

Abstract
Dehydroacetic acid and triacetic acid lactone are known to be versatile substrates for the synthesis of a variety of azaheterocycles. However, their fluorinated analogs were poorly described in the literature. In the present work, we have investigated reactions of trifluorotriacetic acid lactone and hexafluorodehydroacetic acid with primary amines, phenylenediamine, and phenylhydrazine. While hexafluorodehydroacetic acid reacted the same way as non-fluorinated analog giving 2,6-bis(trifluoromethyl)-4-pyridones, trifluorotriacetic acid lactone had different regioselectivity of nucleophilic attack compared to the parent structure, and corresponding 3-amino-6,6,6-trifluoro-5-oxohex-3-eneamides were formed as the products. In the case of binucleophiles, further cyclization took place, forming corresponding benzodiazepine and pyrazoles. The obtained 2,6-bis(trifluoromethyl)-4-pyridones were able to react with active methylene compounds giving fluorinated merocyanine dyes.
Keywords: 4-hydroxy-2-pyrone, 4-pyridone, aminoenone, trifluoromethylated heterocycles, regioselective reactions
1. Introduction
Oxygen heterocycles are common precursors in organic synthesis and have a special place in the synthesis of important nitrogen heterocycles by various methodologies. Among others, pyrones attract significant interest both due to a variety of chemical properties and a wide distribution in nature [1,2,3,4]. Particular attention is paid to 4-hydroxy-2-pyrones, namely, triacetic acid lactone (TAL), dehydroacetic acid (DHA, Figure 1), and their derivatives, which belong to polyketides and occur widely in living organisms [5,6,7,8]. The availability of these compounds and the possibility of their biochemical synthesis contributed to their extensive research and to the development of a wide range of options for their modification, considering them as platform compounds [9,10,11,12].
Figure 1.

Structure of DHA, TAL, DHA-f6, and TAL-f3.
On the other hand, the introduction of fluorine into the structure of molecules often leads to a modification of their chemical properties [13] and is a common strategy in drug design [14,15,16,17,18], pesticides synthesis [19,20], and other areas [21,22,23,24]. However, there are very limited data on fluorine-containing analogs of TAL and DHA in the literature, which practically do not cover their properties. The only synthesis of 4-hydroxy-3-trifluoroacetyl-6-trifluoromethyl-2H-pyran-2-one (hexafluorodehydroacetic acid, DHA-f6, Figure 1) was performed by the heating of trifluoroacetoacetic ester in the presence of P2O5 and proceeds through formation and dimerization of trifluoroacetylketene [25]. Hydrolysis of DHA-f6 in aqueous NaHCO3 leads to 4-hydroxy-6-trifluoromethyl-2H-pyran-2-one (trifluorotriacetic acid lactone, TAL-f3, Figure 1) [25]. The second route for the synthesis of TAL-f3 presented in the literature comprises cyclization of the corresponding trifluorodioxocaproic acid with acetic anhydride [26]. Among the properties of TAL-f3, only modification of the 4-OH group is known, which was methylated with Me2SO4 [27] and triflated with Tf2O [26]. The ring-opening of the O-methyl derivative on treatment with magnesium methylate gave methyl 3-methoxy-5-oxo-6,6,6-trifluoro-5-hexenoate [27] and the 4-OTf derivative reacted with 4-(methylthio)phenylboronic acid to give a Suzuki coupling product [26].
Thus, compounds that are attractive in terms of reactivity and properties of possible products turned out to be completely unexplored, and in this work, we have studied the interaction of DHA-f6 and TAL-f3 with primary amines as typical nucleophiles.
2. Results and Discussion
To begin with, we reproduced the synthesis described by German et al. (Scheme 1) [25], as it gives access both to DHA-f6 and TAL-f3. The results of the first step were quite inconsistent, and the yield ranged from 0% to 75%, presumably due to heating with a flame burner, which is hard to control. Nevertheless, the product was obtained in a satisfactory quantity for the further research. It should be noted that no variation in reaction conditions has improved the yield. Thus, the lowering of P2O5 loading or increasing reaction time resulted in the formation of complex mixtures, either of open-chain condensation products and trimerization ones characterized by the ester methylene quartets in 1H NMR spectra or triads of equal intensity in 19F NMR spectra correspondingly. The hydrolysis of DHA-f6 was more reproducible; however, we could never achieve the literature yield of 95% and had 40% on average (Scheme 1). Apart from NaHCO3, we have used NaOH and Na2CO3 as a base, but the yields were slightly lower. Adjusting pH to 4–5 during hydrolysis as the original procedure claims was quite difficult because it dropped down in time due to trifluoroacetic acid formation, so 2.0 equiv. of NaHCO3 was used instead. Moreover, crystallization of TAL-f3 did not occur until acidification to pH 0–1. We tried, as well, to carry out the detrifluoroacetylation of DHA-f6 by boiling it in water or by treatment with 93% H2SO4 or 70% HClO4 at room temperature, but the conversion was negligible.
Scheme 1.

Synthesis of DHA-f6 (1) and TAL-f3 (2).
At the first step, we studied the reaction of DHA-f6 with aniline. There are three main consecutive products that can be expected based on the literature data (Scheme 2) [28,29]. While a formation of Schiff bases usually proceeds readily in acidic media, no conversion was observed when we attempted the synthesis of intermediate A in aqueous HCl with 1.5 equiv. of PhNH2 (Table 1, entry 1). When EtOH was used as a solvent, spontaneous decarboxylation took place and N-phenylpyridone 3a was isolated in 60% yield (Table 1, entry 2). Formation of intermediate B was also observed and will be discussed later. Increasing the amount of PhNH2 and carrying out the reaction in a less polar solvent or without it as well as raising the temperature did not improve the yield (Table 1, entries 3–10).
Scheme 2.

Reaction of DHA-f6 with aniline.
Table 1.
Optimization of the reaction conditions of DHA-f6 with aniline a.
| Entry | PhNH2, Equiv. | Solvent | Yield 3a, % |
|---|---|---|---|
| 1 | 1.5 | HCl (0.5 M) b | n.r. c |
| 2 | 2.3 | ethanol | 60 |
| 3 | 3.5 | ethanol | 40 |
| 4 | 4.0 | ethanol | 45 |
| 5 | 2.3 | ethanol | 37 d |
| 6 | 4.0 | 1,4-dioxane | 36 |
| 7 | 3.5 | 1,4-dioxane | 27 e |
| 8 | 4.0 | toluene | 17 |
| 9 | 2.3 | toluene | 19 |
| 10 | 2.3 | neat | 25 |
a A solution of PhNH2 in 1 mL of a solvent was added to DHA-f6 (1) (100 mg, 0.36 mmol) and the reaction mixture was stirred at room temperature for 24 h. b Aqueous solution. c No reaction. d The reaction was carried out at 65 °C for 12 h. e The reaction was carried out at reflux for 4 h.
Exploring the scope of the synthesis of 4-pyridones 3 from DHA-f6, we found that the reaction rates and the yields correlate with nucleophilicity of amine. Thus, aniline derivatives bearing π-donor substituents (MeO, F) at the 4-position react considerably faster, whereas those bearing strong acceptor substituents (Ac, CF3) at the 3-position or a weak acceptor substituent (Br) at the 4-position react at a comparable rate but give slightly lower yields (Scheme 3, Table 2). Noteworthy, even extremely weakly nucleophilic 4-nitroaniline was able to react although the conversion was low even after 6 days, and pyridone 3k was formed in only 8% yield. The characteristic signals of pyridones 3 in the NMR spectra acquired in CDCl3 are the singlet of vinylic CH with double intensity at 6.95–7.02 ppm (1H NMR) and the quartets of symmetric carbons at 119.2–119.4 ppm (CF3), 140.8–141.6 ppm (C-2 and C-6), and 119.0–119.6 ppm (C-3 and C-5).
Scheme 3.

Reaction of DHA-f6 with aromatic amines.
Table 2.
The scope of 4-pyridone synthesis from DHA-f6 a.
| Pyridone | Ar | Time, h | Yield, % | Mp, °C |
|---|---|---|---|---|
| 3a | Ph | 24 | 60 | 135–136 |
| 3b | 4-MeOC6H4 | 8 | 56 | 125–126 |
| 3c | 3,4-F2C6H3 | 6 | 57 | 142–143 |
| 3d | 4-BrC6H4 | 24 | 47 | 129–131 |
| 3e | 3-AcC6H4 | 24 | 47 | 118–119 |
| 3f | 3-F3CC6H4 | 24 | 47 | 91–92 |
| 3g | 2,5-F2C6H3 | 48 | 46 | 116–119 |
| 3h | 2-ClC6H4 | 40 | 49 | 132–136 |
| 3i | 2-MeC6H4 | 24 | 65 | Liq. |
| 3j | 2,5-Me2C6H3 | 24 | 48 | 105–107 |
| 3k | 4-O2NC6H4 | 144 | 8 b | – c |
a A solution of ArNH2 (0.83 mmol) in EtOH was added to DHA-f6 (1) (100 mg, 0.36 mmol) and the reaction mixture was stirred at room temperature for a given amount of time. b Based on 1H NMR spectrum. c The product was obtained in the mixture with 4-nitroaniline.
The existence of an ortho-substituent did not affect the initial two steps of the reaction sequence and CO2 evolution occurred during first 1–2 h; however, the cyclization of bisenamines B depends on electron properties of the substituent. In the case of more reactive o-toluidine and 2,5-xylidine, the overall duration was 24 h, whereas 2-chloro- and 2,5-difluoroaniline required twice as much time (Table 2). It allowed us to isolate and characterize the intermediate B′ (Scheme 3), although in the mixture with starting 2-chloroaniline, which did not separate by the column chromatography. Its structure was confirmed by very distinguishable singlets of symmetric groups at 5.78 ppm (vinylic CH) and 11.49 ppm (enaminone NH) in 1H NMR and at 97.8 ppm in 19F NMR spectra in CDCl3. The formation of adducts with the structure of B was observed by TLC in all the cases, but for most of them the simultaneous cyclization to pyridones 3 occurred, so their isolation was impractical.
Surprisingly, aliphatic amines such as butylamine, N,N-dimethylethylenediamine and benzylamine did not react with DHA-f6 under the conditions found, even after several weeks. It can be attributed to their much higher basicity and fixation of the substrate in an anionic form that prevents further nucleophilic attack. This assumption was supported by the formation of the salt 4 (Scheme 3) in the reaction with 2-aminopyridine, which precipitated in nearly quantitative yield and remained unchanged after 8 days at 60 °C in EtOH. The signals in the 19F NMR spectrum of compound 4 in DMSO-d6 almost exactly match the signals of DHA-f6 (91.0 and 89.8 ppm compared to 91.1 and 89.6 ppm, respectively) and the singlet of pyrone CH in the 1H NMR spectrum is shifted upfield by 0.09 ppm (6.15 compared to 6.24 ppm) in agreement with its anionic character.
Reactivity of 4-pyridones 3 is greatly affected by strongly acceptor CF3-groups. Thus, electrophilic bromination of 3a with NBS proceeded poorly compared to the non-fluorinated analog [30] and no distinct product was obtained. On the contrary, Knoevenagel condensation did well with malononitrile, barbituric acid, and indane-1,3-dione in acetic anhydride, leading to fluorinated merocyanine dyes 5 (Scheme 4, Table 3). This reveals that the electron deficiency of a heterocycle plays an important role for this reaction, as 1-aryl-2,6-dimethyl-4-pyridones are less active [31,32] compared both to the CF3-substituted pyridone 3a and to the oxygen analog, 2,6-dimethyl-4-pyrone [33]. The time required for the reaction completion is in accordance with the acidity of an active methylene compound, so one can assume that deprotonation is a rate-limiting step, and a high activity of a substrate is necessary to prevent self-condensation of CH2X2.
Scheme 4.

Reaction of 4-pyridone 3a with active methylene compounds.
Table 3.
Synthesis and structure of adducts 5a–c a.
| Compound | Structure | Time, h | Yield, % | Mp, °C |
|---|---|---|---|---|
| 5a |
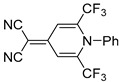
|
10 | 65 | 172–173 |
| 5b |
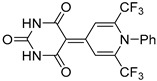
|
4 | 58 | 270–271 |
| 5c |
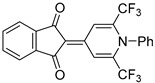
|
6 | 43 | 208–209 |
a A solution of 4-pyridone 3a (100 mg, 0.33 mmol) and the corresponding CH2X2 (0.4 mmol) in Ac2O was stirred at 140 °C for a given amount of time.
The prepared compounds, 5a–c, are found to be yellow solids and exhibit an absorbance major maximum at 382–437 nm in the visible region of the spectra. For barbituric derivative 5b, the high molar extinction coefficient (91406 M−1·cm−1 at 412 nm) is observed and probably indicates the intramolecular charge-transfer described by the aromatic resonance form 5′ (Figure 2). For dihydropyridine 5c bearing the indanedione moiety, additional intensive absorption maxima are observed at 246 and 224 nm. An interesting feature in NMR spectra of compounds 5 is a large difference compared to each other in chemical shifts of CH-groups in the dihydropyridine fragment attributed to the magnetic anisotropy of carbonyl and cyano groups, which have a closer proximity in derivative 5b (δH3 = 9.77 ppm, δC3 = 116.8 ppm) than in 5c (δH3 = 9.10 ppm, δC3 = 113.4 ppm) and in 5a (δH3 = 7.26 ppm, δC3 = 112.7 ppm). Another possible explanation for the difference is a greater contribution of the betaine resonance form 5′ (Figure 2) for the adduct with barbituric acid, but this seems to have a smaller effect as the chemical shifts of the rest of carbon atoms diverge much less (Table S2 in Supplementary Materials).
Figure 2.

Resonance forms of compound 5.
Moving on to the investigation of trifluorotriacetic acid lactone (2) properties, we focused on our previous work on the study of triacetic acid lactone (6) [34] and 2-cyano-6-(trifluoromethyl)-4H-pyran-4-one (7) [35], which gave carbamoylated enaminones 8 (Scheme 5), which proved to be a versatile building blocks for nitrogen heterocycles. In both cases, the substrates were attacked with two equivalents of amine at positions 2 and 6.
Scheme 5.

Reaction of TAL and 2-cyano-6-(trifluoromethyl)-4-pyrone with primary amines.
We started the optimization of the reaction conditions of TAL-f3 (2) with aniline in EtOH using various amount of amine (Scheme 6, Table 4, entries 1–3). The product 9a with unexpected regiochemistry was formed with the best yield of 64% when only little excess of PhNH2 was applied. The reaction performs better in aprotic polar 1,4-dioxane (Table 4, entry 4) and worse in non-polar toluene or without a solvent (Table 4; entries 5, 6). It should be noted that an increase in the reaction temperature did not improve the yield of enaminone 9a.
Scheme 6.

Reaction of TAL-f3 with aniline.
Table 4.
Optimization of the reaction conditions of TAL-f3 with aniline a.
| Entry | PhNH2, Equiv. | Solvent | Yield 9a, % |
|---|---|---|---|
| 1 | 5 | ethanol | 43 |
| 2 | 3.5 | ethanol | 63 |
| 3 | 2.1 | ethanol | 64 |
| 4 | 2.1 | 1,4-dioxane | 72 |
| 5 | 2.1 | toluene | 30 |
| 6 | 2.1 | neat | 12 |
a A solution of PhNH2 was added to TAL-f3 (2) (100 mg, 0.56 mmol). The reaction mixture was stirred at room temperature for 24 h.
The reaction with electron-rich p-anisidine proceeded equally well, whereas electron-poor p-bromoaniline gave lower yield (Scheme 7, Table 5). The reaction with aliphatic amines again has difficulties, probably due to the formation of salts; however, the lower acidity of TAL-f3 compared to DHA-f6 allowed enaminones 9d,e to form in low yields. In the case of butylamine, the heating to 60 °C was also needed. All amines reacted with the same regioselectivity, and no alternative isomers 8 were isolated.
Scheme 7.

Reaction of TAL-f3 with primary amines.
Table 5.
The scope of reaction of TAL-f3 with primary amines a.
| Enaminone | R | Time, h | Yield, % | Z/E ratio b |
|---|---|---|---|---|
| 9a | Ph | 24 | 72 | 59:41 (44:56 c) |
| 9b | 4-MeOC6H4 | 17 | 69 | 65:35 |
| 9c | 4-BrC6H4 | 15 | 41 | 59:41 |
| 9d | Bn | 17 | 24 | 58:42 (67:33 c) |
| 9e | Bu | 72 | 7 d | 65:35 |
a A solution of RNH2 (1.17 mmol) in 1,4-dioxane was added to TAL-f3 (2) (100 mg, 0.56 mmol). The reaction mixture was stirred at room temperature for a given amount of time. b Assessed by 1H and 19F NMR in CDCl3. c Assessed by 1H and 19F NMR in DMSO-d6. d The reaction was carried out at 60 °C.
Although Z-form of enaminones is predominant because of an effective intramolecular hydrogen bonding, the contribution of an E-isomer may be affected by the electronic nature of a substituent at a nitrogen atom [36] or by an alternative hydrogen bond formation [37], which is the case for product 9 (Figure 3). The ratio of isomers is solvent-dependent, and Z-form mostly prevails, except for compound 4a in DMSO-d6 (Table 5). The assignment of isomers was conducted on the basis of 1H and 13C NMR spectra (Table S3 in Supplementary Materials). The key signals are low-field NH-protons of Z-9 isomers at 12.39–12.58 ppm (for N-arylenaminones 9a–c in CDCl3) and at 11.08–11.32 ppm (for N-alkylenaminones 9d,e in CDCl3). The corresponding signals for the E-isomers of 9 are under a much higher field in accordance with the literature [37,38]. Among the other features of the NMR spectra of compounds 9 is the existence of methylene protons and amide NH-protons at a lower field and methyne carbons at a higher field for the E-isomer (Figure 3).
Figure 3.

Equilibrium between Z- and E-isomers of enamides 9 and their characteristic chemical shifts in 1H (in green) and 13C (in magenta) NMR in CDCl3.
For an explanation of the difference in the regioselectivity of the interaction of TAL and TAL-f3 with amines, we propose the following mechanism. The addition of the first equivalent of RNH2 leads to the formation of dioxoamide C (Scheme 8), which was isolated earlier (X = H) [39]. The intermediate C is then attacked by the second amine molecule at the less hindered atom, C-5, in the case of X = H forming product 8. A preference of the attack at C-3 for the fluorinated derivative may be attributed to a high content of cyclic form D (Scheme 8), analogs of which were described in the literature as the only tautomer in CDCl3 and DMSO-d6 solutions [40,41]. The semiaminal carbon atom is less susceptible to nucleophiles than the free keto group that leads to the selective formation of product 9. It also should be noted that a mixture of regioisomers is usually produced when linear aliphatic CF3-diketones react with aniline [42].
Scheme 8.

Proposed mechanism of the reaction of TAL and TAL-f3 with primary amines.
No change in selectivity was observed when bifunctional aromatic amine, o-phenylenediamine, was used in the optimized conditions. Benzodiazepinone 10 was formed as the only isolated product, although the yield was moderate (Scheme 9). Compound 10 was previously obtained from the corresponding dioxoester 11 in the mixture with diazepine 12 [43] (Scheme 9), so our method represents a good alternative with no specific separation needed.
Scheme 9.

Reaction of TAL-f3 and ethyl 6,6,6-trifluoro-3,5-dioxohexanoate with o-phenylenediamine.
Phenylhydrazine also reacted with TAL-f3 (2) regioselectively in 1,4-dioxane, giving pyrazolohydrazide 13, but the yield was poor. Changing the solvent to EtOH substantially increased the outcome, though isomer 14 was also formed and did not separate by the column chromatography (Scheme 10). Compound 14 was previously synthesized from cyanopyrone 7 [35] and its spectral characteristics are in a good correlation with our data. Both isomers appear as two sets of signals corresponding to a major syn- (presented at Scheme 10) and a minor anti-rotamer about the N–N bond. The key differences in the NMR spectra of the regioisomer 13 are the more downfield signal of the CF3 group in 19F NMR (106.2 ppm compared to 101.9 ppm for 14) due to deshielding from the adjacent phenyl substituent, and more downfield signals of NH groups in 1H NMR (9.91 and 7.82 ppm compared to 9.84 and 7.73 ppm for 14) due to additional hydrogen bonding.
Scheme 10.

Reaction of TAL-f3 with phenylhydrazine.
Thus, hexafluorodehydroacetic acid and trifluorotriacetic acid lactone were shown to be active electrophiles and represent interesting fluorinated building blocks, which were transformed to a number of nitrogen heterocycles. Reaction of hexafluorodehydroacetic acid with primary aromatic amines leads to the formation of 2,6-bis(trifluoromethyl)-4-pyridones that are able to undergo Knoevenagel condensation to give merocyanine dyes. Trifluorotriacetic acid lactone undergoes ring-opening transformations with mono- and binucleophilic primary amines at the positions 2 and 4 and differs in regioselectivity compared to the non-fluorinated analog.
3. Materials and Methods
NMR spectra were recorded on Bruker DRX-400 (Bruker BioSpin GmbH, Ettlingen, Germany, work frequencies: 1H–400 MHz, 13C–100 MHz, 19F–376.5 MHz) and Bruker Avance III-500 (Bruker BioSpin GmbH, Rheinstetten, Germany, work frequencies: 1H–500 MHz, 13C–126 MHz, 19F–471 MHz) spectrometers in DMSO-d6 and CDCl3. The chemical shifts (δ) are reported in ppm relative to the internal standard TMS (1H NMR), C6F6 (19F NMR), and residual signals of the solvents (13C NMR). IR spectra were recorded on a Shimadzu IRSpirit-T spectrometer (Shimadzu Corp., Kyoto, Japan) using an attenuated total reflectance (ATR) unit (FTIR mode, ZnSe crystal), and the absorbance maxima (ν) are reported in cm−1. UV-visible spectra were recorded on a Shimadzu UV-1900 spectrophotometer (Shimadzu Corp., Kyoto, Japan) using EtOH as a solvent, the absorbance maxima (λ) are reported in nm, and the molar attenuation coefficients are reported in L·mol−1·cm−1. Elemental analyses were performed on an automatic analyzer PerkinElmer PE 2400 Series II (Perkin Elmer Instruments, Waltham, MA, USA). Melting points were determined using a Stuart SMP40 melting point apparatus (Bibby Scientific Ltd., Stone, Staffordshire, UK). Column chromatography was performed on silica gel (Merck 60, 70–230 mesh). All solvents and reagents were obtained commercially and used without purification.
3.1. Synthesis of Pyrones 1 and 2
4-Hydroxy-3-(2,2,2-trifluoroacetyl)-6-(trifluoromethyl)-2H-pyran-2-one (1). A mixture of ethyl 4,4,4-trifluoroacetoacetate (9.1 g, 0.032 mol) and P2O5 (40 g, 0.141 mol) was heated with a gas burner flame for 10 min. Then, the mixture was distilled in a vacuum (~20 torr), collecting the fraction 90–110 °C. The target fraction was redistilled in the same interval. Yield 4.1 g (75%), yellowish needles, mp 40–42 °C. 1H NMR (500 MHz, DMSO-d6) δ 6.24 (1H, s, H-5), 11.12 (1H, s, OH). 19F NMR (471 MHz, DMSO-d6) δ 89.6 (s, COCF3), 91.1 (s, 6-CF3) [25].
4-Hydroxy-6-(trifluoromethyl)-2H-pyran-2-one (2). A saturated aqueous solution of NaHCO3 (0.5 mL, 0.55 mmol) was added with stirring to an aqueous solution of hexafluorodehydroacetic acid (1) (2.5 mL, 1 M). The mixture was stirred for 10 min and acidified with aqueous HCl (1 mL, 3 M). The precipitate was filtered off and dried. Yield 180 mg (40%), white solid, mp 135–138 °C (lit. mp 135–137 °C [25]). 1H NMR (500 MHz, DMSO-d6) δ 5.59 (1H, d, J = 1.8 Hz, H-3), 6.83 (1H, d, J = 1.7 Hz, H-5), 12.60 (1H, s, OH). 19F NMR (471 MHz, DMSO-d6) δ 92.0 (s, CF3).
3.2. Synthesis of Compounds 3a–k
General Procedure
An aromatic amine (0.83 mmol) was added to a solution of hexafluorodehydroacetic acid (1) (100 mg, 0.36 mmol) in EtOH (1 mL). The reaction mixture was stirred at room temperature for a given amount of time and acidified with aqueous HCl (3 mL, 1 M). The precipitate was filtered off and washed with water. The crude product was recrystallized from hexane.
1-Phenyl-2,6-bis(trifluoromethyl)pyridin-4(1H)-one (3a). The reaction was carried out for 24 h. Yield 66 mg (60%), yellow powder, mp 135–136 °C. 1H NMR (400 MHz, CDCl3) δ 6.97 (2H, s, H-3, H-5), 7.39 (2H, d, J = 7.6 Hz, H Ph), 7.50 (2H, t, J = 7.9 Hz, H Ph), 7.59 (1H, t, J = 7.5 Hz, H Ph). 1H NMR (500 MHz, DMSO-d6) δ 6.97 (2H, s, H-3, H-5), 7.55 (2H, t, J = 7.7 Hz, H Ph), 7.59 (1H, t, J = 7.5 Hz, H Ph), 7.70 (2H, d, J = 7.8 Hz, H Ph). 19F NMR (471 MHz, CDCl3) δ 100.6 (s, CF3). 19F NMR (471 MHz, DMSO-d6) δ 102.5 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 119.1 (2C, q, J = 4.7 Hz, C-3, C-5), 119.4 (2C, q, J = 275.7 Hz, CF3), 128.7 (2C Ph), 130.1 (2C Ph), 131.3 (C Ph), 135.0 (C Ph), 141.2 (2C, q, J = 33.3 Hz, C-2, C-6), 177.4 (CO). IR (ATR) ν 3297, 1700 (C=O), 1658, 1398 (N–C), 1128–1116 (CF3). Anal. Calculated for C13H7F6NO: C 50.83; H 2.30; N 4.56. Found: C 50.77; H 2.38; N 4.46.
1-(4-Methoxyphenyl)-2,6-bis(trifluoromethyl)pyridin-4(1H)-one (3b). The reaction was carried out for 8 h. Yield 68 mg (56%), gray powder, mp 125–126 °C. 1H NMR (400 MHz, CDCl3) δ 3.88 (3H, s, CH3), 6.95 (2H, s, H-3, H-5), 6.95 (2H, d, J = 8.9 Hz, H Ar), 7.28 (2H, d, J = 8.9 Hz, H Ar). 19F NMR (471 MHz, CDCl3) δ 100.5 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 55.6 (OCH3), 113.7 (2C Ar), 119.1 (2C, q, J = 4.7 Hz, C-3, C-5), 119.4 (2C, q, J = 275.6 Hz, CF3), 127.2 (C Ar), 131.2 (2C Ar), 141.6 (2C, q, J = 32.7 Hz, C-2, C-6), 161.3 (C Ar), 177.5 (CO). IR (ATR) ν 3061, 1653 (C=O), 1602, 1510, 1397 (N–C), 1250, 1397, 1158 (CF3). HRMS (ESI) m/z [M + H]+. Calculated for C14H10F6NO2: 338.0616. Found: 338.0613.
1-(3,4-Difluorophenyl)-2,6-bis(trifluoromethyl)pyridin-4(1H)-one (3c). The reaction was carried out for 6 h. Yield 71 mg (57%), gray powder, mp 142–143 °C. 1H NMR (500 MHz, CDCl3) δ 6.96 (2H, s, H-3, H-5), 7.21 (1H, d, J = 8.6 Hz, H Ar), 7.31 (2H, m, H Ar). 19F NMR (471 MHz, CDCl3) δ 28.3 (1F, dt, J = 21.3, 8.8 Hz, F Ar), 31.0 (1F, dddd, J = 21.3, 9.5, 6.7, 3.8 Hz, F Ar), 100.7 (6F, s, CF3). 13C NMR (101 MHz, CDCl3) δ 117.4 (d, J = 18.9 Hz, C Ar), 119.3 (2C, q, J = 275.7 Hz, CF3), 119.4 (2C, q, J = 4.7 Hz, C-3, C-5), 120.2 (d, J = 19.1 Hz, C Ar), 127.3 (C Ar), 130.6 (C Ar), 140.9 (2C, q, J = 33.3 Hz, C-2, C-6), 149.5 (dd, J = 254.1, 13.8 Hz, C Ar), 152.1 (dd, J = 256.4, 12.2 Hz, C Ar), 177.1 (CO). IR (ATR) ν 3091, 3056, 1655 (C=O), 1615, 1519, 1397 (N–C), 1144 (CF3). Anal. Calculated for C13H5F8NO: C 45.50; H 1.47; N 4.08. Found: C 45.33; H 1.46; N 4.09.
1-(4-Bromophenyl)-2,6-bis(trifluoromethyl)pyridin-4(1H)-one (3d). The reaction was carried out for 24 h. Yield 65 mg (47%), yellow powder, mp 129–131 °C (decomp.). 1H NMR (400 MHz, CDCl3) δ 6.97 (2H, s, H-3, H-5), 7.27 (2H, d, J = 8.1 Hz, H Ar), 7.64 (1H, d, J = 8.5 Hz, H Ar). 19F NMR (471 MHz, CDCl3) δ 100.7 (s, CF3). 13C NMR (101 MHz, CDCl3) δ 119.26 (2C, q, J = 4.5 Hz, C-3, C-5), 119.33 (2C, q, J = 275.8 Hz, CF3), 126.0 (C Ar), 131.7 (2C, C Ar), 132.1 (2C, C Ar), 133.9 (C Ar), 141.0 (2C, q, J = 32.8 Hz, C-2, C-6), 177.3 (CO). IR (ATR) ν 3071, 1652 (C=O), 1589, 1480, 1397 (N–C), 1249, 1175 (CF3). HRMS (ESI) m/z [M + H]+. Calculated for C13H7BrF6NO: 385.9615. Found: 385.9606.
1-(3-Acetylphenyl)-2,6-bis(trifluoromethyl)pyridin-4(1H)-one (3e). The reaction was carried out for 24 h. Yield 60 mg (47%), pale pink powder, mp 118–119 °C. 1H NMR (400 MHz, CDCl3) δ 2.65 (3H, s, CH3), 6.99 (2H, s, H-3, H-5), 7.57–7.69 (2H, m, H Ar), 8.00 (1H, s, H Ar), 8.17 (1H, dt, J = 7.4, 1.5 Hz, H Ar). 19F NMR (376 MHz, CDCl3) δ 100.8 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 26.6 (CH3), 119.0 (2C, unres. q, C-3, C-5), 119.2 (2C, q, J = 276.1 Hz, CF3), 129.2 (C Ar), 129.8 (C Ar), 131.2 (C Ar), 134.1 (C Ar), 135.4 (C Ar), 137.7 (C Ar), 141.6 (2C, unres. q, C-2, C-6), 177.2 (4-CO), 195.6 (COCH3). IR (ATR) ν 3068, 1700 (C=O), 1665 (C=O), 1610, 1480, 1402 (N–C), 1138 (CF3). Anal. Calculated for C15H9F6NO2: C 51.59; H 2.60; N 4.01. Found: C 51,50; H 2.63; N 3.99.
1-(3-(Trifluoromethyl)phenyl)-2,6-bis(trifluoromethyl)pyridin-4(1H)-one (3f). The reaction was carried out for 24 h. Yield 63 mg (47%), light-yellow powder, mp 91–92 °C. 1H NMR (600 MHz, CDCl3) δ 7.00 (2H, s, H-3, H-5), 7.64 (1H, d, J = 8.2 Hz, H Ar), 7.69 (1H, t, J = 8.0 Hz, H Ar), 7.72 (1H, s, H Ar), 7.89 (1H, d, J = 7.8 Hz, H Ar). 19F NMR (376 MHz, CDCl3) δ 98.7 (3F, s, CF3 Ar), 100.7 (6F, s, 2-CF3, 6-CF3). 13C NMR (126 MHz, CDCl3) δ 119.3 (2C, q, J = 275.5 Hz, 2-CF3, 6-CF3), 119.4 (2C, q, J = 4.7 Hz, C-3, C-5), 122.9 (q, J = 272.5 Hz, CF3 Ar), 127.6 (C Ar), 128.3 (C Ar), 129.6 (C Ar), 131.8 (q, J = 33.8 Hz, C-3 Ar), 133.6 (C Ar), 135.4 (C Ar), 140.9 (2C, q, J = 33.4 Hz, C-2, C-6), 177.2 (CO). IR (ATR) ν 3091, 1655 (C=O), 1395 (N–C), 1327, 1252, 1126 (CF3). HRMS (ESI) m/z [M + H]+. Calculated for C14H7F9NO: 376.0384. Found: 376.0397.
1-(2,5-Difluorophenyl)-2,6-bis(trifluoromethyl)pyridin-4(1H)-one (3g). The reaction was carried out for 48 h. Yield 58 mg (46%), yellow powder, mp 116–119 °C. 1H NMR (500 MHz, CDCl3) δ 6.99 (2H, s, H-3, H-5), 7.20–7.26 (2H, m, H-3, H-6 Ar), 7.35 (1H, dddd, J = 9.3, 7.4, 3.8, 3.0 Hz, H-4 Ar). 19F NMR (376 MHz, CDCl3) δ 38.6–38.8 (m, F-2 Ar), 46.0 (1F, dtd, J = 15.4, 7.4, 4.7 Hz, F-5 Ar), 98.5 (dd, J = 3.2, 0.9 Hz, CF3). 13C NMR (126 MHz, CDCl3) δ 117.1 (dd, J = 22.4, 8.8 Hz, C Ar), 119.1 (d, J = 25.9 Hz, C Ar), 119.3 (2C, q, J = 275.5 Hz, CF3), 119.5 (2C, q, J = 4.6 Hz, C-3, C-5), 120.8 (dd, J = 23.6, 8.1 Hz, C Ar), 123.4 (dd, J = 16.3, 10.5 Hz, C Ar), 140.8 (2C, q, J = 34.1 Hz, C-2, C-6), 155.9 (dd, J = 252.0, 3.3 Hz, C Ar), 157.4 (dd, J = 247.8, 2.8 Hz, C Ar), 177.2 (CO). IR (ATR) ν 3072, 1666 (C=O), 1615, 1508, 1397 (N–C), 1251, 1136 (CF3). HRMS (ESI) m/z [M + H]+. Calculated for C13H6F8NO: 344.0322. Found: 344.0316.
1-(2-Chlorophenyl)-2,6-bis(trifluoromethyl)pyridin-4(1H)-one (3h). The reaction was carried out for 40 h. Yield is 61 mg (49%), yellow powder, mp 132–136 °C. 1H NMR (500 MHz, CDCl3) δ 6.99 (2H, s, H-3, H-5), 7.41–7.48 (1H, m, H Ar), 7.51–7.58 (3H, m, H Ar). 19F NMR (471 MHz, CDCl3) δ 98.6 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 119.3 (2C, q, J = 275.8 Hz, CF3), 119.6 (2C, q, J = 4.5 Hz, C-3, C-5), 127.2 (C Ar), 130.1 (C Ar), 131.9 (C Ar), 132.7 (C Ar), 132.9 (C Ar), 135.9 (C Ar), 140.9 (2C, q, J = 33.9 Hz, C-2, C-6), 177.6 (CO). IR (ATR) ν 3056, 1743, 1651 (C=O), 1476, 1389 (N–C), 1247, 1136 (CF3). HRMS (ESI) m/z [M + H]+. Calculated for C13H7ClF6NO: 342.0120. Found: 342.0131.
1-(o-Tolyl)-2,6-bis(trifluoromethyl)pyridin-4(1H)-one (3i). The reaction was carried out for 24 h. Yield 74 mg (65%), brown viscous liquid. 1H NMR (400 MHz, CDCl3) δ 2.08 (3H, s, CH3), 7.01 (2H, s, H-3, H-5), 7.35–7.30 (3H, m, H Ar), 7.47 (1H, td, J = 7.5, 1.6 Hz, H Ar). 19F NMR (471 MHz, CDCl3) δ 99.0 (d, J = 1.0 Hz, CF3). 13C NMR (151 MHz, CDCl3) δ 17.0 (CH3), 119.3 (2C, q, J = 275.8 Hz, CF3), 119.6 (2C, q, J = 4.2 Hz, C-3, C-5), 126.2 (C Ar), 130.3 (C Ar), 130.7 (C Ar), 131.5 (C Ar), 134.1 (C Ar), 138.5 (C Ar), 141.3 (2C, q, J = 33.2 Hz, C-2, C-6), 177.9 (CO). IR (ATR) ν 3054, 1696 (C=O), 1620, 1508, 1391 (N–C), 1252, 1135 (CF3). HRMS (ESI) m/z [M + H]+. Calculated for C14H10F6NO: 322.0667. Found: 322.0679.
1-(2,5-Dimethylphenyl)-2,6-bis(trifluoromethyl)pyridin-4(1H)-one (3j). The reaction was carried out for 24 h. Yield 58 mg (48%), brown viscous liquid, crystallizes on standing, mp 105–107 °C. 1H NMR (500 MHz, CDCl3) δ 2.02 (3H, s, CH3) 2.38 (3H, s, CH3), 7.01 (2H, s, H-3, H-5), 7.16 (1H, s, H-2 Ar), 7.18 (1H, d, J = 7.7 Hz, H-5 Ar), 7.27 (1H, d, J = 7.7 Hz, H-4 Ar). 19F NMR (471 MHz, CDCl3) δ 99.0 (s, CF3). 13C NMR (126 MHz, CDCl3) δ 16.6 (CH3), 20.6 (CH3), 119.3 (2C, q, J = 275.8 Hz, CF3), 119.5 (2C, q, J = 4.8 Hz, C-3, C-5), 130.3 (C Ar), 130.6 (C Ar), 132.2 (C Ar), 133.9 (C Ar), 135.2 (C Ar), 136.2 (C Ar), 141.1 (2C, q, J = 33.4 Hz, C-2, C-6), 177.5 (CO). IR (ATR) ν 3091, 1655 (C=O), 1608, 1395 (N–C), 1328, 1251, 1125 (CF3). HRMS (ESI) m/z [M + H]+. Calculated for C15H12F6NO: 336.0823. Found: 336.0802.
1-(4-Nitrophenyl)-2,6-bis(trifluoromethyl)pyridin-4(1H)-one (3k). The reaction was carried out for 6 d. Pale yellow powder, a mixture of 3k (10 mg, 8%) and 4-nitroaniline (32 mg). 1H NMR (500 MHz, CDCl3) δ 7.00 (2H, s, H-3, H-5), 7.64 (2H, d, J = 8.4 Hz, H Ar), 8.39 (2H, d, J = 8.9 Hz, H Ar). 19F NMR (471 MHz, CDCl3) δ 100.9 (s, CF3). HRMS (ESI) m/z [M + H]+. Calculated for C13H7F6N2O3: 353.0361. Found: 353.0357.
3.3. Synthesis of Compound B′
o-Chloroaniline (106 mg, 0.83 mmol) was added to a solution of hexafluorodehydroacetic acid (1) (100 mg, 0.36 mmol) in EtOH (1 mL). The reaction mixture was stirred at room temperature for a 15 h and acidified with aqueous HCl (3 mL, 1 M). The precipitate was filtered off and washed with water. The crude product was purified by column chromatography (CHCl3).
(2Z,5Z)-2,6-Bis((2-chlorophenyl)amino)-1,1,1,7,7,7-hexafluorohepta-2,5-dien-4-one (B′). Dark-yellow solid, a mixture of B′ (108 mg, 64%) and 2-chloroaniline (11 mg), mp 84–85 °C. 1H NMR (400 MHz, CDCl3) δ 5.78 (2H, s, 3-CH, 5-CH), 7.18–7.29 (4H, m, H Ar), 7.33 (2H, d, J = 7.3 Hz, H Ar), 7.44 (2H, dd, J = 7.8, 1.5 Hz, H Ar), 11.49 (2H, s, NH). 19F NMR (376 MHz, CDCl3) δ 97.8 (s, CF3).
3.4. Synthesis of Compound 4
2-Aminopyridine (78 mg, 0.83 mmol) was added to a solution of hexafluorodehydroacetic acid (1) (100 mg, 0.36 mmol) in EtOH (1 mL). The precipitate was filtered and washed with a small amount of EtOH.
2-Aminopyridin-1-ium 2-oxo-3-(2,2,2-trifluoroacetyl)-6-(trifluoromethyl)-2H-pyran-4-olate (4). Yield 127 mg (95%), white solid, mp 205–206 °C. 1H NMR (400 MHz, DMSO-d6) δ 6.15 (1H, s, H-5 pyrone), 6.86 (1H, td, J = 6.8, 1.0 Hz, H-5 pyridine), 6.97 (1H, dd, J = 9.6, 1.0 Hz, H-3 pyridine), 7.92 (4H, m, NH2, H-4, H-6 pyridine), 13.27 (1H, s, NH). 19F NMR (376 MHz, DMSO-d6) δ 89.8 (s, COCF3), 91.1 (s, CF3). 13C NMR (126 MHz, DMSO-d6) δ 98.4 (C-3), 109.7 (q, J = 2.9 Hz, C-5), 112.1 (C Py), 113.4 (C Py), 116.6 (q, J = 292.1 Hz, CF3), 118.9 (q, J = 272.8 Hz, CF3), 136.0 (C Py), 144.1 (C Py), 146.8 (q, J = 37.1 Hz, C-6), 153.9 (C Py), 160.0 (C-2), 176.8 (C-4), 177.7 (q, J = 33.5 Hz, COCF3). IR (ATR) ν 3366, 3310, 3191, 1712 (C=O), 1699 (C=O), 1535, 1372 (N–C), 1134 (CF3). Anal. Calculated for C13H8F6N2O4: C 42.18; H 2.18; N 7.57. Found: C 42.28; H 2.07; N 7.57.
3.5. Synthesis of Compounds 5
General Procedure
A mixture of 1-phenyl-2,6-bis(trifluoromethyl)pyridin-4(1H)-one (100 mg, 0.33 mmol) and a corresponding active methylene compound (0.4 mmol) in Ac2O (1.5 mL) was heated at 140 °C for a given amount of time. The reaction mixture was then cooled and left overnight for crystallization of a product. The precipitate was filtered and washed with EtOH (1 mL). The product was dried at 120 °C for 4 h.
2-(1-Phenyl-2,6-bis(trifluoromethyl)pyridin-4(1H)-ylidene)malononitrile (5a). Synthesized from malononitrile (10 h). Yield 77 mg (65%). Dark-yellow crystals, mp 172–173 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.26 (2H, s, H-3′, H-5′), 7.59 (2H, t, J = 7.8 Hz, H Ph), 7.66 (1H, t, J = 7.6 Hz, H Ph), 7.74 (2H, d, J = 7.8 Hz, H Ph). 19F NMR (471 MHz, DMSO-d6) δ 102.5 (s, CF3). 13C NMR (126 MHz, DMSO-d6) δ 57.1 (C(CN)2), 112.7 (2C, q, J = 5.3 Hz, C-3′, C-5′), 114.8 (2C, CN), 118.7 (2C, q, J = 275.9 Hz, CF3), 128.7 (2C Ph), 130.1 (2C Ph), 131.7 (C Ph), 134.6 (C Ph), 138.2 (2C, q, J = 33.0 Hz, C-2′, C-6′), 154.2 (C-4′). IR (ATR) ν 3058, 2922, 2852, 2215, 1643, 1518, 1302 (N–C), 1275, 1142 (CF3). UV/vis (EtOH): λmax (εmax) = 382 (18222). HRMS (ESI) m/z [M + H]+. Calculated for C16H8F6N3: 356.0622. Found: 356.0624.
5-(1-Phenyl-2,6-bis(trifluoromethyl)pyridin-4(1H)-ylidene)pyrimidine-2,4,6(1H,3H,5H)-trione (5b). Synthesized from barbituric acid (4 h). Yield 81 mg (58%). Dark-yellow powder, mp 270–271 °C (decomp.). 1H NMR (500 MHz, DMSO-d6) δ 7.59 (2H, t, J = 7.7 Hz, H Ph), 7.66 (1H, t, J = 7.5 Hz, H Ph), 7.80 (2H, d, J = 7.9 Hz, H Ph), 9.77 (2H, s, H-3′, H-5′), 10.78 (2H, s, NH). 19F NMR (471 MHz, DMSO-d6) δ 103.2 (s, CF3). 13C NMR (151 MHz, DMSO-d6) δ 95.0 (C-5), 116.8 (2C, q, J = 5.7 Hz, C-3′, C-5′), 119.8 (2C, q, J = 275.5 Hz, CF3), 129.0 (2C Ph), 130.4 (2C Ph), 132.1 (C Ph), 135.6 (C Ph), 138.3 (2C, q, J = 32.3 Hz, C-2′, C-5′), 150.1 (2-CO), 153.0 (C-4′), 165.6 (2C, 4-CO, 6-CO). IR (ATR) ν 3021, 1712, 1673, 1483, 1354 (N–C), 1145 (CF3). UV/vis (EtOH): λmax (εmax) = 412 (91406), 234 (37760). HRMS (ESI) m/z [M + H]+. Calculated for C17H10F6N3O3: 418.0626. Found: 418.0591.
2-(1-Phenyl-2,6-bis(trifluoromethyl)pyridin-4(1H)-ylidene)1,3-indandione (5c). Synthesized from 1,3-indandione (6 h). Yield 62 mg (43%). Dark-yellow crystals, mp 208–209 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.60 (2H, t, J = 7.7 Hz, H Ph), 7.67 (1H, t, J = 7.4 Hz, H Ph), 7.70–7.78 (4H, m, H Ar), 7.80 (2H, d, J = 7.9 Hz, H Ph), 9.10 (2H, s, H-3′, H-5′). 19F NMR (471 MHz, DMSO-d6) δ 102.8 (s, CF3). 13C NMR (126 MHz, DMSO-d6) δ 107.2 (C-2), 113.4 (2C, q, J = 5.9 Hz, C-3′, C-5′), 119.2 (2C, q, J = 275.2 Hz, CF3), 121.3 (2C Ar), 128.5 (2C Ph), 130.0 (2C Ph), 131.6 (C Ph), 134.0 (2C Ar), 135.1 (C Ph), 138.9 (2C, q, J = 32.7 Hz, C-2′, C-6′), 139.8 (2C Ar), 146.0 (C-4′), 191.0 (2C, CO). IR (ATR) ν 3101, 3065, 1693, 1648, 1518, 1319 (N–C), 1141 (CF3). UV/vis (EtOH): λmax (εmax) = 437 (21948), 415 (15988), 246 (74128), 224 (151744). HRMS (ESI) m/z [M + H]+. Calculated for C22H12F6NO2: 436.0772. Found: 436.0779.
3.6. Synthesis of Compounds 9
General Procedure
To a solution of trifluorotriacetic acid (2) (100 mg, 0.56 mmol) in 1,4-dioxane (1 mL), a corresponding amine (1.17 mmol) was added with stirring. The reaction was monitored by TLC (CHCl3:EtOH, 20:1). After completion, the reaction mixture was acidified with aqueous HCl (1 mL, 3 M). The precipitate was filtered and washed with water. The crude product was recrystallized from hexane.
6,6,6-Trifluoro-5-oxo-N-phenyl-3-(phenylamino)hex-3-enamide (9a). The reaction was carried out for 24 h. Yield 139 mg (72%), small yellow crystals, mp 135–137 °C. 1H NMR (500 MHz, DMSO-d6) δ Z-9a (44%): 3.65 (2H, s, 2-CH2), 5.79 (1H, s, 4-CH), 7.05 (2H, t, J = 7.3 Hz, H Ph), 7.30–7.34 (2H, m, H Ph), 7.40 (2H, d, J = 7.5 Hz, H Ph), 7.45 (4H, t, J = 7.0 Hz, H Ph), 10.08 (1H, s, NH amide), 12.56 (1H, s, NH enamine); E-9a (56%): 4.06 (2H, s, 2-CH2), 5.59 (1H, s, 4-CH), 7.25–7.38 (6H, m, H Ph), 7.52 (2H, t, J = 6.8 Hz, H Ph), 7.61 (2H, d, J = 7.4 Hz, H Ph), 10.23 (1H, s, NH), 10.31 (1H, s, NH). 19F NMR (376 MHz, DMSO-d6) δ Z-9a (44%): 87.1 (s, CF3); E-9a (56%): 86.6 (s, CF3). 19F NMR (376 MHz, CDCl3) δ Z-9a (59%): 85.1 (s, CF3); E-9a (41%): 84.8 (s, CF3). 13C NMR (101 MHz, CDCl3) δ Z-9a (59%): 41.5 (CH2), 91.3 (4-CH), 117.2 (q, J = 288.3 Hz, CF3), 120.2 (2C Ph), 125.2 (C Ph), 125.9 (2C Ph), 128.3 (C Ph), 129.1 (2C Ph), 129.82 (2C Ph), 136.4 (C Ph), 136.9 (C Ph) 163.4 (1-CO), 164.0 (C-3), 177.8 (q, J = 33.7 Hz, 5-CO); E-9a (41%): 42.3 (CH2), 88.7 (CH), 117.6 (q, J = 290.2 Hz, CF3), 120.4 (2C Ph), 124.7 (2C Ph), 124.9 (C Ph), 127.8 (C Ph), 129.0 (2C Ph), 129.78 (2C Ph), 136.6 (C Ph), 137.5 (C Ph), 162.5 (1-CO), 166.3 (C-3), 178.5 (q, J = 32.1 Hz, 5-CO). IR (ATR) ν 3299, 3061, 3032, 1658 (C=O), 1578, 1242, 1172, 1116 (CF3). Anal. Calculated for C18H15F3N2O2: C 62.07; H 4.34; N 8.04. Found: C 61.81; H 4.12; N 7.94.
6,6,6-Trifluoro-N-(4-methoxyphenyl)-3-((4-methoxyphenyl)amino)-5-oxohex-3-enamide (9b). The reaction was carried out for 17 h. Yield 160 mg (69%), fine gray crystals, mp 163–165 °C. 1H NMR (400 MHz, CDCl3) δ Z-9b (65%): 3.38 (2H, s, 2-CH2), 3.79 (3H, s, OCH3) 3.80 (3H, s, OCH3), 5.68 (1H, s, 4-CH), 6.85 (2H, d, J = 8.7 Hz, H Ar), 6.91 (2H, d, J = 8.6 Hz, H Ar), 7.16 (1H, d, J = 8.6 Hz, H Ar), 7.29 (1H, d, J = 8.7 Hz, H Ar), 7.12 (1H, s, NH amide), 12.39 (1H, s, NH enamine); E-9b (35%): 3.79 (3H, s, OCH3) 3.83 (3H, s, OCH3), 4.00 (2H, s, 2-CH2), 5.68 (1H, s, 4-CH), 6.84 (2H, d, J = 8.7 Hz, H Ar), 6.90 (2H, d, J = 8.6 Hz, H Ar), 7.11 (1H, d, J = 8.6 Hz, H Ar), 7.42 (1H, d, J = 8.7 Hz, H Ar), 8.74 (1H, s, NH amide), 9.63 (1H, s, NH enamine). 19F NMR (376 MHz, CDCl3) δ Z-9b (65%): 85.1 (s, CF3); E-9b (35%): 84.9 (s, CF3). 13C NMR (126 MHz, CDCl3) δ Z-9b (65%): 41.4 (CH2), 55.47 (OCH3), 55.52 (OCH3), 90.9 (4-CH), 114.3 (2C Ar), 114.88 (2C Ar), 117.3 (q, J = 288.6 Hz, CF3), 122.1 (2C Ar), 127.4 (2C Ar), 129.0 (C Ar), 129.9 (C Ar), 157.1 (C Ar), 159.4 (C Ar), 164.0, 164.2, 177.5 (q, J = 33.6 Hz, 5-CO); E-9b (35%): 42.0 (CH2), 55.47 (OCH3), 55.49 (OCH3), 88.2 (4-CH), 114.1 (2C Ar), 114.91 (2C Ar), 117.7 (q, J = 290.2 Hz, CF3), 122.0 (2C Ar), 126.4 (2C Ar), 129.3 (C Ar), 130.8 (C Ar), 156.7 (C Ar), 159.0 (C Ar), 163.2 (1-CO), 165.9 (C-3), 177.9 (q, J = 31.3 Hz, 5-CO). IR (ATR) ν 3282, 2999, 2842, 1651 (C=O), 1530, 1237, 1169, 1116 (CF3). Anal. Calculated for C20H19F3N2O4·0.25H2O: C 58.18; H 4.76; N 6.79. Found: C 58.25; H 4.48; N 6.54.
N-(4-Bromophenyl)-3-((4-bromophenyl)amino)-6,6,6-trifluoro-5-oxohex-3-enamide (9c). The reaction was carried out for 15 h. Yield 118 mg (41%), fine yellow crystals, mp 209–210 °C (decomp.). 1H NMR (500 MHz, CDCl3) δ Z-9c (58%): 3.41 (2H, s, 2-CH2), 5.70 (1H, s, 4-CH), 7.14 (2H, d, J = 8.4 Hz, H Ar), 7.31 (2H, d, J = 8.6 Hz, H Ar), 7.43 (1H, s, NH amide), 7.46 (2H, d, J = 8.6 Hz, H Ar), 7.54 (2H, d, J = 8.4 Hz, H Ar), 12.42 (1H, s, NH enamine); E-9c (42%): 3.98 (1H, s, 2-CH2), 5.78 (1H, s, 4-CH), 7.09 (2H, d, J = 8.5 Hz, H Ar), 7.41–7.46 (4H, m, H Ar), 7.55 (2H, d, J = 8.5 Hz, H Ar), 8.32 (1H, s, NH amide), 9.62 (1H, s, NH enamine). 19F NMR (471 MHz, CDCl3) δ Z-9c (58%): 85.0 (s, CF3); E-9c (42%): 84.8 (s, CF3). IR (ATR) ν 3262, 3018, 3046, 1664 (C=O), 1589, 1533, 1246, 1111 (CF3). Anal. Calculated for C18H13Br2F3N2O2·0.33H2O: C 42.22; H 2.69; N 5.47. Found: C 42.44; H 2.58; N 5.19.
N-Benzyl-3-(benzylamino)-6,6,6-trifluoro-5-oxohex-3-enamide (9d). The reaction was carried out for 17 h. Yield 51 mg (24%), colorless powder, mp 124–125 °C. 1H NMR (500 MHz, DMSO-d6) δ Z-9d (68%): 3.55 (2H, s, 2-CH2), 4.30 (2H, d, J = 5.8 Hz, NCH2), 4.72 (2H, d, J = 6.1 Hz, NCH2), 5.50 (1H, s, 4-CH), 7.09–7.49 (10H, m, H Ph), 8.76 (1H, t, J = 5.8 Hz, NH amide), 11.29 (1H, t, J = 6.1 Hz, NH enamine); E-9d (32%): 3.84 (2H, s, 2-CH2), 4.31 (2H, d, J = 6.0 Hz, NCH2), 4.46 (2H, d, J = 5.6 Hz, NCH2), 5.27 (1H, s, 4-CH), 7.09–7.49 (10H, m, H Ph), 8.45 (1H, t, J = 5.9 Hz, NH amide), 9.08 (1H, t, J = 5.8 Hz, NH enamine). 1H NMR (500 MHz, CDCl3) δ Z-9d (58%): 3.31 (2H, s, 2-CH2), 4.39 (2H, d, J = 5.7 Hz, NCH2), 4.61 (2H, d, J = 6.1 Hz, NCH2), 5.44 (1H, s, 4-CH), 6.04 (1H, unres. t, NH) 7.20–7.40 (10H, m, H Ph), 11.32 (1H, unres. t, NH); E-9d (42%): 3.79 (2H, s, 2-CH2), 4.32 (2H, d, J = 5.9 Hz, NCH2), 4.33 (2H, d, J = 4.9 Hz, NCH2), 5.41 (1H, s, 4-CH), 7.15 (2H, d, J = 7.0 Hz), 7.20–7.40 (8H, m), 7.90 (2H, unres. t, NH). 19F NMR (471 MHz, CDCl3) δ Z-9d (58%): 85.1 (s, CF3); E-9d (42%): 84.9 (s, CF3). 13C NMR (126 MHz, CDCl3) δ Z-9d (58%): 41.1 (2-CH2), 44.0 (NCH2), 48.0 (NCH2), 90.1 (4-CH), 117.3 (q, J = 288.4 Hz), 127.1 (2C Ph), 127.8 (2C Ph), 127.9 (C Ph), 128.2 (C Ph), 128.9 (2C Ph), 129.2 (2C Ph), 135.7 (C Ph), 137.3 (C Ph), 164.9 (1-CO), 165.3 (C-3), 177.0 (q, J = 33.4 Hz, 5-CO). E-9d (42%): 40.5 (2-CH2), 43.5 (NCH2), 48.3 (NCH2), 86.3 (4-CH), 117.7 (q, J = 290.5 Hz, CF3), 127.2 (2C Ph), 127.4 (C Ph), 127.7 (2C Ph), 128.3 (C Ph), 128.6 (2C Ph), 129.1 (2C Ph), 134.8 (C Ph), 137.6 (C Ph), 163.5 (1-CO), 168.0 (C-3), 176.9 (q, J = 32.1 Hz, 5-CO). IR (ATR) ν 3308, 3062, 3033, 1641 (C=O), 1549, 1264,1165, 1124 (CF3). Anal. Calculated for C20H19F3N2O2·0.33H2O: C 62.82; H 5.18; N 7.33. Found: C 63.01; H 5.10; N 7.09.
N-Butyl-3-(butylamino)-6,6,6-trifluoro-5-oxohex-3-enamide (9e). The reaction was carried out for 72 h at 60 °C. Yield 12 mg (7%), large gray crystals, mp 79–80 °C. 1H NMR (500 MHz, CDCl3) δ Z-9e (65%): 0.93 (3H, t, J = 7.2 Hz, CH3), 0.95 (3H, t, J = 7.2 Hz, CH3), 1.32 (quint, 2H, J = 7.2 Hz, NCH2CH2), 1.43 (2H, sex, J = 7.2 Hz, CH2CH3), 1.49 (2H, sex, J = 7.2 Hz, CH2CH3), 1.63 (2H, quint, J = 7.2 Hz, NCH2CH2), 3.27 (2H, s, 2-CH2), 3.28 (2H, q, J = 6.3 Hz, NCH2), 3.42 (2H, q, J = 6.6 Hz, NCH2), 5.36 (1H, s, 4-CH), 5.65 (1H, s, NH amide), 11.08 (1H, s, NH enamine); E-9e (35%): 0.90 (3H, t, J = 7.2 Hz, CH3), 0.96 (3H, t, J = 7.2 Hz, CH3), 1.34 (quint, 2H, J = 7.2 Hz, NCH2CH2), 1.39–1.53 (4H, masked, CH2CH3), 1.65 (2H, quint, J = 7.2 Hz, NCH2CH2), 3.15–3.24 (4H, m, NCH2), 3.75 (2H, s, 2-CH2), 5.32 (1H, s, 4-CH), 7.10 (1H, s, NH amide), 7.36 (1H, s, NH enamine). 19F NMR (471 MHz, CDCl3) δ Z-9e (65%): 85.04 (s, CF3); E-9e (35%): 84.90 (s, CF3). IR (ATR) ν 3289, 2958, 2872, 1591 (C=O), 1548, 1306, 1184, 1114 (CF3). HRMS (ESI) m/z [M + H]+. Calculated for C14H25F3N2O2: 309.1790. Found: 309.1799.
3.7. Synthesis of Compound 10
1,3,4,5-Tetrahydro-4-(3,3,3-trifluoro-2-oxopropylidene)-2H-1,5-benzodiazepin-2-one (10). Benzene-1,2-diamine (63 mg, 0.58 mmol) was added to a solution of trifluorotriacetic acid (2) (100 mg, 0.56 mmol) in 1,4-dioxane (1 mL) with stirring. The reaction was carried out for 24 h at room temperature. Then, the reaction mixture was acidified with aqueous HCl (1 mL, 3M). The precipitate was filtered and washed with water. The crude product was recrystallized from hexane. Yield 51 mg (33%), fine white powder, mp 244–245 °C (decomp.; lit. mp 243–245 °C [43]). 1H NMR (500 MHz, CDCl3) δ 3.31 (2H, s, CH2), 5.68 (1H, s, CH), 7.12 (1H, dd, J = 7.6, 1.6 Hz, H Ar), 7.24–7.34 (3H, m, H Ar), 8.17 (1H, s, 1-NH), 12.57 (1H, s, 5-NH). 19F NMR (471 MHz, CDCl3) δ 84.8 (s, CF3). HRMS (ESI) m/z [M + H]+. Calculated for C14H25F3N2O2: 271.0694. Found: 271.0673. The analytical data are in consistence with the literature [43].
3.8. Synthesis of Compounds 13 and 14
N’-Phenyl-2-(1-phenyl-5-(trifluoromethyl)-1H-pyrazol-3-yl)acetohydrazide (13) and N’-phenyl-2-(1-phenyl-3-(trifluoromethyl)-1H-pyrazol-5-yl)acetohydrazide (14). Phenylhydrazine (126 mg, 1.17 mmol) was added to a solution of trifluorotriacetic acid (2) (100 mg, 0.56 mmol) in EtOH (1 mL) with stirring. The reaction was carried out for 24 h at room temperature. Then, the reaction mixture was acidified with aqueous HCl (1 mL, 3M). The precipitate was filtered and washed with water. The crude product was recrystallized from hexane. Yield 122 mg (61%), yellow powder, mp 136–138 °C. The mixture of isomers 13 and 14 (82:18) did not separate by column chromatography. IR (ATR) ν 3281, 1657 (C=O), 1603, 1504, 1356, 1083 (CF3). HRMS (ESI) m/z [M + H]+. Calculated for C18H16F3N4O: 361.1276. Found: 361.1276.
13: 1H NMR (500 MHz, DMSO-d6) δ syn-isomer (88%): 3.65 (2H, s, CH2), 6.67–6.71 (1H, m, H Ph), 6.73 (2H, d, J = 7.8 Hz, H Ph), 7.03 (1H, s, H pyraz.), 7.11 (2H, t, J = 7.8 Hz, H Ph), 7.49–7.52 (2H, m, H Ph), 7.56–7.62 (3H, m, H Ph), 7.60–8.03 (br. s, 1H, PhNH), 9.91 (1H, s, CONH); anti-isomer (12%): 3.71 (2H, s, CH2), 6.67–6.71 (2H, m, H Ph), 6.76 (1H, t, J = 7.3 Hz, H Ph), 6.95 (1H, s, H pyraz.), 7.18 (2H, t, J = 7.8 Hz, H Ph), 7.40–7.43 (2H, m, H Ph), 7.52–7.56 (3H, m, H Ph), 8.06 (1H, s, PhNH), 9.22 (1H, s, CONH). 19F NMR (471 MHz, DMSO-d6) δ anti-isomer (12%): 106.18 (s, CF3); syn-isomer (88%): 106.13 (s, CF3). 13C NMR (126 MHz, CDCl3) δ syn-isomer: 34.3 (CH2), 109.0 (C-4 pyraz.), 113.5 (2C Ph), 119.4 (q, J = 269.4 Hz, CF3), 121.3 (C Ph), 125.6 (2C Ph), 129.16 (2C Ph), 129.20 (2C Ph), 129.6 (C Ph), 133.9 (q, J = 39.7 Hz, C-5 pyraz.), 138.7 (C Ph), 146.2 (C Ph), 147.6 (C-3 pyraz.), 168.9 (CO); anti-isomer: most of the signals are masked due to low concentration.
14: 1H NMR (500 MHz, DMSO-d6) δ syn-isomer (71%): 3.78 (2H, s, CH2), 6.55 (2H, d, J = 7.6 Hz, H Ph), 6.69 (1H, t, J = 7.4 Hz, H Ph), 6.87 (1H, s, H pyraz.), 7.10 (2H, t, J = 7.5 Hz, H Ph), 7.56–7.62 (5H, m, H Ph), 7.73 (1H, s, PhNH), 9.83 (1H, s, CONH); anti-isomer (29%): 3.79 (2H, s, CH2), 6.56 (2H, d, J = 7.3 Hz, H Ph), 6.76 (1H, t, J = 7.3 Hz, H Ph), 6.83 (1H, s, H pyraz.), 7.14 (2H, t, J = 7.5 Hz, H Ph), 7.43–7.47 (2H, m, H Ph), 7.51–7.55 (3H, m, H Ph), 7.95 (1H, s, PhNH), 9.25 (1H, s, CONH). 19F NMR (471 MHz, DMSO-d6) δ anti-isomer (29%): 101.91 (s, CF3), syn-isomer (71%): 101.83 (s, CF3). The analytical data are in consistence with the literature [35].
Acknowledgments
Analytical studies were carried out using equipment at the Center for Joint Use ‘Spectroscopy and Analysis of Organic Compounds’ at the Postovsky Institute of Organic Synthesis of the Russian Academy of Sciences (Ural Branch) and the Laboratory of Complex Investigations and Expert Evaluation of Organic Materials of the Center for Joint Use at the Ural Federal University.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27207098/s1. Figure S1: UV/Vis spectra of compounds 5; Table S1: Characteristic chemical shifts in 1H, 13C, and 19F NMR spectra of compounds 3; Table S2: Characteristic chemical shifts in 1H, 13C, and 19F NMR spectra of compounds 5; Table S3: Characteristic chemical shifts in 1H, 13C, and 19F NMR spectra of E- and Z-isomers of enaminones 9; full 1H, 19F, and 13C NMR spectra of all synthesized compounds.
Author Contributions
Conceptualization and methodology were provided by V.Y.S., S.A.U. and D.L.O. conceived and designed the experiments. The experimental work was conducted by V.V.F. and S.A.U., S.A.U. and V.V.F. analyzed the results. S.A.U. studied and systemized the NMR data. S.A.U. and V.Y.S. wrote the paper. Project administration and funding acquisition were carried out by V.Y.S. and D.L.O. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
Funding Statement
This research was funded by the Russian Science Foundation, grant number 18-13-00186 (https://rscf.ru/project/18-13-00186/ accessed on 18 October 2022).
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.McGlacken G.P., Fairlamb I.J.S. 2-Pyrone natural products and mimetics: Isolation, characterisation and biological activity. Nat. Prod. Rep. 2005;22:369–385. doi: 10.1039/b416651p. [DOI] [PubMed] [Google Scholar]
- 2.Goel A., Ram V.J. Natural and synthetic 2H-pyran-2-ones and their versatility in organic synthesis. Tetrahedron. 2009;65:7865–7913. doi: 10.1016/j.tet.2009.06.031. [DOI] [Google Scholar]
- 3.Pratap R., Ram V.J. 2H-Pyran-2-ones and their annelated analogs as multifaceted building blocks for the fabrication of diverse heterocycles. Tetrahedron. 2017;73:2529–2590. doi: 10.1016/j.tet.2017.02.028. [DOI] [Google Scholar]
- 4.Dobler D., Leitner M., Moor N., Reiser O. 2-Pyrone—A privileged heterocycle and widespread motif in nature. Eur. J. Org. Chem. 2021;2021:6180–6205. doi: 10.1002/ejoc.202101112. [DOI] [Google Scholar]
- 5.Hickey A., Pardo L.M., Reen F.J., McGlacken G.P. Pyrones identified as LuxR signal molecules in photorhabdus and their synthetic analogues can alter multicellular phenotypic behavior of Bacillus atropheaus. ACS Omega. 2021;6:33141–33148. doi: 10.1021/acsomega.1c05508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yadav J.S., Ganganna B., Dutta P., Singarapu K.K. Synthesis and determination of absolute configuration of α-pyrones isolated from Penicillium corylophilum. J. Org. Chem. 2014;79:10762–10771. doi: 10.1021/jo5015382. [DOI] [PubMed] [Google Scholar]
- 7.Lee I.K., Yun B.S. Styrylpyrone-class compounds from medicinal fungi Phellinus and Inonotus spp., and their medicinal importance. J. Antibiot. 2011;64:349–359. doi: 10.1038/ja.2011.2. [DOI] [PubMed] [Google Scholar]
- 8.Bhat Z.S., Rather M.A., Maqbool M., Lah H.U., Yousuf S.K., Ahmad Z. α-pyrones: Small molecules with versatile structural diversity reflected in multiple pharmacological activities-an update. Biomed. Pharmacother. 2017;91:265–277. doi: 10.1016/j.biopha.2017.04.012. [DOI] [PubMed] [Google Scholar]
- 9.Aggarwal R., Walia S., Rani C. Dehydroacetic acid and its derivatives as starting synthons for synthesis of heterocyclic compounds. Heterocycles. 2017;94:1197–1244. doi: 10.3987/REV-17-860. [DOI] [Google Scholar]
- 10.Obydennov D.L., El-Tantawy A.I., Sosnovskikh V.Y. Triacetic acid lactone as a bioprivileged molecule in organic synthesis. Mendeleev Commun. 2019;29:1–10. doi: 10.1016/j.mencom.2019.01.001. [DOI] [Google Scholar]
- 11.Prakash O., Kumar A., Singh S.P. Synthesis of heterocyclic compounds from the reactions of dehydroacetic acid (DHA) and its derivatives. Heterocycles. 2004;63:1193–1220. doi: 10.3987/REV-03-572. [DOI] [Google Scholar]
- 12.Moreno-Mañas M., Pleixats R. Dehydroacetic acid, triacetic acid lactone, and related pyrones. Adv. Heterocycl. Chem. 1992;53:1–84. doi: 10.1016/S0065-2725(08)60861-2. [DOI] [Google Scholar]
- 13.Politanskaya L.V., Selivanova G.A., Panteleeva E.V., Tretyakov E.V., Platonov V.E., Nikul’shin P.V., Vinogradov A.S., Zonov Y.V., Karpov V.M., Mezhenkova T.V., et al. Organofluorine chemistry: Promising growth areas and challenges. Russ. Chem. Rev. 2019;88:425–569. doi: 10.1070/RCR4871. [DOI] [Google Scholar]
- 14.Han J., Remete A.M., Dobson L.S., Kiss L., Izawa K., Moriwaki H., Soloshonok V.A., O’Hagan D. Next generation organofluorine containing blockbuster drugs. J. Fluor. Chem. 2020;239:109639. doi: 10.1016/j.jfluchem.2020.109639. [DOI] [Google Scholar]
- 15.Isanbor C., O’Hagan D. Fluorine in medicinal chemistry: A review of anti-cancer agents. J. Fluor. Chem. 2006;127:303–319. doi: 10.1016/j.jfluchem.2006.01.011. [DOI] [Google Scholar]
- 16.O’Hagan D. Fluorine in health care: Organofluorine containing blockbuster drugs. J. Fluor. Chem. 2010;131:1071–1081. doi: 10.1016/j.jfluchem.2010.03.003. [DOI] [Google Scholar]
- 17.Purser S., Moore P.R., Swallow S., Gouverneur V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008;37:320–330. doi: 10.1039/B610213C. [DOI] [PubMed] [Google Scholar]
- 18.Wang J., Sánchez-Roselló M., Aceña J.L., del Pozo C., Sorochinsky A.E., Fustero S., Soloshonok V.A., Liu H. Fluorine in pharmaceutical industry: Fluorine-containing drugs introduced to the market in the last decade (2001–2011) Chem. Rev. 2014;114:2432–2506. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- 19.Fujiwara T., O’Hagan D. Successful fluorine-containing herbicide agrochemicals. J. Fluor. Chem. 2014;167:16–29. doi: 10.1016/j.jfluchem.2014.06.014. [DOI] [Google Scholar]
- 20.Jeschke P. The unique role of fluorine in the design of active ingredients for modern crop protection. ChemBioChem. 2004;5:570–589. doi: 10.1002/cbic.200300833. [DOI] [PubMed] [Google Scholar]
- 21.Berger R., Resnati G., Metrangolo P., Weber E., Hulliger J. Organic fluorine compounds: A great opportunity for enhanced materials properties. Chem. Soc. Rev. 2011;40:3496–3508. doi: 10.1039/c0cs00221f. [DOI] [PubMed] [Google Scholar]
- 22.Granqvist L., Virta P. Characterization of G-quadruplex/hairpin transitions of RNAs by 19F NMR spectroscopy. Chem. Eur. J. 2016;22:15360–15372. doi: 10.1002/chem.201602898. [DOI] [PubMed] [Google Scholar]
- 23.Marsh E.N.G., Suzuki Y. Using 19F NMR to probe biological interactions of proteins and peptides. ACS Chem. Biol. 2014;9:1242–1250. doi: 10.1021/cb500111u. [DOI] [PubMed] [Google Scholar]
- 24.Resnick P.R. Fluorinated Heterocyclic Compounds. John Wiley & Sons, Inc.; Hoboken, NJ, USA: 2009. Practical uses of fluorinated heterocycles; pp. 493–506. [Google Scholar]
- 25.German L.S., Sterlin S.R., Cherstkov V.F. Hexafluorodehydroacetic acid. Bull. Acad. Sci. USSR Div. Chem. Sci. 1982;31:1476–1477. doi: 10.1007/BF00954185. [DOI] [Google Scholar]
- 26.Li C.-S., Lau C.K., Therien M., Prasit P. Pyrones as Inhibitors of Cyclooxygenase-2. 0058690. U.S. Patent. 2002 May 16;
- 27.Hanselmann P., Wenger W. Method for the Production of 6,6,6-Trihalo-3,5-dioxohexanoic Acid Esters. 040087. WO Patent. 2005 May 6;
- 28.Garratt S. The Mechanism of the reaction between dehydroacetic acid and alkylamines. J. Org. Chem. 1963;28:1886–1888. doi: 10.1021/jo01042a038. [DOI] [Google Scholar]
- 29.Cook D. The preparation, properties, and structure of 2,6-bis-(alkylamino)-2,5-heptadien-4-ones. Can. J. Chem. 1963;41:1435–1440. doi: 10.1139/v63-195. [DOI] [Google Scholar]
- 30.Pierce J.B., Ariyan Z.S., Ovenden G.S. Preparation and antiinflammatory activity of 2- and 4-pyridones. J. Med. Chem. 1982;25:131–136. doi: 10.1021/jm00344a008. [DOI] [PubMed] [Google Scholar]
- 31.Schmidt R., Stolte M., Grüne M., Würthner F. Hydrogen-bond-directed formation of supramolecular polymers incorporating head-to-tail oriented dipolar merocyanine dyes. Macromolecules. 2011;44:3766–3776. doi: 10.1021/ma2004184. [DOI] [Google Scholar]
- 32.Würthner F., Schmidt J., Stolte M., Wortmann R. Hydrogen-bond-directed head-to-tail orientation of dipolar merocyanine dyes: A strategy for the design of electrooptical materials. Angew. Chem. Int. Ed. 2006;45:3842–3846. doi: 10.1002/anie.200504581. [DOI] [PubMed] [Google Scholar]
- 33.Eiden F. Untersuchungen an γ-Pyronen. 4. Mitteilung: Die Kondensation von γ-Pyronen mit cyclischen Malonsäureamiden. Arch. Pharm. 1960;293:404–414. doi: 10.1002/ardp.19602930407. [DOI] [PubMed] [Google Scholar]
- 34.Obydennov D.L., El-Tantawy A.I., Sosnovskikh V.Y. Bio-based triacetic acid lactone in the synthesis of azaheterocycles via a ring-opening transformation. New J. Chem. 2018;42:8943–8952. doi: 10.1039/C8NJ01044G. [DOI] [Google Scholar]
- 35.Usachev B.I., Obydennov D.L., Röschenthaler G.-V., Sosnovskikh V.Y. 2-Cyano-6-(trifluoromethyl)-4H-pyran-4-one: A novel versatile CF3-containing building block. J. Fluor. Chem. 2012;137:22–26. doi: 10.1016/j.jfluchem.2012.01.006. [DOI] [Google Scholar]
- 36.Zheglova D.K., Genov D.G., Gribanov A.V., Kol’tsov A.I., Smirnov S.N. NMR spectroscopic study of the (Z)/(E)-isomerism of 1-aryl-3-arylamino-2-propen-1-ones in solution and in the crystalline state. Monatsh. Chem. 1994;125:1443–1446. doi: 10.1007/BF00811094. [DOI] [Google Scholar]
- 37.Gerus I.I., Balabon O.A., Pazenok S.V., Lui N., Kondratov I.S., Tarasenko K.V., Shaitanova E.N., Ivasyshyn V.E., Kukhar V.P. Synthesis and properties of polyfunctional cyclic β-alkoxy-α,β-unsaturated ketones based on 4-methylene-1,3-dioxolanes. Eur. J. Org. Chem. 2018;2018:3853–3861. doi: 10.1002/ejoc.201800786. [DOI] [Google Scholar]
- 38.Wöjcik J., Domalewski W., Kamieńska-Trela K., Stefaniak L., Vdovienko S.I., Gerus I.I., Gorbunova M.G. Conformational behaviour of some trifluoroenaminones by multinuclear magnetic resonance. Magn. Reson. Chem. 1993;31:808–814. doi: 10.1002/mrc.1260310904. [DOI] [Google Scholar]
- 39.Kiang A.K., Tan S.F., Wong W.S. Reactions of acetonedicarboxylic anhydride (tetrahydropyrantrione) and its mono- and di-acetyl derivatives with amines. J. Chem. Soc. C. 1971:2721–2726. doi: 10.1039/j39710002721. [DOI] [Google Scholar]
- 40.Usachev S.A., Usachev B.I., Sosnovskikh V.Y. Synthesis of 6-hydroxy-5,6-dihydro-2-pyrones and -pyridones by reaction of 4-aryl-6-trifluoromethyl-2-pyrones with water, hydrazine, and hydroxylamine. Chem. Heterocycl. Compd. 2017;53:1294–1301. doi: 10.1007/s10593-018-2209-y. [DOI] [Google Scholar]
- 41.Gerus I.I., Tolmachova N.A., Vdovenko S.I., Fröhlich R., Haufe G. A convenient synthesis and chemical properties of 3-acylamino-6-poly-fluoroalkyl-2H-pyran-2-ones. Synthesis. 2005;2005:1269–1278. doi: 10.1055/s-2005-865290. [DOI] [Google Scholar]
- 42.Ye W.-P., Mu H.-L., Shi X.-C., Cheng Y.-X., Li Y.-S. Synthesis and characterization of the titanium complexes bearing two regioisomeric trifluoromethyl-containing enaminoketonato ligands and their behavior in ethylene polymerization. Dalt. Trans. 2009;2009:9452–9465. doi: 10.1039/b906854f. [DOI] [PubMed] [Google Scholar]
- 43.Desens W., Winterberg M., Michalik D., Langer P. Synthesis and solution structure of 1H-benzo-1,5-diazepine derivatives with a perfluoroalkyl side chain. Helv. Chim. Acta. 2016;99:361–372. doi: 10.1002/hlca.201500264. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are contained within the article and Supplementary Materials.