

Abstract
The Collaborative Cross (CC) and the Diversity Outbred (DO) stock mouse panels are the most powerful murine genetics tools available to the genetics community. Together, they combine the strength of inbred animal models with the diversity of outbred populations. Using the 63 CC strains or a panel of DO mice, each derived from the same 8 parental mouse strains, researchers can map genetic contributions to exceptionally complex immunological and infectious disease traits that would require far greater powering if performed by genome-wide association studies (GWAS) in human populations. These tools allow genes to be studied in heterozygous and homozygous states and provide a platform to study epistasis between interacting loci. Most importantly, once a quantitative phenotype is investigated and quantitative trait loci are identified, confirmatory genetic studies can be performed, which is often problematic using the GWAS approach. In addition, novel stable mouse models for immune phenotypes are often derived from studies utilizing the DO and CC mice that can serve as stronger model systems than existing ones in the field. The CC/DO systems have contributed to the fields of cancer immunology, autoimmunity, vaccinology, infectious disease, allergy, tissue rejection, and tolerance but have thus far been greatly underutilized. In this article, we present a recent review of the field and point out key areas of immunology that are ripe for further investigation and awaiting new CC/DO research projects. We also highlight some of the strong computational tools that have been developed for analyzing CC/DO genetic and phenotypic data. Additionally, we have formed a centralized community on the CyVerse infrastructure where immunogeneticists can utilize those software tools, collaborate with groups across the world, and expand the use of the CC and DO systems for investigating immunogenetic phenomena.
Keywords: Collaborative Cross, Diversity Outbred, genetic linkage, immunology, quantitative trait locus
INTRODUCTION
Variation is the blessing and bane of every population geneticist’s existence. Genomic variation within a population is the underlying mechanism that leads to differential genetic phenotypes, and yet the extent of genetic variation within the population is a daunting barrier when studying genetic traits in humans. Genetic researchers must choose their genetic models by balancing between the feasibility of genetically homogeneous populations and highly variable human populations. If a trait exists in low diversity populations, it can be mapped with reasonable numbers of study subjects, but limited diversity populations also lack relevant traits to study. Although highly variable human populations contain highly relevant traits, powering is often insufficient to yield causal variants with confidence. Strategies to facilitate mapping of traits have focused on attempts to limit genetic diversity within study populations: adopting multigenerational human family and twin studies; the study of consanguineous human populations; the adoption of model genetic systems (Drosophila, C elegans, Saccharomyces budding and fission yeast, bacterial and plant models); and inbred mouse models. Mapping traits in model organisms is facilitated by allowing researchers to control the environment of study subjects, to focus on small readily editable genomes, and to reap the cost benefits and fast progress afforded by short life cycles.
Mice are one of the most widely used species for biomedical research worldwide, with inbred BALB/c and C57BL/6 mice being two of the most utilized strains (Festing, 2014; Taylor & Alvarez, 2019). The development of inbred strains of mice pioneered by Clarence Little allowed reproducible analysis of in vivo traits for the first time due to the strain genetic homogeneity. This allowed an unlimited number of subjects that were essentially identical twins. The production of multiple independent inbred strains gives the ability to confirm the heritability of complex traits like disease susceptibility (Anderson & Bluestone, 2005; Little & Tyzzer, 1916). These analyses revealed many single genes with large phenotypic effects such as the endogenous germ line–encoded AKV-1 locus in AKR mice (Chattopadhyay, Rowe, Teich, & Lowy, 1975). Inbred strain information is also cumulative; once the existence of a trait in a strain is noted, it remains constant. Thus, the information accumulates over time with additional investigations.
The strength of inbred strains is also their weakness; any strain only samples a small part of the genetic repertoire of the species and thus limits the number of traits that can be studied at one time. During inbred model development, virtually all loci become homozygous, creating genetically identical mice. The inbreeding process could potentially lead to the selection of genetic backgrounds that are convenient for laboratory use, with favorable breeding and behavior characteristics, at the expense of traits that increase fitness in the wild population (Miller et al., 1999). Several studies have discussed the drawbacks of utilizing inbred mice, particularly if it is only one strain and more generally, the “dogmatic standardization” of animal-based biomedical research (Miller, 2016; Saul, Philip, Reinholdt, Center for Systems Neurogenetics of Addiction, & Chesler, 2019; Tuttle, Philip, Chesler, & Mogil, 2018; Voelkl et al., 2020).
Meanwhile, in human genetics, where formal genetic analysis is difficult, population-based tools such as genome-wide association studies (GWAS) were developed. These can infer association with a gene (or a single nucleotide polymorphism with a trait in a population). However, there are limitations to these analyses as well. When studying genetics in outbred human communities, researchers lack the level of experimental control that is present when studying genetic model systems. In addition, the extensive variation present when studying genetic traits in outbred human populations requires the recruitment of so many study subjects that most research groups lack the funds and infrastructure to research the genetic causes of complex traits. Human GWAS, now being powered at over a million study subjects, are outside the reach of only the largest research consortia (Jansen et al., 2019; Lee et al., 2018). Unlike formal genetics where co-segregation of a marker and a trait can be followed through generations, GWAS at best involve hypothesis generation.
To improve the rigor, reproducibility, translational success of animal research, and better investigate complex traits, the use of mouse models that allow for implementation of controlled heterogenization containing the breadth of mouse genetic diversity should be considered. The difficulty in the identification of quantitative trait loci by conventional genetics spurred efforts to develop new methods to address the problem (Churchill et al., 2004). To meet this need, a panel of recombinant inbred mice, the Collaborative Cross (CC), and a genetically heterogeneous outbred stock, the Diversity Outbred (DO), were created from eight inbred laboratory founder strains. The eight founder strains, A/J, C57BL/6J, 129S1/SvlmJ, NOD, ShiLtJ, NZO/HILtJ, CAST/EiJ, PWK/PhJ, and WSB/EiJ, encompass 37.8 million single nucleotide polymorphisms (SNPs) and 6.9 million insertions/deletions and structural variants, making them an incredibly powerful tool for investigation of complex traits and examining genotype-phenotype interactions (Keane et al., 2011; Saul et al., 2019). The CC and DO founders are fully sequenced and paired with the computation tools needed to map traits without sequencing a single nucleotide. The CC and DO mice represent the ideal experimental balance between genetic homogeneity and variability.
Conceptually, utilizing the CC and DO is equivalent to performing GWAS in which common variants are used to identify genes associated with a trait (phenotype) of interest within a particular genomic region. Humans have a large number (~85 million) of SNPs, and mice are known to have about half of this number. A major advantage of both the CC and the DO models is that the genomes of the parental founder strains have been sequenced and all the SNPs (except de novo mutations) among the founder strains have been identified. Originally, the goal of the CC project was to produce a set of 1000 strains (Collaborative Cross Consortium, 2012); however, challenges with full inbreeding and reproductive capacity have limited this to 63 finished CC lines. Regardless, these genetic models have been able to attack two classes of questions not accessible with GWAS. The first is the ability to replicate a genotype with only homozygous effects with multiple complexes having known genetic backgrounds. The second is the potential to identify interacting genes, which can be attacked using crosses between two different CC stains selected for shared haplotypes, allowing one region to remain homozygous while introducing heterozygosity to much of the remainder of the genome. Although GWAS can quantify genetic contributions to a complex phenotype, replication is difficult. Further, although many alleles have been identified by GWAS that contribute to a phenotype, most have not proven useful due to either a low frequency or relatively small contribution to the phenotype. Two clear examples are type 1 diabetes and Ankylosing spondylitis, where only MHC association has produced actionable information on risk, which was already identified by conventional means. Although this review describes many different and creative uses of the CC and DO populations, the basic roadmap in Figure 1 demonstrates a common theme that many of the studies follow to discover novel genetic influences on complex traits and the creation of new disease models.
Figure 1.
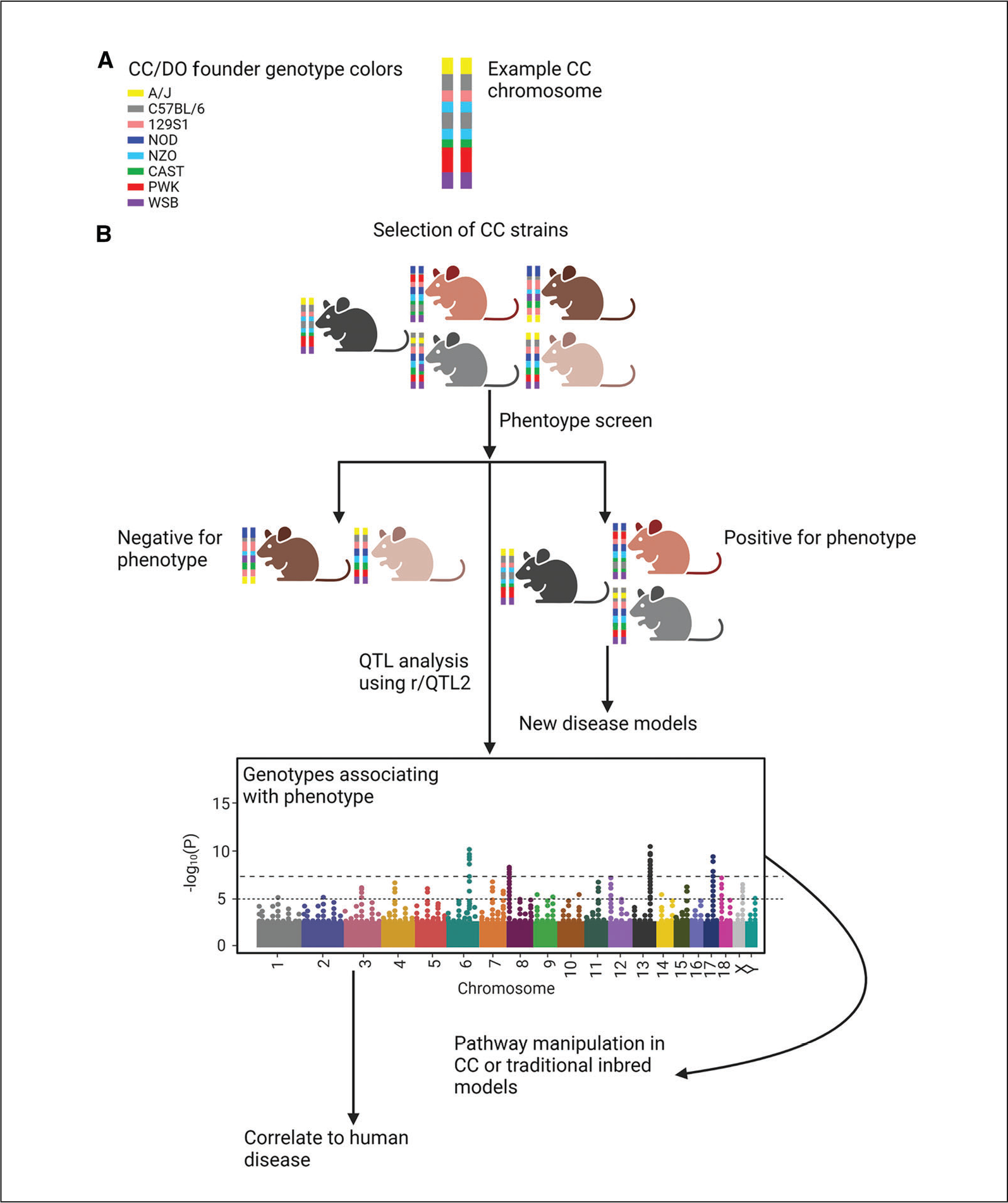
Process map for utilizing CC lines to explore complex phenotypes. (A) Official colors representing the genotypes of DO and CC founders (right) and an example CC line chromosome showing homozygous haplotype blocks with contributions from the CC/DO founder lines. (B) Concept map demonstrating potential usage of CC mice to identify new disease models and dissect genetic underpinning of complex traits. A population of DO mouse could also be used for phenotype screening and subsequent QTL analysis. Created with BioRender.com.
In this review, we will briefly describe the history of the CC and DO mice and set out some of the contributions these models have made to our understanding of the immune system, focusing on four areas summarized in Table 1, namely allergy, autoimmunity, cancer, and infectious disease. Finally, we will describe the overall methodology on how to apply these mouse strains to immunological problems, focusing on the informatics and quantitative trait locus (QTL) mapping. We will not discuss the rapidly growing literature on using these mice in toxicology and pharmacogenetics, where they have already been extraordinarily useful.
Table 1.
Summary of the Studies Reviewed in This Paper
| Disease category | Mice used | Finding/impact | Reference |
|---|---|---|---|
|
| |||
| Allergy | CCa | Identified CC027 as a mouse model of oral allergy response with recapitulation human disease and shows CC027 as having Th2-skewed response | Orgel et al. (2019), Smeekens et al. (2020) |
| Allergy/infectious disease | CC | Linked Sp140 gene to IgE response in the setting of allergy and parasite clearance | Matsushita et al. (2021) |
| Cancer | DOF1b | Demonstrates DOF1 mice can be used to discover germline factors of cancer metastasis that correlated to human diseases; demonstrates roadmap for actionable pathway discovery | Winter et al. (2017) |
| Cancer | CC | Identifies CC036 as model of spontaneous gastric tumorigenesis and identifies NfκB as likely causal gene | Wang et al. (2019) |
| Cancer | CCF1b | Utilized a CCF1 cross to discover loci modifying Apc-driven polyp formation | Dorman et al. (2021) |
| Cancer | CCF1 | Developed a CCF1 model to investigate modifier genes influencing response to asbestos-induced mesothelioma | Behrouzfar et al. (2021) |
| Cancer | DOF1 | Investigated germline modifiers of HER2 targeting breast cancer vaccine utilizing DOF1 mice and identified NK cells as a mediator of vaccine effectiveness | Wei et al. (2020) |
| Cancer | DOF1, CCF1 | Showed that DOF1 and CCF1 mice can accept cross-strain syngeneic tumors and identified prolactin as a germline modifier of immune checkpoint blockade response | Hackett et al. (2022) |
| Infectious disease | CC | Demonstrated that known genetic loci influencing virus-induced paralysis have further unknown modifiers | Perez Gomez et al. (2021) |
| Infectious disease | CCF1,CCF2 | Host genotype regulates anti-influenza-A antibody response | Noll et al. (2020) |
| Infectious disease | CC Founder | Demonstrated that upon infection with respiratory infections, CC mice induce expression of previously unannotated transcripts and strain-specific isoforms | Xiong et al. (2014) |
| Infectious disease | CCF1, CC-RIXc | Discovered baseline immune cell phenotypes to predict disease control of SARS-CoV infection | Graham et al. (2021) |
| Infectious disease | CC, CCF1 | Demonstrated that CC mice recapitulate facets of EBV and can be used to predict human patient outcomes | Price et al. (2020) |
| Infectious disease | CC | Demonstrated that CC mice can serve as a model for Zika virus pathology and that known loci of infectious outcomes have further unknown modifiers | Manet et al. (2020) |
| Infectious disease | CC-RIX | CC-RIX lines can model levels of West Nile Virus infection | Green et al. (2016) |
| Infectious disease | CC | Identified and validated candidate genes influencing P. aeruginosa lung infection using CC mice | Lorè et al. (2020) |
| Infectious disease | CC | Identified CC006 as a model to study NK cell–mediated viral protection | Jensen et al. (2022) |
| Infectious disease | CC | Identified CC046 and CC058 as first immunocompetent mouse models of B. recurrentis | Rogovskyy et al. (2021) |
| Infectious disease | CCF1 | Identified haplotypes associated with influenza A response in CCF1 mice | Maurizio et al. (2018) |
| Infectious disease | DOd | Created model to predict M. tuberculosis outcome using H&E staining from DO mice | Tavolara et al. (2021) |
| Infectious disease | CC | Discovered germline loci that influence host susceptibility to specific pathogens | Durrant et al. (2011), Smith et al. (2022), Vered et al. (2014), Zhang et al. (2018) |
| Inflammatory diseases | DO | Demonstrated large variation of silica-induced autoimmunity in DO mice | Mayeux et al. (2018) |
| Inflammatory diseases | CC | Identified CC011 as a spontaneous model of colitis and identified 4 loci of susceptibility | Rogala et al. (2014) |
| Inflammatory diseases | CC, CCF1, CCN2e | Identified CC003 and CC039 as susceptible to O3-induced inflammatory response and identified differentially basal gene expression in alveolar macrophages | Smith et al. (2021), Tovar et al. (2022) |
CC: Collaborative Cross.
CCF1/DOF1: CC or DO with single cross to other strain.
CC-RIX, Collaborative Cross Recombinant Inbred Cross.
DO: Diversity Outbred.
CCN2: CC with double backcross to CC founder strain.
ORIGINS OF CC AND DO MICE
The CC and DO mice stem from the same origin. Geneticists have struggled to map genes in relatively slow-reproducing eukaryotic species. Since the early days of Morgan’s fly house, the identification of recombination between genes located on the same chromosome by gene mapping has been a priority. The long mammalian generation time and the difficulty in obtaining wide genetic diversity have hobbled mouse genetics in particular. Both the CC and DO models can ameliorate that problem. CC mice are based on the same fundamental concept of traditional recombinant inbred strains. This idea is not new; much of the theoretical groundwork was laid out by Haldane and Waddington (1931). The mathematics of the consequence of inbreeding on recombination and allele fixation provides a critical understanding of the genetics of recombinant inbred lines.
Surprisingly, it took the conflation of two things to allow the idea of the CC and DO models to reach fruition. The first was the establishment and success of the Jackson Laboratory (Jax), now the center of mouse genetics. Spurred on by its founder Little (inbred mice) and pushed by Snell (congenic mice), Jax provided a place where long-term mouse experiments could be pursued. The gold standard for inbreeding is 20 generations, which requires at least 6 years with conventional mouse breeding. When starting from outbred animals that carry many deleterious alleles, many incipient lines become extinct after a few generations of inbreeding. Thus, an institution with both the commitment and facilities for large-scale classical mouse genetics is required. Second, someone has to think of performing the experiment to produce the lines. Don Bailey at Jax achieved this. He produced a panel of recombinant inbred lines made between C57BL/6 and BALB/c. When analyzing the modest number of coat-color genes, the most interesting finding for immunologists was the identification of many minor histocompatibility loci that were instrumental in understanding MHC restriction (Bailey, 1971). A major advantage of RI lines is that the information is permanent and cumulative, unlike F2 mice where each mouse is unique and ephemeral. Each new trait tested adds to the power of the mapping in the RI panel, as each strain can be repeatedly tested for new traits. Because RI lines and CC mice remain heterozygous for several generations, this allows recombination between genetic marks multiple times. These multiple opportunities for detectable recombination result in separation of relatively closely linked genes and provide a much-improved resolution. The outcome of this is an approximately 4-fold expansion of the measured map distance. Two loci that might be mapped as only 5 cm apart (separated in only 5 of 100 F2 progenies tested) would increase to 20 cm or one-fifth in the resulting RI strains.
Ben Taylor joined the Jackson Lab and realized the power of RI lines. He began developing a variety of different sets of RI strains, with different parental lines to take advantage of the known genetic diversity among inbred strains. This resulted in the development of more than a dozen RI strain pairs. Currently, there are 412 RI lines available from Jax including 56 from the CC [https://mice.jax.org/?linkbuilder=1&stockType=Recombinant%20Inbred%20(RI). PubMed lists more than 60,000 references related to RI mice. Clearly, this was a good idea, as noted by Silver (1995).
How are RI lines used? Most simply stated, the pattern of phenotypes among RI lines is compared to known alleles (or SNPs). In other words, any genetic loci where the parental strains differ can be used to probe the locations of the genes encoding them. In principle, any trait where the two parents differ can be mapped to a specific chromosome. This is accomplished by comparing the phenotype of each of the RI strains with the parental phenotype. As is the case with the parents, the RI strains are new inbred strains, but all their genes must come from one or the other parental strain. For simple sets, like the original Bailey CXB lines (of which there are only 9), by inspection, one can compare the phenotype of the trait in question with all other mapped traits. If the pattern is identical to a known trait, then the assumption is that the genes are linked. With a small set of strains, the sweep length (defined as the distance from the known locus) is relatively large; however, using larger sets, the sweep grows smaller, although chance matches remain possible with small strain sets. Indeed, even the distance can be estimated using strains with known recombination points. Currently, most of the existing two-parent RI panels have been extensively genotyped using high-density SNP chips and the recombination points are well defined on each chromosome. Presumed linkage can be further confirmed using consomic mouse strains, as well as by predicting phenotypes of other RI strains from independent panels.
A major limitation of conventional RI models is that the trait of interest must differ between the two parental strains of the RI lines. Many traits cannot be mapped because they do not differ between relevant parental inbred strains. Further, the existing RI panels are derived from relatively closely related strains, and thus the polymorphisms among them are limited. Nearly 20 years ago, a group of mouse geneticists led by Churchill and Threadgill (Churchill et al., 2004) began working on establishing superior RI panels. Here, they envisioned not two but eight parental strains that were not limited to common inbred mice; however, they proposed including strains derived from wild mice of other Mus subspecies. These additions resulted in the ability to capture more than 95% of the natural genetic variation in mice. It is important to stress that this strategy does not allow recessive lethal or sterile alleles, as all input mice are fertile and viable. This approach allows sampling of a much larger area of the mouse genome. The initial plan was to produce 1000 CC strains. However, the appetite for the strains was much larger than the ability to produce them, even with the job distributed over several sites and continents (Asia, North America, and Australia). Difficulties with inbreeding and incompatibilities among the parental strains ultimately led to a much smaller panel. Currently, only 63 strains have made it to completion and are distributed by UNC and Jackson Labs (see: http://www.csbio.unc.edu/Ccstatus/index.py?run=AvailableLines.information).
The advent of rapid low-cost sequencing led the Cambridge mouse genetics group (https://www.sanger.ac.uk/data/mouse-genomes-project/) to provide nearly complete whole-genome sequencing of the eight progenitor strains of the CC. This sequence analysis allowed accurate design of high-density SNP chips (initially by the original mouse universal genotyping array, or MUGA, which contained approximately 10,000 SNPs, but denser now with the GigaMUGA containing 143,259 genetic markers) allowing the assignment of blocks of parental DNA (haplotype maps) in each of the CC strains. Thus, in any CC line, it is possible to assign the origin of each segment of a chromosome to a parent. Like the smaller, simpler RI strain sets, it is the pattern of the phenotypes seen that provides the information. Obviously, this works best with all-or-none phenotypes; however, the major advancement was the ability to map quantitative differences. These QTL are much more challenging to map in a conventional setting, requiring large F2 panels from multiple crosses. R/qtl, an important tool, and R/qtl2, its successor, provide a straightforward way to map such quantitative variations (Broman et al., 2019; Broman, Wu, Sen, & Churchill, 2003). These tools are the basis for much of the current utility of the CC and DO models, and the process for conducting genetic linkage analysis with this software will be discussed below.
A limitation of the CC lines is the confined number of genotypes that can be interrogated due to the relatively few strains completed. This is further complicated by the fact that all combinations of alleles tested must be non-lethal and allow fertility. As the CC developed, many of the same investigators who conceptualized the CC realized that a complementary approach was needed, namely the DO stock (Svenson et al., 2012). These mice are essentially a mirror image of the CC mice. Although derived from the same eight strains by a series of F1 crosses like the initiation of the CC mice, continued outbreeding produces unique individual mice, each with a different genotype. The DO model allows for the interrogation of combinations of alleles not present in the CC mice. These mice can be readily phenotyped in relatively large numbers. The principal disadvantage is that unlike CC mice, each DO mouse is genetically unique; therefore, analyzing replicates of each DO genotype is not possible. Additionally, each mouse must be individually genotyped using a large SNP array such as the GigaMUGA. Fortunately, for the genetics community, the software (R/qtl2) that allows analysis of the CC mice also facilitates analysis of the DO mice.
USING CC MICE TO DESCRIBE NATURAL VARIATION OF IMMUNE PARAMETERS AMONG STRAINS
The CC mice have been used to probe relatively complex normal phenotypes. There is substantial variability among the CC/DO founders in the basal levels of baseline immune cell percentages and quantities (Collin, Balmer, Morahan, & Lesage, 2019; Phillippi et al., 2014). In pre-CC mice, phenotypic differences in the immune cell subset were even more diverse than in the parental strains (Phillippi et al., 2014). Wide ranges in the abundance of splenic T, B, NK, and myeloid populations were detected among strains. Such diversity has long been observed in F2 populations, where individual F2 mice often have more extreme phenotypes than either parent. Intercrossing CC strains results in further immune cell diversity, which is evident in the high variation of T cell subset percentages and differential expression of activation markers and cytokines (Graham et al., 2017). The development and functionality of NK cells were also further investigated in a panel of established CC lines (Dupont et al., 2021). Variation in NK cell numbers, differentiation mechanisms, and functional capacity were observed between CC strains. Genomic loci associated with these phenotypes were identified. Collectively, these studies support the use of CC models to evaluate mechanisms of immune function in different disease contexts.
Allergy
The DO and CC populations have been emerging as useful tools for gene discovery and model development in response to potential allergens. Matsushita et al. (2021) provide a template on how to proceed from phenotype to specific causal gene discovery. The authors investigated mast cell trafficking/function and IgE response, utilizing a screen of 47 CC mouse strains for passive cutaneous anaphylaxis, Strongyloides venezuelensis expulsion, and the corresponding IgE response then a single analysis was used to connect the three related phenotypes. All three phenotypes were correlated, suggesting a shared mechanism, and QTL effects were analyzed for each phenotype to identify shared peaks. This approach improves the rigor of analysis, focusing on QTL peaks that are shared across related mechanisms of interest. The authors confirm that their major significant peak is present in each of the independent genome scans, and critically, they show that the strain effects are shared across phenotypes. QTL effects allow for quick informatic identification of the unique strain-specific SNPs within the locus without the need for comprehensive whole-genome sequencing. They identified SNPs within the Sp140 gene that result in differential expression and RNA splicing in the gene, which contribute to CC strain susceptibility to IgE-mediated allergen response and S. venezuelensis expulsion. Sp140 has previously been shown to be involved in immune functions related to autoimmunity and response to infection, further supporting their results (Ji et al., 2021; Karaky et al., 2018).
The genetics of food allergies have remained poorly understood, beyond a requirement for IgE responses. The CC mice have provided an entrée into the potential mechanisms. Among 47 CC strains tested for development of IgE response induction and passive cutaneous anaphylaxis mentioned above, CC027 showed the most extreme allergy phenotype for both readouts; this is corroborated by additional studies on food allergy susceptibility (Orgel et al., 2019; Smeekens, Orgel, Kesselring, Bagley, & Kulis, 2020). A screen of 16 CC strains identified CC027 as the only strain that reproducibly developed an oral allergy to peanuts, further showing that this strain exhibits a Th2-skewed immune response (Orgel et al., 2019). Interestingly, FcRε (CD23) expression has been suggested to regulate IgE production and lower overall circulating levels of IgE by sequestering IgE via cell surface binding (Ford, Sturgill, & Conrad, 2009). Analysis of CD23 in CC and parental mice showed that IgE levels could be dissociated, suggesting that IgE levels may be regulated by a different mechanism (Phillippi et al., 2014). Additionally, a published abstract from Todd et al. (2020) investigated Th2-associated cytokine response and group 2 innate lymphoid cell (ILC2) recruitment to the lungs after Alternaria extract exposure in 44 CC strains and 6 of the parental lines. Differential immune activity was reported among CC strains, including IL-5, IL-13, and TSLP expression levels. However, there was no observed correlation between thymic stromal lymphopoietin and IL-5/IL-13. From this, they concluded that TSLP expression is not a primary driver of Th2 cytokine production, which had been suggested previously (Soumelis & Liu, 2004; Soumelis et al., 2002).
These studies highlight the consistency of CC mouse models, expanding the potential application of these discovered models to address new questions, collectively emphasizing the cumulative nature of information discovered in CC mice. Even without utilizing the developed tools to determine contributing loci, CC screens can be useful to identify extreme ends of response. For instance, knowledge of Th1 versus Th2 immune dominance may be useful in other settings, such as vaccine development, where one can consider leveraging the published CC models to maximize the broad response range expected in the human population.
Autoimmunity and Inflammatory Disease
The CC and DO mice are an ideal model for the study of autoimmune diseases, which have numerous known genetic susceptibilities (Richard-Miceli & Criswell, 2012). To date, the most prominent discovered genomic regions that associate with disease onset and severity are within MHC [either human leukocyte antigen (HLA) or mouse (H2) loci] (Matzaraki, Kumar, Wijmenga, & Zhernakova, 2017). Most of the autoimmune disease-associated MHC genes have a large influence on phenotypes, and they were discovered without the necessity of complex genetic tools.
Mouse models of autoimmune conditions can vary in their design. Disease can be induced by delivery of an external stimulus, use of genetic models that are fully penetrant, or a combination of heritable susceptibility with a mode of induction. What these models have clearly shown is that genetic background plays a dominant role in autoimmunity onset, and some strains require more manipulation than others to induce autoimmune disease. For example, C57BL/6 mice require multiple manipulations to break self-tolerance compared to single manipulations in BALB/c mice (Peeva, Gonzalez, Hicks, & Diamond, 2006; Peeva et al., 2003). This is compared to the NOD strain that spontaneously develops autoimmune diabetes (Anderson & Bluestone, 2005). Both C57BL/6 and NOD are founders in the DO and CC models and provide an excellent setting for recombination and isolation of susceptibility loci.
Despite their potential for investigating complex traits with genetic influence, to date, there have been limited studies on autoimmunity in the DO and CC mouse models. DO mice have been used to characterize silicosis and silica-induced autoimmunity (Mayeux et al., 2018). The DO mice in this study show a diverse response to the silicosis challenge, in both silicosis pathology within the lungs and subsequent autoimmune development evidenced by glomerulonephritis and auto-antibodies. Interestingly, the authors note that non-silica–exposed mice show a high prevalence of anti-nuclear antibodies (ANA), a common feature of multiple autoimmune diseases including lupus and Sjogren’s syndrome, but no other associated markers of autoimmunity were detected in naive mice. The authors suggest that baseline elevation of ANA levels could be due to the prevalence of lupus susceptibility loci within some of the DO founder strains (C57BL/6, NOD, 129S1). This is consistent with the human population in which a significant portion will have positive ANA without disease (Satoh et al., 2012). However, despite the impressive work presented in this paper, the authors did not report loci or alleles governing susceptibility to either silicosis or silica-induced autoimmunity. This leaves the field open for future researchers to utilize the high phenotypic variation of the DO model to discover genetic loci involved in environmentally induced autoimmunity.
Without the need for large CC or DO screens, CC strains can also be individually investigated as models for autoimmune and inflammation-mediated diseases. During development of the CC strains, it was noted that CC011 was particularly prone to rectal prolapse, and upon necropsy, these mice consistently exhibited signs of colitis. Rogala et al. (2014) established the CC011 strain as a spontaneous colitis mouse model. By backcrossing CC011 with C57BL/6J, they performed high-density SNP mapping using the MegaMUGA genotyping array and identified four susceptibility loci that contributed to the severity of colitis. Interestingly, their analysis also showed that regions of residual heterozygosity in the CC011 strain have a variable impact on the phenotype, which is an important consideration when using CC models.
Screening of CC strains without QTL analysis has also been conducted for nonatopic asthma models. Two recent papers have shown strain-dependent response to O3 (Smith et al., 2021; Tovar et al., 2022). The authors then identified CC003 and CC039 as particularly susceptible to developing an inflammatory response to O3 compared to CC017. The authors performed ATAC-seq and RNA-seq on alveolar macrophages from CC017 and CC003 both pre- and post-O3 exposure and identified 80 genes with basal differences in chromatin accessibility correlated with expression of genes upon O3 exposure. This work highlights that CC strains can be used to tease out gene-environment interactions that depend on genetic background.
Overall, the CC and DO mouse populations have great unrealized potential for the study of autoimmune disease and could be a boon for discovering novel susceptibility loci and related genes/pathways, model development, and development and evaluation of new therapeutic interventions. There is a rich history of recombinant inbred mouse model use for autoimmunity (Vyse & Todd, 1996), and the expanded genetics of the CC and DO models may be useful in locus identification and causal gene discovery.
Cancer
The use of the DO and CC models has been expanding in the fields of cancer development, metastasis, and tumor immunology. Winter et al. (2017) describe a comprehensive study to examine host intrinsic genes that contribute to prostate cancer metastasis. Though the authors did not perform any evaluation of the immune compartment directly, they did lay out a roadmap of how DOF1 models can be utilized for cancer studies. After crossing DO mice with TRAMP mice to obtain a spontaneous prostate cancer model, they used QTL analysis to look for associations with metastasis in 493 DOF1 mice. Although they were unable to identify putative causal non-synonymous SNPs, they were able to utilize expression QTL (eQTL) mapping after performing RNAseq on 195 randomly selected tumors. Utilizing eQTL mapping, the authors identified multiple genes located within metastasis-associated genomic loci that had dysregulated expression. The eQTL results were supported with patient data showing a correlation of prostate cancer gene expression and patient outcome. Finally, the authors validated two of these genes (RWD Domain Containing 4 and Centromere Protein U), showing that their over-expression within prostate cancer cell lines enhanced their metastatic potential.
Immune-regulated development of spontaneous tumorigenesis has also been observed in a CC mouse screen. Wang et al. (2019) performed a screen for spontaneous tumor development in 18 CC lines. The authors found that of these lines, only five remained completely tumor-free over the study period of ~400 days (011, 026, 042, 051, 061). Most interestingly, three CC lines (013, 041, 036) showed a greater than 50% prevalence of spontaneous tumor generation, with thymic or splenic lymphomas being the most common among them and CC036 developing tumors in 100% of mice. The CC036 line also developed gastric tumors in ~40% of mice, with females exhibiting a higher frequency than males. The authors performed QTL analysis using gastric tumor frequency as the phenotype and conducted RNAseq on collected tumors. The QTL analysis indicated a locus on chromosome 3 that likely involves the NfκB gene. RNAseq and IHC on collected tumor tissue highlighted dysregulation of inflammation, particularly in CC036 mice, supporting NfκB as a driver of gastric tumor formation.
Genetic modifiers of polyp formation have also been similarly probed. To examine modifiers of polyp formation in a mouse Familial Adenomatous Polyposis model (Dorman et al., 2021), 49 CC lines were crossed to C57BL/6J-ApcMin/+. A large variation in polyp formation was seen among CC crosses as opposed to consistent detection within each strain. QTL mapping was performed using the polyp count in three regions of the small intestine and colon. Candidate genes were identified using QTL regions and the integrative multi-omics tools BioInfoMiner and Merge analysis.
A recent paper also describes the creation of a CCF1 model to study host regulation of immune response in a mesothelioma model. In this paper, the authors describe the creation of the model by crossing the CC lines with the MexTAg transgenic mouse, which develops mesothelioma upon asbestos exposure (Robinson et al., 2006). With this model, the authors aim to utilize genetic mapping and “omics” data to understand modifier genes influencing immune response to asbestos-induced mesothelioma (Behrouzfar et al., 2021).
F1 crosses with the DO model have also been employed to investigate mechanisms regulating response to cancer therapeutics. DO mice have been crossed with human HER2-transgenic mice to investigate germline loci influencing immune response to human HER2 vaccines (Wei et al., 2020). Susceptibility loci contributing to vaccine-induced anti-HER2 antibody response were identified. A locus shared between HER2-specific total IgG and IgG1 suggested that NK cells are a fundamental component of HER2 vaccine response.
An additional study has also been recently published utilizing DOF1 and CCF1 crosses with C57BL/6 mice to investigate the role of host genetics in therapeutic response to immune checkpoint inhibitors using implanted C57BL/6-syngeneic B16F0 melanoma tumors (Hackett et al., 2022). The use of a single tumor line is intended to limit tumor heterogeneity in this approach. This study shows evidence of germline genetic regulation of checkpoint inhibitor outcomes and multiple associated loci were identified, with further focus on the most prominent locus in chromosome 13. CCF1 crosses were used to validate the genetic drivers that were predicted based on QTL effects. This locus contains the murine prolactin family, and induction of mild hyperprolactinemia in combination with checkpoint inhibitors was shown to increase CD8 T cell infiltration in B16F0-bearing inbred C57BL/6 mice, which are resistant to checkpoint inhibitor therapy alone. The CCF1 models described in this is study may also serve to recapitulate specific immunotherapy response phenotypes with a syngeneic tumor.
Infectious Disease
The CC and DO mouse models have shown exceptional promise in identifying loci that contribute to our understanding of the genetic regulation of the differential outcomes to various infectious diseases caused by viral (influenza, SARS-CoV1, SARS-CoV2, Ebola, Zika, West Nile, TMEV, etc.), bacterial (Pseudomonas, Klebsiella pneumoniae, Salmonella enterica Typhimurium), mycobacterial (TB), fungal (Aspergillus fumigatus, Blastomyces dermatitidis), prion, protist (Plasmodium chabaudi), or helminth parasitic pathogens (S. venezuelensis) (Durrant et al., 2011; Graham et al., 2021; Green et al., 2016; Kamiya, Davis, Greischar, Schneider, & Mideo, 2021; Kohn et al., 2022; Lorè et al., 2020; Manet et al., 2020; Matsushita et al., 2021; Noll et al., 2020; Perez Gomez et al., 2021; Price et al., 2020; Schäfer et al., 2021; Smith et al., 2022; Vered, Durrant, Mott, & Iraqi, 2014; Xiong et al., 2014; Zhang et al., 2018).
The CC and DO panels have the potential to identify major genetic contributors to microbial pathogenesis and resistance, regulators of microbial burden, and/or mechanisms involved in host immune response. The use of the CC and DO mice to interrogate the genetic regulation of infection has been reviewed multiple times (Abu Toamih Atamni, Nashef, & Iraqi, 2018; Noll, Ferris, & Heise, 2019; Sarkar & Heise, 2019). Therefore, we will highlight applicable strategies and particularly impactful outcomes that illustrate the power of the CC and DO panels for modeling the variations seen in infectious phenotypes in the outbred human population. In addition, we will outline select research performed with the CC/DO panels in the interim since those reviews were published, and we will point out additional research areas where we believe the CC and DO have been underutilized but could be leveraged to gain new insight. Although the CC panel continues to be employed to study SARS-CoV2, due to its significant impact on global health, other infections have also been the focus of recent CC research, including advances in the study of mCMV, TMEV, Borrelia, and Pseudomonas, where each study has illustrated creative and useful applications of the CC panel.
NOVEL MODELS FOR CC MICE
The memory NK response to mCMV infection was examined by testing 34 CC lines (Jensen, Martin, Tripathy, & Badovinac, 2022). B6 mice are difficult to use for induction of memory NK responses because they have a high frequency of cells expressing LY49H, the marker of memory NK cells. The authors screened for a CC strain that retained expression of Ly49H at a high level but at a lower frequency, thus allowing the upregulation of the number of memory NK cells to be effectively studied. They confirmed this following mCMV infection, and thus a new effective model for the mechanistic study of NK memory was produced.
A small study interrogated 12 inbred CC strains and 2 CCF1 crosses to investigate infection-associated neurological impairment in Theiler’s murine encephalomyelitis virus (TMEV) infection (Perez Gomez et al., 2021). Viral burden, neurological function (paralysis scaling and postural analysis), and immune response (chemokine and cytokine production) were measured, which identified CC005, CC017, and CC023 as novel models for studying the genetic influences of infection-induced CNS manifestations. Even though these studies were too small to provide any substantive genetic information, these new models may lay the groundwork for an expansion in our knowledge of how viral infections can lead to CNS disease in a genetic background–regulated manner. They also compensate for the lack of CNS phenotypic diversity expressed in pre-existing inbred models of multiple sclerosis, amyotrophic lateral sclerosis, and Parkinson’s disease.
The CC was also recently leveraged to generate a novel small animal model for louse-borne relapsing fever (LBRF), a disease reportedly restricted to humans with no known immunocompetent animal models (Rogovskyy, Rogovska, Taylor, Wiener, & Threadgill, 2021). This understudied disease is a great burden to refugee populations throughout the world, causing high mortality rates. LBRF, caused by the spirochete Borrelia recurrentis, has no known animal reservoir and its vector, the human lice tick (Pediculus humanus), has a limited or an exclusive human host range. Therefore, LBRF is considered a human pathogen with no reasonable immunocompetent small animal model system, which limits comprehensive investigation. By screening 11 CC mouse lines, the researchers discovered that CC strain CC046 showed evidence of B. recurrentis spirochetemia for up to 3 days after inoculation, and CC strain CC058 harbored low, variable levels of spirochete burden for up to 10 days. F1 mice derived from a cross of the CC046 × CC058 lines yielded mice with reproducible infectious loads for 10 days. These results were of sufficient duration for them to detect initial IgM humoral responses to Borrelia. Therefore, the CC panel facilitated the development of a mouse model that will facilitate vaccination studies and therapeutic modalities pertaining to this poorly understood microbe.
In a more classical use of the CC panel to map susceptibility loci, 39 of the CC mouse lines were tested to identify loci that alter the severity of Pseudomonas aeruginosa respiratory infection (Lorè et al., 2020). Survival QTLs associated with the age of initial pseudomonas infection were identified in Dpyd and S1pr1, an observation made clinically relevant by the presence of SNPs in cystic fibrosis (CF) patients within this extended locus. Identifying genetic modifiers in CF patients using classical GWAS techniques has been hampered by the large effect size of CFTR mutations that swamp the effect of modifier loci unless unreasonably large cohorts are studied. This research highlights the strength of the CC system to identify genetic modifiers of infectious phenotypes that cannot be effectively interrogated with other genetic approaches (e.g., GWAS, simple 2 × 2 crosses). In another example of this strength, Maurizio and colleagues used strategies to reveal the loci that have modest effects on the infectious phenotype by controlling for the effects of the Mx1 locus, which has very a large effect size on influenza phenotypes (Maurizio et al., 2018). This may also be applicable in studying OasIb in West Nile infections, IFN pathway genes in mycobacterial MSMD infections, and MHC in multiple infectious settings, among others. Therapeutic strategies have also been used in conjunction with the CC mouse panel to test the influence of the genetic background by removing the effect of a dominant mouse-specific effector pathway to model human viral replication with greater fidelity (Manet et al., 2020). In their work, Manet et al. treated CC mice with monoclonal antibodies to IFNAR, allowing for a sufficient Zika viral load to study symptomatic Zika virus infection. They identified multiple CC strains with extreme differences in plasma viral load, as well as provided novel models for Zika-induced brain pathology.
Interestingly, infectious models using the CC mice have even revealed the host malleability and genetic regulation of the host transcriptome, in addition to the aspects of the infectious model itself. The Katze group showed that influenza and SARS-CoV-1 can induce unique host transcripts. Each mouse background expressed a unique subset of alternatively spliced transcripts in response to infection with the same pathogen. In addition, these infections revealed virus-induced host transcripts yet to be annotated in the genome that also vary with the host genetic background (Xiong et al., 2014). These regions of the genome, including but not limited to those that code for long non-coding RNAs (lncRNAs), have now been shown to be under genetic regulation as interferon-regulated expression modules with considerable effect on infectious outcomes (Josset et al., 2014; Landeras-Bueno & Ortín, 2016; More et al., 2019; Yang, Lin, Wang, Zeng, & Luo, 2021).
COMBINING THE CC/DO MICE WITH OTHER GENETIC TOOLS TO STUDY INFECTIONS
Determining the effect of heterozygosity using CC or DO mice crossed with a susceptible or resistant recombinant inbred line (outside of the parental CC panel) to study gene dosage effects, recessive phenotypes, or dominant negative alleles is also a great strength of the system, as is crossing the CC or DO mice to transgenic or CRISPR/Cas modified mice. CC recombinant inbred inter-crosses (CC RIX) are CCF1 mice that result in outbred mice with genomic heterozygosity like the DO mice, but they allow researchers to duplicate the resulting CCF1 mice by re-mating. This feature lets researchers focus on specific allele mixtures or avoid those alleles that confound further analysis (e.g., controlling for MHC variation). Graham, et al. (2021) recently utilized CCF1 mice to identify that the baseline signature of CD4+ T cells has predictive capacity for viral load and disease severity in SARS-CoV2 infections. In support of this finding, immune cell phenotypes in the regulatory CD4+ T cell compartment have been appreciated in human patients with coronavirus infections as well (Vick et al., 2021). Another omics-based tool to predict infectious outcomes was recently developed using DO mice that demonstrated high tuberculosis susceptibility. Tavolara et al. (2021) used the heterogenous susceptibility of DO mice to tuberculosis to test a potential diagnostic/prognostic tool. This was accomplished by performing deep learning on whole slide scans from hematoxylin and eosin (H&E)–stained lung tissue correlated to gene micro-array results. They found that they could create a model using a standard tissue strain that was predictive to disease outcomes. This highlights the potential for creative use of the DO system to design and test clinical tools that rely on a diverse range of outcomes.
FUTURE OF INFECTIONS IN THE DO/CC SYSTEMS
Although these studies have been fruitful, the CC and DO mice have the potential for identifying the mechanisms of genetic control in a range of host pathogen interactions ranging from commensalism, symbiosis, and mutualism to colonization, pathogenesis, latency, and chronicity. The CC has the potential to elucidate host genetic factors that counter microbial-derived immune-evasion strategies that alter the infectious outcomes that emanate from the host-microbe interface. One key area where the CC can be leveraged is in determining the genetic factors that allow for differentiating between host responses to pathogenic E coli and E coli that are part of the normal gut flora as well as tolerance induction to other gut, skin, oral, and vaginal flora commensals. Other key areas of research where the CC/DO panels can be useful include investigations of gut dysbiosis and the alteration of immunotherapy efficacy observed in probiotic consumption. In addition, latency, chronicity, and reactivation are critical areas of study that can be aided by the CC and DO panels for studying multiple microbes from Adenoviruses, Retroviruses, Herpes viruses (Chicken pox, HSV-1, HSV-2, HSV-3/varicella/Chickenpox/Shingles, EBV/HHV-4, CMV/HHV-5, HSV-6, HHV-7, Kaposi Sarcoma/HHV8), papovaviruses (BK & JC viruses), papillomaviruses (HPV), chronic hepatic infections (HBV, HCV, etc.), parvovirus, as well as fungal, bacterial, and parasitic latency and reactivation (e.g., Toxoplasma gondii, with particular focus on the genetic control of toxo-induced behavioral changes).
Another understudied aspect of chronic infection is the genetic control of granuloma formation and integrity that could be studied using the CC panel. Other fruitful avenues of investigation include microbial induction of autoimmunity (molecular mimicry), tissue destruction and cytokine syndromes, as well as models of infectious alteration of autoimmunity (e.g., segmented filamentous bacteria and arthritis). Microbial-induced oncological (EBV-Burkitt lymphoma, HTLV-T-cell malignancy, KSHV, HBV/HCV-HCC, HPV, HIV, and Merkel Cell polyomavirus) and inflammatory states (CMV and colitis, RSV and asthma, allergy), dissemination and sepsis, vaccine efficacy, age-induced frailty and infection susceptibility, aging effect on vaccine efficacy, antibiotic and antiviral efficacy, microbial influences on shaping the immune repertoire, genetic control of susceptibility to secondary infections, therapeutic immunosuppression-induced microbial susceptibility (autoimmunity and oncology therapeutics), modeling of zoonotic transfer from other hosts, identifying genetic factors that govern longitudinal transfer of infection (maternal transfer) and sexually transmitted diseases, and developing models of PRION disease. The list of applications for using the CC/DO mouse panels is nearly endless, ensuring that these robust genetic tools will be paying research dividends for years to come.
Utilization of the DO and CC Mouse Models
Diving into the CC and DO mouse models is at first glance a seemingly overwhelming jump that requires well-designed experiments, relatively large numbers of animals, and use of computational biology tools. We hope to ease some of the stress of entering this realm with an introduction to the tools required to analyze the genetic data provided by the animals. As mentioned above, the R/qtl2 package developed by Broman et al. (2019) is the primary software package that is currently maintained and has a robust and an active community of members willing to assist users of all levels.
We can make no specific recommendations of computers to use because that will largely be dependent on the size of the dataset. However, some steps in the process can utilize a significant amount of computer memory (RAM) and CPU resources. We would advise using a machine with no less than 8 GB of RAM and at least a modern quad-core processor. All packages are compatible with Windows, MacOS, or Linux-based operating systems.
Before any analysis can be done, you must install the statistical program “R.” The appropriate version to download can be found on the R-Project webpage (https://cran.r-project.org/). Though not necessary, it is highly recommended to also install RStudio, which adds a friendly user interface to R and allows for more intuitive navigation (https://www.rstudio.com/products/rstudio/). All commands can be used without having extensive knowledge of the R programming language, but troubleshooting is much easier with basic R knowledge; therefore, we recommend becoming generally familiar with the R language. There are many resources that can be found for learning the basics of R, but the R package Swirl is free and can provide the user with basic and advanced lessons in utilizing R (https://swirlstats.com/). Finally, the R/qtl2 and R/qtl2convert packages must be installed within R. Installation of the R packages can be performed using the command line or the user interface.
After setting up R and installing the qtl2 and qtl2convert packages, you must determine the appropriate steps for your data analysis. For more detailed explanations of data types that can be used with the qtl2 package, refer to Karl Broman’s qtl2 page on R/qtl2 input file format (https://kbroman.org/qtl2/assets/vignettes/input_files.html). CC and DO experiments are analyzed differently within the qtl2 package, but they unsurprisingly have many similarities. In both CC and DO experiments, the researcher must have the SNP genotypes from founder stains and experimental animals. Fortunately for researchers entering this field, the CC and DO community has deposited founder data in data repositories such as GitHub and figshare.
The CC/DO founder genotypes have been deposited here and have already been processed for direct input into R/qtl2 (https://figshare.com/articles/dataset/GM_processed_files_zip/5404759) (Broman et al., 2019). Analysis of QTL from both CC mice and DO mice without crosses require founder genotypes of the DO founders. For DO mice, the generation number is required for estimation of the haplotype block size and must be supplied in an additional covariate file. The generation number is tracked by Jackson Labs and is supplied with each litter release, and it can also be found on the Jackson Labs website, Breeding Wave (https://www.jax.org/jax-mice-and-services/colony-management/breeding/009376-breeding-wave).
During the creation of the control file, the cross type must be supplied. For DO mice, the “cross type” is “do” and for CC mouse experiments, the “cross type” is “riself8,” as the CC lines are recombinant inbred lines with 8 founders. Additionally important for the analysis of CC data is the specification of the breeding funnel that indicates how the crosses were generated. The CC/DO community has provided the funnel codes and genotyping for the CC lines in a GitHub repository (https://github.com/rqtl/qtl2data/tree/main/CC), derived from Srivastava et al. (2017).
If you are performing F1 cross with the DO mice, you must additionally supply the cross genotypes. This is done by creating a 9th column in the founder files and pasting the genotype calls. If this is a backcross to one of the DO founder strains, you can simply copy this from within the founder files. If this is not a backcross, then you must perform genotyping on the cross strain and obtain the founder genotypes using GigaMUGA markers from your cross strain. This can be accomplished by encoding the genotypes utilizing the same steps necessary for processing experimental DO samples. Karl Broman provides detailed steps and an R script here (https://kbroman.org/qtl2/pages/prep_do_data.html) under “Encoding the DO genotypes.” The result will be a series of CSV files for each chromosome with markers and genotype coded as “A” or “B.” Then, like the backcross, add the new column as the 9th column in the founder genotypes (Fig. 2).
Figure 2.
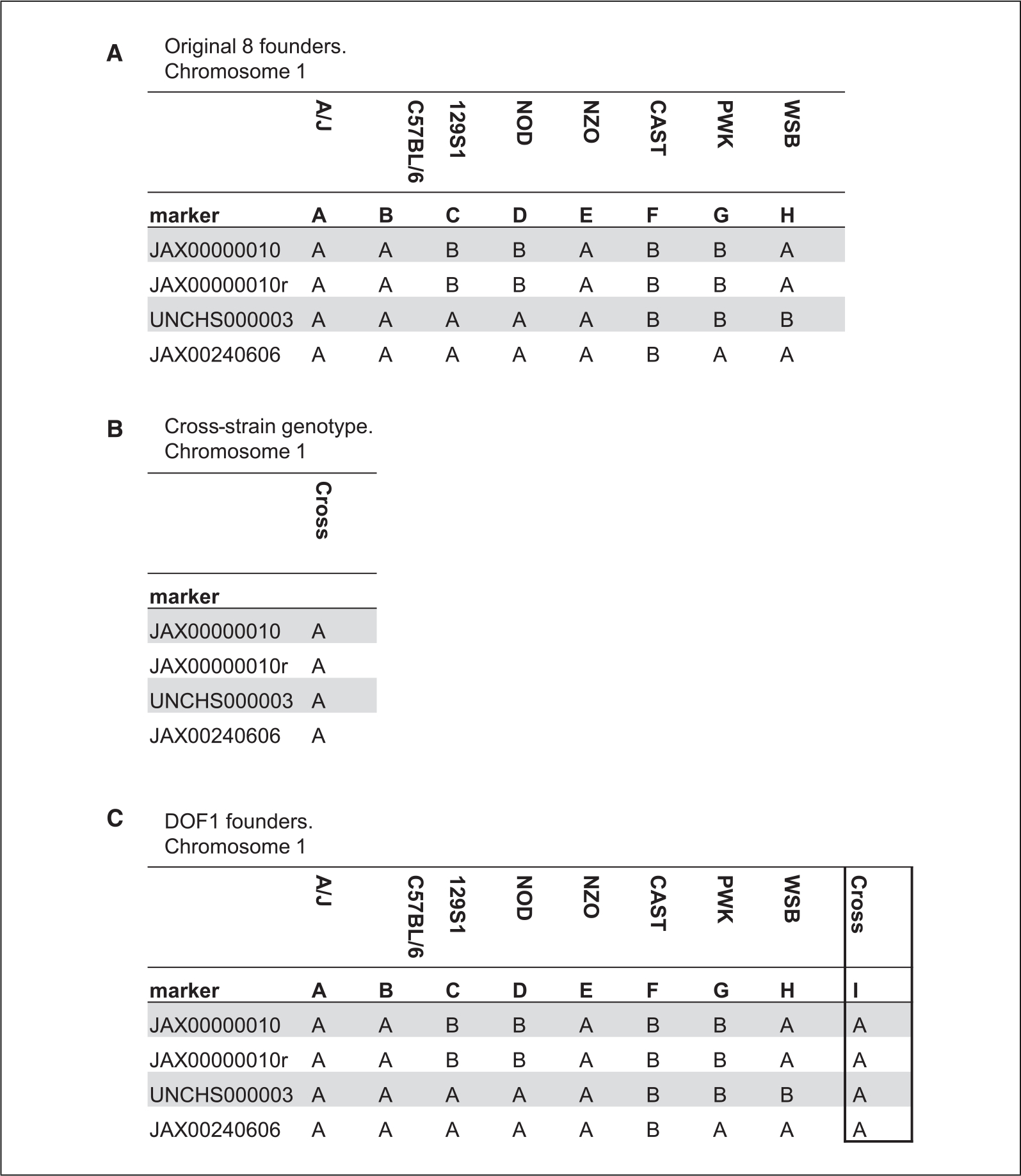
Conceptual demonstration of creation of DOF1 founder files. (A) First 5 genotyped markers of chromosome 1 from the CC/DO founders. (B) First 5 genotyped markers of potential cross strain (C57BL/6). (C) Appearance of first 5 markers in final chromosome 1 file with cross strain as new column “I”. Strain labels added for clarity.
Once genotyping data are correctly formatted and the control file is completed, the commands and steps for performing the analysis are described in detail in Karl Broman’s R/qtl2 user guide (https://kbroman.org/qtl2/assets/vignettes/user_guide.html). The CC/DO community is also very active in helping users troubleshoot in a Google discussion group (https://groups.google.com/forum/#!forum/rqtl2-disc).
Analyzing CC/DO Data on CyVerse
Adopting the CC or DO mice as an experimental model necessitates the use of complex computational and bioinformatic tools that continue to evolve and improve over time. Although experimental benchwork is typically a skill that immunology investigators have acquired during their training, they often lack the computational training needed to adopt complex genetic panels like the CC/DO, and this represents a sizable challenge for many research groups. To minimize the impact of this often daunting task, we have generated an extensible CC/DO computational community within the NSF-funded CyVerse cyberinfrastructure that circumvents the need to navigate complex software installations and the ever-changing software dependencies needed to install and maintain software (Goff et al., 2011; Merchant et al., 2016). Essential genome reference files for analyzing CC data have also been centralized on the CyVerse platform. Placing the CC/DO tool community on CyVerse allows for growth and evolution of resources so that researchers can keep pace with the inevitable software changes that will occur in the field. Extensive high-performance computing (HPC) resources and high-capacity data storage that would be out of reach of many researchers are now easily accessible. The platform provides the ability to manage data security with different access privileges for different members of the research group, which facilitates safe and effective collaboration both in large and small genetics groups and among complex interdisciplinary teams. Researchers can integrate currently available software packages into dynamic pipelines and customized workflows that can take CC and DO data from quality control to analysis and visualization and to publication-ready figures using CyVerse’s DNA Subway. In addition, all CC parental genomes have been incorporated into the CyVerse-associated Comparative Genomics (CoGe) interface for complex comparisons of genomes as well as visualization of genome variation to allow for robust hypothesis testing (Lyons & Freeling, 2008). The platform also allows for comparison of the CC genomes to other genomes to investigate evolutionary correlates and understand synteny of the locus of interest across species, especially when considering disease-relevant mouse-to-man comparisons (Haug-Baltzell, Stephens, Davey, Scheidegger, & Lyons, 2017). The intuitive CoGe interface builds upon, rather than supplants, the existing CC visualization tools like the University of North Carolina’s Collaborative Cross Viewer (http://csbio.unc.edu/CCstatus/index.py?run=CCV) and the Collaborative Cross Graphical Genome (CCGG) (https://devel.csbio.unc.edu/GraphicalGenome/index.html), as well as prior efforts to democratize and simplify the computational aspects of CC projects that include the gQTL web interface (Vered et al., 2014), Churchill’s QTL Viewer (https://churchilllab.jax.org), the Diversity Outbred Database (DODB) (https://dodb.jax.org), and the Mouse Phenome Database (https://phenome.jax.org) at the Jackson Laboratories.
For researchers that require upscaling of computer resources, when HPC needs of additional memory or CPU capacity outstrip a researcher’s current availability, the CyVerse platform offers access to Atmosphere/Jetstream2 cloud computing through the collaborative Extreme Science and Engineering Discovery Environment (XSEDE) portal and the resources of the Texas Advanced Computing Center (TACC) at the University of Texas. All the research input data and output analysis files are stored in the Data Commons area of CyVerse in repositories that can be private, shared among a limited set of investigators, or made publicly accessible. Hundreds of bioinformatics packages are available in the App section of CyVerse; however, key CC-specific software packages like R/qtl2, complete with the availability of onboard RStudio, are accessible in the CC Collection and other CC researchers can be found in the CC Teams sections of CyVerse. Detailed screen grabs showing how to get started using R/qtl2 in CyVerse can be found in the supplement (Fig. S1). Extensive support and educational resources are available on the CyVerse platform, with dedicated personnel tasked to facilitate the research of investigators, including curated guides, manuals, tutorials, webinars, coursework, wiki educational resources, blogs, and direct assistance by trained informaticists (e.g., CoGe Pedia https://genomevolution.org/wiki/index.php/Main_Page).
Studying complex genetics with the powerful DO mouse panel provides the additional challenge of SNP mapping of each mouse studied. GigaMUGA, an Illumina/Neogen Infinium array, provides SNP genotypes of outbred DO mice. Densely mapping SNPs allows researchers to determine the haplotypes of the DO mice they are studying. In other words, the arrays allow researchers to identify the regions of the genomes from each parental mouse that were inherited by each unique DO mouse. Although the GenomeStudio and Beeline software packages are available from Illumina, the HaploQA software is commonly used to infer haplotypes of DO mice (web interface at Jax https://haploqa.jax.org by Keith Sheppard and Dr Gary Churchill) (Morgan et al., 2016; Powers et al., 2020), by running the Python3 program locally (https://github.com/TheJacksonLaboratory/haploqa), or through incorporation into the CyVerse CC Collection.
CONCLUSIONS
The DO and CC mouse populations have shown they are powerful tools to deepen our understanding of the genetic regulation of complex traits and tease out gene-environment interactions. We have described a relatively small body of literature showing the potential these models have to study the immune system in the settings of infection, autoimmunity/inflammation, cancer, and immune development. Within these broad fields, the CC and DO mouse models have primarily served two purposes: identifying genes influencing complex traits and discovering models of disease processes. In the setting of inflammatory diseases and infection, the discovery of this model has been particularly fruitful. Utilizing CC screens, researchers have discovered non-primate models of viral infection and spontaneous models of inflammatory disease. The creation or discovery of these models serves a dual purpose in which researchers can use the models to explore pathology and treatment or discover the genetic underpinnings of the observed traits. We found that within the field of cancer immunology, researchers are increasingly making use of F1 crosses of either CC or DO mice. The F1 models are being used to study spontaneous, implanted, and induced cancers. The models consistently show large variation in measured phenotypes and have led to the identification of genetic loci contributing to the observed phenotype. Immune development and autoimmunity are fields that have particularly underutilized these models. Despite well-known genetic linkages to development and susceptibility in autoimmune disorders, we found limited literature describing QTL analysis in this field. Immune development was also surprisingly understudied in these models. As discussed above, both the CC/DO founder strains and individual CC lines show significant diversity in baseline immune phenotypes. However, there has been little work on determining the genetic regulation of how these differences arise.
Our review has demonstrated that the CC and DO mouse lines have already resulted in impactful discoveries of genetic regulation of immune processes and discovery of novel disease models. We also have highlighted that the CC/DO community is active and welcoming to new researchers and has provided a direction toward making this powerful genetic tool more accessible. The sections for utilizing the models either through the CyVerse system or on a local machine will help reduce difficulties in transitioning to this model and direct the users to the resources that they may need to conduct successful studies.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr Andrew Nelson (Cornell), Dr Tyson Swetnam (UA), and Amanda Cooksey for facilitating the incorporation of the Collaborative Cross genomes and tools into the CyVerse environment, which was made possible through CyVerse’s External Collaborative Partnership program. This material is based upon work supported by the National Science Foundation under award numbers DBI-0735191, DBI-1265383, and DBI-1743442. URL: www.cyverse.org. We received funding from Praespero, an autoimmune research fund (AB), NIH R37 CA220482 (HMG) and NIH T32 CA009531, the RL Kirschstein National Research Service Award Training Grant (JBH).
Footnotes
CONFLICT OF INTEREST
Authors have no conflicts of interest to disclose.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article, as no datasets were generated or analyzed during the current study.
LITERATURE CITED
- Abu Toamih Atamni H, Nashef A, & Iraqi FA (2018). The Collaborative Cross mouse model for dissecting genetic susceptibility to infectious diseases. Mammalian Genome, 29, 471–487. doi: 10.1007/s00335-018-9768-1 [DOI] [PubMed] [Google Scholar]
- Anderson MS, & Bluestone JA (2005). The NOD mouse: A model of immune dysregulation. Annual Review of Immunology, 23, 447–485. doi: 10.1146/annurev.immunol.23.021704.115643 [DOI] [PubMed] [Google Scholar]
- Bailey DW (1971). Recombinant-inbred strains: An aid to finding identity, linkage, and function op histocompatibility and other genes. Transplantation, 11, 325–327. doi: 10.1097/00007890-197103000-00013 [DOI] [PubMed] [Google Scholar]
- Behrouzfar K, Burton K, Mutsaers SE, Morahan G, Lake RA, & Fisher SA (2021). How to better understand the influence of host genetics on developing an effective immune response to thoracic cancers. Frontiers in Oncology, 11, 679609. doi: 10.3389/fonc.2021.679609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman KW, Gatti DM, Simecek P, Furlotte NA, Prins P, Sen Ś, … Churchill GA (2019). R/qtl2: Software for mapping quantitative trait loci with high-dimensional data and multiparent populations. Genetics, 211, 495–502. doi: 10.1534/genetics.118.301595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman KW, Wu H, Sen S, & Churchill GA (2003). R/qtl: QTL mapping in experimental crosses. Bioinformatics, 19, 889–890. doi: 10.1093/bioinformatics/btg112 [DOI] [PubMed] [Google Scholar]
- Chattopadhyay SK, Rowe WP, Teich NM, & Lowy DR (1975). Definitive evidence that the murine C-type virus inducing locus Akv-1 is viral genetic material. Proceedings of the National Academy of Sciences of the United States of America, 72, 906–910. doi: 10.1073/pnas.72.3.906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J, … Complex Trait Consortium. (2004). The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nature Genetics, 36, 1133–1137. doi: 10.1038/ng1104-1133 [DOI] [PubMed] [Google Scholar]
- Collaborative Cross Consortium. (2012). The genome architecture of the Collaborative Cross mouse genetic reference population. Genetics, 190, 389–401. doi: 10.1534/genetics.111.132639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin R, Balmer L, Morahan G, & Lesage S (2019). Common heritable immunological variations revealed in genetically diverse inbred mouse strains of the collaborative cross. Journal of Immunology, 202, 777–786. doi: 10.4049/jimmunol.1801247 [DOI] [PubMed] [Google Scholar]
- Dorman A, Binenbaum I, Abu-Toamih Atamni HJ, Chatziioannou A, Tomlinson I, Mott R, & Iraqi FA (2021). Genetic mapping of novel modifiers for ApcMin induced intestinal polyps’ development using the genetic architecture power of the collaborative cross mice. BMC Genomics, 22, 566. doi: 10.1186/s12864-021-07890-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont MSJ, Guillemot V, Campagne P, Serafini N, Marie S, Montagutelli X, … Vosshenrich CAJ (2021). Host genetic control of natural killer cell diversity revealed in the Collaborative Cross. Proceedings of the National Academy of Sciences of the United States of America, 118, e2018834118. doi: 10.1073/pnas.2018834118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant C, Tayem H, Yalcin B, Cleak J, Goodstadt L, de Villena FP-M, … Iraqi FA (2011). Collaborative Cross mice and their power to map host susceptibility to Aspergillus fumigatus infection. Genome Research, 21, 1239–1248. doi: 10.1101/gr.118786.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festing MFW (2014). Evidence should trump intuition by preferring inbred strains to outbred stocks in preclinical research. ILAR Journal, 55, 399–404. doi: 10.1093/ilar/ilu036 [DOI] [PubMed] [Google Scholar]
- Ford JW, Sturgill JL, & Conrad DH (2009). 129/SvJ mice have mutated CD23 and hyper IgE. Cellular Immunology, 254, 124–134. doi: 10.1016/j.cellimm.2008.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff SA, Vaughn M, McKay S, Lyons E, Stapleton AE, Gessler D, … Stanzione D (2011). The iPlant collaborative: Cyberinfrastructure for plant biology. Frontiers in Plant Science, 2, 34. doi: 10.3389/fpls.2011.00034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham JB, Swarts JL, Leist SR, Schäfer A, Menachery VD, Gralinski LE, … Lund JM (2021). Baseline T cell immune phenotypes predict virologic and disease control upon SARS-CoV infection in Collaborative Cross mice. PLoS Pathogens, 17, e1009287. doi: 10.1371/journal.ppat.1009287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham JB, Swarts JL, Mooney M, Choonoo G, Jeng S, Miller DR, … Lund JM (2017). Extensive homeostatic T cell phenotypic variation within the collaborative cross. Cell Reports, 21, 2313–2325. doi: 10.1016/j.celrep.2017.10.093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green R, Wilkins C, Thomas S, Sekine A, Ireton RC, Ferris MT, … Gale M (2016). Identifying protective host gene expression signatures within the spleen during West Nile virus infection in the collaborative cross model. Genome Data, 10, 114–117. doi: 10.1016/j.gdata.2016.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett JB, Glassbrook JE, Muñiz MC, Bross M, Fielder A, Dyson G, … Gibson HM (2022). A diversity outbred F1 mouse model identifies host-intrinsic genetic regulators of response to immune checkpoint inhibitors. OncoImmunology, 11, 2064958. doi: 10.1080/2162402X.2022.2064958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane JB, & Waddington CH (1931). Inbreeding and linkage. Genetics, 16, 357–374. doi: 10.1093/genetics/16.4.357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haug-Baltzell A, Stephens SA, Davey S, Scheidegger CE, & Lyons E (2017). SynMap2 and SynMap3D: Web-based whole-genome synteny browsers. Bioinformatics, 33, 2197–2198. doi: 10.1093/bioinformatics/btx144 [DOI] [PubMed] [Google Scholar]
- Jansen PR, Watanabe K, Stringer S, Skene N, Bryois J, Hammerschlag AR, … Posthuma D (2019). Genome-wide analysis of insomnia in 1,331,010 individuals identifies new risk loci and functional pathways. Nature Genetics, 51, 394–403. doi: 10.1038/s41588-018-0333-3 [DOI] [PubMed] [Google Scholar]
- Jensen IJ, Martin MD, Tripathy SK, & Badovinac VP (2022). Novel mouse model of murine cytomegalovirus-induced adaptive NK cells. Immunohorizons, 6, 8–15. doi: 10.4049/immunohorizons.2100113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji DX, Witt KC, Kotov DI, Margolis SR, Louie A, Chevée V, … Vance RE (2021). Role of the transcriptional regulator SP140 in resistance to bacterial infections via repression of type I interferons. Elife, 10, e67290. doi: 10.7554/eLife.67290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josset L, Tchitchek N, Gralinski LE, Ferris MT, Eisfeld AJ, Green RR, … Katze MG (2014). Annotation of long non-coding RNAs expressed in collaborative cross founder mice in response to respiratory virus infection reveals a new class of interferon-stimulated transcripts. RNA Biology, 11, 875–890. doi: 10.4161/rna.29442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya T, Davis NM, Greischar MA, Schneider D, & Mideo N (2021). Linking functional and molecular mechanisms of host resilience to malaria infection. Elife, 10, e65846. doi: 10.7554/eLife.65846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaky M, Fedetz M, Potenciano V, Andrés-León E, Codina AE, Barrionuevo C, … Matesanz F (2018). SP140 regulates the expression of immune-related genes associated with multiple sclerosis and other autoimmune diseases by NF-κB inhibition. Human Molecular Genetics, 27, 4012–4023. doi: 10.1093/hmg/ddy284 [DOI] [PubMed] [Google Scholar]
- Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, … Adams DJ (2011). Mouse genomic variation and its effect on phenotypes and gene regulation. Nature, 477, 289–294. doi: 10.1038/nature10413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn EM, Taira C, Dobson H, Dias LDS, Okaa U, Wiesner DL, … Klein BS (2022). Variation in host resistance to blastomyces dermatitidis: Potential use of genetic reference panels and advances in immunophenotyping of diverse mouse strains. mBio, 13, e0340021. doi: 10.1128/mbio.03400-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landeras-Bueno S, & Ortín J (2016). Regulation of influenza virus infection by long non-coding RNAs. Virus Research, 212, 78–84. doi: 10.1016/j.virusres.2015.08.008 [DOI] [PubMed] [Google Scholar]
- Lee JJ, Wedow R, Okbay A, Kong E, Maghzian O, Zacher M, … Cesarini D (2018). Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nature Genetics, 50, 1112–1121. doi: 10.1038/s41588-018-0147-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little CC, & Tyzzer EE (1916). Further experimental studies on the inheritance of susceptibility to a transplantable tumor, carcinoma (JWA) of the Japanese waltzing mouse. The Journal of Medical Research, 33, 393–453. [PMC free article] [PubMed] [Google Scholar]
- Lorè NI, Sipione B, He G, Strug LJ, Atamni HJ, Dorman A, … Bragonzi A (2020). Collaborative cross mice yield genetic modifiers for pseudomonas aeruginosa infection in human lung disease. mBio, 11, e00097–20. doi: 10.1128/mBio.00097-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons E, & Freeling M (2008). How to usefully compare homologous plant genes and chromosomes as DNA sequences. Plant Journal, 53, 661–673. doi: 10.1111/j.1365-313X.2007.03326.x [DOI] [PubMed] [Google Scholar]
- Manet C, Simon-Lorière E, Jouvion G, Hardy D, Prot M, Conquet L, … Montagutelli X (2020). Genetic diversity of collaborative cross mice controls viral replication, clinical severity, and brain pathology induced by zika virus infection, independently of Oas1b. Journal of Virology, 94, e01034–19. doi: 10.1128/JVI.01034-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita K, Li X, Nakamura Y, Dong D, Mukai K, Tsai M, … Galli SJ (2021). The role of Sp140 revealed in IgE and mast cell responses in Collaborative Cross mice. JCI Insight, 6, e146572. doi: 10.1172/jci.insight.146572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzaraki V, Kumar V, Wijmenga C, & Zhernakova A (2017). The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biology, 18, 76. doi: 10.1186/s13059-017-1207-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurizio PL, Ferris MT, Keele GR, Miller DR, Shaw GD, Whitmore AC, … Valdar W (2018). Bayesian diallel analysis reveals Mx1-dependent and Mx1-independent effects on response to influenza A virus in mice. G3 (Bethesda), 8, 427–445. doi: 10.1534/g3.117.300438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayeux JM, Escalante GM, Christy JM, Pawar RD, Kono DH, & Pollard KM (2018). Silicosis and silica-induced autoimmunity in the diversity outbred mouse. Frontiers in Immunology, 9, 874. doi: 10.3389/fimmu.2018.00874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchant N, Lyons E, Goff S, Vaughn M, Ware D, Micklos D, & Antin P (2016). The iPlant collaborative: Cyberinfrastructure for enabling data to discovery for the life sciences. Plos Biology, 14, e1002342. doi: 10.1371/journal.pbio.1002342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA (2016). Not your father’s, or mother’s, rodent: Moving beyond B6. Neuron, 91, 1185–1186. doi: 10.1016/j.neuron.2016.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA, Austad S, Burke D, Chrisp C, Dysko R, Galecki A, … Monnier V (1999). Exotic mice as models for aging research: polemic and prospectus. Neurobiology of Aging, 20, 217–231. doi: 10.1016/s0197-4580(99)00038-x [DOI] [PubMed] [Google Scholar]
- More S, Zhu Z, Lin K, Huang C, Pushparaj S, Liang Y, … Liu L (2019). Long non-coding RNA PSMB8-AS1 regulates influenza virus replication. RNA Biology, 16, 340–353. doi: 10.1080/15476286.2019.1572448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AP, Fu C-P, Kao C-Y, Welsh CE, Didion JP, Yadgary L, … de Villena FP-M (2016). The mouse universal genotyping array: From substrains to subspecies. G3 Genes|Genomes|Genetics, 6, 263–279. doi: 10.1534/g3.115.022087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noll KE, Ferris MT, & Heise MT (2019). The Collaborative Cross: A systems genetics resource for studying host-pathogen interactions. Cell Host & Microbe, 25, 484–498. doi: 10.1016/j.chom.2019.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noll KE, Whitmore AC, West A, McCarthy MK, Morrison CR, Plante KS, … Heise MT (2020). Complex genetic architecture underlies regulation of influenza-A-virus-specific antibody responses in the Collaborative Cross. Cell Reports, 31, 107587. doi: 10.1016/j.celrep.2020.107587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orgel K, Smeekens JM, Ye P, Fotsch L, Guo R, Miller DR, … Kulis MD (2019). Genetic diversity between mouse strains allows identification of the CC027/GeniUnc strain as an orally reactive model of peanut allergy. Journal of Allergy and Clinical Immunology, 143, 1027–1037.e7. doi: 10.1016/j.jaci.2018.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeva E, Gonzalez J, Hicks R, & Diamond B (2006). Cutting edge: Lupus susceptibility interval Sle3/5 confers responsiveness to prolactin in C57BL/6 mice. Journal of Immunology, 177, 1401–1405. doi: 10.4049/jimmunol.177.3.1401 [DOI] [PubMed] [Google Scholar]
- Peeva E, Michael D, Cleary J, Rice J, Chen X, & Diamond B (2003). Prolactin modulates the naive B cell repertoire. Journal of Clinical Investigation, 111, 275–283. doi: 10.1172/JCI200316530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez Gomez AA, Karmakar M, Carroll RJ, Lawley KS, Amstalden K, Young CR, … Brinkmeyer-Langford C (2021). Genetic and immunological contributors to virus-induced paralysis. Brain, Behavior, & Immunity - Health, 18, 100395. 10.1016/j.bbih.2021.100395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillippi J, Xie Y, Miller DR, Bell TA, Zhang Z, Lenarcic AB, … Frelinger JA (2014). Using the emerging Collaborative Cross to probe the immune system. Genes and Immunity, 15, 38–46. doi: 10.1038/gene.2013.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers NR, Dumont BL, Emori C, Lawal RA, Brunton C, Paigen K, … Bhattacharyya T (2020). Sexual dimorphism in the meiotic requirement for PRDM9: A mammalian evolutionary safeguard. Science Advances, 6, eabb6606. doi: 10.1126/sciadv.abb6606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price A, Okumura A, Haddock E, Feldmann F, Meade-White K, Sharma P, … Rasmussen AL (2020). Transcriptional correlates of tolerance and lethality in mice predict ebola virus disease patient outcomes. Cell Reports, 30, 1702–1713.e6. doi: 10.1016/j.celrep.2020.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard-Miceli C, & Criswell LA (2012). Emerging patterns of genetic overlap across autoimmune disorders. Genome Medicine, 4, 6. doi: 10.1186/gm305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson C, van Bruggen I, Segal A, Dunham M, Sherwood A, Koentgen F, … Lake RA (2006). A novel SV40 TAg transgenic model of asbestos-induced mesothelioma: Malignant transformation is dose dependent. Cancer Research, 66, 10786–10794. doi: 10.1158/0008-5472.CAN-05-4668 [DOI] [PubMed] [Google Scholar]
- Rogala AR, Morgan AP, Christensen AM, Gooch TJ, Bell TA, Miller DR, … de Villena FP-M (2014). The Collaborative Cross as a resource for modeling human disease: CC011/Unc, a new mouse model for spontaneous colitis. Mammalian Genome, 25, 95–108. doi: 10.1007/s00335-013-9499-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogovskyy AS, Rogovska YV, Taylor BM, Wiener DJ, & Threadgill DW (2021). The first immunocompetent mouse model of strictly human pathogen, Borrelia recurrentis. Infection and Immunity, 89, e00048–21. doi: 10.1128/IAI.00048-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, & Heise MT (2019). Mouse models as resources for studying infectious diseases. Clinical Therapeutics, 41, 1912–1922. doi: 10.1016/j.clinthera.2019.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh M, Chan EKL, Ho LA, Rose KM, Parks CG, Cohn RD, … Miller FW (2012). Prevalence and sociodemographic correlates of antinuclear antibodies in the United States. Arthritis and Rheumatism, 64, 2319–2327. doi: 10.1002/art.34380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saul MC, Philip VM, Reinholdt LG, & Chesler EJ, Center for Systems Neurogenetics of Addiction. (2019). High-diversity mouse populations for complex traits. Trends in Genetics, 35, 501–514. doi: 10.1016/j.tig.2019.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer A, Gralinski LE, Leist SR, Winkler ES, Hampton BK, Mooney MA, … Baric RS (2021). Common mechanism of SARS-CoV and SARS-CoV-2 pathogenesis across species. bioRxiv, doi: 10.1101/2021.05.14.444205 [DOI] [Google Scholar]
- Silver LM (1995). Mouse genetics: Concepts and applications. Oxford University Press. [Google Scholar]
- Smeekens JM, Orgel KA, Kesselring J, Bagley K, & Kulis MD (2020). Model of walnut allergy in CC027/GeniUnc mice recapitulates key features of human disease. The Yale Journal of Biology and Medicine, 93, 669–673. [PMC free article] [PubMed] [Google Scholar]
- Smith CM, Baker RE, Proulx MK, Mishra BB, Long JE, Park SW, … Sassetti CM (2022). Host-pathogen genetic interactions underlie tuberculosis susceptibility in genetically diverse mice. Elife, 11, e74419. doi: 10.7554/eLife.74419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GJ, Tovar A, McFadden K, Moran TP, Wagner JG, Harkema JR, & Kelada SNP (2021). A murine model of ozone-induced nonatopic asthma from the Collaborative Cross. American Journal of Respiratory Cell and Molecular Biology, 65, 672–674. doi: 10.1165/rcmb.2020-0577LE [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumelis V, & Liu Y -J. (2004). Human thymic stromal lymphopoietin: A novel epithelial cell-derived cytokine and a potential key player in the induction of allergic inflammation. Springer Seminars in Immunopathology, 25, 325–333. doi: 10.1007/s00281-003-0152-0 [DOI] [PubMed] [Google Scholar]
- Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, … Liu Y-J. (2002). Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nature Immunology, 3, 673–680. doi: 10.1038/ni805 [DOI] [PubMed] [Google Scholar]
- Srivastava A, Morgan AP, Najarian ML, Sarsani VK, Sigmon JS, Shorter JR, … de Villena FP-M (2017). Genomes of the mouse collaborative cross. Genetics, 206, 537–556. doi: 10.1534/genetics.116.198838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenson KL, Gatti DM, Valdar W, Welsh CE, Cheng R, Chesler EJ, … Churchill GA (2012). High-resolution genetic mapping using the Mouse Diversity outbred population. Genetics, 190, 437–447. doi: 10.1534/genetics.111.132597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavolara TE, Niazi MKK, Gower AC, Ginese M, Beamer G, & Gurcan MN (2021). Deep learning predicts gene expression as an intermediate data modality to identify susceptibility patterns in Mycobacterium tuberculosis infected Diversity Outbred mice. EBioMedicine, 67, 103388. doi: 10.1016/j.ebiom.2021.103388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor K, & Alvarez LR (2019). An estimate of the number of animals used for scientific purposes worldwide in 2015. Alternatives to Laboratory Animals, 47, 196–213. doi: 10.1177/0261192919899853 [DOI] [PubMed] [Google Scholar]
- Todd M, Malone L, Rusznak M, Stier M, Zhang J, Goleniewska K, … Toki S (2020). Differential Type 2 cytokine responses and group 2 innate lymphoid cell (ILC2) activation among 44 strains of mice in the Collaborative Cross following Alternaria extract challenge. Journal of Allergy and Clinical Immunology, 145, AB2. doi: 10.1016/j.jaci.2019.12.889 [DOI] [Google Scholar]
- Tovar A, Crouse WL, Smith GJ, Thomas JM, Keith BP, McFadden KM, … Kelada SNP (2022). Integrative analysis reveals mouse strain-dependent responses to acute ozone exposure associated with airway macrophage transcriptional activity. American Journal of Physiology Lung Cellular and Molecular Physiology, 322, L33–L49. doi: 10.1152/ajplung.00237.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle AH, Philip VM, Chesler EJ, & Mogil JS (2018). Comparing phenotypic variation between inbred and outbred mice. Nature Methods, 15, 994–996. doi: 10.1038/s41592-018-0224-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vered K, Durrant C, Mott R, & Iraqi FA (2014). Susceptibility to Klebsiella pneumonaie infection in collaborative cross mice is a complex trait controlled by at least three loci acting at different time points. BMC Genomics, 15, 865. doi: 10.1186/1471-2164-15-865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vick SC, Frutoso M, Mair F, Konecny AJ, Greene E, Wolf CR, … Lund JM (2021). A regulatory T cell signature distinguishes the immune landscape of COVID-19 patients from those with other respiratory infections. Science Advances, 7, eabj0274. doi: 10.1126/sciadv.abj0274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelkl B, Altman NS, Forsman A, Forstmeier W, Gurevitch J, Jaric I, … Würbel H (2020). Reproducibility of animal research in light of biological variation. Nature Reviews Neuroscience, 21, 384–393. doi: 10.1038/s41583-020-0313-3 [DOI] [PubMed] [Google Scholar]
- Vyse TJ, & Todd JA (1996). Genetic analysis of autoimmune disease. Cell, 85, 311–318. doi: 10.1016/S0092-8674(00)81110-1 [DOI] [PubMed] [Google Scholar]
- Wang P, Wang Y, Langley SA, Zhou Y-X, Jen K-Y, Sun Q, … Mao J-H (2019). Diverse tumour susceptibility in Collaborative Cross mice: Identification of a new mouse model for human gastric tumourigenesis. Gut, 68, 1942–1952. doi: 10.1136/gutjnl-2018-316691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W-Z, Gibson HM, Jacob JB, Frelinger JA, Berzofsky JA, Maeng H, … Wei K-C (2020). Diversity outbred mice reveal the quantitative trait locus and regulatory cells of HER2 immunity. Journal of Immunology, 205, 1554–1563. doi: 10.4049/jimmunol.2000466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter JM, Gildea DE, Andreas JP, Gatti DM, Williams KA, Lee M, … Crawford NPS (2017). Mapping complex traits in a diversity outbred F1 mouse population identifies germline modifiers of metastasis in human prostate cancer. Cell Systems, 4, 31–45.e6. doi: 10.1016/j.cels.2016.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H, Morrison J, Ferris MT, Gralinski LE, Whitmore AC, Green R, … Katze MG (2014). Genomic profiling of collaborative cross founder mice infected with respiratory viruses reveals novel transcripts and infection-related strain-specific gene and isoform expression. G3 (Bethesda), 4, 1429–1444. doi: 10.1534/g3.114.011759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Lin F, Wang Y, Zeng M, & Luo M (2021). Long noncoding RNAs as emerging regulators of COVID-19. Frontiers in Immunology, 12, 700184. doi: 10.3389/fimmu.2021.700184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Malo D, Mott R, Panthier J-J, Montagutelli X, & Jaubert J (2018). Identification of new loci involved in the host susceptibility to Salmonella Typhimurium in collaborative cross mice. BMC Genomics, 19, 303. doi: 10.1186/s12864-018-4667-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data sharing is not applicable to this article, as no datasets were generated or analyzed during the current study.