

Abstract
Background
Lipoprotein(a) [Lp(a)] is one of the residual risk factors for cardiovascular disease (CVD) in the setting of optimal low-density lipoprotein cholesterol (LDL-C). The association between Lp(a) and CVD is still in the exploratory phase, with few studies indicating a causal connection between Lp(a) and various CVD.
Methods
Lp(a) (n = 377,590) was a genome-wide association study (GWAS) based on European populations from Neale Lab. Large GWAS datasets for CVD, including aortic aneurysm(AA) (n = 209,366), atrial fibrillation(AF) (n = 1,030,836), coronary heart disease(CHD) (n = 361,194), secondary hypertension(HBP) (n = 164,147), heart failure(HF) (n = 208,178), ischemic stroke (IS) (n = 218,792), large artery atherosclerosis stroke(ISL) (n = 150, 765), small vessel stroke(ISS) (n = 198,048), lacunar stroke(LIS) (n = 225,419), and pulmonary embolism(PE) (n = 218,413) were also based on European populations. We performed separate univariate two-sample Mendelian randomization (MR) analysis for Lp(a) and CVD as described above. We evaluated this connection mainly using the random-effects inverse variance weighted technique(IVW1) with a 95% confidence interval (CI) for the odds ratio (OR). This was supplemented by MR-Egger, weighted median, maximum likelihood, penalized weighted median, and fixed-effects inverse variance weighted methods. MR-PRESSO offers another means of statistical detection.
Results
Our two-sample MR, which was predominately based on IVW1, revealed a causal relationship between Lp(a) and AA (OR = 1.005, 95%CI: 1.001–1.010, P = 0.009), CHD (OR = 1.003, 95%CI 1.001–1.004, P = 0.010), and ISL (OR = 1.003, 9 5%CI 1.002–1.004, P = 9.50E−11), in addition, there is no causal association with AF, HBP, HF, IS, ISS, LIS, or PE. Similar conclusions were reached by the MR-PRESSO method.
Conclusion
This MR study suggested a causal relationship between Lp(a) and CHD, AA, and ISL, but not associated with AF, HF, IS, LIS, ISS, HBP, or PE. Our work further verifies the association between Lp(a) and various CVD, resulting in improved Lp(a) management and a reduction in the prevalence of CVD.
Supplementary Information
The online version contains supplementary material available at 10.1186/s40001-022-00825-6.
Keywords: Lipoprotein(a), Mendelian randomization, Cardiovascular disease
Introduction
Cardiovascular disease (CVD), including heart and peripheral vascular disease, is the leading cause of death in the United States and worldwide [1, 2], and the social health and economic burden of its high mortality and disability rates are increasing.
Lipoprotein(a) [Lp(a)] is formed by the covalent binding of apolipoprotein A to apolipoprotein B via disulfide bonds [3], whose concentration is inversely related to the size of apolipoprotein A and whose 90% level is determined genetically [4]. Lp(a) has risen to become a recognized risk factor for CVD, when low-density lipoprotein cholesterol (LDL-C) is 104 mg/dl, Lp(a) is more closely associated with CVD than LDL-C and is a residual risk factor for CVD [5]. Its main physiopathological mechanisms include atherogenesis, promotion of inflammation and thrombosis [6]. Unlike LDL-C, despite the fact that Lp(a) has been linked to CVD, definitive proof of a causal relationship remains to be proven. A large epidemiological study demonstrated that the risk ratio for coronary heart disease (CHD) after adjustment for age and sex was 1.16 (95% CI 1.11–1.22) [7], other epidemiological studies [8, 9] and meta-analyses [10] have confirmed that Lp(a) with the development of CHD was significant. The association between Lp(a) and other CVD is still in dispute. Lp(a) has been identified in observational studies as a risk factor for atrial thrombi in atrial fibrillation (AF) patients, there is no connection between Lp(a) and incident AF events [11]. However, it is limited by the number of AF cases. Lp(a) is an independent risk factor for ischemic stroke(IS) [12], but studies on Lp(a) and stroke type are scarce. We performed Mendelian randomization(MR) analysis of large artery atherosclerosis stroke(ISL), small vessel stroke(ISS), and lacunar stroke(LIS). The relationship between Lp(a) and heart failure(HF) is inconclusive, Lp(a) and HF were not associated in a community study, while another observational study reported an association [13]. Limited evidence suggests a link between Lp(a) and hypertension (HBP), evidence from clinical sources indicates that approximately 30% of hypertensive patients have elevated Lp(a) levels [14]. A study pointed out that Lp(a) plays an important and direct role in thrombosis and reinforcement of the aortic wall of aneurysms [15], however, the correlation between Lp(a) and aortic aneurysm (AA) was not elucidated. The role of Lp(a) in venous embolic events such as pulmonary embolism(PE) is controversial, potential pathogenic mechanisms of Lp(a) include its similarity to fibrinogen, leading to reduced fibrin synthesis and fibrinolysis inhibition, the tendency of Lp(a) to oxidize upon entry into the vessel wall, and the production of highly immunogenic and pro-inflammatory phospholipids [16, 17], and whether these effects play an important role in PE is unclear [18]. Notably, the relationship between Lp(a) and various CVD is controversial, observational studies are susceptible to confounding factors and reverse causality, and the corresponding conclusions can be biased. And few studies on the causal relationship between Lp(a) and various CVD.
Genetic variants are used as instrument variables (IVs) in MR analysis, a strong tool for determining the relationship between exposures and diseases [19]. The confounders of individuals being randomly allocated genetic variants at the moment of conception can be greatly reduced using MR analysis. Furthermore, the possibility of reverse causation is reduced because the presence of the disease has no effect on people’s genotypes [20]. In this study, we performed a univariate MR to explore whether genetic evidence for Lp(a)-related traits were significantly associated with CVD risks.
Materials and methods
This is a univariate two-sample MR study with three major assumptions based on a publicly available GWAS. First, there must be a strong and independent relationship between the chosen instrumental variable, single nucleotide polymorphism, and Lp(a). Second, no correlation should exist between instrumental variables (IVs) and confounders. Third, the relationship between IVs and outcomes can only be through the exposure factor Lp(a). We considered whether the third hypothesis would be influenced by horizontal pleiotropy (IVs directly affecting the outcome) or by other recognized etiologies affecting CVD, excluding these limiting hypotheses (Fig. 1).
Fig. 1.

Schematic diagram of the MR assumptions underpinning an MR analysis of the association between Lipoprotein(a) levels and different CVD. Lp(a) lipoprotein(a), BMI body mass index, MI myocardial infarction, CHD coronary heart disease, MR Mendelian randomization
Study population
Lp(a) GWAS datasets
Single nucleotide polymorphisms (SNPs) were identified as IVs in our study. To obtain exposure group data, we chose SNPs closely associated with Lp(a) levels at the genome-wide significance level (P < 5E−8) from the GWAS including 377,590 European ancestors from Neale Lab. To avoid bias caused by linkage disequilibrium relationships in the analysis, the linkage disequilibrium of SNP, must satisfy the conditions r2 < 0.001 and Kb = 10,000. SNPs associated with CHD, MI, body mass index (BMI), and smoking were identified as multipotent IVs for AA, AF, CHD, and HF (genetic variation associated with multiple variables) [21–25], while SNPs associated with BMI were identified as IVs with pleiotropy for IS, LIS, ISL, ISS, PE, and HBP [26–28]. Extraction of information on IVs related to the exposure factor Lp(a) (Table 1). We estimated the R2 of each instrument and calculated the F statistic overall [29, 30]. The percentage of IVs that referred to exposure factors was what R2 refers to. When F value < 10, a weak instrumental variable was defined by reference to the value of F [31]:
Description: MAF: minor allele frequency; SD = se.exposure ; beta. Exposure; N:no. of samples; K: no. of SNP.
Table 1.
Extraction of information on instrumental variables related to the exposure factor Lp(a)
| SNP | Effect_allele | Other_allele | Beta | eaf | se | P | F |
|---|---|---|---|---|---|---|---|
| rs71565789 | C | T | −5.0482 | 0.021 | 0.473 | 1.364E−26 | 4.772 |
| rs117733303 | G | A | 43.396 | 0.008 | 0.732 | 1E−200 | 57.929 |
| rs146534110 | T | G | 28.147 | 0.013 | 0.597 | 1E−200 | 55.302 |
| rs1086567 | A | G | 2.8444 | 0.626 | 0.138 | 8.983E−94 | 197.929 |
| rs185864730 | C | T | −10.317 | 0.014 | 0.582 | 3.276E−70 | 8.885 |
| rs142709465 | T | C | −4.2508 | 0.021 | 0.471 | 1.683E−19 | 3.332 |
| rs3127580 | T | C | 27.895 | 0.157 | 0.177 | 1E−200 | 6713.695 |
| rs12179053 | T | C | −9.2946 | 0.259 | 0.152 | 1E−200 | 1448.551 |
| rs184158723 | A | G | 18.557 | 0.019 | 0.495 | 1E−200 | 51.219 |
| rs117857195 | T | G | −16.547 | 0.023 | 0.477 | 1E−200 | 54.796 |
| rs112110249 | C | T | −10.644 | 0.043 | 0.329 | 1E−200 | 87.014 |
| rs117881880 | A | T | −13.415 | 0.015 | 0.557 | 5.64E−128 | 17.147 |
| rs494554 | G | C | −9.6137 | 0.027 | 0.426 | 9.89E−113 | 27.003 |
| rs149210101 | A | C | −10.149 | 0.019 | 0.497 | 1.911E−92 | 15.897 |
| rs9355328 | C | T | 6.1439 | 0.975 | 0.432 | 5.955E−46 | 10.051 |
| rs138581538 | T | C | −8.2755 | 0.008 | 0.803 | 6.676E−25 | 1.727 |
| rs10455872 | G | A | 89.361 | 0.072 | 0.204 | 1E−200 | 27,447.45 |
| rs73596816 | A | G | 32.349 | 0.035 | 0.359 | 1E−200 | 552.619 |
| Total | 0.092089 | 118.093 |
Different CVD GWAS datasets
Complete summary of statistical results in genome-wide association studies for AA (n = 209,366), CHD (n = 361,194), ISL (n = 150,765), AF (n = 1,030,836), HBP (n = 164,147), HF (n = 208,178), IS (n = 218,792), and LIS (n = 225,419), ISS (n = 198,048), PE (n = 218,413), and the above outcomes regarding CVD were based on European ancestry. Table 2 and Additional file 5: Table S5 show the SNPs for lipoprotein(a) and the SNPs and sources for each CVD, respectively, as well as the MR framework.
Table 2.
The SNPs for lipoprotein(a) and the SNPs and sources for each cardiovascular disease
| No. of samples | No. of SNPs | Consortium/ID | Ancestry | |
|---|---|---|---|---|
| Lp(a) | 377,590 | 18 | Neale Lab | European |
| AA | 209,366 | 11 | finn-b-I9_STR_EXH | European |
| CHD | 361,194 | 11 | finn-b-I9_CHD | European |
| ISL | 150,765 | 18 | ebi-a-GCST005840 | European |
| AF | 1,030,836 | 14 | ebi-a-GCST006414 | European |
| HBP | 164,147 | 16 | finn-b-I9_HYPTENSEC | European |
| HF | 208,178 | 12 | finn-b-I9_HEARTFAIL | European |
| IS | 218,792 | 16 | finn-b-I9_STR_EXH | European |
| LIS | 225,419 | 13 | ebi-a-GCST90014122 | European |
| ISS | 198,048 | 18 | ebi-a-GCST006909 | European |
| PE | 218,413 | 16 | finn-b-I9_PULMEMB | European |
AA aortic aneurysm, CHD coronary heart disease, ISL large artery atherosclerosis stroke AF atrial fibrillation, HBP secondary hypertension, HF heart failure, IS ischemic stroke, LIS lacunar stroke, ISS small vessel stroke, PE pulmonary embolism
Statistical analysis
To determine the causal relationships between Lp(a) levels and various CVD, MR-Egger, weighted median, random-effects inverse variance weighting (IVW1), maximum likelihood, penalized weighted median, and fixed-effects model inverse variance weighting (IVW2) were performed. For different validity assumptions, different Mendelian estimates can be derived from the aforementioned methods, the most prominent of which is the IVW1 because all of its SNPs are valid IVs and the method produces accurate estimates [32], In the primary analyses, odds ratio (OR) estimates with 95% confidence intervals (CI) were reported. We would have more confidence in inferring causality and opposing multiplicity (or other forms of bias) if all of the above methods were consistent. Mendelian randomization-PRESSO (MR-PRESSO) offers an alternative statistical detection method. Due to the bias of the multieffectiveness (global test) and the provision of an accurate estimate by outlier kick-out [33]. Briefly, MR-PRESSO corrects for horizontal pleiotropy by removing outliers. To assess heterogeneity, we utilized MR-Egger regression and IVW. The MR-Egger interaction method was used to test for horizontal pleiotropy and the leave-one-out method was used to investigate the possibility that this causal relationship was driven by a single SNP. Anderson–Darling normality test and Shapiro–Wilk normality test were used to test for normality. All statistical tests were two-tailed, and a P < 0.05 was considered statistically significant. All data analysis was implemented using R Studio 4.2.1 with the “ Two-Sample-MR (version 0.5.6, Bristol, UK)” “ MR-PRESSO (version 1.0, New York, NY, USA)” and “ Mr. raps” packages for MR analysis.
Results
Selection of instrumental variable
We chose diverse numbers of SNPs as IVs for various CVD. 11, 14, 11, 18, 16, 12, 16,13, 18, and 16 was selected as IVs for AA, AF, CHD, ISL, HBP, HF, IS, LIS, ISS, and PE, respectively. From the scope of the GWAS, they were all associated with Lp(a) levels. When the value of F > 10, a strong IV is defined with reference to the value of F (Table 1).
The causal effect between Lp(a) and different CVD
Patients with high Lp(a) levels has a 0.5-fold increased risk of AA(OR = 1.005, 95% CI 1.001–1.010, P = 0.009), a 0.3-fold increased risk of CHD (OR = 1.003, 95% CI 1.001–1.004, P = 0.010) and a 0.2-fold increased risk of ISL (OR = 1.003, 95% CI 1.002–1.004, P = 9.50E−11) using the IVW1. The IVW1 estimate showed that AF (OR = 1.001, 95% CI 1.000–1.002, P = 0.097), IS(OR = 1.001, 95% CI 1.000–1.001, P = 0.156), LIS(OR = 1.000, 95% CI 1.000–1.001, P = 0.524), PE(OR = 1.000, 95% CI 0.998–1.000, P = 0.210), HBP(OR = 1.000, 95% CI 0.998 1.002, P = 0.927), ISS (OR = 0.999, 95% CI 0.998–1.001, P = 0.430), HF(OR = 0.999, 95% CI 0.997–1.002, P = 0.584) were not associated with Lp(a). The MR-Egger estimate showed that genetically predicted Lp(a) was not significantly associated with the risk of CHD (Table 3). For AA, MR-PRESSO yielded P = 0.081, but no outliers were identified; therefore, The IVW1 method is more reliable. Table 4 displays the MR-PRESSO conclusions. There were no directional pleiotropies for the analysis results (all P > 0.05) (Table 3). Anderson–Darling normality test and Shapiro–Wilk normality test showed that only HF at the Anderson–Darling normality test was not normally distributed (P = 0.041), but the Shapiro–Wilk normality test was predominant.
Table 3.
Association between plasma lipoprotein a levels and cardiovascular diseases in Mendelian randomization analysis (six different methods corresponding to the results), heterogeneity as well as horizontal multiplicity test analysis
| Outcome | MR methods | OR (95% CI) | P for association | P for heterogeneity test | P for MR-Egger intercept |
|---|---|---|---|---|---|
| AA | MR-Egger | 1.010 (1.002–1.019) | 0.038 | 0.623 | 0.220 |
| IVW1 | 1.005 (1.001–1.010) | 0.009 | 0.544 | ||
| Weighted median | 1.006 (1.000–1.011) | 0.039 | |||
| Maximum likelihood | 1.006 (1.001–1.010) | 0.009 | |||
| PWM | 1.006 (1.000–1.011) | 0.042 | |||
| IVW2 | 1.005 (1.001–1.010) | 0.009 | |||
| CHD | MR-Egger | 0.999 (0.992–1.005) | 0.723 | 0.753 | 0.271 |
| IVW1 | 1.003 (1.001–1.004) | 0.010 | 0.703 | ||
| Weighted median | 1.003 (1.001–1.006) | 0.014 | |||
| Maximum likelihood | 1.003 (1.001–1.004) | 0.010 | |||
| PWM | 1.003 (1.001–1.006) | 0.011 | |||
| IVW2 | 1.003 (1.001–1.004) | 0.010 | |||
| ISL | MR-Egger | 1.003 (1.001–1.004) | 3.56E-04 | 0.588 | 0.381 |
| IVW1 | 1.003 (1.002–1.004) | 9.50E-11 | 0.599 | ||
| Weighted median | 1.003 (1.002–1.004) | 1.05E-07 | |||
| Maximum likelihood | 1.003 (1.002–1.004) | 9.10E-11 | |||
| PWM | 1.003 (1.002–1.004) | 2.28E-07 | |||
| IVW2 | 1.003 (1.002–1.004) | 9.50E-11 | |||
| AF | MR-Egger | 1.002 (1.000–1.005) | 0.070 | 0.829 | 0.204 |
| IVW1 | 1.001 (1.000–1.002) | 0.097 | 0.756 | ||
| Weighted median | 1.001 (1.000–1.003) | 0.089 | |||
| Maximum likelihood | 1.001 (1.000–1.002) | 0.098 | |||
| PWM | 1.001 (1.000–1.002) | 0.083 | |||
| IVW2 | 1.001 (1.000–1.002) | 0.097 | |||
| HBP | MR-Egger | 1.000 (0.998–1.002) | 0.997 | 0.457 | 0.900 |
| IVW1 | 1.000 (0.998–1.002) | 0.927 | 0.532 | ||
| Weighted median | 1.000 (0.998–1.002) | 0.892 | |||
| Maximum likelihood | 1.000 (0.998–1.002) | 0.927 | |||
| PWM | 1.000 (0.998–1.002) | 0.894 | |||
| IVW2 | 1.000 (0.998–1.002) | 0.927 | |||
| HF | MR-Egger | 1.000 (0.994–1.005) | 0.883 | 0.060 | 0.909 |
| IVW1 | 0.999 (0.997–1.002) | 0.584 | 0.088 | ||
| Weighted median | 1.000 (0.997–1.003) | 0.917 | |||
| Maximum likelihood | 0.999 (0.997–1.001) | 0.489 | |||
| PWM | 1.000 (0.997–1.003) | 0.921 | |||
| IVW2 | 0.999 (0.997–1.001) | 0.488 | |||
| IS | MR-Egger | 1.001 (1.000–1.002) | 0.152 | 0.197 | 0.514 |
| IVW1 | 1.001 (1.000–1.001) | 0.156 | 0.223 | ||
| Weighted median | 1.001 (1.000–1.001) | 0.069 | |||
| Maximum likelihood | 1.001 (1.000–1.001) | 0.112 | |||
| PWM | 1.001 (1.000–1.001) | 0.698 | |||
| IVW2 | 1.001 (1.000–1.001) | 0.112 | |||
| LIS | MR-Egger | 1.000 (1.000–1.001) | 0.847 | 0.159 | 0.736 |
| IVW1 | 1.000 (1.000–1.001) | 0.524 | 0.205 | ||
| Weighted median | 1.000 (0.999–1.000) | 0.331 | |||
| Maximum likelihood | 1.000 (0.999–1.000) | 0.466 | |||
| PWM | 1.000 (0.999–1.000) | 0.314 | |||
| IVW2 | 1.000 (0.999–1.000) | 0.466 | |||
| ISS | MR-Egger | 0.999 (0.998–1.001) | 0.353 | 0.007 | 0.381 |
| IVW1 | 0.999 (0.998–1.001) | 0.430 | 0.009 | ||
| Weighted median | 0.999 (0.998–1.000) | 0.071 | |||
| Maximum likelihood | 0.999 (0.999–1.000) | 0.265 | |||
| PWM | 0.999 (0.998–1.000) | 0.723 | |||
| IVW2 | 0.999 (0.999–1.000) | 0.265 | |||
| PE | MR-Egger | 1.000 (0.998–1.001) | 0.826 | 0.240 | 0.204 |
| IVW | 1.000 (0.998–1.000) | 0.210 | 0.192 | ||
| Weighted median | 1.000 (0.999–1.001) | 0.835 | |||
| Maximum likelihood | 0.999 (0.998–1.000) | 0.152 | |||
| PWM | 1.000 (0.999–1.001) | 0.879 | |||
| IVW2 | 0.999 (0.998–1.000) | 0.152 |
IVW1 random-effects inverse variance weighting, IVW2 fixed-effects model inverse variance weighting, PWM penalized weighted median, OR odds ratio, CI confidence interval, AA aortic aneurysm CHD coronary heart disease. ISL large artery atherosclerosis stroke, AF atrial fibrillation, HBP secondary hypertension, HF heart failure, IS ischemic stroke, LIS lacunar stroke, ISS small vessel stroke, PE pulmonary embolism
Table 4.
Association of plasma Lp(a) levels with cardiovascular disease in a Mendelian randomization analysis (MR-PRESSO)
| N | RAW OR | 95% CI | Estimates P | outlier N | OR | Corrected 95% CI | Estimates P | |
|---|---|---|---|---|---|---|---|---|
| AA | 11 | 1.005 | 1.000–1.009 | 0.081 | NA | NA | NA | NA |
| CHD | 11 | 1.002 | 1.001–1.005 | 0.028 | NA | NA | NA | NA |
| ISL | 18 | 1.003 | 1.002–1.004 | 1.40E-06 | NA | NA | NA | NA |
| AF | 14 | 1.001 | 1.000–1.002 | 0.081 | NA | NA | NA | NA |
| HBP | 16 | 1.000 | 0.998–1.002 | 0.915 | NA | NA | NA | NA |
| HF | 12 | 0.999 | 0.997–1.002 | 0.564 | NA | NA | NA | NA |
| IS | 16 | 1.000 | 1.000–1.001 | 0.162 | NA | NA | NA | NA |
| LIS | 13 | 1.000 | 0.999–1.000 | 0.547 | NA | NA | NA | NA |
| ISS | 18 | 0.999 | 0.998–1.000 | 0.445 | NA | NA | NA | NA |
| PE | 16 | 0.999 | 0.998–1.000 | 0.228 | NA | NA | NA | NA |
OR odds ratio, CI confidence interval, AA aortic aneurysm, CHD coronary heart disease, ISL large artery atherosclerosis stroke, AF atrial fibrillation, HBP secondary hypertension HF heart failure, IS ischemic stroke, LIS lacunar stroke. ISS small vessel stroke, PE pulmonary embolism
Heterogeneity and sensitivity
MR-Egger regression revealed heterogeneity for ISS (P = 0.007), whereas the IVW revealed heterogeneity for ISS (P = 0.009). The scatter plots and forest plots are displayed in Additional file 1: Figure S1 and Additional file 2: Figure S2. The funnel plots were symmetrical (Additional file 3: Figure S3) and the leave-one-out method indicated that no SNP was substantially driving the association between lipids traits and CVD risks (Figs. 2, 3 and Additional file 4: Figure S4).
Fig. 2.
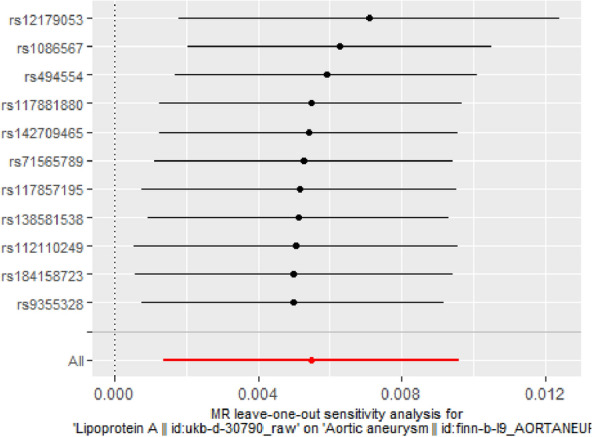
MR leave-one-out sensitivity analysis for Lp(a) on AA
Fig. 3.
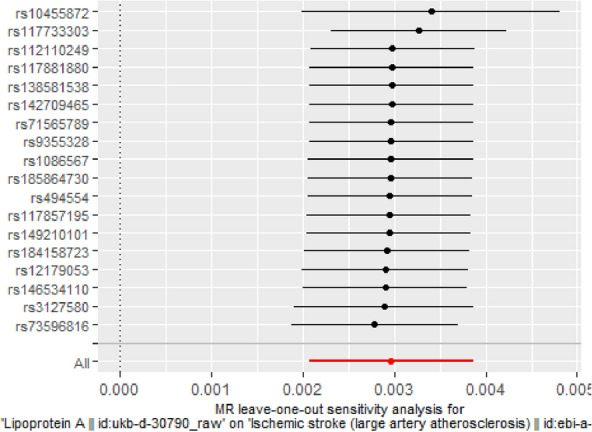
MR leave-one-out sensitivity analysis for Lp(a) on ISL
From the above analysis, it is evident that there is a causal relationship between Lp(a) and AA, CHD, and ISL, but not with other CVDs.
Discussion
This is one of the largest MR analysis to study the effect of Lp(a) on different CVDs such as AA, CHD, ISL, AF, HBP, HF, IS, LIS, ISS, and PE. Our study provided additional evidence that lowering Lp(a) will reduce the prevalence of CVD and may contribute to a better understanding of the genetic impact of Lp(a) on CVD. Our study suggested that patients with high Lp(a) levels has a 0.5-fold increased risk of AA(OR = 1.005, 95% CI 1.001–1.010, P = 0.009), with potential mechanisms including atherosclerosis and promotion of inflammation [34, 35], which all increased the risk of AA. It was demonstrated that elevated Lp(a) concentrations were independently associated with an increased risk of abdominal aortic aneurysms (AAA) in a population-based cohort study [36], and a meta-analysis demonstrated that high levels of Lp(a) may be linked to the presence of AAA and that Lp(a) may be a marker to screen for AAA [37]. Our study may provide more reliable evidence for a causal relationship between Lp(a) and CHD, and the relationship with CHD is similar to the findings previously observed in CHD Exome + (odds ratio rescaled per 50 mg/dL increment of Lp(a) levels, 1.35 [95% CI 1.29–1.41]) [38]. According to a research, Lp(a) is related with rapid advancement of coronary plaques, which may explain the elevated residual risk of MI associated with Lp(a) [39]. Lp(a) was independently associated with ISL [12], and we found a 0.2-fold increased risk of ISL(OR = 1.003, 95% CI 1.002–1.004, P = 9.50E−11). It has been shown that elevated serum Lp(a) levels predict the risk of early stroke recurrence in patients with a first IS [40], but we concluded that there is null association between Lp(a) and IS, LIS and ISS. The unique structure of the Lp(a) may form the link between atherosclerosis (which is partially mediated by the LDL sample) and thrombotic (partially mediated by apolipoprotein B), and contribute to IS [41]. In addition, heterogeneity was observed in the analysis of Lp(a) and ISS, which may be related to age and gender, but we were unable to obtain relevant genome-wide association subgroup information at this time. There was no correlation between Lp(a) and AF, HF, or PE. However, the findings of a previous study suggested that adjusted Lp(a) was predictive of new-onset AF (hazard ratio for 1-SD increase, 2.69; 95% CI 1.00–7.22; P < 0.05) [42]. In contrast, a MR study suggested that Lp(a) may be a potential causative risk factor for AF, which requires confirmation in a large number of future investigations [43]. There have been few studies on Lp(a) and HBP, and it is clear that there is no connection between Lp(a) and HBP, evidence from a clinic indicates that hypertensive cohort's patients had increased Lp(a) levels, indicating that Lp(a) assessment may be helpful in risk stratification [14]. This suggested that more research will be required in this field. There was no link between Lp(a) and HF, and a community research found no correlation between Lp(a) and HF, too; yet, some studies implied that Lp(a) increased the risk of HF [13]; therefore, larger investigations are required. Lp(a) had a structural component comparable to fibrinogen, and its oxidative, pro-inflammatory actions may be related with venous thrombosis leading to PE; nevertheless, our investigation found no association between Lp(a) and the incidence of PE events. Similar investigations also failed to find a link between PE severity and Lp(a) levels [44].
The main advantages of this study were the application of a MR analysis that can resist confounding factors and the use of a large GWAS to reduce the possibility of false negatives. The results of MR investigations, however, were susceptible to pleiotropy [45]. By removing specific genetic variants from the current investigation, we were able to lower the pleiotropy of genetic variants. There were certain restrictions, though. First, MR relies on three main assumptions that were difficult to verify empirically. In addition, we did not investigate the causal relationship between other lipids and CVD because we focused primarily on genetically determined Lp(a), whose causative mechanisms remain controversial and whose effects on a variety of CVD were also debatable. To further justify the use of lipid-lowering drugs, the following study might use a multivariate MR technique to examine the impact of various lipids or lipoproteins on CVD. A MR analysis based on different population groups should be conducted to eliminate racial bias, as Lp(a) concentrations are mostly genetically determined and population-related. Furthermore, because we lacked complete information on our participants' clinical features and numerous sizable GWAS, we were unable to do subgroup analysis. Finally, given that future GWAS will always overlook people who have passed away from exposure or other competing risks for outcomes, our results may be biased due to selection.
Conclusion
This MR study suggested a causal relationship between Lp(a) and CHD, AA, and ISL, but not associated with AF, HF, IS, LIS, ISS, HBP, or PE. Our work further verifies the association between Lp(a) and various CVD, resulting in improved Lp(a) management and a reduction in the prevalence of CVD.
Supplementary Information
Additional file 1: Figure S1. The scatter plots of Lp(a) on CVD.
Additional file 2: Figure S2. The forest plots of Lp(a) on CVD.
Additional file 3: Figure S3. The funnel plots of Lp(a) on CVD.
Additional file 4: Figure S4. The leave-one-out method of Lp(a) on CVD (except AA and ISL).
Additional file 5: The SNPs for each CVD.
Acknowledgements
Not applicable.
Abbreviations
- Lp(a)
Lipoprotein(a)
- CVD
Cardiovascular disease
- LDL-C
Low-density lipoprotein cholesterol
- MR
Mendelian randomization
- GWAS
Genome-wide association study
- IVs
Instrumental variables
- SNPs
Single nucleotide polymorphisms
- AA
Aortic aneurysm
- CHD
Coronary heart disease
- ISL
Large artery atherosclerosis stroke
- AF
Atrial fibrillation
- HBP
Secondary hypertension
- HF
Heart failure
- IS
Ischemic stroke
- LIS
Lacunar stroke
- ISS
Small vessel stroke
- PE
Pulmonary embolism
- IVW1
Random-effects inverse variance weighting
- IVW2
Fixed-effects model inverse variance weighting
- PWM
Penalized weighted median
- MR-PRESSO
Mendelian randomization-PRESSO
- OR
Odds ratio
- CI
Confidence interval
- BMI
Body mass index
- MI
Myocardial infarction
Author contributions
All authors made contribution in writing the manuscript. All authors read and approved the final manuscript.
Funding
Not applicable.
Availability of data and materials
Not applicable.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Shiyue Wang, Email: orangewsydb@163.com.
Li Zha, Email: girlnigela@163.com.
Jian Chen, Email: doctorchensc@163.com.
Dongjie Du, Email: adu1259645770@163.com.
Danyang Liu, Email: liudanyang1121@163.com.
Ming Zhong, Email: zhongming202207@163.com.
Rongfang Shang, Email: s18447112692@163.com.
Dongxue Sun, Email: 15145729872@163.com.
Chang Sun, Email: 18745613698@163.com.
Enze Jin, Email: enzejin@163.com.
References
- 1.Benjamin EJ, et al. Heart disease and stroke statistics-2019 update a report from the american heart association. Circulation. 2019;139(10):E56–E528. doi: 10.1161/CIR.0000000000000659. [DOI] [PubMed] [Google Scholar]
- 2.James SL, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the global burden of disease study 2017. Lancet. 2018;392(10159):1789–1858. doi: 10.1016/S0140-6736(18)32279-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maranhao RC, et al. Lipoprotein (a): structure, pathophysiology and clinical implications. Arq Bras Cardiol. 2014;103(1):76–83. doi: 10.5935/abc.20140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsimikas S, Moriarty PM, Stroes ES. Emerging RNA therapeutics to lower blood levels of Lp(a) JACC focus seminar 2/4. J Am Coll Cardiol. 2021;77(12):1576–1589. doi: 10.1016/j.jacc.2021.01.051. [DOI] [PubMed] [Google Scholar]
- 5.Li C, et al. The correlation between lipoprotein(a) and coronary atherosclerotic lesion is stronger than LDL-C, when LDL-C is less than 104 mg/dL. BMC Cardiovasc Disord. 2021;21(1):7. doi: 10.1186/s12872-021-01861-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reyes-Soffer G, et al. Lipoprotein(a): a genetically determined, causal, and prevalent risk factor for atherosclerotic cardiovascular disease: a scientific statement from the american heart association. Arterioscler Thromb Vasc Biol. 2022;42(1):E48–E60. doi: 10.1161/ATV.0000000000000147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erqou S, et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. Jama-J Am Med Assoc. 2009;302(4):412–423. doi: 10.1001/jama.2009.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Extreme Lipoprotein(a) levels and improved cardiovascular risk prediction. J Am Coll Cardiol. 2013;61(11):1146–1156. doi: 10.1016/j.jacc.2012.12.023. [DOI] [PubMed] [Google Scholar]
- 9.Kamstrup PR, et al. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population. Circulation. 2008;117(2):176–184. doi: 10.1161/CIRCULATIONAHA.107.715698. [DOI] [PubMed] [Google Scholar]
- 10.Erqou S, et al. Apolipoprotein(a) Isoforms and the risk of vascular disease systematic review of 40 studies involving 58,000 participants. J Am Coll Cardiol. 2010;55(19):2160–2167. doi: 10.1016/j.jacc.2009.10.080. [DOI] [PubMed] [Google Scholar]
- 11.Igarashi Y, et al. Elevated serum lipoprotein(a) is a risk factor for left atrial thrombus in patients with chronic atrial fibrillation: a transesophageal echocardiographic study. Am Heart J. 1998;136(6):965–971. doi: 10.1016/s0002-8703(98)70151-6. [DOI] [PubMed] [Google Scholar]
- 12.Arnold M, et al. Lipoprotein(a) is associated with large artery atherosclerosis stroke aetiology and stroke recurrence among patients below the age of 60 years: results from the BIOSIGNAL study. Eur Heart J. 2021;42(22):2186–2196. doi: 10.1093/eurheartj/ehab081. [DOI] [PubMed] [Google Scholar]
- 13.Kamstrup PR, Nordestgaard BG. Elevated Lipoprotein(a) levels, lpa risk genotypes, and increased risk of heart failure in the general population. Jacc-Heart Fail. 2016;4(1):78–87. doi: 10.1016/j.jchf.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 14.Ward NC, et al. Lipoprotein (a) and hypertension. Curr Hypertens Rep. 2021;23(12):9. doi: 10.1007/s11906-021-01161-6. [DOI] [PubMed] [Google Scholar]
- 15.Papagrigorakis E, et al. Lipoprotein(a) in plasma, arterial wall, and thrombus from patients with aortic aneurysm. Clin Genet. 1997;52(5):262–271. doi: 10.1111/j.1399-0004.1997.tb04343.x. [DOI] [PubMed] [Google Scholar]
- 16.von Eckardstein A. Lipoprotein(a) Eur Heart J. 2017;38(20):1530–1532. doi: 10.1093/eurheartj/ehx233. [DOI] [PubMed] [Google Scholar]
- 17.Boffa MB, Koschinsky ML. thematic review series: lipoprotein (a): coming of age at last lipoprotein (a): truly a direct prothrombotic factor in cardiovascular disease? J Lipid Res. 2016;57(5):745–757. doi: 10.1194/jlr.R060582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rijken DC, et al. How significant is the antifibrinolytic effect of lipoprotein(a) for blood clot lysis? Thromb Res. 2021;198:210–212. doi: 10.1016/j.thromres.2020.12.014. [DOI] [PubMed] [Google Scholar]
- 19.Emdin CA, Khera AV, Kathiresan S. Mendelian randomization. Jama-J Am Med Assoc. 2017;318(19):1925–1926. doi: 10.1001/jama.2017.17219. [DOI] [PubMed] [Google Scholar]
- 20.Sheehan NA, et al. Mendelian randomisation and causal inference in observational epidemiology. PLoS Med. 2008;5(8):1205–1210. doi: 10.1371/journal.pmed.0050177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamimura D, et al. Cigarette smoking and incident heart failure: insights from the jackson heart study. Circulation. 2018;137(24):2572–2582. doi: 10.1161/CIRCULATIONAHA.117.031912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Albertsen IE, et al. Smoking, atrial fibrillation, and ischemic stroke: a confluence of epidemics. Curr Opin Cardiol. 2015;30(5):512–517. doi: 10.1097/HCO.0000000000000205. [DOI] [PubMed] [Google Scholar]
- 23.Overvad TF, et al. Body mass index and adverse events in patients with incident atrial fibrillation. Am J Med. 2013;126(7):9. doi: 10.1016/j.amjmed.2012.11.024. [DOI] [PubMed] [Google Scholar]
- 24.Takada M, et al. Body mass index and mortality from aortic aneurysm and dissection. J Atheroscler Thromb. 2021;28(4):338–348. doi: 10.5551/jat.57232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ashton WD, Nanchahal K, Wood DA. Body mass index and metabolic risk factors for coronary heart disease in women. Eur Heart J. 2001;22(1):46–55. doi: 10.1053/euhj.2000.2469. [DOI] [PubMed] [Google Scholar]
- 26.Saini M, et al. Body mass index and acute ischemic stroke outcomes. Int J Stroke. 2014;9(5):618–623. doi: 10.1111/ijs.12168. [DOI] [PubMed] [Google Scholar]
- 27.Robinson RF, et al. Body mass index in primary and secondary pediatric hypertension. Pediatr Nephrol. 2004;19(12):1379–1384. doi: 10.1007/s00467-004-1588-8. [DOI] [PubMed] [Google Scholar]
- 28.Rahmani J, et al. Relationship between body mass index, risk of venous thromboembolism and pulmonary embolism: a systematic review and dose-response meta-analysis of cohort studies among four million participants. Thromb Res. 2020;192:64–72. doi: 10.1016/j.thromres.2020.05.014. [DOI] [PubMed] [Google Scholar]
- 29.Burgess S, Thompson SG, Collaboration CCG. Avoiding bias from weak instruments in mendelian randomization studies. Int J Epidemiol. 2011;40(3):755–764. doi: 10.1093/ije/dyr036. [DOI] [PubMed] [Google Scholar]
- 30.Pierce BL, Ahsan H, VanderWeele TJ. Power and instrument strength requirements for mendelian randomization studies using multiple genetic variants. Int J Epidemiol. 2011;40(3):740–752. doi: 10.1093/ije/dyq151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dan YL, et al. Circulating adiponectin levels and systemic lupus erythematosus: a two-sample mendelian randomization study. Rheumatology. 2021;60(2):940–946. doi: 10.1093/rheumatology/keaa506. [DOI] [PubMed] [Google Scholar]
- 32.Pierce BL, Burgess S. Efficient design for mendelian randomization studies: subsample and 2-sample instrumental variable estimators. Am J Epidemiol. 2013;178(7):1177–1184. doi: 10.1093/aje/kwt084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verbanck M, et al. Detection of widespread horizontal pleiotropy in causal relationships inferred from mendelian randomization between complex traits and diseases. Nat Genet. 2018 doi: 10.1038/s41588-018-0099-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Freestone T, et al. Inflammation and matrix metalloproteinases in the enlarging abdominal aortic-aneurysm. Arterioscler Thromb Vasc Biol. 1995;15(8):1145–1151. doi: 10.1161/01.atv.15.8.1145. [DOI] [PubMed] [Google Scholar]
- 35.Lee AJ, et al. Smoking, atherosclerosis and risk of abdominal aortic aneurysm. Eur Heart J. 1997;18(4):671–676. doi: 10.1093/oxfordjournals.eurheartj.a015314. [DOI] [PubMed] [Google Scholar]
- 36.Kubota Y, et al. Lipoprotein(a) and abdominal aortic aneurysm risk: the atherosclerosis risk in communities study. Atherosclerosis. 2018;268:63–67. doi: 10.1016/j.atherosclerosis.2017.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kotani K, et al. Lipoprotein(a) levels in patients with abdominal aortic aneurysm: a systematic review and meta-analysis. Angiology. 2017;68(2):99–108. doi: 10.1177/0003319716637792. [DOI] [PubMed] [Google Scholar]
- 38.Burgess S, et al. Association of lpa variants with risk of coronary disease and the implications for lipoprotein(a)-lowering therapies a mendelian randomization analysis. JAMA Cardiology. 2018;3(7):619–627. doi: 10.1001/jamacardio.2018.1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaiser Y, et al. Association of lipoprotein(a) with atherosclerotic plaque progression. J Am Coll Cardiol. 2022;79(3):223–233. doi: 10.1016/j.jacc.2021.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hong XW, et al. Lipoprotein (a) as a predictor of early stroke recurrence in acute ischemic stroke. Mol Neurobiol. 2018;55(1):718–726. doi: 10.1007/s12035-016-0346-9. [DOI] [PubMed] [Google Scholar]
- 41.van Capelleveen JC, van der Valk FM, Stroes ESG. Thematic review series: lipoprotein (a): coming of age at last current therapies for lowering lipoprotein (a) J Lipid Res. 2016;57(9):1612–1618. doi: 10.1194/jlr.R053066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li WJ, et al. The prospective effect of lipoprotein(a) on new-onset atrial fibrillation in patients with chronic heart failure. Int J Clin Exp Med. 2016;9(9):18316–18323. [Google Scholar]
- 43.Mohammadi-Shemirani P, et al. elevated lipoprotein(a) and risk of atrial fibrillation an observational and mendelian randomization study. J Am Coll Cardiol. 2022;79(16):1579–1590. doi: 10.1016/S0735-1097(22)02570-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gressenberger P, et al. Lipoprotein(a) and pulmonary embolism severity-a retrospective data analysis. Front Cardiovasc Med. 2022;9:6. doi: 10.3389/fcvm.2022.808605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thompson JR, et al. Mendelian randomization incorporating uncertainty about pleiotropy. Stat Med. 2017;36(29):4627–4645. doi: 10.1002/sim.7442. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. The scatter plots of Lp(a) on CVD.
Additional file 2: Figure S2. The forest plots of Lp(a) on CVD.
Additional file 3: Figure S3. The funnel plots of Lp(a) on CVD.
Additional file 4: Figure S4. The leave-one-out method of Lp(a) on CVD (except AA and ISL).
Additional file 5: The SNPs for each CVD.
Data Availability Statement
Not applicable.