

Abstract
Background:
Mural cells in ascending aortic aneurysms undergo phenotypic changes that promote extracellular matrix destruction and structural weakening. To explore this biology we analyzed the transcriptional features of thoracic aortic tissue.
Methods:
Single nuclear RNA sequencing was performed on 13 samples from human donors, 6 with thoracic aortic aneurysm and 7 without aneurysm. Individual transcriptomes were then clustered based on transcriptional profiles. Clusters were used for between-disease differential gene expression analyses, subcluster analysis, and analyzed for intersection with genetic aortic trait data.
Results:
We sequenced 71,689 nuclei from human thoracic aortas and identified 14 clusters, aligning with 11 cell types, predominantly vascular smooth muscle cells (VSMCs) consistent with aortic histology. With unbiased methodology, we found 7 VSMC and 6 fibroblast subclusters. Differentially expressed genes (DEGs) analysis revealed a VSMC group accounting for the majority of differential gene expression. Fibroblast populations in aneurysm exhibit distinct behavior with almost complete disappearance of quiescent fibroblasts. DEGs were used to prioritize genes at aortic diameter and distensibility GWAS loci highlighting the genes JUN, LTBP4, and IL34 in fibroblasts, ENTPD1, PDLIM5, ACTN4, and GLRX in VSMCs, as well as LRP1 in macrophage populations.
Conclusions:
Using nuclear RNA sequencing we describe the cellular diversity of healthy and aneurysmal human ascending aorta. Sporadic aortic aneurysm is characterized by differential gene expression within known cellular classes rather than by the appearance of novel cellular forms. Single nuclear RNA sequencing of aortic tissue can be used to prioritize genes at aortic trait loci.
Graphical Abstract
INTRODUCTION
Aortic dissection is a life threatening illness associated with a high risk of death and morbidity.1,2 It is well established that the risk of aortic dissection is directly related to aortic diameter, a property easily obtained by noninvasive imaging.3,4 Enlargement of the ascending thoracic aorta is typically referred to as an aneurysm, and the incidental discovery of aneurysms can prompt prophylactic surgical repair to prevent aortic dissection or rupture. Aneurysms can be caused by pathologic processes such as atherosclerosis, hypertension, or genetic variation that affects aortic extracellular matrix homeostasis,5,6 although it is typically a combination of such factors.1,7 Studies of aortic histology note increased pathologic changes including elastin fiber breakage, medial cell disarray, and extracellular mucoid deposition in ascending aortic aneurysms. Furthermore, the degree of such changes correlate with aortic diameter.8 However, up to 50% of aortic dissections occur in patients with diameters smaller than that recommended for prophylactic surgical repair.9 Aortic tissue from such patients show marked pathologic changes despite mild or no aneurysm.8 Therefore, aneurysm size remains an imperfect predictor of aortic dissection risk, and a more complete understanding of aortic biology may assist in better risk stratification for therapeutic applications.
Aortic stability and function are provided by a complex and extensive extracellular matrix. The primary function of cellular constituents of the aorta is to preserve matrix homeostasis.5,10,11 The most abundant cellular class within the aorta are vascular smooth muscle cells (VSMC) that create and maintain matrix components such as amorphous elastin and collagens. As opposed to abdominal aortic aneurysms (AAAs), ascending thoracic aortic aneurysms (TAAs) are typically noninflammatory but are characterized by widespread phenotypic change seen within native aortic cellular classes, most prominently within VSMCs. TAAs can be caused by monogenic mutations in genes that are specific to VSMCs, illustrating the importance of this cellular class in the etiology of aneurysm.12,13 As a result, VSMCs within aortic aneurysms have been extensively studied, primarily in cell culture or with histology, and are known to undergo transcriptional changes that drive extracellular matrix destruction, aneurysm, and eventually aortic failure.14–16 Much of the current mechanistic knowledge about aortic pathology, however, stems from patients and animal models with hereditary TAA or monogenic models. The extent to which the mechanisms and pathways can be extrapolated to sporadic TAAs remains an area with relative paucity of data, despite the sporadic form accounting for greater than 70% of all TAAs.17
Transcriptional analysis is one method for detecting changes in the underlying phenotypes of cells in disease states. The study of transcriptional changes within cells of the aorta have yielded important insights in tissue function and gene expression, despite being predominantly studied in bulk tissue samples. However, advances in single cellular sequencing technologies enabled by microfluidics and barcoding of transcripts allows for more detailed characterization of cellular classes and subdivisions. This method of interrogating disease has thus far enabled identification of new targets for mechanistic and therapeutic investigation and changed our understanding and definition of cells, cell biology and cell identity.18 Furthermore, these technologies allow for direct transcriptional comparisons between healthy and diseased tissues within these cellular groups.19 Coupled with emerging analytic techniques, large-scale genetic, and imaging data-sets with aortic-specific quantitative traits such as diameter and distensibility,20–22 single cellular interrogation of sporadic TAA is particularly equipped help identify new relevant gene targets and clarify the etiologic overlap between hereditary and sporadic TAA.
To this end, we performed single-nucleus RNA sequencing of 71,689 nuclei from the ascending aorta to gain insight into behaviors and phenotypic modulation of human aortic cells in thoracic aortic aneurysms. We define aortic cell types and compositional cell-type shifts in health and disease and highlight relevant differentially expressed genes within 11 major aortic cellular classes. We integrate these findings with recent human genetic studies of aortic diameter and distensibility and identify genes that are both differentially expressed in disease and influence aortic properties in humans.
METHODS
Data Availability
Single nucleus RNA sequencing data are publicly available at the Broad Institute’s Single Cell Portal (https://singlecell.broadinstitute.org/single_cell). The raw dataset is available at the National Center for Biotechnology Information’s Gene Expression Omnibus Database (accession #GSE207784). All other data are contained within the article and its supplementary information, or are available upon reasonable request to the corresponding author.
Human tissue samples
Adult human ascending aortic samples were collected from deceased organ donors by MAGNet (the Myocardial Applied Genetics Network; www.med.upenn.edu/magnet) and the MGH rapid autopsy study. These patients had tricuspid valves, no clinical diagnosis or radiographic evidence of aortic aneurysm, or family history of aneurysm disease. Patients who underwent open aortic reconstruction for sporadic ascending aortic aneurysm underwent tissue collection during the index operation. Patients were excluded if they had bicuspid valves, aortic dissection or heritable aortopathy. All tissues were collected within 4 hours of death for Rapid Autopsy participants and during transplant or aortic replacement for all other participants. Tissues were transported to the laboratory on ice and immediately frozen with liquid nitrogen. Written informed consent for research use of donated tissue was obtained for all participants. Research use of tissues was approved by the institutional review boards at the Gift-of-Life Donor Program, the University of Pennsylvania, Massachusetts General Hospital, and the Broad Institute. The protocol developed to process frozen aortic tissue for single nuclei data, described below, afforded the opportunity to compare banked tissue and a larger number of human aorta than previously described. Aortic tissue from both sexes was obtained in this proof-of-concept, exploratory endeavor, with acknowledgement of potential limitations and biases in data interpretation.
Staining validation
To image fibroblast, and vascular smooth muscle subclusters, and differentially expressed genes (DEGs), colorimetric RNAscope was carried out on 10 um fresh frozen tissue sections using the RNAscope 2.5 HD Duplex kit (ACDbio: 322430) following manufacturers protocols. We used a combination of two probes to identify each of the populations, one which is specific to either fibroblasts (DCN: 589521-C2), or vascular smooth muscle cells (ITGA8: 417951-C2), and one which was highly specific to each subcluster. The probes used to identify the fibroblast subclusters were FB-S1: PLIN2 (508741), FB-S2: ERBB4 (407831) FB-S1: ADAMTS4 (537341), and those used to identify the vascular smooth muscle cell subclusters were VSMC-1: CNTN4 (438491) VSMC-2:CFH (428731). Prioritized DEGs from GWAS were similarly validated through staining of cluster-specific probes: fibroblasts (DCN: 589521-C2) macrophages (CD163: 417061-C1); and prioritized DEGs: JUN (470541-C1), LTBP4 (893621-C1), LRP1 (836621-C2). Slides were imaged on a Zeiss Observer Z1 microscope using 20x objective for tiles and 40x for single images.
Single nuclei RNA sequencing
Single nucleus suspensions were generated as previously described with some modifications.20,23 In brief, approximately 30 × 100 um thick cryosections of aorta were placed into 1 mL of ice-cold digestion buffer (250 mM sucrose, 10 mM Tris pH 8.0, 25 mM KCL, 5 mM MGCl2, 0.01% IGELPAL, 0.05% BSA, 1:100 Rnase inhibitor, 1 ug/ul elastase) mixed gently, and left on ice for 10 min, with gentle mixing at 5 min. Samples were then transferred into a 7 mL dounce containing 3 mL of Lysis buffer (250 mM sucrose, 10 mM Tris pH 8.0, 25 mM KCL, 5 mM MGCl2, 0.01% IGELPAL, 0.05% BSA, 1:100 Rnase inhibitor, 1uM DTT) and were homogenized on ice 10 x with the loose pestle, then left for 5 min on ice. Samples were homogenized on ice 10 x with the tight pestle, left to sit for 5 min on ice then transferred into a 15 mL tube. Large debris was pelleted by centrifugation at 40 x g at 4 degrees for 1 min. The supernatant was filtered through stacked 40 um and 10 um filters. Filters were washed with 6 mL of PBS wash buffer (0.05% BSA, 5 mM MgCl2, PBS, 1:1000 RNase inhibitor) and the nuclei pelleted by centrifugation at 550 x g at 4 degrees for 5 min. The nuclei pellet was washed by removal of the supernatant, and addition of 6 mL of PBS wash buffer, followed by centrifugation at 550 x g at 4 degrees for 5 min. The supernatant was removed and nuclei resuspended in 100 uL of nuclei resuspension buffer (0.05% BSA, 5 mM MgCl2, 0.4 U/uL RNase inhibitor), and then counted using a hemocytometer. Approximately 6000 nuclei per sample were loaded onto a 10x genomics microfluidic chip and samples were processed according to the 10x genomics protocol (Single cell 3’ solution, v3.1) with the following changes. First, nuclei were incubated at 4C for 15 minutes after emulsion generation to promote nuclear lysis. Second, the reverse transcription protocol was modified for one of the two replicates to be 42C for 20 minutes then 53C for 120 minutes. Libraries were multiplexed at an average of 16 libraries per flow cell on an Illumina Novaseq S1 at the Broad Institute’s Genomics Platform.
Global single nuclei map construction
Single nuclei RNA sequencing libraries were processed with 10x Genomics toolkit CellRanger 4.0.0. First, libraries were demultiplexed using cellranger mkfastq with default settings. Resulting FASTQ files were trimmed to remove the template switch oligo (TSO) and homopolymer repeats using cutadapt with parameters (max_error_rate=0.07, min_overlap=10) and (max_error_rate=0.1, min_overlap=20), respectively.24 Trimmed reads were aligned to GRCh38 pre-mRNA human reference (v2020-A) using cellranger count with --expect-cells set to 6000 and all other parameters as default. To identify non-empty droplets and remove ambient counts from droplets, we used CellBender v0.2 (https://github.com/broadinstitute/CellBender)25 with the following parameters: --expected-cells 6000, --z-dim 100, --total-droplets-included 20000, --epochs 150, --learning_rate 1e-4, and --fpr 0.01. For one sample, C3, (Supplemental Table 1) --expect-cells was set to 2000 and --learning-rate was set to 5e-5 to ensure model convergence (Supplemental Table 2). Finally, we applied the tool scR-Invex (https://github.com/broadinstitute/scrinvex) to calculate the proportion of reads in droplet mapping exclusively to exonic regions as a measure of how much cytoplasmic material is captured in each droplet.
CellRanger quality control metrics were inspected for each sample (Supplemental Table 2, Supplemental Figure 1). Additionally, we plotted the total unique molecular identifiers (UMIs) in each droplet against the respective rank to ensure a sufficient ambient plateau was detected to differentiate empty and non-empty droplets (Supplemental Figure 2). Finally, for each sample we quantified the total UMIs derived from Y chromosome genes as well as the total UMIs from the highly female specific X-chromosome gene XIST. We plotted these values against the phenotypic sex of each sample to ensure there were no sample swaps in library generation (Supplemental Figure 3). No samples were removed during these quality control steps.
To construct a global map of all cell types, we first aggregated the 123,578 nuclei determined as non-empty droplets by CellBender. All analyses were performed in scanpy 1.7.2 unless otherwise stated.26 We selected the top 2000 most highly variable genes using the strategy introduced in Seurat V3 as implemented in scanpy.27 We then log-normalized the count data by dividing the UMI for each gene in a droplet by the total number of UMI in the droplet, multiplying by 10,000, and taking the natural log. After subsetting our data to the top 2000 most highly variable genes, we scaled the data to unit variance and zero mean for each gene and performed principal component (PC) analysis to estimate the first 50 PCs. To account for biological heterogeneity between samples and technical differences between libraries, we adjusted these PCs using Harmony as implemented in harmony-pytorch v0.1.4 (https://github.com/lilab-bcb/harmony-pytorch) with each sample as their own batch.28 We then constructed a neighborhood graph on these adjusted PCs with 15 neighbors using Euclidean distance followed by Uniform Manifold Approximation and Projection (UMAP) for visualization with min_dist=0.2. Leiden clustering at high resolution (2.0) was used to over-cluster the nuclei into 51 groups. We looked at the distribution of four quality control metrics for each cluster: 1. Proportion of exonic reads (‘exon_prop’), 2. Percent of reads mapping to mitochondrial genes (‘percent_mito’), 3. A doublet score as calculated by Scrublet (‘doublet_score’),29 4. A measure of gene entropy calculated with the ndd python library (https://pypi.org/project/ndd/1.6.3/). All outlier clusters on these metrics were removed.
In addition to removing clusters of low-quality nuclei with increased cytoplasmic material, we also performed a per-cluster, per-sample quality control step. In brief, for each cluster/sample combination, we removed nuclei with extremely low, first quartile (Q1) minus 1.5 times the interquartile range (IQR), or extremely high, third quartile (Q3) plus 1.5 times IQR, values for the following metrics: 1. number of UMI (nUMI), 2. number of unique genes (nGenes), 3. entropy, and 4. log(nGenes)*entropy. Similarly, we removed nuclei with extremely high values, Q3 + 1.5 times IQR, for: 1. exon_prop, 2. percent_mito, and 3. doublet_score. Additional hard cutoffs for removal were set at nGenes < 150, nUMI < 150, or percent_mito > 5%. When a given cluster/sample combination had less than 30 nuclei, we removed nuclei with nUMI > 15000 or nUMI ≤ 150, nGenes > 6000 or nGenes ≤ 150, percent_mito > 5%, entropy < 8, exon_prop > 0.18, doublet_score > 0.3, or log(nGenes)*entropy > 75.
We then re-clustered all remaining nuclei as described above using cosine distance instead of Euclidean distance in the neighborhood graph estimation. Leiden clustering resolution was set at 0.3. As a final cleaning step, we performed sub-clustering within each cell type by selecting only nuclei in that population, recalculating the neighborhood graph with the Harmony-adjusted PCs, and iterating over Leiden resolution of 0.05 to 1.0 in increments of 0.05. At each resolution, marker genes were calculated on the log-normalized expression of each gene in the given cluster compared to all other clusters using an area under the receiver operating characteristic curve (AUC). When a sub-cluster emerged with no genes having AUC > 0.6, the previous resolution was retained. Nuclei were then scored for the top 50 marker genes of the global cell types, based on AUC, using sc.tl.score_genes() as implemented in scanpy. Sub-clusters of each cell type with elevated scores of another cell type were deemed misclassified or contaminated and removed. Finally, after pruning misclassified sub-clusters one cluster of low complexity (median nUMI=389 and median nGenes=355) with no distinct marker genes was removed. After quality control 71,689 nuclei (average of 5,515 per sample) remained for downstream analysis.
Marker gene detection
To assign cell type labels to each cluster we identified upregulated genes in each population using two approaches. (Supplemental Table 3) First, using the log-normalized expression in each nucleus, we calculated the AUC comparing the expression of each gene in each cluster compared to all other clusters. Second, to account for the fact that nuclei are derived from distinct biological individuals and will be correlated within a sample, we performed a formal differential expression test on the summed gene counts across all nuclei of a given cluster/sample combination. Only cluster/sample combinations with at least 20 nuclei were included. Lowly expressed genes were identified with filterByExpr as implemented in edgeR30 and removed. DESeq231 normalization was applied with the function estimateSizeFactorsForMatrix() from the R package DESeq2, and differential expression testing carried out with the limma-voom32 framework as implemented in the R package limma. We fit a linear model in limma including fixed effects for cluster and individual, and excluding an intercept (Expression=β1(cluster)+β2(individual)+ε; R notation: ‘~0+cluster+individual’). We then extracted contrasts comparing each cluster versus the average of all other clusters to estimate log fold-changes and P-values. Multiple testing was accounted for using the Benjamini-Hochberg correction to control the false discovery rate (FDR) at 0.05.
Marker genes were defined as protein coding genes expressed in at least 20% of nuclei of the given cluster with AUC > 0.60, log fold-change > 2, and FDR-adjusted P-value < 0.01 from the limma-voom model. For one rare cell type, adipocytes, there were insufficient nuclei to test the limma-voom model and marker genes were selected as protein coding genes expressed in at least 20% of nuclei of the given cluster with AUC > 0.80.
To compare the expression profiles in our clusters to previously published single cell RNA-sequencing, we scored our nuclei based on published single cell profiles using sc.tl.score_genes() in scanpy with default settings. A maximum of the top 50 markers were used for each cell type as previously described.33–35 Only the top 10 marker genes were reported and used for scoring. Genes from mouse data were linked to human identifiers using biomaRt.36
Composition analysis
To compare the cellular composition of aneurysm samples from control samples, we used the method scCODA v 0.1.2.post1.37 We selected the endothelial I population as the reference group as it was present in a similar proportion of all samples, regardless of disease status. For VSMC sub-clustering we selected VSMC-S5 as the reference group and for fibroblast sub-clustering we selected FB-S2 as the reference group. For the Hamiltonian Monte Carlo process, we used 20,000 iterations with 5,000 iterations as burn-in.
Aneurysm versus control differential expression analysis
To identify up- and down-regulated genes in aneurysm cases compared to controls in each cell type we summed the gene counts across all nuclei for each sample and used the limma-voom framework as in the marker gene testing.32 We removed genes expressed in less than 1% of nuclei in both aneurysm and control samples in the given cell type, as well as those identified as too lowly expressed by filterByExpr implemented in edgeR.30 Our primary model for differential expression included a fixed effect for disease and sex (Expression=β0+β1(disease)+β2(sex)+ε; R notation: ‘~1+ disease+sex’), including duplicateCorrelation(), as implemented in the R package limma, for study of origin to control for the fact that control samples came from two sources. We applied the Benjamini-Hochberg correction to account for multiple testing and set a significance threshold at FDR = 0.05. We performed differential expression testing using both CellBender-adjusted counts and raw CellRanger counts. Only genes significant in both tests that were expressed in at least 5% of nuclei from the aneurysm or control group were considered robust. (Supplemental Table 4)
As sensitivity differential expression tests on both CellBender-adjusted and raw CellRanger counts, we also fit two additional limma-voom linear models: 1. A test for differences in expression between studies, restricted to controls, including a fixed effect for sex (Expression=β0+β1(study)+β2(sex)+ε; R notation: ‘~1+study+sex’), and 2. A test for differences between aneurysm cases and controls controlling for study as a fixed effect (Expression=β0+β1(disease)+β2(sex)+β3(study)+ε; R notation: ‘~1+disease+sex+study’). We filtered out genes with FDR < 0.05 for either the CellBender or CellRanger results in model 1 as this suggests these genes are affected by technical differences between the control studies. Genes with FDR ≥ 0.10 in either the CellBender or CellRanger results for model 2 were not considered significant to focus on genes that showed consistent effects.
Background contamination heuristic
In addition to controlling for technical effects in our differential expression modeling, we also controlled for spurious differential expression results driven by ambient background counts by calculating a background contamination probability heuristic for each gene, in each cell type. This heuristic consists of two pieces of information. First, we estimate the likelihood that counts from a given gene are derived from background (“bkg_prob”) by dividing the total counts for each gene by the total counts in the dataset and estimating the value for each gene along the cumulative distribution. Second, we estimate the likelihood that counts from each gene come from a different cell type other than the cell type of interest (e.g. the cell type where the differential expression test was performed). To do this, we calculate normalized positive predictive values (PPV) standardized for the prevalence of each cell type as:
where sensitivity and specificity are calculated by constructing a 2×2 table predicting inclusion of each nucleus in the cell type of interest based on having >0 count (PPV0) or >1 count (PPV1). We then took the 1 minus the average of PPV0 and PPV1 to represent a likelihood that counts for the given gene are derived from background from a different cell type (“nontarget_prob”). We then used the product of bkg_prob and nontarget_prob as a heuristic estimate of whether the differential expression result for a given gene in a given cell type is driven by background contamination of that gene from another cell type. We filtered out genes with a value ≥ 0.4 in the target cell type to help minimize the chances of identifying background expressed genes.
Pathway enrichment analysis
Gene ontology enrichment for biological processes was performed using the R package GOStats v2.46.0.38 (Supplemental Table 5) Genes were mapped to Entrez identifiers using org.Hs.eg.db v3.12.0 in R and removed if no matching identifier existed. For marker gene pathway enrichment, a hypergeometric test was used to identify biological processes where marker genes were overrepresented compared to the background of all genes tested (n=23,744). Only ontologies with more than 10 genes and less than 1000 genes were considered. Multiple testing across all cell types was accounted for using the Benjamini-Hochberg correction at FDR=0.05.
Genes up- and down-regulated in aneurysm patients compared to controls were tested for enrichment in biological process enrichment as described above. For each cell type, genes were separated into those significantly up- and down-regulated in aneurysm patients and compared for enrichment against the background of all genes tested after applying the filters described in Aneurysm versus control differential expression analysis (nVSMC1=5257; nFibroblast=6179; nEndothelial1=5238; nVSMC2=6839; nMacrophage=6014; nPericyte=6242). Multiple testing across all cell types was accounted for using the Benjamini-Hochberg correction at FDR=0.05.
Vascular smooth muscle cell (VSMC) and fibroblast sub-clustering analysis
Cellular heterogeneity within the VSMC and fibroblast cell types was investigated in a secondary clustering analysis. For VSMCs, we selected all nuclei from the VSMC1, VSMC2, and VSMC3, populations (n=46,106) and repeated global clustering procedures restricted to these nuclei. In brief, we estimated the top 2000 most highly variable genes, log-normalized and scaled the count data, regressed out technical variation including the percent of reads mapping to mitochondrial genes and total UMI,39 estimated the top 50 principal components on scaled data, aligned nuclei across samples using Harmony, estimated the neighborhood graph based on 15 nearest neighbors, and ran UMAP to visualize the data. Leiden clustering was iterated from 0.1 to 2.0 in increments of 0.1 until a sub-cluster emerged that did not have any genes with AUC > 0.60 and the previous resolution was selected (resolution=1.2; nclusters=14).
Transcriptionally similar sub-clusters were merged using hierarchical clustering of sub-cluster centroids based on the top 2000 most highly variable genes, Euclidean distance, and the Ward method. Sub-clusters with distance less than 0.5x the max distance were merged.
The same procedure was used on all fibroblasts from the global map (n=8,925) with a couple modifications. First, in the initial round of sub-clustering, we identified a small population (n=247) of misclassified/contaminated nuclei which we removed before repeating the sub-clustering procedure. Second, when merging sub-clusters, a distance cutoff was set at 0.4x the max distance. Finally, two fibroblast populations were very small (FB-S5, n=18 and FB-S6, n=8) and were not included in marker gene detection testing.
Sub-cluster marker genes were calculated as in the global map and were defined as protein coding genes expressed in at least 15% of nuclei of the given cluster with AUC > 0.50, log fold-change > 0, and FDR-adjusted P-value < 0.01 from the limma-voom model.32 More lenient cutoffs were used here as sub-clusters tend to have less distinction than global cell types. Ontology enrichment was performed for sub-cluster marker genes as described previously.
Intersection of aortic transcriptome with medical traits
To intersect our snRNA-seq data with previously published GWAS summary statistics we used two strategies. First, we performed stratified LD-score regression to assess for enriched GWAS heritability around genes with cell-type specific expression.40,41 We only considered protein coding genes with at least 5 UMI across all nuclei. For each cell type, we took the top 10% of up-regulated genes (n=1425) based on the t-statistic from the limma-voom differential expression model. We then annotated any single nucleotide polymorphism within 100 kilobases of these genes for each cell type and performed stratified LD-score regression as previously described for GWAS of ascending and descending aortic diameter and aortic distensibility.20,21 We corrected for multiple testing per GWAS by using Bonferroni significance across the number of cell types tested.
Second, we prioritized potentially causal GWAS genes from the same GWAS of ascending and descending aortic diameter and aortic distensibility20,21 and intersected these with differentially expressed genes from our snRNA-seq data. Of the 334 DEGs (aneurysm versus control) identified in aortic tissues, we first identified how many were protein coding genes that fell within plus or minus 500 kilobases of a GWAS locus. We then further limited these genes by considering only those ranked as the top candidate through the polygenic priority score (PoPS), a method that integrates gene expression, biological pathway, and predicted protein-protein interaction data to nominate potentially causal genes.42 This unbiased strategy prioritized seven genes at aortic GWAS loci that were also DEGs in aortic cell types (Figure 7b).
RESULTS
Tissue Characteristics of Ascending Aortic Aneurysms
Ascending aortic aneurysms demonstrate stereotypical pathologic changes including degeneration of the medial aortic layer, increased collagen deposition, fragmentation of elastin fibers, and deposition of glycosoaminoglycans.8,43,44 Histologically, aneurysms are characterized by disarray of vascular smooth muscle cells, including intracellular abnormalities such as loss of and dysfunction of actin stress fibers and focal adhesions. (Figure 1a)13,15,45 To better characterize the composition and cell-specific transcriptome differences between the aneurysmal and non-aneurysmal ascending aorta, we performed snRNA-seq on human aortic samples with particular focus on the VSMC population. (Figure 1b)
Figure 1. Study overview.
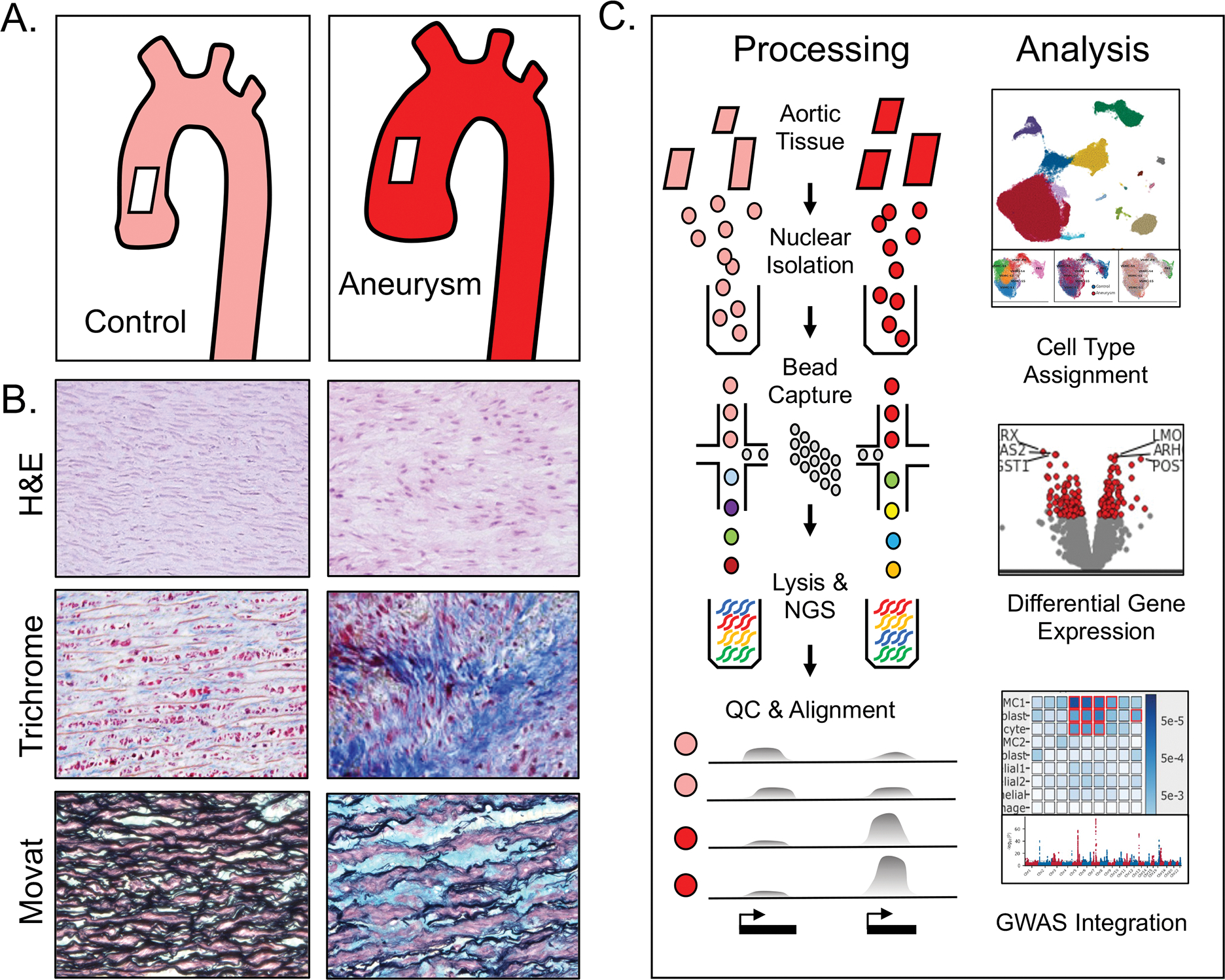
a. Aortic aneurysm tissue is characterized by smooth muscle cell disorganization (H&E), deposition of collagen (Trichrome), and elastin fiber breaks with deposition of glycosoaminoglycans (Movat’s pentachrome). b. Aortic samples from patients with and without ascending aortic aneurysm were subjected to mechanical disruption and collection of cellular nuclei.
Eleven Major Cell Types Identified in Human Aortic Tissue
We obtained ascending aortic tissue from 6 patients with and 7 patients without thoracic aortic aneurysm and normal tricuspid valves. Baseline characteristics of participants are available in Supplemental Table 1. In order to enable accurate comparisons of transcriptional profiles across multiple samples, tissues underwent immediate cryopreservation and storage at −80C until use. Nuclear isolation of aortic tissue required additional mechanical disruption and digestion with elastase compared to other cardiovascular tissues and isolation methods previously described.20,33,46,47 Isolated nuclei were manually counted and inspected for debris prior to proceeding to droplet generation. Approximately 6000 nuclei per sample were loaded and processed according to the 10x Genomics protocol (Single cell 3’ solution, v3.1), with some modifications. (Methods) 123,578 nuclei underwent analytic quality control by removal of outliers based on exonic reads, mitochondrial genes, doublet score, gene entropy and cytoplasmic material, as well as by per-cluster and per-sample analysis. (Methods, Supplemental Figure 1)
A total of 71,689 nuclei (average of 5,515 per sample) from 13 libraries passed quality control and underwent unsupervised Leiden clustering. Using canonical marker and ontology analysis, 11 major conventional cell types were defined from 14 clusters. (Figure 2a, Figure 3a, Supplemental Figure 4) Assignment of nuclei to conventional cell types involved identification of marker genes using two complementary methods. First, using the log-normalized expression in each nucleus, we calculated the area under the receiver operating characteristic curve (AUC) comparing the expression of each gene in each cluster compared to all other clusters. Second, to account for the fact that nuclei are derived from distinct biological individuals and will be correlated within a sample, we performed a formal differential expression test on the summed gene counts across all nuclei of a given cluster/sample combination. We highlight the importance of sample and data quality control metrics to account for biological heterogeneity and technical differences in computational analysis,49,50 and provide an illustration of result variability based on sample alignment and correction in Supplemental Figure 5. Cell types were then designated by marker genes as defined by protein coding genes expressed in at least 20% of nuclei of the given cluster with AUC > 0.60, log fold-change > 2, and FDR-adjusted P-value < 0.01. (Methods)
Figure 2. Aortic cell type assignment.
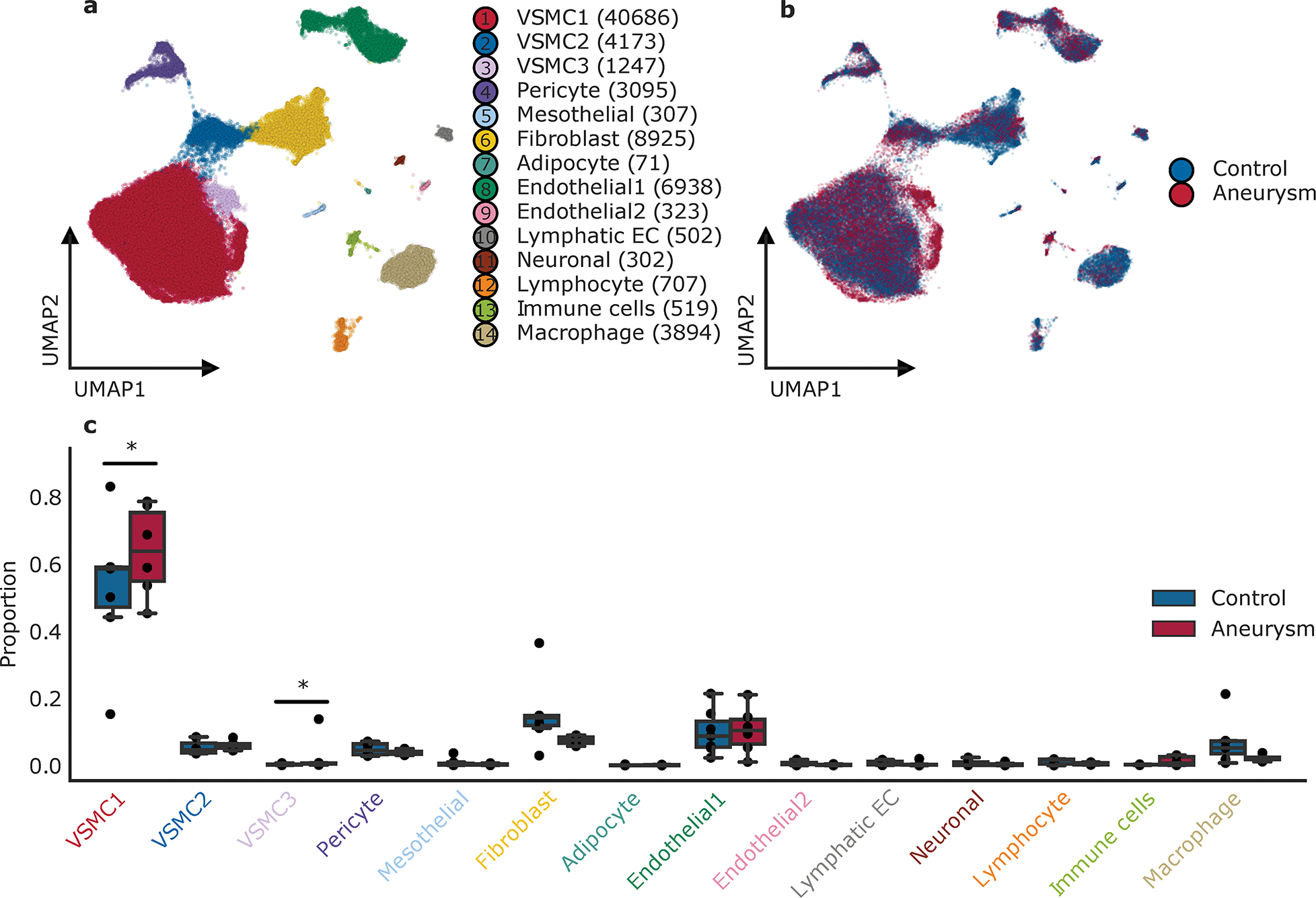
Observed cell types in the ascending aorta. a. Uniform manifold approximation and projection plot displaying cellular diversity across 71,689 nuclei from the ascending aorta of aneurysm and control patients. Each dot represents an individual nucleus. Colors correspond to the cell cluster labels. b. Combined uniform manifold approximation and projection plot contrasting 6 individuals with aneurysm and 7 control aortic samples. Aneurysm nuclei represented in magenta, control aortic nuclei are colored in blue. c. Distribution of cell clusters across individuals, stratified by disease status (aneurysm=6, control=7). Statistically credible shifts in proportions as tested using scCODA (see Methods) are denoted with a *. Center line, median; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range.
Figure 3. Definitions of observed cell clusters.
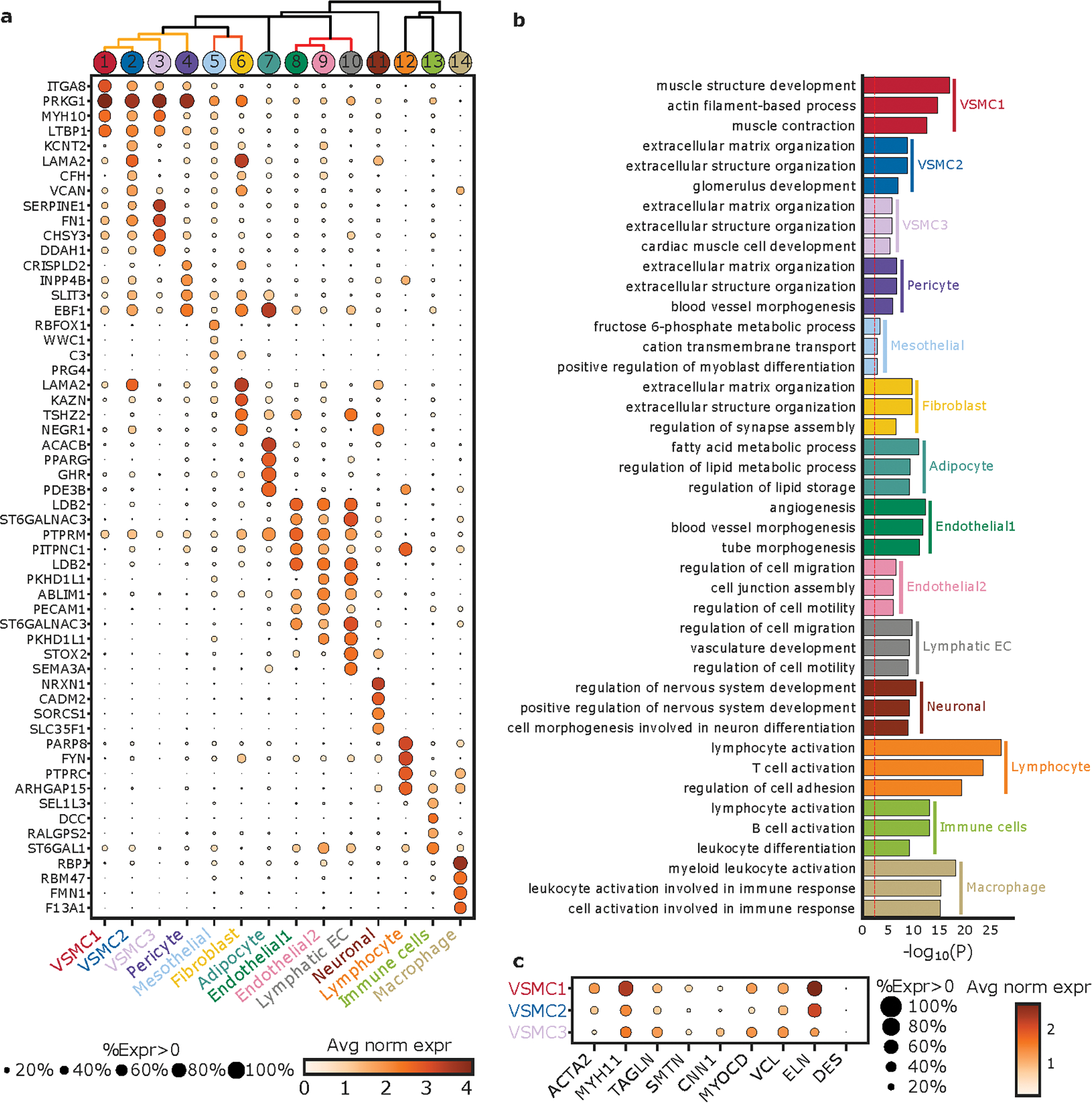
a. Top 6 marker genes for each cluster listed in the left panel. Size of the dot represents the percentage of cells within the cluster where the marker gene is detected. Gradation corresponds to the mean log2 of the counts normalized by total counts per cell ×10 000. b. The top 3 gene ontologies for each cell cluster as determined by gene ontology enrichment analysis by GOStats of marker genes (see Methods). The red dotted line indicates a Bonferroni statistical significance threshold. c. Expression of marker genes typically used to characterize VSMCs amongst the three subgroups of VSMCs identified through unbiased cell clustering. %Expr > 0, Percent of nuclei in a given sub-cluster that express the gene at non-zero levels; Avg norm expr, Average log-normalized expression.
Distributions of cell type by uniform manifold approximation and projection representations are shown in Figure 2a–b. We describe three distinct populations of VSMCs, three endothelial populations, pericytes, fibroblasts, adipocytes, neuronal cells, mesothelial cells, and three immune lineages. Significant overlap in cell population distribution, expression, and diversity is observed between control and aneurysmal aortic tissue. (Figure 2b–c, Supplemental Figure 6) The most striking compositional difference between normal and aneurysmal aortas was an increase in the relative proportion of the VSMC1 group. Other cell type proportions were largely similar between samples, with the exception of the VSMC3 group which was derived mostly from one sample (774). (Supplemental Table 1) In contrast to studies of abdominal aortic aneurysm cellular composition,47,48 there were no significant increases in immune or inflammatory cell lineages, (Figure 2c) consistent with the described non-inflammatory nature of ascending aortic histology.8
The most common cell type observed in aortas were VSMCs, which comprised approximately ~65% of nuclei in our samples (clusters 1, 2, 3), consistent with observations from aortic histology. (Figure 2c, Supplemental Table 3) Clusters 1, 2, 3, and 4 all expressed canonical markers of VSMCs including MYH11 and ACTA2, and included VSMC subtypes as well as pericytes. (Figure 3a, Supplemental Figure 4) Fibroblast cluster 6 was the next most common cell type after VSMCs and expressed typical but less specific gene markers such as LAMA2 and TSHZ2. As expected for vascular tissue, endothelial cells expressing PECAM1 (CD31) were also numerous and were observed in three separate clusters 8, 9, 10. (Figure 2a, Figure 3a, Supplemental Figure 4) Pathway analysis of individual cell groups was performed for independent verification of cluster identity. (Figure 3b) Gene expression within the major VSMC groups was notable for differences between subtypes of cellular classes. (Figure 3a) For instance, the VSMC2 group, which represents ~10% of VSMC content, is differentiated by higher expression of SERPINE1 and FN1, when compared to the VSMC1 group, with the rare VSMC3 group showing even higher expression. These genes encode proteins PAI-1 and fibronectin, both known to be associated with extracellular matrix production. The VSMC2 group also expresses smooth muscle marker genes including MYH11, ACTA2, CNN, and SMTN at intermediate levels between VSMC1 and fibroblasts. (Figure 3c) Pathway analysis supports these observations with a shift of implicated pathways in these groups from ‘muscle structure development’ and ‘muscle contraction’ pathways in VSMC1 to ‘extracellular matrix organization’ and ‘extracellular structure organization’ in VSMC2 group. (Figure 3b) Therefore ~10% of VSMCs in both healthy and aneurysmal aortas exhibit a phenotype encompassing lower expression of contractile elements and increased extracellular matrix production, but the presence of these cells does not correlate with aneurysm status.
Differentially Expressed Genes (DEGs)
Despite the global similarity in major cell classes between aneurysmal and normal ascending aortic tissue, we further explored the transcriptional differences within cell populations that may underlie aortic aneurysm. Principal component analysis of the data can differentiate normal ascending aortic tissue compared to aneurysmal tissue as well as by sex of the patient. This supports the notion that the global snRNAseq data by specimen is unique to the participant and share similarities and differences in gene expression by pathology and sex. (Figure 4a) Additional principal component analysis by cell type revealed similar patterns to that of the combined analysis. (Supplemental Figure 7) Analysis of differential gene expression between aneurysm and non-aneurysmal specimens within cell groups identified potentially biologically relevant genes in disease. (Figure 4b, Supplemental Table 4)
Figure 4. Differentially Expressed Genes.
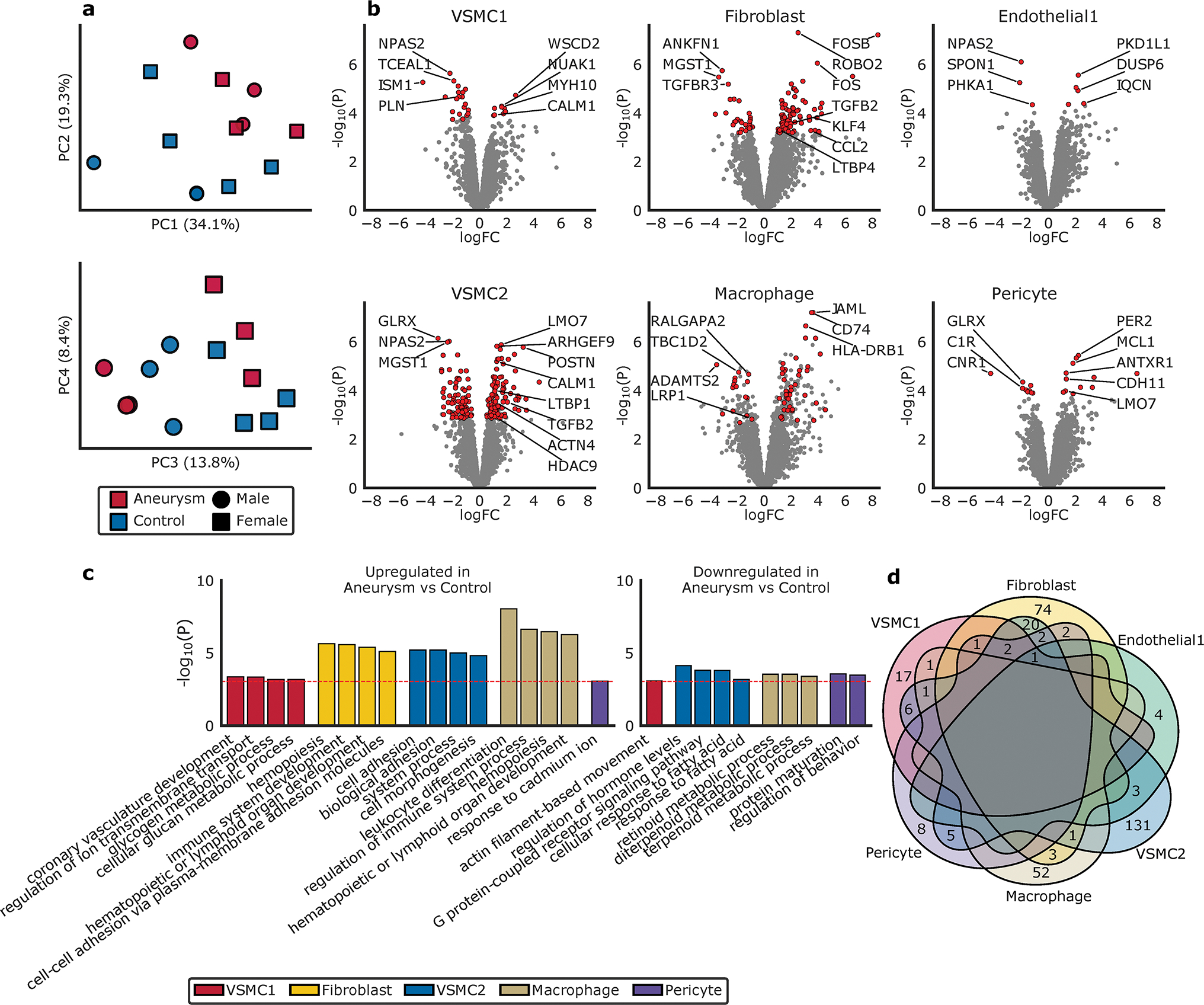
a. Principal components demonstrate clustering differences dependent on aneurysm status and genetic sex. b. Volcano plot highlighting the top differentially expressed genes in aneurysm compared to normal aorta, based on cellular groups, as tested using limma-voom. The X-axis represents the log fold-change (logFC) and the Y-axis represents the −log10 (P value). Genes colored red are significantly differentially expressed whereas genes colored grey are not significant (see Methods). c. The top gene ontologies for significantly differentially expressed genes between aneurysm and control in each cell type based on gene ontology enrichment analysis by GOStats (see Methods). The red dotted line indicates a Bonferroni statistical significance threshold. d. Venn diagram depicting the overlap of significantly differentially expressed genes by cell cluster.
Overall, 334 genes were found to be differentially expressed between aneurysm and non-aneurysm tissue. Several identified DEGs have been implicated in prior genetic and molecular studies. These include HDAC9,15,51 ACTN4,20,52,53 PLN,54 CALM1,55 TGFB2,56,57 TGFBR3,58 KLF4,59 LTBP1,60,61 LTBP4,62 CCL2,63,64 POSTN,52,65 CDH11,66 and LMO7.67 (Figure 4b) The VSMC2 cluster was notable for having the largest number of unique DEGs (131). (Figure 4b,d) In contrast, the VSMC1 (17), endothelial (4), fibroblast (74) and macrophage (52) cell groups had fewer unique DEGs based on aneurysm status. Pathway analysis of these DEGs implicate the VSMC2 cell population as a group of interest due to its relative upregulation of genes involved in cell morphogenesis and adhesion, (Figure 4c, Supplemental Table 5) consistent with altered cellular phenotype and previous molecular findings in aneurysm.15,68–70 RNAscope of selected DEGs confirmed non-uniform spatial distributions of VSMC1-enriched transcript CNTN4 and VSMC2 enriched transcript CFH in the aortic media. (Supplemental Figure 8)
Determination and definition of VSMC sub-classes
Based on the distinct differences in gene expression amongst VSMC1 and VSMC2 cell populations in aneurysm compared to non-aneurysm tissue, further investigation of the VSMC cell compartment was explored by subcluster analysis. The process of cluster and subcluster analysis in single-cell RNA sequencing remains in evolution and the computational method of defining subgroups is critical in the interpretation of biologically significant molecular profiling and differences in expression amongst biologically heterogeneous samples.71–76 In general, additional subpopulations can be defined depending on the nature of the analysis protocol. Whether the identified subpopulations are relevant to disease, or plausible as distinct groups with biological implications, however, is difficult to determine. Furthermore, the use of bespoke integrative methodologies, sometimes buried in the supplemental data of a manuscript can make data interpretation between studies and datasets difficult, while systematic benchmarking is still in development.
To address this challenge, we provide our approach to interrogate the VSMC population, identify subpopulations and reconcile molecular variability with phenotypic diversity. Through subcluster methodology as described in Figure 5a, seven VSMC subclusters were defined by representative marker genes. (Supplemental Table 6) To emphasize the ongoing challenges of subcluster characterization and analysis, we illustrate the differences in subcluster results based on resolution. (Supplemental Figure 9a) Pathway analysis of upregulated genes in each of our final subclusters suggested distinct functional phenotypes of individual VSMC subclusters enriched across the UMAP visualization. (Figure 5f, Supplemental Figure 9b) Gene ontology analysis and the subsequent pathways associated with each subcluster did not appear to completely align with the classically described VSMC phenotypes.59 (Supplemental Table 7) Further analysis of each subcluster by classical VSMC phenotype markers also did not categorize the subclusters according to a VSMC phenotype. (Figure 5e)
Figure 5. VSMC Cell Subclustering.
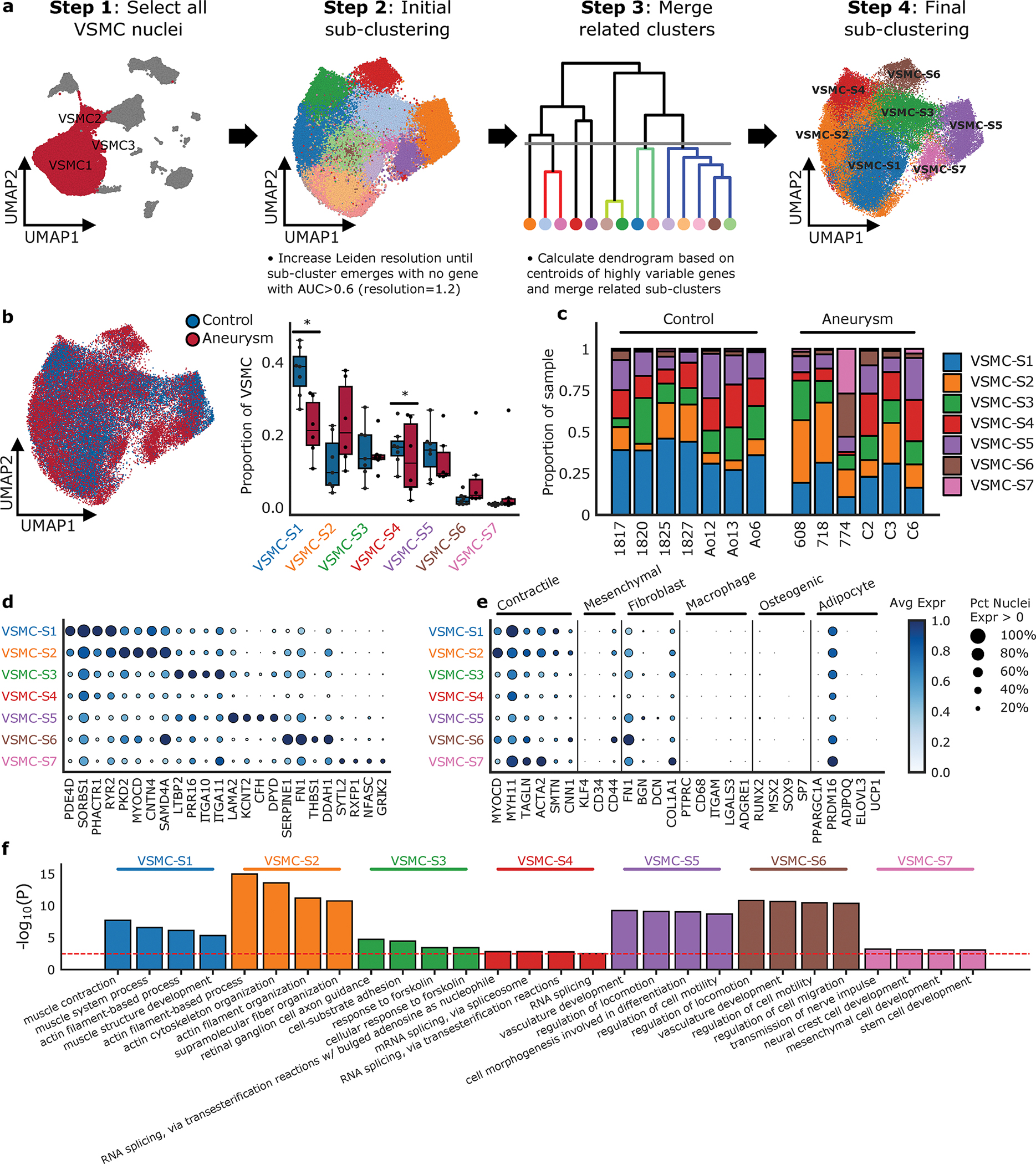
a. Subclustering technologic approach; Nuclei clustered as VSMC1, VSMC2, VSMC3 were selected to identify clusters enriched for marker genes of other global cell types. Subcluster methodology includes re-estimation of the top 2000 most highly variable genes and recalculation of the top principal components using log-normalized and scaled expression. Harmonizing the principal components, UMAP construction and Leiden clustering at increasing resolutions until subclusters with no marker genes (AUC > 0.6) compared to all other clusters emerge. Transcriptional similar subclusters are merged to get final sub-clustering results. b. Overlap of aneurysm and control nuclei on VSMC sub-clustering and the relative proportion of each subcluster between control and aneurysmal aortic tissue. Statistically credible shifts in proportions as tested using scCODA (see Methods) are denoted with a *. Center line, median; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range. c. The relative proportion of each subcluster type by sample. d. Selected marker genes that define each subcluster. e. Classic VSMC markers grouped by VSMC phenotype and the expression of each marker in each VSMC subcluster. f. Enrichment analysis of the VSMC subclusters identified significant pathways of interest based on marker genes of the subclusters. The red dotted line represents a Benjamini-Hochberg FDR corrected threshold of 0.05. Avg Expr, Average log-normalized expression scaled to the maximum expression in any sub-cluster; Pct Nuclei Expr > 0, Percent of nuclei in a given sub-cluster that express the gene at non-zero levels.
Previous studies have identified VSMC groups specific to disease, named mod-VSMCs or fibromyocytes in models of cardiovascular disease including mouse models of atherosclerosis and aneurysm.34,35,59,77 With our clustering methodology, we were unable to identify a novel cellular class present within human thoracic aortic aneurysm tissue that was not present in control aortic tissue. We systematically compared our clusters to data from available studies and could not identify a matching population. (Supplemental Figure 10)The absence of a new infiltrative cell class reflected in this analysis is consistent with long-standing histological studies,69 and more recent single cell investigations in mouse aortic tissue.78,79 Similarly, the relative proportions of VSMC subclusters were similar between aneurysm tissue and control aortic tissue, with the exception of a drop in VSMC-S1 from healthy to aneurysmal tissue. (Figure 5b–c) This subcluster had the highest expression of contractile markers and showed pathway enrichment for “muscle contraction”. To approach the hypothesis that one of these subclusters may share identity with mod-VSMCs or ‘fibromyocytes’ found in previous studies, we performed additional subcluster analysis in the fibroblast population. (Figure 6) This analysis did not find a novel cell population within the fibroblast population that could account for a shift from VSMCs to fibroblasts, however there was disease specific diversity.
Figure 6. Fibroblast Subclustering.
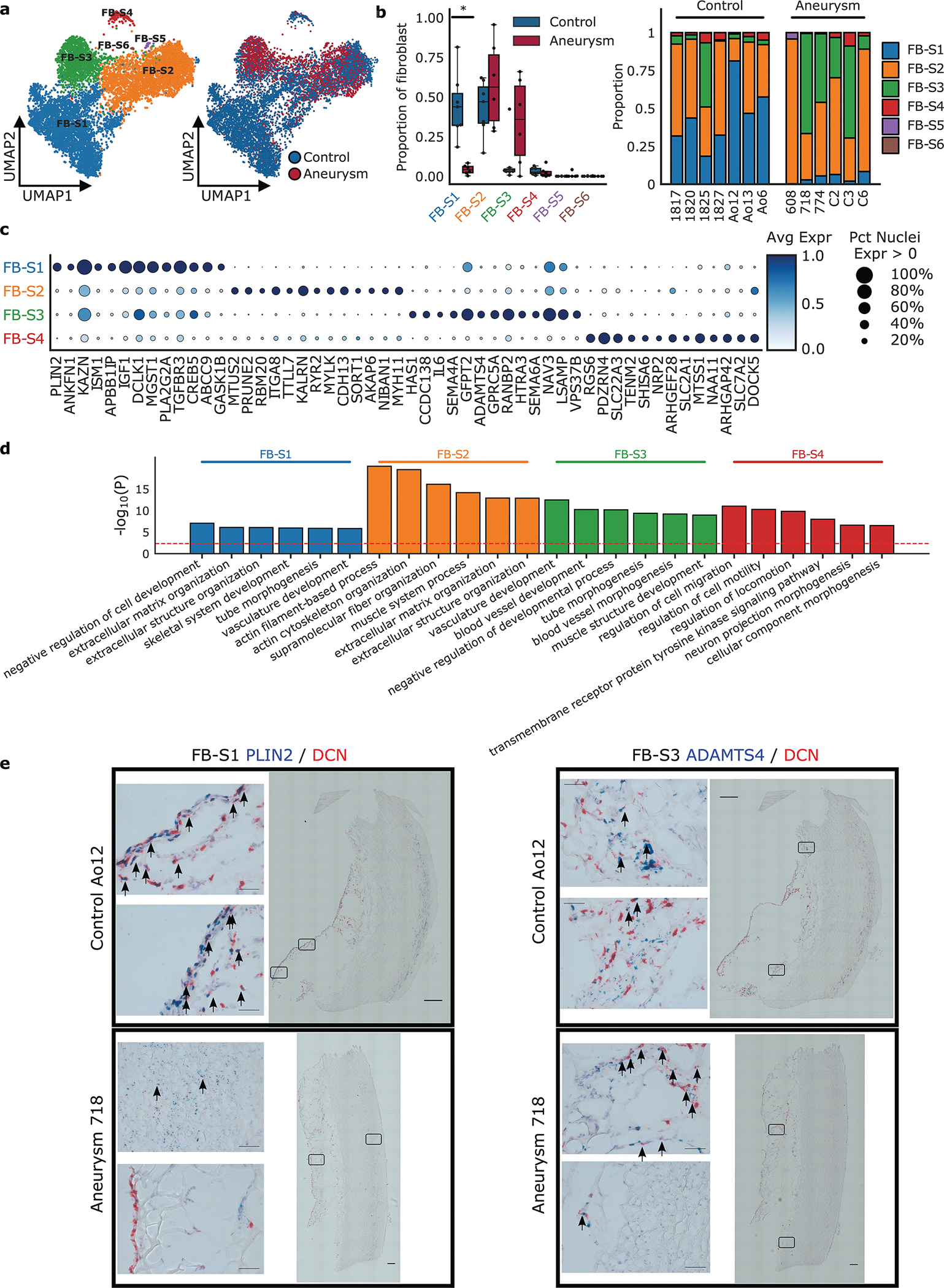
a. Nuclei clustered as fibroblast were selected to identify clusters enriched for marker genes of other global cell types. Results of subcluster analysis showing six fibroblast sub-clusters. b. The relative proportion of each subcluster between control and aneurysmal aortic tissue, and the relative proportion of each subcluster by sample. Statistically credible shifts in proportions as tested using scCODA (see Methods) are denoted with a *. Center line, median; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range. c. Selected marker genes that define each subcluster. d. Gene ontology pathway enrichment based on marker genes for each fibroblast sub-cluster. The red line indicates FDR < 0.05. Avg Expr, Average log-normalized expression scaled to the maximum expression in any sub-cluster; Pct Nuclei Expr > 0, Percent of nuclei in a given sub-cluster that express the gene at non-zero levels. e. RNA labeling of aortic tissue using RNAscope in situ hybridization. Control and aneurysm aortic tissue section labeled with DCN (red, global fibroblast marker), PLIN2 (blue, FB-S1 marker) and ADAMTS4 (blue, FB-S3 marker). Identified FB-S1 and FB-S3 populations appear blue and are indicated by arrows. Images show lack of FB-S1 in the aneurysm tissue, presence of FB-S3 in the aneurysm tissue, both in the adventitial layer. 40x single images = 200 um20x tiled images = 1000 um
Fibroblast population shift in disease
We identified 4 appreciably sized fibroblast sub-clusters with distinct gene expression profiles. (Figure 6a) FB-S1 was prevalent in control patients and largely absent in the aneurysmal aorta, a finding secondarily confirmed with RNAscope staining of FB-S1 marker gene PLIN2. (Figure 6b,e) The FB-S1 subcluster is enriched in genes that comprise “extracellular matrix and structure” pathways and shows enriched expression of disease relevant genes such as TGFBR3, a membrane bound inhibitor of TGF-β signaling, and ABCC9, an ATP-sensitive potassium channel associated with Cantu syndrome and aneurysm. (Figure 6c–d) This population is similar to previously described “quiescent” populations.
The FB-S2 subcluster was found in similar proportions between control and aneurysm patients (Figure 6b) and appears to show a myofibroblast-like phenotype as evidenced by enriched expression of genes involved in pathways of “actin filament-based process” and “actin cytoskeleton organization”. (Figure 6d) Additionally, this population showed elevated expression of several genes (AEBP1, FN1, and ACTA2) that have been previously used to characterize an activated fibroblast population in aortic tissue.33 (Supplemental Figure 11a, Supplemental Table 8) Given the dramatic shifts observed in the fibroblast sub-clusters between aneurysm and healthy aortas, additional sensitivity analysis was performed. The FB-S2 subcluster was removed during testing for differentially expressed genes between cases and controls due to the subcluster’s expression of significant classical VSMC markers and inability to exclude the possibility of a doublet population of combined fibroblasts and VSMCs. Removal of the subcluster identified significantly more differentially expressed genes and correlated with the main analysis. (Supplemental Figure 12)
The FB-S3 sub-cluster represents a distinct population of fibroblasts that is notably more prevalent in aneurysm samples. (Figure 6b) This population shows enriched expression of ATP1B3, PGAP1, NAMPT, and ADAMTS4 (Figure 6c, Supplemental Figure 11b, Supplemental Table 8); a pattern previously observed in a profibrotic cardiac fibroblast population.46 RNAscope showed increased staining of fibroblasts expressing of ADAMTS4 in the aortic adventitia of aneurysm samples, consistent with snRNASeq clustering (Figure 6e). Interestingly, this sub-cluster also expresses higher levels of KLF4 and MYC, both markers of a specialized multipotent adventitial cellular class derived from VSMCs.59,80,81(Supplemental Table 8).
Finally, we observed a fourth sub-cluster of fibroblasts, FB-S4, which represents an overall low proportion of nuclei in our samples. This population shows upregulation of genes involved in cell migration and motility (Figure 6d). These nuclei possess some neuron-like features including an elevated expression of genes involved in “neuron projection morphogenesis” including NRP2, MAP2, and MATN2 (Figure 6c–d, Supplemental Table 8).
Intersection of aortic transcriptome with medical traits
Recently, the quantification of aortic traits such as diameter and distensibility in large imaging datasets such as the UK Biobank has allowed for the identification of hundreds of novel genetic loci associated with these medically important traits.20,21 We choose to examine the relationship between transcriptional data identified through snRNA-Seq and genetic loci associated with medically-relevant traits including aortic diameter, aortic distensibility, and aortic dissection. First, to examine the effects of cellular groups on aortic traits, we performed cell-type specific LD-score regression. Cell-type profiles were generated by identifying cell-type specific genes and examining for enriched GWAS heritability near these genes. VSMC1, VSMC2, and pericyte groups were enriched at Bonferroni significance for ascending aortic diameter with these groups plus the additional involvement of fibroblasts for the descending aortic diameter. (Figure 7a)
Figure 7. Transcriptomic Intersection with Aortic Genetic Traits.
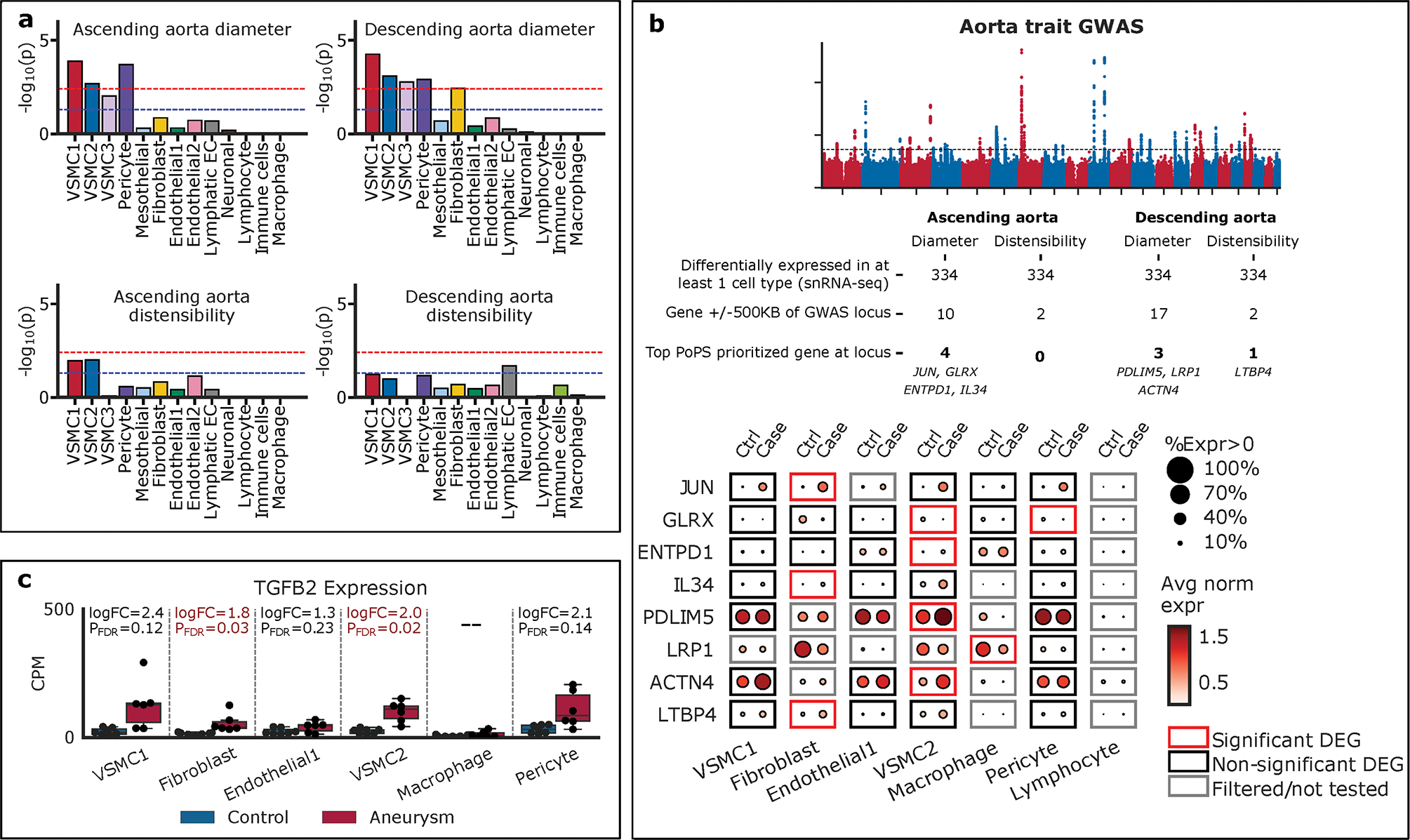
a. Cell-type specific LD-score regression. The blue line represents nominal significance (P<0.05) and the red line represents Bonferroni significance (P<0.05/13=0.0038). VSMC1, VSMC2, and pericyte groups were enriched at Bonferroni significance for ascending and descending aortic diameter. b. 334 differentially expressed genes were intersected with GWAS of ascending and descending aortic diameter and distensibility and eight genes were prioritized. The identified genes were displayed by their expression in each cell-type and by control compared to aneurysm tissue. c. Examination of TGFB2 showed differential expression in VSMC1, VSMC2, and fibroblast cellular groups. PFDR, Benjamini-Hochberg adjusted P-value.
Next we sought to identify genes that may be associated with genetic loci identified from GWAS of aortic traits. We identified all genes in the dataset differentially expressed between aneurysm versus control in at least 1 cell type, which resulted in 334 genes. We then reduced this list to protein coding genes falling within plus or minus 500 kilobases of genetic loci for ascending and descending aortic diameter and aortic distensibility.21 (Figure 7b) Finally, we limited genes by only considering those with the highest polygenic priority score (PoPs),42 at a given locus, an unbiased strategy integrating gene expression, pathway data, and predicted protein-protein interaction data to prioritize putative causal GWAS genes. Overall, eight DEGs in aortic cell types were prioritized at aortic GWAS loci. (Figure 7b, Supplemental Figure 13) Ascending aortic diameter was associated with the genes JUN, GLRX, ENTPD1, and IL34. In the descending aorta, diameter was associated with the genes PDLIM5, LRP1, and ACTN4, while descending aortic distensibility was associated with LTBP4. Individually identified genes were most commonly differentially expressed in the VSMC2 cell group, consistent with aggregate heritability. (Figure 7)
Finally, we investigated the expression profiles of genes associated with Mendelian cases of aortic aneurysm and dissection.82 Of the 23 genes analyzed (COL3A1, FBN1, SMAD3, TGFB2, TGFBR1, TGFBR2, ACTA2, MYH11, MYLK, LOX, PRKG1, EFEMP2, ELN, FBN2, FLNA, NOTCH1, SLC2A10, SMAD4, SKI, CBS, COL4A5, PKD1, PKD2), only TGFB2 showed significant differential expression in VSMC1, VSMC2, and fibroblast cellular groups (Figure 7c).
DISCUSSION
We have developed a comprehensive map of the transcriptional landscape of aneurysmal and healthy human ascending aorta comprising snRNA-seq of 71,689 nuclei. This study provides several novel advances that enhance our understanding of cardiovascular biology and demonstrates the feasibility of using banked, frozen human aortic tissue for high-quality single nuclei analysis. With the largest collection of single nuclear transcriptomes from human aortas to date, we defined 11 major conventional cell types and 7 distinct subclusters of VSMCs. We identified dozens of DEGs with high confidence between healthy and aneurysmal aortas in multiple cell classes. Finally, we linked specific cell types to common and rare genetic variants underlying cardiovascular diseases.
In contrast to previous scRNA-seq analyses of human aortas, this study more accurately encompasses the known cellular composition of the human aorta with ~60–70% of the cellular mass representing smooth muscle populations.33,35,47,76 The difference likely represents the technical advantages of nuclei isolation from frozen tissue samples without regard to cell viability, limiting dissociation bias,83 and bypass of flow cytometry sorting which may confer additional bias by cell size and viability.84–86 Unlike prior single cell33,65 and histologic studies87,88 of human aortic tissue, we did not identify a significant portion of immune cells. Our data provides support for non-immune mediated aneurysmal generation in sporadic TAA.8,44 Furthermore, classic molecular studies were limited in the number of simultaneous probes available for staining and interpretation, and single markers such as CD68, were frequently used to identify immune cell populations within aortic tissue.87,88 More recent studies23,59,89 and our data, along with computational analysis enabling the differential expression analysis of multiple genes, support that some previously identified immune cell populations may have been VSMCs of a different cluster or subcluster.
Interestingly, the only compositional change between the major cellular groups was an increase in the proportion of VSMC1 cluster in aneurysm samples versus controls. This change was modest and may reflect aortic wall thickening in aneurysm or the availability of nuclei to be released from disease versus healthy tissue. Overall, the results are consistent with models of ascending aneurysm development that emphasize altered VSMC behavior rather than invasive cell types or VSMC death or loss.69,90,91 In contrast to recent reports from animal and human disease, there were no infiltrative or novel cellular subclasses seen in this analysis of control and aneurysm tissue. This may relate to the nature of sporadic aortic aneurysm. Unlike aggressive forms of hereditary TAA such as seen in Marfan syndrome and related aortopathies, sporadic aneurysm growth is slow and indolent. One patient sample in particular helps illustrate this biology. Patient sample 774 (Supplemental Table 1) contributed the bulk of the nuclei seen in the main VSMC cluster (VSMC3) as well as subcluster VSMC-S6 and -S7 (Supplemental Figure 6 & Figure 5c), which were the most immature appearing VSMCs in the main clusters. VSMC3 expressed the lowest level of VSMC identity markers such as ACTA2 and MYH11, and showed higher expression of extracellular matrix genes such as SERPINE1 and FN1. (Figure 3a) These are characteristics of typical classically modulated “synthetic” VSMCs.59,92,93 After surgery, tissue pathology determined the aneurysm represented “resolved aortitis” although the immune cellular component was not elevated. (Figure 2c, Supplemental Figure 6) Therefore, while a highly modulated VSMC population may be seen in some aneurysms, no other aneurysm sample had more than ~1% VSMC3 cells, making them an unlikely “culprit” cell class. Future studies in larger numbers of genetically-triggered aortic disease samples may determine if this type of cellular model is appropriate for aggressive or genetically triggered aortic disease, as previously demonstrated in animal models of Marfan syndrome.35 Despite a well powered sample of over 70K nuclei, we were unable to identify a subcluster of VSMCs that showed characteristics similar to the previously identified novel subpopulations unique to diseased tissues–variably labeled fibromyocytes or modulated VSMCs (modVSMCs).33–35 (Supplemental Figure 10) Instead, shifts in gene expression were observed amongst subpopulations of VSMCs of aneurysm tissue compared to control tissue (Figure 4b). Furthermore, VSMC subclusters did not group into classical VSMC phenotypes.59 Instead, most subclusters expressed a range of contractile and fibroblast marker genes. (Figure 5e, Supplemental Table 6) The absence of subclusters expressing mesenchymal, macrophage, or osteogenic markers may be due to the relative decrease in the classic atherosclerotic etiology of ascending aortic aneurysm compared to descending or infrarenal aortic aneurysms.34,94 Taken together, these findings emphasize the distinct nature of sporadic aortic aneurysm biology.
Exploration of the fibroblast subclusters revealed some of the most significant phenotypic diversity between aneurysm and control tissue, not previously appreciated. The significant disappearance in the FB-S1 population and substantial increase in FB-S3 (activated fibroblast) populations in aneurysm tissue may implicate these fibroblast subpopulations as an area of interest in the etiology or progression of aortic aneurysm pathophysiology. In the aorta, fibroblasts primarily reside in the aortic adventitia, the outermost layer of the aorta, responsible for aortic tensile strength and resistance to longitudinal strain.95,96 Aortic adventitial remodeling is known to accompany pathologic arterial disorders such as hypertension, aging, and aneurysm.97,98 In experimental models, increased expression of type 1 collagens and other matrix components typically occurs through an expansion of myofibroblast populations and macrophages, and can at least partially be inhibited through angiotensin type 2 receptor inhibition.99,100 However, in our samples the expansion of a myofibroblast population was mild and instead an increase in the FB-S3 subcluster was seen. This cluster is most similar to previously described KLF4-expressing “activated” fibroblast populations.81 However, the relationship of KLF4 expression in various cell populations and disease states remains an active area of exploration, with opposing roles depending on cell type and disease process.20,89,101–103 FB-S3 also showed strong up-regulation of IL6, HAS1, and HAS2 (Figure 6c, Supplemental Table 8). IL-6 has been shown to induce HAS1 and HAS2 in cardiac fibroblasts which contribute to a hyaluronan-rich matrix post-acute myocardial infarction,104 and a snp (rs10111085) near the HAS2 has been implicated in human aortic phenotypes including dimension and distensibility.20,21 This suggests that FB-S3 may contribute to pathogenic changes in the matrix of aortic aneurysm patients. Interruption of adventitial remodeling has been proposed as a strategy to limit arterial stiffening during hypertension, and further work may reveal whether this represents a viable strategy to control aneurysm growth or as a biomarker to monitor progression.
Multiple DEGs were identified between cellular groups with the VSMC2 cellular class having the largest number of unique genes at 131. Included in these are multiple genes known to play prominent roles in vascular biology such as the epigenetic regulator HDAC9, known to modulate VSMC behavior by altering the expression of contractile protein elements such as those encoded by ACTA2, MYH11, and TAGLN.15 Additionally, multiple modulators of the TGF-β signaling pathway were identified including LMO7 which has been shown to negatively regulate TGF-β expression in VSMCs,67 and LTBP1 which directly binds and sequesters all three TGF-β isoforms.105 There was also increased expression of the aneurysm associated gene TGFB2, which encodes TGF-β isoform 2.56 Intriguingly, several DEGs are known to associate with cardiomyopathy, including RBM20,106 CALM1,107 and EYA4.108 This likely reflects the ancestral biological connection between cardiac and smooth muscle contraction, and supports that disturbances in contractile function cause disease in either cell type.109
Finally, we used our snRNA-Seq data to prioritize individual genes at GWAS loci associated with human aortic traits such as diameter and distensibility. A closer look at the 8 genes implicated from GWAS on aortic diameter and from single cell analysis of aortic and control tissue suggests specific cell compartments for further investigation. (Figure 7b, Supplemental Figure 13) JUN has been implicated with increased ascending aortic diameter20 and is significantly differentially expressed in the fibroblast compartment, with a similar magnitude and directionality in VSMC2 compartment, of aortic aneurysms compared to normal aortic tissue. JUN, a classic immediate early gene, has been described to regulate gene expression through epigenetic modifications, with specific impact on VSMCs in coronary artery disease and VSMC growth in arterial injury.110,111 Increased expression of IL34 was also seen in the fibroblast cell population in aneurysm tissue compared to controls. IL34 has been extensively implicated in acute and chronic inflammatory conditions, usually associated with active expansion of immune cell populations. Our data suggests that there is increased IL34 expression without a proportional increase in immune cells.112 We were also able to provide support for the role of LTBP4 in descending aortic distensibility21. Given the role of LTBP4 in matrix homeostasis and elastogenesis,113 our data provide support for further investigation in fibroblasts, specifically, where it is significantly differentially expressed.
Within the VSMC2 cell population, four genes were significantly differentially expressed and implicated in GWAS. GLRX has been associated with VSMC proliferation, along with Serpine1, which also shows increased expression in the VSMC2 compartment in aneurysm tissue, and VSMC-S6, in particular.114 ENTPD1 is also significantly differentially expressed in VSMC2 cells in aneurysm tissue. It is known to regulate VSMC migration and primes proliferation.115,116 PDLIM5, a cytoskeletal binding protein that regulates cell proliferation and cell fate also has increased expression in aneurysm tissue in the VSMC2 cell population. Over expression of PDLIM5 inhibits the TGF-β/Smad signal pathway, which has been implicated in the development of cardiovascular disease.117 Finally, ACTN4, a cytoskeletal α-actinin involved in cell motility and contractility,118,119 with known change in function of VSMCs with ACTN4 upregulation,120 also had increased expression in aneurysm tissue, specifically the VSMC2 population. Although we did not identify a significant portion of immune cells in aortic tissue.33,65 We did, however, identify and prioritize LRP1 as a gene of interest from GWAS. It is differentially expressed in the macrophage cell population, with decreased expression in aneurysm tissue. This pattern is consistent with prior literature demonstrating that LRP1 knockout mice undergo accelerated plaque formation, exhibit aortic dilation and influence vessel integrity.121–124
LIMITATIONS
Our study has several limitations. This study presents the largest single cell data on human normal and aneurysm aorta to date. Despite this, the overall number of participants is modest, reflecting economic burdens of single cell studies and the importance of collecting surgical tissue. One participant with aortic aneurysm (774) had quiescent inflammatory aortitis on further chart review. The nuclei from the tissue had corresponding shifts in the VSMC population and were included to provide further support that even in the setting of potential inflammatory origins of aortopathy, the predominant cell population remains VSMCs. Exclusion of this patient sample did not significantly change overall findings or interpretation. (Supplemental Figure 14) The study was underpowered to compare differences in expression by sex; however, principal component analysis did suggest sex-based differences on a global scale, which supports existing literature on sex differences in aortic aneurysm.125 The inclusion of both sex in the transcriptomic may introduce some bias in interpretation, however, given the limited data from human aortic tissue, both sexes were included. Furthermore, all participants of the study except for one were non-Hispanic White, potentially limiting interpretations to other races and ethnicities. The public availability of the transcriptomic data, however, may allow for future analysis of potential biases by sex or race. Finally, nuclear transcriptomes are a small percentage of total cellular mRNA. Despite prior studies showing concordance of whole cell and nuclear transcriptomes,83,126 further studies investigating the difference between the two methods and the transcriptomes are needed.
CONCLUSION
Using single nuclear RNA sequencing we describe the cellular diversity of healthy and aneurysmal human ascending aorta with emphasis on the vascular smooth muscle cell population, the described culprit cell class in thoracic aortic disease. Typical, nonsyndromic ascending aortic aneurysm appears to be a disease of abnormal behavior of known cell classes rather than infiltrating or novel cellular forms. Single nuclear RNA sequencing of aortic tissue can be used to prioritize genes at aortic-specific quantitative trait loci.
Supplementary Material
Highlights:
This study provides the largest collection of single nuclear transcriptomes from healthy and sporadic aneurysmal human aortas to date
Contrary to prior single cell studies of human aorta, no predominant immune cell population or novel infiltrative cell population was identified in aneurysm tissue
Amongst 334 differentially expressed genes found between healthy and diseased states, 8 were prioritized by aortic diameter and distensibility GWAS loci: ENTPD1, PDLIM5, ACTN4, and GLRX in vascular smooth muscle cells, JUN, LTBP4 and IL34 in fibroblasts, and LRP1 in macrophages
Findings emphasize the distinct nature of sporadic aortic aneurysm biology and identify new genetic targets for future investigation
ACKNOWLEDGEMENTS
We would like to thank the patients and families who agreed to participate in this research.
DISCLOSURES
A-D.A. and C.K. are employees of Bayer US LLC (a subsidiary of Bayer AG) and may own stock in Bayer AG. P.T.E has received sponsored research support from Bayer AG and IBM Health, and he has served on advisory boards or consulted for Bayer AG, MyoKardia and Novartis. K.B.M. has research grant funding from Sanofi-Aventis, USA and has also served on advisory boards for MyoKardia and Pfizer. M.E.L. has received salary support from Bayer AG.
SOURCES OF FUNDNG
The Precision Cardiology Laboratory is a joint effort between the Broad Institute and Bayer AG. This work was supported by grants from the National Institutes of Health to P.T.E. (1RO1HL092577), K.B.M. (1R01HL105993). P.T.E is also supported by a grant from the American Heart Association Strategically Focused Research Networks (18SFRN34110082) and by the European Union MAESTRIA project (965286) M.E.L. is supported by the Fredman Fellowship for Aortic Disease, the Toomey Fund for Aortic Dissection Research, and NIH (1RO1HL1301). E.L.C. and C.L.L were supported by NIH (T32HL007208) and C.L.L. by NIH (K01HL164687). The rapid autopsy effort was funded by the Susan Eid Tumor Heterogeneity Initiative.
Footnotes
REFERENCES
- 1.Bossone E, Eagle KA. Epidemiology and management of aortic disease: aortic aneurysms and acute aortic syndromes. Nat Rev Cardiol. 2021;18(5):331–348. doi: 10.1038/s41569-020-00472-6 [DOI] [PubMed] [Google Scholar]
- 2.Vilacosta I, San Román JA, di Bartolomeo R, Eagle K, Estrera AL, Ferrera C, Kaji S, Nienaber CA, Riambau V, Schäfers HJ, Serrano FJ, Song JK, Maroto L. Acute Aortic Syndrome Revisited: JACC State-of-the-Art Review. J Am Coll Cardiol. 2021;78(21):2106–2125. doi: 10.1016/j.jacc.2021.09.022 [DOI] [PubMed] [Google Scholar]
- 3.Erbel R, Aboyans V, Boileau C, Bossone E, Bartolomeo RD, Eggebrecht H, Evangelista A, Falk V, Frank H, Gaemperli O, Grabenwöger M, Haverich A, Iung B, Manolis AJ, Meijboom F, Nienaber CA, Roffi M, Rousseau H, Sechtem U, Sirnes PA, Allmen RS von, Vrints CJM, ESC Committee for Practice Guidelines. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: Document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(41):2873–2926. doi: 10.1093/eurheartj/ehu281 [DOI] [PubMed] [Google Scholar]
- 4.Kim JB, Spotnitz M, Lindsay ME, MacGillivray TE, Isselbacher EM, Sundt TM. Risk of Aortic Dissection in the Moderately Dilated Ascending Aorta. J Am Coll Cardiol. 2016;68(11):1209–1219. doi: 10.1016/j.jacc.2016.06.025 [DOI] [PubMed] [Google Scholar]
- 5.Jana S, Hu M, Shen M, Kassiri Z. Extracellular matrix, regional heterogeneity of the aorta, and aortic aneurysm. Exp Mol Med. 2019;51(12):160. doi: 10.1038/s12276-019-0286-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin CJ, Lin CY, Stitziel NO. Genetics of the Extracellular Matrix in Aortic Aneurysmal Diseases. Matrix Biol. 2018;71–72:128–143. doi: 10.1016/j.matbio.2018.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nienaber CA, Clough RE, Sakalihasan N, Suzuki T, Gibbs R, Mussa F, Jenkins MP, Thompson MM, Evangelista A, Yeh JSM, Cheshire N, Rosendahl U, Pepper J. Aortic dissection. Nat Rev Dis Primers. 2016;2(1):16053. doi: 10.1038/nrdp.2016.53 [DOI] [PubMed] [Google Scholar]
- 8.Amemiya K, Mousseaux E, Ishibashi-Ueda H, Achouh P, Ochiai M, Bruneval P. Impact of histopathological changes in ascending aortic diseases. Int J Cardiol. 2020;311:91–96. doi: 10.1016/j.ijcard.2020.04.011 [DOI] [PubMed] [Google Scholar]
- 9.Pape LA, Tsai TT, Isselbacher EM, Oh JK, O’Gara PT, Evangelista A, Fattori R, Meinhardt G, Trimarchi S, Bossone E, Suzuki T, Cooper JV, Froehlich JB, Nienaber CA, Eagle KA, International Registry of Acute Aortic Dissection I. Aortic diameter >or = 5.5 cm is not a good predictor of type A aortic dissection: observations from the International Registry of Acute Aortic Dissection (IRAD). Circulation. 2007;116(10):1120–1127. doi: 10.1161/CIRCULATIONAHA.107.702720 [DOI] [PubMed] [Google Scholar]
- 10.Quintana RA, Taylor WR. Cellular Mechanisms of Aortic Aneurysm Formation. Circ Res. 2019;124(4):607–618. doi: 10.1161/CIRCRESAHA.118.313187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michel JB, Jondeau G, Milewicz DM. From genetics to response to injury: vascular smooth muscle cells in aneurysms and dissections of the ascending aorta. Cardiovasc Res. 2018;114(4):578–589. doi: 10.1093/cvr/cvy006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pannu H, Tran-Fadulu V, Papke CL, Scherer S, Liu Y, Presley C, Guo D, Estrera AL, Safi HJ, Brasier AR, Vick GW, Marian AJ, Raman CS, Buja LM, Milewicz DM. MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum Mol Genet. 2007;16(20):2453–2462. doi: 10.1093/hmg/ddm201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, Amor D, Ades L, McConnell V, Willoughby CE, Abuelo D, Willing M, Lewis RA, Kim DH, Scherer S, Tung PP, Ahn C, Buja LM, Raman CS, Shete SS, Milewicz DM. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39(12):1488–1493. doi: 10.1038/ng.2007.6 [DOI] [PubMed] [Google Scholar]
- 14.Bunton TE, Biery NJ, Myers L, Gayraud B, Ramirez F, Dietz HC. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ Res. 2001;88(1):37–43. doi: 10.1161/01.res.88.1.37 [DOI] [PubMed] [Google Scholar]
- 15.Lino Cardenas CL, Kessinger CW, Cheng Y, MacDonald C, MacGillivray T, Ghoshhajra B, Huleihel L, Nuri S, Yeri AS, Jaffer FA, Kaminski N, Ellinor P, Weintraub NL, Malhotra R, Isselbacher EM, Lindsay ME. An HDAC9-MALAT1-BRG1 complex mediates smooth muscle dysfunction in thoracic aortic aneurysm. Nat Commun. 2018;9(1):1009. doi: 10.1038/s41467-018-03394-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clément M, Chappell J, Raffort J, Lareyre F, Vandestienne M, Taylor AL, Finigan A, Harrison J, Bennett MR, Bruneval P, Taleb S, Jørgensen HF, Mallat Z. Vascular Smooth Muscle Cell Plasticity and Autophagy in Dissecting Aortic Aneurysms. Arterioscler Thromb Vasc Biol. 2019;39(6):1149–1159. doi: 10.1161/ATVBAHA.118.311727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen YH, LeMaire SA. Molecular pathogenesis of genetic and sporadic aortic aneurysms and dissections. Curr Probl Surg. 2017;54(3):95–155. doi: 10.1067/j.cpsurg.2017.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quake SR. The cell as a bag of RNA. Trends in Genetics. 2021;37(12):1064–1068. doi: 10.1016/j.tig.2021.08.003 [DOI] [PubMed] [Google Scholar]
- 19.Paik DT, Cho S, Tian L, Chang HY, Wu JC. Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nat Rev Cardiol. 2020;17(8):457–473. doi: 10.1038/s41569-020-0359-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pirruccello JP, Chaffin MD, Chou EL, Fleming SJ, Lin H, Nekoui M, Khurshid S, Friedman SF, Bick AG, Arduini A, Weng LC, Choi SH, Akkad AD, Batra P, Tucker NR, Hall AW, Roselli C, Benjamin EJ, Vellarikkal SK, Gupta RM, Stegmann CM, Juric D, Stone JR, Vasan RS, Ho JE, Hoffmann U, Lubitz SA, Philippakis AA, Lindsay ME, Ellinor PT. Deep learning enables genetic analysis of the human thoracic aorta. Nat Genet. 2022;54(1):40–51. doi: 10.1038/s41588-021-00962-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pirruccello JP, Choi SH, Chaffin MD, Nekoui M, Chou EL, Jurgens SJ, Friedman SF, Juric D, Stone JR, Batra P, Ng K, Philippakis AA, Lindsay ME, Ellinor PT. The Genetic Determinants of Aortic Distension. Published online October 25, 2021:2021.10.16.21265089. doi: 10.1101/2021.10.16.21265089 [DOI] [Google Scholar]
- 22.Nekoui M, Pirruccello JP, Achille PD, Choi SH, Friedman SN, Nauffal V, Ng K, Batra P, Ho JE, Philippakis AA, Lubitz SA, Lindsay ME, Ellinor PT. Using Machine Learning to Elucidate the Spatial and Genetic Complexity of the Ascending Aorta; 2021:2021.11.01.21265701. doi: 10.1101/2021.11.01.21265701 [DOI] [Google Scholar]
- 23.Chou EL, Lino Cardenas CL, Chaffin M, Arduini AD, Juric D, Stone JR, LaMuraglia GM, Eagleton MJ, Conrad MF, Isselbacher EM, Ellinor PT, Lindsay ME. Vascular smooth muscle cell phenotype switching in carotid atherosclerosis. JVS Vasc Sci. 2022;3:41–47. doi: 10.1016/j.jvssci.2021.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;17(1):10–12. doi: 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- 25.Fleming SJ, Marioni JC, Babadi M. CellBender remove-background: a deep generative model for unsupervised removal of background noise from scRNA-seq datasets. Published online October 3, 2019:791699. doi: 10.1101/791699 [DOI] [Google Scholar]
- 26.Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19(1):1–5. doi: 10.1186/s13059-017-1382-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177(7):1888–1902.e21. doi: 10.1016/j.cell.2019.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh P ru, Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16(12):1289–1296. doi: 10.1038/s41592-019-0619-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolock SL, Lopez R, Klein AM. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. cels. 2019;8(4):281–291.e9. doi: 10.1016/j.cels.2018.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y, Lun ATL, Smyth GK. From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. Published online August 2, 2016. doi: 10.12688/f1000research.8987.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):1–21. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Law CW, Chen Y, Shi W, Smyth GK. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15(2):1–17. doi: 10.1186/gb-2014-15-2-r29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y, Ren P, Dawson A, Vasquez HG, Ageedi W, Zhang C, Luo W, Chen R, Li Y, Kim S, Lu HS, Cassis LA, Coselli JS, Daugherty A, Shen YH, LeMaire SA. Single-Cell Transcriptome Analysis Reveals Dynamic Cell Populations and Differential Gene Expression Patterns in Control and Aneurysmal Human Aortic Tissue. Circulation. 2020;142(14):1374–1388. doi: 10.1161/CIRCULATIONAHA.120.046528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wirka RC, Wagh D, Paik DT, Pjanic M, Nguyen T, Miller CL, Kundu R, Nagao M, Coller J, Koyano TK, Fong R, Woo YJ, Liu B, Montgomery SB, Wu JC, Zhu K, Chang R, Alamprese M, Tallquist MD, Kim JB, Quertermous T. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med. 2019;25(8):1280–1289. doi: 10.1038/s41591-019-0512-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pedroza AJ, Tashima Y, Shad R, Cheng P, Wirka R, Churovich S, Nakamura K, Yokoyama N, Cui JZ, Iosef C, Hiesinger W, Quertermous T, Fischbein MP. Single-Cell Transcriptomic Profiling of Vascular Smooth Muscle Cell Phenotype Modulation in Marfan Syndrome Aortic Aneurysm. Arterioscler Thromb Vasc Biol. 2020;40(9):2195–2211. doi: 10.1161/ATVBAHA.120.314670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc. 2009;4(8):1184–1191. doi: 10.1038/nprot.2009.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Büttner M, Ostner J, Müller CL, Theis FJ, Schubert B. scCODA is a Bayesian model for compositional single-cell data analysis. Nat Commun. 2021;12(1):6876. doi: 10.1038/s41467-021-27150-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23(2):257–258. doi: 10.1093/bioinformatics/btl567 [DOI] [PubMed] [Google Scholar]
- 39.Buettner F, Natarajan KN, Casale FP, Proserpio V, Scialdone A, Theis FJ, Teichmann SA, Marioni JC, Stegle O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat Biotechnol. 2015;33(2):155–160. doi: 10.1038/nbt.3102 [DOI] [PubMed] [Google Scholar]
- 40.Finucane HK, Reshef YA, Anttila V, Slowikowski K, Gusev A, Byrnes A, Gazal S, Loh PR, Lareau C, Shoresh N, Genovese G, Saunders A, Macosko E, Pollack S, Consortium TB, Perry JRB, Buenrostro JD, Bernstein BE, Raychaudhuri S, McCarroll S, Neale BM, Price AL. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nature genetics. 2018;50(4):621. doi: 10.1038/s41588-018-0081-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh PR, Anttila V, Xu H, Zang C, Farh K, Ripke S, Day FR, Purcell S, Stahl E, Lindstrom S, Perry JRB, Okada Y, Raychaudhuri S, Daly MJ, Patterson N, Neale BM, Price AL. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet. 2015;47(11):1228–1235. doi: 10.1038/ng.3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weeks E, Ulirsch J, Cheng N, Trippe B, Fine R, Miao J, Patwardhan T, Kanai M, Nasser J, Fulco C, Tashman K, Aguet F, Li T, Ordovas-Montanes J, Smillie C, Biton M, Shalek A, Ananthakrishnan A, Xavier R, Regev A, Gupta R, Lage K, Ardlie K, Hirschhorn J, Lander E, Engreitz J, Finucane H. Leveraging polygenic enrichments of gene features to predict genes underlying complex traits and disease. Published online 2020. doi: 10.1101/2020.09.08.20190561 [DOI] [PMC free article] [PubMed]
- 43.Leone O, Pacini D, Foà A, Corsini A, Agostini V, Corti B, Di Marco L, Leone A, Lorenzini M, Reggiani LB, Di Bartolomeo R, Rapezzi C. Redefining the histopathologic profile of acute aortic syndromes: Clinical and prognostic implications. J Thorac Cardiovasc Surg. 2018;156(5):1776–1785.e6. doi: 10.1016/j.jtcvs.2018.04.086 [DOI] [PubMed] [Google Scholar]
- 44.Halushka MK, Angelini A, Bartoloni G, Basso C, Batoroeva L, Bruneval P, Buja LM, Butany J, d’Amati G, Fallon JT, Gallagher PJ, Gittenberger-de Groot AC, Gouveia RH, Kholova I, Kelly KL, Leone O, Litovsky SH, Maleszewski JJ, Miller DV, Mitchell RN, Preston SD, Pucci A, Radio SJ, Rodriguez ER, Sheppard MN, Stone JR, Suvarna SK, Tan CD, Thiene G, Veinot JP, van der Wal AC. Consensus statement on surgical pathology of the aorta from the Society for Cardiovascular Pathology and the Association For European Cardiovascular Pathology: II. Noninflammatory degenerative diseases - nomenclature and diagnostic criteria. Cardiovasc Pathol. 2016;25(3):247–257. doi: 10.1016/j.carpath.2016.03.002 [DOI] [PubMed] [Google Scholar]
- 45.Liu Z, Chang AN, Grinnell F, Trybus KM, Milewicz DM, Stull JT, Kamm KE. Vascular disease-causing mutation, smooth muscle α-actin R258C, dominantly suppresses functions of α-actin in human patient fibroblasts. PNAS. 2017;114(28):E5569–E5578. doi: 10.1073/pnas.1703506114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tucker NR, Chaffin M, Fleming SJ, Hall AW, Parsons VA, Bedi KC Jr, Akkad AD, Herndon CN, Arduini A, Papangeli I, Roselli C, Aguet F, Choi SH, Ardlie KG, Babadi M, Margulies KB, Stegmann CM, Ellinor PT. Transcriptional and Cellular Diversity of the Human Heart. Circulation. 2020;142(5):466–482. doi: 10.1161/CIRCULATIONAHA.119.045401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davis FM, Tsoi LC, Melvin WJ, denDekker A, Wasikowski R, Joshi AD, Wolf S, Obi AT, Billi AC, Xing X, Audu C, Moore BB, Kunkel SL, Daugherty A, Lu HS, Gudjonsson JE, Gallagher KA. Inhibition of macrophage histone demethylase JMJD3 protects against abdominal aortic aneurysms. J Exp Med. 2021;218(6). doi: 10.1084/jem.20201839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Silvestro M, Hadi T, Cayne NS, Maldonado TS, Gelb BE, Jacobowitz GR, Ramkhelawon B. AAA 25. Cell-Specific Profiling of Transcriptional Landscape in Human Abdominal Aortic Aneurysm by Single-Cell RNA Sequencing. Journal of Vascular Surgery. 2019;70(5):e142–e143. doi: 10.1016/j.jvs.2019.08.081 [DOI] [Google Scholar]
- 49.Squair JW, Gautier M, Kathe C, Anderson MA, James ND, Hutson TH, Hudelle R, Qaiser T, Matson KJE, Barraud Q, Levine AJ, La Manno G, Skinnider MA, Courtine G. Confronting false discoveries in single-cell differential expression. Nat Commun. 2021;12(1):5692. doi: 10.1038/s41467-021-25960-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zimmerman KD, Espeland MA, Langefeld CD. A practical solution to pseudoreplication bias in single-cell studies. Nat Commun. 2021;12(1):738. doi: 10.1038/s41467-021-21038-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lino Cardenas CL, Kessinger CW, MacDonald C, Jassar AS, Isselbacher EM, Jaffer FA, Lindsay ME. Inhibition of the methyltranferase EZH2 improves aortic performance in experimental thoracic aortic aneurysm. JCI Insight. 2018;3(5). doi: 10.1172/jci.insight.97493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saddic L, Orosco A, Guo D, Milewicz DM, Troxlair D, Heide RV, Herrington D, Wang Y, Azizzadeh A, Parker SJ. Proteomic analysis of descending thoracic aorta identifies unique and universal signatures of aneurysm and dissection. JVS-Vascular Science. 2022;0(0). doi: 10.1016/j.jvssci.2022.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kjellqvist S, Maleki S, Olsson T, Chwastyniak M, Branca RMM, Lehtiö J, Pinet F, Franco-Cereceda A, Eriksson P. A Combined Proteomic and Transcriptomic Approach Shows Diverging Molecular Mechanisms in Thoracic Aortic Aneurysm Development in Patients with Tricuspid- And Bicuspid Aortic Valve. Molecular & Cellular Proteomics : MCP. 2013;12(2):407. doi: 10.1074/mcp.M112.021873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perisic Matic L, Rykaczewska U, Razuvaev A, Sabater-Lleal M, Lengquist M, Miller CL, Ericsson I, Röhl S, Kronqvist M, Aldi S, Magné J, Paloschi V, Vesterlund M, Li Y, Jin H, Diez MG, Roy J, Baldassarre D, Veglia F, Humphries SE, de Faire U, Tremoli E, Odeberg J, Vukojević V, Lehtiö J, Maegdefessel L, Ehrenborg E, Paulsson-Berne G, Hansson GK, Lindeman JHN, Eriksson P, Quertermous T, Hamsten A, Hedin U. Phenotypic Modulation of Smooth Muscle Cells in Atherosclerosis Is Associated With Downregulation of LMOD1, SYNPO2, PDLIM7, PLN, and SYNM. Arterioscler Thromb Vasc Biol. 2016;36(9):1947–1961. doi: 10.1161/ATVBAHA.116.307893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang L, Guo DC, Cao J, Gong L, Kamm KE, Regalado E, Li L, Shete S, He WQ, Zhu MS, Offermanns S, Gilchrist D, Elefteriades J, Stull JT, Milewicz DM. Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet. 2010;87(5):701–707. doi: 10.1016/j.ajhg.2010.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boileau C, Guo DC, Hanna N, Regalado ES, Detaint D, Gong L, Varret M, Prakash SK, Li AH, d’Indy H, Braverman AC, Grandchamp B, Kwartler CS, Gouya L, Santos-Cortez RL, Abifadel M, Leal SM, Muti C, Shendure J, Gross MS, Rieder MJ, Vahanian A, Nickerson DA, Michel JB, National Heart L, Blood Institute Go Exome Sequencing P, Jondeau G, Milewicz DM. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet. 2012;44(8):916–921. doi: 10.1038/ng.2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lindsay ME, Schepers D, Bolar NA, Doyle JJ, Gallo E, Fert-Bober J, Kempers MJ, Fishman EK, Chen Y, Myers L, Bjeda D, Oswald G, Elias AF, Levy HP, Anderlid BM, Yang MH, Bongers EM, Timmermans J, Braverman AC, Canham N, Mortier GR, Brunner HG, Byers PH, Van Eyk J, Van Laer L, Dietz HC, Loeys BL. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet. 2012;44(8):922–927. doi: 10.1038/ng.2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.MacFarlane EG, Haupt J, Dietz HC, Shore EM. TGF-β Family Signaling in Connective Tissue and Skeletal Diseases. Cold Spring Harb Perspect Biol. 2017;9(11):a022269. doi: 10.1101/cshperspect.a022269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yap C, Mieremet A, de Vries CJM, Micha D, de Waard V. Six Shades of Vascular Smooth Muscle Cells Illuminated by KLF4 (Krüppel-Like Factor 4). Arterioscler Thromb Vasc Biol. 2021;41(11):2693–2707. doi: 10.1161/ATVBAHA.121.316600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chaudhry SS, Cain SA, Morgan A, Dallas SL, Shuttleworth CA, Kielty CM. Fibrillin-1 regulates the bioavailability of TGFβ1. Journal of Cell Biology. 2007;176(3):355–367. doi: 10.1083/jcb.200608167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zilberberg L, Phoon CKL, Robertson I, Dabovic B, Ramirez F, Rifkin DB. Genetic analysis of the contribution of LTBP-3 to thoracic aneurysm in Marfan syndrome. Proc Natl Acad Sci USA. 2015;112(45):14012–14017. doi: 10.1073/pnas.1507652112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Francis CM, Futschik ME, Huang J, Bai W, Sargurupremraj M, Petretto E, Ho AS, Amouyel P, Engelter ST, Ware JS, Debette S, Elliott P, Dehghan A, Matthews PM. GENOME-WIDE ASSOCIATIONS OF AORTIC DISTENSIBILITY SUGGEST CAUSAL RELATIONSHIPS WITH AORTIC ANEURYSMS AND BRAIN WHITE MATTER HYPERINTENSITIES. Published online 2021:2021.09.01.21262202. doi: 10.1101/2021.09.01.21262202 [DOI] [Google Scholar]
- 63.Da Ros F, Carnevale R, Cifelli G, Bizzotto D, Casaburo M, Perrotta M, Carnevale L, Vinciguerra I, Fardella S, Iacobucci R, Bressan GM, Braghetta P, Lembo G, Carnevale D. Targeting Interleukin-1β Protects from Aortic Aneurysms Induced by Disrupted Transforming Growth Factor β Signaling. Immunity. 2017;47(5):959–973.e9. doi: 10.1016/j.immuni.2017.10.016 [DOI] [PubMed] [Google Scholar]
- 64.Ishida Y, Kuninaka Y, Nosaka M, Kimura A, Taruya A, Furuta M, Mukaida N, Kondo T. Prevention of CaCl2-induced aortic inflammation and subsequent aneurysm formation by the CCL3–CCR5 axis. Nat Commun. 2020;11(1):5994. doi: 10.1038/s41467-020-19763-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dawson A, Li Y, Li Y, Ren P, Vasquez HG, Zhang C, Rebello KR, Ageedi W, Azares AR, Mattar AB, Sheppard MB, Lu HS, Coselli JS, Cassis LA, Daugherty A, Shen YH, LeMaire SA. Single-Cell Analysis of Aneurysmal Aortic Tissue in Patients with Marfan Syndrome Reveals Dysfunctional TGF-β Signaling. Genes (Basel). 2021;13(1):95. doi: 10.3390/genes13010095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Row S, Liu Y, Alimperti S, Agarwal SK, Andreadis ST. Cadherin-11 is a novel regulator of extracellular matrix synthesis and tissue mechanics. Journal of Cell Science. Published online January 1, 2016:jcs.183772. doi: 10.1242/jcs.183772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xie Y, Ostriker AC, Jin Y, Hu H, Sizer AJ, Peng G, Morris AH, Ryu C, Herzog EL, Kyriakides T, Zhao H, Dardik A, Yu J, Hwa J, Martin KA. LMO7 Is a Negative Feedback Regulator of Transforming Growth Factor β Signaling and Fibrosis. Circulation. 2019;139(5):679–693. doi: 10.1161/CIRCULATIONAHA.118.034615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li Y, Gao S, Han Y, Song L, Kong Y, Jiao Y, Huang S, Du J, Li Y. Variants of Focal Adhesion Scaffold Genes Cause Thoracic Aortic Aneurysm. Circ Res. 2021;128(1):8–23. doi: 10.1161/CIRCRESAHA.120.317361 [DOI] [PubMed] [Google Scholar]
- 69.Tang PCY, Coady MA, Lovoulos C, Dardik A, Aslan M, Elefteriades JA, Tellides G. Hyperplastic cellular remodeling of the media in ascending thoracic aortic aneurysms. Circulation. 2005;112(8):1098–1105. doi: 10.1161/CIRCULATIONAHA.104.511717 [DOI] [PubMed] [Google Scholar]
- 70.Wang L, Zhang J, Fu W, Guo D, Jiang J, Wang Y. Association of smooth muscle cell phenotypes with extracellular matrix disorders in thoracic aortic dissection. Journal of Vascular Surgery. 2012;56(6):1698–1709.e1. doi: 10.1016/j.jvs.2012.05.084 [DOI] [PubMed] [Google Scholar]
- 71.Stuart T, Satija R. Integrative single-cell analysis. Nat Rev Genet. 2019;20(5):257–272. doi: 10.1038/s41576-019-0093-7 [DOI] [PubMed] [Google Scholar]
- 72.Satija R, Shalek AK. Heterogeneity in immune responses: from populations to single cells. Trends in Immunology. 2014;35(5):219–229. doi: 10.1016/j.it.2014.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bacher R, Kendziorski C. Design and computational analysis of single-cell RNA-sequencing experiments. Genome Biol. 2016;17:63. doi: 10.1186/s13059-016-0927-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhao J, Jaffe A, Li H, Lindenbaum O, Sefik E, Jackson R, Cheng X, Flavell RA, Kluger Y. Detection of differentially abundant cell subpopulations in scRNA-seq data. PNAS. 2021;118(22). doi: 10.1073/pnas.2100293118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Burkhardt DB, Stanley JS, Tong A, Perdigoto AL, Gigante SA, Herold KC, Wolf G, Giraldez AJ, van Dijk D, Krishnaswamy S. Quantifying the effect of experimental perturbations at single-cell resolution. Nat Biotechnol. 2021;39(5):619–629. doi: 10.1038/s41587-020-00803-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li Y, LeMaire SA, Shen YH. Molecular and Cellular Dynamics of Aortic Aneurysms Revealed by Single-Cell Transcriptomics. Arterioscler Thromb Vasc Biol. 2021;41(11):2671–2680. doi: 10.1161/ATVBAHA.121.315852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Iyer D, Zhao Q, Wirka R, Naravane A, Nguyen T, Liu B, Nagao M, Cheng P, Miller CL, Kim JB, Pjanic M, Quertermous T. Coronary artery disease genes SMAD3 and TCF21 promote opposing interactive genetic programs that regulate smooth muscle cell differentiation and disease risk. PLoS Genet. 2018;14(10):e1007681. doi: 10.1371/journal.pgen.1007681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kalluri AS, Vellarikkal SK, Edelman ER, Nguyen L, Subramanian A, Ellinor PT, Regev A, Kathiresan S, Gupta RM. Single-Cell Analysis of the Normal Mouse Aorta Reveals Functionally Distinct Endothelial Cell Populations. Circulation. 2019;140(2):147–163. doi: 10.1161/CIRCULATIONAHA.118.038362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lukowski SW, Patel J, Andersen SB, Sim SL, Wong HY, Tay J, Winkler I, Powell JE, Khosrotehrani K. Single-Cell Transcriptional Profiling of Aortic Endothelium Identifies a Hierarchy from Endovascular Progenitors to Differentiated Cells. Cell Reports. 2019;27(9):2748–2758.e3. doi: 10.1016/j.celrep.2019.04.102 [DOI] [PubMed] [Google Scholar]
- 80.Lu S, Jolly AJ, Strand KA, Dubner AM, Mutryn MF, Moulton KS, Nemenoff RA, Majesky MW, Weiser-Evans MC. Smooth muscle-derived progenitor cell myofibroblast differentiation through KLF4 downregulation promotes arterial remodeling and fibrosis. JCI Insight. 2020;5(23):139445. doi: 10.1172/jci.insight.139445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Majesky MW, Horita H, Ostriker A, Lu S, Regan JN, Bagchi A, Dong XR, Poczobutt J, Nemenoff RA, Weiser-Evans MCM. Differentiated Smooth Muscle Cells Generate a Subpopulation of Resident Vascular Progenitor Cells in the Adventitia Regulated by Klf4. Circ Res. 2017;120(2):296–311. doi: 10.1161/CIRCRESAHA.116.309322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Renard M, Francis C, Ghosh R, Scott AF, Witmer PD, Ades LC, Andelfinger GU, Arnaud P, Boileau C, Callewaert BL, Guo D, Hanna N, Lindsay ME, Morisaki H, Morisaki T, Pachter N, Robert L, Van Laer L, Dietz HC, Loeys BL, Milewicz DM, De Backer J. Clinical Validity of Genes for Heritable Thoracic Aortic Aneurysm and Dissection. J Am Coll Cardiol. 2018;72(6):605–615. doi: 10.1016/j.jacc.2018.04.089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu H, Kirita Y, Donnelly EL, Humphreys BD. Advantages of Single-Nucleus over Single-Cell RNA Sequencing of Adult Kidney: Rare Cell Types and Novel Cell States Revealed in Fibrosis. JASN. 2019;30(1):23–32. doi: 10.1681/ASN.2018090912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Davey HM, Kell DB. Flow cytometry and cell sorting of heterogeneous microbial populations: the importance of single-cell analyses. Microbiol Rev. 1996;60(4):641–696. doi: 10.1128/mr.60.4.641-696.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Baron CS, Barve A, Muraro MJ, van der Linden R, Dharmadhikari G, Lyubimova A, de Koning EJP, van Oudenaarden A. Cell Type Purification by Single-Cell Transcriptome-Trained Sorting. Cell. 2019;179(2):527–542.e19. doi: 10.1016/j.cell.2019.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Williams JW, Winkels H, Durant CP, Zaitsev K, Ghosheh Y, Ley K. Single Cell RNA Sequencing in Atherosclerosis Research. Circ Res. 2020;126(9):1112–1126. doi: 10.1161/CIRCRESAHA.119.315940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.He R, Guo DC, Sun W, Papke CL, Duraisamy S, Estrera AL, Safi HJ, Ahn C, Buja LM, Arnett FC, Zhang J, Geng YJ, Milewicz DM. Characterization of the inflammatory cells in ascending thoracic aortic aneurysms in patients with Marfan syndrome, familial thoracic aortic aneurysms, and sporadic aneurysms. J Thorac Cardiovasc Surg. 2008;136(4):922–929, 929.e1. doi: 10.1016/j.jtcvs.2007.12.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ejiri J, Inoue N, Tsukube T, Munezane T, Hino Y, Kobayashi S, Hirata K ichi, Kawashima S, Imajoh-Ohmi S, Hayashi Y, Yokozaki H, Okita Y, Yokoyama M. Oxidative stress in the pathogenesis of thoracic aortic aneurysm: Protective role of statin and angiotensin II type 1 receptor blocker. Cardiovascular Research. 2003;59(4):988–996. doi: 10.1016/S0008-6363(03)00523-6 [DOI] [PubMed] [Google Scholar]
- 89.Haskins RM, Nguyen AT, Alencar GF, Billaud M, Kelly-Goss MR, Good ME, Bottermann K, Klibanov AL, French BA, Harris TE, Peirce SM, Isakson BE, Owens GK. Klf4 has an unexpected protective role in perivascular cells within the microvasculature. Am J Physiol Heart Circ Physiol. 2018;315(2):H402–H414. doi: 10.1152/ajpheart.00084.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kan H, Zhang K, Mao A, Geng L, Gao M, Feng L, You Q, Ma X. Single-cell transcriptome analysis reveals cellular heterogeneity in the ascending aortas of normal and high-fat diet-fed mice. Exp Mol Med. 2021;53(9):1379–1389. doi: 10.1038/s12276-021-00671-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dobnikar L, Taylor AL, Chappell J, Oldach P, Harman JL, Oerton E, Dzierzak E, Bennett MR, Spivakov M, Jørgensen HF. Disease-relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels. Nat Commun. 2018;9(1):4567. doi: 10.1038/s41467-018-06891-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brown DJ, Schermerhorn ML, Powell RJ, Fillinger MF, Rzucidlo EM, Walsh DB, Wyers MC, Zwolak RM, Cronenwett JL. Mesenteric stenting for chronic mesenteric ischemia. J Vasc Surg. 2005;42(2):268–274. doi: 10.1016/j.jvs.2005.03.054 [DOI] [PubMed] [Google Scholar]
- 93.Liu M, Gomez D. Smooth Muscle Cell Phenotypic Diversity. Arterioscler Thromb Vasc Biol. 2019;39(9):1715–1723. doi: 10.1161/ATVBAHA.119.312131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vapnik JS, Kim JB, Isselbacher EM, Ghoshhajra BB, Cheng Y, Sundt TM, MacGillivray TE, Cambria RP, Lindsay ME. Characteristics and Outcomes of Ascending Versus Descending Thoracic Aortic Aneurysms. Am J Cardiol. 2016;117(10):1683–1690. doi: 10.1016/j.amjcard.2016.02.048 [DOI] [PubMed] [Google Scholar]
- 95.Majesky MW, Dong XR, Hoglund V, Mahoney WM, Daum G. The adventitia: a dynamic interface containing resident progenitor cells. Arterioscler Thromb Vasc Biol. 2011;31(7):1530–1539. doi: 10.1161/ATVBAHA.110.221549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiol Rev. 2009;89(3):957–989. doi: 10.1152/physrev.00041.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Morrison TM, Choi G, Zarins CK, Taylor CA. Circumferential and longitudinal cyclic strain of the human thoracic aorta: age-related changes. J Vasc Surg. 2009;49(4):1029–1036. doi: 10.1016/j.jvs.2008.11.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bersi MR, Bellini C, Wu J, Montaniel KRC, Harrison DG, Humphrey JD. Excessive Adventitial Remodeling Leads to Early Aortic Maladaptation in Angiotensin-Induced Hypertension. Hypertension. 2016;67(5):890–896. doi: 10.1161/HYPERTENSIONAHA.115.06262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Han WQ, Wu LY, Zhou HY, Zhang J, Che ZQ, Wu YJ, Liu JJ, Zhu DL, Gao PJ. Changes in the composition of the thoracic aortic wall in spontaneously hypertensive rats treated with losartan or spironolactone. Clin Exp Pharmacol Physiol. 2009;36(5–6):583–588. doi: 10.1111/j.1440-1681.2008.05116.x [DOI] [PubMed] [Google Scholar]
- 100.Kuang SQ, Geng L, Prakash SK, Cao JM, Guo S, Villamizar C, Kwartler CS, Peters AM, Brasier AR, Milewicz DM. Aortic remodeling after transverse aortic constriction in mice is attenuated with AT1 receptor blockade. Arterioscler Thromb Vasc Biol. 2013;33(9):2172–2179. doi: 10.1161/ATVBAHA.113.301624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chandran RR, Xie Y, Gallardo-Vara E, Adams T, Garcia-Milian R, Kabir I, Sheikh AQ, Kaminski N, Martin KA, Herzog EL, Greif DM. Distinct roles of KLF4 in mesenchymal cell subtypes during lung fibrogenesis. Nat Commun. 2021;12(1):7179. doi: 10.1038/s41467-021-27499-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ghaleb AM, Yang VW. Krüppel-like factor 4 (KLF4): What we currently know. Gene. 2017;611:27–37. doi: 10.1016/j.gene.2017.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AAC, Greene ES, Straub AC, Isakson B, Randolph GJ, Owens GK. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21(6):628–637. doi: 10.1038/nm.3866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Müller J, Gorressen S, Grandoch M, Feldmann K, Kretschmer I, Lehr S, Ding Z, Schmitt JP, Schrader J, Garbers C, Heusch G, Kelm M, Scheller J, Fischer JW. Interleukin-6-dependent phenotypic modulation of cardiac fibroblasts after acute myocardial infarction. Basic Res Cardiol. 2014;109(6):440. doi: 10.1007/s00395-014-0440-y [DOI] [PubMed] [Google Scholar]
- 105.Pottie L, Adamo CS, Beyens A, Lütke S, Tapaneeyaphan P, De Clercq A, Salmon PL, De Rycke R, Gezdirici A, Gulec EY, Khan N, Urquhart JE, Newman WG, Metcalfe K, Efthymiou S, Maroofian R, Anwar N, Maqbool S, Rahman F, Altweijri I, Alsaleh M, Abdullah SM, Al-Owain M, Hashem M, Houlden H, Alkuraya FS, Sips P, Sengle G, Callewaert B. Bi-allelic premature truncating variants in LTBP1 cause cutis laxa syndrome. Am J Hum Genet. 2021;108(6):1095–1114. doi: 10.1016/j.ajhg.2021.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.van den Hoogenhof MMG, Beqqali A, Amin AS, van der Made I, Aufiero S, Khan MAF, Schumacher CA, Jansweijer JA, van Spaendonck-Zwarts KY, Remme CA, Backs J, Verkerk AO, Baartscheer A, Pinto YM, Creemers EE. RBM20 Mutations Induce an Arrhythmogenic Dilated Cardiomyopathy Related to Disturbed Calcium Handling. Circulation. 2018;138(13):1330–1342. doi: 10.1161/CIRCULATIONAHA.117.031947 [DOI] [PubMed] [Google Scholar]
- 107.Nyegaard M, Overgaard MT, Søndergaard MT, Vranas M, Behr ER, Hildebrandt LL, Lund J, Hedley PL, Camm AJ, Wettrell G, Fosdal I, Christiansen M, Børglum AD. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet. 2012;91(4):703–712. doi: 10.1016/j.ajhg.2012.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schönberger J, Wang L, Shin JT, Kim SD, Depreux FFS, Zhu H, Zon L, Pizard A, Kim JB, Macrae CA, Mungall AJ, Seidman JG, Seidman CE. Mutation in the transcriptional coactivator EYA4 causes dilated cardiomyopathy and sensorineural hearing loss. Nat Genet. 2005;37(4):418–422. doi: 10.1038/ng1527 [DOI] [PubMed] [Google Scholar]
- 109.Pinard A, Jones GT, Milewicz DM. Genetics of Thoracic and Abdominal Aortic Diseases. Circ Res. 2019;124(4):588–606. doi: 10.1161/CIRCRESAHA.118.312436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhao Y, Hasse S, Zhao C, Bourgoin SG. Targeting the autotaxin - Lysophosphatidic acid receptor axis in cardiovascular diseases. Biochem Pharmacol. 2019;164:74–81. doi: 10.1016/j.bcp.2019.03.035 [DOI] [PubMed] [Google Scholar]
- 111.Khachigian LM, Fahmy RG, Zhang G, Bobryshev YV, Kaniaros A. c-Jun regulates vascular smooth muscle cell growth and neointima formation after arterial injury. Inhibition by a novel DNA enzyme targeting c-Jun. J Biol Chem. 2002;277(25):22985–22991. doi: 10.1074/jbc.M200977200 [DOI] [PubMed] [Google Scholar]
- 112.Lelios I, Cansever D, Utz SG, Mildenberger W, Stifter SA, Greter M. Emerging roles of IL-34 in health and disease. J Exp Med. 2020;217(3):e20190290. doi: 10.1084/jem.20190290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Noda K, Dabovic B, Takagi K, Inoue T, Horiguchi M, Hirai M, Fujikawa Y, Akama TO, Kusumoto K, Zilberberg L, Sakai LY, Koli K, Naitoh M, von Melchner H, Suzuki S, Rifkin DB, Nakamura T. Latent TGF-β binding protein 4 promotes elastic fiber assembly by interacting with fibulin-5. Proceedings of the National Academy of Sciences. 2013;110(8):2852–2857. doi: 10.1073/pnas.1215779110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Huhtinen A, Hongisto V, Laiho A, Löyttyniemi E, Pijnenburg D, Scheinin M. Gene expression profiles and signaling mechanisms in α2B-adrenoceptor-evoked proliferation of vascular smooth muscle cells. BMC Syst Biol. 2017;11(1):65. doi: 10.1186/s12918-017-0439-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Helenius MH, Vattulainen S, Orcholski M, Aho J, Komulainen A, Taimen P, Wang L, de Jesus Perez VA, Koskenvuo JW, Alastalo TP. Suppression of endothelial CD39/ENTPD1 is associated with pulmonary vascular remodeling in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2015;308(10):L1046–1057. doi: 10.1152/ajplung.00340.2014 [DOI] [PubMed] [Google Scholar]
- 116.Behdad A, Sun X, Khalpey Z, Enjyoji K, Wink M, Wu Y, Usheva A, Robson SC. Vascular smooth muscle cell expression of ectonucleotidase CD39 (ENTPD1) is required for neointimal formation in mice. Purinergic Signal. 2009;5(3):335–342. doi: 10.1007/s11302-009-9158-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Huang X, Qu R, Ouyang J, Zhong S, Dai J. An Overview of the Cytoskeleton-Associated Role of PDLIM5. Front Physiol. 2020;11:975. doi: 10.3389/fphys.2020.00975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shao H, Wang JHC, Pollak MR, Wells A. α-Actinin-4 Is Essential for Maintaining the Spreading, Motility and Contractility of Fibroblasts. PLoS One. 2010;5(11):e13921. doi: 10.1371/journal.pone.0013921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Quick Q, Skalli O. Alpha-actinin 1 and alpha-actinin 4: contrasting roles in the survival, motility, and RhoA signaling of astrocytoma cells. Exp Cell Res. 2010;316(7):1137–1147. doi: 10.1016/j.yexcr.2010.02.011 [DOI] [PubMed] [Google Scholar]
- 120.Beauchamp NJ, van Achterberg TAE, Engelse MA, Pannekoek H, de Vries CJM. Gene expression profiling of resting and activated vascular smooth muscle cells by serial analysis of gene expression and clustering analysis. Genomics. 2003;82(3):288–299. doi: 10.1016/s0888-7543(03)00127-7 [DOI] [PubMed] [Google Scholar]
- 121.Mueller PA, Zhu L, Tavori H, Huynh K, Giunzioni I, Stafford JM, Linton MF, Fazio S. Deletion of Macrophage Low-Density Lipoprotein Receptor-Related Protein 1 (LRP1) Accelerates Atherosclerosis Regression and Increases C-C Chemokine Receptor Type 7 (CCR7) Expression in Plaque Macrophages. Circulation. 2018;138(17):1850–1863. doi: 10.1161/CIRCULATIONAHA.117.031702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Muratoglu SC, Belgrave S, Lillis AP, Migliorini M, Robinson S, Smith E, Zhang L, Strickland DK. Macrophage LRP1 suppresses neo-intima formation during vascular remodeling by modulating the TGF-β signaling pathway. PLoS One. 2011;6(12):e28846. doi: 10.1371/journal.pone.0028846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Au DT, Ying Z, Hernández-Ochoa EO, Fondrie WE, Hampton B, Migliorini M, Galisteo R, Schneider MF, Daugherty A, Rateri DL, Strickland DK, Muratoglu SC. LRP1 (Low-Density Lipoprotein Receptor-Related Protein 1) Regulates Smooth Muscle Contractility by Modulating Ca2+ Signaling and Expression of Cytoskeleton-Related Proteins. Arterioscler Thromb Vasc Biol. 2018;38(11):2651–2664. doi: 10.1161/ATVBAHA.118.311197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Strickland DK, Au DT, Cunfer P, Muratoglu SC. Low-density lipoprotein receptor-related protein-1: role in the regulation of vascular integrity. Arterioscler Thromb Vasc Biol. 2014;34(3):487–498. doi: 10.1161/ATVBAHA.113.301924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Prakash SK, Milewicz DM. ‘X’ marks the spot: the profound impact of sex on aortic disease. Arterioscler Thromb Vasc Biol. 2018;38(1):9–11. doi: 10.1161/ATVBAHA.117.310433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ding J, Adiconis X, Simmons SK, Kowalczyk MS, Hession CC, Marjanovic ND, Hughes TK, Wadsworth MH, Burks T, Nguyen LT, Kwon JYH, Barak B, Ge W, Kedaigle AJ, Carroll S, Li S, Hacohen N, Rozenblatt-Rosen O, Shalek AK, Villani AC, Regev A, Levin JZ. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat Biotechnol. 2020;38(6):737–746. doi: 10.1038/s41587-020-0465-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single nucleus RNA sequencing data are publicly available at the Broad Institute’s Single Cell Portal (https://singlecell.broadinstitute.org/single_cell). The raw dataset is available at the National Center for Biotechnology Information’s Gene Expression Omnibus Database (accession #GSE207784). All other data are contained within the article and its supplementary information, or are available upon reasonable request to the corresponding author.