

Abstract
Introduction.
Trauma patients with abnormal fibrinolysis have increased morbidity and mortality. Knowledge of mechanisms differentiating fibrinolytic phenotypes is important to optimize treatment. We hypothesized that subjects with abnormal fibrinolysis identified by whole blood viscoelastometry can also be distinguished by plasma thrombin generation, clot structure, fibrin formation, and plasmin generation measurements.
Methods.
Platelet-poor plasma (PPP) from an observational cross-sectional trauma cohort with fibrinolysis shutdown (% lysis at 30 minutes [LY30]<0.9, N=11) or hyperfibrinolysis (LY30>3%, N=9) defined by whole blood thromboelastography were studied. Non-injured control subjects provided comparative samples. Thrombin generation, fibrin structure and formation, and plasmin generation were measured by fluorescence, confocal microscopy, turbidity, and a fluorescence-calibrated plasmin assay, respectively, in the absence/presence of tissue factor or tissue plasminogen activator (tPA).
Results.
Whereas spontaneous thrombin generation was not detected in PPP from control subjects, PPP from hyperfibrinolysis or shutdown patients demonstrated spontaneous thrombin generation, and the lag time was shorter in hyperfibrinolysis versus shutdown. Addition of tissue factor masked this difference but revealed increased thrombin generation in hyperfibrinolysis samples. Compared to shutdown, hyperfibrinolysis PPP formed denser fibrin networks. In the absence of tPA, the fibrin formation rate was faster in shutdown than hyperfibrinolysis, but hyperfibrinolysis clots lysed spontaneously; these differences were masked by addition of tPA. TPA-stimulated plasmin generation was similar in hyperfibrinolysis and shutdown samples. Differences in LY30, fibrin structure, and lysis correlated with pH.
Conclusions.
This exploratory study using PPP-based assays identified differences in thrombin generation, fibrin formation and structure, and lysis in hyperfibrinolysis and shutdown subgroups. These groups did not differ in their ability to promote tPA-triggered plasmin generation. The ability to characterize these activities in PPP facilitates studies to identify mechanisms that promote adverse outcomes in trauma.
Keywords: Trauma, fibrinolysis, thrombin, fibrin, clot structure
BACKGROUND
Trauma is associated with substantial morbidity and mortality. Worldwide, traumatic injury leads to 9% of deaths, and in the United States is the leading cause of death in ages 1–44 (1). Traumatic injury induces rapid changes in blood and vascular function that can subvert normal hemostatic mechanisms. These changes are further complicated during treatment and resuscitation by transfusion of blood products that alter blood composition and therefore biochemical mechanisms that regulate hemostasis (2–5). The spectrum of coagulation changes induced by trauma, so-called “trauma-induced coagulopathy” (TIC), is associated with hypoperfusion, acidosis, and tissue injury (2–5). These complications increase risk of both hemorrhage and thromboembolism which complicates clinical management. Increased understanding of trauma-induced changes in blood function may improve treatment and outcomes in these patients.
In normal physiology (hemostasis), clot formation and dissolution (fibrinolysis) are intimately related. Following vascular injury, exposure of blood to extravascular tissue factor (TF) triggers production of thrombin, which converts soluble fibrinogen to insoluble fibrin. This fibrin network stabilizes the cellular plug to seal the wound. Fibrin also initiates reparative processes by stimulating tissue plasminogen activator (tPA)-mediated conversion of plasminogen to plasmin, which promotes fibrinolysis to facilitate wound healing. Abnormalities in each of these processes have been associated with the extent and type of injury, as well as clinical outcomes (2–5). Efforts to characterize trauma-induced abnormalities in coagulation and fibrinolysis have largely focused on viscoelastic assays (rotational thromboelastometry [ROTEM] and thromboelastography [TEG]). These point-of-care methods can rapidly assess clot formation and lysis in whole blood and have revealed a spectrum of trauma-induced phenotypes defined by the extent of detectable fibrinolysis. The “hyperfibrinolysis” phenotype, defined as >3% spontaneous lysis at 30 minutes (LY30>3%) (6), correlates with severity of shock and hypoperfusion, and patients in this subgroup typically have high risk of bleeding (7, 8). Hyperfibrinolysis has been attributed to multiple mechanisms, including release of tPA without compensatory release of the tPA inhibitor, plasminogen activator inhibitor-1 (PAI-1), and formation of irregular clots that have increased susceptibility to fibrinolysis (8–11). The “fibrinolysis shutdown” phenotype, defined as <0.9% lysis at 30 minutes (LY30<0.9%) (12), is associated with increased risk of venous thromboembolism, stroke, and end organ damage (3). Fibrinolysis shutdown is thought to stem from excessive release of PAI-1 (13) and to a lesser degree, activation of the thrombin activable fibrinolysis inhibitor (TAFI), which limit endogenous fibrinolysis. Shutdown has been associated with formation of dense fibrin that may restrict access of fibrinolytic enzymes to the clot (11). Assignment into these subgroups has therapeutic implications (12). Some studies support treating hyperfibrinolytic states with antifibrinolytics (e.g., tranexamic acid [TXA]) (14–17), although concerns have been raised regarding the potential that administering TXA to patients with shutdown may enhance risk of thromboembolic complications (18–20). Characterizing mechanisms driving fibrinolytic phenotypes is important for defining the underlying etiology and improving trauma-related treatment and outcomes.
To gain insight into the pathophysiology underlying these phenotypes, we used established and new assays to characterize procoagulant and fibrinolytic mechanisms in patients with viscoelastometry-defined hyperfibrinolysis or shutdown. We hypothesized that these phenotypes can be distinguished by plasma-based assays that assess the capacity to generate thrombin, fibrin, and/or plasmin.
MATERIALS AND METHODS
Study design and setting.
This was an exploratory, observational, cross-sectional study (following STROBE guidelines) using blood samples collected at a single academic trauma center. Trauma samples were collected in the emergency department, prior to resuscitation with blood products, between February 2018-February 2020 with IRB approval (COMIRB#13-3087). Non-injured control subjects provided comparison samples and were collected between May-September 2014 with IRB approval (COMIRB#14-0366).
Sample collection.
Whole blood was collected into citrate (3.2%, Becton-Dickinson, Franklin Lakes, NJ). Platelet-poor plasma (PPP) was prepared from citrated whole blood within 30 minutes of phlebotomy by centrifuging blood at 1000xg (4°C, 15 minutes) to yield platelet-rich plasma and then centrifuging the platelet-rich plasma (12,600xg, 4°C, 6 minutes) to yield PPP. PPP was frozen in liquid nitrogen and stored at −80°C. All samples remained frozen until they were thawed once for analysis.
Study subject selection.
Whole blood was re-calcified and assayed by TEG with the TEG 5000 Hemostatic Analyzer (Haemonetics, Braintree, MA). Patients with hyperfibrinolysis were defined by LY30>3; patients with shutdown were defined by LY30<0.9. Inclusion criteria for the trauma cohort were patients qualifying for the trauma activation protocol in the emergency department (Supplemental Methods), age >18, and LY30 within the respective ranges for each subgroup. Of 406 patients initially enrolled in the trauma cohort, 125 were categorized as shutdown (LY30<0.9%) and 63 as hyperfibrinolysis (LY30>3%). Representative PPP from each subgroup (n=9 hyperfibrinolysis, n=11 shutdown) were selected to include samples representing the range of LY30s identified by TEG. Supplemental Figure 1 shows LY30 values for included subjects.
Thrombin generation (TG).
TG was assessed as described (21). Briefly, plasma was thawed at 37°C in the presence of corn trypsin inhibitor (CTI, 0.1 mg/mL final) and incubated with the thrombin substrate Z-Gly-Gly-Arg 7-amido-4-methylcoumarin hydrochloride (0.42 mM final, Bachem AG, Switzerland) and CaCl2 (15 mM final) (3 minutes, 37°C). Reactions were initiated by the addition of relipidated TF1–242 (gift from Dr. R. Lundblad, Baxter Healthcare Corp) and synthetic lipid vesicles (80% phosphatidylcholine, 20% phosphatidylserine, Haematologic Technologies, Essex Junction, VT). Final concentrations of TF and phospholipids were 6.5 pM and 20 μM, respectively. Fluorescence was measured (λex=370 nm, λem=460 nm) for 1 hour with a Cytation 3 imaging reader (BioTek, Winooski, VT). Changes in fluorescence were converted to thrombin concentrations using a calibration curve generated from sequential dilutions of human thrombin. If no change in fluorescence was noted after 60 minutes, the lag time and time to peak were defined as 60 minutes, and the velocity, peak, and area under the curve (AUC) were defined as zero.
Fibrin structure.
Fibrin structure was measured using a modification of published method (22). Plasma was thawed at 37°C in the presence of CTI (0.1 mg/mL final) and then diluted with 20 mM HEPES pH 7.4, 150 mM NaCl (HEPES-buffered saline, HBS) in a 1:1 ratio. Diluted plasma (88.8 μL) was then spiked with AlexaFluor-488 labeled fibrinogen (Thermo Fisher Scientific, Waltham, MA), CaCl2, and TF with phospholipids. Final concentrations were: AlexaFluor-488 labeled fibrinogen (220 nM), CaCl2 (5 mM), TF (8.7 pM), and phospholipids (20 μM). Plasmas were pipetted onto glass chamber slides (Thermo Fisher Scientific, Waltham, MA) and clots were allowed to form for 30 minutes. Clots were then fixed with HistoChoice (MilliporeSigma, Burlington, MA) and treated with SlowFade Diamond anti-photobleaching agent (Thermo Fisher Scientific, Waltham, MA). Three-dimensional (20 μm) “Z-stack” images series were acquired at 0.25 μm intervals using a Nikon A1R confocal microscope (Nikon Corporation, Tokyo, Japan) at 60×2.0 magnification. Clot density was quantified using multi-slice 2-dimensional Euclidian distance mapping with custom software. After deconvolving each image series, fibers were identified, thresholded (~2.5 standard deviations above background) and converted to coordinate-based 3D objects. The volume of fiber objects in the image stack was calculated as total fiber density (% total volume). Object perimeters ere demarcated, and the Euclidian distance mapping routine was used to determine the proximity of each pixel in the spaces between fibers to the closest fiber perimeter.
Fibrin formation and lysis.
Fibrin formation and lysis were measured by turbidity, as described (23). Briefly, TF, phospholipids, and tPA (10 μL, total) were added to a 96-well plate. Plasma (40 μL of 1:2 dilution [20 μL plasma plus 40 μL HBS]) was added to the 96-well plate and incubated at 37°C for 10 minutes. Reactions were initiated by addition of CaCl2. Final concentrations were: TF (0.5 pM), phospholipids (4 μM), rtPA (0.31 μg/mL), and CaCl2 (16.6 mM). Clot formation and lysis were monitored at 405 nm (SpectraMax 384Plus plate reader, Molecular Devices, Sunnyvale, CA) for 90 minutes at 37°C. The clot lysis time was the time from assay initiation to the time turbidity returned to baseline.
Plasmin generation (PG).
PG was measured in plasma using a calibrated, automated method based on cleavage of a plasmin-specific fluorogenic substrate (24). Briefly, two measurements were collected for each sample: one in which endogenous PG was triggered (reaction), and one in which α2-macroglobulin-plasmin complex was added (calibrator). To trigger PG, 10 μL of solution containing TF, phospholipids, and tPA were added to reaction wells. The α2-macroglobulin-plasmin complex calibrator (10 μL) was added to separate wells. Diluted plasma (40 μL of 1:2 dilution [20 μL of plasma plus 40 μL of HBS]) was added to each well and plates were warmed for 10 minutes at 37°C. CaCl2 and fluorogenic substrate (in 10 μL) were dispensed and plates were mixed by shaking for 10 seconds. Final concentrations were: TF (0.5 pM), phospholipids (4 μM), tPA (0.31 μg/mL), CaCl2 (16.6 mM), and fluorogenic substrate (0.5 mM). Reactions were monitored every 20 seconds with a fluorometer (Fluoroskan Ascent, Thrombinoscope, Maastricht, the Netherlands) equipped with a dispenser and 390/460 filter set (λex/λem).
Statistical methods.
Data show mean±standard deviation. Since control samples were collected separately, these were not included in statistical comparisons, but are shown for context. Data from hyperfibrinolysis and shutdown patients were analyzed using an unpaired t-test or Mann-Whitney test depending on data normality determined by GraphPad Prism (version 9.3.0). P <0.05 was considered significant.
RESULTS
Both hyperfibrinolysis and shutdown subgroups had high risk of morbidity and mortality.
The study cohort consisted of 20 individuals who met the criteria for trauma activation: 9 with hyperfibrinolysis (LY30>3) and 11 with shutdown (LY30<0.9) (Supplemental Figure 1). Characteristics of individual subjects and tPA sensitivity by TEG are shown in Supplemental Table 1 and Supplemental Results, respectively. Briefly, the population included 20-to-66 year-olds across a range of injury severity; hyperfibrinolysis patients were more severely injured than shutdown patients, with elevated injury severity scores (ISS, 17–75 vs 1–43) and higher mortality (7/9 vs 1/11 deaths) (Supplemental Table 2). Hyperfibrinolysis patients also had elevated lactate and international normalized ratios, and reduced systolic and diastolic blood pressures, pH, and fibrinogen compared to shutdown patients (Supplemental Table 2).
Patients in the hyperfibrinolysis subgroup have enhanced plasma TG potential.
We first compared TG potential in PPP from hyperfibrinolysis and shutdown subgroups. Since TF and factor XIa(a) have been detected in plasma from trauma patients (21, 25), we initially measured TG in the absence of exogenous initiators and the presence of CTI to block additional contact pathway activation. Both trauma subgroups spontaneously generated thrombin, whereas control (non-injured) subjects did not. Compared to patients with shutdown, hyperfibrinolysis patients had significantly shorter lag time (20.6±10.1 vs 40.5±16.8 minutes) and time to peak thrombin (29.2±15.6 vs 45.1±15.0 minutes), but no significant difference in peak, rate, or total thrombin generated (AUC) (Figure 1A–D, and data not shown). When we triggered reactions with exogenous TF to mask endogenous initiating activity, differences in lag time and time to peak between subgroups were lost, but hyperfibrinolysis plasmas showed enhanced peak thrombin (297.9±64.0 vs 233.6±53.0 nM, Figure 1E–H). Together, these results show preservation of TG in both subgroups, but differences in subgroup ability to initiate and propagate TG.
Figure 1. Thrombin generation (TG) potential differs in plasma from patients with hyperfibrinolysis and fibrinolysis shutdown.
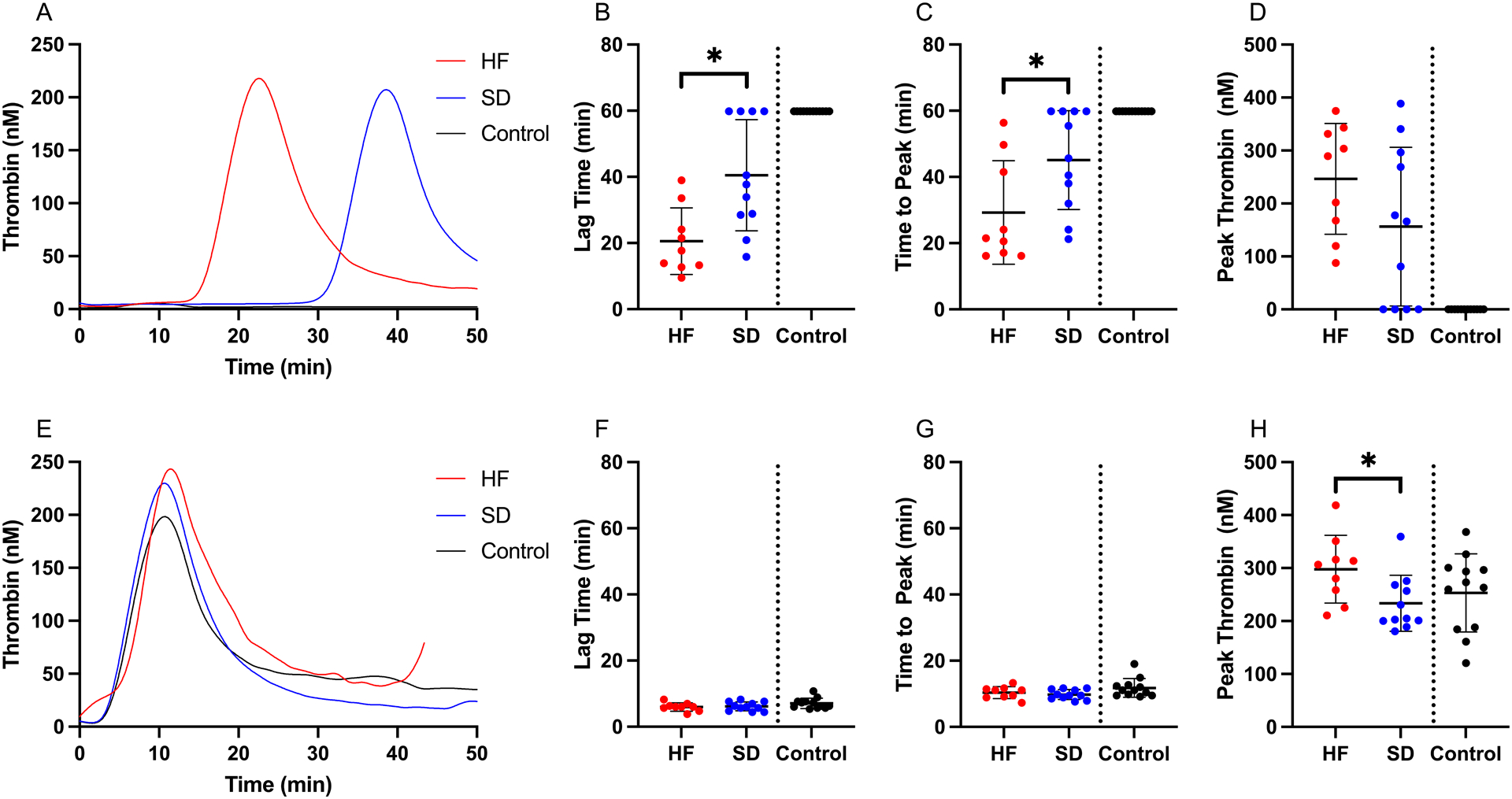
TG potential was measured in the absence of exogenous TF: (A) Representative TG curves, (B) lag time (Unpaired t-test), (C) time to peak thrombin (Mann-Whitney), and (D) peak thrombin (Unpaired t-test). TG potential was also measured in the presence of exogenous TF: (E) representative TG curves, (F) lag time (Unpaired t-test), (G) time to peak thrombin (Unpaired t-test), and (H) peak (Mann-Whitney). Healthy controls are shown for comparison, but were not included in the statistical analysis. Dots show individual patients, lines show mean ± standard deviation. *P<0.05
Hyperfibrinolysis plasma form clots that are more dense and less porous than shutdown.
The thrombin concentration present during fibrin formation dictates fibrin network structure (26). Given enhanced TG in the hyperfibrinolysis plasmas (Figure 1), we examined fibrin structure by initiating clot formation with exogenous TF to synchronize the clot formation onset between groups, and imaged clots using confocal microscopy after 30 minutes (Figure 2A). All three groups produced fibrin networks within this time frame. However, clots formed from hyperfibrinolysis PPP had increased network density (20.6±6.6 vs 10.7±2.4 A.U.) and smaller pores (0.60±0.29 vs 1.07±0.26 μm) compared to clots from shutdown PPP (Figure 2B–C).
Figure 2. Patients with the hyperfibrinolysis phenotype produce abnormal fibrin networks.
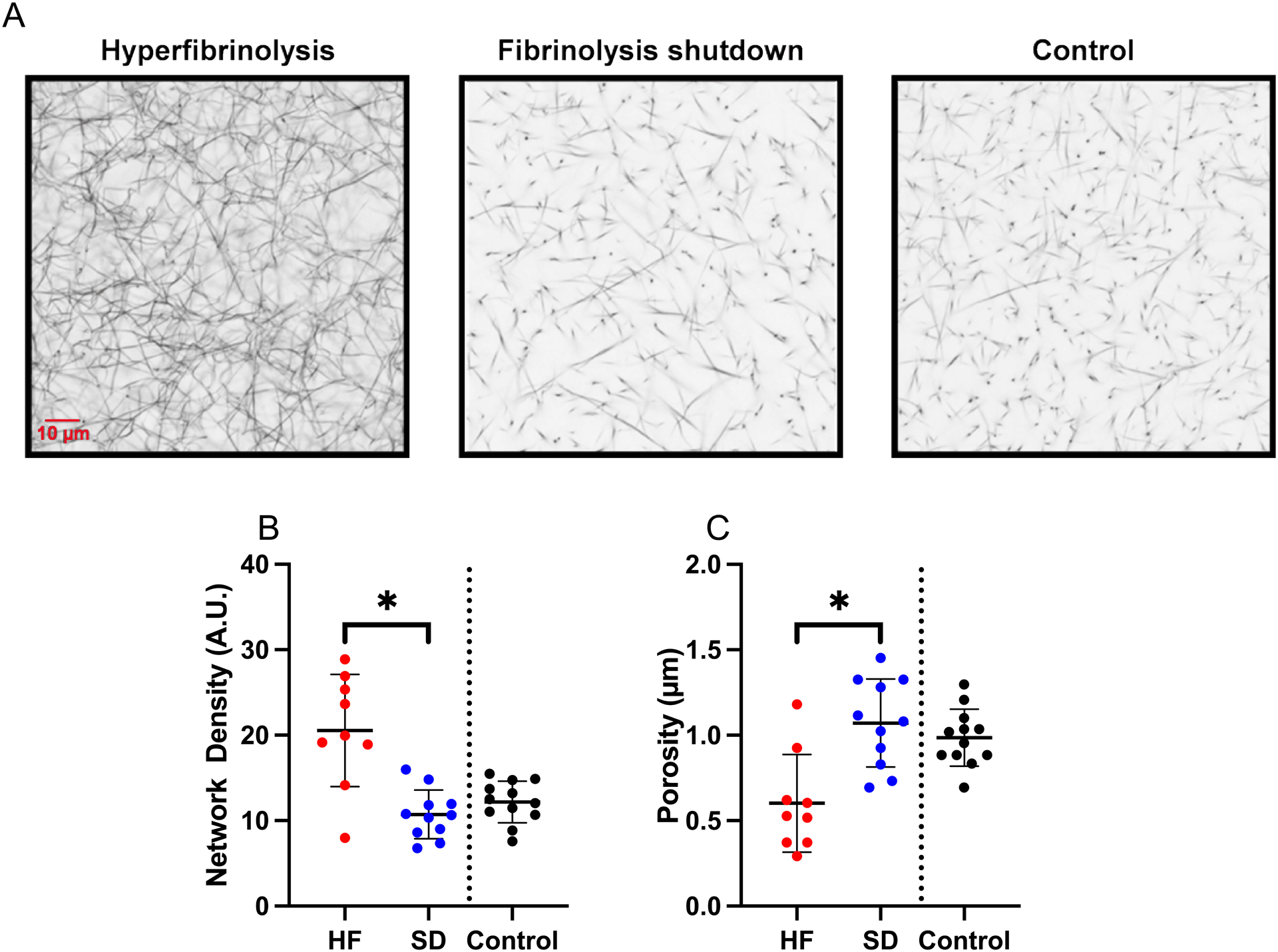
Clot formation was triggered by addition of TF to recalcified plasma and clots were analyzed by confocal microscopy. (A) Representative images showing cross-sections from a three-dimensional Z-stacks of 40 images captured from each clot. (B) Fibrin network density (total fluorescence per clot, Unpaired t-test). (C) Size of pores between fibrin fibers (Unpaired t-test). Healthy controls are shown for comparison, but were not included in statistical analysis. Dots show individual patients, lines show mean ± standard deviation. *P<0.05
PPP clots from the hyperfibrinolysis subgroup lyse spontaneously.
To characterize differences in fibrin formation that may have led to differences in the clot structures, we initiated clotting assays with TF and followed fibrin formation by turbidity. Neither the lag time nor time to plateau differed between groups (Figure 3A–C). However, compared to the shutdown subgroup, the hyperfibrinolysis subgroup had reduced fibrin formation rates (84.3±37.9 vs 103.0±53.8 mOD/minute) and smaller turbidity increase (0.079±0.047 vs 0.101±0.052) during clot formation (Figure 3A, D, E). Following clot formation, 8 of 9 clots formed by the hyperfibrinolysis subgroup, but only 1 of 11 clots formed by the shutdown subgroup, underwent spontaneous lysis (detected as a decrease in turbidity). Lysis began in hyperfibrinolysis samples 37.3±12.6 minutes after clotting initiation (median 31.9 [range 21.6–56.8]) and was complete (full return to baseline turbidity) within 90 minutes (Figure 3F). Spontaneous lysis of PPP clots was consistent with lysis detected in whole blood by TEG (Supplemental Figure 1) and showed that mechanisms predisposing these clots to fibrinolysis are present in the plasma compartment and not isolated to the cellular fraction.
Figure 3. Clot formation differs in plasma from patients with hyperfibrinolysis and fibrinolysis shutdown.
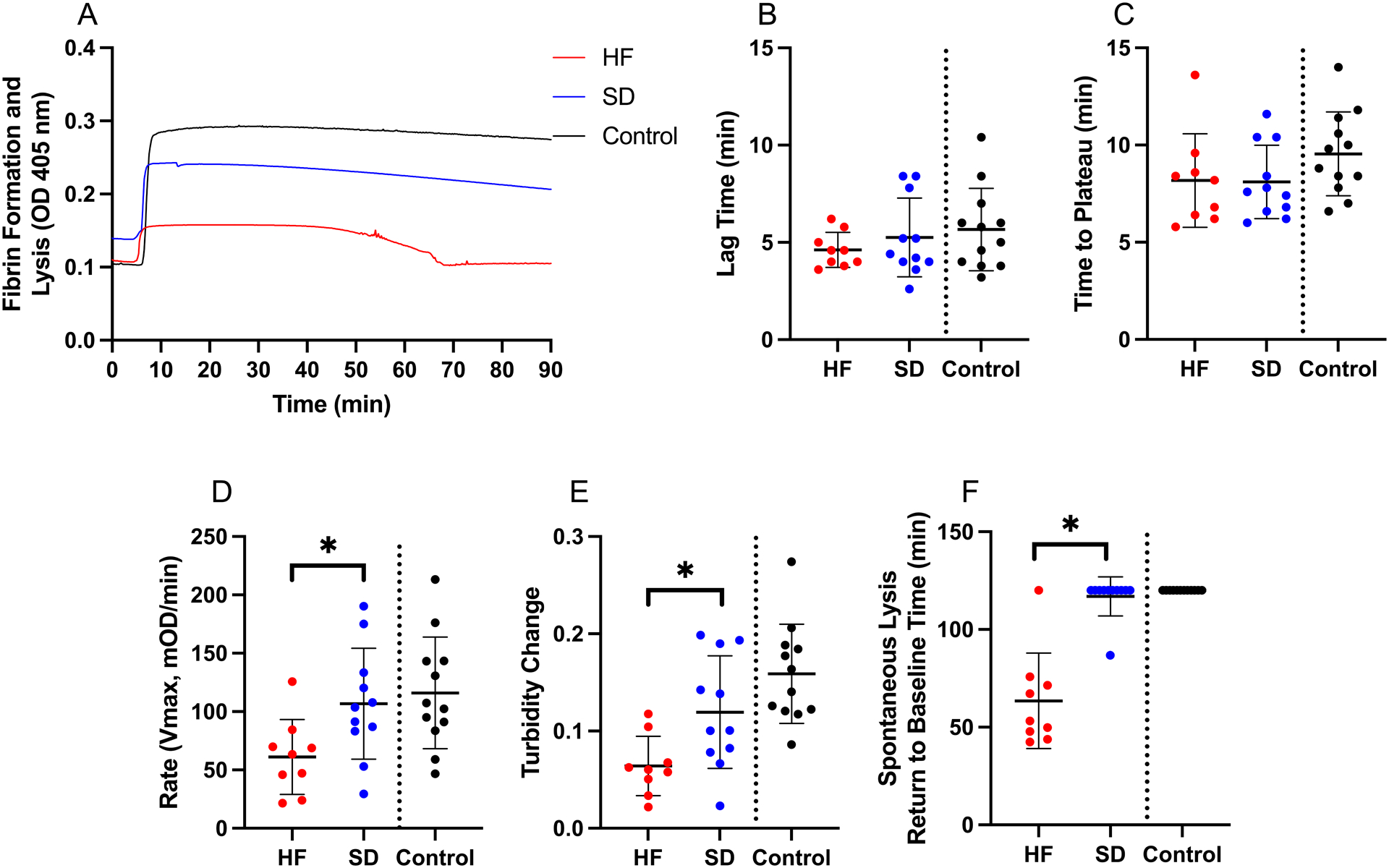
Clot formation was triggered by addition of TF to recalcified plasma and followed by turbidity. (A) Representative clot formation curves, (B) lag time (Unpaired t-test), (C) time to plateau (Unpaired t-test), (D) rate (Unpaired t-test), (E) turbidity change (Unpaired t-test), and (F) spontaneous lysis time (Mann-Whitney). Healthy controls are shown for comparison, but were not included in statistical analysis. Dots show individual patients, lines show mean ± standard deviation. *P<0.05
Plasma from patients with fibrinolysis shutdown can support tPA-mediated PG.
To test the hypothesis that PPP from the shutdown subgroup lacked biochemical machinery to support fibrinolysis, we employed two experimental approaches. First, we measured PG kinetics using a novel assay that is sensitive to concentrations of plasminogen, α2-antiplasmin, fibrinogen, and TAFI (24, 27). By triggering this assay with exogenous TF and relatively high tPA, we bypassed potential differences in endogenous coagulation initiators, tPA, or PAI-1, enabling us to directly test plasminogen conversion to plasmin. There were no differences in the onset, time to peak, rate, or peak plasmin generated between the trauma subgroups, and surprisingly, the shutdown group generated more total plasmin (higher endogenous plasmin potential) than the hyperfibrinolysis group (Figure 4). Second, we confirmed these findings in TF and tPA-initiated fibrinolysis assays in which we followed fibrin formation and lysis by turbidity. Consistent with the PG data, fibrin formation and fibrinolysis parameters from these assays were similar in both trauma subgroups (Supplemental Figure 2). These data indicate both subgroups can support PG and dissolve fibrin and suggest spontaneous fibrinolysis detected by TEG (Supplemental Figure 1) and turbidity (Figure 3F) was due to differences in endogenous fibrinolytic initiators, but not a lack of fibrinolytic potential.
Figure 4. Plasmin generation (PG) parameters are similar in the trauma subgroups.
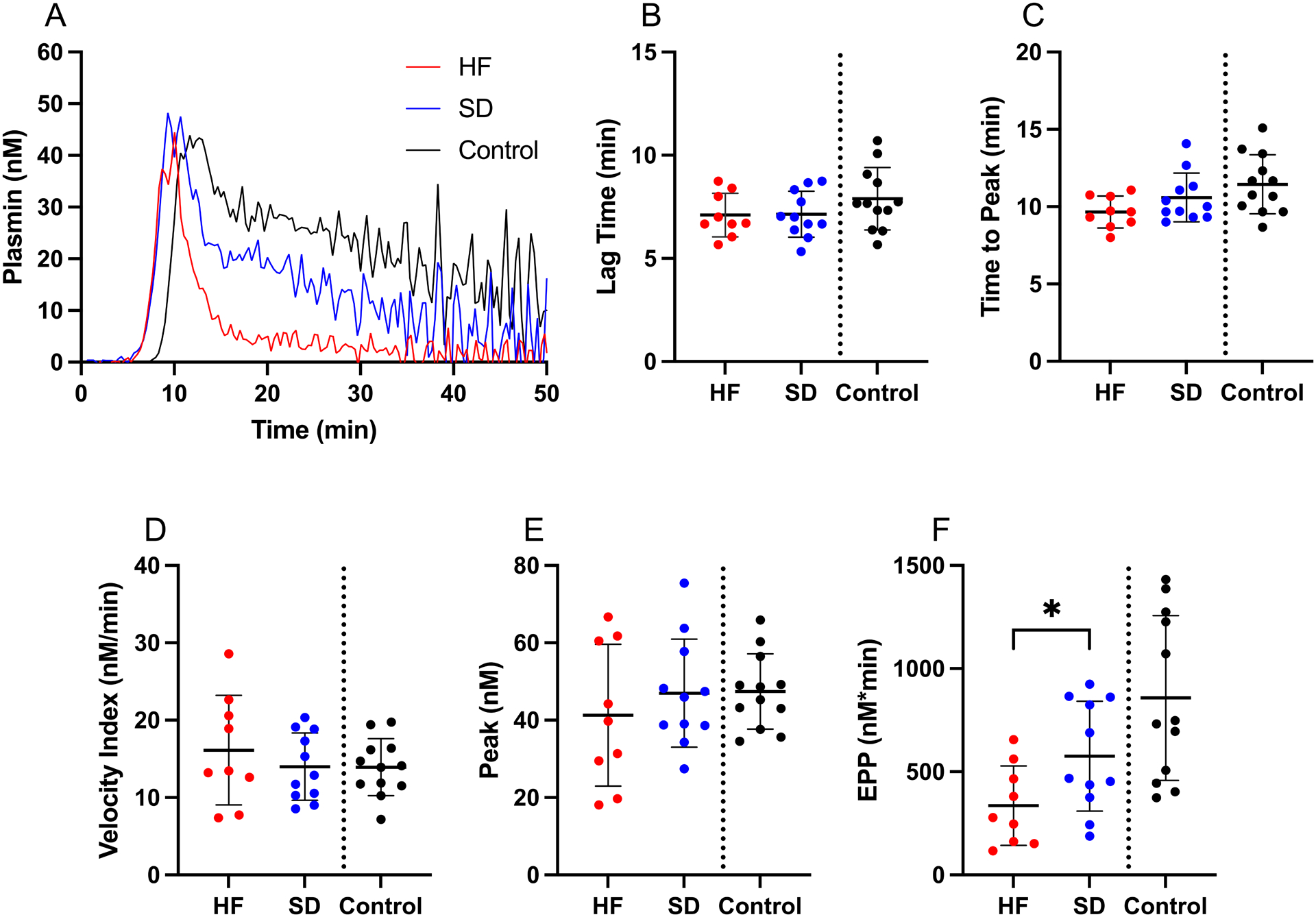
PG was triggered by addition of TF and tPA to recalcified plasma in the presence of a plasmin-cleavable fluorescent substrate. (A) Representative PG curves, (B) lag time (Unpaired t-test), (C) time to peak (Unpaired t-test), (D) velocity index (Unpaired t-test), (E) peak (Unpaired t-test), and (F) endogenous plasmin potential (EPP) (Unpaired t-test). Healthy controls are shown for comparison, but were not included in statistical analysis. Dots show individual patients, lines show mean ± standard deviation. *P<0.05
Significant correlations between injury severity, LY30, clinical indices, and global assay measurements in trauma patients.
Finally, to identify relationships between the global assay parameters and subgroups, we performed two types of analyses. We first calculated a ratio of TG-to-PG parameters (27, 28) to identify imbalances in procoagulant and fibrinolytic function in the fibrinolysis subgroups. Although ratios for lag time, time to peak, and velocity were similar between subgroups (data not shown), ratios for thrombin-to-plasmin peak (8.4±3.6 vs 5.3±1.8) and total potential (21.1±11.9 vs 12.2±5.8) demonstrated higher net procoagulant activity in the hyperfibrinolysis subgroup relative to the shutdown subgroup (Figure 5A–B). These data suggest clot instability in the hyperfibrinolysis subgroup does not stem from a failure of these plasmas to support TG relative to PG.
Figure 5. Injury severity correlates with parameters from plasma-based assays of coagulation and fibrinolytic potential.
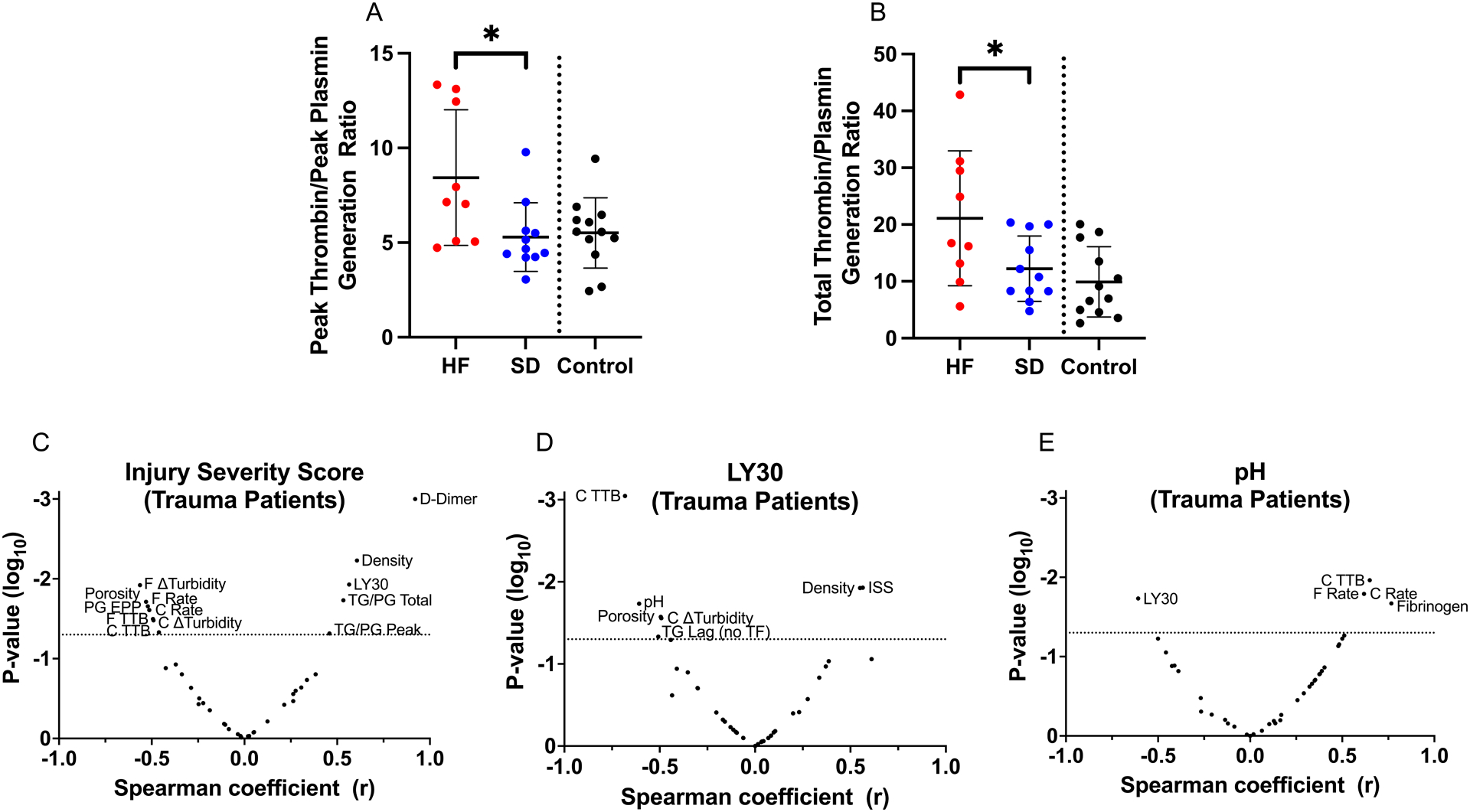
(A-B) Ratios of measurements from TG and PG assays were calculated by dividing each TG parameter by the corresponding PG parameter. Panels show ratios for (A) peak and (B) total TG-to-PG. (C-E) Volcano plots showing Spearman correlations (x-axis) and significance (y-axis) for selected parameters: (C) Injury severity score, (D) LY30 for trauma patients, and (E) pH for trauma patients.
Second, we characterized relationships across a broader range of key clinical readouts and the global assay parameters measured. For this analysis, we constructed Spearman correlation matrices to survey multiple readouts simultaneously (Supplemental Figure 3). In trauma patients the ISS correlated positively and significantly with LY30 (r=0.5646, P=0.0118, Supplemental Figure 3, Figure 5C). The ISS also correlated with D-dimer, fibrin formation and fibrinolysis parameters (fibrin density, fibrin formation rate, turbidity change, time to return to baseline), the endogenous plasmin potential, and the thrombin-to-plasmin ratios for peak and total enzyme produced (Figure 5C). No measurements correlated significantly with LY30 in control subjects (Supplemental Figure 4). However, several indices from the trauma subgroups correlated with LY30, including ISS, TG lag time in the absence of TF, fibrin density and porosity, and fibrinolysis measurements (turbidity change and time to return to baseline turbidity, Figure 5D). Interestingly, pH, a marker of tissue hypoxia and lactic acid accumulation, was negatively and significantly associated with LY30 (r=−0.6079, P=0.0185, Supplemental Figure 3, Figure 5D–E), as well as with fibrinogen, fibrin formation and lysis rates, and time to return to baseline turbidity (Figure 5E). Collectively, these data associate tissue hypoperfusion and/or injury and acidosis with functional effects in whole blood and plasma-based assays that differentiate trauma subgroups.
DISCUSSION
Clinical outcomes in trauma patients have been linked to abnormal fibrinolysis. However, mechanisms mediating fibrinolytic potential and susceptibility of clots to fibrinolysis in trauma patients have not been fully characterized. Efforts have been limited, in part, from challenges in analyzing whole blood in real-time in emergency settings. Our panel of assays using stored PPP effectively differentiates hyperfibrinolysis and shutdown phenotypes defined by whole blood thromboelastography, validating the assignment of these phenotypes by viscoelastometry. Our data demonstrate abnormalities in multiple pathways, including the presence of functional coagulation and fibrinolysis initiators, differences in mechanisms that propagate TG, and abnormal fibrin formation and structure. These findings suggest derangements in both coagulation and fibrinolysis contribute to the subgroup phenotypes and potential outcomes of trauma patients.
Both hyperfibrinolysis and shutdown subgroups showed spontaneous TG, consistent with prior studies that detected TF-bearing extracellular vesicles and/or factor XIa in trauma samples shortly after injury (21, 25). Interestingly, although addition of exogenous TF normalized the onset of TG in our samples, significant differences in the rate of TG emerged, suggesting additional mechanisms modify procoagulant activity in hyperfibrinolysis and shutdown subgroups. Our experiments did not identify this activity, but several candidates have been proposed. For example, endothelial damage or dysfunction from tissue injury or hypoperfusion may induce release of factor VIII or thrombomodulin. Simultaneous release of these proteins with potentially competing activities (29, 30) could alter net procoagulant activity in complex ways. Thrombomodulin can also stabilize clots by facilitating TAFI activation. The relative contribution of thrombomodulin to anticoagulant or antifibrinolytic pathways depend, in part, on its concentration (31–33), which may change dynamically during trauma and subsequent treatment. Plasmin generated in response to trauma may activate factor V and enhance procoagulant activity (34). Which, if any, of these or other proposed mechanisms (e.g., release of myosin, histones, or other molecules) contributes to plasma procoagulant activity will require further study.
Differences in procoagulant activity have implications for the quality of the ensuing clot. In particular, the thrombin concentration that converts fibrinogen to fibrin determines fibrin network formation and structure; high thrombin concentrations produce dense networks of thin fibrin strands in clots that are typically resistant to fibrinolysis, whereas low thrombin concentrations produce porous, turbid clots composed of thick fibrin fibers and these clots are typically susceptible to fibrinolysis (26, 35). Our data showing both enhanced TG and increased network density in clots from hyperfibrinolysis PPP are consistent with these observations. Although we previously reported dense, poorly-resolved fibrin in PPP clots from patients with shutdown (11), our present analysis did not corroborate this finding. The discord may stem from different inclusion criteria in the two studies; the earlier study included shutdown patients with severe trauma in which the densest clots were detected in the subgroup with tPA-resistant shutdown, whereas shutdown patients in the current study were generally less severely injured and only 1 of 9 exhibited tPA resistance. Other potential differences in the degree of hypoperfusion and tissue injury between the two patient groups may also differently alter fibrin network structure.
In addition to abnormal clot structure, mechanisms that regulate the fibrinolytic pathway are also compromised in trauma patients. Previous studies have documented increased tPA and a higher tPA-to-PAI-1 ratio in hyperfibrinolysis plasmas, providing one mechanism to explain the phenotype of these samples (8–10, 36). Few studies have tested contributions of abnormalities downstream of initiation (e.g., altered plasminogen and/or plasmin inhibitors) to fibrinolytic function in these subgroups. The development of a plasma-based PG assay (24, 27) enabled us to directly test these mechanisms. Our data demonstrated similar PG kinetics in hyperfibrinolysis and shutdown samples, suggesting the trauma subgroups do not differ in their potential ability to convert plasminogen to active plasmin. This finding refocuses efforts to understand trauma-induced pathology on events that initiate fibrinolysis, and illuminate the need to understand the origin of tPA and PAI-1 synthesis and release in response to hypoperfusion and tissue damage.
A comprehensive mechanism(s) to unite the observed changes in TG, fibrin formation, and fibrinolysis are currently unclear, but an exploratory correlation analysis of clinical and laboratory parameters from our assays identified an intriguing relationship between pH and several key readouts, including fibrin formation and fibrinolysis. An extensive literature has previously correlated acidosis with poor outcome in trauma patients and animal models of trauma (37–39). Several mechanisms mediating this relationship have been proposed, including pH-dependent changes in the activity of coagulation factors (e.g., factors VIIa, Xa), inhibition rate of procoagulant enzymes (e.g., antithrombin inhibition of thrombin), and conversion of fibrinogen to fibrin and assembly of the fibrin network (40–45). pH also directly influences fibrinolysis; compared to control human blood (pH 7.37), induction of severe acidosis (pH 6.9) enhances tPA-induced hyperfibrinolysis (46, 47). This pattern is consistent with our data in which hyperfibrinolysis patients had significantly lower pH (7.02±0.16) than patients with shutdown (7.30±0.14). Collectively, these data suggest even small changes in pH within the physiologic range may alter clot formation and stability in a complex biochemical milieu that includes multiple enzymes with different susceptibilities to changes in pH. While lactate and/or base deficit are typically used in lieu of pH to measure shock and predict clinical trajectory in settings of hypoperfusion, pH may drive the biochemical abnormalities in coagulation and/or fibrinolysis detected by TEG and the global assays used here. Alternatively, effects of pH may be related to anaerobic metabolites or hemolysis, whereby pH is a surrogate for shock that generates these mediators.
Our study has limitations. First, as an exploratory study, our sample size was small (N=20 patients), which limited our statistical power. Our cohort included subjects with tPA-resistant shutdown, but lacked subjects from other fibrinolysis subgroups, including “physiologic fibrinolysis.” However, use of a small cohort enabled us to apply a series of assays to measure plasma coagulation and fibrinolytic function. Notably, our data show that plasma from patients with early onset shutdown retains the ability to generate plasmin. Whether these results can be extrapolated to other groups of patients with differential sensitivity to tPA or patients with later onset shutdown warrant further investigation (48). Second, similar to other studies of trauma, our study included patients with diverse injuries; patients in the hyperfibrinolysis subgroup were generally more severely injured and had greater mortality than shutdown samples. However, inclusion of these patients with clearly distinguished phenotypes increased our ability to identify differences between groups. Moreover, by stratifying patients according to ISS, we were able to relate injury to global assay parameters independent of assignment into trauma subgroups. Third, our plasma-based assays lacked potential contributions from cellular components. However, correlations between findings in whole blood and PPP suggest the detected differences in fibrinolytic potential between these groups are preserved – and therefore available for study – in frozen/thawed plasma samples. This finding simplifies the workflow needed to characterize mechanisms leading to trauma-induced abnormalities. Finally, preclinical variables can significantly influence coagulation parameters (49). Since some blood draws in the trauma patients were performed invasively, we lacked a well-matched control group. Additionally, our control samples differed in length of time frozen which could introduce variability. Thus, we used controls to establish baseline characteristics, but did not include these individuals in our statistical comparisons.
In summary, we describe an exploratory study of plasma from trauma patients manifesting hyperfibrinolysis and shutdown defined by TEG. Our data reveal differences in TG and fibrin formation and structure between subgroups and show differences in fibrinolysis do not stem from altered ability of these plasmas to support PG. Preliminary analysis suggests the degree of acidosis may influence fibrin formation and susceptibility to fibrinolysis. The correlation between data from whole blood thromboelastography (LY30) and plasma-based assays supports the use of stored plasma samples to define the operant mechanisms in future studies.
Supplementary Material
Supplemental Table 1. Demographic, clinical, and biochemical assay data from individual study subjects. HF, hyperfibrinolysis; SD, fibrinolysis shutdown.
Supplemental Figure 1. Patient selection criteria based on percent lysis at 30 minutes (LY30) in thromboelastography. Citrated blood was recalcified and analyzed by thromboelastography. Patients were separated into fibrinolysis subgroups based on LY30: hyperfibrinolysis (HF) and shutdown (SD). LY30 of healthy (non-injured) controls subjects is also shown for comparison. Data were compared using an Unpaired t-test. *P<0.05
Supplemental Figure 2. TPA-initiated fibrinolysis does not differ in the trauma subgroups. Clot formation and fibrinolysis were triggered by addition of TF and tPA to recalcified plasma and followed by turbidity. (A) Representative fibrinolysis curves, (B) lag time (Mann-Whitney), (C) time to peak (Mann-Whitney), (D) rate (Unpaired t-test), (E) turbidity change (Unpaired t-test), and (F) spontaneous lysis time (Mann-Whitney). Healthy controls are shown for comparison, but were not included in statistical analysis. Dots show individual patients, lines show mean ± standard deviation. *P<0.05
Supplemental Figure 3. Spearman correlations between clinical metrics and procoagulant and fibrinolytic parameters. Spearman rank correlations were performed for clinical metrics, plasma biomarkers, and coagulation and fibrinolytic parameters measured in trauma patients. (A) Significant correlations (P<0.05) are indicated in yellow shading. (B) Positive correlations are indicated in blue and negative correlations are indicated in red. Abbreviations are: ISS, injury severity score; TG Lag, thrombin generation lag time; TG TTP, thrombin generation time to peak thrombin; TG Velocity, thrombin generation velocity; TG Peak, thrombin generation peak thrombin; TG AUC, thrombin generation area under the curve; density, fibrin network density; porosity, size of pores between fibrin fibers; C Lag, clotting lag time; C TTP, clotting time to peak; C Rate, clotting Rate; C Δturbidity, clotting Δturbidity; C TTB, clotting time to baseline; F Lag, fibrinolysis lag time; F TTP, fibrinolysis time to peak; F Rate, fibrinolysis Rate; F Δturbidity, fibrinolysis Δturbidity; F TTB, fibrinolysis time to baseline; PG Lag, plasmin generation lag time; PG TTP, plasmin generation time to peak; PG Velocity, plasmin generation velocity; PG peak, plasmin generation peak; PG EPP, plasmin generation endogenous plasmin potential; TG/PG Lag, thrombin-to-plasmin generation lag time ratio; TG/PG TTP, thrombin-to-plasmin generation time to peak ratio; TG/PG Velocity, thrombin-to-plasmin generation velocity ratio; TG/PG Peak, thrombin-to-plasmin generation peak ratio; TG/PG Total, total thrombin-to-plasmin generation ratio (AUC).
Supplemental Figure 4. LY30 in healthy controls does not significantly correlate with parameters from plasma-based assays of coagulation and fibrinolytic potential. Volcano plot showing Spearman correlations (x-axis) and significance (y-axis) for LY30 for controls.
Supplemental Table 2. Demographic characteristics and clinical data collected for hyperfibrinolysis and fibrinolysis shutdown patient groups.
ACKNOWLEDGEMENTS
The authors thank Arsen Ghasabyan for sample coordination.
FUNDING
Research reported in this publication was supported by grants from the National Institutes of Health (National Institute of General Medical Sciences (R35-GM-144099-01 to K.F.), National Heart, Lung, and Blood Institute (R01HL126974 to A.S.W. and 1OT2HL156812-01 to K.F.); the Totman Medical Research Trust (to K.F.); the Department of Defense/The Henry M. Jackson Foundation for the Advancement of Military Medicine (HU001-18-2-0016 to K.F.); and Department of Defense (DM190028 to E.E.M).
Footnotes
CONFLICT OF INTEREST DISCLOSURE
B.dL. is employed by Synapse Research Institute, a not-for-profit member of the STAGO Diagnostic group that produces calibrated automated thrombography for thrombin generation measurements in plasma. Synapse Research Institute holds the patent on calibrated plasmin generation.
SUPPLEMENTAL DIGITAL CONTENT
Supplemental files include additional methods, table, and figures.
REFERENCES
- 1.Kauvar DS, Wade CE. The epidemiology and modern management of traumatic hemorrhage: US and international perspectives. Crit Care. 2005;9 Suppl 5:S1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dobson GP, Morris JL, Davenport LM, Letson HL. Traumatic-Induced Coagulopathy as a Systems Failure: A New Window into Hemostasis. Semin Thromb Hemost. 2020;46(2):199–214. [DOI] [PubMed] [Google Scholar]
- 3.Moore EE, Moore HB, Kornblith LZ, Neal MD, Hoffman M, Mutch NJ, et al. Trauma-induced coagulopathy. Nat Rev Dis Primers. 2021;7(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moore HB, Moore EE, Neal MD. Trauma Induced Coagulopathy. Second ed: Springer Nature; 2021 12/June/2021. 794 p. [Google Scholar]
- 5.Cardenas JC. Thrombin Generation Following Severe Trauma: Mechanisms, Modulators, and Implications for Hemostasis and Thrombosis. Shock. 2021;56(5):682–90. [DOI] [PubMed] [Google Scholar]
- 6.Chapman MP, Moore EE, Ramos CR, Ghasabyan A, Harr JN, Chin TL, et al. Fibrinolysis greater than 3% is the critical value for initiation of antifibrinolytic therapy. J Trauma Acute Care Surg. 2013;75(6):961–7; discussion 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brohi K, Cohen MJ, Ganter MT, Schultz MJ, Levi M, Mackersie RC, et al. Acute coagulopathy of trauma: hypoperfusion induces systemic anticoagulation and hyperfibrinolysis. J Trauma. 2008;64(5):1211–7; discussion 7. [DOI] [PubMed] [Google Scholar]
- 8.Chapman MP, Moore EE, Moore HB, Gonzalez E, Gamboni F, Chandler JG, et al. Overwhelming tPA release, not PAI-1 degradation, is responsible for hyperfibrinolysis in severely injured trauma patients. J Trauma Acute Care Surg. 2016;80(1):16–23; discussion −5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schleef RR, Higgins DL, Pillemer E, Levitt LJ. Bleeding diathesis due to decreased functional activity of type 1 plasminogen activator inhibitor. J Clin Invest. 1989;83(5):1747–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cardenas JC, Matijevic N, Baer LA, Holcomb JB, Cotton BA, Wade CE. Elevated tissue plasminogen activator and reduced plasminogen activator inhibitor promote hyperfibrinolysis in trauma patients. Shock. 2014;41(6):514–21. [DOI] [PubMed] [Google Scholar]
- 11.Dow N, Coleman JR, Moore H, Osborn ZT, Sackheim AM, Hennig G, et al. Dense and dangerous: The tissue plasminogen activator-resistant fibrinolysis shutdown phenotype is due to abnormal fibrin polymerization. J Trauma Acute Care Surg. 2020;88(2):258–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez E, Moore EE, Moore HB. Management of Trauma-Induced Coagulopathy with Thrombelastography. Crit Care Clin. 2017;33(1):119–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore HB, Moore EE, Neal MD, Sheppard FR, Kornblith LZ, Draxler DF, et al. Fibrinolysis Shutdown in Trauma: Historical Review and Clinical Implications. Anesth Analg. 2019;129(3):762–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pabinger I, Fries D, Schochl H, Streif W, Toller W. Tranexamic acid for treatment and prophylaxis of bleeding and hyperfibrinolysis. Wien Klin Wochenschr. 2017;129(9–10):303–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khan M, Jehan F, Bulger EM, O’Keeffe T, Holcomb JB, Wade CE, et al. Severely injured trauma patients with admission hyperfibrinolysis: Is there a role of tranexamic acid? Findings from the PROPPR trial. J Trauma Acute Care Surg. 2018;85(5):851–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harvin JA, Peirce CA, Mims MM, Hudson JA, Podbielski JM, Wade CE, et al. The impact of tranexamic acid on mortality in injured patients with hyperfibrinolysis. J Trauma Acute Care Surg. 2015;78(5):905–9; discussion 9–11. [DOI] [PubMed] [Google Scholar]
- 17.Gall LS, Brohi K, Davenport RA. Diagnosis and Treatment of Hyperfibrinolysis in Trauma (A European Perspective). Semin Thromb Hemost. 2017;43(2):224–34. [DOI] [PubMed] [Google Scholar]
- 18.Richards JE, Fedeles BT, Chow JH, Morrison JJ, Renner C, Trinh AT, et al. Is Tranexamic Acid Associated With Mortality or Multiple Organ Failure Following Severe Injury? Shock. 2021;55(1):55–60. [DOI] [PubMed] [Google Scholar]
- 19.Moore EE, Moore HB, Gonzalez E, Sauaia A, Banerjee A, Silliman CC. Rationale for the selective administration of tranexamic acid to inhibit fibrinolysis in the severely injured patient. Transfusion. 2016;56 Suppl 2:S110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walsh M, Thomas S, Moore E, Moore H, Piscoya A, Hake D, et al. Tranexamic Acid for Trauma Resuscitation in the United States of America. Semin Thromb Hemost. 2017;43(2):213–23. [DOI] [PubMed] [Google Scholar]
- 21.Prior SM, Cohen MJ, Conroy AS, Nelson MF, Kornblith LZ, Howard BM, et al. Correlation between factor (F)XIa, FIXa and tissue factor and trauma severity. J Trauma Acute Care Surg. 2017;82(6):1073–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campbell RA, Overmyer KA, Selzman CH, Sheridan BC, Wolberg AS. Contributions of extravascular and intravascular cells to fibrin network formation, structure, and stability. Blood. 2009;114(23):4886–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zucker M, Seligsohn U, Salomon O, Wolberg AS. Abnormal plasma clot structure and stability distinguish bleeding risk in patients with severe factor XI deficiency. J Thromb Haemost. 2014;12(7):1121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miszta A, Ahmadzia HK, Luban NLC, Li S, Guo D, Holle LA, et al. Application of a plasmin generation assay to define pharmacodynamic effects of tranexamic acid in women undergoing cesarean delivery. J Thromb Haemost. 2021;19(1):221–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gando S, Nanzaki S, Sasaki S, Kemmotsu O. Significant correlations between tissue factor and thrombin markers in trauma and septic patients with disseminated intravascular coagulation. Thromb Haemost. 1998;79(6):1111–5. [PubMed] [Google Scholar]
- 26.Wolberg AS. Thrombin generation and fibrin clot structure. Blood Rev. 2007;21(3):131–42. [DOI] [PubMed] [Google Scholar]
- 27.Miszta A, Kopec AK, Pant A, Holle LA, Byrnes JR, Lawrence DA, et al. A high-fat diet delays plasmin generation in a thrombomodulin-dependent manner in mice. Blood. 2020;135(19):1704–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bouck EG, Denorme F, Holle LA, Middelton EA, Blair AM, de Laat B, et al. COVID-19 and Sepsis Are Associated With Different Abnormalities in Plasma Procoagulant and Fibrinolytic Activity. Arterioscler Thromb Vasc Biol. 2021;41(1):401–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Esmon CT. The protein C pathway. Chest. 2003;124(3 Suppl):26S–32S. [DOI] [PubMed] [Google Scholar]
- 30.Samuelson Bannow B, Recht M, Negrier C, Hermans C, Berntorp E, Eichler H, et al. Factor VIII: Long-established role in haemophilia A and emerging evidence beyond haemostasis. Blood Rev. 2019;35:43–50. [DOI] [PubMed] [Google Scholar]
- 31.Wang W, Boffa MB, Bajzar L, Walker JB, Nesheim ME. A study of the mechanism of inhibition of fibrinolysis by activated thrombin-activable fibrinolysis inhibitor. J Biol Chem. 1998;273(42):27176–81. [DOI] [PubMed] [Google Scholar]
- 32.Bajzar L, Morser J, Nesheim M. TAFI, or plasma procarboxypeptidase B, couples the coagulation and fibrinolytic cascades through the thrombin-thrombomodulin complex. J Biol Chem. 1996;271(28):16603–8. [DOI] [PubMed] [Google Scholar]
- 33.Mosnier LO, Meijers JC, Bouma BN. Regulation of fibrinolysis in plasma by TAFI and protein C is dependent on the concentration of thrombomodulin. Thromb Haemost. 2001;85(1):5–11. [PubMed] [Google Scholar]
- 34.Lee CD, Mann KG. Activation/inactivation of human factor V by plasmin. Blood. 1989;73(1):185–90. [PubMed] [Google Scholar]
- 35.Collet JP, Woodhead JL, Soria J, Soria C, Mirshahi M, Caen JP, et al. Fibrinogen Dusart: electron microscopy of molecules, fibers and clots, and viscoelastic properties of clots. Biophys J. 1996;70(1):500–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ilich A, Kumar V, Ferrara MJ, Henderson MW, Noubouossie DF, Jenkins DH, et al. Euglobulin clot lysis time reveals a high frequency of fibrinolytic activation in trauma. Thromb Res. 2021;204:22–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Darlington DN, Kheirabadi BS, Delgado AV, Scherer MR, Martini WZ, Dubick MA. Coagulation changes to systemic acidosis and bicarbonate correction in swine. J Trauma. 2011;71(5):1271–7. [DOI] [PubMed] [Google Scholar]
- 38.Martini WZ, Dubick MA, Wade CE, Holcomb JB. Evaluation of tris-hydroxymethylaminomethane on reversing coagulation abnormalities caused by acidosis in pigs. Crit Care Med. 2007;35(6):1568–74. [DOI] [PubMed] [Google Scholar]
- 39.Martini WZ, Pusateri AE, Uscilowicz JM, Delgado AV, Holcomb JB. Independent contributions of hypothermia and acidosis to coagulopathy in swine. J Trauma. 2005;58(5):1002–9; discussion 9–10. [DOI] [PubMed] [Google Scholar]
- 40.Gissel M, Brummel-Ziedins KE, Butenas S, Pusateri AE, Mann KG, Orfeo T. Effects of an acidic environment on coagulation dynamics. J Thromb Haemost. 2016;14(10):2001–10. [DOI] [PubMed] [Google Scholar]
- 41.Meng ZH, Wolberg AS, Monroe DM 3rd, Hoffman M. The effect of temperature and pH on the activity of factor VIIa: implications for the efficacy of high-dose factor VIIa in hypothermic and acidotic patients. J Trauma. 2003;55(5):886–91. [DOI] [PubMed] [Google Scholar]
- 42.Nair CH, Shah GA, Dhall DP. Effect of temperature, pH and ionic strength and composition on fibrin network structure and its development. Thromb Res. 1986;42(6):809–16. [DOI] [PubMed] [Google Scholar]
- 43.Ferry JD, Morrison PR. Preparation and properties of serum and plasma proteins; the conversion of human fibrinogen to fibrin under various conditions. J Am Chem Soc. 1947;69(2):388–400. [DOI] [PubMed] [Google Scholar]
- 44.Shenkman B, Budnik I, Einav Y, Hauschner H, Andrejchin M, Martinowitz U. Model of trauma-induced coagulopathy including hemodilution, fibrinolysis, acidosis, and hypothermia: Impact on blood coagulation and platelet function. J Trauma Acute Care Surg. 2017;82(2):287–92. [DOI] [PubMed] [Google Scholar]
- 45.Mitrophanov AY, Rosendaal FR, Reifman J. Mechanistic Modeling of the Effects of Acidosis on Thrombin Generation. Anesth Analg. 2015;121(2):278–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dirkmann D, Radu-Berlemann J, Gorlinger K, Peters J. Recombinant tissue-type plasminogen activator-evoked hyperfibrinolysis is enhanced by acidosis and inhibited by hypothermia but still can be blocked by tranexamic acid. J Trauma Acute Care Surg. 2013;74(2):482–8. [DOI] [PubMed] [Google Scholar]
- 47.Tappy L, Hauert J, Bachmann F. Effects of hypoxia and acidosis on vascular plasminogen activator release in the pig ear perfusion system. Thromb Res. 1984;33(2):117–24. [DOI] [PubMed] [Google Scholar]
- 48.Rossetto A, Vulliamy P, Lee KM, Brohi K, Davenport R. Temporal Transitions in Fibrinolysis after Trauma: Adverse Outcome Is Principally Related to Late Hypofibrinolysis. Anesthesiology. 2022;136(1):148–61. [DOI] [PubMed] [Google Scholar]
- 49.Dargaud Y, Wolberg AS, Luddington R, Regnault V, Spronk H, Baglin T, et al. Evaluation of a standardized protocol for thrombin generation measurement using the calibrated automated thrombogram: an international multicentre study. Thromb Res. 2012;130(6):929–34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. Demographic, clinical, and biochemical assay data from individual study subjects. HF, hyperfibrinolysis; SD, fibrinolysis shutdown.
Supplemental Figure 1. Patient selection criteria based on percent lysis at 30 minutes (LY30) in thromboelastography. Citrated blood was recalcified and analyzed by thromboelastography. Patients were separated into fibrinolysis subgroups based on LY30: hyperfibrinolysis (HF) and shutdown (SD). LY30 of healthy (non-injured) controls subjects is also shown for comparison. Data were compared using an Unpaired t-test. *P<0.05
Supplemental Figure 2. TPA-initiated fibrinolysis does not differ in the trauma subgroups. Clot formation and fibrinolysis were triggered by addition of TF and tPA to recalcified plasma and followed by turbidity. (A) Representative fibrinolysis curves, (B) lag time (Mann-Whitney), (C) time to peak (Mann-Whitney), (D) rate (Unpaired t-test), (E) turbidity change (Unpaired t-test), and (F) spontaneous lysis time (Mann-Whitney). Healthy controls are shown for comparison, but were not included in statistical analysis. Dots show individual patients, lines show mean ± standard deviation. *P<0.05
Supplemental Figure 3. Spearman correlations between clinical metrics and procoagulant and fibrinolytic parameters. Spearman rank correlations were performed for clinical metrics, plasma biomarkers, and coagulation and fibrinolytic parameters measured in trauma patients. (A) Significant correlations (P<0.05) are indicated in yellow shading. (B) Positive correlations are indicated in blue and negative correlations are indicated in red. Abbreviations are: ISS, injury severity score; TG Lag, thrombin generation lag time; TG TTP, thrombin generation time to peak thrombin; TG Velocity, thrombin generation velocity; TG Peak, thrombin generation peak thrombin; TG AUC, thrombin generation area under the curve; density, fibrin network density; porosity, size of pores between fibrin fibers; C Lag, clotting lag time; C TTP, clotting time to peak; C Rate, clotting Rate; C Δturbidity, clotting Δturbidity; C TTB, clotting time to baseline; F Lag, fibrinolysis lag time; F TTP, fibrinolysis time to peak; F Rate, fibrinolysis Rate; F Δturbidity, fibrinolysis Δturbidity; F TTB, fibrinolysis time to baseline; PG Lag, plasmin generation lag time; PG TTP, plasmin generation time to peak; PG Velocity, plasmin generation velocity; PG peak, plasmin generation peak; PG EPP, plasmin generation endogenous plasmin potential; TG/PG Lag, thrombin-to-plasmin generation lag time ratio; TG/PG TTP, thrombin-to-plasmin generation time to peak ratio; TG/PG Velocity, thrombin-to-plasmin generation velocity ratio; TG/PG Peak, thrombin-to-plasmin generation peak ratio; TG/PG Total, total thrombin-to-plasmin generation ratio (AUC).
Supplemental Figure 4. LY30 in healthy controls does not significantly correlate with parameters from plasma-based assays of coagulation and fibrinolytic potential. Volcano plot showing Spearman correlations (x-axis) and significance (y-axis) for LY30 for controls.
Supplemental Table 2. Demographic characteristics and clinical data collected for hyperfibrinolysis and fibrinolysis shutdown patient groups.