

Abstract
BACKGROUND & AIMS:
Mechanisms contributing to the onset and progression of Barrett’s (BE)-associated esophageal adenocarcinoma (EAC) remain elusive. Here, we interrogated the major signaling pathways deregulated early in the development of Barrett’s neoplasia.
METHODS:
Whole-transcriptome RNA sequencing analysis was performed in primary BE, EAC, normal esophageal squamous, and gastric biopsy tissues (n = 89). Select pathway components were confirmed by quantitative polymerase chain reaction in an independent cohort of premalignant and malignant biopsy tissues (n = 885). Functional impact of selected pathway was interrogated using transcriptomic, proteomic, and pharmacogenetic analyses in mammalian esophageal organotypic and patient-derived BE/EAC cell line models, in vitro and/or in vivo.
RESULTS:
The vast majority of primary BE/EAC tissues and cell line models showed hyperactivation of EphB2 signaling. Transcriptomic/proteomic analyses identified EphB2 as an endogenous binding partner of MYC binding protein 2, and an upstream regulator of c-MYC. Knockdown of EphB2 significantly impeded the viability/proliferation of EAC and BE cells in vitro/in vivo. Activation of EphB2 in normal esophageal squamous 3-dimensional organotypes disrupted epithelial maturation and promoted columnar differentiation programs, notably including MYC. EphB2 and MYC showed selective induction in esophageal submucosal glands with acinar ductal metaplasia, and in a porcine model of BE-like esophageal submucosal gland spheroids. Clinically approved inhibitors of MEK, a protein kinase that regulates MYC, effectively suppressed EAC tumor growth in vivo.
CONCLUSIONS:
The EphB2 signaling is frequently hyperactivated across the BE-EAC continuum. EphB2 is an upstream regulator of MYC, and activation of EphB2-MYC axis likely precedes BE development. Targeting EphB2/MYC could be a promising therapeutic strategy for this often refractory and aggressive cancer.
Keywords: EFNB, SOX9, P63, FOXA2, MUC1
Graphical Abstract
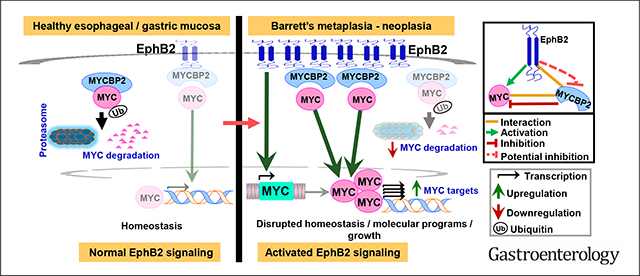
Esophageal adenocarcinoma (EAC) is an aggressive malignancy with increasing incidence rates even in younger populations within the United States.1,2 Five-year survival in EAC remains dismal,2,3 and the vast majority of EACs are refractory to standard-of-care treatments.4 Effective targeted therapies are virtually nonexistent,3,4 owing to a dearth in the knowledge of actionable EAC drivers. EACs typically originate in the setting of Barrett’s esophagus (BE), a precursor lesion in which the squamous epithelia (SQ) in distal esophagus is replaced by intestinal-type columnar metaplasia.5 BE, in turn, develops as a reparative response to chronic reflux-mediated injury to esophageal mucosa.6,7 Although several models have been put forth regarding the cell-of-origin giving rise to BE,5,7 molecular determinants regulating cell fate/commitment in BE progenitors and/or sustaining the BE-EAC phenotype, however, remain elusive. Deciphering the unifying mechanisms contributing to BE-EAC pathobiology would thus enable developing effective prognostic biomarkers, and early chemopreventive/treatment strategies against vulnerable BE-EAC molecular networks.
Here, using a unique systems biology approach combined with genome-scale transcriptomic profiles derived from treatment-naïve premalignant and EAC biopsy tissues, we attempted to identify the most frequently deregulated networks, on a pathway level, early in Barrett’s neoplasia. Subsequent molecular and phenotypic studies were performed to dissect the functional significance of select network components in relevant nonmalignant, premalignant, and malignant mammalian model systems. Overall, our study uncovered Eph receptor B2 (EphB2) as an upstream regulator of MYC, and the EphB2-MYCBP2-MYC as a major determinant axis contributing to BE-EAC pathobiology.
Methods
Detailed methods are provided as Supplementary Methods.
Patient Samples
A discovery set of treatment-naïve nondysplastic stable Barrett’s metaplasia (NDSBM), EAC, paired normal esophageal squamous biopsy (nSQ) matching respective EACs, and random normal gastric (GAST) biopsy samples (n = 89) was compiled for whole-transcriptome RNA sequencing (RNAseq) under an approved Institutional Review Board for Human Subjects Investigation protocol, as previously described.8 An independent cohort of treatment-naïve EAC, nondysplastic Barrett’s metaplasia with no follow-up, BE with high-grade dysplasia, normal gastric, and normal squamous (with the vast majority matching respective EAC, nondysplastic Barrett’s metaplasia with no follow-up, BE with high-grade dysplasia cases) biopsies (n = 885) were used for quantitative polymerase chain reaction (qPCR)-based validation studies.
InFlo Systems Biology Framework for Assessing Signaling-Network Activities
Genome-scale signaling-network activities in NDSBM/EAC vs nSQ/GAST were estimated using a unique systems biology framework (InFlo) that we developed.9,10 Networks significantly and consistently activated (InFlo Activity Index > 0; Wilcoxon P value ≤ .05; Benjamini-Hochberg false discovery rate ≤ 0.2) in the vast majority of NDSBM and EAC, compared with both nSQ and GAST samples, were considered for further studies.
Cell Culture
Human EAC cell lines were cultured in either Dulbecco’s minimal essential medium (FLO-1) or Roswell Park Memorial Institute medium (SKGT4, EsoAd1, OE19, OE33, and Eso26) supplemented with 10% fetal bovine serum. Nondysplastic (CP-A) and dysplastic (CP-B, CP-C, CP-D) human BE cell lines were cultured as we previously described.9 Human esophageal squamous esophageal cell line (EPC2) was kindly provided by Dr Anil Rustgi, Columbia University, New York, NY. Cell lines were tested for authenticity using short tandem repeat genotyping and were screened periodically for mycoplasma contamination. Cell lines were assessed for baseline levels of EphB2 expression.
Generation of Cells With Stable Knockdown or Reconstituted EPHB2
For stable knockdown of EphB2, cells were infected with a lentiviral Tet-On 3G Doxycycline (Dox)-inducible vector, containing either the nontargeting or EPHB2-targeting short hairpin RNA (shRNA), and were subsequently selected using puromycin. For stable expression of EphB2, cells were infected with V5-tagged full-length EPHB2 coding transcript, or vector control carrying a stuffer fragment, and were subsequently selected using G418.
Identification of Effector/Downstream Pathways of EphB2 signaling
EAC and BE cells, transfected with either EPHB2- or nontargeting/control small interfering RNA (siRNA), were subjected to RNAseq. Resulting transcriptomic profiles were processed using InFlo by comparing the profiles of each of the siEPHB2-treated samples against the set of nontargeting controls to identify signaling networks deregulated on EphB2 silencing.
Mass Spectrometry and Proteomic Analysis
To identify EphB2-interacting partners, total cell lysates from parental SKGT4 in addition to SKGT4 cells stably expressing V5-tagged wild-type EphB2 or stuffer-control were immunoprecipitated (IP) using anti-V5, anti-EphB2, or immunoglobulin (Ig)G isotype controls. Immunoprecipitates were analyzed using liquid chromatography–mass spectrometry by the Proteomics and Metabolomics Core at Cleveland Clinic Lerner Research Institute.
In Vitro Phenotypic Assays
Colony growth assessments were quantified as previously described.9 Cell viability assessments were performed using the CellTiter-Glo 2.0 Cell Viability Assay. Significant differences in cell growth/clonogenicity between test vs control groups were estimated using a Student t test assuming unequal variances.
Esophageal Squamous 3-Dimensional Organotypic Culture Model
EPC2 SQ organotypes were generated as previously described.11,12 Briefly, human esophageal fibroblasts were embedded in a 3-dimensional (3D) matrix of Geltrex and collagen and incubated for 7 days at 37°C. Human esophageal squamous cells (EPC2), transduced with lentiviral particles (LV-EPHB2 or LV-control), were seeded onto the fibroblast matrix. Cultures were incubated for an additional 10 days, fixed with formalin and embedded in paraffin (FFPE) for subsequent histologic and immunohistochemistry (IHC) analyses.
In Vivo Tumor Growth Assay
Xenograft tumor growth assays were performed as previously described.9 Briefly, subcutaneous tumor xenografts were established by injecting EAC cells (4 × 106 cells per flank), suspended in 50% Geltrex, bilaterally into the flanks of 4- to 5-week old female athymic Crl:NU(NCr)-Foxn1nu mice. For shRNA xenograft studies, mice were switched to a 625-mg/kg Dox diet 24 hours after inoculation for the duration of the study. For MEK inhibitor studies, after the xenograft tumors reached a minimum size of 50 to 60 mm3, mice were randomized and treated via oral gavage with vehicle (0.5% methylcellulose) or with vehicle containing 10 mg/kg of cobimetinib, once daily. Tumor volumes were estimated 2 to 3 times weekly. A 2-sided Student t test, assuming unequal variances, was used to determine significant differences in tumor volumes across comparisons. All animal procedures were approved by the Case Western Reserve University Institutional Animal Care and Use Committee and followed National Institutes of Health guidelines.
Results
The EphB2 Signaling Sub-network Is Hyperactivated in Near-all BE and EACs
We first determined the most frequently altered pathway-networks, consistent across BE-EAC continuum, using our InFlo systems biology framework9,10 on the discovery RNAseq data derived from pretreatment EAC/BE, and nonmalignant (normal SQ and gastric) biopsy tissues (Supplementary Table 1). Besides identifying known BE-associated pathways (see Supplementary Methods), our analyses revealed the EphB2 tyrosine kinase signaling to be hyperactivated in 100% of BE and 88% of EAC (Figure 1A, Supplementary Figure 1). Consistent with our InFlo-based predictions, RNAseq-based gene expression analyses revealed the EPHB2 receptor to be selectively induced in BE/EAC lesions together with a modest increase in EphB ligands (EFNB1 and EFNB2), when compared with normal SQ and gastric tissues (Figure 1B). Because the role and significance of ephrin pathway in Barrett’s neoplasia is unknown, we prioritized characterizing EphB2 for subsequent studies.
Figure 1.

Hyperactivation of EphB2 signaling in BE and EACs. (A) (Left) InFlo-based analysis of RNAseq data identifying EphB2 signaling sub-network activation in the vast majority of NDSBM and EAC tissues, compared with nSQ and GAST samples. (Right) InFlo-derived EphB2 activity levels are plotted along the X-axis. Activities (−2 to +2) associated with the control/comparators (nSQ/GAST) are shaded gray. Pie charts indicate the percentage of NDSBM (n = 18) and EAC (n = 49) samples exhibiting EphB2 hyperactivation (EphB2 high). (B) Gene expression (Y-axis, log2FPKM) of EPHB2 receptor and its associated ligands (EFNB1, EFNB2, and EFNB3) in respective primary tissues, included in the discovery RNAseq cohort. Error bars indicate mean ± SEM. (C) EPHB2 qPCR analysis in an independent cohort of treatment-naïve biopsy tissues. Y-axis shows the log2 fold-changes in EPHB2 gene expression across tissues, relative to the median expression value in nSQ samples. Horizontal lines within each tissue type indicate the median ± interquartile range. (B, C) ***P < .0005 indicates statistical significance between NDSBM/EAC vs nSQ/GAST tissues, estimated using a Student t test assuming unequal variances. (D) Representative IHC images in FFPE tissue sections demonstrating EphB2 protein (brown staining), overexpressed in BE/EAC lesions compared with normal esophageal SQ or gastric tissues (Scale bar, 150 μm). A total of 5 random treatment-naïve samples per nSQ, BE, EAC, and GAST were analyzed by IHC. (E) Western blot images depicting protein levels of EphB2 and its ligands (EFNB1/B2) across EAC, BE-dysplasia, BE-metaplasia (BM), and normal SQ cell lines. β-actin was used as a loading control.
To test the generality of EPHB2 induction in BE and EAC, we performed qPCR-based assessments in an independent cohort (Supplementary Table 2) of treatment-naïve nonmalignant, premalignant, and EAC biopsy tissues (n = 885). Reprising our findings in RNAseq data (Figure 1A and B), we found a marked (>8-fold) and significant (P < .0005) induction of EPHB2 in BE metaplasia/dysplasia/adenocarcinoma, as compared with normal esophageal SQ and gastric tissues (Figure 1C). Consistent with this, IHC analyses in primary tissues confirmed the selective induction of EphB2 protein in BE and EAC lesions, compared with SQ and gastric tissues that showed minimal/no EphB2 positivity (Figure 1D). Immunoblot analysis in patient-derived cell line models revealed higher EphB2 protein expression in BE-EAC cells, compared with the normal esophageal SQ cell line (EPC2) that showed no detectable EphB2 protein expression (Figure 1E). Also, in line with our RNAseq-based findings (Figure 1B), we found the EFNB1/B2 ligands to be expressed across SQ, BE, and EAC, albeit with higher protein levels in EAC cells (Figure 1E). Of note, the lack of robust EFNB1/B2 antibodies for IHC analyses precluded us from further assessing this observation in primary tissues.
EphB2 Regulates the Proto-oncogene MYC
To understand the mediators and effector pathways downstream of EphB2 signaling, we performed RNAseq and InFlo-based analyses in representative EAC and BE cell lines following EPHB2 knockdown (see Methods). Besides previously implicated pathways in BE/EAC, such as NF-κB/RelA and Wnt/β-catenin13,14 (Supplementary Figure 2), we particularly noted the c-MYC activity and associated transcriptional network to be significantly and positively regulated by EphB2 (Figure 2A). Interestingly, RNAseq-based gene expression analyses revealed that EphB2 did not modulate MYC RNA expression (Figure 2B). These findings were further assessed by qPCR (RNA) and Western blot (protein) analyses in EAC (SKGT4, OE33) and BE models (CP-A, CP-D), following EPHB2 knockdown (Figure 2C). Consistent with our observations in RNAseq data (Figure 2B), we found no changes in qPCR-based MYC RNA expression in SKGT4 and CP-D on EPHB2 knockdown (Figure 2C). OE33 and CP-A, on the other hand, showed a modest relative decrease in MYC RNA following EPHB2 knockdown (Figure 2C). Our subsequent protein analyses nonetheless showed a pronounced suppression of MYC protein following EPHB2 knockdown across all BE and EAC cell lines tested (Figure 2C), and EphB2 induction correlated with MYC protein positivity in primary BE/EAC lesions (Figure 2D). Notably, the preceding findings further suggest a direct regulatory effect of EphB2 on MYC protein per se as one of the major modes of regulation. Consistent with this observation, knockdown of EPHB2 in the representative EAC (SKGT4) cell line accelerated MYC-degradation (Figure 2E), adding further support to such posttranslational regulation of MYC by EphB2. We nonetheless note that our findings in OE33 and CP-A models do suggest additional modes of regulation of MYC (possibly at the transcriptional level) by EphB2.
Figure 2.

MYC is a downstream target of EphB2 signaling. (A) Representative EAC (SKGT4) and BE (CP-D) cells were subjected to RNAseq following EPHB2-siRNA or control-siRNA transfections. Subsequent InFlo-based analyses identified a significant reduction in the activities of MYC and associated protein complexes/transcriptional targets on EPHB2 knockdown in both BE/EAC cells. (B) RNAseq-based gene expression (Y-axis, log2FPKM) of EPHB2 receptor and MYC in EAC (SKGT4) and BE (CP-D) cells on EPHB2-siRNA or control-siRNA transfections. Error bars indicate mean ± SEM. (C) (Top) qPCR-based gene expression (Y-axis, log2 fold-change) of EPHB2 receptor and MYC on EPHB2-siRNA treatment, relative to the gene expression in control-siRNA transfection, in EAC and BE cells. Error bars indicate mean ± SEM. Representative images of qPCR products on agarose gel depicting EPHB2 and MYC RNA expression with B2M RNA used as loading controls. (Bottom) Western blot (WB) images depicting EphB2 and MYC protein levels upon EPHB2-siRNA (+) or Control-siRNA (−) treatment. β-actin was used as a loading control. (D) Representative IHC images showing overexpression of EphB2 along with nuclear-MYC positivity (brown staining) in serial sections of human BE and EAC FFPE tissues, compared with minimal/lack of expression of either in normal SQ and gastric tissues (scale bar, 150 μm). A total of 5 random treatment-naïve samples per nSQ, BE, EAC, and GAST were analyzed by IHC. (E) Representative EPHB2-knockdown EAC (SKGT4) cells, were incubated with 50 μg/mL cycloheximide (chx) at regular time intervals up to 90 minutes. WB analyses for EphB2 and MYC were performed at indicated time points (in minutes). The colored bar graphs below WB images are a 3D-rendered depiction of the actual band intensities of MYC, obtained using Image Lab software. Both the height and color intensity (yellow; low to black; high) of a bar jointly depict the signal strength of respective bands in the WB. The numbers beneath each solid bar are the normalized MYC-signal values, relative to time zero (0′), within respective treatment groups, from 2 independent experiments.
EphB2 Interacts With MYC Binding Protein 2, and Regulates MYC Via the Ubiquitin-Proteasome Pathway
To further interrogate the mechanisms underlying EphB2-regulation of MYC protein, we performed IP with native EphB2 or control-IgG antibodies, as well as with and without stable reconstitution of EphB2, in a representative EAC line (SKGT4). Subsequent mass spectrometry analyses identified MYC binding protein 2 (MYCBP2) as the top endogenous EphB2-binding partner (Figure 3A, Supplementary Table 3), which we orthogonally confirmed using forward and reverse IP-Western blot analysis (Figure 3B). Of note, we observed no detectable interaction between EphB2 and MYC in our IP–mass spectrometry studies.
Figure 3.

EphB2 interacts with MYCBP2. (A) Mass spectrometry analysis identifies MYCBP2 as an interacting partner of EphB2. Endogenous EphB2 and V5-tagged wild-type EphB2 were IP from parental SKGT4 and SKGT4 cells engineered to stably express V5-tagged EphB2 respectively and analyzed independently. IgG isotype-matched antibody immunoprecipitates and SKGT4 cells engineered to stably express a V5-tagged stuffer fragment (st-Control) served as independent experimental controls. (B) (Left) Protein lysates from EAC (SKGT4) cells, stably expressing V5-tagged EphB2 or V5-tagged stuffer-control (st-Con), were subjected to immunoprecipitation (ip) using anti-V5, anti-MYCBP2, or IgG isotype-matched antibodies. Subsequent Western blot (WB) analyses were performed using anti-EphB2 and -MYCBP2 antibodies. (Right) Relevant MYCBP2 and V5-tagged EphB2 input (inp) protein levels in corresponding whole cell lysates. (C) MYCBP2 interacts with MYC. Parental SKGT4 cells were subjected to ip using anti-MYCBP2, anti-MYC, or IgG isotype-matched antibodies, and subsequent WB analyses against native proteins. (D) MYCBP2 negatively regulates MYC. WB images depicting the protein levels of MYCBP2 and MYC in representative EAC (SKGT4, OE33), non-dysplastic BE (CP-A), and dysplastic BE (CP-D) cells, transfected with siRNAs targeting MYCBP2 for 48 hours. (E) MYC does not regulate EphB2 or MYCBP2. WB images depicting the protein levels of MYC, MYCBP2, and EphB2 in the above cell line models, transfected with siRNAs targeting MYC for 48 hours. (F) EphB2 regulates MYC protein levels potentially via MYCBP2. WB images depicting protein levels of MYC, MYCBP2 and EphB2 in the above cell line models, transfected with control-siRNA (−) or siRNAs targeting EPHB2, MYCBP2, or both (+) for 48 hours. (D–F) β-actin was used as loading control for all WBs. (G) EphB2 modulates MYC ubiquitination. SKGT4-EAC cells were transfected with control-siRNA (−) or siRNAs targeting EPHB2, MYCBP2, or both (+) for 48 hours, followed by treatment with proteasome inhibitor, MG-132 (5 μM), for 6 hours. Protein lysates from above cells were subjected to immunoprecipitation (ip) with an anti-Ubiquitin (Ub) antibody. WB analysis was subsequently performed using antibodies against Ub and MYC. EphB2, MYCBP2 and MYC input (inp) protein levels in corresponding whole cell lysates are shown below.
MYCBP2 is a large (molecular weight, 510 kDA) atypical E3 ubiquitin-protein ligase that mediates ubiquitination of threonine/serine residues on target proteins.15,16 Notably, MYCBP2 interacts with the transactivation domain of MYC and is speculated to either facilitate or otherwise regulate MYC activity.15 Co-IP studies in parental EAC cells indeed confirmed endogenous MYCBP2-MYC interactions (Figure 3C). Furthermore, in contrast to our findings with EphB2 (Figure 2B and C), knockdown of MYCBP2 was associated with increased MYC protein across EAC, BE-metaplasia, and dysplasia cell lines (Figure 3D), pointing its function as a negative regulator of MYC in Barrett’s neoplasia. Conversely, knockdown of MYC did not affect EphB2 or MYCBP2, indicating that both EphB2 and MYCBP2 are upstream of MYC in the regulatory cascade (Figure 3E).
To further interrogate the functional relationship among EphB2, MYCBP2, and MYC, we performed knockdown of EPHB2, MYCBP2, or both in EAC/BE cells. Intriguingly, MYCBP2-knockdown concurrent with EPHB2-silencing was unable to fully restore MYC levels observed in MYCBP2-knockdown alone (Figure 3F). Given that MYCBP2 is an E3 ubiquitin ligase, we further assessed for alterations in MYC ubiquitination following single or dual knockdown of EPHB2/MYCBP2 in representative EphB2-high EAC cells. Silencing of EPHB2 or MYCBP2 led to an increase or near-all suppression of MYC ubiquitination, respectively (Figure 3G). In line with our observations (Figure 3F), loss of MYCBP2 concurrent with EPHB2 silencing was unable to fully suppress MYC ubiquitination, when compared with MYCBP2 knockdown alone (Figure 3G). Collectively, these findings strongly suggest that EphB2 posttranslationally modulates MYC stability/activity, in part, via its interaction with MYCBP2 and the ubiquitin-proteasome pathway.
EphB2 Signaling Affects the Growth/Viability of EAC and BE Cells
We next evaluated the phenotypic consequences of EphB2 signaling by performing transient siRNA-based knockdown of EPHB2 in distinct EAC cell line models. In particular, we observed that suppression of EphB2 signaling significantly impeded the colony-forming ability of multiple EAC models (Figure 4A). To further confirm these siRNA-based observations, we generated EAC cells (SKGT4) stably expressing Dox-inducible EPHB2 shRNA and repeated the analysis. Here again, we found the knockdown of EPHB2 to markedly reduce EAC colony growth (Figure 4B). Moreover, knockdown of EPHB2 significantly decreased MYC protein levels and impeded EAC tumor xenograft growth in vivo (Figure 4B).
Figure 4.

EphB2 impacts the growth characteristics of EAC and BE cells. (A) Assessment of clonogenicity/colony-forming ability (CFA) on siRNA-based EPHB2 knockdown in EAC cell lines. (Top) Western blot (WB) images depicting EphB2 protein expression, 48 hours following transfection with either siRNA targeting EPHB2 (+) or nontargeting control-siRNA (−). (Below) Representative colony images, 10 to 14 days following EPHB2-siRNA or control-siRNA transfection. The bar graphs depict CFA fold-changes (CFA-FC) observed in respective EPHB2-siRNA, compared with control-siRNA groups. (B) Assessment of colony growth in representative EAC (SKGT4) cells, stably expressing either Dox-inducible EPHB2-targeting shRNA, or control-shRNA vector. (Left) WB images depicting EphB2 protein expression 120 hours following Dox treatment, and representative colony images and CFA-FC bar graphs in EPHB2-shRNA, relative to control-shRNA group. (Right) In vivo tumor growth kinetics of SKGT4 xenografts, expressing EPHB2-shRNA or control-shRNA, in immune-deficient mice fed with Dox diet. Y-axis depicts tumor volume in mm3, over time (X-axis). Data are plotted as mean ± SEM, obtained from at least 20 established xenograft tumors at day zero in respective arms. Also shown below are photographic images of harvested tumors from mice at the final time point in respective EPHB2-shRNA and control-shRNA arms. WB images depict EphB2 and MYC protein levels in representative tumor xenografts harvested 1 week following Dox treatment. Bar graphs show body-weight assessments in mice at the beginning and at the end of the study. (C) Assessment of cell viability in representative metaplastic (CP-A) and dysplastic (CP-D) BE cell line models. Y-axis depicts cell viability fold-change (FC) in respective EPHB2-siRNA vs control-siRNA transfected cells. WB images depict EphB2 protein expression 48 hours following siRNA transfections. (A–C) Data in bar graphs are plotted as mean ± SEM, obtained from at least 3 independent replicate experiments. *P < .05, **P < .005, and ***P < .0005 indicate significant differences between EphB2 knockdown vs nontargeting control-siRNA/-shRNA group, estimated using a Student t test assuming unequal variances. β-actin was used as a loading control in all WB analyses.
To test if EphB2-associated growth dependencies are specific to the malignant stage, or whether they are evident in antecedent BE lesions, we performed EPHB2 siRNA knockdown studies in representative BE-metaplasia (CP-A) and BE-dysplasia (CP-D) cells. Similar to our observations in EACs, knockdown of EPHB2 significantly reduced (albeit to a lesser degree) the viabilities of both BE-metaplasia and -dysplasia cells (Figure 4C). Of note, besides MYC (Figure 2C), we assessed the impact of EphB2 knockdown on additional BE-associated markers, including MUC1, FOXA2, and SOX917–19 (Supplementary Figure 3). Of these, we found EphB2 to positively regulate MUC1 and FOXA2 proteins in BE metaplasia (CP-A) as well as dysplasia (CP-D) cells (Supplementary Figure 3), with EphB2 regulation of FOXA2 likely being dependent on MYC (Supplementary Figure 4). Taken together, these findings provide further insights into the role of EphB2-MYC axis in regulating key BE-associated columnar differentiation programs that contribute to BE-EAC pathobiology.
Activation of EphB2 Disrupts the Maturation of Normal Esophageal SQ Epithelia and Promotes BE-associated Molecular Programs
Esophageal 3D-organotypic culture (OTC) comprises a matrix-type culture system, in which SQ epithelial cells are seeded on top of a fibroblast feeder layer and when exposed to liquid-air interface can differentiate and recapitulate the in vivo epithelial architecture.20 In particular, hTERT-immortalized esophageal SQ cells (EPC2)21 form stratified squamous layers when grown as 3D-OTC cultures, mimicking the esophageal morphology and homeostasis in vivo.22 Importantly, the propensity of EPC2-organotypes to undergo SQ to columnar/BE-like transition enables interrogation of potential trans-differentiation mechanisms suspected in BE pathogenesis,22,23 as we have previously described.24
We accordingly posited whether EphB2 activation could impact the morphologic and molecular dynamics of EPC2 SQ-OTCs, with inherently undetectable EphB2 expression (Figure 1E). Immortalized EPC2 cells were stably reconstituted with EPHB2 or stuffer-control vectors, and were allowed to grow and differentiate as 3D-OTCs, as previously described.24 Although control EPC2 cells differentiated and matured into a durable stratified SQ epithelium as anticipated, reconstitution of EphB2 disrupted the morphology and normal maturation of the SQ-OTCs, albeit with no impact on cell proliferation (Figure 5A). Assessment of BE-associated columnar differentiation markers showed increased expression of SOX9 and FOXA2 in EphB2-reconstituted EPC2 OTC and monolayer cultures (Figure 5A and B), with FOXA2 being modulated by EphB2 consistently in both EPC2-SQ and BE cells (Figure 5B, Supplementary Figure 3). EphB2 activation also led to a modest decrease in P63 (Figure 5B), a transcription factor critical for the development and homeostasis of squamous epithelia.25 Importantly, consistent with the strong association between EphB2 and MYC activity observed in EAC/BE (Figure 2), reconstitution of EphB2 in EPC2-SQ cells enhanced MYC protein in OTC and monolayer cultures (Figure 5A and B), again with no significant impact on MYC RNA levels (Figure 5C). In line with this, EphB2 also modestly affected MYC-degradation (Figure 5C) and showed interaction with MYCBP2 in the SQ cells (Figure 5D), as observed in EAC and BE cells (Figure 2).
Figure 5.

Activation of EphB2 disrupts maturation of esophageal SQ epithelia. (A) FFPE sections of esophageal EPC2-SQ OTC cultures, stably expressing V5-tagged EPHB2 or stuffer-control (st-Con) vector, were subjected to hematoxylin-eosin (H&E) and IHC analyses with indicated marker proteins (scale bar, 150 μm). As shown in the st-Control H&E panel (first row), SQ cells grown as 3D-OTCs mature and differentiate into stratified squamous epithelial layer (Sq) on top of stroma. Note the disruption in the differentiation/maturation of SQ epithelial layer in EphB2-reconstituted EPC2 cells. IHC analyses of the V5 epitope (brown staining) demonstrates the specific expression and membrane localization of EphB2 protein in the SQ epithelia (second row). Also shown are IHC-based assessments of respective P63, SOX9, FOXA2, and MYC proteins in serial sections. (B) Western blot (WB) analyses of whole cell lysates (WCLs), derived from EPC2-SQ cells grown as monolayers. (Left) WB analysis performed with a global anti-phospho tyrosine (p-Tyr) antibody. The bar graph below denotes normalized total lane volume (NTV, Y-axis) of p-Tyr signal in EphB2-reconstituted, relative to st-Control cells. Note the increase in EphB2 tyrosine-phosphorylation (arrow) and broader increase in p-Tyr levels (*) on EphB2 reconstitution in EPC2-SQ cells. (Right) WB analysis of indicated proteins in EphB2-reconstituted EPC2 cells, compared with st-Control. β-actin was used as loading control for WBs. (C) (Top) qPCR-based gene expression analysis (Y-axis, log2 fold-change) of MYC in EPHB2-reconstitued EPC2-SQ cells. Error bars indicate mean ± SEM from at least 3 independent replicate experiments. Representative images of qPCR products on agarose gel depicting MYC RNA expression with B2M RNA used as loading controls. (Bottom) Assessment of MYC-degradation kinetics in EPHB2-reconstitued EPC2-SQ cells using the same strategy outlined in Figure 2E. (D) EphB2 interacts with MYCBP2. Protein lysates from EPC2-SQ cells, engineered to express V5-tagged EphB2 or V5-tagged stuffer-control (st-Con), were subjected to immunoprecipitation (ip) using anti-V5 or anti-MYCBP2 antibodies. Subsequent WB analyses were performed using anti-EphB2 and -MYCBP2 antibodies. (Right) Relevant MYCBP2 and V5-tagged EphB2 input (inp) protein levels in corresponding WCLs. (E) MYCBP2 interacts with MYC. EPC2-SQ cells were subjected to ip using anti-MYCBP2, anti-MYC, or IgG control antibodies, and subsequent WB analyses against native proteins.
EphB2 Is Present in EAC-associated Esophageal Submucosal Glands, Another Potential Source of BE Progenitors
Given the preceding observations, we next evaluated EphB2 and MYC in esophageal submucosal glands (ESMGs), a cluster of progenitor cells involved in esophageal injury-repair.26 ESMGs can differentiate into BE-like columnar rather than SQ-like epithelial cells on abnormal repair following injury,26 and serve as another potential progenitor source of BE.27 Importantly, acinar ductal metaplasia (ADM) of ESMG has been shown to be associated with the etiology of Barrett’s neoplasia as well as with carcinogenesis of stomach and pancreas in the gastrointestinal tract.28,29 We therefore first evaluated EphB2 status in ESMGs, obtained from surgically resected esophageal tissues from patients with EAC before undergoing chemoradiation treatment (n = 10). Using IHC, we observed a robust pattern of EphB2 and MYC selectively in ESMGs associated with EAC (Figure 6A). As an additional control, esophageal tissue blocks were obtained from surgical resections of treatment-naïve non-EAC esophagectomy/trauma patients (n = 7), and generally showed absence of EphB2 and MYC; although one esophagectomy case for fistula with incidental BE showed positive staining in an ESMG with ADM, as well as in an ESMG with ADM in a patient with achalasia (data not shown). Next, using a companion spheroid culture model of ESMGs, derived from porcine esophagus,26 we assessed EphB2-MYC activities by single-cell RNAseq followed by InFlo analyses (see Supplementary Methods). We observed significantly higher EphB2 and MYC activities in BE-like, compared with SQ-like ESMG spheroids, independent of cell-cycle phase (Figure 6B). These findings, taken together with our observations in SQ cells (Figure 5), further suggest that induction of EphB2-MYC activity in progenitor cells is potentially a key step in BE development.
Figure 6.
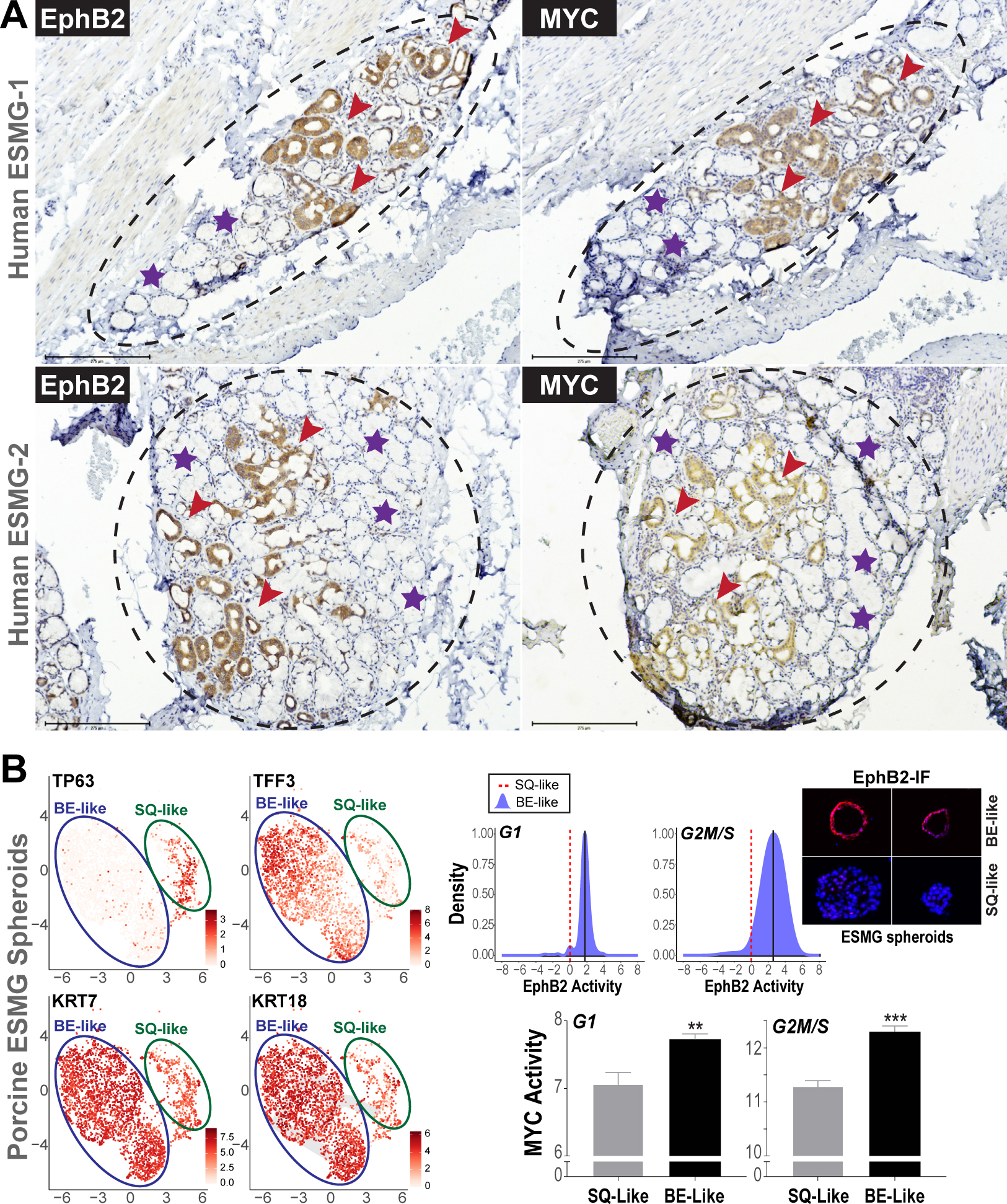
Activation of EphB2-MYC in ESMG-associated ADM, and in porcine BE-like ESMG spheroids. (A) Representative IHC images of EphB2 (left) and corresponding MYC (right) in ESMGs (dashed circles) derived from 2 random patients with EAC. Note the strong induction of EphB2 and MYC in ADMs (brown staining, red arrowheads). Normal mucinous acini adjacent to ADM within the ESMG (violet stars) show absence of EphB2 and MYC. (B) UMAP plots of single-cell RNAseq (scRNAseq) on the left showing 2 distinct ESMG cell clusters, exhibiting differential expression of SQ marker (TP63) and BE/columnar markers (TFF3, CK7, CK18). To the right are density plots (blue) denoting distributions of InFlo-derived EphB2 signaling activities (X-axis) in the BE-like cells (TP63-low, TFF3/KRT7/KRT18-high), compared with respective SQ-like (TP63-high, TFF3/KRT7/KRT18-low) manifolds (red dotted line), in either G1 or G2M/S cell phases. We note that that the levels of EphB2 RNA in these porcine scRNAseq clusters were generally low to obtain robust estimation of differential gene expression status. We therefore assessed EphB2 protein levels in porcine ESMG spheroid OCT-sections by immunofluorescence (IF), using the same antibody used in (A). Provided to the right are representative images of 2 individual hollow/BE-like spheroids showing higher EphB2 (red) protein expression, compared with solid/SQ-like spheroids. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). A total of 10 solid and 10 hollow spheroid sections, selected randomly, were assessed for EphB2. We, however, express a note of caution with the IF analysis because the anti-EphB2 antibody is not optimized for porcine tissues/cells. We also note that InFlo-derived differential EphB2 activity estimates in BE-like clusters could stem from neighboring nodes within the EphB2 sub-network (Supplementary Figure 1), as well as effects of protein/posttranslational modifications of EphB2 in this ESMG-based progenitor model. Bar graphs below denote MYC activity in BE-like vs SQ-like cells within the G1 and G2M/S populations. All data are plotted as mean ± SEM, with **P < .005 and ***P < .0005 indicating significant differences estimated using a Student t test assuming unequal variances.
Small Molecule Inhibitors of MEK1 Suppress MYC Activity and Impede EAC Tumor Growth In Vivo
Our phenotypic assessments thus far suggest the EphB2-MYC axis as a potentially vulnerable therapeutic target in EACs. However, given that there are no clinically approved inhibitors specifically against EphB2 or c-MYC, we sought surrogate druggable targets involved in upstream regulation of MYC activity. The MAPK signaling cascade emerged as a potential target given its known role in regulating MYC stability and activity,30,31 and because EphB2 also modulated the activity of multiple components of the MAPK signaling cascade in BE/EAC cells (Supplementary Figure 2). In particular, cobimetinib, a potent clinically approved MEK1 inhibitor (MEKi) showed significant cytotoxicity across EAC cell lines, with half maximal inhibitory concentration values ranging between 100 nM and 1 μM (data not shown). We then evaluated the in vivo antitumor efficacy of cobimetinib in multiple preclinical tumor xenograft models. Cobimetinib treatment per se resulted in strong suppression and/or regression of EAC tumor growth in vivo (Figure 7). Consistent with this, we found a marked reduction in MYC protein in the tumors on cobimetinib treatment in vivo (Figure 7). Of note, further induction of EphB2 in a representative, EphB2-high, EAC line did not significantly alter the antitumor efficacy of cobimetinib (Supplementary Figure 5), suggesting that the threshold of response to MEKi may likely be dependent on relative/joint levels of ERK, MYC, and/or additional downstream effectors. Nonetheless, these findings suggest that targeting MYC-associated upstream signaling axes could be a beneficial new therapeutic strategy in EACs.
Figure 7.

MEK inhibition suppresses MYC activity and EAC tumor growth in vivo. Assessment of in vivo tumor growth kinetics in distinct EAC xenograft models treated with MEK inhibitor, cobimetinib (COBI), or vehicle control, via oral gavage. Y-axis of the line graphs depict tumor volume in mm3 over time (X-axis) in respective EAC xenografts. Data are plotted as mean ± SEM estimated using at least 10 established xenograft tumors at day zero in respective arms per EAC cell line model. *P < .05, **P < .005, and ***P < .0005 indicate significant differences in tumor volumes at the final time point between COBI vs vehicle groups, estimated using a Student t test assuming unequal variances. Photographic images of harvested xenograft tumors at the final time point from respective COBI and vehicle control groups are provided below. Western blot images (right) depicting protein levels of pharmacodynamic marker, phospho (p)-ERK, total ERK, and MYC in representative tumor xenografts harvested after 1 week of treatment with either COBI or vehicle control. Bar graphs depict body-weight assessments of COBI- and vehicle-treated mice at the beginning and at the end of the study.
Discussion
EAC management remains one of the most pressing unmet medical needs, being heterogeneous and highly refractory to standard-of-care treatments, lack of effective targeted therapies, and dismal 5-year survival, coupled with increasing incidence rates. There is, therefore, a clear need to decipher critical vulnerable networks driving the onset and progression of BE-EAC. Here, we identify EphB2 signaling as a major network central to BE-EAC pathobiology.
EphB2 is a member of the largest subgroup of receptor tyrosine kinases that affect diverse cellular processes during development and tissue homeostasis,32 and exhibit complex/paradoxical functions in cancers based on the tissue context.33,34 To date, 2 exploratory studies evaluating EphB2 expression, broadly across 138 tumor types35 or in the context of stem cell markers associated with intestinal metaplasia,36 reported EphB2 positivity in Barrett’s metaplasia and/or in esophageal adenocarcinoma, consistent with our findings. To our knowledge, this is the first report to now functionally and broadly implicate EphB2 signaling in the BE-EAC disease context. Furthermore, our observation of persistent EphB2 activation across progressive stages of Barrett’s neoplasia (Figure 1) is in stark contrast to other gastrointestinal malignancies, such as colorectal cancer, in which EPHB2 is lost during the adenoma-carcinoma transition and induction of EPHB2 markedly suppresses colorectal cancer progression.35,37 These observations particularly underscore the distinct, context-specific, function of EphB2 signaling along the length of the gastrointestinal tract.
Our in-depth molecular characterizations reveal EphB2 as a potential master regulator of MYC (Figure 2). Although EphB2’s regulation of MYC has not been previously reported, prior studies have found other Eph members to modulate MYC levels, via unknown/translational mechanisms in specific cancer contexts,38,39 suggesting MYC as a potential key downstream effector of ephrin signaling in general. That EphB2 is a potent and positive regulator of MYC has significant implications in the BE-EAC context, given the likely role of MYC as an early promoter of BE pathogenesis. For example, activation of MYC in esophageal SQ-OTCs has been shown to promote trans-differentiation of SQ toward a columnar phenotype.40 Moreover, deoxycholate, a key bile acid in gastroesophageal refluxate and a risk factor for BE, is shown to induce MYC protein expression.41 Furthermore, a recent study using molecular profiles and organoid models of gastroesophageal tissues proposed that BE originates from gastric cardia through MYC-driven transcriptional programs.42 These findings also indicate that cell-intrinsic/extrinsic factors that induce and/or sustain MYC activity likely play a determinant role in BE pathogenesis, and our study connects this important proto-oncogene to an upstream regulator, EphB2, in BE-EAC pathobiology. Moreover, although several cellular origins of BE have been proposed,7 it is plausible that there exists a degree of plasticity where any of the suggested progenitors can undergo reprogramming in response to specific molecular cues, and give rise to BE. In this regard, the ability of EphB2 to disrupt esophageal SQ maturation (Figure 5), combined with its positive regulation of MYC and columnar differentiation markers such as FOXA2 and/or SOX9 in esophageal SQ and BE cells (Figure 5, Supplementary Figure 3), argues for such an opportunistic model. Moreover, our observations of EphB2 and MYC in select ESMGs (that are potential sources of BE and associated carcinogenesis26–29), and in BE-like differentiated state of ESMGs (Figure 6), strongly suggest that EphB2-MYC activation likely precedes the development of BE.
Proteomic studies revealed MYCBP2, an E3 ubiquitin ligase,43 as a key endogenous binding partner of EphB2 and negative regulator of MYC (Figure 3). To our knowledge, this is also the first report identifying a direct interaction of MYC ubiquitin ligase to a receptor tyrosine kinase, and posit similar associations to likely exist between MYCBP2 and other ephrin family members. Also, given that the regulation of MYC by MYCBP2 per se is, in part, dependent on EphB2 status (Figure 3), we additionally speculate EphB2 to play a rate-limiting role in the EphB2-MYCBP2-MYC functional hierarchy. This is further supported by our incidental (yet repeated) observation that EphB2, besides interacting with MYCBP2, also appears to control its steady-state levels (Figure 3F and G). Exactly how the regulatory loop between EphB2 and MYCBP2 intersect, and the precise interaction sites of EphB2 and MYCBP2, warrant further in-depth biochemical characterization. Nonetheless, our current study reveals an important and relevant functional axis, connecting a receptor tyrosine kinase (EphB2), E3 ubiquitin ligase (MYCBP2), and a proto-oncogene (MYC) in BE-EAC pathobiology.
Our phenotypic studies reveal yet another important function of EphB2 signaling as a positive regulator of EAC proliferation/growth in vitro and in vivo (Figure 4). From a clinical perspective, particularly given their broader roles in health/disease and our current findings (Figure 4), it is compelling to exploit the Eph receptors for potential therapeutic and/or biomarker utility.44,45 To date, however, there are no approved inhibitors specifically against EphB2, or MYC. Our preclinical studies with a clinically approved MEK1 inhibitor, demonstrating significant antitumor efficacy concomitant with MYC inhibition (Figure 7), suggest that targeting MYC-associated signaling axes could be an alternative/viable therapeutic approach in EAC. Nonetheless, emerging strategies for targeting EphB2 signaling, including peptides, antibodies, and nanoliposomes,46 can be further explored as potential therapeutic approaches in the future.
In summary, our investigations identify EphB2 as a potential key determinant of BE-EAC pathobiology, and the convergence of EphB2 signaling on MYC offers new opportunities for cancer prevention and therapy in this increasingly prevalent and aggressive esophageal malignancy.
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT
Unifying mechanisms contributing to the complex Barrett’s esophagus–esophageal adenocarcinoma pathobiology remain elusive.
NEW FINDINGS
In a transcriptome analysis of esophageal adenocarcinoma, Barrett’s esophagus, and normal esophageal squamous/gastric tissues, we found the EphB2 tyrosine kinase signaling to be hyperactivated at the onset and during progression of Barrett’s neoplasia. EphB2 regulates proto-oncogene MYC, promotes Barrett’s esophagus–associated molecular programs, and affects the growth of esophageal adenocarcinoma and Barrett’s esophagus.
LIMITATIONS
Further in-depth studies are needed to determine whether EphB2 is indispensable for the origination of Barrett’s esophagus, and what mechanisms drive EphB2 activity.
IMPACT
Chemopreventive/therapeutic agents that block EphB2 signaling might be effective in patients with Barrett’s esophagus and esophageal adenocarcinoma.
Acknowledgments
We thank Lakshmeswari Ravi and Aruna Kumar Chelluboyina for their technical input on the experimental methodologies used in this study. We thank City Packing in Burlington, NC, in particular Thomas McGarity and Jamie Corbett, for their generous contributions of porcine esophagus for this research. We thank Dr DunFa Peng and Dr Wael El-Rifai for sharing their technical expertise in performing 3-dimensional organotypic culture experiments. We thank Dr Belinda Willard of the Proteomics and Metabolomics Core at Lerner Research Institute, Cleveland Clinic, for providing guidance and assistance on mass spectrometric analysis. We thank Dr Vamsi Muthavarapu for his kind assistance in logistics, enabling us to continue conducting the animal studies uninterrupted during the pandemic. This work was also supported by the Animal Resource Center and Tissue Resources Core at Case Western Reserve University. We also acknowledge the assistance of the Duke Molecular Physiology Institute Molecular Genomics Core at Duke University for the generation of single-cell RNA sequencing data included in this article.
Funding
This research was supported by PHS awards R01 CA204549 (K. Guda), U01 CA152756 (K. Guda), Case BETRNet U54 CA163060 (J.E. Willis, A. Chak, K. Guda), Case GI SPORE P50 CA150964 (J.E. Willis, A. Chak, K. Guda, A. Blum), K08 DK098528 (K.S. Garman), R01 DK118022 (K.S. Garman), 3T32-DK007568–28S2 (O. Martinez-Uribe), K25 DK115904 (V. Varadan), P30 CA043703 (V. Varadan, K. Guda); CDA-2 1IK2CX001831 US Department of Veterans Affairs, Biomedical Laboratory Research and Development Service (A. Blum); T32-DK083251 (S. Venkitachalam); the DeGregorio Family Foundation, the Savone Family, and the Esophageal Cancer Awareness Association (K. Guda); and the Torrey Coast Foundation GEMINI network (K. Guda). The contents do not represent the views of the U.S. Department of Veterans Affairs or the U.S. government. Pertinent high-throughput sequencing and integrative analytics in cell line models, where applicable, will be deposited in GitHub and made freely available to the general public.
Abbreviations used in this paper:
- 3D
3-dimensional
- ADM
acinar ductal metaplasia
- BE
Barrett’s esophagus
- BM
Barrett’s metaplasia
- Dox
Doxycycline
- EAC
esophageal adenocarcinoma
- ESMG
esophageal submucosal gland
- FFPE
formalin fixed paraffin embedded
- GAST
normal gastric cardia
- Ig
immunoglobulin
- IHC
immunohistochemistry
- IP
immunoprecipitated
- MYCBP2
MYC binding protein 2
- NDSBM
nondysplastic stable BM
- nSQ
normal esophageal squamous biopsy
- OTC
organotypic culture
- qPCR
quantitative polymerase chain reaction
- RNAseq
RNA sequencing
- shRNA
short hairpin RNA
- siRNA
small interfering RNA
- SQ
normal esophageal squamous
Footnotes
Conflict of Interest
The authors disclose no conflicts.
CRediT Authorship Contributions
Order of Authors (with Contributor Roles):
Srividya Venkitachalam, PhD (Formal analysis: Equal; Investigation: Equal; Supervision: Supporting; Writing – original draft: Equal; Writing – review & editing: Equal)
Deepak Babu, PhD (Formal analysis: Equal; Investigation: Equal; Methodology: Equal; Writing – review & editing: Supporting)
Durgadevi Ravillah, PhD (Formal analysis: Equal; Investigation: Equal; Methodology: Equal)
Ramachandra M. Katabathula, PhD (Data curation: Lead; Formal analysis: Equal; Methodology: Supporting; Software: Lead)
Peronne Joseph, MS (Data curation: Supporting; Investigation: Supporting; Methodology: Supporting)
Salendra Singh, MS (Data curation: Lead; Formal analysis: Lead; Methodology: Equal; Software: Supporting)
Bhavatharini Udhayakumar, MS (Formal analysis: Supporting; Methodology: Supporting)
Yanling Miao, MD (Investigation: Supporting; Methodology: Supporting)
Omar Martinez-Uribe, MD (Investigation: Supporting; Methodology: Supporting; Resources: Supporting)
Joyce A. Hogue, BS (Investigation: Supporting; Methodology: Supporting; Resources: Supporting)
Adam M. Kresak, MS (Formal analysis: Supporting; Methodology: Equal; Resources: Supporting)
Dawn Dawson, MD (Formal analysis: Supporting; Investigation: Supporting;Methodology: Supporting)
Thomas LaFramboise, PhD (Formal analysis: Supporting; Methodology: Supporting; Software: Supporting)
Joseph Willis, MD (Data curation: Equal; Formal analysis: Equal; Methodology: Equal; Resources: Equal)
Amitabh Chak, MD (Conceptualization: Supporting; Formal analysis: Supporting; Investigation: Supporting; Supervision: Equal; Writing – review & editing: Equal)
Katherine Garman, MD (Conceptualization: Supporting; Formal analysis: Supporting; Investigation: Supporting; Methodology: Equal; Resources: Equal; Writing – review & editing: Supporting)
Andrew Blum, MD, PhD (Conceptualization: Equal; Data curation: Equal; Formal analysis: Equal; Funding acquisition: Supporting; Investigation: Equal; Methodology: Equal; Supervision: Equal; Validation: Equal; Writing – review & editing: Equal)
Vinay Varadan, PhD (Conceptualization: Equal; Data curation: Equal; Formal analysis: Equal; Funding acquisition: Equal; Investigation: Equal; Methodology: Equal; Software: Lead; Supervision: Equal; Writing – review & editing: Equal)
Kishore Guda, DVM, PhD (Conceptualization: Lead; Data curation: Equal; Formal analysis: Lead; Funding acquisition: Lead; Investigation: Lead; Methodology: Equal; Project administration: Equal; Resources: Equal; Software: Supporting; Supervision: Lead; Validation: Equal; Writing – original draft: Equal; Writing – review & editing: Equal)
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at https://doi.org/10.1053/j.gastro.2022.07.045.
References
- 1.Reid BJ, Li X, Galipeau PC, et al. Barrett’s oesophagus and oesophageal adenocarcinoma: time for a new synthesis. Nat Rev Cancer 2010;10:87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Codipilly DC, Sawas T, Dhaliwal L, et al. Epidemiology and outcomes of young-onset esophageal adenocarcinoma: an analysis from a population-based database. Cancer Epidemiol Biomarkers Prev 2021;30:142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smyth EC, Lagergren J, Fitzgerald RC, et al. Oesophageal cancer. Nat Rev Dis Primers 2017;3:17048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abdo J, Agrawal DK, Mittal SK. “Targeted” chemotherapy for esophageal cancer. Front Oncol 2017;7:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rhee H, Wang DH. Cellular origins of Barrett’s esophagus: the search continues. Curr Gastroenterol Rep 2018;20:51. [DOI] [PubMed] [Google Scholar]
- 6.Fitzgerald RC. Molecular basis of Barrett’s oesophagus and oesophageal adenocarcinoma. Gut 2006;55:1810–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Que J, Garman KS, Souza RF, et al. Pathogenesis and cells of origin of Barrett’s esophagus. Gastroenterology 2019;157:349–364.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Purkayastha BPD, Chan ER, Ravillah D, et al. Genome-scale analysis identifies novel transcript-variants in esophageal adenocarcinoma. Cell Mol Gastroenterol Hepatol 2020;10:652–654.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blum AE, Venkitachalam S, Ravillah D, et al. Systems biology analyses show hyperactivation of transforming growth factor-beta and JNK signaling pathways in esophageal cancer. Gastroenterology 2019;156:1761–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dimitrova N, Nagaraj AB, Razi A, et al. InFlo: a novel systems biology framework identifies cAMP-CREB1 axis as a key modulator of platinum resistance in ovarian cancer. Oncogene 2017;36:2472–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Z, Hu T, Zhu S, et al. Glutathione peroxidase 7 suppresses cancer cell growth and is hypermethylated in gastric cancer. Oncotarget 2017;8:54345–54356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalabis J, Wong GS, Vega ME, et al. Isolation and characterization of mouse and human esophageal epithelial cells in 3D organotypic culture. Nat Protoc 2012;7:235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abdel-Latif MM, O’Riordan J, Windle HJ, et al. NF-kappaB activation in esophageal adenocarcinoma: relationship to Barrett’s metaplasia, survival, and response to neoadjuvant chemoradiotherapy. Ann Surg 2004;239:491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bian YS, Osterheld MC, Bosman FT, et al. Nuclear accumulation of beta-catenin is a common and early event during neoplastic progression of Barrett esophagus. Am J Clin Pathol 2000;114:583–590. [DOI] [PubMed] [Google Scholar]
- 15.Guo Q, Xie J, Dang CV, et al. Identification of a large Myc-binding protein that contains RCC1-like repeats. Proc Natl Acad Sci U S A 1998;95:9172–9177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pao KC, Wood NT, Knebel A, et al. Activity-based E3 ligase profiling uncovers an E3 ligase with esterification activity. Nature 2018;556:381–385. [DOI] [PubMed] [Google Scholar]
- 17.Clemons NJ, Wang DH, Croagh D, et al. Sox9 drives columnar differentiation of esophageal squamous epithelium: a possible role in the pathogenesis of Barrett’s esophagus. Am J Physiol Gastrointest Liver Physiol 2012;303:G1335–G1346. [DOI] [PubMed] [Google Scholar]
- 18.Wang DH, Tiwari A, Kim ME, et al. Hedgehog signaling regulates FOXA2 in esophageal embryogenesis and Barrett’s metaplasia. J Clin Invest 2014;124:3767–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arul GS, Moorghen M, Myerscough N, et al. Mucin gene expression in Barrett’s oesophagus: an in situ hybridisation and immunohistochemical study. Gut 2000;47:753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whelan KA, Muir AB, Nakagawa H. Esophageal 3D culture systems as modeling tools in esophageal epithelial pathobiology and personalized medicine. Cell Mol Gastroenterol Hepatol 2018;5:461–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harada H, Nakagawa H, Oyama K, et al. Telomerase induces immortalization of human esophageal keratinocytes without p16INK4a inactivation. Mol Cancer Res 2003;1:729–738. [PubMed] [Google Scholar]
- 22.Kosoff RE, Gardiner KL, Merlo LM, et al. Development and characterization of an organotypic model of Barrett’s esophagus. J Cell Physiol 2012;227:2654–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kong J, Nakagawa H, Isariyawongse BK, et al. Induction of intestinalization in human esophageal keratinocytes is a multistep process. Carcinogenesis 2009;30:122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fecteau RE, Kong J, Kresak A, et al. Association between germline mutation in VSIG10L and familial Barrett neoplasia. JAMA Oncol 2016;2:1333–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glickman JN, Yang A, Shahsafaei A, et al. Expression of p53-related protein p63 in the gastrointestinal tract and in esophageal metaplastic and neoplastic disorders. Hum Pathol 2001;32:1157–1165. [DOI] [PubMed] [Google Scholar]
- 26.von Furstenberg RJ, Li J, Stolarchuk C, et al. Porcine esophageal submucosal gland culture model shows capacity for proliferation and differentiation. Cell Mol Gastroenterol Hepatol 2017;4:385–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garman KS. Origin of Barrett’s epithelium: esophageal submucosal glands. Cell Mol Gastroenterol Hepatol 2017;4:153–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garman KS, Kruger L, Thomas S, et al. Ductal metaplasia in oesophageal submucosal glands is associated with inflammation and oesophageal adenocarcinoma. Histopathology 2015;67:771–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mills JC, Sansom OJ. Reserve stem cells: differentiated cells reprogram to fuel repair, metaplasia, and neoplasia in the adult gastrointestinal tract. Sci Signal 2015;8:re8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sears R, Leone G, DeGregori J, et al. Ras enhances Myc protein stability. Mol Cell 1999;3:169–179. [DOI] [PubMed] [Google Scholar]
- 31.Sears R, Nuckolls F, Haura E, et al. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev 2000;14:2501–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lisabeth EM, Falivelli G, Pasquale EB. Eph receptor signaling and ephrins. Cold Spring Harb Perspect Biol 2013;5:a009159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pasquale EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer 2010;10:165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathot L, Kundu S, Ljungstrom V, et al. Somatic ephrin receptor mutations are associated with metastasis in primary colorectal cancer. Cancer Res 2017;77:1730–1740. [DOI] [PubMed] [Google Scholar]
- 35.Lugli A, Spichtin H, Maurer R, et al. EphB2 expression across 138 human tumor types in a tissue microarray: high levels of expression in gastrointestinal cancers. Clin Cancer Res 2005;11:6450–6458. [DOI] [PubMed] [Google Scholar]
- 36.Jang BG, Lee BL, Kim WH. Intestinal stem cell markers in the intestinal metaplasia of stomach and Barrett’s esophagus. PLoS One 2015;10:e0127300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cortina C, Palomo-Ponce S, Iglesias M, et al. EphB-ephrin-B interactions suppress colorectal cancer progression by compartmentalizing tumor cells. Nat Genet 2007;39:1376–1383. [DOI] [PubMed] [Google Scholar]
- 38.McCall JL, Gehring D, Clymer BK, et al. KSR1 and EPHB4 regulate Myc and PGC1beta to promote survival of human colon tumors. Mol Cell Biol 2016;36:2246–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song W, Hwang Y, Youngblood VM, et al. Targeting EphA2 impairs cell cycle progression and growth of basal-like/triple-negative breast cancers. Oncogene 2017;36:5620–5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stairs DB, Nakagawa H, Klein-Szanto A, et al. Cdx1 and c-Myc foster the initiation of transdifferentiation of the normal esophageal squamous epithelium toward Barrett’s esophagus. PLoS One 2008;3:e3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tselepis C, Morris CD, Wakelin D, et al. Upregulation of the oncogene c-myc in Barrett’s adenocarcinoma: induction of c-myc by acidified bile acid in vitro. Gut 2003;52:174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nowicki-Osuch K, Zhuang L, Jammula S, et al. Molecular phenotyping reveals the identity of Barrett’s esophagus and its malignant transition. Science 2021;373:760–767. [DOI] [PubMed] [Google Scholar]
- 43.Holland S, Scholich K. Regulation of neuronal functions by the E3-ubiquitinligase protein associated with MYC (MYCBP2). Commun Integr Biol 2011;4:513–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barquilla A, Pasquale EB. Eph receptors and ephrins: therapeutic opportunities. Annu Rev Pharmacol Toxicol 2015;55:465–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saha N, Robev D, Mason EO, et al. Therapeutic potential of targeting the Eph/ephrin signaling complex. Int J Biochem Cell Biol 2018;105:123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hughes RM, Virag JAI. Harnessing the power of Eph/ephrin biosemiotics for theranostic applications. Pharmaceuticals (Basel) 2020;13:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.