophosphorus compounds, which are molecules with high academic and industrial interest (such as lubricants, nerve agents, insecticides, fuel additives, and drugs).103 In particular, employing Mn-based precursors, this type of reactivity has been documented for alkenes but including isolated examples with alkynes as substrates (Figure 19).
Figure 19.

Mn-based catalytic precursors for the hydrophosphorylation of alkenes and alkynes.
The first of these reports was made in 2004 by Ishii’s group, where Mn-30 was employed as a precatalyst for the synthesis of organophosphonates from alkenes and diethyl phosphites under an air atmosphere (Figure 19).104 Interestingly, Mn(OAc)3 showed similar catalytic activity and even slight conversion in the absence of air, while this last condition inhibited the process when Mn-30 was used. On the basis of this information, the authors proposed an air-assisted in-situ oxidation of Mn(II) to Mn(III), which apparently is the catalytically active species, but no further information was presented for a mechanistic proposal. It should be noted that under these reaction conditions, an aliphatic terminal alkyne was successfully hydrophosphorylated, although with modest yield and stereoselectivity (51%, 69:31 Z/E).
Some years later, in 2014, Montchamp and co-workers disclosed the addition of H-phosphinates into alkenes with Mn-30 as the precatalyst (Figure 19).105 Among the highlights of this work are the incorporation of a new kind of organophosphorus reagent (which was used in almost stoichiometric amounts) and the formation of 1,2-bisphosphinates when using terminal alkynes as substrates. Once again, this system was reported to be efficient only under an air atmosphere, although phosphinate oxidation was identified as a considerable side reaction. Finally, the authors inferred that the reaction proceeded via a radical pathway (which could be supported by other types of products obtained under similar conditions), but a mechanistic proposal remains lacking.
Finally, the latest report regarding the hydrophosphorylation of alkenes in the presence of Mn-30 was presented by Reznikov’s group in 2016.106 Here, they employed reaction conditions like those indicated by Ishii in 2004 (Figure 19) for the hydrofunctionalization of unsaturated adamantane derivates.
As already mentioned, an explicit mechanistic proposal was not presented in any of the discussed reports. Nevertheless, there exists a consensus for a radical pathway. In this way, considering what was reported for the alkene dimerization with a relatively similar system (Scheme 28), it is likely that the Mn(II) is in-situ oxidated to Mn(III), which later reacts with the P-containing substrate to yield a P-centered radical intermediate that triggers the reaction with the alkene. However, if the Mn precursor only participates as an initiator upon oxidation, it is still unclear why a Mn(III)-based precatalyst only led to low conversions in the absence of oxygen. Hence, a more detailed mechanistic explanation would be desirable to unveil some key points for the development of more active catalytic systems with higher atom efficiency.
Scheme 28. Mechanistic Proposal for the Dimerization of Methoxystyrenes with Mn-30 as a Catalytic Precursor.

2.10. Hydrogermylation
As mentioned in the alkyne hydrosilylation section, Zhang, Zhang, and co-workers reported the hydrogermylation of terminal alkynes with Mn-12 as a precatalyst and under blue light irradiation (Figure 20).51 Although this reactivity was briefly explored, it seems to be applicable to both aromatic and aliphatic (complex) alkynes with good stereoselectivities toward the Z-vinylgermanium products. Moreover, even though it was not demonstrated, it is likely that, based on the nature of the reagents and catalyst, this process proceeds through a mechanism analogous to that of its congener with Si (Scheme 9B).
Figure 20.

Mn-based catalytic precursor for the hydrogermylation of alkynes.
2.11. Hydroalkylation
The Mn-catalyzed hydroalkylation of unsaturated C–C bonds has only been documented for alkenes, with most of these contributions made by Ishii and co-workers in the early 2000s. In general, these methodologies operate with a similar strategy, but they vary in the aliphatic coupling partner. Therefore, in Figure 21 the substrates to make the differences evident are also depicted .
Figure 21.

Mn-based catalytic precursor for the hydroalkylation of alkenes.
Briefly, the first report of this type of reactivity in catalytic amounts was documented in 1995 by Linker and co-workers, with Mn-30 and the assistance of KMnO4 and KOAc (Figure 21).107 In this work, the authors afforded the acetone incorporation to a few aliphatic alkene substrates. By the continuous oxidation of the unactive Mn(II) species with KMnO4, they could turn a previously Mn(III)-mediated reaction into a catalytic process. Furthermore, in this new catalytic regime, a more active and selective reaction was observed in comparison to the stoichiometric version. On the basis of the nature of the reagents and products, they proposed that the process is initiated by the Mn(II) oxidation into Mn(III), which causes a radical reaction between the substrates. This methodology was later expanded by the same group to include dimethyl malonate as a coupling partner (Figure 21), providing highly consistent results.108
In 2000, Ishii’s group disclosed the hydroarylation of alkenes in the presence of Mn-30 and with Co(II)/O2 as cocatalysts.109 In this work, the authors achieved the radical addition of ketones (with the bond formed at the α position) to alkenes under a N2/O2 atmosphere; for terminal alkenes, the anti-Markovnikov product was observed to be favored. By independent experiments, they showed that under air (without Co(II)), Mn(OAc)2 and Mn(OAc)3 yielded similar and modest conversions. In contrast, under a N2 atmosphere, only the latter promoted the reaction (although in low yield). Thus, they proposed that Mn(II) can be continually reoxidized to Mn(III), which is responsible for triggering the reaction. In this regard, addition of Co(II) enhanced the oxidation of Mn(II) but not the oxidation of the substrate. Finally, the catalytic activity was also observed to be dependent on the oxygen concentration. All of these observations set the basis for the successive contributions in the field.
Subsequently, under similar conditions, the same group expanded the process toward the addition of enolizable carbonyl compounds, such as malonate derivates110 and acid anhydrides111 (Figure 21). In general, the observations for these processes are consistent with a system involving ketones. It is noteworthy that the reaction with the acid anhydrides did not occur when the respective carboxylic acids were added instead. Moreover, the products reported for this reactivity are carboxylic acids, which are formed upon treatment with aqueous sulfuric acid.
The last report related to this strategy was presented also by the Ishii group, although with slightly different reaction conditions.112 Here, malonate derivates (mainly, cyanoacetates) were also incorporated into alkenes but this time in the presence of various bases. With the addition of a base, the reaction can be carried out at room temperature for some substrates, which represents a beneficial advantage in terms of industrial applications. Nevertheless, the variety of bases also revealed that (although good to excellent conversions were obtained) the chemoselectivity of the process was strongly modified according to the type of base; among these, potassium acetate was chosen due to its good result and its easier accessibility. For the role of the base in this catalytic system, the authors proposed that it might be related to a deprotonation step for the formation of an enolate intermediate, which is likely to be the rate-determining step of the reaction.
Finally, considering all of the observations already discussed, a general and simplified mechanistic proposal for the Mn-catalyzed hydroalkylation of alkenes with Mn-30 is depicted in Scheme 25. Manganese was crucial for the interaction with the substrates since no conversions were detected in the absence of Mn-based precursors, although other oxidants were also present in the reaction mixtures. As mentioned, the different groups proposed that Mn(II) is in-situ converted into a Mn(III) species, capable of reacting with the enolizable (or resonance-stabilized congeners) aliphatic substrate to generate a carbon-centered radical that reacts with the olefin and yields the final product upon H-atom abstraction. At the same time, the Mn(II) species formed in the early stage of the process is reoxidized with either KMnO4 (added in 30–40 mol % amounts) or a catalytic Co(II)/Co(III) system in the presence of oxygen.
Scheme 25. Mechanistic Proposal for the Alkene Hydroalkylations with Mn-30.

2.12. Hydrofluorocarbofunctionalization
A novel type of Mn-catalyzed alkene derivatization was presented very recently by Xie’s group, consisting of the incorporation of a variety of (poly)fluorine-containing segments into a plethora of simple and complex alkenes under visible light irradiation (Scheme 26).113 This process was achieved in the presence of stoichiometric amounts of HSi(TMS)3 and catalytic loads of both [Mn2(CO)10] and ligand (Figure 22); while the system with dppbz (Mn-31) functioned better for the incorporation of fluoroalkyl bromides, cis-1,2-bis(diphenylphosphino)ethene (Mn-32) was a more suitable ligand for the reaction with polyfluoroaryl iodides. An impressive fine tuning of the reaction conditions was presented, where a high dependence on the solvent (even in amount), silane, and ligand was observed. The closest catalytic performances were attained with THF (solvent), 2-(diphenylphosphino)biphenyl (ligand), dppp (ligand), N,N-bis(diphenylphosphino)isopropylamine (ligand), bis(bicyclohexylphosphinophenyl) ether (ligand), and [Mn(CO)5Br] (catalytic precursor); conditions other than these led to considerably lower yields.
Scheme 26. Mn-Catalyzed Hydrofluorocarbofunctionalization of Alkenes.

Figure 22.

Mn-based catalytic precursors for the hydrofluorocarbofunctionalization of alkenes; X-RFn = (poly)fluoroalkyl/aryl halide.
Subsequently, several mechanistic insights were obtained by experimental and computational studies. The authors demonstrated that (1) the process has a radical mechanism involving a carbon-centered radical intermediate, (2) the ligand was crucial for the bromine-atom transfer (BAT) process, (3) both [Mn(dppbz)(CO)3H] and [Mn(dppbz)(CO)3Br] served as precatalysts, with relatively similar catalytic activity, (4) the in-situ formation of a [Mn2(dppbz)2(CO)6] species might occur that generates the [Mn(dppbz)(CO)3]• radical upon light irradiation, and (5) there are two plausible and potentially concomitant routes for this process where a HAT step is likely to be the rate-determining step. Considering this information, the authors proposed the mechanisms depicted in Scheme 27 as the possible routes. From these, mechanism B is theoretically more energetically favorable than mechanism A; nonetheless, the latter cannot be omitted since both are viable under the experimental conditions, and [Mn(dppbz)(CO)3H] was found to have catalytic activity.
Scheme 27. Mechanisms Proposed for the Hydrofluorocarbofunctionalization of Alkenes with Either Mn-31 or Mn-32 as the Catalytic Precursor: (A) HAT Occurs First; (B) Halogen-Atom Transfer Occurs First.

2.13. Dimerization
Only one report has been found for the Mn-catalyzed dimerization of either alkenes or alkynes. For the alkene’s case, in 2012, Xiang and co-workers disclosed that methoxystyrenes are dimerized in the presence of Mn-30, Scheme 28, in a head-to-tail manner and with Co(OAc)2 and an alkyl phenylphosphinate as cocatalysts (Figure 23).114 For this system, the oxygen was indispensable since the reaction did not proceed under a nitrogen atmosphere. In addition, the process was also inhibited in the presence of a radical scavenger (namely, p-hydroquinone), which is indicative of a radical mechanism. From the substrate scope, the authors observed that the alkene structure had a significant impact on the reaction. Good performance was only detected with methoxystyrenes; otherwise, there were null conversions. On the basis of previous reports about the Mn/Co/O2 system, they proposed that it has a role as a radical initiator upon reaction with the phosphinate, which is proposed to then react with the substrate. Despite there being a mechanistic proposal (Scheme 28), this remains speculative because no more evidence was presented.
Figure 23.

Mn-based catalytic precursor for the dimerization of methoxystyrenes.
The Mn-catalyzed dimerization (and cross-coupling) of terminal alkynes was documented recently by Kirchner’s group employing Mn-2 as a catalytic precursor (Figure 24).115 In this work, the authors presented the catalytic synthesis of a series of enynes, where the regioselectivity of the addition is determined by the nature of the alkynes involved. In general, aromatic alkynes led to head-to-head additions and Z-configured products, while geminal alkenes (from head-to-tail processes) were afforded with either aliphatic alkynes or a mixture of aromatic and aliphatic substrates. From the optimization of the reaction with aryl alkynes, the authors observed that the conversion and the stereoselectivity of the process are highly influenced by the electronic and steric properties of the diphosphine ligand, whereas the change of the anionic ligand and the presence of water can even suppress the catalytic activity.
Figure 24.

Mn-based catalytic precursor for the dimerization of terminal alkynes.
In addition, other solvents led to decreased yields but not lower stereoselectivities. Further experiments revealed the homogeneity of the process, an inner-sphere pathway, and the C–H bond cleavage as the rate-determining step. Finally, the mechanistic proposal for this reaction (supported by DFT calculations) is presented in Scheme 29, where the regioselectivity appeared to be determined by the orientation of the η2-acetylene coordination, which at the same time is influenced by the electronic properties of the substrate.
Scheme 29. Mechanistic Proposal for the Dimerization of Terminal Alkynes with Mn-2 as a Catalytic Precursor.

3. Conclusions
In recent years, Mn-catalyzed processes have been developed as powerful alternatives to precious metal-based processes for a wide variety of reactions of both academic and industrial interest. A large extension of the field of the homogeneous hydrofunctionalizations of alkenes and alkynes has been covered, not only disclosing the versatility of the reactions but also inquiring into their mechanisms for their rationalization. For certain cases, this approach has led to refining the methodologies to the point where they can be used in complex substrates with high chemo-, regio-, and stereoselectivities, highlighting their synthetic potential, even for the late-stage production of fine chemicals.
Among the reactions here described, hydroarylation appeared to be the most explored (based on the number of contributions), followed by hydrogenation and hydrosilylation. Notably, some of the transformations have been observed to be manganese-exclusive types of reactivity, surpassing the commonly more active catalytic systems based on noble transition metals.
Regarding the catalyst design, this was observed to be a function of the strategy followed and the substrates involved. In general, radical pathways have been found to be applicable with simple manganese precursors, such as [Mn(CO)5Br], [Mn2(CO)10], [Mn(dpm)3], and Mn(OAc)2. In particular, most of these reactions proceed via atom transfer methodologies (H, Br, and F) with silanes having the main role as the propagation agent and the H source to complete the respective hydrofunctionalization reaction. In contrast, as can be expected, the presence of ligands usually led to mechanisms with classic organometallic reactions and tuned types of reactivities and selectivities; both inner- and outer-sphere pathways have been documented for these well-defined Mn-based catalysts.
Nevertheless, despite the enormous attention on this subject, there are still opportunity areas and goals that are expected to be fulfilled in the research to come. For instance, more detailed mechanism descriptions, broader substrate scopes, more robust reaction conditions, new site reactions, and clearer selectivities are highly desirable features.
Acknowledgments
We thank CONACyT (A1-S-7657) and DGAPA-UNAM (IN- 200119) for financial support. A.T.-C. also thanks CONACyT for a grant (972053).
The authors declare no competing financial interest.
References
- Chirik P.; Morris R. Getting Down to Earth: The Renaissance of Catalysis with Abundant Metals. Acc. Chem. Res. 2015, 48, 2495–2495. 10.1021/acs.accounts.5b00385. [DOI] [PubMed] [Google Scholar]
- Zweig J. E.; Kim D. E.; Newhouse T. R. Methods Utilizing First-Row Transition Metals in Natural Product Total Synthesis. Chem. Rev. 2017, 117, 11680–11752. 10.1021/acs.chemrev.6b00833. [DOI] [PubMed] [Google Scholar]
- Rajesh N.; Barsu N.; Sundararaju B. Recent Advances in C(sp3)–H bond Carbonylation by First Row Transition Metals. Tetrahedron Lett. 2018, 59, 862–868. 10.1016/j.tetlet.2018.01.065. [DOI] [Google Scholar]
- Gandeepan P.; Müller T.; Zell D.; Cera G.; Warratz S.; Ackermann L. 3d Transition Metals for C–H Activation. Chem. Rev. 2019, 119, 2192–2452. 10.1021/acs.chemrev.8b00507. [DOI] [PubMed] [Google Scholar]
- Alig L.; Fritz M.; Schneider S. First-Row Transition Metal (De)Hydrogenation Catalysis Based On Bifunctional Pincer Ligands. Chem. Rev. 2019, 119, 2681–2751. 10.1021/acs.chemrev.8b00555. [DOI] [PubMed] [Google Scholar]
- Wen J.; Wang F.; Zhang X. Asymmetric hydrogenation catalyzed by first-row transition metal complexes. Chem. Soc. Rev. 2021, 50, 3211–3237. 10.1039/D0CS00082E. [DOI] [PubMed] [Google Scholar]
- a Carney J. R.; Dillon B. R.; Thomas S. P. Recent Advances of Manganese Catalysis for Organic Synthesis. Eur. J. Org. Chem. 2016, 2016, 3912–3929. 10.1002/ejoc.201600018. [DOI] [Google Scholar]; b Sortais J.-B.Manganese Catalysis in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2022. [Google Scholar]
- Khusnutdinov R. I.; Bayguzina A. R.; Dzhemilev U. M. Manganese Compounds in the Catalysis of Organic Reactions. Russ. J. Org. Chem. 2012, 48, 309–348. 10.1134/S1070428012030013. [DOI] [Google Scholar]
- Valyaev D. A.; Lavigne G.; Lugan N. Manganese organometallic compounds in homogeneous catalysis: Past, present, and prospects. Coord. Chem. Rev. 2016, 308, 191–235. 10.1016/j.ccr.2015.06.015. [DOI] [Google Scholar]
- Garbe M.; Junge K.; Beller M. Homogeneous Catalysis by Manganese-Based Pincer Complexes. Eur. J. Org. Chem. 2017, 2017, 4344–4362. 10.1002/ejoc.201700376. [DOI] [Google Scholar]
- Mukherjee A.; Milstein D. Homogeneous Catalysis by Cobalt and Manganese Pincer Complexes. ACS Catal. 2018, 8, 11435–11469. 10.1021/acscatal.8b02869. [DOI] [Google Scholar]
- Liu W.; Ackermann L. Manganese-Catalyzed C-H Activation. ACS Catal. 2016, 6, 3743–3752. 10.1021/acscatal.6b00993. [DOI] [Google Scholar]
- Aneeja T.; Neetha M.; Afsina C. M. A.; Anilkumar G. Recent advances and perspectives in manganese-catalyzed C-H activation. Catal. Sci. Technol. 2021, 11, 444–458. 10.1039/D0CY02087G. [DOI] [Google Scholar]
- Wang Y.; Wang M.; Li Y.; Liu Q. Homogeneous manganese-catalyzed hydrogenation and dehydrogenation reactions. Chem. 2021, 7, 1180–1223. 10.1016/j.chempr.2020.11.013. [DOI] [Google Scholar]
- Das K.; Barman M. K.; Maji B. Advancements in multifunctional manganese complexes for catalytic hydrogen transfer reactions. Chem. Commun. 2021, 57, 8534–8549. 10.1039/D1CC02512K. [DOI] [PubMed] [Google Scholar]
- Azouzi K.; Valyaev D. A.; Bastin S.; Sortais J.-B. Manganese—New prominent actor in transfer hydrogenation catalysis. Curr. Opin. Green Sustainable Chem. 2021, 31, 100511. 10.1016/j.cogsc.2021.100511. [DOI] [Google Scholar]
- Schlichter P.; Werlé C. The Rise of Manganese-Catalyzed Reduction Reactions. Synthesis 2022, 54, 517–534. 10.1055/a-1657-2634. [DOI] [Google Scholar]
- Anaya de Parrodi C.; Walsh P. J. All Kinds of Reactivity: Recent Breakthroughs in Metal-Catalyzed Alkyne Chemistry. Angew. Chem., Int. Ed. 2009, 48, 4679–4682. 10.1002/anie.200900900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anannikov V. P.; Tanaka M.. Hydrofunctionalization; Springer: Germany, 2013. [Google Scholar]
- Trotuş I.-T.; Zimmermann T.; Schüth F. Catalytic Reactions of Acetylene: A Feedstock for the Chemical Industry Revisited. Chem. Rev. 2014, 114, 1761–1782. 10.1021/cr400357r. [DOI] [PubMed] [Google Scholar]
- Crossley S. W. M.; Obradors C.; Martinez R. M.; Shenvi R. A. Mn-, Fe-, and Co-Catalyzed Radical Hydrofunctionalizations of Olefins. Chem. Rev. 2016, 116, 8912–9000. 10.1021/acs.chemrev.6b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Lu Z. Asymmetric Hydrofunctionalization of Minimally Functionalized Alkenes via Earth Abundant Transition Metal Catalysis. Org. Chem. Front. 2018, 5, 260–272. 10.1039/C7QO00613F. [DOI] [Google Scholar]
- Das K.; Waiba S.; Jana A.; Maji B. Manganese-catalyzed hydrogenation, dehydrogenation, and hydroelementation reactions. Chem. Soc. Rev. 2022, 51, 4386–4464. 10.1039/D2CS00093H. [DOI] [PubMed] [Google Scholar]
- a Sweany R. L.; Halpern J. Hydrogenation of.alpha.-methylstyrene by hydridopentacarbonylmanganese (I). Evidence for a free-radical mechanism. J. Am. Chem. Soc. 1977, 99, 8335–8337. 10.1021/ja00467a045. [DOI] [Google Scholar]; b Nalesnik T. E.; Freudenberger J. H.; Orchin M. Stoichiometric radical hydrogenations with HCo(CO)4, HMn(CO)5 and their deuterio analogs. J. Mol. Catal. 1982, 16, 43–49. 10.1016/0304-5102(82)80062-X. [DOI] [Google Scholar]; c Nalesnik T. E.; Freudenberger J. H.; Orchin M. Radical hydroformylation and hydrogenation of cyclopropenes with HCo(CO)4 and HMn(CO)5. J. Organomet. Chem. 1982, 236, 95–100. 10.1016/S0022-328X(00)86753-4. [DOI] [Google Scholar]
- Magnus P.; Waring M. J.; Scott D. A. Conjugate reduction of α,β-unsaturated ketones using an MnIII catalyst, phenylsilane and isopropyl alcohol. Tetrahedron Lett. 2000, 41, 9731–9733. 10.1016/S0040-4039(00)01728-7. [DOI] [Google Scholar]
- Iwasaki K.; Wan K. K.; Oppedisano A.; Crossley S. W. M.; Shenvi R. A. Simple, Chemoselective Hydrogenation with Thermodynamic Stereocontrol. J. Am. Chem. Soc. 2014, 136, 1300–1303. 10.1021/ja412342g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obradors C.; Martinez R. M.; Shenvi R. A. Ph(i-PrO)SiH2: An Exceptional Reductant for Metal-Catalyzed Hydrogen Atom Transfers. J. Am. Chem. Soc. 2016, 138, 4962–4971. 10.1021/jacs.6b02032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber S.; Stöger B.; Veiros L. F.; Kirchner K. Rethinking Basic Concepts—Hydrogenation of Alkenes Catalyzed by Bench-Stable Alkyl Mn(I) Complexes. ACS Catal. 2019, 9, 9715–9720. 10.1021/acscatal.9b03963. [DOI] [Google Scholar]
- Rahaman S. M. W.; Pandey D. K.; Rivada-Wheelaghan O.; Dubey A.; Fayzullin R. R.; Khusnutdinova J. R. Hydrogenation of Alkenes Catalyzed by a Non-pincer Mn Complex. ChemCatChem. 2020, 12, 5912–5918. 10.1002/cctc.202001158. [DOI] [Google Scholar]
- a Zou Y.-Q.; Chakraborty S.; Nerush A.; Oren D.; Diskin-Posner Y.; Ben-David Y.; Milstein D. Highly Selective, Efficient Deoxygenative Hydrogenation of Amides Catalyzed by a Manganese Pincer Complex via Metal-Ligand Cooperation. ACS Catal. 2018, 8, 8014–8019. 10.1021/acscatal.8b02902. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Das U. K.; Kumar A.; Ben-David Y.; Iron M. A.; Milstein D. Manganese Catalyzed Hydrogenation of Carbamates and Urea Derivatives. J. Am. Chem. Soc. 2019, 141, 12962–12966. 10.1021/jacs.9b05591. [DOI] [PubMed] [Google Scholar]
- Vielhaber T.; Topf C. Manganese-catalyzed homogeneous hydrogenation of ketones and conjugate reduction of α,β-unsaturated carboxylic acid derivatives: A chemoselective, robust, and phosphine-free in situ-protocol. Appl. Catal., A 2021, 623, 118280. 10.1016/j.apcata.2021.118280. [DOI] [Google Scholar]
- Bruneau-Voisine A.; Wang D.; Dorcet V.; Roisnel T.; Darcel C.; Sortais J.-B. Transfer Hydrogenation of Carbonyl Derivatives Catalyzed by an Inexpensive Phosphine-Free Manganese Precatalyst. Org. Lett. 2017, 19, 3656–3659. 10.1021/acs.orglett.7b01657. [DOI] [PubMed] [Google Scholar]
- Zhou Y.-P.; Mo Z.; Luecke M.-P.; Driess M. Stereoselective Transfer Semi-Hydrogenation of Alkynes to E-Olefins with N-Heterocyclic Silylene-Manganese Catalysts. Chem.—Eur. J. 2018, 24, 4780–4784. 10.1002/chem.201705745. [DOI] [PubMed] [Google Scholar]
- Brzozowska A.; Azofra L. M.; Zubar V.; Atodiresei I.; Cavallo L.; Rueping M.; El-Sepelgy O. Highly Chemo- and Stereoselective Transfer Semihydrogenation of Alkynes Catalyzed by a Stable, Well-Defined Manganese(II) Complex. ACS Catal. 2018, 8, 4103–4109. 10.1021/acscatal.8b00983. [DOI] [Google Scholar]
- Garbe M.; Budweg S.; Papa V.; Wei Z.; Hornke H.; Bachmann S.; Scalone M.; Spannenberg A.; Jiao H.; Junge K.; Beller M. Chemoselective semihydrogenation of alkynes catalyzed by manganese(i)-PNP pincer complexes. Catal. Sci. Technol. 2020, 10, 3994–4001. 10.1039/D0CY00992J. [DOI] [Google Scholar]
- Zubar V.; Sklyaruk J.; Brzozowska A.; Rueping M. Chemoselective Hydrogenation of Alkynes to (Z)-Alkenes Using an Air-Stable Base Metal Catalyst. Org. Lett. 2020, 22, 5423–5428. 10.1021/acs.orglett.0c01783. [DOI] [PubMed] [Google Scholar]
- Sklyaruk J.; Zubar V.; Borghs J. C.; Rueping M. Methanol as the Hydrogen Source in the Selective Transfer Hydrogenation of Alkynes Enabled by a Manganese Pincer Complex. Org. Lett. 2020, 22, 6067–6071. 10.1021/acs.orglett.0c02151. [DOI] [PubMed] [Google Scholar]
- Farrar-Tobar R. A.; Weber S.; Csendes Z.; Ammaturo A.; Fleissner S.; Hoffmann H.; Veiros L. F.; Kirchner K. E-Selective Manganese-Catalyzed Semihydrogenation of Alkynes with H2 Directly Employed or In Situ-Generated. ACS Catal. 2022, 12, 2253–2260. 10.1021/acscatal.1c06022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Calis A.; García J. J. Manganese-catalyzed transfer semihydrogenation of internal alkynes to E-alkenes with iPrOH as hydrogen source. Catal. Sci. Technol. 2022, 12, 3004–3015. 10.1039/D2CY00246A. [DOI] [Google Scholar]
- a Nakajima Y.; Shimada S. Hydrosilylation reaction of olefins: recent advances and perspectives. RSC Adv. 2015, 5, 20603–20616. 10.1039/C4RA17281G. [DOI] [Google Scholar]; b Zaranek M.; Pawluc P. Markovnikov Hydrosilylation of Alkenes: How an Oddity Becomes the Goal. ACS Catal. 2018, 8, 9865–9876. 10.1021/acscatal.8b03104. [DOI] [Google Scholar]
- Pratt S. L.; Faltynek R. A. Hydrosilation catalysis via silylmanganese carbonyl complexes: Thermal vs. photochemical activation. J. Organomet. Chem. 1983, 258, C5–C8. 10.1016/0022-328X(83)89515-1. [DOI] [Google Scholar]
- Hilal H. S.; Abu-Eid M.; Al-Subu M.; Khalaf S. Hydrosilylation reactions catalysed by decacarbonyldimanganese(0). J. Mol. Catal. 1987, 39, 1–11. 10.1016/0304-5102(87)80042-1. [DOI] [Google Scholar]
- Docherty J. H.; Peng J.; Dominey A. P.; Thomas S. P. Activation and discovery of earth-abundant metal catalysts using sodium tert-butoxide. Nat. Chem. 2017, 9, 595–600. 10.1038/nchem.2697. [DOI] [PubMed] [Google Scholar]
- Carney J. R.; Dillon B. R.; Campbell L.; Thomas S. P. Manganese-Catalyzed Hydrofunctionalization of Alkenes. Angew. Chem., Int. Ed. 2018, 57, 10620–10624. 10.1002/anie.201805483. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay T. K.; Flores M.; Groy T. L.; Trovitch R. J. A β-diketiminate manganese catalyst for alkene hydrosilylation: substrate scope, silicone preparation, and mechanistic insight. Chem. Sci. 2018, 9, 7673–7680. 10.1039/C8SC02768D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.; Wang C. Diverse Fates of β-Silyl Radical under Manganese Catalysis: Hydrosilylationand Dehydrogenative Silylation of Alkenes. Chin. J. Chem. 2018, 36, 1047–1051. 10.1002/cjoc.201800367. [DOI] [Google Scholar]
- Dong J.; Yuan X.-A.; Yan Z.; Mu L.; Ma J.; Zhu C.; Xie J. Manganese-catalysed divergent silylation of alkenes. Nat. Chem. 2021, 13, 182–190. 10.1038/s41557-020-00589-8. [DOI] [PubMed] [Google Scholar]
- Vivien A.; Veyre L.; Mirgalet R.; Camp C.; Thieuleux C. Mn2(CO)10 and UV light: a promising combination for regioselective alkene hydrosilylation at low temperature. Chem. Commun. 2022, 58, 4091–4094. 10.1039/D2CC00377E. [DOI] [PubMed] [Google Scholar]
- Yang X.; Wang C. Dichotomy of Manganese Catalysis via Organometallic or Radical Mechanism: Stereodivergent Hydrosilylation of Alkynes. Angew. Chem., Int. Ed. 2018, 57, 923–928. 10.1002/anie.201710206. [DOI] [PubMed] [Google Scholar]
- Li Q.; Huo S.; Meng L.; Li X. Mechanism and origin of the stereoselectivity of manganese-catalyzed hydrosilylation of alkynes: a DFT study. Catal. Sci. Technol. 2022, 12, 2649–2658. 10.1039/D1CY02340C. [DOI] [Google Scholar]
- Liang H.; Ji Y.-X.; Wang R.-H.; Zhang Z.-H.; Zhang B. Visible-Light-Initiated Manganese-Catalyzed E-Selective Hydrosilylation and Hydrogermylation of Alkynes. Org. Lett. 2019, 21, 2750–2754. 10.1021/acs.orglett.9b00701. [DOI] [PubMed] [Google Scholar]
- Obligacion J. V.; Chirik P. J. Earth-abundant transition metal catalysts for alkene hydrosilylation and hydroboration. Nat. Rev. Chem. 2018, 2, 15–34. 10.1038/s41570-018-0001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Zeng H.; Wu J.; Yin Z.; Zheng S.; Fettinger J. C. Highly Selective Hydroboration of Alkenes, Ketones and Aldehydes Catalyzed by a Well-Defined Manganese Complex. Angew. Chem., Int. Ed. 2016, 55, 14369–14372. 10.1002/anie.201607579. [DOI] [PubMed] [Google Scholar]
- Macaulay C. M.; Gustafson S. J.; Fuller J. T. III; Kwon D.-H.; Ogawa T.; Ferguson M. J.; McDonald R.; Lumsden M. D.; Bischof S. M.; Sydora O. L.; Ess D. H.; Stradiotto M.; Turculet L. Alkene Isomerization-Hydroboration Catalyzed by First-Row Transition-Metal (Mn, Fe, Co, and Ni) N-Phosphinoamidinate Complexes: Origin of Reactivity and Selectivity. ACS Catal. 2018, 8, 9907–9925. 10.1021/acscatal.8b01972. [DOI] [Google Scholar]
- Garhwal S.; Kroeger A. A.; Thenarukandiyil R.; Fridman N.; Karton A.; de Ruiter G. Manganese-Catalyzed Hydroboration of Terminal Olefins and Metal-Dependent Selectivity in Internal Olefin Isomerization-Hydroboration. Inorg. Chem. 2021, 60, 494–504. 10.1021/acs.inorgchem.0c03451. [DOI] [PubMed] [Google Scholar]
- Weber S.; Zobernig D.; Stoger B.; Veiros L. F.; Kirchner K. Hydroboration of Terminal Alkenes and trans-1,2-Diboration of Terminal Alkynes Catalyzed by a Manganese(I) Alkyl Complex. Angew. Chem., Int. Ed. 2021, 60, 24488–24492. 10.1002/anie.202110736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzozowska A.; Zubar V.; Ganardi R.-C.; Rueping M. Chemoselective Hydroboration of Propargylic Alcohols and Amines Using a Manganese(II) Catalyst. Org. Lett. 2020, 22, 3765–3769. 10.1021/acs.orglett.0c00941. [DOI] [PubMed] [Google Scholar]
- a Cano R.; Mackey K.; McGlacken G. P. Recent advances in manganese-catalysed C-H activation: scope and mechanism. Catal. Sci. Technol. 2018, 8, 1251–1266. 10.1039/C7CY02514A. [DOI] [Google Scholar]; b Hu Y.; Zhou B.; Wang C. Inert C-H Bond Transformations Enabled by Organometallic Manganese Catalysis. Acc. Chem. Res. 2018, 51, 816–827. 10.1021/acs.accounts.8b00028. [DOI] [PubMed] [Google Scholar]
- Zhou B.; Ma P.; Chen H.; Wang C. Amine-accelerated manganese-catalyzed aromatic C-H conjugate addition to α,β-unsaturated carbonyls. Chem. Commun. 2014, 50, 14558–14561. 10.1039/C4CC07598F. [DOI] [PubMed] [Google Scholar]
- Wang C.; Wang A.; Rueping M. Manganese-Catalyzed C-H Functionalizations: Hydroarylations and Alkenylations Involving an Unexpected Heteroaryl Shift. Angew. Chem., Int. Ed. 2017, 56, 9935–9938. 10.1002/anie.201704682. [DOI] [PubMed] [Google Scholar]
- Chen S.-Y.; Li Q.; Wang H. Manganese(I)-Catalyzed Direct C-H Allylation of Arenes with Allenes. J. Org. Chem. 2017, 82, 11173–11181. 10.1021/acs.joc.7b02220. [DOI] [PubMed] [Google Scholar]
- Liu T.; Yang Y.; Wang C. Manganese-Catalyzed Hydroarylation of Unactivated Alkenes. Angew. Chem., Int. Ed. 2020, 59, 14256–14260. 10.1002/anie.202003830. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Wang C. Manganese/NaOPh co-catalyzed C2-selective C-H conjugate addition of indoles to α,β-unsaturated carbonyls. Green Synthesis and Catalysis 2021, 2, 66–69. 10.1016/j.gresc.2021.01.010. [DOI] [Google Scholar]
- Ghosh S.; Khandelia T.; Patel B. K. Solvent-Switched Manganese(I)-Catalyzed Regiodivergent Distal vs Proximal C-H Alkylation of Imidazopyridine with Maleimide. Org. Lett. 2021, 23, 7370–7375. 10.1021/acs.orglett.1c02536. [DOI] [PubMed] [Google Scholar]
- Liu S.-L.; Li Y.; Guo J. R.; Yang G.-C.; Li X.-H.; Gong J.-F.; Song M.-P. An Approach to 3-(Indol-2-yl)succinimide Derivatives by Manganese-Catalyzed C-H Activation. Org. Lett. 2017, 19, 4042–4045. 10.1021/acs.orglett.7b01795. [DOI] [PubMed] [Google Scholar]
- Wang D.; Dong J.; Fan W.; Yuan X.-A.; Han J.; Xie J. Dimeric Manganese-Catalyzed Hydroarylation and Hydroalkenylation of Unsaturated Amides. Angew. Chem., Int. Ed. 2020, 59, 8430–8434. 10.1002/anie.201916305. [DOI] [PubMed] [Google Scholar]
- Wang D.; He Y.; Dai H.; Huang C.; Yuan X.-A.; Xie J. Manganese-Catalyzed Hydrocarbofunctionalization of Internal Alkenes. Chin. J. Chem. 2020, 38, 1497–1502. 10.1002/cjoc.202000376. [DOI] [Google Scholar]
- He Y.; Du C.; Han J.; Han J.; Zhu C.; Xie J. Manganese-Catalyzed Anti-Markovnikov Hydroarylation of Enamides: Modular Synthesis of Arylethylamines. Chin. J. Chem. 2022, 40, 1546–1552. 10.1002/cjoc.202200202. [DOI] [Google Scholar]
- Zhou B.; Chen H.; Wang C. Mn-Catalyzed Aromatic C-H Alkenylation with Terminal Alkynes. J. Am. Chem. Soc. 2013, 135, 1264–1267. 10.1021/ja311689k. [DOI] [PubMed] [Google Scholar]
- Shi L.; Zhong X.; She H.; Lei Z.; Li F. Manganese catalyzed C-H functionalization of indoles with alkynes to synthesize bis/trisubstituted indolylalkenes and carbazoles: the acid is the key to control selectivity. Chem. Commun. 2015, 51, 7136–7139. 10.1039/C5CC00249D. [DOI] [PubMed] [Google Scholar]
- Wang H.; Pesciaioli F.; Oliveira J. C. A.; Warratz S.; Ackermann L. Synergistic Manganese(I) C-H Activation Catalysis in Continuous Flow: Chemoselective Hydroarylation. Angew. Chem., Int. Ed. 2017, 56, 15063–15067. 10.1002/anie.201708271. [DOI] [PubMed] [Google Scholar]
- Wang C.; Rueping M. Rhenium- and Manganese-Catalyzed Selective Alkenylation of Indoles. ChemCatChem. 2018, 10, 2681–2685. 10.1002/cctc.201800553. [DOI] [Google Scholar]
- Jia T.; Wang C. Manganese-Catalyzed ortho-Alkenylation of Aromatic Amidines with Alkynes via C-H Activation. ChemCatChem. 2019, 11, 5292–5295. 10.1002/cctc.201900387. [DOI] [Google Scholar]
- Cembellín S.; Dalton T.; Pinkert T.; Schäfers F.; Glorius F. Highly Selective Synthesis of 1,3-Enynes, Pyrroles, and Furans by Manganese(I)-Catalyzed C-H Activation. ACS Catal. 2020, 10, 197–202. 10.1021/acscatal.9b03965. [DOI] [Google Scholar]
- Wan S.; Luo Z.; Xu X.; Yu H.; Li J.; Pan Y.; Zhang X.; Xu L.; Cao R. Manganese(I)-Catalyzed Site-Selective C6-Alkenylation of 2-Pyridones Using Alkynes via C-H Activation. Adv. Synth. Catal. 2021, 363, 2586–2593. 10.1002/adsc.202100056. [DOI] [Google Scholar]
- Kaplaneris N.; Kaltenhäuser F.; Sirvinskaite G.; Fan S.; De Oliveira T.; Conradi L.-C.; Ackermann L. Late-stage stitching enabled by manganese-catalyzed C-H activation: Peptide ligation and access to cyclopeptides. Sci. Adv. 2021, 7, eabe6202 10.1126/sciadv.abe6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplaneris N.; Son J.; Mendive-Tapia L.; Kopp A.; Barth N. D.; Maksso I.; Vendrell M.; Ackermann L. Chemodivergent manganese-catalyzed C-H activation: modular synthesis of fluorogenic probes. Nat. Commun. 2021, 12, 3389. 10.1038/s41467-021-23462-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jei B. B.; Yang L.; Ackermann L. Selective Labeling of Peptides with o-Carboranes via Manganese(I)-Catalyzed C-H Activation. Chem. Eur. J. 2022, 28, e202200811 10.1002/chem.202200811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das K. K.; Ghosh A. K.; Hajra A. Late-stage ortho-C-H alkenylation of 2-arylindazoles in aqueous medium by Manganese(i)-catalysis. RSC Adv. 2022, 12, 19412–19416. 10.1039/D2RA03547B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yahaya N. P.; Appleby K. M.; Teh M.; Wagner C.; Troschke E.; Bray J. T. W.; Duckett S. B.; Hammarback L. A.; Ward J. S.; Milani J.; Pridmore N. E.; Whitwood A. C.; Lynam J. M.; Fairlamb I. J. S. Manganese(I)-Catalyzed C-H Activation: The Key Role of a 7-Membered Manganacycle in H-Transfer and Reductive Elimination. Angew. Chem., Int. Ed. 2016, 55, 12455–12459. 10.1002/anie.201606236. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hammarback L. A.; Clark I. P.; Sazanovich I. V.; Towrie M.; Robinson A.; Clarke F.; Meyer S.; Fairlamb I. J. S.; Lynam J. M. Mapping out the key carbon-carbon bond-forming steps in Mn-catalysed C-H functionalization. Nat. Catal. 2018, 1, 830–840. 10.1038/s41929-018-0145-y. [DOI] [Google Scholar]; c Hammarback L. A.; Robinson A.; Lynam J. M.; Fairlamb I. J. S. Delineating the critical role of acid additives in Mn-catalysed C-H bond functionalisation processes. Chem. Commun. 2019, 55, 3211–3214. 10.1039/C9CC00257J. [DOI] [PubMed] [Google Scholar]; d Hammarback L. A.; Robinson A.; Lynam J. M.; Fairlamb I. J. S. Mechanistic Insight into Catalytic Redox-Neutral C-H Bond Activation Involving Manganese(I) Carbonyls: Catalyst Activation, Turnover, and Deactivation Pathways Reveal an Intricate Network of Steps. J. Am. Chem. Soc. 2019, 141, 2316–2328. 10.1021/jacs.8b09095. [DOI] [PubMed] [Google Scholar]
- Yang X.; Jin X.; Wang C. Manganese-Catalyzed ortho-C-H Alkenylation of Aromatic N-H Imidates with Alkynes: Versatile Access to Mono-Alkenylated Aromatic Nitriles. Adv. Synth. Catal. 2016, 358, 2436–2442. 10.1002/adsc.201600128. [DOI] [Google Scholar]
- Kumar A.; Muniraj N.; Prabhu K. R. Manganese-Catalysed C-H Activation: A Regioselective C-H Alkenylation of Indoles and other (hetero)aromatics with 4-Hydroxy-2-Alkynoates Leading to Concomitant Lactonization. Adv. Synth. Catal. 2019, 361, 4933–4940. 10.1002/adsc.201900678. [DOI] [Google Scholar]
- Yan Z.; Yuan X.-A.; Zhao Y.; Zhu C.; Xie J. Selective Hydroarylation of 1,3-Diynes Using a Dimeric Manganese Catalyst: Modular Synthesis of Z-Enynes. Angew. Chem., Int. Ed. 2018, 57, 12906–12910. 10.1002/anie.201807851. [DOI] [PubMed] [Google Scholar]
- Yuan X.-A.; Huang C.; Wang X.; Liu P.; Bi S.; Li D. Computational Study on the Mechanisms and Origins of Selectivity in Hydroarylation of 1,3-Diyne Alcohol Catalyzed by Di- and Mononuclear Manganese Complexes. Organometallics 2021, 40, 3124–3135. 10.1021/acs.organomet.1c00370. [DOI] [Google Scholar]
- Pang Y.; Liu G.; Huang C.; Yuan X.-A.; Li W.; Xie J. A Highly Efficient Dimeric Manganese-Catalyzed Selective Hydroarylation of Internal Alkynes. Angew. Chem., Int. Ed. 2020, 59, 12789–12794. 10.1002/anie.202004950. [DOI] [PubMed] [Google Scholar]
- Huang C.; Pang Y.; Yuan X.-A.; Jiang Y.-Y.; Wang X.; Liu P.; Bi S.; Xie J. Noncovalent Interaction- and Steric Effect-Controlled Regiodivergent Selectivity in Dimeric Manganese-Catalyzed Hydroarylation of Internal Alkynes: A Computational Study. J. Org. Chem. 2022, 87, 4215–4225. 10.1021/acs.joc.1c03058. [DOI] [PubMed] [Google Scholar]
- a Guo J.; Teo P. Anti-Markovnikov oxidation and hydration of terminal olefins. Dalton Trans. 2014, 43, 6952–6964. 10.1039/c3dt53600a. [DOI] [PubMed] [Google Scholar]; b Salvio R.; Bassetti M. Sustainable hydration of alkynes promoted by first row transition metal complexes. Background, highlights and perspectives. Inorg. Chim. Acta 2021, 522, 120288. 10.1016/j.ica.2021.120288. [DOI] [Google Scholar]
- Inoki S.; Kato K.; Isayama S.; Mukaiyama T. A New and Facile Method for the Direct Preparation of α-Hydroxycarboxylic Acid Esters from α,β-Unsaturated Carboxylic Acid Esters with Molecular Oxygen and Phenylsilane Catalyzed by Bis(dipivaloylmethanato)manganese(II) Complex. Chem. Lett. 1990, 19, 1869–1872. 10.1246/cl.1990.1869. [DOI] [Google Scholar]
- Magnus P.; Payne A. H.; Waring M. J.; Scott D. A.; Lynch V. Conversion of α,β-unsaturated ketones into α-hydroxy ketones using an MnIII catalyst, phenylsilane and dioxygen: acceleration of conjugate hydride reduction by dioxygen. Tetrahedron Lett. 2000, 41, 9725–9730. 10.1016/S0040-4039(00)01727-5. [DOI] [Google Scholar]
- Magnus P.; Scott D. A.; Fielding M. R. Direct conversion of α,β-unsaturated nitriles into cyanohydrins using Mn(dpm)3 catalyst, dioxygen and phenylsilane. Tetrahedron Lett. 2001, 42, 4127–4129. 10.1016/S0040-4039(01)00693-1. [DOI] [Google Scholar]
- Sato M.; Gunji Y.; Ikeno T.; Yamada T. Stereoselective Preparation of α-Hydroxycarboxamide by Manganese Complex Catalyzed Hydration of α,β-Unsaturated Carboxamide with Molecular Oxygen and Phenylsilane. Chem. Lett. 2004, 33, 1304–1305. 10.1246/cl.2004.1304. [DOI] [Google Scholar]
- Cassayre J.; Winkler T.; Pitterna T.; Quaranta L. Application of Mn(III)-catalysed olefin hydration reaction to the selective functionalisation of avermictin B1. Tetrahedron Lett. 2010, 51, 1706–1709. 10.1016/j.tetlet.2010.01.080. [DOI] [Google Scholar]
- Donnelly P. S.; North A. J.; Radjah N. C.; Ricca M.; Robertson A.; White J. M.; Rizzacasa M. A. An effective cis-β-octahedral Mn(III) SALPN catalyst for the Mukaiyama-Isayama hydration of α,β-unsaturated esters. Chem. Commun. 2019, 55, 7699–7702. 10.1039/C9CC03921J. [DOI] [PubMed] [Google Scholar]
- Tachinami T.; Nishimura T.; Ushimaru R.; Noyori R.; Naka H. Hydration of Terminal Alkynes Catalyzed by Water-Soluble Cobalt Porphyrin Complexes. J. Am. Chem. Soc. 2013, 135, 50–53. 10.1021/ja310282t. [DOI] [PubMed] [Google Scholar]
- a Severin R.; Doye S. The catalytic hydroamination of alkynes. Chem. Soc. Rev. 2007, 36, 1407–1420. 10.1039/b600981f. [DOI] [PubMed] [Google Scholar]; b Huo J.; He G.; Chen W.; Hu X.; Deng Q.; Chen D. A minireview of hydroamination catalysis: alkene and alkyne substrate selective, metal complex design. BMC Chem. 2019, 13, 89. 10.1186/s13065-019-0606-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y.-X.; Li J.; Li C.-M.; Qu S.; Zhang B. Manganese-Catalyzed N-F Bond Activation for Hydroamination and Carboamination of Alkenes. Org. Lett. 2021, 23, 207–212. 10.1021/acs.orglett.0c03916. [DOI] [PubMed] [Google Scholar]
- Duarte de Almeida L.; Bourriquen F.; Junge K.; Beller M. Catalytic Formal Hydroamination of Allylic Alcohols Using Manganese PNP-Pincer Complexes. Adv. Synth. Catal. 2021, 363, 4177–4181. 10.1002/adsc.202100081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das K.; Sarkar K.; Maji B. Manganese-Catalyzed Anti-Markovnikov Hydroamination of Allyl Alcohols via Hydrogen-Borrowing Catalysis. ACS Catal. 2021, 11, 7060–7069. 10.1021/acscatal.1c01199. [DOI] [Google Scholar]
- Li F.; Long L.; He Y.-M.; Li Z.; Chen H.; Fan Q.-H. Manganese-Catalyzed Asymmetric Formal Hydroamination of Allylic Alcohols: A Remarkable Macrocycle Ligand Effect. Angew. Chem., Int. Ed. 2022, 61 (26), e202202972 10.1002/anie.202202972. [DOI] [PubMed] [Google Scholar]
- a Tšupova S.; Mäeorg U. Hydrazines and Azo-Compounds in the Synthesis of Heterocycles Comprising N-N Bond. Heterocycles 2014, 88, 129–173. 10.3987/REV-13-SR(S)3. [DOI] [Google Scholar]; b de Oliveira Carneiro Brum J.; Franca T. C. C.; LaPlante S. R.; Villar J. D. F. Synthesis and Biological Activity of Hydrazones and Derivatives: A Review. Mini-Rev. Med. Chem. 2020, 20, 342–368. 10.2174/1389557519666191014142448. [DOI] [PubMed] [Google Scholar]
- Waser J.; Carreira E. M. Catalytic Hydrohydrazination of a Wide Range of Alkenes with a Simple Mn Complex. Angew. Chem., Int. Ed. 2004, 43, 4099–4102. 10.1002/anie.200460811. [DOI] [PubMed] [Google Scholar]
- Sato M.; Gunji Y.; Ikeno T.; Yamada T. Stereoselective α-Hydrazination of α,β-Unsaturated Carboxylates Catalyzed by Manganese(III) Complex with Dialkylazodicarboxylate and Phenylsilane. Chem. Lett. 2005, 34, 316–317. 10.1246/cl.2005.316. [DOI] [Google Scholar]
- Richardson R. J.; Makhaeva G. F.. Encyclopedia of Toxicology, 3rd ed.; Wexler P., Ed.; Academic Press: Oxford, 2014; pp 714–719. [Google Scholar]
- Tayama O.; Nakano A.; Iwahama T.; Sakaguchi S.; Ishii Y. Hydrophosphorylation of Alkenes with Dialkyl Phosphites Catalyzed by Mn(III) under Air. J. Org. Chem. 2004, 69, 5494–5496. 10.1021/jo049657j. [DOI] [PubMed] [Google Scholar]
- Fisher H. C.; Berger O.; Gelat F.; Montchamp J.-L. Manganese-Catalyzed and Promoted Reactions of H-Phosphinate Esters. Adv. Synth. Catal. 2014, 356, 1199–1204. 10.1002/adsc.201301157. [DOI] [Google Scholar]
- Reznikov A. N.; Klimochkin Y. N. Manganese(II) Acetate-Catalyzed Hydrophosphorylation of Unsaturated Adamantane Derivates. Russ. J. Gen. Chem. 2016, 86, 2710–2712. 10.1134/S1070363216120252. [DOI] [Google Scholar]
- Linker U.; Kersten B.; Linker T. Potassium permanganate-mediated radical reactions: chemoselective addition of acetone to olefins. Tetrahedron 1995, 51, 9917–9926. 10.1016/0040-4020(95)00563-N. [DOI] [Google Scholar]
- Linker T.; Kersten B.; Linker U.; Peters K.; Peters E.-M.; von Schnering H. G. Manganese(III)-Mediated Radical Additions of Dimethyl Malonate to Olefins. The Chemoselective Synthesis of Diesters and Lactones. Synlett 1996, 1996, 468–470. 10.1055/s-1996-5451. [DOI] [Google Scholar]
- Iwahama T.; Sakaguchi S.; Ishii Y. Catalytic radical addition of ketones to alkenes by a metal-dioxygen redox system. Chem. Commun. 2000, 2317–2318. 10.1039/b007182j. [DOI] [Google Scholar]
- Hirase K.; Iwahama T.; Sakaguchi S.; Ishii Y. Catalytic Radical Addition of Carbonyl Compounds to Alkenes by Mn(II)/Co(II)/O2 System. J. Org. Chem. 2002, 67, 970–973. 10.1021/jo0162282. [DOI] [PubMed] [Google Scholar]
- Hirase K.; Sakaguchi S.; Ishii Y. Addition of Carboxyalkyl Radicals to Alkenes through a Catalytic Process, Using a Mn(II)/Co(II)/O2 Redox System. J. Org. Chem. 2003, 68, 5974–5976. 10.1021/jo034584+. [DOI] [PubMed] [Google Scholar]
- Kagayama T.; Fuke T.; Sakaguchi S.; Ishii Y. A Remarkable Effect of Bases on the Catalytic Radical Addition of Cyanoacetates to Alkenes Using a Mn(II)/Co(II)/O2 Redox System. Bull. Chem. Soc. Jpn. 2005, 78, 1673–1676. 10.1246/bcsj.78.1673. [DOI] [Google Scholar]
- Han J.; Han J.; Chen S.; Zhong T.; He Y.; Yang X.; Wang G.; Zhu C.; Xie J. Photoinduced manganese-catalysed hydrofluorocarbofunctionalization of alkenes. Nat. Synth. 2022, 1, 475–486. 10.1038/s44160-022-00074-9. [DOI] [Google Scholar]
- Liu K.; Wang Y.; Li P.; Cai W.; Xiang J. Dimerization of styrenes catalyzed by PhP(O)HOR/Mn(II)/Co(II)/O2. Catal. Commun. 2012, 20, 107–110. 10.1016/j.catcom.2012.01.010. [DOI] [Google Scholar]
- Weber S.; Veiros L. F.; Kirchner K. Selective Manganese-Catalyzed Dimerization and Cross-Coupling of Terminal Alkynes. ACS Catal. 2021, 11, 6474–6483. 10.1021/acscatal.1c01137. [DOI] [PMC free article] [PubMed] [Google Scholar]