

Abstract
Short tandem repeats, a class of DNA elements composed of 2–12 bp motifs repeated in tracts of variable lengths, are abundant throughout the human genome. Expansions of these repeats underlie over four dozen human diseases, the first of which was described 30 years ago. This paradigm-shifting discovery radically changed how we conceive DNA, RNA, and protein dynamics and their interactions across human disease states. Here, we dive into the four major modes by which short tandem repeat expansions cause disease: loss of function through transcriptional silencing, RNA-mediated gain of function through gelation and RNA-binding protein sequestration, gain of function of AUG-initiated repeat-harboring native proteins, and repeat-associated non-AUG (RAN) translation of toxic repetitive peptides. Somatic repeat instability, driven by replication- and repair-dependent processes, titrates toxicity with age and across tissues. We focus on the crosstalk between these mechanisms, with particular examples which emphasize that targeting a single pathway alone may be insufficient to fully address disease pathogenesis. We also discuss the emerging native functions of repeat elements and how their dynamics might contribute to disease at a larger scale than currently appreciated. Lastly, we use these molecular insights to suggest holistic approaches to known and novel repeat expansion disorders as a method for better understanding disease mechanisms and expediting therapeutic development.
INTRODUCTION:
Short tandem repeats [G] (STRs, or microsatellites) are stretches of repeated 2–12 bp units of DNA found within both coding and non-coding regions of the genome. STRs make up about 3% of the human genome and are intrinsically unstable, showing a high degree of variation across species and within human populations1,2. Small fluctuations in length enable STRs to function as important adaptive regulators of gene expression by modulating DNA methylation, alternative splicing efficiency, and transcription factor binding3,4—yet, this instability comes at a cost.
In 1991, four groups independently identified instability and expansion of an STR composed of CGG repeats in the 5’ untranslated region (UTR) of the FMR1 gene as the underlying cause of Fragile X syndrome5–8. In parallel, a fifth group described a CAG repeat expansion encoding a polyglutamine stretch within the androgen receptor AR as the underlying cause of spinal and bulbar muscular atrophy (SBMA)9. These seminal findings introduced nucleotide repeat expansions as a cause of human disease and redefined how gene mutations can occur and contribute to neurological dysfunction. Moreover, these two disease-associated repeats highlight distinct modes of pathogenesis, opening the door to a wide spectrum of mechanisms by which expanded repeats lead to disease.
Since those initial discoveries, there has been significant growth in our understanding of both how repeat expansions arise and how they contribute to more than fifty human disorders. Disease-causing expansions can reside within gene promoters, protein-coding exons, 5’ and 3’ UTRs, or introns. Expansion motifs range from trinucleotides to dodecameric units [Table 1], and pathogenic expansion lengths vary from a few repeat units to thousands. These repeats have had an outsized influence on our understanding of molecular and cellular biology and neuroscience, providing novel insights into chromatin biology, DNA repair, RNA-protein condensates, non-canonical translation [G], and protein aggregation. Furthermore, although more than 1.5 million STRs have been identified in the human genome to date4, our knowledge of their native functions at non-pathogenic lengths remains in its infancy10–12.
Table 1:
Repeat expansion diseases by location and pathomechanism.
| Disease | Abbrev. | Inheritance | Host gene | Repeat motif | Location | Somatic instability | LOF | RBP sequestration | AUG-initiated protein GOF | RAN translation | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Unverricht-Lundborg disease | EPM1 | AR | CSTB | CCCCGCCCCGCG | promoter | + | |||||
| Baratela-Scott syndrome | BSS | AR | XYLT1 | CGG | promoter/5’ UTR | + | + | ||||
| Glutaminase deficiency | GAD | AR | GLS | CAG | 5’ UTR | + | + | ||||
| Spinocerebellar ataxia type 12 | SCA12 | AD | PPP2R2B | CAG | 5’ UTR | + | |||||
| Fragile XE syndrome | FRAXE | XL | AFF2 | CCG | 5’ UTR | + | + | ||||
| Jacobsen syndrome | FRA11B | AD | CBL2 | CCG | 5’ UTR | + a | |||||
| Intellectual disability, associated with fragile site FRA2A | FRA2A | AD | AFF3 | CGG | 5’ UTR | + | + | ||||
| Intellectual disability, associated with fragile site FRA12A | FRA12A | AD | DIP2B | CGG | 5’ UTR | + | |||||
| Fragile X syndrome | FXS | XL | FMR1 | CGG | 5’ UTR | + | + | ||||
| Fragile X-associated primary ovarian insufficiency | FXPOI | XL | FMR1 | CGG | 5’ UTR | + | + | ||||
| Fragile X-associated tremor/ataxia syndrome | FXTAS | XL | FMR1 | CGG | 5’ UTR | + | + | + | |||
| Neuronal intranuclear inclusion disease | NIID | AD | NOTCH2NLC | CGG | 5’ UTR | ||||||
| Oculopharyngodistal myopathy 1 | OPDM1 | AD | LRP12 | CGG | 5’ UTR | + | |||||
| Oculopharyngodistal myopathy 2 | OPDM2 | AD | GIPC1 | CGG | 5’ UTR | ||||||
| Oculopharyngeal myopathy with leukoencephalopathy | OPML | AD | LOC642361/NUTM2B-AS1 | CGG/CCG | lncRNA | + | |||||
| Cerebellar ataxia, neuropathy, vestibular areflexia syndrome | CANVAS | AR | RFC1 | AAGGG | intron | ||||||
| Spinocerebellar ataxia type 10 | SCA10 | AD | ATXN10 | ATTCT | intron | + | + | ||||
| X-linked dystonia-parkinsonism | XDP | XL | TAF1 | CCCTCT | intron | + | + | ||||
| Myotonic dystrophy type 2 | DM2 | AD | CNBP | CCTG | intron | + | + | + | |||
| Autism spectrum disorder, associated with fragile site FRA7A | FRA7A | AD | ZNF713 | CGG | intron | + | + | ||||
| Fuchs endothelial corneal dystrophy | FECD | AD | TCF4 | CTG | intron | + | + | + | |||
| Friedreich’s ataxia | FA | AR | FXN | GAA | intron | + | + | ||||
| Spinocerebellar ataxia type 36 | SCA36 | AD | NOP56 | GGCCTG | intron | + | + | ||||
| C9ORF72 amyotrophic lateral sclerosis/frontotemporal dementia | C9 ALS/FTD | AD | C9ORF72 | GGGGCC | intron | + | + | + | + | ||
| Spinocerebellar ataxia type 31 | SCA31 | AD | BEAN1/TK2 | TGGAA/TTCCA | intron | + | + | ||||
| Familial adult myoclonic epilepsy 1 | FAME1 | AD | SAMD12 | TTTCA | intron | + | |||||
| Familial adult myoclonic epilepsy 2 | FAME2 | AD | STARD7 | TTTCA | intron | ||||||
| Familial adult myoclonic epilepsy 3 | FAME3 | AD | MARCH6 | TTTCA | intron | + | |||||
| Familial adult myoclonic epilepsy 4 | FAME4 | AD | YEATS2 | TTTCA | intron | ||||||
| Familial adult myoclonic epilepsy 6 | FAME6 | AD | TNRC6A | TTTCA | intron | ||||||
| Familial adult myoclonic epilepsy 7 | FAME7 | AD | RAPGEF2 | TTTCA | intron | ||||||
| Spinocerebellar ataxia type 37 | SCA37 | AD | DAB1 | TTTCA | intron | ||||||
| Dentatorubral-pallidoluysian atrophy | DRPLA | AD | ATN1 | CAG | CDS | + | + | + | |||
| Huntington’s disease | HD | AD | HTT | CAG | CDS | + | + | + | + | ||
| Spinal and bulbar muscular atrophy | SBMA | XL | AR | CAG | CDS | + | + | + | |||
| Spinocerebellar ataxia type 1 | SCA1 | AD | ATXN1 | CAG | CDS | + | + | + | |||
| Spinocerebellar ataxia type 2 | SCA2 | AD | ATXN2 | CAG | CDS | + | + | + | |||
| Spinocerebellar ataxia type 3 | SCA3 | AD | ATXN3 | CAG | CDS | + | + | + | |||
| Spinocerebellar ataxia type 6 | SCA6 | AD | CACNA1A | CAG | CDS | + | |||||
| Spinocerebellar ataxia type 7 | SCA7 | AD | ATXN7 | CAG | CDS | + | + | ||||
| Spinocerebellar ataxia type 17 | SCA17 | AD | TBP | CAG | CDS | + | + | + | |||
| Pseudoachondroplasia and multiple epiphyseal dysplasia | PSACH/MED | AD | COMP | GAC | CDS | + | + | ||||
| Blepharophimosis, ptosis, and epicanthus inversus syndrome | BPES | AD | FOXL2 | GCN | CDS | + | |||||
| Cleidocranial dysplasia | CCD | AD | RUNX2 | GCN | CDS | + | |||||
| Congenital central hypoventilation syndrome | CCHS | AD | PHOX2B | GCN | CDS | + c | + | ||||
| Hand-foot-genital syndrome | HFGS | AD | HOXA13 | GCN | CDS | + c | + | ||||
| Holoprosencephaly 5 | HPE | AD | ZIC2 | GCN | CDS | + c | + | ||||
| Oculopharyngeal muscular dystrophy | OPMD | AD | PABPN1 | GCN | CDS | + | + | ||||
| Synpolydactyly 1 | SPD | AD | HOXD13 | GCN | CDS | + | |||||
| X-linked hypopituitarism | XH | XL | SOX3 | GCN | CDS | + | |||||
| X-linked intellectual disability b | XLID | XL | ARX | GCN | CDS | + c | + | ||||
| Spinocerebellar ataxia type 8 | SCA8 | AD | ATXN8OS/ATXN8 | CTG/CAG | 3’ UTR/CDS | + | + | + | |||
| Myotonic dystrophy type 1 | DM1 | AD | DMPK | CTG | 3’ UTR | + | + | + | |||
| Huntington’s disease-like 2 | HDL2 | AD | JPH3 | CTG | 3’ UTR | + | + |
FRA11B expansion causes hypermethylation and is associated with chromosomal breakage and deletion of the telomeric end of 11q.271
Loss-of-function mutations in ARX are associated with a spectrum of clinical phenotypes, including X-linked infantile spasm syndrome, X-linked lissencephaly with ambiguous genitalia, X-linked myoclonic epilepsy and intellectual disability, Partington syndrome, Ohtahara syndrome, and Proud syndrome.272
STR expansions induce architectural changes in DNA and elicit a series of concurrent molecular processes through either loss-of-function (LOF) or gain-of-function (GOF) mechanisms at the DNA, RNA, and/or protein levels [Figure 1]. The toxicity of each of these mechanisms is modulated by somatic instability [G] of the repeat, a stochastic process that titrates repeat length differentially across a patient’s tissues and cells over their lifetime13–15.
Figure 1: Molecular mechanisms driving nucleotide repeat expansion pathogenesis.
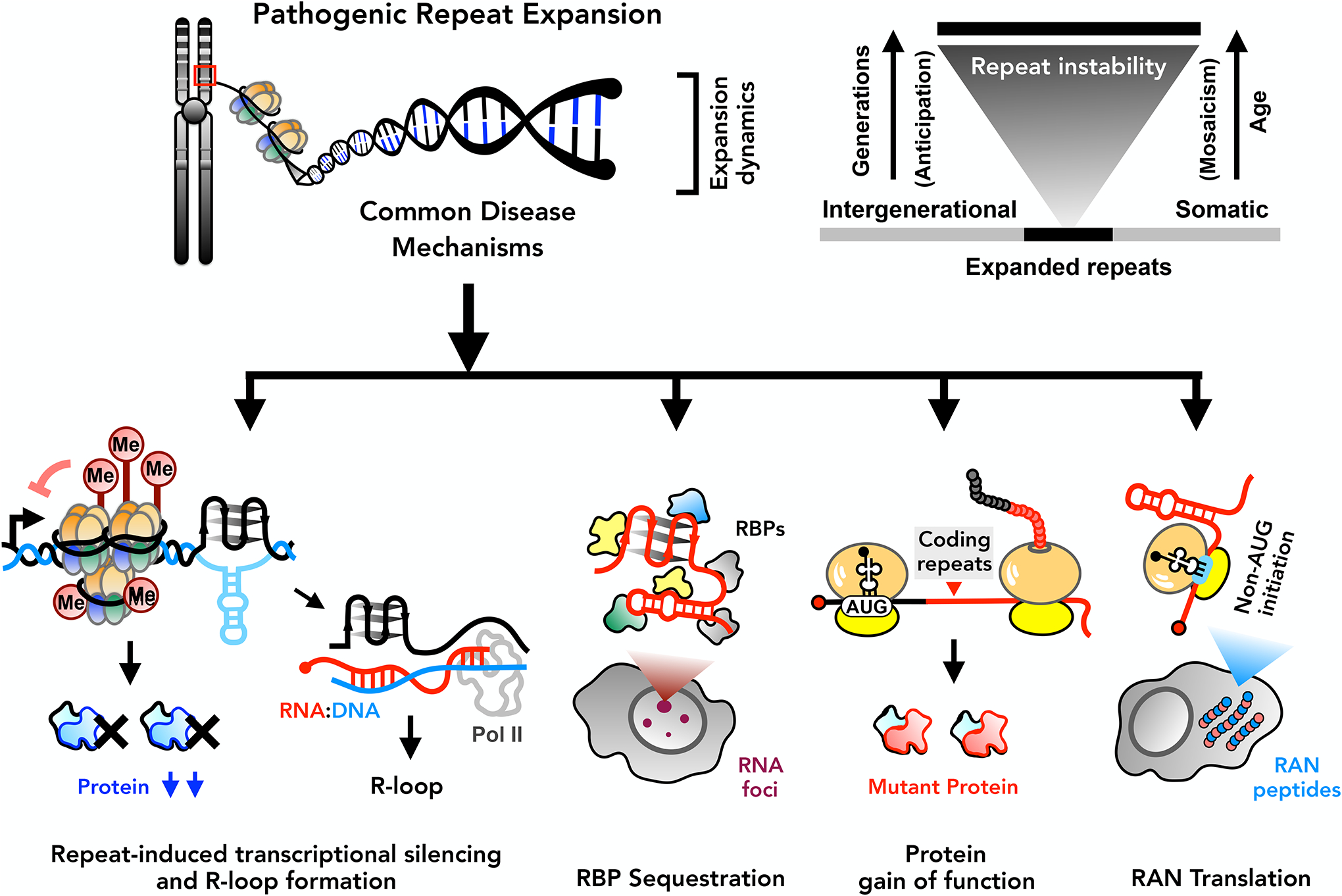
Hyper-methylation of promoter regions can lead to transcriptional silencing, resulting in partial or complete loss of the native protein harboring the repeats. In contrast, active transcription through the repeats can trigger formation of R-loops (RNA:DNA hybrids that lead to DNA damage response pathway activation) and potentially exacerbate somatic repeat instability. Transcribed repeat RNAs fold into complex structures, which aberrantly interact with and sequester cellular RNA-binding proteins (RBPs). Trinucleotide repeat expansions in protein-coding sequences generate mutant proteins that elicit gain-of-function toxicity. Finally, the coding and non-coding repeat RNAs are translated in the absence of canonical AUG-mediated initiation through repeat-associated non-AUG (RAN) translation, producing toxic polymeric peptides.
In this review, we detail each of the core molecular mechanisms of pathogenesis in repeat expansion diseases, and we discuss how synergy between mechanisms produces the unique and complex pathology for each disease. Given the breadth of research in the field over the past three decades, comprehensive coverage of all important discoveries within a single article is not feasible, as attested to by multi-chapter books on the topic16,17. As such, we for the most part do not discuss the downstream sequelae of these events, which are myriad, but instead focus on understanding the mechanisms most proximal to the underlying cause of these disorders. We largely focus on a subset of disorders (fragile X-associated disorders, myotonic dystrophies, and C9ORF72 amyotrophic lateral sclerosis and/or frontotemporal dementia (C9 ALS/FTD)) which our own groups study and which we feel offer a representative through-line in the field. We place a particular emphasis on emerging roles for how repetitive RNAs generate aberrant ribonucleoprotein (RNP) complex [G] formations and trigger RAN translation, given recent advances in these spaces. We also highlight critical points of synergy across disease mechanisms, potentially revealing novel therapeutic targets. Throughout, we include references to more detailed recent reviews of specific topics and disorders, and we apologize in advance to the many investigators whose excellent work we were not able to highlight.
Pathomechanisms of repeat expansions
The proximal pathomechanisms of repeat expansion diseases can be broadly classified into four interrelated categories [Figure 1]: (1) DNA-based mechanisms, including LOF via repeat-induced transcriptional silencing and GOF via R-loop [G] formation and DNA damage response activation; (2) toxic RNA-mediated GOF through gelation and RNA-binding protein (RBP) sequestration; (3) GOF of AUG-initiated repeat-harboring native proteins; and (4) repeat-associated non-AUG (RAN) translation of toxic repetitive peptides. Here, we unravel each of these categories, providing historical foundations and highlighting the current state of research in the contexts of representative diseases.
Transcriptional silencing, R-loop formation and somatic instability
Fragile X syndrome (FXS) was originally described in the 1940s as a form of intellectual disability and autism in young males18, and it was linked to the rare (familial) eponymous folate-sensitive fragile site [G] on the X chromosome in the late 1960s19. The causative mutation was mapped to a CGG STR in the 5’ UTR of the FMRP translational regulator 1 (FMR1) gene5–8. Almost all mammals have a repeat element at this locus, with an average size of around 30 repeats in humans20. In FXS, this repeat expands to >200 CGG units, inducing hyper-methylation of the repeat tract and the neighboring FMR1 promoter and epigenetic silencing of the FMR1 locus (Figure 2B). The end result of these transformations is the loss of FMR1 transcription and the absence of the FMRP protein21,22.
Figure 2: Repeat-induced transcriptional silencing, R-loops and somatic instability.
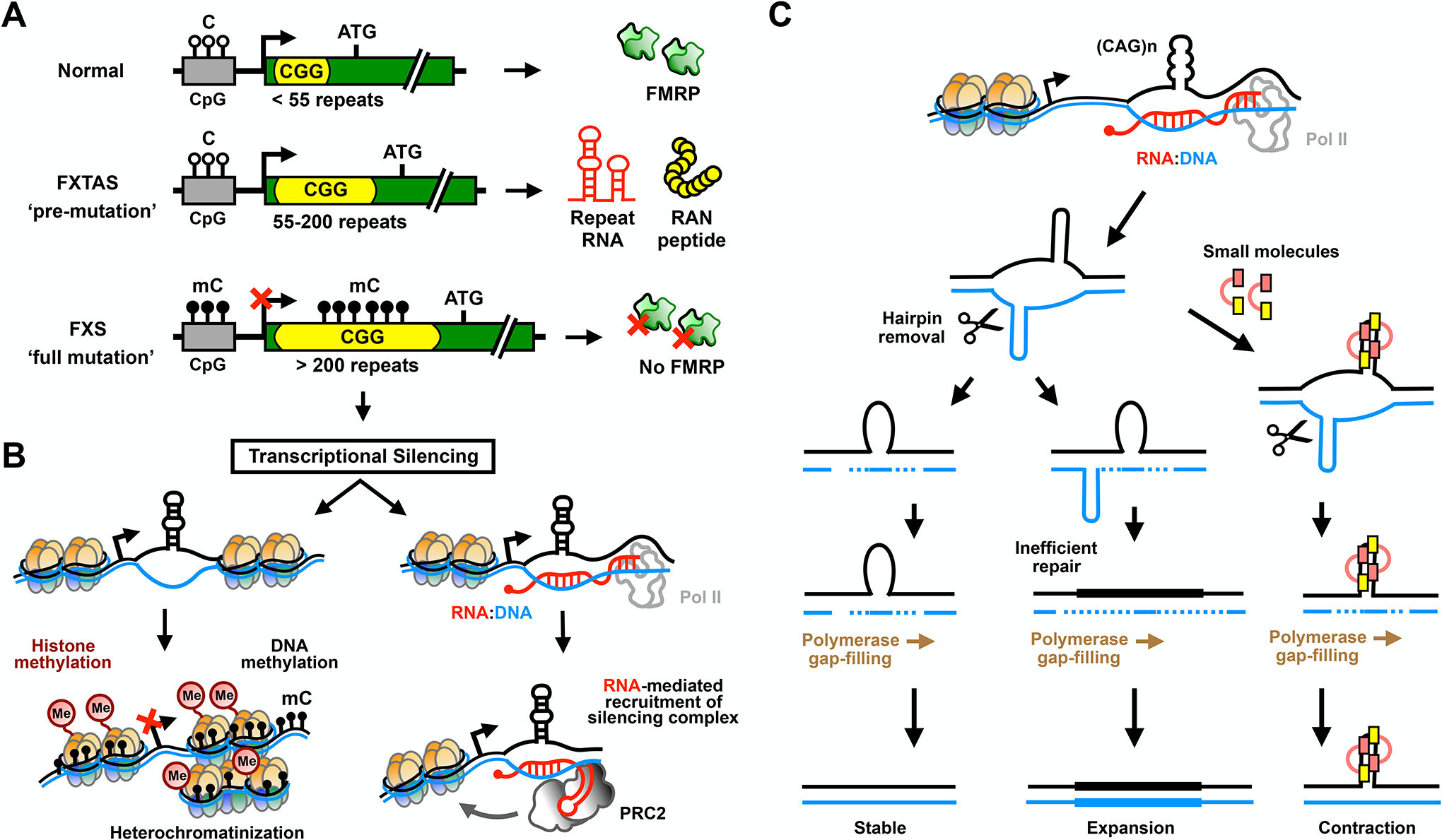
(A) Allelic classes of the FMR1 gene containing normal to pathogenic CGG repeats. FMR1 normally has ~30 CGG repeats in its 5’ UTR that are not included as a part of the mature protein product FMRP. Pre-mutation (55–200 repeats) expansions result in production of large CGG repeat-containing RNAs that underlie the age-related neurodegenerative disorder fragile X-associated tremor/ataxia syndrome (FXTAS). Full mutation (>200 repeats) expansions in subsequent generations lead to silencing of the FMR1 locus and fragile X syndrome (FXS). (B) CGG repeat methylation (mC) may direct transcriptional silencing by favoring histone methylation and heterochromatin formation through mechanisms similar to those typically active at CpG islands (left panel). Alternatively or cooperatively, nascent RNA may trigger epigenetic silencing by hybridizing to the complementary CGG-repeat DNA to form RNA:DNA duplexes that recruit polycomb repressive complex 2 (PRC2) (right panel). (C) Transcription-induced R-loops also support formation of DNA slip-out structures that contribute to repeat instability. For CAG/CTG trinucleotide repeat expansions, extended stable hairpins form in both strands. Normally, mismatch repair (MMR) pathways keep the repeat tract length stable by melting the slip-outs, followed by gap-filling by DNA polymerase across the region. Inefficient repair or formation of multiple slip-outs leads to retention of the slip-out structures and expansion of the repeat region by incorporation of the looped DNA. Small molecules that target slip-out structures in CAG repeat DNA inhibit repeat expansion and bias instability toward contraction.
Loss of FMRP drives pathology in FXS. Disease severity correlates with repeat methylation and transcriptional silencing [G], and FMR1 mutations that disrupt FMRP function replicate the clinical features of FXS in patients and animal models23–25. However, the exact mechanism by which the repeat expansion triggers transcriptional silencing remains unresolved26. One proposal is that expanded FMR1 RNA instigates formation of R-loops, stalling RNA polymerase II (Pol II) transcription complexes and triggering recruitment of the histone methyltransferase polycomb repressive complex 2 (PRC2)27,28 (Figure 2B). PRC2 then drives epigenetic changes at the locus through H3K27 histone methylation that favor stable silencing of FMR1. Consistent with this hypothesis, embryo-derived full mutation hESCs often exhibit active FMR1 transcription and a lack of DNA methylation prior to neuronal differentiation, but FMR1 is silenced later in development29. Interestingly, depletion of nascent FMR1 mRNA favors an open chromatin state in FXS hESCs during neuronal differentiation and maintenance of FMR1 expression, suggesting a role for either R-loops or RNA-induced transcriptional silencing in the heterochromatinization process27,28,30. However, this model largely ignores a direct role for DNA methylation in the silencing of the locus. Delivery of demethylating agents or targeting of DNA methyltransferases to FMR1 in patient iPSCs is sufficient to reactivate FMR1 expression28,31,32. Moreover, methylation of CGG repeats in vitro favors heterochromatin formation and nucleosome assembly22,33, suggesting that DNA methylation itself plays an active role in transcriptional silencing. As such, CGG repeat-mediated transcriptional silencing is likely triggered by a combination of DNA hyper-methylation and histone methylation elicited by R-loops and Pol II stalling, leading to heterochromatinization of the locus and loss of FMRP expression26. Similar methylation-mediated silencing mechanisms may contribute to reduced C9ORF72 expression at GGGGCC repeats in C9 ALS/FTD34–38, as well as other disease-causing repeats containing CpG elements39,40. Key next steps are to define the exact interplay of these epigenetic processes and determine the proximal trigger for repeat DNA methylation.
Other repeats trigger transcriptional silencing and LOF phenotypes largely in the absence of DNA methylation. Most prominent among these is Friedreich’s ataxia, the most common autosomal recessive ataxia, which is typically caused by a homozygous GAA repeat expansion in the first intron of FXN41. This expansion triggers heterochromatin protein (HP1)-sensitive silencing of the gene42–44. At least three proposed mechanisms appear to contribute to FXN silencing. First, the expanded GAA/TTC repeats form “sticky” triplex H-DNA structures that directly impede transcription45. Second, an FXN antisense transcript (FAST-1) is upregulated in the disease and triggers a loss of CTCF binding in the FXN 5’ UTR, which in turn favors heterochromatization46. Interestingly, FAST-1 expression in isolation is sufficient to suppress FXN transcription47. Third, R-loops similar to those discussed above for FMR1 are also thought to form at expanded GAA repeats, where they can trigger RNA-induced transcriptional silencing48,49. Once heterochromatinized, transcription elongation through the repeat is markedly diminished50–52. Importantly—and regardless of the etiology of silencing—direct targeting of heterochromatinization by histone deacetylase and methyltransferase inhibitors appears sufficient to reactivate FXN transcription in many contexts and is the basis for multiple ongoing clinical trials42.
While epigenetic silencing causes loss of expression of the repeat-harboring gene, active transcription across repeats can encourage R-loop formation that does not ultimately lead to transcriptional shutdown. These R-loops trigger DNA damage response (DDR) cascades in cells that may contribute to pathogenesis53. In Fragile X-associated tremor/ataxia syndrome (FXTAS), an age-related neurodegenerative disorder caused by moderately sized (50–200 CGGs) unmethylated repeat expansions in FMR1, R-loop formation rate increases with repeat length and is associated with DDR activation in both patient cells and mouse models54–56 (Figure 2A). R-loops are also observed at other disease-causing repeat expansions, including the GGGGCC repeat implicated in C9 ALS/FTD and the CAG repeat in HTT that causes Huntington disease [G] 57–59. Once activated, these DNA damage response cascades can trigger mitochondrial dysfunction and apoptosis60,61. While Huntington disease cells and other cells expressing CAG repeat expansions are more sensitive to DNA damage62,63, it is not clear whether resolution of these events is sufficient to preclude repeat toxicity.
DNA mismatch repair (MMR) [G] pathways that typically resolve abnormal DNA-DNA and DNA-RNA structures formed during transcription can exacerbate instability and cause further expansion of repeat elements in somatic cells15,64,65 (Figure 2C). This somatic instability is observed in the majority of repeat expansion diseases [Table 1] and produces variation in repeat size and toxicity across tissues within the same patient. This process notably does not require DNA replication, as somatic expansions are observed in terminally differentiated cells such as neurons and myofibers66–69. The precise mechanisms of somatic instability in repeat expansion diseases remain under intensive study and have been recently reviewed15. A role for somatic instability in disease pathogenesis is supported by genetic studies implicating MMR proteins as modifiers of age of onset in Huntington disease70,71 and spinocerebellar ataxias (SCAs)72. In rodent models of Huntington disease, modulation of MMR is sufficient to suppress somatic instability and can reduce toxicity73,74. Indeed, small-molecule targeting of slip-out structures in CAG repeat DNA, which recruit MMR, induces contractions in disease models (Figure 2C)75. These results suggest that these processes may serve as cross-platform therapeutic targets.
RNA multivalency and RNA-binding protein interactions
Most repeat expansion diseases do not involve significant transcriptional silencing; in these cases, they are often inherited in a dominant fashion [Table 1]. In some contexts, RNA expressed from expanded repeats accumulates within cells into complexes commonly referred to as RNA foci [G] [Figure 3a]. These foci were first observed in myotonic dystrophy type 1 (DM1) patient fibroblasts and myofibers, where nuclear clumps of DMPK mRNA with expanded CUG repeats (CUGexp) were detected by fluorescence in situ hybridization76. Since this discovery, RNA foci have been identified as a hallmark of many repeat expansion diseases, including DM2, C9 ALS/FTD, Fuch’s endothelial corneal dystrophy (FECD), FXTAS, and many SCAs77. These RNA foci are often retained in the nucleus, but cytoplasmic foci are also detected in some cases, such as in SCA10 fibroblasts78 and C9 ALS/FTD neurons79–81. Bidirectional transcription of expanded repeats produces both sense and antisense transcripts, leading to formation of both sense and antisense foci82. In C9 ALS/FTD, GGGGCC and CCCCGG foci can coexist within the same nuclei and have even been observed to colocalize79. In some diseases, including C9 ALS/FTD79 and DM183, the presence or number of RNA foci is directly correlated with onset or pathology84.
Figure 3: Mechanisms of RNA toxicity in repeat expansion diseases.
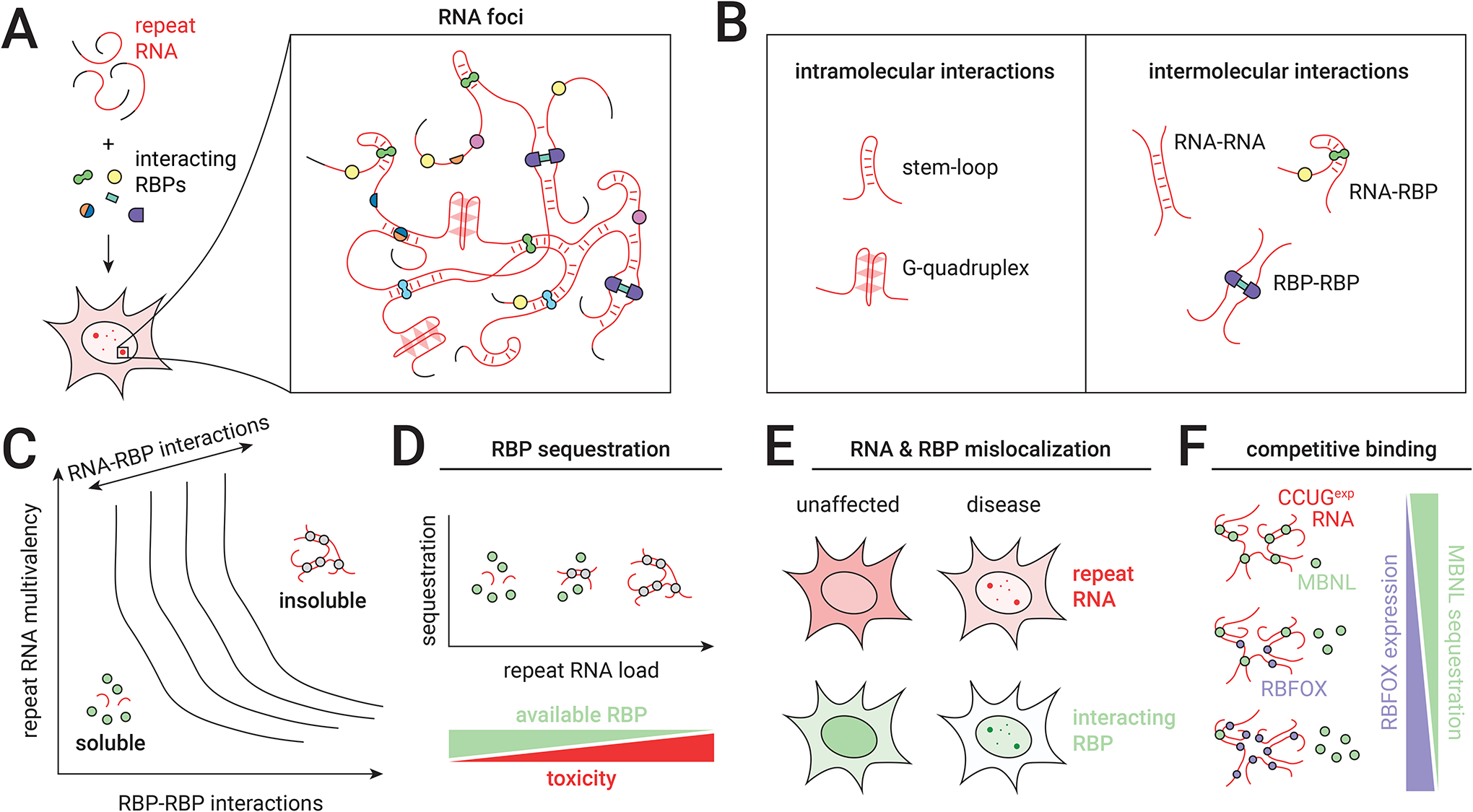
(A) Long repetitive RNAs and RNA-binding proteins (RBPs) interact to form complex nuclear-retained RNA foci. (B) RNA foci are formed and maintained through a stochastic combination of intramolecular and intermolecular interactions. (C) A conceptual phase diagram [G] describes the thermodynamics of RNA foci in repeat expansion diseases. The transition from soluble RNA to RNA-protein phase separation is defined by the sum of RNA-RNA, protein-protein, and RNA-protein interactions (phase boundary isolines drawn as solid lines). (D) RNA processing is impaired by sequestration of RBPs on repetitive RNA, the extent of which is a cell-specific function of repeat length, host gene expression, and RBP expression. (E) Effects of nuclear retention on RBP localization can be exacerbated by autoregulatory dynamics, which may additionally disrupt cytoplasmic processes mediated by RBPs. (F) Competition between RBPs at RNA foci may modulate disease-associated sequestration. In DM2, both MBNL and RBFOX proteins bind the expanded CCUG repeat RNA, and overexpression of RBFOX partially displaces MBNL from RNA foci in muscle cells.
RNA foci in repeat expansion diseases are likely formed and maintained by a combination of intramolecular and intermolecular interactions [Figure 3b]. Through Watson-Crick and non-canonical base pairing, repetitive RNAs can form stable intramolecular secondary structures [G] in vitro and in vivo, including via imperfect stem-loops [G]85 and G-quadruplexes [G]86–88. For example, short (CUG)6 RNA folds into a single stable A-form double helix [G] with U-U mismatches89, while longer CUG RNAs likely explore an ensemble of stem-loop orientations90,91. Intermolecular interactions may also drive repeat RNA aggregation [Figure 3b]. Through multivalent base pairing alone, (CUG)47, (CAG)47, and (GGGGCC)5 RNAs undergo sol-gel transition to form phase-separated [G] RNA droplets in vitro92. Similar liquid-like behaviors are also observed in cell models: in C2C12 mouse myoblasts expressing (CUG)145 RNA, spontaneous division and coalescence of foci have been detected93. In addition, (CAG)47 RNA foci in transfected U2OS cells are solubilized when treated with inhibitors of RNA base pairing, including ammonium acetate and doxorubicin92.
Repetitive RNAs are not the only inhabitants of these structures. Expanded RNAs attract RNA-binding proteins [G] (RBPs) with expected motif and/or structure preferences, and these RBPs can coat the mutant transcripts, resulting in high local concentrations. In DM1, CUG foci co-localize with the muscleblind-like (MBNL) family of RBPs94. In C2C12 cells, knockdown of Mbnl1 by RNAi reduces aggregation of (CUG)145 RNA93, suggesting that MBNL proteins are necessary to stabilize intermolecular CUGexp RNA interactions in a cellular context. RBPs with multiple RNA-binding domains may contribute to RNP granule stability through multivalent protein-RNA interactions as well as higher order protein-protein interactions95. This concept, when combined with intermolecular base pairing of long repetitive RNAs, evokes a model in which RNA-RNA, RNA-protein, and protein-protein interactions each play an important part in RNA foci formation [Figure 3c]. The effects of biopolymer multivalency on phase separation dynamics are well appreciated96–99, and RNA acts as a scaffold for RNP phase separation in many contexts100. Thus, gelation of repeat RNA and RBPs in repeat expansion diseases appears to be an exacerbation of the general thermodynamic processes that normally regulate RNP granule self-assembly.
Repeat expansion RNAs can be toxic if recruitment of an RBP sufficiently depletes the protein from the nucleoplasm [Figure 3d]. The most notable example of this phenomenon is myotonic dystrophy, in which MBNL proteins are sequestered by CUGexp or CCUGexp RNA in the nucleus94. MBNL proteins are global alternative splicing factors, and their sequestration and inactivation in myotonic dystrophy produces a transcriptome-wide spliceopathy101. In mice, knockout of Mbnl1 produces a phenotype of myotonia, myopathy, and cataracts102. Mice expressing ~250 CUG repeats in a human skeletal actin transgene (HSALR) develop myotonia103 that is ameliorated by overexpression of Mbnl1 administered by AAV to skeletal muscle104.
Impaired splicing has also been observed in other disease contexts, such as in SCA10 fibroblasts via sequestration of hnRNP K78. In C9 ALS/FTD, GGGGCC RNAs associate with many proteins, including Pur-α, ALYREF, SRSF2, RanGAP1, and hnRNPs105–108, and sequestration of hnRNP H causes mis-splicing88. Antisense CCCCGG RNAs also interact with SRSF2, ALYREF, and hnRNPs80. In FXTAS neurons, CGG repeat RNA accumulates within large ubiquitinated inclusions and co-localizes with DGCR8, hnRNP A2/B1, and Pur-α, and overexpression of any of these factors reduces neurodegeneration in a Drosophila model expressing (CGG)90 RNA109–111. In addition to splicing, miRNA biogenesis109 and alternative polyadenylation pathways112,113 controlled by RBPs can be undermined by sequestration.
Besides regulating RNA maturation in the nucleus, RBPs play important roles in the cytoplasm as modulators of mRNA stability114 and mediators of a broad RNA transport program that enables the cell to shuttle RNAs to their proper locations115–117. Indeed, abundance and subcellular localization of RBPs are often autoregulated to confer robustness in a dynamic and stochastic cellular environment118. However, upon sequestration, these autoregulatory loops can shift RBP localization away from the cytoplasm to compensate for reduced nuclear activity, exacerbating the effects of RNA toxicity119. Mis-localization of RBPs into the nucleus could therefore disrupt both nuclear and cytoplasmic RBP functions [Figure 3e].
By inactivating a small number of nodes in the RNA processing network, non-coding repeat expansions can have potent and entangled consequences on a large number of cellular processes. However, it is important to note that the presence of RNA foci alone does not confirm a central role in disease pathology; for example, while sense RNA foci have been observed in Huntington disease120 and SCA3121, polyglutamine proteotoxicity (see below) seems to dominate as a pathomechanism16. In addition, RNA foci may play protective roles by reducing RAN translation of toxic peptide repeats (see below) through inhibition of nuclear export122–124.
What makes RBP sequestration such a defining mechanism of toxicity in DM1, and is it unique among repeat expansion diseases? Clearly, stoichiometry of RBP binding is a function of the number of available target sites on the RNA, and somatic instability can produce hundreds or thousands of tandem repeats in DM1 muscle and neurons125,126. Furthermore, the toxic CUG repeats are present in mature mRNA, which likely enhances their stability relative to intronic expansions, such as the GGGGCC repeats in C9 ALS/FTD or the UUUCA repeats in familial adult myoclonic epilepsy (FAME) [Table 1]. In diseases where repeat lengths are commonly shorter or where expression of the repeat element is low across most cell types, the impact of RBP sequestration may be limited. Competition between RBPs for binding sites on expanded RNAs may also limit exhaustion of any particular protein. In DM2, RBFOX proteins also bind to the CCUGexp RNA and actively compete with MBNL, and overexpression of RBFOX1 in C2C12 cells expressing (CCUG)1000 RNA partially restores Mbnl-directed splicing127 [Figure 3f]. Perhaps this phenomenon plays a role in other diseases as well, including C9 ALS/FTD and FXTAS, in which the repeated RNA motif attracts a large number of proteins. Finally, the lack of severe pathology in mice upon knockout of Dmpk suggests that haploinsufficiency is not a substantial contributor to DM1128; in contrast, diseases caused by expansions in essential genes, such as the SCA17-linked CAG repeat in TBP, naturally exhibit another layer of complexity, as inhibition of host gene expression is likely to produce a phenotype129.
Protein-mediated gain of function and the role of native gene context
The formation of insoluble neuronal protein aggregates is a common pathological feature across many neurodegenerative disorders. In repeat expansion diseases, aggregated proteins play a direct role in pathology13,16. At least nine disorders, including HD, multiple SCAs, SBMA, and dentatorubro-pallidoluysian atrophy (DRPLA), are caused by CAG repeat expansions in protein-coding sequences, resulting in expression of polyglutamine proteins (polyQ)130 [Table 1]. Although short glutamine stretches are present throughout the proteome, larger polyQ-containing proteins undergo conformational changes to form insoluble aggregates131–134, and these aggregates can induce cellular proteotoxicity independent of the functions of their host proteins135. For example, in mice, expression of either HTT exon 1 containing an expanded CAG repeat or insertion of a polyQ repeat in the unrelated gene HPRT is sufficient to produce intranuclear inclusions, neuritic aggregates and neurological phenotypes, which are not observed at normal repeat sizes135–138.
Most polyQ disease genes encode multifunctional proteins involved in various stages of gene expression, RNA metabolism, and proteostatic pathways139. Therefore, although polyQ toxicity is shared among these conditions, disease-specific phenomena are modulated by the functions of the protein containing the expansion. This concept is best exemplified by SBMA, which is caused by a CAG expansion in the androgen receptor gene AR. AR is a transcription factor whose nuclear entry is ligand-activated. In SBMA, androgens promote translocation of the polyQ-containing AR to the nucleus, where it aggregates and triggers transcriptional dysregulation and cytotoxicity140,141. Indeed, genetic or pharmacological blockade of androgen binding causes cytoplasmic retention of mutant AR, enhancing clearance by autophagy and ameliorating disease phenotypes142. Similarly, phosphorylation of mutant AR that inhibits ligand-activated nuclear translocation also suppresses disease-relevant phenotypes143.
Other diseases also demonstrate how native protein context can modulate repeat toxicity. For example, SCA1 is caused by expanded CAG repeats in ATXN1, resulting in polyQ expression. ATXN1 normally shuttles between the nucleus and cytoplasm and plays active roles in gene regulation144. When expanded, polyQ ATXN1 is enriched in the nucleus, where it can interact with nascent RNAs and protein regulators of transcription, including the repressor Capicua (CIC), a critical mediator of toxicity145. Similarly to SBMA, mice with mutations in the NLS domain of polyQ ATXN1 do not develop disease phenotypes, suggesting that nuclear localization is critical for pathogenesis146. Furthermore, mutation of phosphorylation site S776 to a phosphomimetic aspartic acid impedes its interactions with the 14-3-3 chaperone in the cytosol, resulting in nuclear translocation and toxicity147. Conversely, replacement of S766 to a phospho-dead alanine prevents neuronal toxicity148,149.
Ultimately, while native protein context importantly influences pathology unique to each disease, symptoms and clinical outcomes of polyQ diseases begin to converge at longer expansion lengths, with more prominent pathology and earlier onset of motor dysfunction, dystonia, parkinsonism and dementia16,150,151. Accordingly, transgenic polyQ disease models with large repeats outside of their normal protein context tend to show diffuse patterns of neurodegeneration135. It thus appears that at larger repeat sizes, disease-specific contributions of repeat-harboring genes are overwhelmed by proteotoxic impacts of polyQ expression.
Repeat-associated non-AUG (RAN) translation
Dominantly inherited diseases caused by repeat expansions located outside protein-coding regions were initially thought to manifest solely via haploinsufficiency or RNA gain of function. However, the seminal discovery of non-AUG-initiated translation of repetitive elements raised the possibility of yet another mechanism of pathology152 [Figure 4]. During study of CAG repeats in SCA8, a serendipitous observation was made that an AUG start codon was not required to generate polyQ protein from an ATXN8 minigene, even in the presence of multiple stop codons upstream of the repeat. Repeat-associated non-AUG (RAN) translation [G] was observed in all three reading frames to produce three homopolymeric proteins: polyQ, polyS, and polyA152. CUG repeat RNA also supported RAN translation from reporter constructs, but a CAA repeat did not, suggesting that secondary structure of the repeat RNA may be required152. Subsequently, RAN translation was described at repeat loci associated with FXTAS, C9 ALS/FTD, FECD, DM1, DM2, Huntington disease and multiple SCAs82,153–159. For many of these diseases, RAN translation occurs on both sense and antisense transcripts, and RAN peptides accumulate in patient tissues82,122,157,158,160,161.
Figure 4: Mechanisms of repeat-associated non-AUG (RAN) translation.
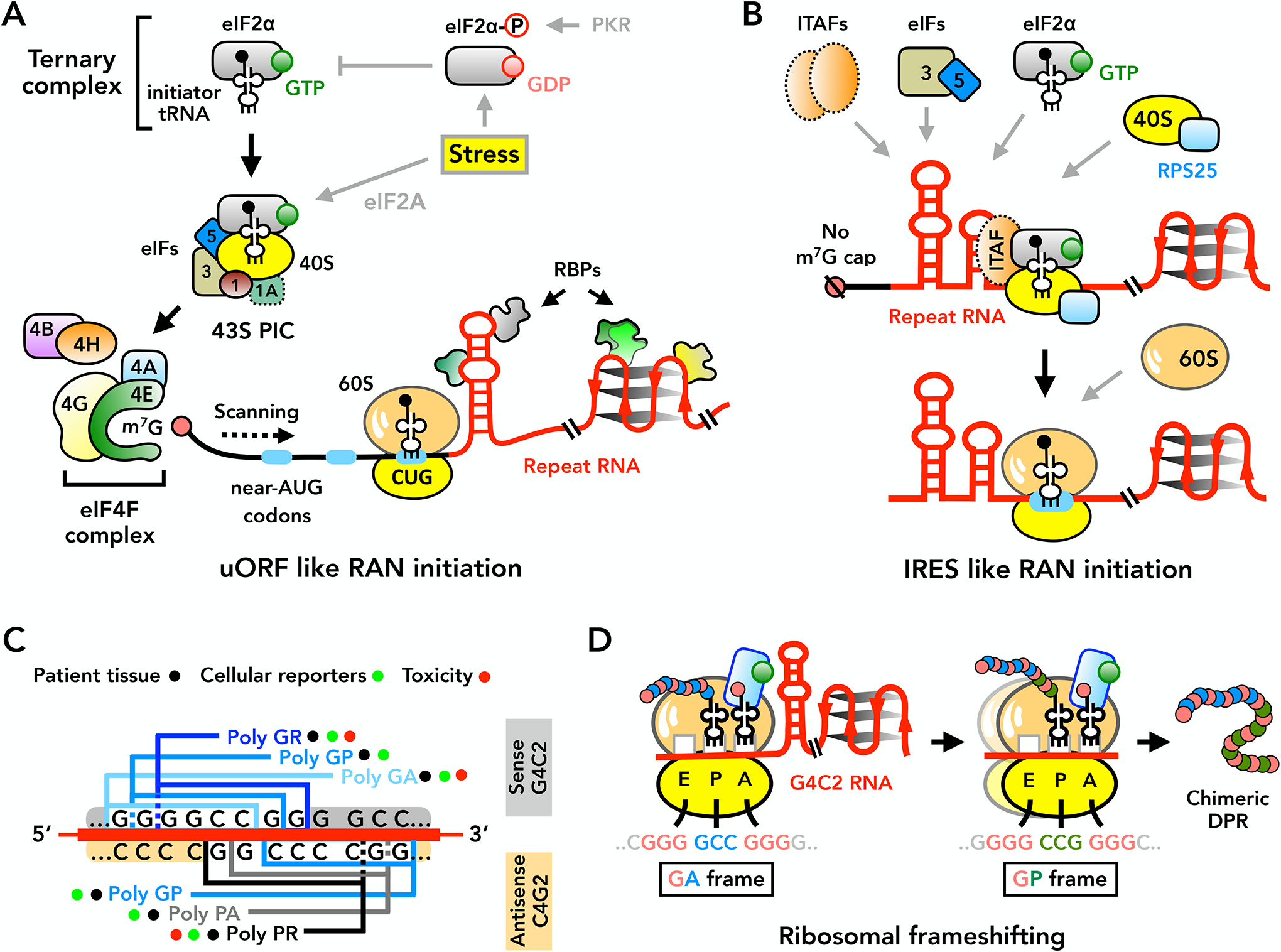
(A) Canonical AUG-mediated initiation and some forms of RAN translation require binding of eIF4F complex (eIF4E, eIF4G and eIF4A) to the 5’ m7G cap with eIF4B and/or eIF4H. After assembly, the 43S pre-initiation complex (PIC) scans 5’ to 3’ along the mRNA until selecting an AUG or near-AUG codon (for example: CUG) for initiation. eIF2α phosphorylation (eIF2α-P) under stress blocks ternary complex recycling and inhibits canonical translation, but allows for continued RAN translation. RBPs regulate RAN initiation by binding and altering repeat RNA structures. Known RAN-associated factors are depicted with solid lines, while canonical and IRES initiation factors involved in RAN translation are depicted with dashed lines. (B) RAN translation may also initiate through IRES-like mechanisms in a cap-independent manner, supported by RPS25 and other IRES-trans acting factors (ITAFs). (C) RAN translation from the C9ORF72 GGGGCC sense and CCCCGG antisense transcripts generates multiple dipeptide repeats (DPRs). While all DPRs are detected in patient tissues or generated by cellular reporters, arginine-containing DPRs show the highest intrinsic toxicity in model systems. (D) Stable RNA secondary structures formed by GGGGCC repeats induce ribosomal frameshifting during RAN translation, leading to production of chimeric DPRs. 40S and 60S = ribosomal subunits, eIF = eukaryotic initiation factor, IRES = internal ribosome entry site, m7G = 7-methylguanosine, PKR = Protein kinase R, RBPs = RNA-binding proteins, uORF = upstream open reading frame.
Initial studies of RAN translation suggested functional overlap with canonical mechanisms of translation initiation [Figure 4a]. Translation is typically a highly regulated step-wise process that starts with binding of the eukaryotic initiation factor complex 4F (consisting eIF4E, eIF4G, and eIF4A) to the 5′ m7G cap of the mRNA, recruitment of the small 40S ribosomal subunit, eIF2 binding to methionine tRNA (Met-tRNAMeti) to form the preinitiation complex (PIC), and finally scanning of the assembled PIC along the mRNA162. Recognition of the AUG start codon is promoted by eIF5, which triggers eIF2-GTP hydrolysis and initiation factor release, coupled with 60S subunit recruitment and formation of the first peptide bond. RAN translation at FMR1 CGG repeats proceeds efficiently in two of the three reading frames and is largely cap-dependent, requiring eIF4A in both cell-free systems and transfected cells163. In the GGC (polyG) reading frame, initiation occurs predominantly at near-cognate start codons [G] (ACG or GUG) just 5’ of the repeat163,164. In the GCG (polyA) reading frame, initiation occurs within the repeat itself, akin to observations at SCA8 CAG repeats152. Initiation in both reading frames is suppressed by overexpression of eIF1, which favors AUG codon usage, and enhanced by overexpression of eIF5, which relaxes the stringency of start codon selection165. Thus, RAN translation at CGG repeats mimics upstream open reading frames (uORFs) [G] that are ubiquitous in human genomes166 and results predominantly from repeat-induced decrements in start codon fidelity [Figure 4A].
However, at other repeats, findings suggest contributions from alternative initiation mechanisms. Sense and antisense transcripts from expanded C9ORF72 are translated into five different dipeptide repeat (DPR) peptides: polyGA, polyGR, polyGP, polyPR, and polyPA122,154,155,160 [Figure 4C]. As in the FMR1 CGG repeat, RAN translation of GGGGCC repeats within monocistronic mRNA reporters shows strong dependence on the 5’ m7G cap and on eIF4A-dependent scanning167,168. Similarly, a near-cognate CUG codon just 5’ of the repeat is critical for initiation in the polyGA reading frame167–170. However, studies using bicistronic reporters support RAN translation in all reading frames in a repeat length-dependent fashion169,171, suggesting that cap-independent internal ribosomal entry site [G] (IRES)-like initiation can occur within GGGGCC RNAs. Classically described in viral mRNAs, IRES-mediated translation bypasses the need for a 5’ cap by directly recruiting initiation factors and ribosomal components onto a structured mRNA sequence172. In some cases, the mRNA itself can mimic the initiator tRNA to enable initiation in the absence of any AUG or near-AUG codons. Such a mechanism may explain how intronic repeats could be translated in C9 ALS/FTD patient neurons. In support of this model, knockdown of RPS25, an IRES-associated 40S ribosomal subunit, strongly and selectively modifies RAN translation of GGGGCC and CAG repeats173 [Figure 4B].
RAN peptides generated from CGG, CAG, GGGGCC and CCCCGG repeats cause toxicity in various model systems153,156,160,174–176. Overexpression of pathogenic CGG repeats that support RAN translation of FMRpolyG leads to toxic phenotypes in cultured neurons, fly, and mouse models of FXTAS153,164,177. This toxicity depends on the ability of these repeats to be translated, as mutation of the near-AUG codons that support translation strongly suppresses phenotypes in flies and transgenic mice, while mutating these near-cognate start codons to AUG boosts FMRpolyG production and toxicity153,164,178,179. Moreover, antisense oligonucleotides (ASOs) [G] that selectively impede RAN translation of FMRpolyG enhance survival in human neurons with expanded CGG repeats180.
In C9 ALS/FTD, AUG-initiated expression of DPRs alone in the absence of the native repeat RNA sequence (accomplished via use of alternative codons for the DPR protein sequence) elicits toxicity that is equivalent to or often greater than that of the pure tandem repeats themselves175,176,181,182. Arginine-rich DPRs (polyGR and polyPR) in particular are highly toxic, inducing cell death in cultured primary and iPSC-derived neurons as well as eye degeneration and early mortality in Drosophila175,176,183–191. In mice, expression of polyGR or polyPR in isolation leads to early-onset and severe neurodegeneration with motor dysfunction and memory impairment189–191. PolyGA DPRs may also be important in disease, as they form filamentous amyloid-like structures and are moderately toxic in mammalian neurons176,183,192–194, while polyGP and polyPA exhibit limited toxicity in model systems. The mechanisms by which DPRs elicit toxicity is extensively discussed in recent reviews195–197.
Despite observations that DPRs and other RAN-generated proteins are sufficient to elicit toxicity, most of the evidence demonstrating their roles in disease pathogenesis to date have relied on overexpression systems, which may not faithfully recapitulate actual disease states. The relative toxicity of DPRs do not correlate with their relative rates of production in reporter systems167,168,171 or, for the most part, their relative abundance and sites of accumulation in pathological analyses198–201. Thus, it is not yet clear whether repeat RNA or DPRs alone are sufficient to explain human disease phenotypes. Studies selectively expressing DPRs at levels equivalent to the endogenous state or selective blockade of RAN translation in model systems are still needed to delineate these roles.
Modifiers of RAN translation
Multiple translation initiation factors, including orthologs of eIF4E, eIF4B, eIF4H, eIF5, eIF3D1 and eIF3I, modulate GGGGCC RAN translation in Drosophila202, and eIF3F modifies RAN translation of CAG and GGGGCC repeat reporters in mammalian cells203 [Figure 4A–B]. The eIF4A helicase co-stimulatory factors eIF4B and eIF4H also suppress CGG RAN translation in flies and mammalian reporter systems165, suggesting convergence around the activity of this helicase. Consistent with this, DDX3X, a DEAD-box helicase that binds CGG and GGGGCC repeat RNA, emerged as a key modulator of RAN translation from two independent screens165,204. However, the effect of DDX3X on RAN translation is complex: knockdown of DDX3X or its homolog in Drosophila inhibits CGG RAN translation and repeat-associated toxicity165, but it enhances GGGGCC RAN translation and toxicity in multiple systems, including patient iPSC-derived neurons204. These differences may reflect dependence on sequence context surrounding the repeats. The FMR1 5’ UTR is highly GC-rich outside the CGG repeat region; thus, DDX3X helicase activity is likely required to facilitate initiation at the canonical FMRP start codon even in the absence of expanded repeats. In contrast, if an IRES-like mechanism is critical for RAN initiation on GGGGCC repeats, then the loss of DDX3X may induce formation of RNA structures that support IRES mediated-ribosomal recruitment.
External stimuli, such as ER stress, viral infection and amino acid starvation, activate the integrated stress response (ISR), which leads to stress granule [G] formation and inhibition of global translation through phosphorylation of eIF2α205–207 [Figure 4A]. A subset of transcripts that initiate using non-AUG codons or via IRES-dependent mechanisms escape this translational suppression. Accordingly, ISR activation significantly enhances RAN translation of both CGG and GGGGCC reporters in a process dependent on eIF2α phosphorylation167,169–171,208. In C9ORF72, stress-induced escalation of GGGGCC RAN translation occurs with both cap-dependent and cap-independent bicistronic constructs167,169–171,208. Repeat RNAs and DPR proteins can independently activate the ISR, creating a potential positive feedback loop where cellular stress enhances RAN translation, which in turn elicits further stress167. Consistent with this concept, pharmacological or genetic suppression of PKR, which phosphorylates eIF2α to activate the ISR, reduces RAN translation in cells and improves disease phenotypes in a RAN mouse model of C9 ALS/FTD208. Similarly, knockout of the alternative initiation factor eIF2A, which allows for initiation when eIF2α is phosphorylated, partially suppresses RAN-initiated polyGA expression in C9 ALS/FTD models169 [Figure 4a].
Mechanistic synergy may drive disease
As alluded to in several cases above, multifactorial pathomechanisms may perhaps be the rule rather than the exception. However, each of the four mechanisms previously described has most commonly been studied separately, either due to limited availability of appropriate disease models and reagents, or to simplify interpretation by separating confounding variables. Indeed, the pathomechanisms described above are roughly chronological in their discovery, and newer mechanisms often have not been thoroughly investigated in diseases for which earlier mechanisms have provided reasonable explanations for pathology. More recently, new animal models and combinations of models have facilitated deliberate investigation of combinatorial effects. Several examples of multifactorial interactions among disease pathways are described below [Figure 5].
Figure 5: Synergy across pathogenic mechanisms in repeat expansion diseases.
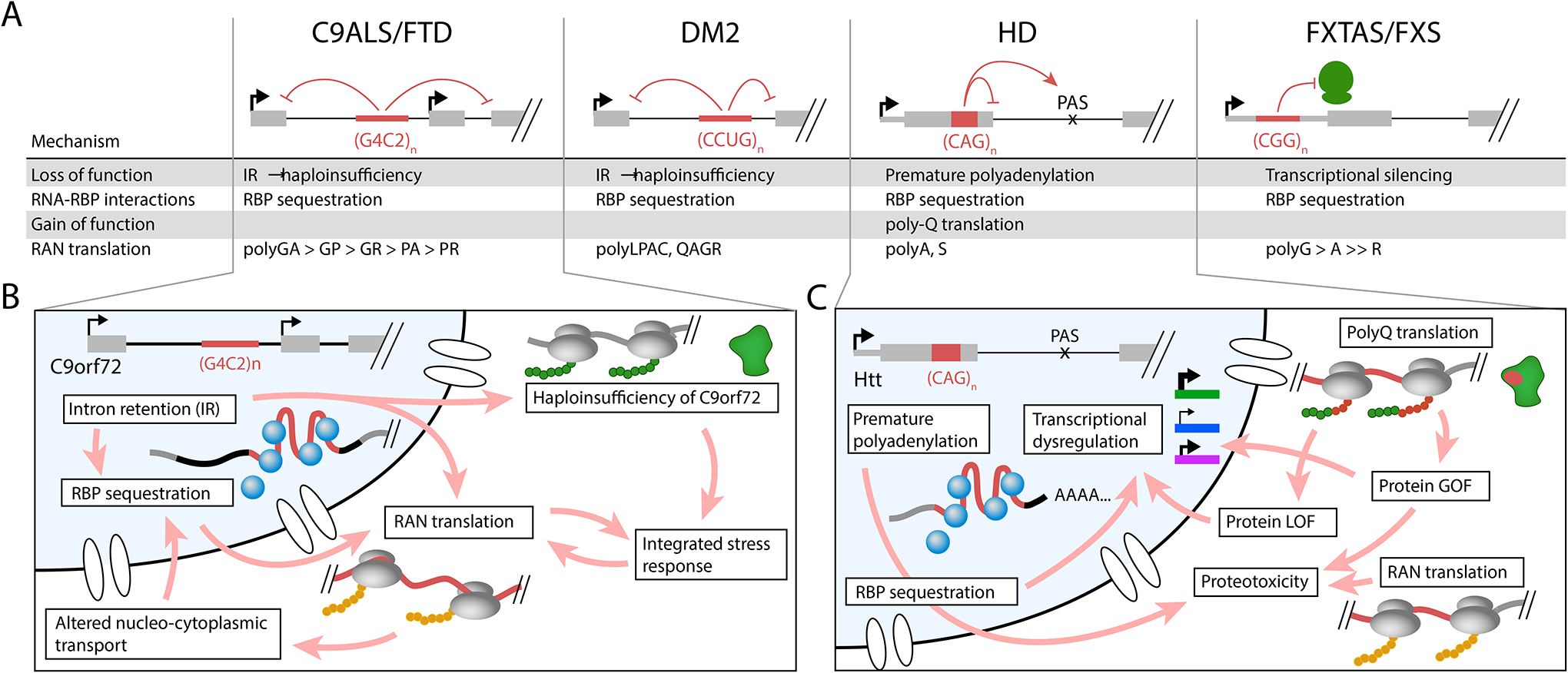
(A) In multiple diseases, the four major mechanisms detailed in this review can co-exist and/or synergize to drive complex pathology. For example, in C9 ALS/FTD, expanded GGGGCC repeats can induce intron retention, which leads to haploinsufficiency of C9ORF72, as well as exacerbates RBP sequestration by increasing the half-life of the repeat RNA. In addition, intron retention may increase the production of dipeptide repeats that activate numerous downstream pathogenic pathways. In DM2, expanded CCTG repeats lead to intron retention, which also results in reduction of mRNA available to generate full-length CNBP protein. RAN translation products can be generated from the intron-retained mRNA. In Huntington disease (HD), expanded CAG repeats alter RNA processing to impair recognition of the exon 1 donor splice site; this results in the formation of a truncated polyQ-containing HTT protein that is more toxic than full-length polyQ-containing HTT. RAN translation can also occur across the CAG repeat. In FXTAS/FXS, the CGG repeat can not only sequester RBPs, but can also enhance RAN translation of the uORF such that translation initiation for FMRP is reduced. (B) A more detailed view of pathways activated in C9 ALS/FTD shows that some pathogenic mechanisms can exacerbate or feed into other mechanisms. A complex network of cause and effect, including feed-forward loops, may synergize to drive disease pathology. (C) A more detailed view of pathways activated in HD also similarly reveals feedback loops in both the nucleus and cytoplasm.
Loss or gain of protein function due to alterations in RNA processing
Repeat expansions can alter RNA processing, and this phenomenon occurs in multiple repeat expansion diseases, resulting in intron retention209,210, changes in alternative transcription start site usage211,212, or premature polyadenylation213. Alterations to RNA processing can change the repertoire of transcribed isoforms, with some leading to LOF and others leading to production of proteins with GOF activity. For example, repeats can trigger intron retention, allowing for efficient export of repeat RNA into the cytoplasm171, where it may elicit toxicity directly and/or undergo RAN translation. Simultaneously, intron retention can trigger depletion of the full-length protein product. As such, haploinsufficiency can potentially compound the effects of expanded repeat expression, even when haploinsufficiency alone may be benign. In C9 ALS/FTD, multiple studies indicate that loss of C9ORF72 can synergize with GGGGCC expression to exacerbate symptoms in both C9BAC mice214,215 as well as an AAV-based model216. In contrast, in Huntington disease, expanded CAG repeats impair recognition of the donor splice site of HTT exon 1, leading to premature polyadenylation and translation of a truncated polyQ-containing HTT peptide213. This peptide may be much more toxic than full-length HTT containing expanded polyQ. Whether alterations to RNA processing and protein gain- or loss-of-function effects modulate symptoms in other repeat expansion diseases remains to be fully explored.
RNA gain of function and RAN translation
Although DM1 and DM2 have served as excellent examples in which repeat RNA acquires new functions to sequester RBPs, a clear pathogenic role for RBP sequestration has not been fully established in many other diseases. Many RBPs associate with GGGGCC, CAG, CGG, and other repeat RNAs, but in these contexts, the repetitive RNAs also generate pathogenic peptides. Efforts have been made to separate these effects, for example by using alternative codons to preserve protein sequence yet disrupt the simple tandem RNA repeats. In BACHD mice, a model of Huntington disease, a clear role for polyQ toxicity is suggested by the observation of progressive neurodegeneration upon CAGCAA repeat expression217. However, other studies highlight a role for RNA toxicity independent from protein218. For example, targeting CAG RNA with locked nucleic acid ASOs can ameliorate phenotypes in the R6/2 mouse model of Huntington disease, even when Htt protein is not perturbed219. In DM2, in which a role for RNA toxicity is well established, RAN peptides polyLPAC and polyQAGR also occur in various brain regions157 and may be modulated by the extent to which MBNL associates with CCUG RNA and prevents it from exiting the nucleus. However, clear pathogenic roles for these peptides remain to be defined. In C9 ALS/FTD, GGGGCC RNAs bind to hnRNP H and SRSF proteins and trigger splicing changes88,106,220, but it remains unclear whether RBP sequestration synergizes or competes with the effects of DPRs. A potential interaction between RNA sequestration and RAN translation has been proposed, in which GGGGCC RNAs associate with nucleolin58, which can also associate with DPRs175. In contrast, SRSF protein binding to GGGGCC repeats appears to influence the nucleocytoplasmic transport of repeat RNA and thus titrates its ability to undergo RAN translation221.
Production of multiple proteins from a single repeat-containing message
Because RAN translation can initiate in a variety of reading frames222, the contribution of each potential peptide repeat in disease has been challenging to separate. In C9 ALS/FTD, studies of each potential DPR have suggested that some DPRs are more toxic than others195. However, additional mechanisms such as frameshifting [G]168 may generate chimeric species and further complicate pathomechanisms [Figure 4D]. In vitro, mutation of the near-cognate CUG start codon that initiates polyGA translation modulates translation efficiency of not only polyGA, but of other reading frames as well168. Indeed, polyGA:polyGP chimeric peptides, presumably produced by frameshifting events, accumulate in C9 ALS/FTD and may account for differential toxicity compared to polyGP produced in SCA36223. Even in HD, the presence of non-canonical protein species from both sense and antisense transcripts156, 224–227 raises questions about whether polyQ is the sole protein-based driver. Sorely needed are basic studies to probe fundamental mechanisms of RAN translation initiation and frameshifting, as well as careful studies using disease samples to characterize the distribution and abundance of each potential species, to gain clarity on pathogenesis.
Further work is needed to determine how, and if, RAN proteins contribute to pathogenesis in humans. Most studies to date have relied on overexpression systems or peptides generated from AUG-initiated constructs. This ignores key elements related to the inefficiency of RAN translation from different repeats and the endogenous stoichiometry of their protein products in patient tissues. For example, while polyGR and polyPR are more toxic in isolation, polyGA is more efficiently translated in the absence of an AUG start codon and is more abundant in human patient brains155,167–171,198. PolyGA may act as a seeding factor for aggregation of other C9ORF72 RAN peptides such as polyGP and polyPA, which are otherwise soluble, or as a suppressor of polyGR toxicity186. In FXTAS, the absolute abundance of FMRpolyG in patients may be low, and its presence does not always correlate with relevant phenotypes in mice228,229. Moreover, both repeat RNA and RAN proteins are found in FXTAS inclusions, and these molecules may directly interact230. Thus, for each disease and RAN product, their relative contributions to toxicity will need to be correlated with their abundance and interaction with relevant pathways.
Mechanisms of tissue-specific pathogenesis
Despite often shared repeat sequences, repeat expansion diseases are largely syndromic, with incomplete overlap for a given repeat unit and its clinical presentation or its cell-type specific dysfunction16. In DM1, for example, while myotonia is a widely recognized characteristic symptom, many patients report that neurological symptoms, such as fatigue, hypersomnolence, and cognitive difficulties, are more debilitating231. Differences in pathogenicity of repeat expansions across tissues emerge from multiple interacting variables, including differential rates of somatic instability, host gene expression, and expression of trans factors. Somatic instability of the CTG repeat in DM1 produces alleles in skeletal muscle and brain as much as 13 times larger than in leukocytes125,232, likely exacerbating RNA toxicity in those tissues. Further, although neuromuscular symptoms are often more severe in DM1 than in DM2, alternative splicing biomarkers in blood are more pronounced in DM2, likely as a result of higher expression of CNBP in this tissue than DMPK233.
Recently, six of the seven known subtypes of familial adult myoclonic epilepsy (FAME, or BAFME), a slowly progressing disease primarily exhibiting myoclonic seizures and cortical tremor, were mapped to long intronic TTTCA expansions in various genes234–237. While much remains unknown about the pathogenesis of FAME, RNA toxicity has been proposed to play a role, as host gene expression appears generally unaffected and nuclear accumulations of UUUCA RNA have been observed in FAME1 patient brain234. Interestingly, an intronic TTTCA expansion is also associated with SCA37, an entirely different disease characterized by gait instability, limb ataxia, and nystagmus238. If GOF mechanisms of a TTTCA expansion drive pathogenesis in both diseases, how do they differ so extensively in disease symptoms? A leading hypothesis is that host gene expression patterns may partially explain this phenomenon, as while the FAME-linked genes are generally expressed throughout the brain, expression of the SCA37-linked DAB1 gene is more specific to the cerebellum235.
Still, much about the precise mechanisms of tissue-specific pathology remains unclear. For example, in Huntington disease, HTT is expressed in most tissues, and while its expression is indeed highest in the nervous system, HTT protein is not confined to areas susceptible to neurodegeneration239. In fact, while many other polyQ diseases cause pronounced cerebellar ataxia, this symptom is considered rare in HD, even though mutant HTT inclusions are observed in the cerebellum240.
A broader role for short tandem repeats
Most of the >50 disease-causing repeat expansions identified to date were found in association with highly penetrant clinical syndromes, which allowed for careful genetic analysis and exclusion of other mutations as potential causes. However, these relatively rare syndromic conditions may represent the tip of a much larger iceberg. STRs account for ~3% of genomic DNA, and a significant fraction reside within genes and their regulatory regions1,241 [Figure 6a]. As such, STRs can impact the structure and function of DNA, RNA, and proteins, with a range of molecular and cellular consequences. While such sequences have traditionally been viewed as “junk” DNA, evolutionary genomics suggests that STRs and other classes of DNA repeats have evolved under tight selective constraints242.
Figure 6: Roles of repeats in human disease and neuronal function.
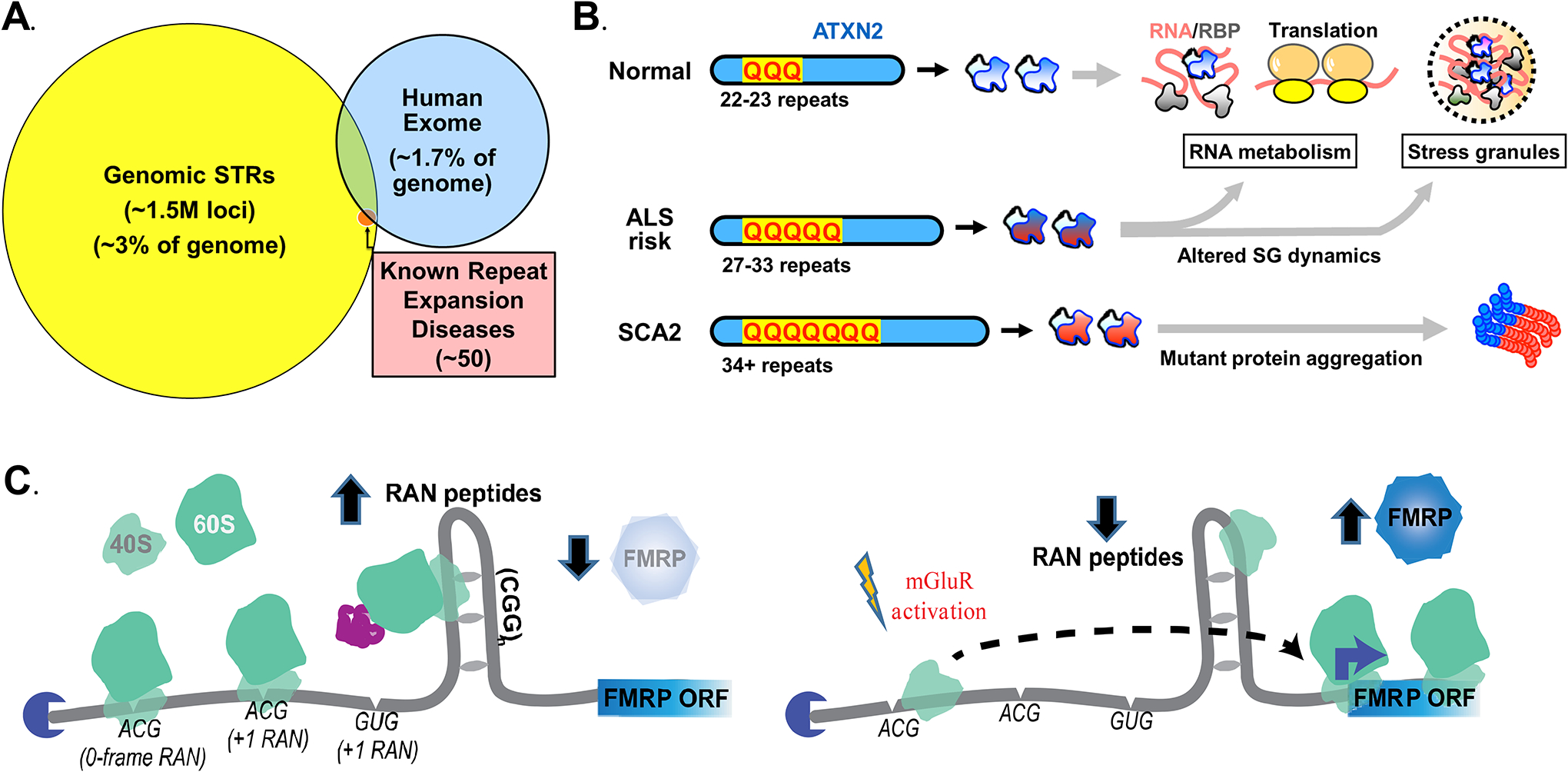
(A) Short tandem repeats represent ~3% of the human genome, with enrichment of specific elements within 5’ UTRs, ORFs, and introns. STR mutation rates are orders of magnitude higher than single nucleotide polymorphisms and their size can influence gene expression. (B) CAG repeats in ATXN2, which when fully expanded cause spinocerebellar ataxia type 2 (SCA2), act as risk alleles for development of ALS and other neurodegenerative disorders when the repeats are of intermediate size. Loss of ATXN2 suppresses ALS phenotypes in model systems. (C) The normal-length CGG repeat in FMR1, which when expanded causes FXS and FXTAS, serves to regulate translation of the FMR1 gene product, FMRP, in response to synaptic stimuli.
All of these factors suggest that variations in STRs could serve as risk alleles for non-Mendelian human disorders10,11. Consistent with this concept, some disease-associated repeats have been identified as risk alleles for other neurological conditions at smaller expansion sizes (Figure 6B). For example, intermediate-length CAG expansions in the ATXN2 gene, which at larger sizes are associated with SCA2, serve as a common risk allele for development of ALS and other neurodegenerative disorders243. A similar relationship with ALS and FTD phenotypes was recently identified for intermediate CAG expansions in ATXN1244–246 and for both intermediate and full mutations in HTT247. Recently, polyalanine expansions in NIPA1 and a more complex intronic tandem repeat in WDR7 were also associated with ALS with incomplete penetrance248,249, as was a CGG repeat expansion in NOTCH2NLC250.
While it is possible that these disease loci are unique examples for how variation in STR length might contribute to multiple diseases, recent studies suggest that other STRs broadly contribute to multiple diseases and biological phenotypes. First, improved measurement of STR length and instability genome-wide with standard next generation sequencing platforms4,251 suggests that a significant fraction of the signal for SNP markers used in genome-wide association (GWAS) studies may derive from tight linkage with STRs that modulate neighboring gene expression and RNA processing4. Second, gene-associated tandem repeat expansions at 2,588 loci are more prevalent among individuals with autism than their siblings or controls, particularly in exons and near splice junctions of genes related to nervous system development252,253. Future studies aimed at linking STR variation to disease risk across a broad spectrum of human disease are likely to be fruitful.
Emerging data suggests that STRs likely perform important native functions in the genes in which they reside, implying that their loss may also contribute to human disease10–12. The majority of repeat expansion disorders identified to date impact the nervous system, suggesting that STRs may have particular roles in these specialized cell types. STRs mutate more rapidly than single nucleotides, enabling them to hasten evolutionary adaptation by acting as tunable regulators of gene expression and function254. For example, yeast take advantage of the relative instability of repeats to allow proteins to have pleiotropic behaviors across a population of cells in response to environmental stressors255,256.
As such, the native roles of repeats in humans remain understudied, even in the context of known disease-causing loci. At normal sizes, CAG-encoded polyQ repeats in proteins such as HTT serve as flexible hinges that link functional domains257. This flexibility enables the multifunctional proteins to play key roles in events mediated by large biomolecular complexes, such as transcriptional regulation. As an example, elimination of CAG repeats in ATXN3, a deubiquitinase with roles in autophagy, impedes its normal function in cells258. The CGG repeat in FMR1 plays an active role in regulating FMRP translation at normal repeat sizes180. This CGG repeat serves as a RAN-translated uORF in the FMR1 5’ UTR that suppresses basal FMRP translation, while allowing for upregulation of FMRP expression in response to specific stimuli (Figure 6C). This finding is particularly intriguing, as nearly 100 other human genes, many involved in neuronal function, have CGG or CCG repeat elements in their 5’ UTRs.
Approaching newly discovered diseases
As a result of continuous technological improvements in recognition and detection, 28% of known disease-associated repeat expansions were mapped in the last four years alone, and more will undoubtedly be discovered. As new associations emerge, the advancements made over the past three decades should enable rapid evaluation of potential disease mechanisms and streamlining of therapeutic development. Initial assessments should draw comparisons to similar repeat loci and to diseases with similar clinical syndromes and pathologies. As an example, the discovery of a CCTG repeat expansion as the cause of DM2259 was rapidly followed by assessment of the association of MBNL proteins with CCUG RNA and identification of similar MBNL splicing abnormalities between DM1 and DM2260. However, the two repeats exhibit differential affinities for RBPs and reside in different genomic contexts. These aspects may help to explain clinical differences between the conditions and the lack of a congenital phenotype in DM2127.
Mechanistically, the first key question to answer for each new disease-associated repeat is whether dysfunction occurs primarily through GOF or LOF mechanisms, while recognizing that both modes may act in concert or in competition. This step is critical, as the emergence of modular technologies aimed at knockdown (eg. ASOs and siRNAs) or upregulation (eg. gene therapy or CRISPR-mediated activation) of specific genes can enable rapid movement toward therapies once this separator is defined. However, even when a primary loss of function is clear, it is important to consider the potential negative impacts of gene reactivation, as upregulation of repeat mRNA or RAN-translated proteins might elicit additional toxicity. In certain contexts, combinatorial approaches may be needed to target simultaneous pathomechanisms261.
Initial assessments in simple model systems, such as human and rodent cell lines, Drosophila, and C. elegans, have reliably generated valuable insights into these modes of toxicity and are useful for rapid screens of genetic modifiers and suppressors of relevant phenotypes. These preliminary observations are often robust and conserved across phylogenetic lines, but a rigorous approach must be taken to confirm such findings in more complex in vivo models, including rodents, larger mammals and human iPSC-derived neurons or organoids, which express the repeats from endogenous loci. Appropriate validation of the relative contributions from each branch of the repeat toxicity tree (DNA, RNA, AUG-initiated translation, and RAN translation) early in the therapeutic pipeline will significantly streamline development.
As an example of how this approach might be applied, we can examine a recently discovered GGC repeat expansion in the 5’ UTR of NOTCH2NLC262–264 [Table 1]. Expansion of this repeat from normal lengths (~20 GGCs) to >50 GGCs causes neuronal intranuclear inclusion disease (NIID) in East Asian populations, and this expansion has been linked to a number of other neurological disorders. Even prior to the discovery of a shared repeat motif, its pathological, radiological and clinical phenotypes were noted to overlap with FXTAS265. This suggests shared pathomechanisms between these two conditions, with specific differences likely dependent on the properties of the repeat-harboring genes. In silico analysis of the sequence just 5’ of the repeat in NOTCH2NLC reveals an AUG codon that could generate a polyG protein, resembling the FMRpolyG that is produced by RAN translation in FXTAS. Given that FMRpolyG readily forms inclusions in FXTAS patient tissue and that AUG-initiated translation is more efficient than RAN translation, this observation suggests that polyG proteotoxicity may be a major pathomechanism in NIID. If this is the case, then therapeutic strategies in development for FXTAS could be explored early on for NIID to determine whether mechanistic convergence might lead to a clinical breakthrough.
Future perspective
After three decades of research on nucleotide repeat expansion disorders, we now have a roadmap for many of the central mechanisms that drive disease pathogenesis. Yet, it remains important to stay humble in the face of what we do not know. A salient example is cerebellar ataxia, neuropathy, and vestibular areflexia syndrome (CANVAS), caused by a recently discovered biallelic expansion of intronic AAGGG repeats in RFC1266,267. This condition is unusual in at least two ways. First, the expansion often accompanies a shift in repeat sequence from AAAAG or AAAGG to AAGGG, but the exact repeat motif and structure varies geographically among patient populations268–270. Interestingly, the repeat tract falls within the poly(A) tail of an AluSx3 element, raising the possibility that genomic instability engendered by a retrotransposon may drive pathogenic expansion in RFC1. Second, despite the autosomal recessive inheritance pattern of CANVAS, the repeat does not appear to impact expression of RFC1 protein266, making loss of function less likely. Yet, to date, studies of pathology have not revealed evidence of RNA foci or aggregated proteins in affected tissues, drawing classical gain-of-function mechanisms into question as well. The pathomechanisms that drive CANVAS, a disease caused by a unique and complex repeat expansion, remain undefined and yearn to be understood. Based on the past three decades, we expect that the solution to this newest conundrum will again change the way we think about expanded repeats and human disease.
Acknowledgements:
This work was supported by National Institute of Health grants NS099280, NS086810, and P50HD104463 and VA BLRD BX004842 to PKT, and AG058636, R01NS112291, and R01NS114253 to ETW. IM was supported by an Alzheimer’s Association Research Fellowship (AARF), AARF-20-684648. CPK is supported by the National Science Foundation Graduate Research Fellowship Program (NSF GRFP).
References
- 1.Lander ES et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Kruglyak S, Durrett RT, Schug MD & Aquadro CF Equilibrium distributions of microsatellite repeat length resulting from a balance between slippage events and point mutations. Proc. Natl. Acad. Sci. U. S. A 95, 10774–10778 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quilez J et al. Polymorphic tandem repeats within gene promoters act as modifiers of gene expression and DNA methylation in humans. Nucleic Acids Res 44, 3750–3762 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fotsing SF et al. The impact of short tandem repeat variation on gene expression. Nat. Genet 51, 1652–1659 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fu YH et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 67, 1047–1058 (1991). [DOI] [PubMed] [Google Scholar]
- 6.Oberlé I et al. Instability of a 550-Base Pair DNA Segment and Abnormal Methylation in Fragile X Syndrome. Science 252, 1097–1102 (1991). [DOI] [PubMed] [Google Scholar]
- 7.Verkerk AJ et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914 (1991). [DOI] [PubMed] [Google Scholar]
- 8.Kremer EJ et al. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science 252, 1711–1714 (1991). [DOI] [PubMed] [Google Scholar]
- 9.La Spada AR, Wilson EM, Lubahn DB, Harding AE & Fischbeck KH Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352, 77–79 (1991). [DOI] [PubMed] [Google Scholar]
- 10.Hannan AJ Tandem repeats mediating genetic plasticity in health and disease. Nat. Rev. Genet 19, 286–298 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Gymrek M A genomic view of short tandem repeats. Curr. Opin. Genet. Dev 44, 9–16 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Balzano E, Pelliccia F & Giunta S Genome (in)stability at tandem repeats. Semin. Cell Dev. Biol (2020) doi: 10.1016/j.semcdb.2020.10.003. [DOI] [PubMed] [Google Scholar]
- 13.La Spada AR & Taylor JP Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat. Rev. Genet 11, 247–258 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nussbacher JK, Tabet R, Yeo GW & Lagier-Tourenne C Disruption of RNA Metabolism in Neurological Diseases and Emerging Therapeutic Interventions. Neuron 102, 294–320 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khristich AN & Mirkin SM On the wrong DNA track: Molecular mechanisms of repeat-mediated genome instability. J. Biol. Chem 295, 4134–4170 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paulson H Repeat expansion diseases. Handb. Clin. Neurol 147, 105–123 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wells RD & Ashizawa T Genetic Instabilities and Neurological Diseases (Elsevier, 2006). [Google Scholar]
- 18.Martin JP & Bell J A PEDIGREE OF MENTAL DEFECT SHOWING SEX-LINKAGE. J. Neurol. Psychiatry 6, 154–157 (1943). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lubs HA A marker X chromosome. Am. J. Hum. Genet 21, 231–244 (1969). [PMC free article] [PubMed] [Google Scholar]
- 20.Eichler EE et al. Evolution of the cryptic FMR1 CGG repeat. Nat. Genet 11, 301–308 (1995). [DOI] [PubMed] [Google Scholar]
- 21.Sutcliffe JS et al. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet 1, 397–400 (1992). [DOI] [PubMed] [Google Scholar]
- 22.Coffee B, Zhang F, Ceman S, Warren ST & Reines D Histone modifications depict an aberrantly heterochromatinized FMR1 gene in fragile x syndrome. Am. J. Hum. Genet 71, 923–932 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gedeon AK et al. Fragile X syndrome without CCG amplification has an FMR1 deletion. Nat. Genet 1, 341–344 (1992). [DOI] [PubMed] [Google Scholar]
- 24.De Boulle K et al. A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat. Genet 3, 31–35 (1993). [DOI] [PubMed] [Google Scholar]
- 25.Santoro MR, Bray SM & Warren ST Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu. Rev. Pathol 7, 219–245 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Usdin K & Kumari D Repeat-mediated epigenetic dysregulation of the FMR1 gene in the fragile X-related disorders. Front. Genet 6, 192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Colak D et al. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 343, 1002–1005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumari D & Usdin K Polycomb group complexes are recruited to reactivated FMR1 alleles in Fragile X syndrome in response to FMR1 transcription. Hum. Mol. Genet 23, 6575–6583 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eiges R et al. Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell 1, 568–577 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Kumari D, Sciascia N & Usdin K Small Molecules Targeting H3K9 Methylation Prevent Silencing of Reactivated FMR1 Alleles in Fragile X Syndrome Patient Derived Cells. Genes 11, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu XS et al. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 172, 979–992.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This group used Tet1-dCas9 targeted to the CGG repeat to drive demethylation of the FMR1 locus and gene reactivation in iPSC-derived neurons, resulting in phenotypic correction.
- 32.Chiurazzi P, Pomponi MG, Willemsen R, Oostra BA & Neri G In vitro reactivation of the FMR1 gene involved in fragile X syndrome. Hum. Mol. Genet 7, 109–113 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Godde JS, Kass SU, Hirst MC & Wolffe AP Nucleosome assembly on methylated CGG triplet repeats in the fragile X mental retardation gene 1 promoter. J. Biol. Chem 271, 24325–24328 (1996). [DOI] [PubMed] [Google Scholar]
- 34.Liu EY et al. C9orf72 hypermethylation protects against repeat expansion-associated pathology in ALS/FTD. Acta Neuropathol. (Berl.) 128, 525–541 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xi Z et al. Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am. J. Hum. Genet 92, 981–989 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Russ J et al. Hypermethylation of repeat expanded C9orf72 is a clinical and molecular disease modifier. Acta Neuropathol. (Berl.) 129, 39–52 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xi Z et al. The C9orf72 repeat expansion itself is methylated in ALS and FTLD patients. Acta Neuropathol. (Berl.) 129, 715–727 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Gijselinck I et al. The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol. Psychiatry 21, 1112–1124 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gu Y, Shen Y, Gibbs RA & Nelson DL Identification of FMR2, a novel gene associated with the FRAXE CCG repeat and CpG island. Nat. Genet 13, 109–113 (1996). [DOI] [PubMed] [Google Scholar]
- 40.Gecz J, Gedeon AK, Sutherland GR & Mulley JC Identification of the gene FMR2, associated with FRAXE mental retardation. Nat. Genet 13, 105–108 (1996). [DOI] [PubMed] [Google Scholar]
- 41.Campuzano V et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271, 1423–1427 (1996). [DOI] [PubMed] [Google Scholar]
- 42.Gottesfeld JM Molecular Mechanisms and Therapeutics for the GAA·TTC Expansion Disease Friedreich Ataxia. Neurother. J. Am. Soc. Exp. Neurother 16, 1032–1049 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bidichandani SI, Ashizawa T & Patel PI The GAA triplet-repeat expansion in Friedreich ataxia interferes with transcription and may be associated with an unusual DNA structure. Am. J. Hum. Genet 62, 111–121 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodden LN et al. Methylated and unmethylated epialleles support variegated epigenetic silencing in Friedreich ataxia. Hum. Mol. Genet 29, 3818–3829 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sakamoto N et al. Sticky DNA: self-association properties of long GAA.TTC repeats in R.R.Y triplex structures from Friedreich’s ataxia. Mol. Cell 3, 465–475 (1999). [DOI] [PubMed] [Google Scholar]
- 46.De Biase I, Chutake YK, Rindler PM & Bidichandani SI Epigenetic silencing in Friedreich ataxia is associated with depletion of CTCF (CCCTC-binding factor) and antisense transcription. PloS One 4, e7914 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mikaeili H, Sandi M, Bayot A, Al-Mahdawi S & Pook MA FAST-1 antisense RNA epigenetically alters FXN expression. Sci. Rep 8, 17217 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eimer H et al. RNA-Dependent Epigenetic Silencing Directs Transcriptional Downregulation Caused by Intronic Repeat Expansions. Cell 174, 1095–1105.e11 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Li L, Matsui M & Corey DR Activating frataxin expression by repeat-targeted nucleic acids. Nat. Commun 7, 10606 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Punga T & Bühler M Long intronic GAA repeats causing Friedreich ataxia impede transcription elongation. EMBO Mol. Med 2, 120–129 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim E, Napierala M & Dent SYR Hyperexpansion of GAA repeats affects post-initiation steps of FXN transcription in Friedreich’s ataxia. Nucleic Acids Res 39, 8366–8377 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kumari D, Biacsi RE & Usdin K Repeat expansion affects both transcription initiation and elongation in friedreich ataxia cells. J. Biol. Chem 286, 4209–4215 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reddy K et al. Determinants of R-loop formation at convergent bidirectionally transcribed trinucleotide repeats. Nucleic Acids Res 39, 1749–1762 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Loomis EW, Sanz LA, Chédin F & Hagerman PJ Transcription-Associated R-Loop Formation across the Human FMR1 CGG-Repeat Region. PLOS Genet 10, e1004294 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abu Diab M et al. The G-rich Repeats in FMR1 and C9orf72 Loci Are Hotspots for Local Unpairing of DNA. Genetics 210, 1239–1252 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robin G et al. Calcium dysregulation and Cdk5-ATM pathway involved in a mouse model of fragile X-associated tremor/ataxia syndrome. Hum. Mol. Genet 26, 2649–2666 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Farg MA, Konopka A, Soo KY, Ito D & Atkin JD The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum. Mol. Genet 26, 2882–2896 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Haeusler AR et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507, 195–200 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lin Y, Dent SYR, Wilson JH, Wells RD & Napierala M R loops stimulate genetic instability of CTG.CAG repeats. Proc. Natl. Acad. Sci. U. S. A 107, 692–697 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Massey TH & Jones L The central role of DNA damage and repair in CAG repeat diseases. Dis. Model. Mech 11, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Askeland G et al. Increased nuclear DNA damage precedes mitochondrial dysfunction in peripheral blood mononuclear cells from Huntington’s disease patients. Sci. Rep 8, 9817 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moshell AN, Tarone RE, Barrett SF & Robbins JH Radiosensitivity in Huntington’s disease: implications for pathogenesis and presymptomatic diagnosis. Lancet Lond. Engl 1, 9–11 (1980). [DOI] [PubMed] [Google Scholar]
- 63.Xiao H et al. A polyglutamine expansion disease protein sequesters PTIP to attenuate DNA repair and increase genomic instability. Hum. Mol. Genet 21, 4225–4236 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.López Castel A, Cleary JD & Pearson CE Repeat instability as the basis for human diseases and as a potential target for therapy. Nat. Rev. Mol. Cell Biol 11, 165–170 (2010). [DOI] [PubMed] [Google Scholar]
- 65.Reddy K et al. Processing of double-R-loops in (CAG)·(CTG) and C9orf72 (GGGGCC)·(GGCCCC) repeats causes instability. Nucleic Acids Res 42, 10473–10487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pearson CE, Ewel A, Acharya S, Fishel RA & Sinden RR Human MSH2 binds to trinucleotide repeat DNA structures associated with neurodegenerative diseases. Hum. Mol. Genet 6, 1117–1123 (1997). [DOI] [PubMed] [Google Scholar]
- 67.Keogh N, Chan KY, Li G-M & Lahue RS MutSβ abundance and Msh3 ATP hydrolysis activity are important drivers of CTG•CAG repeat expansions. Nucleic Acids Res 45, 10068–10078 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Neil AJ et al. Replication-independent instability of Friedreich’s ataxia GAA repeats during chronological aging. Proc. Natl. Acad. Sci 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gonitel R et al. DNA instability in postmitotic neurons. Proc. Natl. Acad. Sci 105, 3467–3472 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease. Cell 162, 516–526 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee J-M et al. A modifier of Huntington’s disease onset at the MLH1 locus. Hum. Mol. Genet 26, 3859–3867 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bettencourt C et al. DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases: DNA Repair Pathways Modify polyQ Disease Onset. Ann. Neurol 79, 983–990 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kovalenko M et al. Msh2 acts in medium-spiny striatal neurons as an enhancer of CAG instability and mutant huntingtin phenotypes in Huntington’s disease knock-in mice. PloS One 7, e44273 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pinto RM et al. Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington’s disease mice: genome-wide and candidate approaches. PLoS Genet 9, e1003930 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nakamori M et al. A slipped-CAG DNA-binding small molecule induces trinucleotide-repeat contractions in vivo. Nat. Genet 52, 146–159 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This group identified small molecule binders to CTG/CAG slipouts that form during repeat transcription and replication that favored MMR-dependent repeat contraction in patient cells.
- 76.Taneja KL, McCurrach M, Schalling M, Housman D & Singer RH Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol 128, 995–1002 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang N & Ashizawa T RNA toxicity and foci formation in microsatellite expansion diseases. Curr. Opin. Genet. Dev 44, 17–29 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.White MC et al. Inactivation of hnRNP K by Expanded Intronic AUUCU Repeat Induces Apoptosis Via Translocation of PKCδ to Mitochondria in Spinocerebellar Ataxia 10. PLOS Genet 6, e1000984 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mizielinska S et al. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. (Berl.) 126, 845–857 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cooper-Knock J et al. Antisense RNA foci in the motor neurons of C9ORF72-ALS patients are associated with TDP-43 proteinopathy. Acta Neuropathol. (Berl.) 130, 63–75 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Burguete AS et al. GGGGCC microsatellite RNA is neuritically localized, induces branching defects, and perturbs transport granule function. eLife 4, e08881 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mori K et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. (Berl.) 126, 881–893 (2013). [DOI] [PubMed] [Google Scholar]
- 83.Botta A et al. The CTG repeat expansion size correlates with the splicing defects observed in muscles from myotonic dystrophy type 1 patients. J. Med. Genet 45, 639–646 (2008). [DOI] [PubMed] [Google Scholar]
- 84.Wojciechowska M & Krzyzosiak WJ Cellular toxicity of expanded RNA repeats: focus on RNA foci. Hum. Mol. Genet 20, 3811–3821 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Krzyzosiak WJ et al. Triplet repeat RNA structure and its role as pathogenic agent and therapeutic target. Nucleic Acids Res 40, 11–26 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fratta P et al. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci. Rep 2, 1016 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Reddy K, Zamiri B, Stanley SYR, Macgregor RB & Pearson CE The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J. Biol. Chem 288, 9860–9866 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Conlon EG et al. The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. eLife 5, e17820 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mooers BHM, Logue JS & Berglund JA The structural basis of myotonic dystrophy from the crystal structure of CUG repeats. Proc. Natl. Acad. Sci. U. S. A 102, 16626–16631 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tian B et al. Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-dependent protein kinase PKR. RNA N. Y. N 6, 79–87 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.van Cruchten RTP, Wieringa B & Wansink DG Expanded CUG repeats in DMPK transcripts adopt diverse hairpin conformations without influencing the structure of the flanking sequences. RNA N. Y. N 25, 481–495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jain A & Vale RD RNA phase transitions in repeat expansion disorders. Nature 546, 243–247 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This group demonstrated that repeat RNAs form phase-separated droplets by gelation in vitro and in cellular nuclei, with different properties dependent on the repeat length and sequence.
- 93.Querido E, Gallardo F, Beaudoin M, Ménard C & Chartrand P Stochastic and reversible aggregation of mRNA with expanded CUG-triplet repeats. J. Cell Sci 124, 1703–1714 (2011). [DOI] [PubMed] [Google Scholar]
- 94.Miller JW et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J 19, 4439–4448 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Van Treeck B & Parker R Emerging Roles for Intermolecular RNA-RNA Interactions in RNP Assemblies. Cell 174, 791–802 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li P et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336–340 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Banjade S & Rosen MK Phase transitions of multivalent proteins can promote clustering of membrane receptors. eLife 3, e04123 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lin Y, Protter DSW, Rosen MK & Parker R Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Mol. Cell 60, 208–219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Loughlin FE et al. Tandem RNA binding sites induce self-association of the stress granule marker protein TIA-1. Nucleic Acids Res (2021) doi: 10.1093/nar/gkab080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rhine K, Vidaurre V & Myong S RNA Droplets. Annu. Rev. Biophys 49, 247–265 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang ET et al. Transcriptome alterations in myotonic dystrophy skeletal muscle and heart. Hum. Mol. Genet 28, 1312–1321 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kanadia RN et al. A muscleblind knockout model for myotonic dystrophy. Science 302, 1978–1980 (2003). [DOI] [PubMed] [Google Scholar]
- 103.Mankodi A et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 289, 1769–1773 (2000). [DOI] [PubMed] [Google Scholar]
- 104.Kanadia RN et al. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc. Natl. Acad. Sci 103, 11748–11753 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper and earlier work (ref. 102) by the same group established in mice that muscleblind protein sequestration by CUG repeat RNA was sufficient to explain most of the muscle phenotypes observed in myotonic dystrophy type 1.
- 105.Cooper-Knock J et al. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain J. Neurol 137, 2040–2051 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee Y-B et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep 5, 1178–1186 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang K et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mori K et al. hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. (Berl.) 125, 413–423 (2013). [DOI] [PubMed] [Google Scholar]
- 109.Sellier C et al. Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep 3, 869–880 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sofola OA et al. RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron 55, 565–571 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jin P et al. Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron 55, 556–564 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Batra R et al. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol. Cell 56, 311–322 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Prudencio M et al. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci 18, 1175–1182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Masuda A et al. CUGBP1 and MBNL1 preferentially bind to 3′ UTRs and facilitate mRNA decay. Sci. Rep 2, 209 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Farina KL, Huttelmaier S, Musunuru K, Darnell R & Singer RH Two ZBP1 KH domains facilitate beta-actin mRNA localization, granule formation, and cytoskeletal attachment. J. Cell Biol 160, 77–87 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang ET et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell 150, 710–724 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Taliaferro JM et al. Distal Alternative Last Exons Localize mRNAs to Neural Projections. Mol. Cell 61, 821–833 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Müller-McNicoll M, Rossbach O, Hui J & Medenbach J Auto-regulatory feedback by RNA-binding proteins. J. Mol. Cell Biol 11, 930–939 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Konieczny P, Stepniak-Konieczna E & Sobczak K MBNL expression in autoregulatory feedback loops. RNA Biol 15, 1–8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.de Mezer M, Wojciechowska M, Napierala M, Sobczak K & Krzyzosiak WJ Mutant CAG repeats of Huntingtin transcript fold into hairpins, form nuclear foci and are targets for RNA interference. Nucleic Acids Res 39, 3852–3863 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mykowska A, Sobczak K, Wojciechowska M, Kozlowski P & Krzyzosiak WJ CAG repeats mimic CUG repeats in the misregulation of alternative splicing. Nucleic Acids Res 39, 8938–8951 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gendron TF et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. (Berl.) 126, 829–844 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kino Y et al. Nuclear localization of MBNL1: splicing-mediated autoregulation and repression of repeat-derived aberrant proteins. Hum. Mol. Genet 24, 740–756 (2015). [DOI] [PubMed] [Google Scholar]
- 124.Tran H et al. Differential Toxicity of Nuclear RNA Foci versus Dipeptide Repeat Proteins in a Drosophila Model of C9ORF72 FTD/ALS. Neuron 87, 1207–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Thornton CA, Johnson K & Moxley RT Myotonic dystrophy patients have larger CTG expansions in skeletal muscle than in leukocytes. Ann. Neurol 35, 104–107 (1994). [DOI] [PubMed] [Google Scholar]
- 126.Otero BA et al. Transcriptome alterations in myotonic dystrophy frontal cortex. Cell Rep 34, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sellier C et al. rbFOX1/MBNL1 competition for CCUG RNA repeats binding contributes to myotonic dystrophy type 1/type 2 differences. Nat. Commun 9, 2009 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This group established that RBFOX proteins compete with MBNL for binding sites on CCUG repeat RNAs, proposing a novel mechanism by which RBP sequestration and toxicity can be attenuated by competition between proteins.
- 128.Carrell ST et al. Dmpk gene deletion or antisense knockdown does not compromise cardiac or skeletal muscle function in mice. Hum. Mol. Genet 25, 4328–4338 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hsu T-C et al. Deactivation of TBP contributes to SCA17 pathogenesis. Hum. Mol. Genet 23, 6878–6893 (2014). [DOI] [PubMed] [Google Scholar]
- 130.Lieberman AP, Shakkottai VG & Albin RL Polyglutamine Repeats in Neurodegenerative Diseases. Annu. Rev. Pathol 14, 1–27 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bäuerlein FJB et al. In Situ Architecture and Cellular Interactions of PolyQ Inclusions. Cell 171, 179–187.e10 (2017). [DOI] [PubMed] [Google Scholar]
- 132.Peskett TR et al. A Liquid to Solid Phase Transition Underlying Pathological Huntingtin Exon1 Aggregation. Mol. Cell 70, 588–601.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Scherzinger E et al. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90, 549–558 (1997). [DOI] [PubMed] [Google Scholar]
- 134.Paulson HL et al. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 19, 333–344 (1997). [DOI] [PubMed] [Google Scholar]
- 135.Ordway JM et al. Ectopically expressed CAG repeats cause intranuclear inclusions and a progressive late onset neurological phenotype in the mouse. Cell 91, 753–763 (1997). [DOI] [PubMed] [Google Scholar]
- 136.Mangiarini L et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506 (1996). [DOI] [PubMed] [Google Scholar]
- 137.Schilling G et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum. Mol. Genet 8, 397–407 (1999). [DOI] [PubMed] [Google Scholar]
- 138.Yamamoto A, Lucas JJ & Hen R Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell 101, 57–66 (2000). [DOI] [PubMed] [Google Scholar]
- 139.Riley BE & Orr HT Polyglutamine neurodegenerative diseases and regulation of transcription: assembling the puzzle. Genes Dev 20, 2183–2192 (2006). [DOI] [PubMed] [Google Scholar]
- 140.Katsuno M et al. Reversible Disruption of Dynactin 1-Mediated Retrograde Axonal Transport in Polyglutamine-Induced Motor Neuron Degeneration. J. Neurosci 26, 12106–12117 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Katsuno M et al. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 35, 843–854 (2002). [DOI] [PubMed] [Google Scholar]
- 142.Montie HL et al. Cytoplasmic retention of polyglutamine-expanded androgen receptor ameliorates disease via autophagy in a mouse model of spinal and bulbar muscular atrophy. Hum. Mol. Genet 18, 1937–1950 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Palazzolo I et al. Akt blocks ligand binding and protects against expanded polyglutamine androgen receptor toxicity. Hum. Mol. Genet 16, 1593–1603 (2007). [DOI] [PubMed] [Google Scholar]
- 144.Irwin S et al. RNA association and nucleocytoplasmic shuttling by ataxin-1. J. Cell Sci 118, 233–242 (2005). [DOI] [PubMed] [Google Scholar]
- 145.Lam YC et al. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell 127, 1335–1347 (2006). [DOI] [PubMed] [Google Scholar]
- 146.Klement IA et al. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell 95, 41–53 (1998). [DOI] [PubMed] [Google Scholar]
- 147.Lai S, O’Callaghan B, Zoghbi HY & Orr HT 14–3-3 Binding to Ataxin-1(ATXN1) Regulates Its Dephosphorylation at Ser-776 and Transport to the Nucleus. J. Biol. Chem 286, 34606–34616 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Emamian ES et al. Serine 776 of Ataxin-1 Is Critical for Polyglutamine-Induced Disease in SCA1 Transgenic Mice. Neuron 38, 375–387 (2003). [DOI] [PubMed] [Google Scholar]
- 149.Duvick L et al. SCA1-like disease in mice expressing wild-type ataxin-1 with a serine to aspartic acid replacement at residue 776. Neuron 67, 929–935 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This group established that altering the phosphorylation state of ataxin-1 was sufficient to elicit toxicity even in the absence of a polyglutamine expansion, confirming that aberrant native protein function plays a role in its pathogenesis.
- 150.Williams AJ & Paulson HL Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci 31, 521–528 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Klockgether T, Mariotti C & Paulson HL Spinocerebellar ataxia. Nat. Rev. Dis. Primer 5, 24 (2019). [DOI] [PubMed] [Google Scholar]
- 152.Zu T et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. U. S. A 108, 260–265 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This group discovered that repeat RNAs can be translated in the absence of an AUG start codon, leading to recognition of this event in many repeat expansion disorders.
- 153.Todd PK et al. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 78, 440–455 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ash PEA et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mori K et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338 (2013). [DOI] [PubMed] [Google Scholar]
- 156.Bañez-Coronel M et al. RAN Translation in Huntington Disease. Neuron 88, 667–677 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zu T et al. RAN Translation Regulated by Muscleblind Proteins in Myotonic Dystrophy Type 2. Neuron 95, 1292–1305.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Soragni E et al. Repeat-Associated Non-ATG (RAN) Translation in Fuchs’ Endothelial Corneal Dystrophy. Invest. Ophthalmol. Vis. Sci 59, 1888–1896 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ishiguro T et al. Regulatory Role of RNA Chaperone TDP-43 for RNA Misfolding and Repeat-Associated Translation in SCA31. Neuron 94, 108–124.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zu T et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. U. S. A 110, E4968–4977 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Krans A, Kearse MG & Todd PK Repeat-associated non-AUG translation from antisense CCG repeats in fragile X tremor/ataxia syndrome. Ann. Neurol 80, 871–881 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Jackson RJ, Hellen CUT & Pestova TV The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol 11, 113–127 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kearse MG et al. CGG Repeat-Associated Non-AUG Translation Utilizes a Cap-Dependent Scanning Mechanism of Initiation to Produce Toxic Proteins. Mol. Cell 62, 314–322 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sellier C et al. Translation of Expanded CGG Repeats into FMRpolyG Is Pathogenic and May Contribute to Fragile X Tremor Ataxia Syndrome. Neuron 93, 331–347 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Linsalata AE et al. DDX3X and specific initiation factors modulate FMR1 repeat-associated non-AUG-initiated translation. EMBO Rep 20, e47498 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kearse MG & Wilusz JE Non-AUG translation: a new start for protein synthesis in eukaryotes. Genes Dev 31, 1717–1731 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Green KM et al. RAN translation at C9orf72-associated repeat expansions is selectively enhanced by the integrated stress response. Nat. Commun 8, 2005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Tabet R et al. CUG initiation and frameshifting enable production of dipeptide repeat proteins from ALS/FTD C9ORF72 transcripts. Nat. Commun 9, 152 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Sonobe Y et al. Translation of dipeptide repeat proteins from the C9ORF72 expanded repeat is associated with cellular stress. Neurobiol. Dis 116, 155–165 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Westergard T et al. Repeat-associated non-AUG translation in C9orf72-ALS/FTD is driven by neuronal excitation and stress. EMBO Mol. Med 11, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Cheng W et al. C9ORF72 GGGGCC repeat-associated non-AUG translation is upregulated by stress through eIF2α phosphorylation. Nat. Commun 9, 51 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kwan T & Thompson SR Noncanonical Translation Initiation in Eukaryotes. Cold Spring Harb. Perspect. Biol 11, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yamada SB et al. RPS25 is required for efficient RAN translation of C9orf72 and other neurodegenerative disease-associated nucleotide repeats. Nat. Neurosci 22, 1383–1388 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.May S et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. (Berl.) 128, 485–503 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wen X et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84, 1213–1225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mizielinska S et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345, 1192–1194 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Oh SY et al. RAN translation at CGG repeats induces ubiquitin proteasome system impairment in models of fragile X-associated tremor ataxia syndrome. Hum. Mol. Genet 24, 4317–4326 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hukema RK et al. Reversibility of neuropathology and motor deficits in an inducible mouse model for FXTAS. Hum. Mol. Genet 24, 4948–4957 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Castro H et al. Selective rescue of heightened anxiety but not gait ataxia in a premutation 90CGG mouse model of Fragile X-associated tremor/ataxia syndrome. Hum. Mol. Genet 26, 2133–2145 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Rodriguez CM et al. A native function for RAN translation and CGG repeats in regulating fragile X protein synthesis. Nat. Neurosci 23, 386–397 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This group showed that selective targeting of RAN translation initiation with non-cleaving ASOs suppresses repeat toxicity while boosting FMRP production in human neurons, establishing a native role for RAN translation in neuronal protein synthesis regulation.
- 181.Jovičić A et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci 18, 1226–1229 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lee K-H et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 167, 774–788.e17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zhang Y-J et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. (Berl.) 128, 505–524 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Yamakawa M et al. Characterization of the dipeptide repeat protein in the molecular pathogenesis of c9FTD/ALS. Hum. Mol. Genet 24, 1630–1645 (2015). [DOI] [PubMed] [Google Scholar]
- 185.Tao Z et al. Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum. Mol. Genet 24, 2426–2441 (2015). [DOI] [PubMed] [Google Scholar]
- 186.Yang D et al. FTD/ALS-associated poly(GR) protein impairs the Notch pathway and is recruited by poly(GA) into cytoplasmic inclusions. Acta Neuropathol. (Berl.) 130, 525–535 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kanekura K et al. Poly-dipeptides encoded by the C9ORF72 repeats block global protein translation. Hum. Mol. Genet 25, 1803–1813 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Boeynaems S et al. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci. Rep 6, 20877 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zhang Y-J et al. Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat. Med 24, 1136–1142 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hao Z et al. Motor dysfunction and neurodegeneration in a C9orf72 mouse line expressing poly-PR. Nat. Commun 10, 2906 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zhang Y-J et al. Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science 363, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Flores BN et al. Distinct C9orf72-Associated Dipeptide Repeat Structures Correlate with Neuronal Toxicity. PloS One 11, e0165084 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ohki Y et al. Glycine-alanine dipeptide repeat protein contributes to toxicity in a zebrafish model of C9orf72 associated neurodegeneration. Mol. Neurodegener 12, 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Guo Q et al. In Situ Structure of Neuronal C9orf72 Poly-GA Aggregates Reveals Proteasome Recruitment. Cell 172, 696–705.e12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Freibaum BD & Taylor JP The Role of Dipeptide Repeats in C9ORF72-Related ALS-FTD. Front. Mol. Neurosci 10, 35 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Nguyen L, Cleary JD & Ranum LPW Repeat-Associated Non-ATG Translation: Molecular Mechanisms and Contribution to Neurological Disease. Annu. Rev. Neurosci 42, 227–247 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Odeh HM & Shorter J Arginine-rich dipeptide-repeat proteins as phase disruptors in C9-ALS/FTD. Emerg. Top. Life Sci 4, 293–305 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Mackenzie IRA et al. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol. (Berl.) 130, 845–861 (2015). [DOI] [PubMed] [Google Scholar]
- 199.Davidson YS et al. Brain distribution of dipeptide repeat proteins in frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol. Commun 2, 70 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Davidson Y et al. Neurodegeneration in frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9orf72 is linked to TDP-43 pathology and not associated with aggregated forms of dipeptide repeat proteins. Neuropathol. Appl. Neurobiol 42, 242–254 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Quaegebeur A, Glaria I, Lashley T & Isaacs AM Soluble and insoluble dipeptide repeat protein measurements in C9orf72-frontotemporal dementia brains show regional differential solubility and correlation of poly-GR with clinical severity. Acta Neuropathol. Commun 8, 184 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Goodman LD et al. eIF4B and eIF4H mediate GR production from expanded G4C2 in a Drosophila model for C9orf72-associated ALS. Acta Neuropathol. Commun 7, 62 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Ayhan F et al. SCA8 RAN polySer protein preferentially accumulates in white matter regions and is regulated by eIF3F. EMBO J 37, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Cheng W et al. CRISPR-Cas9 Screens Identify the RNA Helicase DDX3X as a Repressor of C9ORF72 (GGGGCC)n Repeat-Associated Non-AUG Translation. Neuron 104, 885–898.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Harding HP et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633 (2003). [DOI] [PubMed] [Google Scholar]
- 206.Pakos-Zebrucka K et al. The integrated stress response. EMBO Rep 17, 1374–1395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Harding HP et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108 (2000). [DOI] [PubMed] [Google Scholar]
- 208.Zu T et al. Metformin inhibits RAN translation through PKR pathway and mitigates disease in C9orf72 ALS/FTD mice. Proc. Natl. Acad. Sci. U. S. A 117, 18591–18599 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Tiscornia G & Mahadevan MS Myotonic dystrophy: the role of the CUG triplet repeats in splicing of a novel DMPK exon and altered cytoplasmic DMPK mRNA isoform ratios. Mol. Cell 5, 959–967 (2000). [DOI] [PubMed] [Google Scholar]
- 210.Sznajder ŁJ et al. Intron retention induced by microsatellite expansions as a disease biomarker. Proc. Natl. Acad. Sci. U. S. A 115, 4234–4239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Sirp A et al. The Fuchs corneal dystrophy-associated CTG repeat expansion in the TCF4 gene affects transcription from its alternative promoters. Sci. Rep 10, 18424 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Sareen D et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med 5, 208ra149 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Neueder A et al. The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Sci. Rep 7, 1307 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Shi Y et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med 24, 313–325 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Shao Q et al. C9orf72 deficiency promotes motor deficits of a C9ALS/FTD mouse model in a dose-dependent manner. Acta Neuropathol. Commun 7, 32 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Zhu Q et al. Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat. Neurosci 23, 615–624 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Gray M et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci. Off. J. Soc. Neurosci 28, 6182–6195 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Li L-B, Yu Z, Teng X & Bonini NM RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature 453, 1107–1111 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Rué L et al. Targeting CAG repeat RNAs reduces Huntington’s disease phenotype independently of huntingtin levels. J. Clin. Invest 126, 4319–4330 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wang Q, Conlon EG, Manley JL & Rio DC Widespread intron retention impairs protein homeostasis in C9orf72 ALS brains. Genome Res 30, 1705–1715 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Hautbergue GM et al. SRSF1-dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits. Nat. Commun 8, 16063 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Cleary JD, Pattamatta A & Ranum LPW Repeat-associated non-ATG (RAN) translation. J. Biol. Chem 293, 16127–16141 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.McEachin ZT et al. Chimeric Peptide Species Contribute to Divergent Dipeptide Repeat Pathology in c9ALS/FTD and SCA36. Neuron 107, 292–305.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Toulouse A et al. Ribosomal frameshifting on MJD-1 transcripts with long CAG tracts. Hum. Mol. Genet 14, 2649–2660 (2005). [DOI] [PubMed] [Google Scholar]
- 225.Wills NM & Atkins JF The potential role of ribosomal frameshifting in generating aberrant proteins implicated in neurodegenerative diseases. RNA N. Y. N 12, 1149–1153 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Stochmanski SJ et al. Expanded ATXN3 frameshifting events are toxic in Drosophila and mammalian neuron models. Hum. Mol. Genet 21, 2211–2218 (2012). [DOI] [PubMed] [Google Scholar]
- 227.Saffert P, Adamla F, Schieweck R, Atkins JF & Ignatova Z An Expanded CAG Repeat in Huntingtin Causes +1 Frameshifting. J. Biol. Chem 291, 18505–18513 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Ma L et al. Composition of the Intranuclear Inclusions of Fragile X-associated Tremor/Ataxia Syndrome. Acta Neuropathol. Commun 7, 143 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Haify SN et al. Lack of a Clear Behavioral Phenotype in an Inducible FXTAS Mouse Model Despite the Presence of Neuronal FMRpolyG-Positive Aggregates. Front. Mol. Biosci 7, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Asamitsu S et al. CGG repeat RNA G-quadruplexes interact with FMRpolyG to cause neuronal dysfunction in fragile X-related tremor/ataxia syndrome. Sci. Adv 7, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Heatwole C et al. Patient-reported impact of symptoms in myotonic dystrophy type 1 (PRISM-1). Neurology 79, 348 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Ashizawa T, Dubel JR & Harati Y Somatic instability of CTG repeat in myotonic dystrophy. Neurology 43, 2674–2678 (1993). [DOI] [PubMed] [Google Scholar]
- 233.Sznajder ŁJ et al. Loss of MBNL1 induces RNA misprocessing in the thymus and peripheral blood. Nat. Commun 11, 2022 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Ishiura H et al. Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat. Genet 50, 581–590 (2018). [DOI] [PubMed] [Google Scholar]
- 235.Corbett MA et al. Intronic ATTTC repeat expansions in STARD7 in familial adult myoclonic epilepsy linked to chromosome 2. Nat. Commun 10, 4920 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Florian RT et al. Unstable TTTTA/TTTCA expansions in MARCH6 are associated with Familial Adult Myoclonic Epilepsy type 3. Nat. Commun 10, 4919 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Yeetong P et al. TTTCA repeat insertions in an intron of YEATS2 in benign adult familial myoclonic epilepsy type 4. Brain J. Neurol 142, 3360–3366 (2019). [DOI] [PubMed] [Google Scholar]
- 238.Seixas AI et al. A Pentanucleotide ATTTC Repeat Insertion in the Non-coding Region of DAB1, Mapping to SCA37, Causes Spinocerebellar Ataxia. Am. J. Hum. Genet 101, 87–103 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Saudou F & Humbert S The Biology of Huntingtin. Neuron 89, 910–926 (2016). [DOI] [PubMed] [Google Scholar]
- 240.Rüb U et al. Degeneration of the Cerebellum in Huntington’s Disease (HD): Possible Relevance for the Clinical Picture and Potential Gateway to Pathological Mechanisms of the Disease Process. Brain Pathol 23, 165–177 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Shortt JA, Ruggiero RP, Cox C, Wacholder AC & Pollock DD Finding and extending ancient simple sequence repeat-derived regions in the human genome. Mob. DNA 11, 11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Pheasant M & Mattick JS Raising the estimate of functional human sequences. Genome Res 17, 1245–1253 (2007). [DOI] [PubMed] [Google Scholar]
- 243.Elden AC et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This group found that ataxin-2 can modify TDP-43 toxicity in yeast and flies and that intermediate repeats in ataxin-2 serve as a common risk allele in ALS.
- 244.Tazelaar GHP et al. ATXN1 repeat expansions confer risk for amyotrophic lateral sclerosis and contribute to TDP-43 mislocalization. Brain Commun 2, fcaa064 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Conforti FL et al. Ataxin-1 and ataxin-2 intermediate-length PolyQ expansions in amyotrophic lateral sclerosis. Neurology 79, 2315–2320 (2012). [DOI] [PubMed] [Google Scholar]
- 246.Lattante S et al. ATXN1 intermediate-length polyglutamine expansions are associated with amyotrophic lateral sclerosis. Neurobiol. Aging 64, 157.e1–157.e5 (2018). [DOI] [PubMed] [Google Scholar]
- 247.R D et al. Pathogenic Huntingtin Repeat Expansions in Patients with Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Neuron (2020) doi: 10.1016/j.neuron.2020.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Blauw HM et al. NIPA1 polyalanine repeat expansions are associated with amyotrophic lateral sclerosis. Hum. Mol. Genet 21, 2497–2502 (2012). [DOI] [PubMed] [Google Scholar]
- 249.Course MM et al. Evolution of a Human-Specific Tandem Repeat Associated with ALS. Am. J. Hum. Genet 107, 445–460 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Yuan Y et al. Identification of GGC repeat expansion in the NOTCH2NLC gene in amyotrophic lateral sclerosis. Neurology 95, e3394–e3405 (2020). [DOI] [PubMed] [Google Scholar]
- 251.Willems T et al. Genome-wide profiling of heritable and de novo STR variations. Nat. Methods 14, 590–592 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Trost B et al. Genome-wide detection of tandem DNA repeats that are expanded in autism. Nature 586, 80–86 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper established that repeat expansions are a common feature in autism genomes and occur at a much higher frequency than previously appreciated.
- 253.Mitra I et al. Patterns of de novo tandem repeat mutations and their role in autism. Nature 589, 246–250 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Jansen A, Gemayel R & Verstrepen KJ Unstable microsatellite repeats facilitate rapid evolution of coding and regulatory sequences. Genome Dyn 7, 108–125 (2012). [DOI] [PubMed] [Google Scholar]
- 255.Vinces MD, Legendre M, Caldara M, Hagihara M & Verstrepen KJ Unstable tandem repeats in promoters confer transcriptional evolvability. Science 324, 1213–1216 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Verstrepen KJ, Jansen A, Lewitter F & Fink GR Intragenic tandem repeats generate functional variability. Nat. Genet 37, 986–990 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Caron NS, Desmond CR, Xia J & Truant R Polyglutamine domain flexibility mediates the proximity between flanking sequences in huntingtin. Proc. Natl. Acad. Sci (2013) doi: 10.1073/pnas.1301342110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Ashkenazi A et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 545, 108–111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Liquori CL et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293, 864–867 (2001). [DOI] [PubMed] [Google Scholar]
- 260.Ranum LPW & Day JW Myotonic dystrophy: clinical and molecular parallels between myotonic dystrophy type 1 and type 2. Curr. Neurol. Neurosci. Rep 2, 465–470 (2002). [DOI] [PubMed] [Google Scholar]
- 261.Kim G, Gautier O, Tassoni-Tsuchida E, Ma XR & Gitler AD ALS Genetics: Gains, Losses, and Implications for Future Therapies. Neuron 108, 822–842 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Ishiura H et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat. Genet 51, 1222–1232 (2019). [DOI] [PubMed] [Google Scholar]
- 263.Sone J et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat. Genet 51, 1215–1221 (2019). [DOI] [PubMed] [Google Scholar]
- 264.Tian Y et al. Expansion of Human-Specific GGC Repeat in Neuronal Intranuclear Inclusion Disease-Related Disorders. Am. J. Hum. Genet 105, 166–176 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Gelpi E et al. Neuronal intranuclear (hyaline) inclusion disease and fragile X-associated tremor/ataxia syndrome: a morphological and molecular dilemma. Brain J. Neurol 140, e51 (2017). [DOI] [PubMed] [Google Scholar]
- 266.Cortese A et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat. Genet 51, 649–658 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This group discovered a new recessive repeat expansion whose mechanisms of both generation and pathogenesis remain a mystery.
- 267.Rafehi H et al. Bioinformatics-Based Identification of Expanded Repeats: A Non-reference Intronic Pentamer Expansion in RFC1 Causes CANVAS. Am. J. Hum. Genet 105, 151–165 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Scriba CK et al. A novel RFC1 repeat motif (ACAGG) in two Asia-Pacific CANVAS families. Brain J. Neurol 143, 2904–2910 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Tsuchiya M et al. RFC1 repeat expansion in Japanese patients with late-onset cerebellar ataxia. J. Hum. Genet 65, 1143–1147 (2020). [DOI] [PubMed] [Google Scholar]
- 270.Beecroft SJ et al. A Māori specific RFC1 pathogenic repeat configuration in CANVAS, likely due to a founder allele. Brain J. Neurol 143, 2673–2680 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Jones C et al. Association of a chromosome deletion syndrome with a fragile site within the proto-oncogene CBL2. Nature 376, 145–149 (1995). [DOI] [PubMed] [Google Scholar]
- 272.Friocourt G & Parnavelas JG Mutations in ARX Result in Several Defects Involving GABAergic Neurons. Front. Cell. Neurosci 4, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Parodi S et al. Parental origin and somatic mosaicism of PHOX2B mutations in Congenital Central Hypoventilation Syndrome. Hum. Mutat 29, 206–206 (2008). [DOI] [PubMed] [Google Scholar]
- 274.Owens KM et al. Analysis of De Novo HOXA13 Polyalanine Expansions Supports Replication Slippage Without Repair in Their Generation. Am. J. Med. Genet. A 161, 1019–1027 (2013). [DOI] [PubMed] [Google Scholar]
- 275.Brown LY et al. Holoprosencephaly due to mutations in ZIC2: alanine tract expansion mutations may be caused by parental somatic recombination. Hum. Mol. Genet 10, 791–796 (2001). [DOI] [PubMed] [Google Scholar]
- 276.Poirier K et al. Maternal mosaicism for mutations in the ARX gene in a family with X linked mental retardation. Hum. Genet 118, 45–48 (2005). [DOI] [PubMed] [Google Scholar]