

Abstract
Binswanger’s disease (BD) is the small vessel form of vascular cognitive impairment and dementia (VCID). Deposition of Alzheimer’s Disease (AD) proteins can begin in midlife and progress slowly, while aging of the vasculature also can begin in midlife, continuing to progress into old age, making mixed dementia (MX) the most common type of dementia. Biomarkers facilitate the early diagnosis of dementias. It is possible to diagnose MX prior to autopsy with biomarkers for vascular disease derived from diffusor tensor images (DTI) on MRI and AD proteins, amyloid β (Aβ), and phosphorylated tau, in CSF or in brain with PET. The presence of vascular disease accelerates cognitive decline. Both misfolded proteins and vascular disease promote inflammation, which can be detected in CSF by the presence of matrix metalloproteinases (MMPs), angiogenic growth factors and cytokines. MMPs disrupt the blood-brain barrier and break down myelin, producing BD’s two main pathological features. Advances in detecting biomarkers in plasma will provide early detection of dementia and aided by machine learning and artificial intelligence, will enhance diagnosis and form the basis for early treatments.
Introduction
The 1904 report by Otto Binswanger of a patient with white matter disease associated with arteriolosclerosis went largely unappreciated for many years due to the paucity of autopsies performed in the elderly1. In addition, interest in dementia was focused on gray matter by the discovery of amyloid in AD plaques2. With the advent of neuroimaging in the 1970s, revealing the extent of white matter disease, interest in vascular causes of dementia was stimulated and has continued to grow. This revival of interest in chronic vascular disease has led to attempts to agree on terminology3. The presence of vascular disease accelerates the cognitive loss in patients with AD pathology4, making mixed dementia (MX) the most common form of dementia5. This has led to a focus on treating vascular risk factors in midlife, such as hypertension, diabetes and sleep apnea, to slow the later onset of dementia6.
An understanding of the pathophysiology of white matter disease has emerged from autopsy studies in chronic vascular disease augmented with animal studies. This review will focus on pathophysiology since there are excellent papers on the clinical aspects of BD7–9.
Evolving concepts of white matter disease in dementia
Prior to modern neuroimaging, hardening of the arteries was considered the main cause of dementia. However, the importance of vascular disease waned until the “Nun study” demonstrated that cognitive loss in AD was accelerated in those with both AD and vascular pathology4. Subsequently, the importance of dual pathology was confirmed in a community-based cohort5 and in the AD research centers’ brain bank10.
In the 1970s computed tomography (CT) and magnetic resonance imaging (MRI) revealed a high incidence of white matter change that was unexpected. Prior to MRI showing the high incidence of white matter disease, multi-infarct dementia was considered the leading vascular cause of dementia11. Interest in vascular causes of dementia was further enhanced by the decline of the amyloid hypothesis with amyloid considered mainly important in the early stages rather than the sole cause12.
Misfolded proteins, such as amyloid β (Aβ), phosphorylated Tau (pTau) and α-synuclein induce an inflammatory response that occurs at a later age when both Aβ and pTau are present in the brain13. Vascular disease adds to the inflammation and promotes the disruption of the blood-brain barrier (BBB) in vascular dementia14–16, and AD17,18. The recognition of inflammation in dementia was aided by research into the role of inflammation in other organs.
Binswanger’s disease
There is controversy surrounding the use of the term, BD, with some advising against it19,20, while others found it useful as an indicator of white matter damage, particularly for research in vascular dementia21. AD research has benefited from well-defined diagnostic criteria based on clinical symptomatology, known rates of progression, and biomarkers in CSF and positron emission tomography (PET) to detect amyloid and tau in the brain22,23. This has not been true of research and clinical trials in vascular dementia because it is a heterogeneous disease ranging from multiple strokes to diffuse white matter injury. For many years the concept of multi-infarct dementia with a step-wise course was considered the main form of vascular dementia11. This changed when a consensus report recommended that subcortical ischemic vascular disease or BD and lacunar state were optimal for research studies21.
Neuroimaging caused a so-called “epidemic” of BD based on over diagnosis of white matter hyperintensities (WMH) on MRI24. Quantifying the WMHs by computer was an essential step in understanding their significance25,26. The neutral term leukoaraiosis (LA) was introduced to describe WMHs without pathological implications27.
The first advanced MRI method used to show white matter injury more precisely than fluid attenuated inversion recovery (FLAIR) was proton magnetic resonance spectroscopic imaging (1H-MRSI), which showed that the high concentration of N-acetylaspartate (NAA) in the brain was reduced in ischemic tissue, making it an excellent biomarker to separate ischemic white matter from leukoaraiosis28,29. This method was technically difficult and was rapidly replaced by diffusor tensor imaging (DTI) which was as accurate and more available on all MRI instruments. Several DTI methods have been adopted for research studies: peak width of skeletonized water mean diffusivity (PSMD) and mean free water (mFW)30,31. Abnormal DTI metrics are associated with neuropsychological tests for executive function, validating their usefulness as biomarkers of white matter injury31.
A genetic disease with similarities to BD is the inherited vascular disease, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) with Notch3 protein deposited in smooth muscle cells around capillaries. CADASIL is caused by mutations in the NOTCH3 gene which is important for the normal function and survival of vascular smooth muscle cells. CADASIL and BD have symptoms that are similar32.
High prevalence of mixed dementia
The development of multimodal biomarkers has made diagnosis of MX possible during life. Biomarkers were originally found for AD after the discovery of amyloid in the plaques with extension to the cerebrospinal fluid (CSF)2. Measurement of amyloid and tau in CSF or PET are biological biomarkers for diagnosis of AD in research studies; they are part of the ATN formula with amyloid (A), tau (T) and neurodegeneration (N)23. The authors left open the possibility of adding other biomarkers to the formula, particularly vascular disease (V)33, 34. MX can be identified during life by combining AD biomarkers obtained from CSF or PET to construct a composite AD score (ADS). Similarly, vascular injury to the white matter can be identified by DTI and formed into a composite vascular disease score (VDS). Plotting the ADS on the y-axis and VDS on the x-axis forms four quadrants: AD, vascular cognitive impairment and dementia (VCID), MX and leukoaraiosis (LA) (FIGURE 1). This approach is called “double dichotomy” and the graph reduces the complexity of the clustering problem to two dimensions35.
Figure 1:
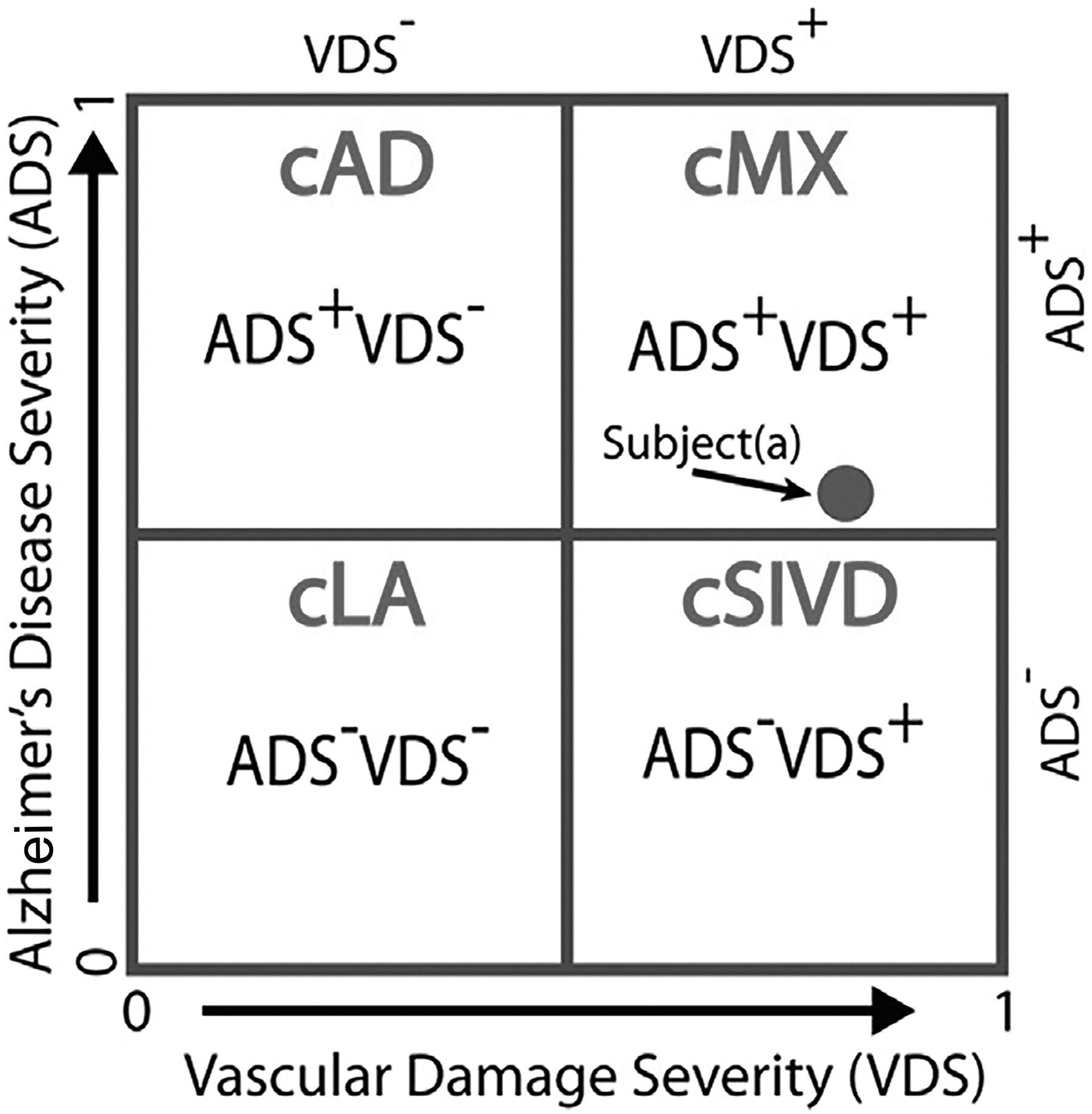
The double-dichotomy clustering method is a two-dimensional scatter plot with the x-axis being the VDS score and the y-axis being the ADS score, and a cut-off of 0.5 for each axis gives four quadrants, with each quadrant describing subjects with different disease characteristics. The four cluster (c)-based patient groups are cLA = VDS− ADS−, cAD = VDS− ADS+, cSIVD = VDS+ ADS−, and cMX = VDS+ ADS+. The location of a subject on this plot describes disease severity. For example, subject(a) is classified as cMX, but being close to the ADS boundary, we know that its ADS factors are similar to some of the SIVD subjects. (Reprinted from Caprihan et al35 © 2021 by the Authors. Published by Elsevier B.V. This is an open access article under the CC BY-NC-ND license).
Patients dying with dementia often have more than one pathological protein identified in their brains36. In addition, autopsy series of subjects with dementia indicate that a high percentage of patients have MX5,10,37. Autopsies of elderly patients dying with diagnoses other than dementia also show multiple misfolded proteins38; MX, which is found in up to 70% of dementia cases at autopsy, is the most common10. This is a strong impetus to find a way to diagnose these patients during life to avoid treating one pathological process while leaving others untreated, which may help explain the high failure rate of AD trials12,13.
Pathophysiology of white matter injury
BD has characteristic imaging and pathological findings. The MRI shows large areas of WMHs (Figure 2A). The arterioles have arteriolosclerosis with narrowed lumens and fibrotic, thickened outer walls. White matter has demyelination extending into the sulci; a characteristic finding is the sparing of the U-fibers joining cortical regions (FIGURE 2B). Sparing the U-fibers argues against strokes as the etiology and favors other causes such as inflammation for the loss of myelin39. Gray matter is generally unaffected except in patients with dual pathology. Cerebral amyloid angiopathy is seen in patients with AD but less frequently in small vessel disease40.
Figure 2:
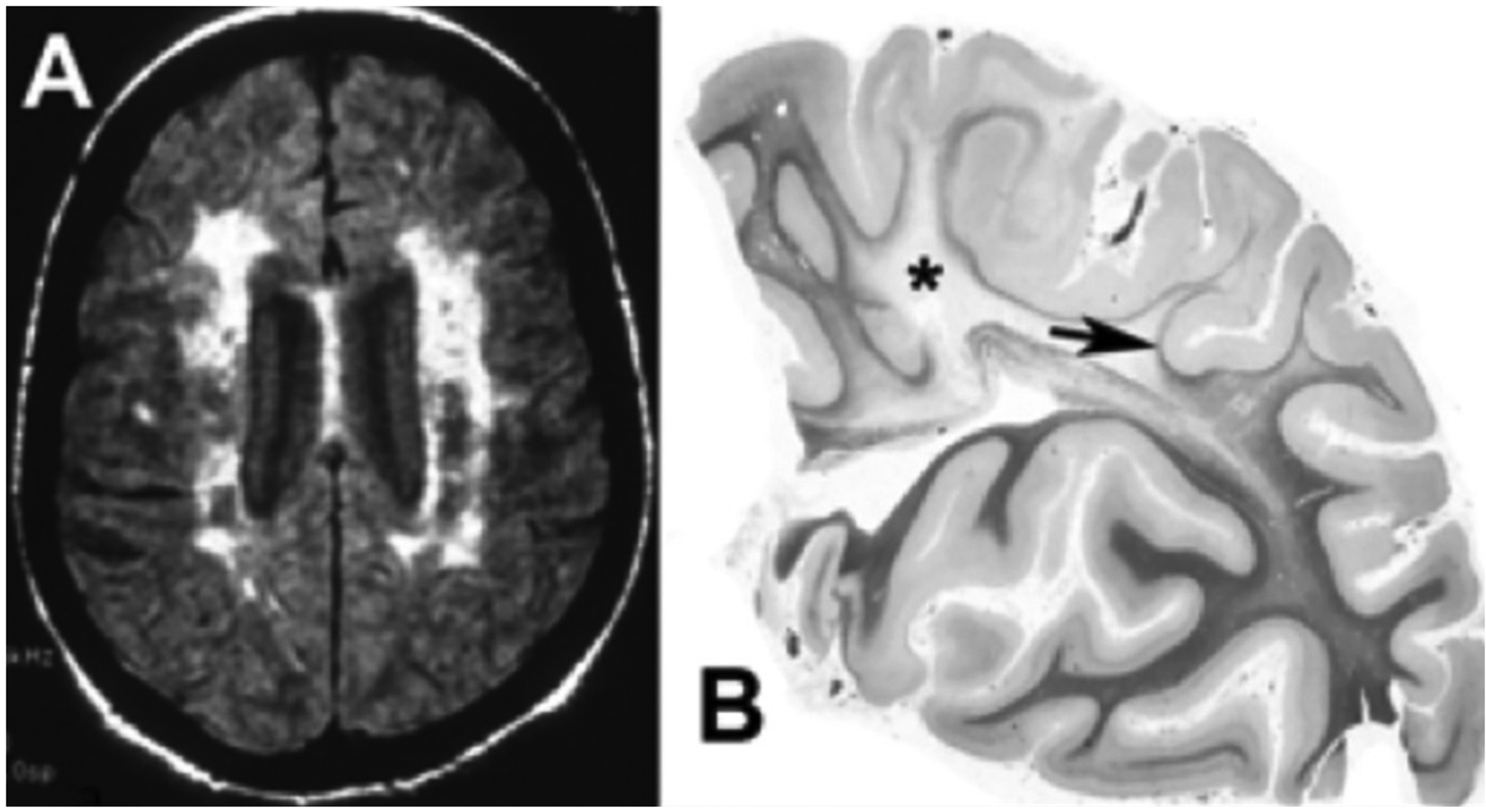
A) FLAIR MRI from a patient with Binswanger’s disease showing extensive white matter involvement in the subcortical region. There is relative preservation of the cortex and lack of ventricular enlargement. B) A hemisphere section from a patient with long-standing hypertension. In this myelin stain, there is loss of myelination (Asterix) in the centrum semiovale extending into the sulci and sparing U-fibers (arrow). (Figure 2B is from46 with permission. Copyright © 2001, Wolters Kluwer Health).
Impaired cerebral blood flow causing “malnutrition of the white matter” was suspected by Binswanger. Early PET studies of blood flow and tissue oxygenation showed a reduction in both in patients with BD41. Although many studies have confirmed the reduction in cerebral blood flow, particularly in the white matter, its role as a cause of the white matter damage has remained unresolved42.
Enlargement of perivascular spaces can be seen pathologically and on MRI. Ventricular enlargement can occur because of normal aging or pathologically in normal pressure hydrocephalus (NPH)43. There is a high incidence of hypertension in patients with NPH, and the clinical symptomatology of NPH and BD overlap44,45. Clinically separating the two diseases can be difficult.
Glial fibrillary acidic protein (GFAP) is often present, suggesting astrogliosis with astrocytic hypertrophy. The presence of inflammation is indicated by the presence of matrix metalloproteinases (MMPs) around blood vessels; activated microglia/macrophages are prominent, particularly around the hypertensive blood vessels46.
Blood-brain barrier disruption (BBB) has been observed pathologically by the extravasation of plasma proteins47. Brains of patients with BD showed extravasation of serum proteins, including immunoglobulins, complements and fibrinogen. However, pathological studies are of limited use in understanding the evolving brain damage since the end stage has multiple pathologies. Animal models are necessary to reveal the progression of molecular events involved in white matter injury.
Animal models in vascular dementia
Bilateral carotid occlusion (BCAO) was one of the first animal models used to study vascular cognitive impairment48. These animals had hypoxic hypoperfusion with the production of MMP-249. The limitations of BCAO were the ischemia produced at the onset of the occlusion because of the sudden restriction of cerebral blood flow and the lack of vascular risk factors such as hypertension.
An animal model closer to the human disease is the stroke-prone spontaneously hypertensive rat (SPSHR). These animals typically develop white matter disease with strokes and hemorrhages over one to two years; the time course of injury can be accelerated by feeding them a low protein, high salt diet and occluding one carotid50. We reduced white matter edema and improved behavior by treatment with the tetracycline derivative, minocycline, an inhibitor of MMPs with anti-inflammatory actions51.
A mouse model has been developed with the use of ameroid constrictors that imbibe fluid and slowly occlude the carotids, avoiding the initial ischemia, allowing them to survive long enough to produce white matter damage and behavioral changes52,53. Hypoxic hypoperfusion is the basis for the injury in the animal models; it is thought to be similar to the events occurring in hypertensive patients. The fall in oxygen levels initiates the expression of hypoxia-inducible factor-1α (HIF-1α), the central event in the subsequent molecular injury cascade (FIGURE 3). HIF-1α leads to the transcription of a cassette of molecules to restore oxygen to the tissues. The molecules induced by HIF-1α include erythropoietin to form red blood cells, angiogenic growth factors to facilitate vessel sprouting, and activation of mechanisms to promote glucose uptake54. Other molecular events promote the expression of proteins that perpetuate the injury. Among these are the genes involved in the expression of convertases, such as fur, which leads to the transcription of Furin, an activator of MMPs that plays a key role in injury and repair55.
Figure 3:
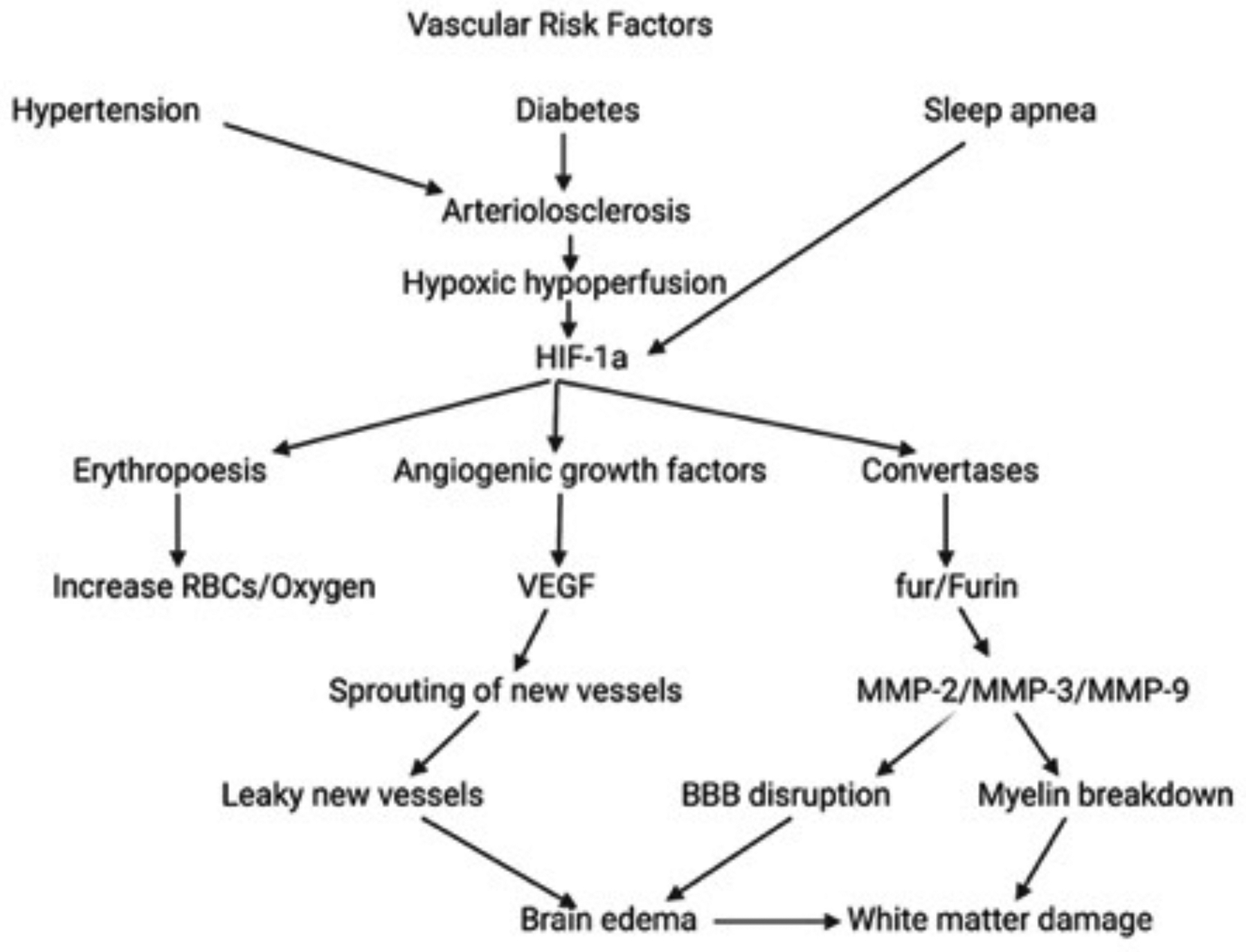
Illustration of the molecular cascade involved in the white matter injury in the Binswanger-type of vascular dementia. Vascular risk factors lead to arteriolosclerosis. Hypoxic hypoperfusion activates hypoxia inducible factor-1α (HIF-1α), which leads to erythropoiesis, angiogenesis, and convertases. The result is opening of the BBB with brain edema and white matter damage.
The MMPs are produced in latent forms that require activation. Constitutive MMPs, such as MMP-2, are produced normally to enable ongoing remodeling of the extracellular matrix. On the other hand, inducible MMPs, such as MMP-3 and MMP-9, only appear during inflammation in a latent form requiring activation by Furin and other enzymes. Furin initiates the activation cascade by activating pro-membrane-type metalloproteinase (proMT-MMP) (FIGURE 4). Activated MT-MMP activates proMMP-2 by forming a trimolecular complex that includes tissue inhibitor to metalloproteinases 1 (TIMP-1). Other molecules contribute to the expression and activation of these molecules. For example, MMPs and plasminogen activators work together; tissue plasminogen activator and urokinase plasminogen form plasmin, an activator of other enzymes.
Figure 4:
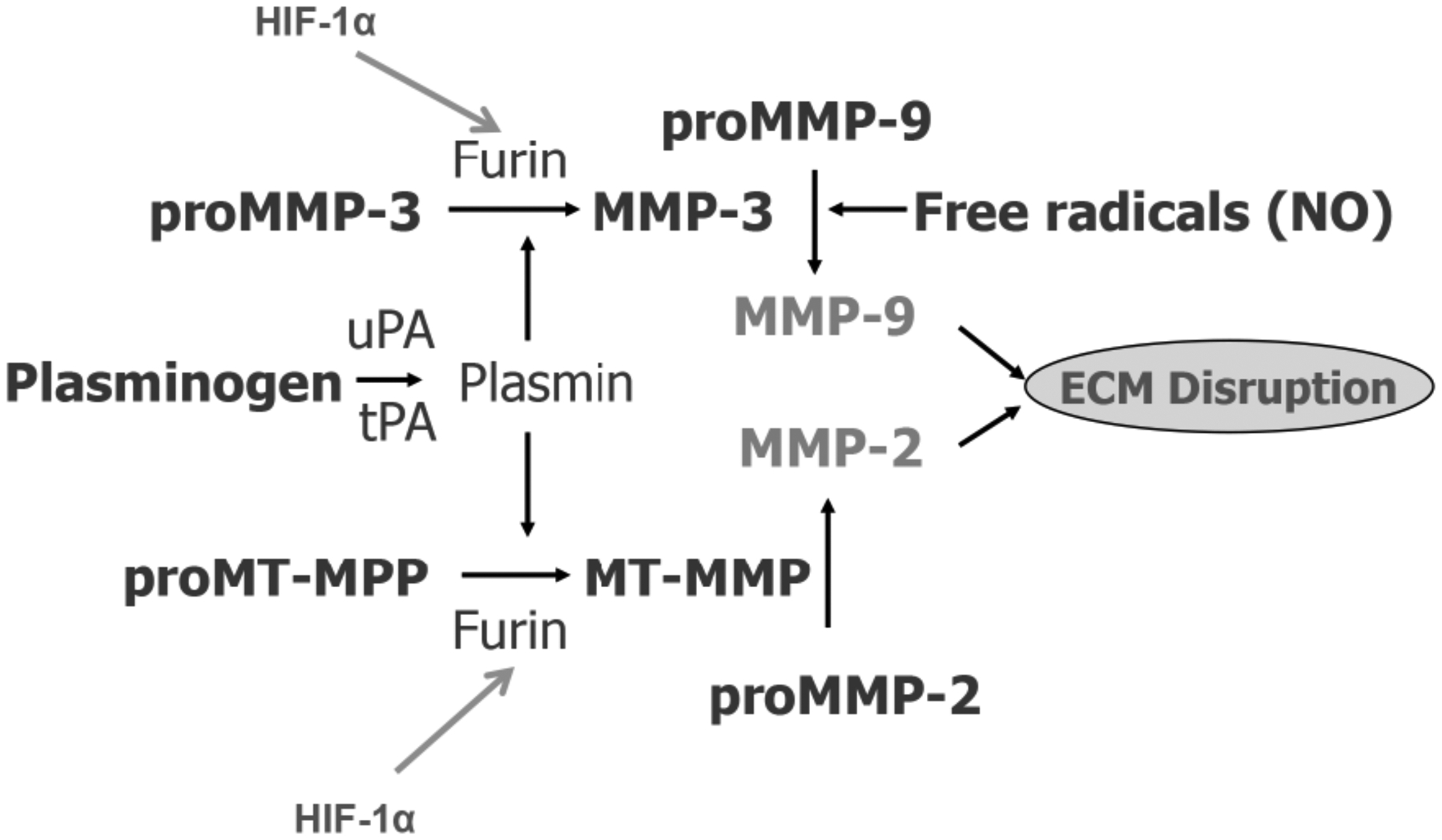
Mechanisms of activation of the matrix metalloproteinases (MMPs), which are normally latent enzymes. When the tissue is hypoxic and HIF-1α is released, Furin is formed. This convertase activates proMT-MMP and proMMP-3. Active MT-MMP activates proMMP-2 and MMP-3 activates proMMP-9 along with free radicals such as nitric oxide (NO). The plasminogen activators (PA), tissue PA and urokinase PA form plasmin, which is an activator of proMMP-3 and proMT-MMP. The result of these active enzymes is to disrupt the extracellular matrix (ECM). MT is membrane-type.
Blood vessels have junctions held together by several tight junction proteins, such as occludin, claudin and zona occludin56. Around the blood vessels is a basal lamina composed of type IV collagen, chondroitin sulfate and other matrix proteins. In the MMP family are gelatinases (MMP-2 and MMP-9) that attack extracellular matrix type IV collagen and tight junction proteins, increasing blood vessel permeability57.
Chronic inflammation and progressive white matter injury
MMP-9 is involved in acute ischemia, as shown by the reduced injury after stroke in MMP-9 knock-out mice58. Different MMPs are involved in vascular dementia where chronic ischemia induces MMP-2 in astrocytes (FIGURE 5A and 5B) and MMP-3 in inflammatory cells, such as microglia/macrophages and pericytes (FIGURE 5C)46. Astrocytic foot processes that surround blood vessels contain MMP-2 (FIGURE 5D). The close proximity to the blood vessels of the astrocytes and the pericytes suggests that they play a role in the proteolytic disruption of the BBB. Pericytes have characteristics of macrophages and smooth muscle cells and are in the basal lamina around the blood vessels (FIGURE 5E). Other molecules found in CSF in vascular dementia, in addition to MMPs, include angiogenic growth factors, vascular endothelial growth factor (VEGF-C) and placental growth factor (PlGF), and cytokines, interleukin-6 and -8 (IL-6 and IL-8), along with tumor necrosis factor-α (TNF-α), which contribute to the inflammatory damage59. Differentiation of resident microglia and infiltrating macrophages is difficult without special staining methods; it is best to refer to these cells a microglia/macrophage. These inflammatory cells are the source of the cytokines that are involved in the inflammatory damage.
Figure 5:
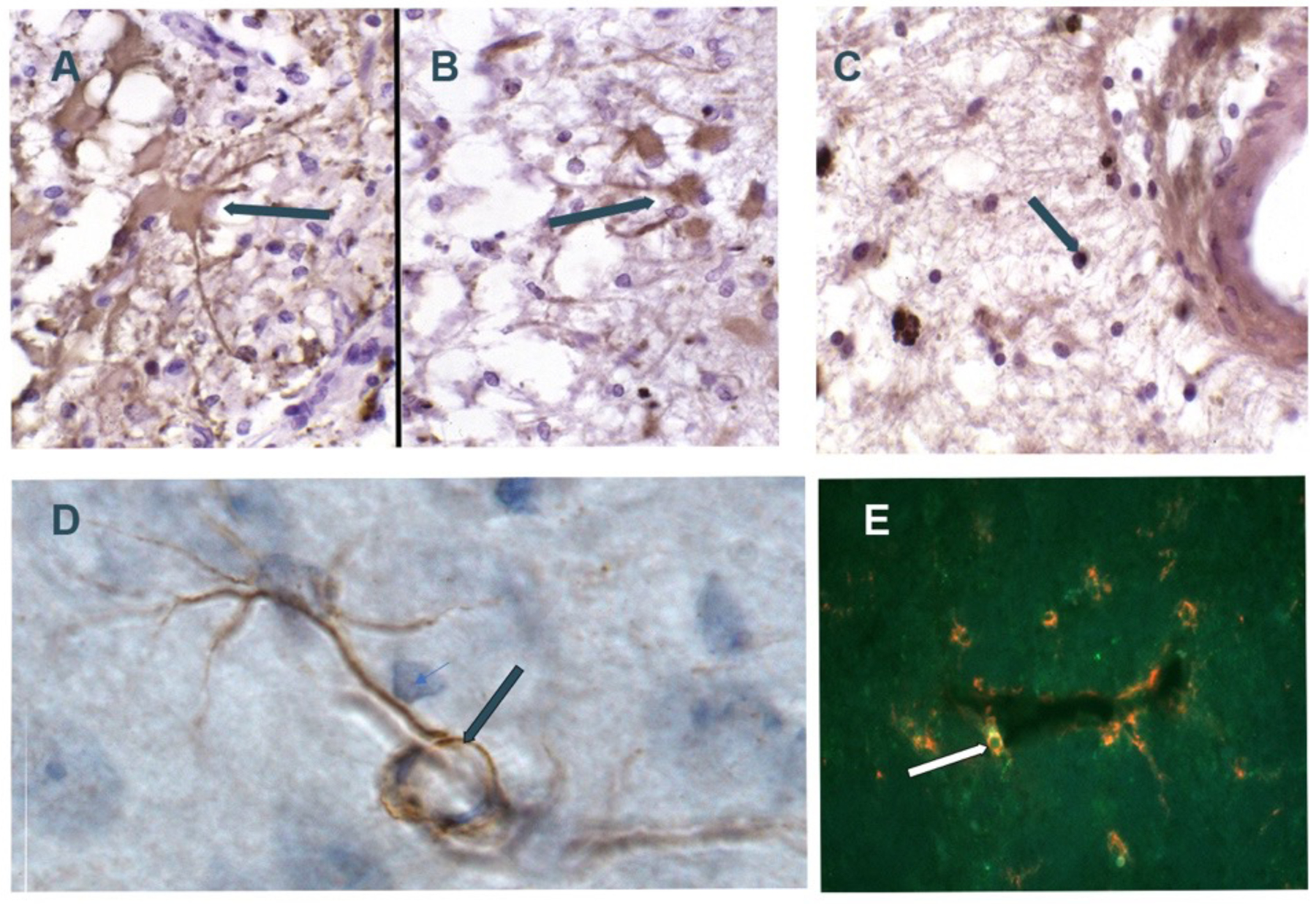
Composite of MMP immunostaining. A) GFAP+ astrocyte (arrow) in patient with vascular dementia; B) Similar region as in A with astrocyte stained with MMP-2 antibody; C) PG-1+ macrophage/microglia (arrow) in patient with vascular dementia (5A,5B,5C are from Rosenberg et al46 Copyright © 2001, Wolters Kluwer Health); D) normal rat brain with MMP-2-staining astrocyte. Astrocyte foot process (arrow) wrapped around vessel; E) Rat brain dual labelled fluorescent Ox-42+ pericyte (green) and MMP-3 (red) next to vessel (arrow) (5D and 5E from Rosenberg et al70 Copyright © 2001 Elsevier Science B.V.).
Multiple processes are involved in the disruption of the BBB through the action of the MMPs and angiogenic growth factors. In addition to the direct attack on vessels by MMPs in inflammation, stimulation of new vessel growth by angiogenic growth factors leads to permeable blood vessels. Angiogenesis involves the combined action of MMPs to break down the extracellular matrix and VEGF to induce the sprouting of vessels from existing ones60. When proteases and angiogenic factors disrupt the blood vessels, brain edema is formed that contributes to the white matter injury as seen with DTI.
Inflammation is an important component of the pathology of chronic forms of ischemia. The presence of misfolded proteins, Aβ, pTau, and α-synuclein activate microglia/macrophages; these microglia/macrophages contribute to ongoing tissue damage in chronic conditions where they continue to produce toxic products long after the acute insult61. The continuity of the CSF with the interstitial fluid allows molecules produced in the brain to be detected in CSF. Measurements in the blood of molecules produced in the brain is more difficult due to very low concentrations and interference with plasma proteins. We identified MMP-1, MMP-2, MMP-3, MMP-9 and MMP-10 in CSF from patients with various types of dementia with a multiplex assay kit, including those with Binswanger-type changes in the white matter on FLAIR MRI. The MMPs that were significantly elevated in the CSF in the dementia patients were MMP-2, MMP-3 and MMP-10, but not MMP-9, which is consistent with the dichotomy between the acute and chronic response to injury59. MMP-3 and MMP-10 are stromelysins (1 and 2, respectively), which can attack all the components of the extracellular matrix and the tight junction proteins.
Inflammation involves the production of inflammasomes in dementia. In AD, microglia/macrophages secrete ASC specks, a micrometer-sized structure formed by the inflammasome adaptor protein ASC (apoptosis-associated speck-like protein containing a CARD) that is a major component of the inflammasome complex. ASC binds to Aβ, forming a molecule that can seed other cells62. PET studies with ligands for microglia/macrophages show an increase in inflammation as Aβ increases in the early stages, followed by a second increase as pTau increases63. There is microglia/macrophages-mediated inflammation in vascular dementia, as shown by PET64.
In a study that provides insight into mechanisms involved in progressive white matter damage, microglia that had ingested neurons containing tau entered a hypofunctional, senescent state and continually secreted MMP-3, which could be related to the progressive white matter injury65. Biomarkers relate to cognitive function establishing their relevance: we found that many biomarkers correlated with neuropsychological test results (FIGURE 6).
Figure 6:
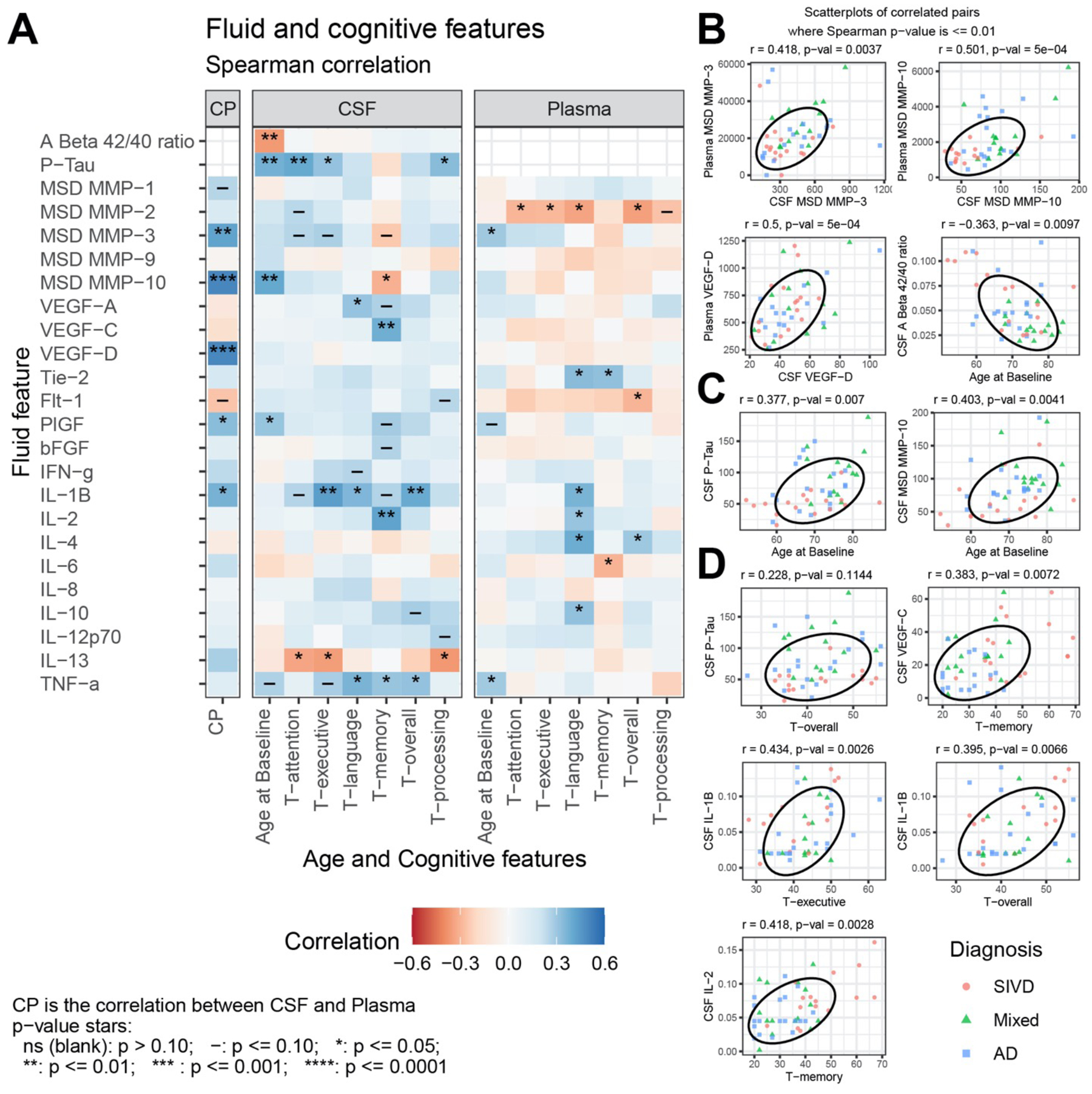
(A) Spearman correlation of CSF and Plasma features with Age and Cognitive features for subcortical ischemic vascular disease (SIVD), Mixed dementia, and AD patients. Selected scatterplots illustrate relationships at the p ≤ 0.01 significance level: (B) same fluid features CSF vs. plasma, (C) fluid features with Age, and (D) fluid features with cognitive features. (From Erhardt et al59 Open Access Permission Copyright © 2021 Erhardt et al).
The pathophysiology of BD is the breakdown of myelin and disruption of the BBB. Both of these mechanisms of injury can be explained by a non-immunological mechanism of demyelination referred to as “bystander” demyelination66,67. Stimulated macrophages released the protease, plasmin, which degraded myelin into myelin basic protein fragments. Microglia/macrophages release a similar mix of enzymes. This suggests that microglia/macrophages could release plasmin and MMP-3, which would lead to BBB and myelin damage (FIGURE 7).
Figure 7:
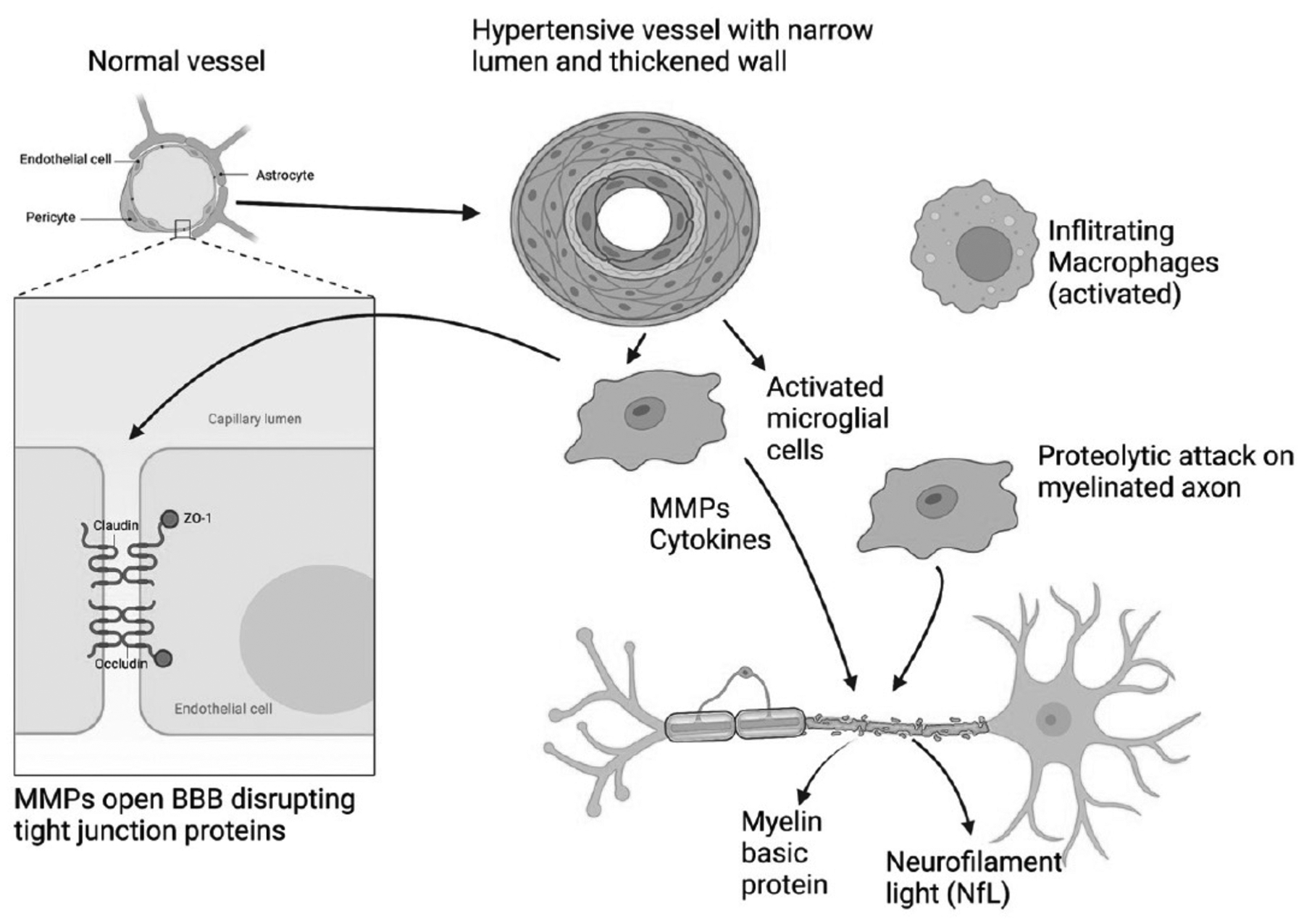
Schematic illustration of by-stander demyelination mechanism in BD. A normal vessel is shown in the upper left. There are tight junction proteins between the endothelial cells that include claudin, occludin and zona occluden (zo-1). The vessel in the top center shows the effects of long-standing hypertension with narrowing of the lumen and thickening of the outer wall. Hypertensive vessels attract microglia/macrophages that attempt to remodel the vessel. These activated inflammatory cells secrete inflammatory factors, namely, MMPs and cytokines. These activated cells destroy the myelinated axons resulting in the release of myelin basic protein and neurofilament light (NfL), which can be measured in CSF and blood as a biomarker of inflammatory injury. Infiltrating macrophages (top right) can add to the proteolytic damage. (Figure created with BioRender with permission).
Biomarkers, machine learning, artificial intelligence, and precision medicine
The pathology that characterizes AD and VCID begins in midlife and slowly progresses over 20 years before its full impact is apparent. This is a major impetus to identify disease processes at an early stage, which will be critical for treatments to be effective. Early identification will require biomarkers that can be used as surrogates for early disease. This process has begun in AD where Aβ and pTau measured in CSF and with PET have been used for research studies in AD23. Similar biomarkers for VCID remain to be identified. An early attempt to diagnose VCID was the use of lists of symptoms, like the Hachinski Ischemic Score, which gave a high score to those with symptoms and signs suggesting vascular disease11,68. We have refined this approach by adding other diagnostic modalities to construct a 10-point BD score and used statistical analysis with exploratory factor analysis (EFA) to select patients with BD. Patients were followed for an average of 2 years to obtain clinical consensus diagnoses to improve accuracy69.
The rapid growth in measurements of multiple biomarkers has resulted in large data sets that can only be analyzed by a “big data” approach59. Further refinements led to the inclusion of additional biomarkers, improving the classification of vascular and neurodegenerative causes. These statistical programs identify the most important features from several biomarkers, improving the accuracy of diagnosis and delineating the optimal biomarkers.
Learning machine programs with artificial intelligence can improve diagnostic accuracy by incorporating diagnoses from experts and outcomes of treatments; as the size of the database increases, diagnostic accuracy grows. This approach can also incorporate responses to future treatment trials.
There has been a major increase in the understanding of the role of plasma biomarkers that will impact the way we detect and treat patients with dementia. The early use of biomarkers was dominated by studies in AD, culminating in the biological diagnosis of AD based on the amyloid (A), tau (T), and neurodegeneration (N) formula with A and T obtained from CSF proteins or PET scanning and N from MRI measurements23. A new formulation proposes to add other features, including vascular disease and other misfolded proteins, expanding the original formula to ATX(N) with X indicating future biomarkers33.
In conclusion, along with the increase in the number of elderly individuals with dementia, there has been a parallel growth in information on the role of vascular disease in dementia, recognizing the role of small vessel disease, making MX the most common form of dementia. The major small vessel form of vascular dementia is BD because the gradually progressive white matter changes can be monitored by MRI, revealing the natural history, and allowing the effects of treatments to be studied. Hypoxic hypoperfusion due to hypertensive narrowing of the arterioles initiates a cascade of molecular events that culminate in the opening of the BBB and the breakdown of the myelinated white matter. Matrix metalloproteinases, which are induced under hypoxic conditions, are an important cause of the neuroinflammation driving the opening of the BBB and myelin damage.
Supplementary Material
Acknowledgments:
Funding for these studies came from NIH grants to GAR: RO1 NS052305 and RO1 NS068048, UH3 NS100598 (MarkVCID), and P20 AD068077. This project was supported by an award from the National Center for Advancing Translational Sciences, National Institutes of Health under grant number UL1TR001449. The PRISMA 3T upgrade at The Mind Research Network was supported by NIH award S10OD025313.
Non-standard Abbreviations and Acronyms
- BD
Binswanger’s disease
- AD
Alzheimer’s disease
- VCID
vascular cognitive impairment and dementia
- MX
mixed dementia
- DTI
diffusor tensor images
- Aβ
amyloid β
- MMPs
matrix metalloproteinases
- CT
computed tomography
- MRI
magnetic resonance imaging
- pTau
phosphorylated Tau
- BBB
blood-brain barrier
- PET
positron emission tomography
- WMH
white matter hyperintensities
- LA
leukoaraiosis
- FLAIR
fluid attenuated inversion recovery
- PSMD
peak width of skeletonized water mean diffusivity
- mFW
mean free water
- CADASIL
cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy
- NPH
normal pressure hydrocephalus
- proMT-MMP
pro-membrane-type metalloproteinase
- TNF-α
tumor necrosis factor-α
- ASC
apoptosis-associated speck-like protein containing a CARD
Footnotes
Disclosures: Nothing to disclose.
REFERENCES
- 1.Román GC. Senile dementia of the Binswanger type. A vascular form of dementia in the elderly. Jama. 1987;258:1782–1788. doi: 10.1001/jama.1987.03400130096040 [DOI] [PubMed] [Google Scholar]
- 2.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4 [DOI] [PubMed] [Google Scholar]
- 3.Hachinski V, Iadecola C, Petersen RC, Breteler MM, Nyenhuis DL, Black SE, Powers WJ, DeCarli C, Merino JG, Kalaria RN, et al. National Institute of Neurological Disorders and Stroke-Canadian Stroke Network vascular cognitive impairment harmonization standards. Stroke. 2006;37:2220–2241. doi: 10.1161/01.Str.0000237236.88823.47 [DOI] [PubMed] [Google Scholar]
- 4.Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study [see comments]. JAMA. 1997;277:813–817. doi: [PubMed] [Google Scholar]
- 5.Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007;69:2197–2204. doi: 10.1212/01.wnl.0000271090.28148.24 10.1212/01.wnl.0000271090.28148.24. Epub 2007 Jun 13. [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg A, Ngandu T, Rusanen M, Antikainen R, Bäckman L, Havulinna S, Hänninen T, Laatikainen T, Lehtisalo J, Levälahti E, et al. Multidomain lifestyle intervention benefits a large elderly population at risk for cognitive decline and dementia regardless of baseline characteristics: The FINGER trial. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2018;14:263–270. doi: 10.1016/j.jalz.2017.09.006 [DOI] [PubMed] [Google Scholar]
- 7.Caplan LR, Schoene WC. Clinical features of subcortical arteriosclerotic encephalopathy (Binswanger disease). Neurology 1978;28:1206–1215. doi: 10.1212/wnl.28.12.1206 [DOI] [PubMed] [Google Scholar]
- 8.Fisher CM. Binswanger’s encephalopathy: a review. J Neurol 1989;236:65–79. doi: 10.1007/bf00314400 [DOI] [PubMed] [Google Scholar]
- 9.Bennett DA, Wilson RS, Gilley DW, Fox JH. Clinical diagnosis of Binswanger’s disease. J Neurol Neurosurg Psychiatry. 1990;53:961–965. doi: 10.1136/jnnp.53.11.961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, Grossman M, Monsell SE, Kukull WA, Trojanowski JQ. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain 2013;136:2697–2706. doi: 10.1093/brain/awt188 10.1093/brain/awt188. Epub 2013 Jul 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hachinski VC, Lassen NA, Marshall J. Multi-infarct dementia. A cause of mental deterioration in the elderly. Lancet 1974;2:207–210. doi: 10.1016/s0140-6736(74)91496-2 [DOI] [PubMed] [Google Scholar]
- 12.Long JM, Holtzman DM. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell. 2019;179:312–339. doi: 10.1016/j.cell.2019.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 2016;8:595–608. doi: 10.15252/emmm.201606210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wallin A, Blennow K, Fredman P, Gottfries CG, Karlsson I, Svennerholm L. Blood brain barrier function in vascular dementia. Acta Neurol Scand 1990;81:318–322. doi: 10.1111/j.1600-0404.1990.tb01562.x [DOI] [PubMed] [Google Scholar]
- 15.Wardlaw JM. Blood-brain barrier and cerebral small vessel disease. Journal of the neurological sciences. 2010;299:66–71. doi: 10.1016/j.jns.2010.08.042 [DOI] [PubMed] [Google Scholar]
- 16.Taheri S, Gasparovic C, Huisa BN, Adair JC, Edmonds E, Prestopnik J, Grossetete M, Shah NJ, Wills J, Qualls C, et al. Blood-brain barrier permeability abnormalities in vascular cognitive impairment. Stroke. 2011;42:2158–2163. doi: 10.1161/STROKEAHA.110.611731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua L, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296–302. doi: 10.1016/j.neuron.2014.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nation DA, Sweeney MD, Montagne A, Sagare AP, D’Orazio LM, Pachicano M, Sepehrband F, Nelson AR, Buennagel DP, Harrington MG, et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med 2019;25:270–276. doi: 10.1038/s41591-018-0297-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hachinski V Binswanger’s disease: neither Binswanger’s nor a disease. Journal of the neurological sciences. 1991;103:1. doi: 10.1016/0022-510x(91)90274-b [DOI] [PubMed] [Google Scholar]
- 20.Pantoni L, Garcia JH. The significance of cerebral white matter abnormalities 100 years after Binswanger’s report. A review. Stroke. 1995;26:1293–1301. doi: 10.1161/01.str.26.7.1293 [DOI] [PubMed] [Google Scholar]
- 21.Erkinjuntti T, Inzitari D, Pantoni L, Wallin A, Scheltens P, Rockwood K, Roman GC, Chui H, Desmond DW. Research criteria for subcortical vascular dementia in clinical trials. J Neural Transm Suppl 2000;59:23–30. doi: 10.1007/978-3-7091-6781-6_4 [DOI] [PubMed] [Google Scholar]
- 22.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr., Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2018;14:535–562. doi: 10.1016/j.jalz.2018.02.018 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Binswanger’s encephalopathy. Lancet. 1981;1:923. doi: [PubMed] [Google Scholar]
- 25.DeCarli C, Murphy DG, Tranh M, Grady CL, Haxby JV, Gillette JA, Salerno JA, Gonzales-Aviles A, Horwitz B, Rapoport SI, et al. The effect of white matter hyperintensity volume on brain structure, cognitive performance, and cerebral metabolism of glucose in 51 healthy adults. Neurology 1995;45:2077–2084. doi: 10.1212/wnl.45.11.2077 [DOI] [PubMed] [Google Scholar]
- 26.Au R, Massaro JM, Wolf PA, Young ME, Beiser A, Seshadri S, D’Agostino RB, DeCarli C. Association of white matter hyperintensity volume with decreased cognitive functioning: the Framingham Heart Study. Arch Neurol 2006;63:246–250. doi: 10.1001/archneur.63.2.246 [DOI] [PubMed] [Google Scholar]
- 27.Hachinski VC, Potter P, Merskey H. Leuko-araiosis. Archives of Neurology 1987;44:21–23. doi: [DOI] [PubMed] [Google Scholar]
- 28.Brooks WM, Wesley MH, Kodituwakku PW, Garry PJ, Rosenberg GA. 1H-MRS differentiates white matter hyperintensities in subcortical arteriosclerotic encephalopathy from those in normal elderly. Stroke 1997;28:1940–1943. doi: [DOI] [PubMed] [Google Scholar]
- 29.Sappey-Marinier D, Calabrese G, Hetherington HP, Fisher SN, Deicken R, Van Dyke C, Fein G, Weiner MW. Proton magnetic resonance spectroscopy of human brain: applications to normal white matter, chronic infarction, and MRI white matter signal hyperintensities. Magnetic resonance in medicine. 1992;26:313–327. doi: 10.1002/mrm.1910260211 [DOI] [PubMed] [Google Scholar]
- 30.Baykara E, Gesierich B, Adam R, Tuladhar AM, Biesbroek JM, Koek HL, Ropele S, Jouvent E, Chabriat H, Ertl-Wagner B, et al. A Novel Imaging Marker for Small Vessel Disease Based on Skeletonization of White Matter Tracts and Diffusion Histograms. Annals of neurology 2016;80:581–592. doi: 10.1002/ana.24758 [DOI] [PubMed] [Google Scholar]
- 31.Maillard P, Fletcher E, Singh B, Martinez O, Johnson DK, Olichney JM, Farias ST, DeCarli C. Cerebral white matter free water: A sensitive biomarker of cognition and function. Neurology 2019;92:e2221–e2231. doi: 10.1212/WNL.0000000000007449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jolly AA, Nannoni S, Edwards H, Morris RG, Markus HS. Prevalence and Predictors of Vascular Cognitive Impairment in Patients With CADASIL. Neurology 2022. doi: 10.1212/WNL.0000000000200607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hampel H, Cummings J, Blennow K, Gao P, Jack CR Jr., Vergallo A Developing the ATX(N) classification for use across the Alzheimer disease continuum. Nat Rev Neurol 2021;17:580–589. doi: 10.1038/s41582-021-00520-w [DOI] [PubMed] [Google Scholar]
- 34.Wilcock D, Jicha G, Blacker D, Albert MS, D’Orazio LM, Elahi FM, Fornage M, Hinman JD, Knoefel J, Kramer J, et al. MarkVCID cerebral small vessel consortium: I. Enrollment, clinical, fluid protocols. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2021;17:704–715. doi: 10.1002/alz.12215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caprihan A, Raja R, Hillmer LJ, Erhardt EB, Prestopnik J, Thompson J, Adair JC, Knoefel JE, Rosenberg GA. A double-dichotomy clustering of dual pathology dementia patients. Cereb Circ Cogn Behav 2021;2. doi: 10.1016/j.cccb.2021.100011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karanth S, Nelson PT, Katsumata Y, Kryscio RJ, Schmitt FA, Fardo DW, Cykowski MD, Jicha GA, Van Eldik LJ, Abner EL. Prevalence and Clinical Phenotype of Quadruple Misfolded Proteins in Older Adults. JAMA Neurol 2020. doi: 10.1001/jamaneurol.2020.1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Snyder HM, Corriveau RA, Craft S, Faber JE, Greenberg SM, Knopman D, Lamb BT, Montine TJ, Nedergaard M, Schaffer CB, et al. Vascular contributions to cognitive impairment and dementia including Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2015;11:710–717. doi: 10.1016/j.jalz.2014.10.008 10.1016/j.jalz.2014.10.008. Epub 2014 Dec 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sonnen JA, Santa Cruz K, Hemmy LS, Woltjer R, Leverenz JB, Montine KS, Jack CR, Kaye J, Lim K, Larson EB, et al. Ecology of the aging human brain. Arch Neurol 2011;68:1049–1056. doi: 10.1001/archneurol.2011.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tomimoto H Subcortical vascular dementia. Neuroscience research. 2011;71:193–199. doi: 10.1016/j.neures.2011.07.1820 [DOI] [PubMed] [Google Scholar]
- 40.Chui HC, Zarow C, Mack WJ, Ellis WG, Zheng L, Jagust WJ, Mungas D, Reed BR, Kramer JH, Decarli CC, et al. Cognitive impact of subcortical vascular and Alzheimer’s disease pathology. Annals of neurology 2006;60:677–687. doi: 10.1002/ana.21009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yao H, Sadoshima S, Kuwabara Y, Ichiya Y, Fujishima M. Cerebral blood flow and oxygen metabolism in patients with vascular dementia of the Binswanger type. Stroke. 1990;21:1694–1699. doi: 10.1161/01.str.21.12.1694 [DOI] [PubMed] [Google Scholar]
- 42.Iadecola C, Duering M, Hachinski V, Joutel A, Pendlebury ST, Schneider JA, Dichgans M. Vascular Cognitive Impairment and Dementia: JACC Scientific Expert Panel. J Am Coll Cardiol 2019;73:3326–3344. doi: 10.1016/j.jacc.2019.04.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bethlehem RAI, Seidlitz J, White SR, Vogel JW, Anderson KM, Adamson C, Adler S, Alexopoulos GS, Anagnostou E, Areces-Gonzalez A, et al. Brain charts for the human lifespan. Nature. 2022;604:525–533. doi: 10.1038/s41586-022-04554-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koto A, Rosenberg G, Zingesser LH, Horoupian D, Katzman R. Syndrome of normal pressure hydrocephalus: possible relation to hypertensive and arteriosclerotic vasculopathy. J Neurol Neurosurg Psychiatry 1977;40:73–79. doi: 10.1136/jnnp.40.1.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Graff-Radford NR, Godersky JC. Idiopathic normal pressure hydrocephalus and systemic hypertension. Neurology 1987;37:868–871. doi: 10.1212/wnl.37.5.868 [DOI] [PubMed] [Google Scholar]
- 46.Rosenberg GA, Sullivan N, Esiri MM. White matter damage is associated with matrix metalloproteinases in vascular dementia. Stroke. 2001;32:1162–1168. doi: 10.1161/01.str.32.5.1162 [DOI] [PubMed] [Google Scholar]
- 47.Akiguchi I, Tomimoto H, Suenaga T, Wakita H, Budka H. Blood-brain barrier dysfunction in Binswanger’s disease; an immunohistochemical study. Acta Neuropathol 1998;95:78–84. doi: 10.1007/s004010050768 [DOI] [PubMed] [Google Scholar]
- 48.Wakita H, Tomimoto H, Akiguchi I, Matsuo A, Lin JX, Ihara M, McGeer PL. Axonal damage and demyelination in the white matter after chronic cerebral hypoperfusion in the rat. Brain Res 2002;924:63–70. doi: 10.1016/s0006-8993(01)03223-1 [DOI] [PubMed] [Google Scholar]
- 49.Ihara M, Tomimoto H, Kinoshita M, Oh J, Noda M, Wakita H, Akiguchi I, Shibasaki H. Chronic cerebral hypoperfusion induces MMP-2 but not MMP-9 expression in the microglia and vascular endothelium of white matter. JCerebBlood Flow Metab 2001;21:828–834. doi: [DOI] [PubMed] [Google Scholar]
- 50.Jalal FY, Yang Y, Thompson J, Lopez AC, Rosenberg GA. Myelin loss associated with neuroinflammation in hypertensive rats. Stroke. 2012;43:1115–1122. doi: 10.1161/STROKEAHA.111.643080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jalal FY, Yang Y, Thompson JF, Roitbak T, Rosenberg GA. Hypoxia-induced neuroinflammatory white-matter injury reduced by minocycline in SHR/SP. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2015;35:1145–1153. doi: 10.1038/jcbfm.2015.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hainsworth AH, Allan SM, Boltze J, Cunningham C, Farris C, Head E, Ihara M, Isaacs JD, Kalaria RN, Lesnik Oberstein SA, et al. Translational models for vascular cognitive impairment: a review including larger species. BMC Med 2017;15:16. doi: 10.1186/s12916-017-0793-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hattori Y, Kitamura A, Nagatsuka K, Ihara M. A novel mouse model of ischemic carotid artery disease. PloS one. 2014;9:e100257. doi: 10.1371/journal.pone.0100257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Semenza GL. Hypoxia-inducible factor 1 and cardiovascular disease. Annual review of physiology. 2014;76:39–56. doi: 10.1146/annurev-physiol-021113-170322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rosenberg GA. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet neurology. 2009;8:205–216. doi: 10.1016/S1474-4422(09)70016-X S1474–4422(09)70016-X [pii] 10.1016/S1474–4422(09)70016-X. [DOI] [PubMed] [Google Scholar]
- 56.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. PharmacolRev 2005;57:173–185. doi: [DOI] [PubMed] [Google Scholar]
- 57.Candelario-Jalil E, Yang Y, Rosenberg GA. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience. 2009;158:983–994. doi: S0306–4522(08)00895–6 [pii] 10.1016/j.neuroscience.2008.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2001;21:7724–7732. doi: 10.1523/jneurosci.21-19-07724.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Erhardt EB, Adair JC, Knoefel JE, Caprihan A, Prestopnik J, Thompson J, Hobson S, Siegel D, Rosenberg GA. Inflammatory Biomarkers Aid in Diagnosis of Dementia. Frontiers in aging neuroscience. 2021;13:717344. doi: 10.3389/fnagi.2021.717344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rundhaug JE. Matrix metalloproteinases and angiogenesis. J Cell Mol Med 2005;9:267–285. doi: 10.1111/j.1582-4934.2005.tb00355.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med 2017;23:1018–1027. doi: 10.1038/nm.4397 [DOI] [PubMed] [Google Scholar]
- 62.Tejera D, Mercan D, Sanchez-Caro JM, Hanan M, Greenberg D, Soreq H, Latz E, Golenbock D, Heneka MT. Systemic inflammation impairs microglial Aβ clearance through NLRP3 inflammasome. Embo j 2019;38:e101064. doi: 10.15252/embj.2018101064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ismail R, Parbo P, Madsen LS, Hansen AK, Hansen KV, Schaldemose JL, Kjeldsen PL, Stokholm MG, Gottrup H, Eskildsen SF, et al. The relationships between neuroinflammation, beta-amyloid and tau deposition in Alzheimer’s disease: a longitudinal PET study. Journal of neuroinflammation. 2020;17:151. doi: 10.1186/s12974-020-01820-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Walsh J, Tozer DJ, Sari H, Hong YT, Drazyk A, Williams G, Shah NJ, O’Brien JT, Aigbirhio FI, Rosenberg G, et al. Microglial activation and blood-brain barrier permeability in cerebral small vessel disease. Brain. 2021;144:1361–1371. doi: 10.1093/brain/awab003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brelstaff JH, Mason M, Katsinelos T, McEwan WA, Ghetti B, Tolkovsky AM, Spillantini MG. Microglia become hypofunctional and release metalloproteases and tau seeds when phagocytosing live neurons with P301S tau aggregates. Sci Adv 2021;7:eabg4980. doi: 10.1126/sciadv.abg4980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cammer W, Bloom BR, Norton WT, Gordon S. Degradation of basic protein in myelin by neutral proteases secreted by stimulated macrophages: a possible mechanism of inflammatory demyelination. Proceedings of the National Academy of Sciences of the United States of America. 1978;75:1554–1558. doi: 10.1073/pnas.75.3.1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Norton WT, Cammer W, Bloom BR, Gordon S. Neutral proteinases secreted by macrophages degrade basic protein: a possible mechanism of inflammatory demyelination. Adv Exp Med Biol 1978;100:365–381. doi: 10.1007/978-1-4684-2514-7_26 [DOI] [PubMed] [Google Scholar]
- 68.Hachinski V, Oveisgharan S, Romney AK, Shankle WR. Optimizing the Hachinski Ischemic Scale. Arch Neurol 2012;69:169–175. doi: 10.1001/archneurol.2011.1698 [DOI] [PubMed] [Google Scholar]
- 69.Rosenberg GA, Prestopnik J, Adair JC, Huisa BN, Knoefel J, Caprihan A, Gasparovic C, Thompson J, Erhardt EB, Schrader R. Validation of biomarkers in subcortical ischaemic vascular disease of the Binswanger type: approach to targeted treatment trials. J Neurol Neurosurg Psychiatry. 2015;86:1324–1330. doi: 10.1136/jnnp-2014-309421 10.1136/jnnp-2014–309421. Epub 2015 Jan 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rosenberg GA, Cunningham LA, Wallace J, Alexander S, Estrada EY, Grossetete M, Razhagi A, Miller K, Gearing A. Immunohistochemistry of matrix metalloproteinases in reperfusion injury to rat brain: activation of MMP-9 linked to stromelysin-1 and microglia in cell cultures. Brain Res 2001;893:104–112. doi: 10.1016/s0006-8993(00)03294-7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.