

Abstract
AKT is an important target for cancer therapeutics. Significant advancements have been made in developing ATP-competitive and allosteric AKT inhibitors. Recently, several AKT proteolysis targeting chimeras (PROTACs) derived from ATP-competitive AKT inhibitors have been reported, including MS21. While MS21 potently degraded AKT and inhibited the growth in tumor cells harboring PI3K/PTEN pathway mutation, it was largely ineffective in degrading AKT in KRAS/BRAF mutated cells as a single agent. To overcome the AKT degradation resistance in KRAS/BRAF mutated cells, we developed novel AKT PROTACs derived from an AKT allosteric inhibitor, including degrader 62 (MS15). 62 displayed potent and selective AKT degradation activity and potent anti-proliferative activity in KRAS/BRAF mutated cancer cells, in addition to PI3K/PTEN mutated cancer cells. Furthermore, 62 was bioavailable in mice through intraperitoneal administration. Overall, 62 is a valuable chemical tool to degrade AKT in cells harboring KRAS/BRAF mutation and expands the tool box for pharmacologically modulating AKT.
Table of Contents Graphic
Introduction
AKT (protein kinase B) is a serine/threonine kinase that belongs to AGC protein kinase family. As a central node of the most commonly dysregulated PI3K/AKT/m-TOR signaling cascade in human cancers, AKT plays critical roles in regulating cancer hallmarks, such as cell proliferation, metabolism, metastasis, invasiveness and survival of tumor cells.1–5 AKT comprises three highly homologous isoforms, AKT1/2/3, all of which share three evolutionarily conserved domains: an N-terminal pleckstrin-homology (PH) domain, a central kinase catalytic domain, and a C-terminal regulatory domain.6–8 Hyperactivation and overexpression of AKT have been implicated in many human cancers with overall poor prognosis.9, 10 Pharmacological inhibition of the AKT kinase activity has been extensively pursued as a potential therapeutic strategy for treating cancers, and many AKT inhibitors, including ATP-competitive inhibitors GSK690693,11 GDC-0068,12 AZD5363,13 and allosteric inhibitors MK-220614–16, ARQ-09217 and TAS-11718, have been advanced into clinical investigation.6, 10, 19–21 However, ATP-competitive AKT inhibitors are associated with severe side-effects, including hyperglycemia, thrombocytopenia and infections, and AKT allosteric inhibitors have shown limited efficacy in human clinical trials.22
Targeting protein degradation (TPD) using proteolysis targeting chimeras (PROTACs) or molecular glues represents an appealing therapeutic strategy for treating cancers by depleting disease-causing proteins.23–29 The ubiquitin-proteasome system (UPS)-mediated degradation induced by PROTACs or molecular glues can target both the catalytic and non-catalytic functions of oncoproteins, leading to potentially greater efficacy than enzymatic inhibitors. A number of AKT PROTAC degraders have been recently developed from ATP-competitive AKT inhibitors (Figure 1). You et al. reported the first GDC-0068-derived cereblon (CRBN)-recruiting AKT degrader, INY-03–041, which induced potent AKT degradation in breast cancer cells.30 We reported a CRBN-recruiting AKT degrader, MS170, and a von Hippel-Lindau (VHL)-recruiting AKT degrader, MS98, both of which are based on GDC-0068.31 We also discovered a couple of AZD5363-derived VHL-recruiting AKT degraders, MS21 and MS143, and a CRBN-recruiting degrader MS5033, which is also derived from AZD5363.32, 33 Recently, Dong’s lab reported the discovery of a CRBN-recruiting AKT degrader B4 utilizing an in silico modeling approach, which induced AKT degradation in hematological cancer cells.34 Among these reported degraders, MS21 is the most potent and effective AKT degrader. MS21 selectively degraded AKT and exerted superior cell growth inhibition compared to the parent AKT inhibitor AZD5363, which is in clinical development with very high potency and selectivity for AKT, in a large panel of cancer cells with PI3K/PTEN pathway mutation.32 However, MS21 was unable to effectively induce AKT degradation in cancer cells harboring KRAS or BRAF mutation.32
Figure 1.

Chemical structures of representative reported AKT PROTAC degraders.
To overcome this AKT degradation resistance of the AKT PROTACs derived from ATP-competitive AKT inhibitors in cancer cells harboring KRAS/BRAF mutations, we sought to develop AKT PROTACs from allosteric inhibitors of AKT. Here, we report the discovery of novel AKT PROTAC degraders derived from the AKT allosteric inhibitor ARQ-092. Our lead compound, 62 (MS15), which is VHL-recruiting, potently and selectively degraded AKT and effectively suppressed the proliferation in cancer cells harboring KRAS/BRAF mutations. 62 also effectively degraded AKT in cancer cells harboring PTEN/PI3K pathway mutation. Furthermore, 62 was bioavailable in a mouse pharmacokinetic (PK) study via intraperitoneal (IP) administration. Our extensive structure-activity relationship (SAR) study that led to the discovery of 62 and detailed biological characterization of 62 are described below.
Results and Discussion
Design and Biological Evaluation of ARQ-092-based AKT PROTAC Degraders.
ATP-competitive AKT inhibitors preferentially bind the activated AKT (phosphorylated AKT, p-AKT) over inactivated AKT.35 In our previous study, we demonstrated that MS21, which contains the ATP-competitive inhibitor AZD5363 as the binding moiety to AKT, did not effectively engage AKT in KRAS/BRAF mutated cells due to the low p-AKT levels in these cells.32 Different from ATP-competitive AKT inhibitors, AKT allosteric inhibitors bind and stabilize the inactivation form of AKT.36, 37 Therefore, degraders derived from AKT allosteric inhibitors may have the potential to engage the inactivated AKT in cancer cells harboring KRAS/BRAF mutations and could induce effective AKT degradation in these MS21-resistant cells.
ARQ-092 is a potent and selective allosteric AKT inhibitor with high enzymatic potency against AKT1, AKT2 and AKT3 with IC50 values of 5.0, 4.5 and 16 nM, respectively.38 ARQ-092 binds AKT at a unique interdomain region between the kinase and the PH domains, and stabilizes AKT in the inactive “PH-in” conformation. By examining the co-crystal structure of AKT1 in complex with ARQ-092 (PDB ID: 5KCV), we identified that the left-hand side phenyl ring of ARQ-092 was exposed to solvent (Figure 2A). To facilitate the linker attachment, two short spacers were installed at the 3-position of this phenyl moiety to generate precursors 1 and 2 (Figure 2B). The commonly used VHL ligand VHL-1, and CRBN ligand pomalidomide (POM) were attached to the aforementioned precursors through various linkers to generate two series of AKT putative PROTACs (Figure 3).
Figure 2.

Design of PROTAC precursors derived from the AKT allosteric inhibitor ARQ-092. (A) Co-crystal structure (left) of AKT1 (in grey) in complex with ARQ-092 (in cyan and blue) (PDB ID: 5KCV) and chemical structure of ARQ-092 (right). The phenyl group of ARQ-092 (indicated by the orange circle) is solvent-exposed. (B) Chemical structures of designed precursors 1 and 2.
Figure 3.

Chemical structures of ARQ-092-derived putative AKT degraders.
The AKT degradation and downstream signaling inhibition activities of these putative AKT PROTAC degraders were evaluated in PC3 cells using an immunoblotting assay. As shown in Figure 4, compared to the dimethyl sulfoxide (DMSO) control, the parent inhibitor ARQ-092 had no effect on total AKT (T-AKT) protein level, but potently inhibited the phosphorylation of AKT, PRAS40, and S6. For the precursor 1-derived VHL-recruiting degraders, compounds 3–7 with short polyethylene glycol (PEG) linkers (1–3 PEGs) effectively reduced T-AKT levels and inhibited the phosphorylation of AKT. However, compounds 8–10 with long PEG linkers (3–5 PEGs) displayed decreased AKT degradation and downstream signaling inhibition activities. Different from the SAR trend with PEG linkers, long alkylene linkers are preferred for effective AKT degradation and downstream signaling inhibition. Among the compounds with alkylene linkers, compounds 11–12 with short alkylene linkers were completely inactive. The AKT degradation activity was improved with the increase of the linker length. Notably, compounds 17–20 containing long alkylene linkers (7–10 methylene units) exhibited the most effective AKT degradation and downstream signaling (P-AKT, P-PRAS40 and P-S6) inhibition. Similar SAR trends were observed for CRBN-recruiting degraders 21–32. However, these CRBN-recruiting compounds were much less effective than VHL-recruiting analogues in reducing T-AKT levels in general. Next, we explored precursor 2-derived VHL-recruiting degraders. Compounds 33 and 34 with short ether linkers effectively induced T-AKT degradation, while the AKT degradation activity of compounds 35–39 with longer linkers decreased. However, further extending the linker length to five PEG units (compounds 40 and 41) restored effective AKT degradation activity. For alkylene linkers, compounds bearing a long linker, such as heptylene (47), octylene (48), and nonylene (49), were particularly effective in T-AKT degradation and downstream signaling (P-AKT and P-PRAS40) inhibition. In addition, we also evaluated the CRBN-recruiting compounds 50–61. However, no prominent AKT degradation was achieved by these compounds. Overall, the SAR trends on the linker moieties are consistent for precursor 1- and precursor 2-conjugated degraders. In general, VHL-recruiting compounds induced more effective AKT degradation than CRBN-recruiting ones. Alkylene linkers are preferred compared to PEG linkers. Compounds possessing long alkylene linkers induced AKT degradation more effectively than compounds bearing short alkylene linkers. Through this comprehensive SAR study on various linkers and E3 ligase recruiting ligands, we discovered several highly effective AKT degraders such as 20, 48 and 49.
Figure 4.

Effects of compounds 3 – 61 on reducing the AKT protein level and inhibiting the downstream signaling. PC3 cells were treated with DMSO, ARQ-092 (1 μM), or the indicated compound at 1 μM for 24 h. The cell lysates were analyzed by western blotting to examine the protein levels of P-AKT (S473), T-AKT, P-PRAS40 (T246) and P-S6 (S240/244). β-Actin was used as the loading control.
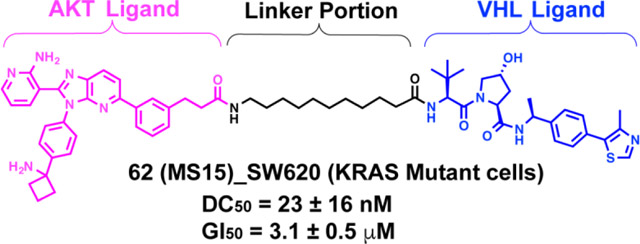
Next, we designed and synthesized a close analog (62, MS15) of compound 20 by replacing VHL-1 with a reported higher affinity VHL ligand, (S,R,S)-AHPC-Me (VHL-2) (Figure 5).31, 39–41 We then compared the effect of compound 62 with that of 20, 47, and 48 in a colony formation assay in PC3 cells. In this MS21-sensitive cell line32, these compounds displayed similar potency in inhibiting colony formation (Figure S1). We next evaluated the effect of these compounds on inducing AKT degradation in MS21-resistant KRAS mutant cell lines: colorectal SW620 cells and pancreatic Mia PaCa-2 cells (Figure S2). Interestingly, only compound 62 was able to effectively and completely depleted T-AKT in SW620 cells (Figure S2A). 62 also modestly induced AKT degradation in Mia PaCa-2 cells, but its effectiveness was much weaker than that in SW620 cells (Figure S2A). We next confirmed that 62 was much more effective than MS21 in degrading AKT in SW620 cells (Figure S2B). Based on these results, we selected 62 for further biological characterization. We also designed two structurally similar analogs, 63 (MS15N1) and 64 (MS15N2), as the negative controls of 62 (Figure 5). 63 contains a diastereomer of VHL-2 to abolish the VHL binding,31, 41 and 64 (MS15N2) was designed to abrogate its binding to the VHL E3 ligase by introducing a benzyl group on the hydroxyl proline moiety of VHL-2.33, 42
Figure 5.

Chemical structures of degrader 62 and its negative controls 63 and 64.
Compounds 62, 63 and 64 Inhibit the AKT Kinase Activity.
We next assessed the AKT kinase inhibitory activity of these compounds in an in vitro biochemical assay. As shown in Figure 6, ARQ-092 displayed excellent potency in inhibiting the AKT1, 2, and 3 activities with IC50 values of 1.3 ± 0.3 nM, 1.4 ± 0.1 nM and 9.9 ± 0.3 nM, respectively, which are in line with the previously reported data.43 Compared to ARQ-092, degrader 62 showed decreased potency (798 ± 190 nM (AKT1), 90 ± 2.8 nM (AKT2) and 544 ± 2.9 nM (AKT3)) for all three AKT isoforms. The negative control 63, which contains a diastereomer of VHL-2, exhibited similar potency for three AKT isoforms as degrader 62, whereas the negative control 64 with the attachment of a benzyl group to the hydroxyl proline moiety of 62 showed decreased inhibition potency for AKT1/2/3 by 1.5-, 6-, and 3.6-fold, respectively, compared to 62. We next assessed the effect of 62 on degrading AKT1, 2 and 3 in SW620 cells (Figure S3). 62 potently induced AKT1 and AKT2 degradation, while the effect of 62 on AKT3 degradation is not clear because of the low expression level of AKT3 in SW620 cell. Interestingly, although 62 displayed weaker AKT kinase inhibition potency than the parental inhibitor ARQ-092, 62 was still capable of inducing robust AKT degradation in cells.
Figure 6.

Compounds 62, 63 and 64 inhibit the kinase activity of AKT1 (A), AKT2 (B) and AKT3 (C). The AKT kinase activity inhibition was determined in a biochemical assay in duplicate. The IC50 values were determined using 10-concentration points (the top concentration of 10 μM) with 3-fold dilution mode in the presence of 10 μM of ATP with DMSO as control.
Compound 62 Induces Robust AKT Degradation in MS21-Resistant KRAS/BRAF Mutant Cells in a Concentration-, Time- and UPS-dependent Manner.
We next further evaluated the effect of 62 on AKT degradation in SW620 cells. As shown in Figure 7, compound 62 effectively induced T-AKT degradation in a concentration-dependent manner with a DC50 value of 23 ± 16 nM. The substantial T-AKT depletion was observed at 30 nM and nearly complete AKT degradation was achieved at a concentration of 100 nM (Figure 7A–B). Notably, no “hooker effect” was observed at high concentrations up to 10 μM. As expected, both negative control compounds, 63 and 64, had no effect on AKT protein levels in SW620 cells at concentrations up to 10 μM, demonstrating that the induced AKT degradation was dependent on recruiting the VHL E3 ligase (Figure 7C–D). Consistent with our previous findings,32 we confirmed that MS21, which is derived from the ATP-competitive AKT inhibitor AZD5363, was largely ineffective in degrading AKT in this KRAS mutant cell line (Figure 7E).
Figure 7.

Compound 62 potently induces AKT degradation in SW620 cells. SW620 cells were treated with 62 (A), 63 (C), 64 (D) or MS21 (E) at indicated concentration for 24 h, and the cell lysates were analyzed by western blotting to examine the protein levels of T-AKT. β-Actin was used as the loading control. (B) DC50 curve of 62, generated using the data in (A).
Encouraged by these findings, we next compared the AKT degradation effect of 62 with that of MS21 in a few additional cancer cell types bearing KRAS or BRAF mutation (Figure 8). We found that 62 was more effective than MS21 in inducing AKT degradation at 1 μM in BRAF mutant cell lines, such as Colo205, HT-29 and SKMEL 239 cells (Figure 8A). Notably, in more aggressive KRAS mutant PANC-1 cells, 62 was highly effective in degrading AKT with near-complete AKT degradation at 1 μM, whereas MS21 was unable to reduce the AKT protein level at the same concentration (Figure 8A). Furthermore, in PANC-1 cells, 62 potently induced T-AKT degradation in a concentration-dependent manner, with complete degradation achieved at 100 nM and no “hooker effect” was observed at high concentrations up to 10 μM (Figure 8B). Taken together, these data indicate that 62, which is derived from the AKT allosteric inhibitor ARQ-092, is much superior to MS21, which is derived from the ATP-competitive AKT inhibitor AZD5363, in inducing AKT degradation in KRAS/BRAF mutant cells.
Figure 8.

Compound 62 effectively induces AKT degradation in MS21-resistant KRAS/BRAF mutant cells. (A) Colo205, HT-29, SKMEL 239 and PANC-1 cells were treated with DMSO, MS21 or 62 at 1 μM for 24 h, and the cell lysates were analyzed by western blotting to examine the protein levels of T-AKT. β-Actin was used as the loading control. (B) PANC-1 cells were treated with 62 at the indicated concentration for 24 h, and the cell lysates were analyzed by western blotting to examine the protein levels of T-AKT. β-Actin was used as the loading control.
Next, we performed time-course studies to examine the kinetics of 62-mediated AKT degradation and downstream signaling pathway inhibition in SW620 cells with the parental inhibitor ARQ-092 as a control (Figure 9A). As expected, ARQ-092 was inactive in decreasing the AKT protein level but potently inhibited the phosphorylation of PRAS40. On the other hand, compound 62 at 1 μM time-dependently reduced T-AKT protein levels. Significant AKT degradation was observed at 4 h, demonstrating a fast degradation kinetic profile. Near complete AKT degradation was achieved at 12 h, and this effect was maintained for at least another 12 h.
Figure 9.

Compound 62 mediates AKT degradation in a time- and UPS-dependent manner. (A) SW620 cells were treated with ARQ-092 (1 μM) or 62 (1 μM) for 24 h, and the cell lysates were harvested at the indicated time and then analyzed by western blotting to examine the protein levels of T-AKT, P-PRAS40 and T-PRAS40. β-Actin was used as the loading control. (B) SW620 cells were pretreated with VHL ligand VHL-2 (1 μM), MLN4924 (1 μM), MG132 (20 μM) or inhibitor ARQ-092 (1 μM) for 2 h before the treatment of 62 for additional 24 h. The cell lysates were harvested and analyzed by western blotting to examine the protein levels of T-AKT, P-PRAS40 and T-PRAS40. β-Actin was used as the loading control.
We also conducted a series of rescue experiments to explore the mechanism of action (MOA) of 62-induced AKT degradation (Figure 9B). Pretreatment with the VHL E3 ligase ligand VHL-2 partially rescued the T-AKT level and downstream signaling P-PRAS40 inhibition in SW620 cells, indicating a VHL-dependent mechanism for 62-induced AKT degradation. Pretreatment with the NEDD8-activating enzyme (NAE) inhibitor MLN492444 or the proteasome inhibitor MG132 also rescued the AKT degradation and P-PRAS40 inhibition, supporting the requirement of the active cullin-ring ubiquitin ligase complex and proteasome for AKT degradation. Moreover, pretreatment with inhibitor ARQ-092 completely blocked the AKT degradation induced by 62, suggesting that 62 occupies the same allosteric binding site as ARQ-092. Altogether, these data demonstrated that 62 is a bona fide AKT degrader, mediating AKT degradation in an AKT binding-, VHL- and UPS-dependent manner.
Compound 62 is a Selective AKT Degrader.
To delineate the proteome-wide degradation selectivity, we performed a global tandem mass tag (TMT) mass spectrometry (MS)-based quantitative proteomic study with the SW620 cells treated with DMSO, degrader 62 or negative control 63 at 1 μM for 18 h (Figure 10). Compared to DMSO, compound 62 significantly reduced the T-AKT level and inhibited downstream P-PRAS40, while negative control 63 had no effect on the AKT protein level and P-PRAS40 inhibition (Figure S4). In the TMT-labeled proteomic study, among approximately 6433 proteins detected, AKT1 is one of a very small set of proteins whose levels were reduced significantly by compound 62 (Figure 10A–C). The AKT2 protein level was also reduced when comparing 62 to 63 (Figure 10C), albeit the reduction in the AKT2 protein level is less profound compared to that in the AKT1 protein level. AKT3 was not detected in the proteomic study due to the low expression level of AKT3 in SW620 cells as illustrated in Figure S3. Overall, these global proteomic results suggest that compound 62 is a generally selective AKT degrader.
Figure 10.

Compound 62 selectively induce AKT degradation in SW620 cells in an unbiased global tandem mass tagged (TMT) MS-based proteomic study. SW620 cells were treated with 62 (A, C) or 63 (B, C) at 1 μM for 18 h before the cell lysates were harvested for MS analysis. The log2 fold change is indicated on the x axis and the negative log10 p value is indicated in the y axis for two independent biological replicates of each treatment.
Degrader 62 Exhibits Strong Anti-Proliferative Activity in SW620 Cells.
Next, we further evaluate the anti-proliferative effects of degrader 62, negative control 64 and parent inhibitor ARQ-092 in KRAS mutant SW620 cells using an IncuCyte assay32 (Figure 11A). 62 (GI50 = 3.1 ± 0.3 μM) displayed slightly better antiproliferative activity than ARQ-092 (GI50 = 5.2 ± 0.4 μM) in SW620 cells, and exhibited much more potent cell growth inhibition than 64, which was inactive in this assay, indicating that AKT degradation is an important contributor to the observed cell growth inhibition by 62 (Figure 11A). Furthermore, 62 induced cell apoptosis most effectively among these tested compounds (Figure 11B). In addition, we conducted colony formation experiments in SW620 cells treated with 62, 64, ARQ-092 or MS21 (Figure 11C). We found that degrader 62, but not negative control 64, significantly inhibited colony formation, suggesting this cell growth inhibition effect of 62 is partially dependent on AKT degradation. In consistent with the IncuCyte assay data, ARQ-092 also displayed comparative ability in inhibiting colony formation. However, MS21 had no effect on cell colony formation, which is consistent to its lack of the effect on AKT degradation in this cell line (Figure 7E). Overall, these results suggest that AKT degraders such as 62, which effectively induced AKT degradation in KRAS/BRAF mutant cells, could provide a potential strategy to target MS21-resistant KRAS/BRAF mutant cancers.
Figure 11.

Compound 62 inhibits the proliferation of KRAS mutant SW620 cells. (A) SW620 cells were treated with DMSO or indicated compound at indicated concentrations for 5 days. Cell growth was determined using calculations derived from phase-contrast images in IncuCyte. Error bars indicate the standard errors in three independent experiments. (B) SW620 cells were treated with DMSO or 5 μM of indicated compound using DRAQ7 to monitor percent cell death at 0, 24, 36, 48 and 72 h. Error bars indicate the standard errors in three independent experiments. (C) SW620 cells were treated with DMSO or indicated compound at indicated concentrations for 2 weeks. Cells were stained with crystal violet.
Compound 62 is Bioavailable in Mice via IP Administration.
Next, we performed a PK study to evaluate the bioavailability of 62 in mice (Figure 12). 62 was administrated through IP injection at a single dose of 75 mg/kg. No clinical signs were observed in the tested mice during the PK study. The maximum plasma concentration (Cmax = 1 μM) was achieved at 0.5 h post-treatment, and plasma concentrations were maintained above 100 nM for at least 12 h. These results suggested that 62 could achieve enough plasma exposure for effective AKT degradation (DC50 = 23 ± 16 nM) in in vivo studies.
Figure 12.

Plasma concentrations of 62 over 12 h after male Swiss albino mice were administrated of 62 at a single dose of 75 mg/kg via IP injection. Experiments were performed in biological triplicates. Each point represents mean concentration ± SEM.
Chemical Synthesis.
The synthesis of key intermediates 1 and 2 is depicted in Scheme 1. The intermediate 65 was prepared following the reported procedures.38 Suzuki-Miyaura coupling reaction between 65 and (3-(3-ethoxy-3-oxopropyl)phenyl)boronic acid (66) and subsequent hydrolysis of the ethyl ester group afforded precursor 1. Similarly, intermediate 2 was yielded from Suzuki-Miyaura coupling reaction between 65 and (3-(cyanomethyl)phenyl)boronic acid (67), followed by cyanide reduction under hydrogenation reaction.
Scheme 1. Syntheses of Precursors 1 and 2.a.

aReagents and conditions: a) Pd(PPh3)4, K2CO3, dioxane / H2O (5 : 3), MW, 120 oC, 30 min; b) LiOH, THF / H2O (2 : 1), rt, 12 h; c) Raney nickel, H2, MeOH / NH3.H2O (5 : 1), rt, 1 h.
VHL-1 conjugated linkers 68a-h, 69a-j, 72a-i and 73a-h, and POM-tethered linkers 70a-e, 71a-g, 74a-g and 75a-e were prepared according to our previously reported procedures.31, 33 As shown in Scheme 2, compounds 2–32 were synthesized by amide coupling reactions between intermediate 1 and linkers 68a-h, 69a-j, 70a-e, and 71a-g, followed by Boc-deprotection. Following the same reaction sequence, compounds 33–61 were synthesized from intermediate 2 and linkers 72a-i, 73a-h, 74a-g and 75a-e as outlined in Scheme 3.
Scheme 2. Synthetic Routes for Compounds 3–32.a.

aReagents and conditions: a) 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI), 1-hydroxy-7-azabenzo-triazole (HOAt), N-methylmorpholine (NMM), DMSO, rt, 12 h; b) trifluoroacetic acid (TFA), dichloromethane (DCM), rt, 30 min.
Scheme 3. Syntheses of Compounds 33–61.a.

aReagents and conditions: a) EDCI, HOAt, NMM, DMSO, rt, 12 h; b) TFA, DCM, rt, 30 min.
Synthetic routes for preparing compound 62 and its two negative control compounds 63 and 64 are described in Scheme 4. The synthesis of 62 started from amide coupling between a dipeptide intermediate 76 and the commercially available dodecanedioc acid 77. Another amide coupling between the resulting intermediate 78 and intermediate 1, followed by Boc group deprotection, afforded the desired product 62. Using the same reaction sequence, 63 was synthesized from dipeptide intermediate 79. The synthesis of 64 started from amide coupling between the Boc protected amino acid 81 and amine 82. After Boc deprotection, intermediate 83 was obtained. Amide coupling between 83 and Boc-protected amino acid 84, followed by Boc-deprotection, afforded intermediate 85, which was then coupled with dodecanedioc acid (77) to yield 86. Following the same reaction sequence of preparing compounds 62 and 63, negative control compound 64 was synthesized from intermediates 1 and 86.
Scheme 4. Syntheses of Compounds 62, Negative Controls 63 and 64.a.

aReagents and conditions: a) EDCI, HOAt, NMM, DMSO, rt, 12 h; b) 1, EDCI, HOAt, NMM, DMSO, rt, 12 h; c) TFA, DCM, rt, 30 min; d) (S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoic acid (84), EDCI, HOAt, NMM, DMSO, rt, 12 h; e) dodecanedioc acid (77), EDCI, HOAt, NMM, DMSO, rt, 12 h.
Conclusion
PI3K/AKT/m-TOR signaling, one of the most frequently dysregulated pathways, is implicated in a wide variety of human cancers. As a central node of the PI3K/AKT/m-TOR pathway, AKT has been an attractive therapeutic target for decades. Although several inhibitors have been investigated in clinic, none of them showed the desired balance between efficacy and toxicity. Using the PROTAC strategy, we previously developed several ATP-competitive inhibitor-derived AKT PROTAC degraders, such as MS21. However, MS21 was largely ineffective in degrading AKT in cancer cells with KRAS/BRAF mutation. In this study, we used an AKT allosteric inhibitor, ARQ-092, to develop AKT degraders that could overcome this degradation resistance issue. We discovered a potent and selective AKT degrader, 62, through an extensive SAR study on linkers and E3 recruiting ligands. We also developed two negative control compounds 63 and 64, which were unable to recruit the VHL E3 ligase. Importantly, compound 62, but not 63 or 64, effectively induced AKT degradation in a concentration-, time- and UPS-dependent manner in MS21-resistant KRAS/BRAF mutant cells. Furthermore, degrader 62, but not MS21, significantly suppressed the proliferation and effectively induced cell apoptosis in MS21-resistant KRAS/BRAF mutant cancer cells. Moreover, 62 was bioavailable in a mouse PK study via IP administration. In summary, we generated a novel AKT PROTAC degrader, 62, which can effectively degrade AKT in MS21-resistant KRAS/BRAF mutant cells. Compound 62 is a useful chemical tool and further expands the tool box for the scientific community to pharmacologically modulate AKT.
Experimental Section
Chemistry General Procedures.
All commercially available solvents and reagents were used without further purification. Microwave-heated reactions were performed on a Discover SP microwave system with an Explorer 12 Hybrid Autosampler by CEM (Buckingham, UK). Normal and reverse phase flash chromatography were performed using a Teledyne ISCO CombiFlash instrument and HP C18 RediSep Rf reverse phase silica columns, respectively. Final compounds for biological evaluation were purified with preparative high-performance liquid chromatography (HPLC) on an Agilent Prep 1200 series with the UV detector set to 254 nm with solvent A (0.1% of TFA in water) and solvent B (acetonitrile) as eluents with a flow rate of 40 mL/min at room temperature. Purities of the final compounds were assessed > 95% by HPLC under following conditions: Agilent 1200 series system, 2.1 mm x 150 mm Zorbax 300SB-C18 5 μm column, 1–99% gradient of 0.1% trifluoroacetic acid in water, and 0.1% trifluoroacetic acid in acetonitrile; flow rate, 0.4 mL/min; High-resolution mass spectra (HRMS) data was acquired on an Agilent G1969A API-TOF with an electrospray ionization (ESI) source. Proton (1H NMR) and Carbon (13C NMR) nuclear magnetic resonance spectra were recorded on either Bruker DXI 800 MHz or AVANCE NEO 600 MHz NMR spectrometer, and were reported in parts per million (ppm) on the δ scale in the following format: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quarter, m = multiplet), coupling constant (J, Hz), and integration. Linkers 68a-h, 69a-j, 70a-e, 71a-g, 72a-i, 73a-h, 74a-g, 75a-e, 78 and 80 were synthesized following our previously reported procedures.31, 33
3-(3-(2-(2-Aminopyridin-3-yl)-3-(4-(1-((tert-butoxycarbonyl)amino)cyclobutyl)phenyl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)propanoic acid (1).
To a suspension of intermediate 6538 (152 mg, 0.31 mmol) and (3-(3-ethoxy-3-oxopropyl)phenyl)boronic acid (66, 137 mg, 0.62 mmol) in 2.5 mL of dioxane and 1.5 mL of H2O was added potassium carbonate (128 mg, 0.93 mmol). The resulting mixture was degassed for 5 min, before Pd(PPh3)4 (18 mg, 5 mol %) was added. After the reaction mixture was stirred at 120 oC for 30 min under microwave irradiation, the solvent was removed and the mixture was purified by reverse phase C18 column (10% – 100% methanol / 0.1% TFA in H2O) to afford the desired product as white solid (206 mg, yield 97%). After this product was dissolved in THF (5 mL) and H2O (2 mL), lithium hydroxide (15 mg, 0.64 mmol) was added. After the reaction mixture was stirred at rt overnight, the solvent was removed and the resulting residue was purified by reverse phase C18 column (10% – 100% methanol / 0.1% TFA in H2O) to afford compound 1 as white solid (193 mg, yield 99%). 1H NMR (600 MHz, DMSO-d6) δ 8.31 (d, J = 8.4 Hz, 1H), 8.05 (dd, J = 5.8, 1.7 Hz, 1H), 8.01 (d, J = 8.5 Hz, 1H), 7.87 (d, J = 1.7 Hz, 1H), 7.86 – 7.83 (m, 1H), 7.64 – 7.57 (m, 3H), 7.57 – 7.51 (m, 4H), 7.50 – 7.45 (m, 1H), 2.87 (t, J = 7.7 Hz, 2H), 2.57 (t, J = 7.7 Hz, 2H), 2.07 – 1.91 (m, 1H), 1.84 – 1.77 (m, 1H), 1.33 (s, 9H), 1.13 – 1.08 (m, 4H). ESI-MS (m/z) [M + H]+: 605.3.
tert-Butyl (1-(4-(5-(3-(2-aminoethyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-3-yl)phenyl)cyclobutyl)carbamate (2).
To a suspension of intermediate 6538 (156.4 mg, 0.32 mmol) and (3-(cyanomethyl)phenyl)boronic acid (67, 102 mg, 0.64 mmol) in dioxane (2.5 mL) and H2O (1.5 mL) was added potassium carbonate (144 mg, 0.93 mmol). The mixture was degassed for 5 min, before Pd(PPh3)4 (19 mg, 5 mol %) was added. After the reaction mixture was stirred at 120 oC for 30 min under microwave irradiation, the solvent was removed and the resulting mixture was purified by reverse phase C18 column (10% – 100% methanol / 0.1% TFA in H2O) to afford the desired product as white solid (206 mg, yield 97%). After this product was dissolved in 5 mL of methanol, Raney nickel (20 mg, 10% w/w) and NH3·H2O (1 mL) were added. The reaction was stirred at rt for 1 h, before the mixture was filtered through Celite. After the filtrate was concentrated, the resulting residue was purified by reverse phase C18 column (10% – 100% methanol / 0.1% TFA in H2O) to afford compound 2 as yellow solid (272.5 mg, yield 43%). 1H NMR (600 MHz, CD3OD) δ 8.31 (d, J = 8.4 Hz, 1H), 8.09 – 7.93 (m, 5H), 7.81 – 7.68 (m, 2H), 7.53 (d, J = 8.1 Hz, 2H), 7.46 (t, J = 7.7 Hz, 1H), 7.38 – 7.34 (m, 1H), 6.77 (t, J = 6.9 Hz, 1H), 3.25 (t, J = 7.7 Hz, 2H), 3.06 (t, J = 7.7 Hz, 2H), 2.64 – 2.58 (m, 2H), 2.55 (d, J = 13.8 Hz, 2H), 2.20 (s, 1H), 2.00 (s, 1H), 1.56 – 1.06 (m, 9H). ESI-MS (m/z) [M + H]+: 576.4.
(2S,4R)-1-((S)-2-(2-(2-(3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)propanamido)ethoxy)acetamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (3).
To a solution of intermediate 1 (12 mg, 0.02 mmol) in DMSO (1 mL) were added 68a (11.4 mg, 0.02 mmol, 1.0 equiv), EDCI (5.8 mg, 0.03 mmol, 1.5 equiv), HOAt (4.1 mg, 0.03 mmol, 1.5 equiv), and NMM (6.1 mg, 0.06 mmol, 3.0 equiv). After being stirred overnight at rt, the resulting mixture was purified by preparative HPLC (10%−100% methanol / 0.1% TFA in H2O) to afford the corresponding product. After this product was dissolved in DCM (1 mL), the reaction mixture was treated with TFA (1 mL) for 30 min. After the solvent was removed, the resulting residue was purified by preparative HPLC (10%−100% methanol / 0.1% TFA in H2O) to afford compound 3 as white solid (7.8 mg, yield 38% for two steps). 1H NMR (800 MHz, CD3OD) δ 8.88 (s, 1H), 8.30 (d, J = 8.3 Hz, 1H), 8.03 (d, J = 17.2 Hz, 1H), 7.99 (d, J = 8.3 Hz, 1H), 7.91 – 7.78 (m, 5H), 7.72 (d, J = 8.0 Hz, 2H), 7.42 (d, J = 7.9 Hz, 2H), 7.38 (d, J = 7.8 Hz, 2H), 7.33 (t, J = 7.7 Hz, 1H), 7.27 (d, J = 7.6 Hz, 1H), 6.83 (s, 1H), 4.70 (s, 1H), 4.60 (t, J = 8.3 Hz, 1H), 4.56 – 4.47 (m, 2H), 4.32 (d, J = 15.4 Hz, 1H), 3.93 – 3.86 (m, 2H), 3.86 – 3.78 (m, 2H), 3.48 (dd, J = 10.1, 5.3 Hz, 1H), 3.40 (dt, J = 9.7, 5.2 Hz, 1H), 3.35 – 3.29 (m, 2H), 3.00 (t, J = 7.6 Hz, 2H), 2.93 – 2.89 (m, 2H), 2.73 – 2.67 (m, 2H), 2.58 (t, J = 7.6 Hz, 2H), 2.44 (s, 3H), 2.42 – 2.30 (m, 1H), 2.26 (dd, J = 13.2, 7.6 Hz, 1H), 2.15 – 2.03 (m, 2H), 1.03 (s, 9H). HRMS (m/z) for C56H64N11O6S+ [M + H]+: calcd: 1018.4756, found, 1018.4762.
(2S,4R)-1-((S)-2-(3-(2-(3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)propanamido)ethoxy)propanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (4).
Compound 4 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 68b (11.9 mg, 0.02 mmol, 1.0 equiv). Compound 4 was obtained as white solid (14.6 mg, yield 71%). 1H NMR (800 MHz, CD3OD) δ 8.96 (s, 1H), 8.31 (dd, J = 8.5, 4.3 Hz, 1H), 8.05 – 7.97 (m, 2H), 7.91 – 7.85 (m, 2H), 7.84 (d, J = 7.6 Hz, 1H), 7.81 (d, J = 8.0 Hz, 2H), 7.72 (d, J = 8.1 Hz, 2H), 7.42 (d, J = 7.9 Hz, 2H), 7.37 (dd, J = 23.2, 7.8 Hz, 3H), 7.29 (d, J = 7.5 Hz, 1H), 6.84 (t, J = 6.9 Hz, 1H), 4.64 (s, 1H), 4.63 – 4.54 (m, 1H), 4.52 – 4.44 (m, 2H), 4.33 (d, J = 15.4 Hz, 1H), 3.88 (d, J = 10.9 Hz, 1H), 3.79 (dd, J = 11.2, 4.0 Hz, 1H), 3.54 – 3.50 (m, 1H), 3.49– 3.47 (m, 1H), 3.40 – 3.35 (m, 2H), 3.30 (t, J = 5.3 Hz, 2H), 2.98 (t, J = 7.6 Hz, 2H), 2.94 – 2.91 (m, 2H), 2.77 – 2.67 (m, 2H), 2.56 (t, J = 7.6 Hz, 2H), 2.47 (s, 3H), 2.41 – 2.31 (m, 3H), 2.24 (dd, J = 13.3, 7.6 Hz, 1H), 2.10 – 2.06 (m, 2H), 1.01 (s, 9H). HRMS (m/z) for C57H66N11O6S+ [M + H]+: calcd: 1032.4913, found, 1032.4912.
(2S,4R)-1-((S)-15-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-2-(tert-butyl)-4,13-dioxo-6,9-dioxa-3,12-diazapentadecanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (5).
Compound 5 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 68c (12.2 mg, 0.02 mmol, 1.0 equiv). Compound 5 was obtained as white solid (10.5 mg, yield 49%). 1H NMR (800 MHz, CD3OD) δ 8.94 (s, 1H), 8.31 (dd, J = 8.3, 3.7 Hz, 1H), 8.02 (dd, J = 18.9, 7.4 Hz, 2H), 7.93 – 7.86 (m, 2H), 7.82 (dd, J = 20.7, 8.0 Hz, 3H), 7.72 (d, J = 8.0 Hz, 2H), 7.49 – 7.32 (m, 5H), 7.28 (d, J = 7.7 Hz, 1H), 6.84 (t, J = 6.8 Hz, 1H), 4.73 (s, 1H), 4.62 – 4.56 (m, 1H), 4.54 – 4.45 (m, 2H), 4.40 – 4.32 (m, 1H), 4.01 – 3.93 (m, 1H), 3.93 – 3.86 (m, 2H), 3.82 (dd, J = 11.2, 4.1 Hz, 1H), 3.66 – 3.54 (m, 2H), 3.52 – 3.36 (m, 5H), 3.27 – 3.24 (m, 1H), 3.02 – 2.95 (m, 2H), 2.90 (dd, J = 12.7, 7.3 Hz, 2H), 2.75 – 2.69 (m, 2H), 2.60 – 2.57 (m, 1H), 2.55 – 2.52 (m, 1H), 2.47 (s, 3H), 2.35 – 2.33 (m, 1H), 2.25 (dd, J = 13.4, 7.7 Hz, 1H), 2.11 – 2.07 (m, 2H), 1.02 (s, 9H). HRMS (m/z) for C58H68N11O7S+ [M + H]+: calcd: 1062.5018, found, 1062.5023.
(2S,4R)-1-((S)-16-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-2-(tert-butyl)-4,14-dioxo-7,10-dioxa-3,13-diazahexadecanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (6).
Compound 6 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 68d (16.3 mg, 0.02 mmol, 1.0 equiv). Compound 6 was obtained as white solid (12.6 mg, yield 58%). 1H NMR (800 MHz, CD3OD) δ 8.95 (d, J = 7.0 Hz, 1H), 8.31 (d, J = 8.4 Hz, 1H), 8.03 (t, J = 6.4 Hz, 2H), 7.92 – 7.86 (m, 2H), 7.83 (dd, J = 20.3, 7.9 Hz, 3H), 7.73 (d, J = 8.0 Hz, 2H), 7.48 – 7.38 (m, 4H), 7.40 – 7.33 (m, 1H), 7.29 (d, J = 7.6 Hz, 1H), 6.84 (t, J = 7.0 Hz, 1H), 4.65 (s, 1H), 4.61 – 4.56 (m, 1H), 4.56 – 4.49 (m, 2H), 4.40 – 4.34 (m, 1H), 3.89 (d, J = 10.9 Hz, 1H), 3.80 (dd, J = 11.1, 4.1 Hz, 1H), 3.72 – 3.62 (m, 2H), 3.52 (t, J = 4.5 Hz, 2H), 3.47 (t, J = 5.1 Hz, 2H), 3.40 (t, J = 5.4 Hz, 2H), 3.29 (t, J = 5.4 Hz, 2H), 2.99 (t, J = 7.6 Hz, 2H), 2.93– 2.89 (m, 2H), 2.74 – 2.70 (m, 2H), 2.53 (s, 3H), 2.49 – 2.42 (m, 4H), 2.36 – 2.30 (m, 1H), 2.23 (dd, J = 13.4, 7.6 Hz, 1H), 2.12 – 2.03 (m, 2H), 1.03 (s, 9H). HRMS (m/z) for C59H70N11O7S+ [M + H]+: calcd: 1076.5175, found, 1076.5164.
(2S,4R)-1-((S)-18-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-2-(tert-butyl)-4,16-dioxo-6,9,12-trioxa-3,15-diazaoctadecanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (7).
Compound 7 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 68e (12.2 mg, 0.02 mmol, 1.0 equiv). Compound 7 was obtained as white solid (16.4 mg, yield 74%). 1H NMR (800 MHz, CD3OD) δ 8.93 (s, 1H), 8.31 (d, J = 8.3 Hz, 1H), 8.03 (t, J = 6.9 Hz, 2H), 7.91 (s, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.83 (dd, J = 19.6, 7.8 Hz, 3H), 7.73 (d, J = 5.3 Hz, 2H), 7.46 (d, J = 8.1 Hz, 2H), 7.41 (d, J = 7.9 Hz, 2H), 7.37 (t, J = 7.7 Hz, 1H), 7.29 (d, J = 7.5 Hz, 1H), 6.84 (t, J = 6.9 Hz, 1H), 4.70 (s, 1H), 4.59 (t, J = 8.6 Hz, 1H), 4.52 (d, J = 4.3 Hz, 2H), 4.37 (d, J = 15.4 Hz, 1H), 4.06 – 3.96 (m, 4H), 3.88 (d, J = 11.0 Hz, 1H), 3.81 (dd, J = 11.2, 4.0 Hz, 1H), 3.69 – 3.62 (m, 4H), 3.57 (q, J = 4.5 Hz, 2H), 3.47 (t, J = 4.7 Hz, 2H), 3.38 (t, J = 5.5 Hz, 2H), 3.27 (t, J = 5.4 Hz, 2H), 2.99 (t, J = 7.6 Hz, 2H), 2.91 (dt, J = 14.3, 8.7 Hz, 2H), 2.76 – 2.68 (m, 2H), 2.53 (t, J = 7.6 Hz, 2H), 2.47 (s, 3H), 2.38 – 2.29 (m, 1H), 2.25 (dd, J = 12.8, 7.9 Hz, 1H), 2.14 – 2.04 (m, 2H), 1.04 (s, 9H). HRMS (m/z) for C60H72N11O8S+ [M + H]+: calcd: 1106.5281, found, 1106.5268.
(2S,4R)-1-((S)-19-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-2-(tert-butyl)-4,17-dioxo-7,10,13-trioxa-3,16-diazanonadecanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (8).
Compound 8 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 68f (12.2 mg, 0.02 mmol, 1.0 equiv). Compound 8 was obtained as white solid (12.2 mg, yield 54%). 1H NMR (800 MHz, CD3OD) δ 8.95 (s, 1H), 8.32 (d, J = 8.4 Hz, 1H), 8.04 (t, J = 6.5 Hz, 2H), 7.92 (s, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.85 (d, J = 7.6 Hz, 1H), 7.82 (d, J = 8.0 Hz, 2H), 7.73 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 7.8 Hz, 2H), 7.42 (d, J = 7.8 Hz, 2H), 7.37 (t, J = 7.7 Hz, 1H), 7.30 (d, J = 7.5 Hz, 1H), 6.84 (t, J = 6.9 Hz, 1H), 4.65 (s, 1H), 4.58 (t, J = 8.5 Hz, 1H), 4.54 (d, J = 15.4 Hz, 1H), 4.52 – 4.50 (m, 1H), 4.40 – 4.34 (m, 1H), 3.90 (d, J = 10.9 Hz, 1H), 3.81 (dd, J = 11.1, 4.1 Hz, 1H), 3.74 – 3.66 (m, 2H), 3.63 – 3.55 (m, 4H), 3.53 (t, J = 4.7 Hz, 2H), 3.44 (t, J = 4.7 Hz, 2H), 3.39 (d, J = 5.4 Hz, 2H), 3.28 (t, J = 5.5 Hz, 2H), 3.00 (t, J = 7.6 Hz, 2H), 2.93 – 2.88 (m, 2H), 2.74 – 2.72 (m, 2H), 2.60 – 2.51 (m, 3H), 2.51 – 2.43 (m, 4H), 2.37 – 2.32 (m, 1H), 2.24 (dd, J = 13.4, 7.6 Hz, 1H), 2.11 – 2.08 (m, 2H), 1.04 (s, 9H). HRMS (m/z) for C61H74N11O8S+ [M + H]+: calcd: 1120.5437, found, 1120.5423.
(2S,4R)-1-((S)-22-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-2-(tert-butyl)-4,20-dioxo-7,10,13,16-tetraoxa-3,19-diazadocosanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (9).
Compound 9 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 68g (14.2 mg, 0.02 mmol, 1.0 equiv). Compound 9 was obtained as white solid (12.2 mg, yield 52%). 1H NMR (800 MHz, CD3OD) δ 8.92 (s, 1H), 8.31 (d, J = 8.3 Hz, 1H), 8.08 – 8.00 (m, 2H), 7.92 (s, 1H), 7.89 (d, J = 7.8 Hz, 1H), 7.83 (dd, J = 12.6, 7.9 Hz, 3H), 7.73 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 7.9 Hz, 2H), 7.42 (d, J = 7.9 Hz, 2H), 7.37 (t, J = 7.7 Hz, 1H), 7.30 (d, J = 7.6 Hz, 1H), 6.83 (t, J = 6.9 Hz, 1H), 4.65 (s, 1H), 4.62 – 4.47 (m, 3H), 4.37 (d, J = 15.4 Hz, 1H), 3.90 (d, J = 10.9 Hz, 1H), 3.81 (dd, J = 11.1, 4.1 Hz, 1H), 3.73 – 3.69 (m, 2H), 3.64 – 3.55 (m, 8H), 3.52 (t, J = 4.6 Hz, 2H), 3.44 (t, J = 4.7 Hz, 2H), 3.39 (t, J = 5.5 Hz, 2H), 3.28 (t, J = 5.4 Hz, 2H), 3.00 (t, J = 7.6 Hz, 2H), 2.95 – 2.87 (m, 2H), 2.77 – 2.68 (m, 2H), 2.59 – 2.51 (m, 3H), 2.51 – 2.44 (m, 4H), 2.33 (dd, J = 13.4, 8.2 Hz, 1H), 2.24 (dd, J = 13.2, 7.7 Hz, 1H), 2.12 – 2.04 (m, 2H), 1.04 (s, 9H). HRMS (m/z) for C63H78N11O9S+ [M + H]+: calcd: 1164.5699, found, 1164.5673.
(2S,4R)-1-((S)-25-(3-(3-(4-(1-aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-2-(tert-butyl)-4,23-dioxo-7,10,13,16,19-pentaoxa-3,22-diazapentacosanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (10).
Compound 10 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 68h (14.2 mg, 0.02 mmol, 1.0 equiv). Compound 10 was obtained as white solid (13.9 mg, yield 58%). 1H NMR (800 MHz, CD3OD) δ 8.91 (s, 1H), 8.32 (d, J = 8.4 Hz, 1H), 8.04 (d, J = 8.0 Hz, 2H), 7.92 (s, 1H), 7.89 (d, J = 7.8 Hz, 1H), 7.83 (dd, J = 12.5, 7.9 Hz, 3H), 7.73 (d, J = 8.0 Hz, 2H), 7.48 (d, J = 7.8 Hz, 2H), 7.42 (d, J = 7.8 Hz, 2H), 7.38 (t, J = 7.7 Hz, 1H), 7.30 (d, J = 7.5 Hz, 1H), 6.83 (t, J = 6.9 Hz, 1H), 4.66 (s, 1H), 4.63 – 4.53 (m, 2H), 4.51 (s, 1H), 4.37 (d, J = 15.4 Hz, 1H), 3.90 (d, J = 10.9 Hz, 1H), 3.81 (dd, J = 11.1, 4.1 Hz, 1H), 3.74 – 3.71 (m, 2H), 3.62 – 3.56 (m, 12H), 3.52 (t, J = 4.6 Hz, 2H), 3.44 (t, J = 4.6 Hz, 2H), 3.39 (t, J = 5.5 Hz, 2H), 3.27 (t, J = 5.5 Hz, 2H), 3.00 (t, J = 7.6 Hz, 2H), 2.95 – 2.88 (m, 2H), 2.78 – 2.69 (m, 2H), 2.60 – 2.51 (m, 3H), 2.51 – 2.41 (m, 4H), 2.36 – 2.33 (m, 1H), 2.24 (dd, J = 13.1, 7.7 Hz, 1H), 2.13 – 2.03 (m, 2H), 1.04 (s, 9H). HRMS (m/z) for C65H82N11O10S+ [M + H]+: calcd: 1208.5961, found, 1208.5948.
(2S,4R)-1-((S)-2-(2-(3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)propanamido)acetamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (11).
Compound 11 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 69a (14.3 mg, 0.02 mmol, 1.0 equiv). Compound 11 was obtained as white solid (11.7 mg, yield 60%). 1H NMR (800 MHz, CD3OD) δ 8.88 (s, 1H), 8.30 (d, J = 8.4 Hz, 1H), 8.07 – 7.98 (m, 2H), 7.92 (s, 1H), 7.87 (d, J = 7.8 Hz, 1H), 7.83 – 7.80 (m, 3H), 7.73 (s, 2H), 7.48 (d, J = 7.9 Hz, 2H), 7.41 (d, J = 7.8 Hz, 2H), 7.36 (t, J = 7.6 Hz, 1H), 7.31 (d, J = 7.5 Hz, 1H), 6.83 (t, J = 6.9 Hz, 1H), 4.64 (s, 1H), 4.61 – 4.53 (m, 2H), 4.52 (d, J = 4.4 Hz, 1H), 4.36 (d, J = 15.4 Hz, 1H), 3.89 – 3.79 (m, 4H), 3.03 (t, J = 7.8 Hz, 2H), 2.92 – 2.88 (m, 2H), 2.74 – 2.70 (m, 2H), 2.63 (t, J = 7.8 Hz, 2H), 2.46 (s, 3H), 2.34 (dd, J = 10.8, 6.3 Hz, 1H), 2.25 (dd, J = 13.3, 7.6 Hz, 1H), 2.16 – 2.02 (m, 2H), 1.03 (s, 9H). HRMS (m/z) for C54H60N11O5S+ [M + H]+: calcd: 974.4494, found, 974.4486.
(2S,4R)-1-((S)-2-(3-(3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)propanamido)propanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (12).
Compound 12 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 69b (14.6 mg, 0.02 mmol, 1.0 equiv). Compound 12 was obtained as white solid (13.5 mg, yield 68%). 1H NMR (800 MHz, CD3OD) δ 8.93 (s, 1H), 8.30 (d, J = 8.4 Hz, 1H), 8.02 (dd, J = 11.7, 7.3 Hz, 2H), 7.94 – 7.79 (m, 5H), 7.72 (d, J = 8.0 Hz, 2H), 7.45 (d, J = 7.9 Hz, 2H), 7.40 (d, J = 7.9 Hz, 2H), 7.38 – 7.35 (m, 1H), 7.29 (d, J = 7.6 Hz, 1H), 6.84 (t, J = 6.9 Hz, 1H), 4.61 – 4.46 (m, 4H), 4.35 (d, J = 15.4 Hz, 1H), 3.91 (d, J = 10.9 Hz, 1H), 3.80 (dd, J = 11.1, 4.1 Hz, 1H), 3.42 – 3.35 (m, 2H), 2.99 (t, J = 7.6 Hz, 2H), 2.95 – 2.88 (m, 2H), 2.76 – 2.64 (m, 2H), 2.51 (t, J = 7.6 Hz, 2H), 2.47 (s, 3H), 2.40 – 2.28 (m, 3H), 2.23 (dd, J = 13.3, 7.5 Hz, 1H), 2.11 – 2.07 (m, 2H), 1.01 (s, 9H). HRMS (m/z) for C55H62N11O5S+ [M + H]+: calcd: 988.4651, found, 988.4638.
(2S,4R)-1-((S)-2-(4-(3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)propanamido)butanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (13).
Compound 13 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 69c (14.8 mg, 0.02 mmol, 1.0 equiv). Compound 13 was obtained as white solid (13.8 mg, yield 69%). 1H NMR (800 MHz, CD3OD) δ 8.94 (s, 1H), 8.31 (d, J = 8.4 Hz, 1H), 8.03 (dd, J = 11.1, 7.1 Hz, 2H), 7.92 (s, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.85 (d, J = 7.7 Hz, 1H), 7.81 (d, J = 8.1 Hz, 2H), 7.73 (d, J = 8.1 Hz, 2H), 7.48 (d, J = 7.8 Hz, 2H), 7.42 (d, J = 7.9 Hz, 2H), 7.37 (q, J = 7.9 Hz, 1H), 7.30 (d, J = 7.7 Hz, 1H), 6.84 (t, J = 7.0 Hz, 1H), 4.60 – 4.56 (m, 2H), 4.55 (s, 1H), 4.52 (d, J = 3.2 Hz, 1H), 4.37 (d, J = 15.4 Hz, 1H), 3.90 (d, J = 10.9 Hz, 1H), 3.81 (dd, J = 11.1, 4.1 Hz, 1H), 3.16 – 3.10 (m, 2H), 3.02 – 2.98 (m, 2H), 2.94 – 2.89 (m, 2H), 2.74 – 2.73 (m, 2H), 2.53 (t, J = 7.5 Hz, 2H), 2.48 (s, 3H), 2.39 – 2.29 (m, 1H), 2.28 – 2.21 (m, 1H), 2.17 – 2.02 (m, 4H), 1.73 – 1.64 (m, 2H), 1.03 (s, 9H). HRMS (m/z) for C56H64N11O5S+ [M + H]+: calcd: 1002.4807, found, 1002.4804.
(2S,4R)-1-((S)-2-(5-(3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)propanamido)pentanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (14).
Compound 14 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 69d (11.3 mg, 0.02 mmol, 1.0 equiv). Compound 14 was obtained as white solid (15.5 mg, yield 76%). 1H NMR (800 MHz, CD3OD) δ 8.93 (s, 1H), 8.32 (d, J = 8.4 Hz, 1H), 8.03 (dd, J = 8.4, 4.5 Hz, 2H), 7.92 (s, 1H), 7.88 (d, J = 8.0 Hz, 1H), 7.85 (d, J = 7.5 Hz, 1H), 7.81 (d, J = 8.0 Hz, 2H), 7.77 – 7.67 (m, 2H), 7.47 (d, J = 7.8 Hz, 2H), 7.44 – 7.33 (m, 3H), 7.32 – 7.25 (m, 1H), 6.84 (t, J = 7.0 Hz, 1H), 4.60 (s, 1H), 4.60 – 4.49 (m, 3H), 4.37 (d, J = 15.4 Hz, 1H), 3.90 (d, J = 10.9 Hz, 1H), 3.81 (dd, J = 11.1, 4.1 Hz, 1H), 3.11 (t, J = 6.9 Hz, 2H), 3.00 (t, J = 7.5 Hz, 2H), 2.92 (dt, J = 14.5, 8.6 Hz, 2H), 2.72 (dt, J = 13.2, 8.0 Hz, 2H), 2.56 – 2.50 (m, 2H), 2.48 (s, 3H), 2.37 – 2.30 (m, 1H), 2.26 – 2.21 (m, 1H), 2.19 – 2.14 (m, 1H), 2.13 – 2.05 (m, 2H), 1.50 – 1.41 (m, 2H), 1.40 – 1.32 (m, 2H), 1.03 (s, 9H). HRMS (m/z) for C57H66N11O5S+ [M + H]+: calcd: 1016.4964, found, 1016.4976.
(2S,4R)-1-((S)-2-(6-(3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)propanamido)hexanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (15).
Compound 15 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 69e (11.6 mg, 0.02 mmol, 1.0 equiv). Compound 15 was obtained as white solid (13.6 mg, yield 66%). 1H NMR (800 MHz, CD3OD) δ 8.97 (s, 1H), 8.31 (d, J = 8.4 Hz, 1H), 8.03 (t, J = 6.8 Hz, 2H), 7.92 (s, 1H), 7.88 (dd, J = 18.0, 7.8 Hz, 2H), 7.83 – 7.79 (m, 2H), 7.74 (d, J = 8.1 Hz, 2H), 7.48 (d, J = 7.8 Hz, 2H), 7.43 (d, J = 7.8 Hz, 2H), 7.38 (t, J = 7.7 Hz, 1H), 7.30 (d, J = 7.6 Hz, 1H), 6.85 (t, J = 7.0 Hz, 1H), 4.62 (s, 1H), 4.61 – 4.57 (m, 1H), 4.55 (d, J = 15.4 Hz, 1H), 4.52 (s, 1H), 4.38 (d, J = 15.4 Hz, 1H), 3.91 (d, J = 10.9 Hz, 1H), 3.81 (dd, J = 11.1, 4.1 Hz, 1H), 3.11 – 3.08 (m, 2H), 2.99 (t, J = 7.5 Hz, 2H), 2.94 – 2.90 (m, 2H), 2.77 – 2.69 (m, 2H), 2.52 (d, J = 7.5 Hz, 2H), 2.48 (s, 3H), 2.39 – 2.30 (m, 1H), 2.24 (dd, J = 13.4, 7.6 Hz, 1H), 2.19 – 2.16 (m, 1H), 2.15 – 2.01 (m, 3H), 1.48 – 1.44 (m, 2H), 1.38 – 1.35 (m, 2H), 1.20 – 1.14 (m, 2H), 1.03 (s, 9H). HRMS (m/z) for C58H68N11O5S+ [M + H]+: calcd: 1030.5120, found,1030.5111.
(2S,4R)-1-((S)-2-(7-(3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)propanamido)heptanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (16).
Compound 16 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 69f (11.8 mg, 0.02 mmol, 1.0 equiv). Compound 16 was obtained as white solid (8.4 mg, yield 40%). 1H NMR (800 MHz, CD3OD) δ 8.95 (s, 1H), 8.32 (d, J = 8.3 Hz, 1H), 8.04 (q, J = 3.7 Hz, 2H), 7.93 – 7.87 (m, 2H), 7.86 (d, J = 7.6 Hz, 1H), 7.82 (d, J = 8.0 Hz, 2H), 7.74 (d, J = 8.0 Hz, 2H), 7.48 (d, J = 7.8 Hz, 2H), 7.43 (d, J = 7.8 Hz, 2H), 7.41 – 7.35 (m, 1H), 7.29 (d, J = 7.5 Hz, 1H), 6.85 (t, J = 6.9 Hz, 1H), 4.63 (s, 1H), 4.60 (t, J = 8.0 Hz, 1H), 4.57 – 4.49 (m, 2H), 4.38 (d, J = 15.4 Hz, 1H), 3.92 (d, J = 10.9 Hz, 1H), 3.82 (dd, J = 11.1, 4.1 Hz, 1H), 3.08 (t, J = 7.1 Hz, 2H), 2.99 (t, J = 7.5 Hz, 2H), 2.93 – 2.90 (m, 2H), 2.76 – 2.67 (m, 2H), 2.52 (d, J = 7.5 Hz, 2H), 2.48 (s, 3H), 2.35 – 2.33 (m, 1H), 2.28 – 2.03 (m, 5H), 1.53 – 1.44 (m, 2H), 1.35 – 1.32 (m, 2H), 1.21 – 1.12 (m, 4H), 1.04 (s, 9H). HRMS (m/z) for C59H70N11O5S+ [M + H]+: calcd: 1044.5277, found, 1044.5284.
(2S,4R)-1-((S)-2-(8-(3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)propanamido)octanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (17).
Compound 17 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 69g (16 mg, 0.02 mmol, 1.0 equiv). Compound 17 was obtained as white solid (9.6 mg, yield 45%). 1H NMR (800 MHz, CD3OD) δ 8.96 (s, 1H), 8.32 (d, J = 8.3 Hz, 1H), 8.07 – 8.00 (m, 2H), 7.92 (s, 1H), 7.89 (d, J = 7.8 Hz, 1H), 7.86 (d, J = 7.6 Hz, 1H), 7.82 (d, J = 8.0 Hz, 2H), 7.73 (d, J = 8.1 Hz, 2H), 7.48 (d, J = 7.8 Hz, 2H), 7.43 (d, J = 7.8 Hz, 2H), 7.37 (t, J = 7.7 Hz, 1H), 7.29 (d, J = 7.6 Hz, 1H), 6.84 (t, J = 6.9 Hz, 1H), 4.64 (s, 1H), 4.59 (t, J = 8.3 Hz, 1H), 4.56 (d, J = 15.4 Hz, 1H), 4.52 (d, J = 4.2 Hz, 1H), 4.38 (d, J = 15.4 Hz, 1H), 3.91 (d, J = 11.0 Hz, 1H), 3.82 (dd, J = 11.1, 4.1 Hz, 1H), 3.08 (q, J = 6.7 Hz, 2H), 2.99 (t, J = 7.4 Hz, 2H), 2.92 (tt, J = 9.3, 5.8 Hz, 2H), 2.75 – 2.68 (m, 2H), 2.52 (t, J = 7.4 Hz, 2H), 2.49 (s, 3H), 2.37 – 2.31 (m, 1H), 2.28 – 2.21 (m, 2H), 2.19 – 2.16 (m, 1H), 2.14 – 2.04 (m, 2H), 1.57 – 1.45 (m, 2H), 1.33 (p, J = 7.1 Hz, 2H), 1.24 – 1.09 (m, 6H), 1.04 (s, 9H). HRMS (m/z) for C60H72N11O5S+ [M + H]+: calcd: 1058.5433, found, 1058.5425.
(2S,4R)-1-((S)-2-(9-(3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)propanamido)nonanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (18).
Compound 18 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 69h (12.4 mg, 0.02 mmol, 1.0 equiv). Compound 18 was obtained as white solid (8.6 mg, yield 40%). 1H NMR (800 MHz, CD3OD) δ 8.92 (s, 1H), 8.32 (d, J = 8.4 Hz, 1H), 8.07 – 8.00 (m, 2H), 7.92 (s, 1H), 7.89 (d, J = 7.7 Hz, 1H), 7.83 (dd, J = 14.8, 7.8 Hz, 3H), 7.77 – 7.69 (m, 2H), 7.48 (d, J = 7.8 Hz, 2H), 7.43 (d, J = 7.8 Hz, 2H), 7.37 (t, J = 7.7 Hz, 1H), 7.29 (d, J = 7.6 Hz, 1H), 6.83 (t, J = 6.9 Hz, 1H), 4.65 (s, 1H), 4.60 (t, J = 8.1 Hz, 1H), 4.57 – 4.50 (m, 2H), 4.38 (d, J = 15.4 Hz, 1H), 3.92 (d, J = 10.9 Hz, 1H), 3.82 (dd, J = 11.1, 4.1 Hz, 1H), 3.08 (t, J = 7.1 Hz, 2H), 2.99 (t, J = 7.4 Hz, 2H), 2.94 – 2.92 (m, 2H), 2.76 – 2.67 (m, 2H), 2.52 (t, J = 7.6 Hz, 2H), 2.48 (s, 3H), 2.36 – 2.33 (m, 1H), 2.25 – 2.20 (m, 3H), 2.12 – 2.09 (m, 2H), 1.55 – 1.52 (m, 2H), 1.33 – 1.20 (m, 2H), 1.27 – 1.09 (m, 8H), 1.05 (s, 9H). HRMS (m/z) for C61H74N11O5S+ [M + H]+: calcd: 1072.5590, found, 1072.5598.
(2S,4R)-1-((S)-2-(10-(3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)propanamido)decanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (19).
Compound 19 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 69i (16.5 mg, 0.02 mmol, 1.0 equiv). Compound 19 was obtained as white solid (7.3 mg, yield 34%). 1H NMR (800 MHz, CD3OD) δ 8.92 (s, 1H), 8.32 (d, J = 8.3 Hz, 1H), 8.09 – 8.00 (m, 2H), 7.92 (s, 1H), 7.89 (d, J = 7.8 Hz, 1H), 7.82 (t, J = 7.9 Hz, 3H), 7.73 (d, J = 8.1 Hz, 2H), 7.48 (d, J = 7.8 Hz, 2H), 7.43 (d, J = 7.9 Hz, 2H), 7.37 (t, J = 7.7 Hz, 1H), 7.30 (d, J = 7.4 Hz, 1H), 6.83 (t, J = 6.9 Hz, 1H), 4.65 (s, 1H), 4.62 – 4.51 (m, 3H), 4.38 (d, J = 15.4 Hz, 1H), 3.92 (d, J = 10.9 Hz, 1H), 3.83 (dd, J = 11.1, 4.1 Hz, 1H), 3.08 (t, J = 7.1 Hz, 2H), 2.99 (t, J = 7.5 Hz, 2H), 2.92 (q, J = 9.8, 7.7 Hz, 2H), 2.75 – 2.70 (m, 2H), 2.54 – 2.46 (m, 5H), 2.38 – 2.31 (m, 1H), 2.30 – 2.18 (m, 3H), 2.15 – 2.02 (m, 2H), 1.58 – 1.56 (m, 2H), 1.32 (t, J = 7.1 Hz, 2H), 1.29 – 1.23 (m, 2H), 1.23 – 1.19 (m, 2H), 1.18 – 1.14 (m, 2H), 1.14 – 1.11 (m, 4H), 1.06 (s, 9H). HRMS (m/z) for C62H76N11O5S+ [M + H]+: calcd: 1086.5746, found, 1086.5721.
(2S,4R)-1-((S)-2-(11-(3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)propanamido)undecanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (20).
Compound 20 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 69j (16.5 mg, 0.02 mmol, 1.0 equiv). Compound 20 was obtained as white solid (8.7 mg, yield 37%). 1H NMR (800 MHz, CD3OD) δ 8.92 (s, 1H), 8.31 (d, J = 8.3 Hz, 1H), 8.05 – 8.01 (m, 2H), 7.93 (s, 1H), 7.89 (d, J = 7.7 Hz, 1H), 7.82 (t, J = 6.9 Hz, 3H), 7.75 – 7.68 (m, 2H), 7.48 (d, J = 7.9 Hz, 2H), 7.43 (d, J = 7.9 Hz, 2H), 7.37 (t, J = 7.6 Hz, 1H), 7.30 (d, J = 7.4 Hz, 1H), 6.83 (t, J = 6.9 Hz, 1H), 4.65 (s, 1H), 4.62 – 4.58 (m, 1H), 4.56 (d, J = 15.5 Hz, 1H), 4.52 (d, J = 3.8 Hz, 1H), 4.41 – 4.35 (m, 1H), 3.92 (d, J = 10.9 Hz, 1H), 3.85 – 3.79 (m, 1H), 3.08 (t, J = 7.1 Hz, 2H), 3.00 (t, J = 7.5 Hz, 2H), 2.94 – 2.90 (m, 2H), 2.77 – 2.68 (m, 2H), 2.52 (t, J = 7.5 Hz, 2H), 2.49 (s, 3H), 2.36 – 2.34 (m, 1H), 2.30 – 2.28 (m, 1H), 2.26 – 2.23 (m, 2H), 2.14 – 2.02 (m, 2H), 1.64 – 1.52 (m, 2H), 1.37 – 1.23 (m, 6H), 1.22 – 1.10 (m, 8H), 1.05 (s, 9H). HRMS (m/z) for C63H78N11O5S+ [M + H]+: calcd: 1110.5903, found, 1110.5912.
3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-N-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethyl)propenamide (21).
Compound 21 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 70a (9.4 mg, 0.02 mmol, 1.0 equiv). Compound 21 was obtained as yellow solid (10.9 mg, yield 64%). 1H NMR (800 MHz, CD3OD) δ 8.22 (d, J = 8.4 Hz, 1H), 8.03 (d, J = 6.2 Hz, 1H), 8.02 – 7.96 (m, 1H), 7.92 (s, 1H), 7.86 (d, J = 7.8 Hz, 1H), 7.79 (t, J = 8.0 Hz, 3H), 7.77 – 7.67 (m, 2H), 7.45 (t, J = 7.8 Hz, 1H), 7.32 (q, J = 7.8 Hz, 1H), 7.27 (d, J = 7.5 Hz, 1H), 6.98 – 6.93 (m, 1H), 6.88 (d, J = 8.5 Hz, 1H), 6.82 (t, J = 7.0 Hz, 1H), 4.95 – 4.92 (m, 1H), 3.48 – 3.37 (m, 3H), 3.35 – 3.32 (m, 1H), 3.33 – 3.23 (m, 2H), 3.27 – 3.25 (m, 2H), 3.03 – 2.98 (m, 2H), 2.92 – 2.89 (m, 2H), 2.72 – 2.68 (m, 3H), 2.67 – 2.53 (m, 4H), 2.34 – 2.31 (m, 1H), 2.09 – 2.06 (m, 1H), 1.96 (dd, J = 12.5, 6.4 Hz, 1H). HRMS (m/z) for C47H47N10O6+ [M + H]+: calcd: 847.3605, found, 847.3668.
3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-N-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethyl)propenamide (22).
Compound 22 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 70b (10.3 mg, 0.02 mmol, 1.0 equiv). Compound 22 was obtained as yellow solid (13.4 mg, yield 75%). 1H NMR (800 MHz, CD3OD) δ 8.28 (d, J = 8.4 Hz, 1H), 8.01 (dd, J = 14.4, 7.2 Hz, 2H), 7.92 – 7.85 (m, 2H), 7.80 (t, J = 7.8 Hz, 3H), 7.71 (d, J = 8.0 Hz, 2H), 7.46 (t, J = 7.8 Hz, 1H), 7.35 (t, J = 7.7 Hz, 1H), 7.27 (d, J = 7.5 Hz, 1H), 6.98 (t, J = 8.3 Hz, 2H), 6.81 (t, J = 6.9 Hz, 1H), 5.01 (dd, J = 12.8, 5.4 Hz, 1H), 3.64 (t, J = 5.2 Hz, 2H), 3.55 (t, J = 4.4 Hz, 2H), 3.48 (t, J = 4.5 Hz, 2H), 3.41 (dt, J = 10.5, 5.4 Hz, 4H), 3.30 (q, J = 5.8 Hz, 2H), 2.98 (t, J = 7.5 Hz, 2H), 2.94 – 2.87 (m, 2H), 2.84 – 2.79 (m, 1H), 2.75 – 2.67 (m, 3H), 2.68 – 2.62 (m, 1H), 2.52 (t, J = 7.5 Hz, 2H), 2.35 – 2.33 (m, 1H), 2.09 – 2.05 (m, 2H). HRMS (m/z) for C49H51N10O7+ [M + H]+: calcd: 891.3937, found, 891.3945.
3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-N-(2-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethoxy)ethyl)propenamide (23).
Compound 23 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 70c (11.2 mg, 0.02 mmol, 1.0 equiv). Compound 23 was obtained as yellow solid (11.9 mg, yield 64%). 1H NMR (800 MHz, CD3OD) δ 8.28 (d, J = 8.4 Hz, 1H), 8.02 (dd, J = 14.8, 7.2 Hz, 2H), 7.90 (s, 1H), 7.87 (d, J = 7.8 Hz, 1H), 7.81 (d, J = 7.9 Hz, 3H), 7.71 (d, J = 8.0 Hz, 2H), 7.47 (t, J = 7.8 Hz, 1H), 7.36 (t, J = 7.7 Hz, 1H), 7.28 (d, J = 7.6 Hz, 1H), 7.07 – 6.94 (m, 2H), 6.81 (t, J = 6.9 Hz, 1H), 5.01 (dd, J = 12.7, 5.3 Hz, 1H), 3.66 (t, J = 5.2 Hz, 2H), 3.64 – 3.60 (m, 4H), 3.55 (t, J = 4.5 Hz, 2H), 3.46 (t, J = 4.5 Hz, 2H), 3.44 – 3.38 (m, 4H), 3.27 (t, J = 5.4 Hz, 2H), 2.99 (t, J = 7.6 Hz, 2H), 2.94 – 2.87 (m, 2H), 2.83 – 2.81 (m, 1H), 2.74 – 2.62 (m, 4H), 2.54 (t, J = 7.6 Hz, 2H), 2.35 – 2.32 (m, 1H), 2.08 – 2.05 (m, 2H). HRMS (m/z) for C51H55N10O8+ [M + H]+: calcd: 935.4199, found, 935.4187.
3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-N-(14-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3,6,9,12-tetraoxatetradecyl)propenamide (24).
Compound 24 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 70d (11.3 mg, 0.02 mmol, 1.0 equiv). Compound 24 was obtained as yellow solid (9.5 mg, yield 49%). 1H NMR (800 MHz, CD3OD) δ 8.29 (s, 1H), 8.02 (d, J = 8.0 Hz, 2H), 7.91 (s, 1H), 7.87 (d, J = 7.9 Hz, 1H), 7.79 (d, J = 8.0 Hz, 2H), 7.71 (t, J = 11.1 Hz, 3H), 7.49 (t, J = 7.9 Hz, 1H), 7.37 (t, J = 7.8 Hz, 1H), 7.29 (d, J = 7.6 Hz, 1H), 7.04 – 6.99 (m, 2H), 6.76 (s, 1H), 5.01 (dd, J = 12.6, 5.3 Hz, 1H), 3.71 – 3.59 (m, 8H), 3.59 – 3.56 (m, 2H), 3.52 (t, J = 4.6 Hz, 2H), 3.43 (q, J = 4.9 Hz, 4H), 3.38 (t, J = 5.4 Hz, 2H), 3.27 (t, J = 5.4 Hz, 2H), 2.99 (t, J = 7.6 Hz, 2H), 2.91 (dd, J = 13.1, 7.1 Hz, 2H), 2.86 – 2.78 (m, 1H), 2.73 – 2.65 (m, 4H), 2.54 (t, J = 7.6 Hz, 2H), 2.33 (d, J = 10.2 Hz, 1H), 2.10 – 2.03 (m, 2H). HRMS (m/z) for C53H59N10O9+ [M + H]+: calcd: 979.4461, found, 979.4463.
3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-N-(17-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3,6,9,12,15-pentaoxaheptadecyl)propenamide (25).
Compound 25 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 70e (12.2 mg, 0.02 mmol, 1.0 equiv). Compound 25 was obtained as yellow solid (11.3 mg, yield 55%). 1H NMR (800 MHz, CD3OD) δ 8.30 (d, J = 8.3 Hz, 1H), 8.08 – 7.97 (m, 2H), 7.91 (s, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.81 (d, J = 8.3 Hz, 3H), 7.72 (d, J = 8.0 Hz, 2H), 7.50 (t, J = 7.8 Hz, 1H), 7.37 (t, J = 7.7 Hz, 1H), 7.29 (d, J = 7.5 Hz, 1H), 7.02 (dd, J = 17.2, 7.9 Hz, 2H), 6.82 (t, J = 6.9 Hz, 1H), 5.02 (dd, J = 12.6, 5.4 Hz, 1H), 3.68 (t, J = 5.1 Hz, 2H), 3.66 – 3.54 (m, 12H), 3.52 (q, J = 4.6 Hz, 2H), 3.46 – 3.40 (m, 4H), 3.38 (t, J = 5.4 Hz, 2H), 3.26 (t, J = 5.4 Hz, 2H), 2.99 (t, J = 7.5 Hz, 2H), 2.93 – 2.89 (m, 2H), 2.83 – 2.80 (m, 1H), 2.75 – 2.64 (m, 4H), 2.54 (t, J = 7.6 Hz, 2H), 2.34 (tt, J = 12.1, 6.4 Hz, 1H), 2.10 – 2.07 (m, 2H). HRMS (m/z) for C55H63N10O10+ [M + H]+: calcd: 1023.4723, found, 1023.4738.
3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-N-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethyl)propenamide (26).
Compound 26 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 71a (8.6 mg, 0.02 mmol, 1.0 equiv). Compound 26 was obtained as yellow solid (10.5 mg, yield 65%). 1H NMR (800 MHz, CD3OD) δ 8.22 (d, J = 8.4 Hz, 1H), 8.03 (d, J = 6.2 Hz, 1H), 7.99 – 7.93 (m, 1H), 7.91 (s, 1H), 7.85 (d, J = 7.8 Hz, 1H), 7.80 (t, J = 7.1 Hz, 3H), 7.66 (d, J = 8.0 Hz, 2H), 7.38 – 7.33 (m, 1H), 7.31 – 7.25 (m, 2H), 6.92 – 6.87 (m, 1H), 6.82 (t, J = 6.9 Hz, 1H), 6.70 (d, J = 8.5 Hz, 1H), 5.01 (dd, J = 12.7, 5.4 Hz, 1H), 3.30 – 3.25 (m, 2H), 3.16 – 3.10 (m, 2H), 3.01 (t, J = 7.2 Hz, 2H), 2.95 – 2.90 (m, 2H), 2.84 – 2.81 (m, 1H), 2.75 – 2.63 (m, 4H), 2.53 (t, J = 7.3 Hz, 2H), 2.35 – 2.32 (m, 1H), 2.10 – 2.07 (m, 2H). HRMS (m/z) for C45H43N10O5+ [M + H]+: calcd: 803.3412, found, 803.3417.
3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-N-(3-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)propyl)propenamide (27).
Compound 27 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 71b (8.9 mg, 0.02 mmol, 1.0 equiv). Compound 27 was obtained as yellow solid (16.3 mg, yield 99%). 1H NMR (800 MHz, CD3OD) δ 8.20 (d, J = 8.4 Hz, 1H), 8.03 (d, J = 6.2 Hz, 1H), 7.93 (s, 1H), 7.92 – 7.86 (m, 1H), 7.84 – 7.80 (m, 4H), 7.68 (d, J = 8.0 Hz, 2H), 7.39 (t, J = 7.7 Hz, 1H), 7.35 – 7.28 (m, 2H), 6.87 (d, J = 7.1 Hz, 1H), 6.83 (q, J = 6.6 Hz, 1H), 6.62 (d, J = 8.5 Hz, 1H), 5.00 (dd, J = 12.8, 5.5 Hz, 1H), 3.20 – 3.16 (m, 2H), 3.03 (t, J = 7.1 Hz, 2H), 2.95 – 2.90 (m, 2H), 2.87 – 2.79 (m, 3H), 2.77 – 2.66 (m, 4H), 2.56 (t, J = 7.0 Hz, 2H), 2.34 (dtd, J = 16.2, 10.3, 6.5 Hz, 1H), 2.16 – 2.13 (m, 1H), 2.09 – 2.06 (m, 1H), 1.57 – 1.53 (m, 2H). HRMS (m/z) for C46H45N10O5+ [M + H]+: calcd: 817.3569, found, 817.3578.
3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-N-(4-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)butyl)propenamide (28).
Compound 28 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 71c (9.2 mg, 0.02 mmol, 1.0 equiv). Compound 28 was obtained as yellow solid (10.5 mg, yield 63%). 1H NMR (800 MHz, CD3OD) δ 8.23 (d, J = 8.3 Hz, 1H), 8.02 (d, J = 6.3 Hz, 1H), 7.97 (d, J = 8.3 Hz, 1H), 7.90 – 7.85 (m, 2H), 7.78 (dd, J = 21.0, 7.8 Hz, 3H), 7.66 (d, J = 8.0 Hz, 2H), 7.38 (t, J = 7.7 Hz, 2H), 7.31 (d, J = 7.6 Hz, 1H), 6.97 – 6.91 (m, 1H), 6.82 (q, J = 9.2, 6.9 Hz, 1H), 6.67 (d, J = 8.4 Hz, 1H), 5.09 – 5.03 (m, 1H), 3.10 (t, J = 6.6 Hz, 2H), 3.01 (t, J = 7.1 Hz, 2H), 2.94 – 2.83 (m, 5H), 2.80 – 2.67 (m, 4H), 2.54 (t, J = 7.1 Hz, 2H), 2.35 – 2.31 (m, 1H), 2.16 – 2.12 (m, 1H), 2.07 – 2.05 (m, 1H), 1.34 – 1.29 (m, 2H), 1.24 (q, J = 7.6 Hz, 2H). HRMS (m/z) for C47H47N10O5+ [M + H]+: calcd: 831.3725, found, 831.3721.
3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-N-(5-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)pentyl)propenamide (29).
Compound 29 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 71d (9.7 mg, 0.02 mmol, 1.0 equiv). Compound 29 was obtained as yellow solid (9.7 mg, yield 66%). 1H NMR (800 MHz, CD3OD) δ 8.27 (d, J = 8.4 Hz, 1H), 8.01 (dd, J = 10.1, 7.0 Hz, 2H), 7.94 – 7.88 (m, 2H), 7.79 (d, J = 8.1 Hz, 2H), 7.71 (d, J = 7.7 Hz, 1H), 7.65 (d, J = 8.1 Hz, 2H), 7.41 (t, J = 7.7 Hz, 1H), 7.38 (t, J = 7.7 Hz, 1H), 7.31 (d, J = 7.6 Hz, 1H), 6.94 – 6.88 (m, 1H), 6.79 (t, J = 7.0 Hz, 1H), 6.76 (d, J = 8.5 Hz, 1H), 5.02 (dd, J = 12.7, 5.4 Hz, 1H), 3.14 – 3.08 (m, 2H), 3.01 (t, J = 7.2 Hz, 2H), 2.97 (t, J = 7.3 Hz, 2H), 2.93 – 2.87 (m, 2H), 2.86 – 2.79 (m, 1H), 2.77 – 2.64 (m, 4H), 2.54 (t, J = 7.2 Hz, 2H), 2.35 – 2.32 (m, 1H), 2.10 – 2.06 (m, 2H), 1.38 – 1.29 (m, 4H), 1.11 (q, J = 7.8 Hz, 2H). HRMS (m/z) for C48H49N10O5+ [M + H]+: calcd: 845.3882, found, 845.3867.
3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-N-(6-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)hexyl)propenamide (30).
Compound 30 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 71e (9.9 mg, 0.02 mmol, 1.0 equiv). Compound 30 was obtained as yellow solid (10.9 mg, yield 63%). 1H NMR (800 MHz, CD3OD) δ 8.28 (d, J = 8.3 Hz, 1H), 8.05 – 7.99 (m, 2H), 7.93 – 7.88 (m, 2H), 7.80 (d, J = 8.0 Hz, 2H), 7.70 (dd, J = 27.6, 7.9 Hz, 3H), 7.46 (t, J = 7.8 Hz, 1H), 7.37 (t, J = 7.7 Hz, 1H), 7.30 (d, J = 7.6 Hz, 1H), 6.95 (d, J = 7.1 Hz, 1H), 6.86 (d, J = 8.5 Hz, 1H), 6.78 (t, J = 6.9 Hz, 1H), 5.07 (dd, J = 12.6, 5.4 Hz, 1H), 3.09 (t, J = 6.8 Hz, 2H), 3.04 (t, J = 7.3 Hz, 2H), 3.01 (t, J = 7.2 Hz, 2H), 2.94 – 2.83 (m, 3H), 2.80 – 2.68 (m, 4H), 2.54 (t, J = 7.2 Hz, 2H), 2.35 – 2.33 (m, 1H), 2.14 – 2.11 (m, 1H), 2.09 – 2.06 (m, 1H), 1.40 – 1.36 (m, 2H), 1.31 (q, J = 7.3 Hz, 2H), 1.18 – 1.14 (m, 2H), 1.08 (q, J = 7.9 Hz, 2H). HRMS (m/z) for C49H51N10O5+ [M + H]+: calcd: 859.4038, found, 859.4043.
3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-N-(7-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)heptyl)propenamide (31).
Compound 31 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 71f (10.0 mg, 0.02 mmol, 1.0 equiv). Compound 31 was obtained as yellow solid (15.2 mg, yield 87%). 1H NMR (800 MHz, CD3OD) δ 8.29 (d, J = 8.4 Hz, 1H), 8.03 (d, J = 8.4 Hz, 1H), 8.00 (d, J = 6.3 Hz, 1H), 7.93 – 7.88 (m, 2H), 7.81 (d, J = 8.1 Hz, 2H), 7.74 (d, J = 7.6 Hz, 1H), 7.69 (d, J = 8.0 Hz, 2H), 7.50 (t, J = 7.8 Hz, 1H), 7.37 (t, J = 7.6 Hz, 1H), 7.30 (d, J = 7.5 Hz, 1H), 7.01 – 6.96 (m, 1H), 6.92 (d, J = 8.5 Hz, 1H), 6.78 (t, J = 6.9 Hz, 1H), 5.05 (dd, J = 12.7, 5.4 Hz, 1H), 3.14 – 3.05 (m, 4H), 3.00 (t, J = 7.2 Hz, 2H), 2.94 – 2.82 (m, 3H), 2.78 – 2.67 (m, 4H), 2.53 (t, J = 7.2 Hz, 2H), 2.35 – 2.32 (m, 1H), 2.13 – 2.03 (m, 2H), 1.46 – 1.43 (m, 2H), 1.31 – 1.28 (m, 2H), 1.21 – 1.18 (m, 2H), 1.13 – 1.03 (m, 4H). HRMS (m/z) for C50H53N10O5+ [M + H]+: calcd: 873.4195, found, 873.4186.
3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-N-(8-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)octyl)propenamide (32).
Compound 32 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 71g (10.2 mg, 0.02 mmol, 1.0 equiv). Compound 32 was obtained as yellow solid (11.1 mg, yield 63%). 1H NMR (800 MHz, CD3OD) δ 8.32 – 8.26 (m, 1H), 8.05 – 7.98 (m, 2H), 7.92 – 7.87 (m, 2H), 7.85 – 7.78 (m, 2H), 7.76 (d, J = 7.7 Hz, 1H), 7.70 (d, J = 8.1 Hz, 2H), 7.50 (t, J = 7.8 Hz, 1H), 7.40 – 7.33 (m, 1H), 7.30 (d, J = 7.6 Hz, 1H), 7.02 – 6.97 (m, 1H), 6.94 (d, J = 8.4 Hz, 1H), 6.79 (t, J = 7.0 Hz, 1H), 5.06 (dd, J = 12.8, 5.3 Hz, 1H), 3.17 (t, J = 7.2 Hz, 2H), 3.09 (t, J = 7.0 Hz, 2H), 3.00 (t, J = 7.3 Hz, 2H), 2.94 – 2.82 (m, 3H), 2.79 – 2.66 (m, 4H), 2.53 (t, J = 7.3 Hz, 2H), 2.36 – 2.34 (m, 1H), 2.13 – 2.04 (m, 2H), 1.52 – 1.47 (m, 2H), 1.34 – 1.28 (m, 2H), 1.26 (q, J = 7.4 Hz, 2H), 1.15 (q, J = 7.3 Hz, 2H), 1.13 – 1.08 (m, 4H). HRMS (m/z) for C51H55N10O5+ [M + H]+: calcd: 887.4351, found, 887.4367.
(2S,4R)-1-((S)-2-(2-(2-((3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)amino)-2-oxoethoxy)acetamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (33).
Compound 33 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 72a (8.3 mg, 0.02 mmol, 1.0 equiv). Compound 32 was obtained as white solid (7.8 mg, yield 38%). 1H NMR (800 MHz, CD3OD) δ 8.88 (d, J = 25.3 Hz, 1H), 8.43 – 7.64 (m, 10H), 7.61 – 7.20 (m, 6H), 6.83 (t, J = 25.0 Hz, 1H), 4.77 – 4.27 (m, 5H), 4.20 – 3.78 (m, 6H), 3.70 – 3.43 (m, 2H), 2.92 (t, J = 21.5 Hz, 6H), 2.56 – 1.98 (m, 7H), 1.03 (s, 9H). HRMS (m/z) for C55H62N11O6S+ [M + H]+: calcd: 1004.4600, found, 1004.4613.
(2S,4R)-1-((S)-2-(3-(3-((3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)amino)-3-oxopropoxy)propanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (34).
Compound 34 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 72b (9.3 mg, 0.02 mmol, 1.0 equiv). Compound 34 was obtained as white solid (9.1 mg, yield 55%). 1H NMR (800 MHz, CD3OD) δ 8.95 (s, 1H), 8.32 (d, J = 8.3 Hz, 1H), 8.04 (d, J = 8.1 Hz, 2H), 7.96 (s, 1H), 7.88 (dd, J = 22.4, 7.7 Hz, 2H), 7.82 (d, J = 8.0 Hz, 2H), 7.73 (d, J = 8.0 Hz, 2H), 7.48 – 7.35 (m, 5H), 7.30 (d, J = 7.5 Hz, 1H), 6.85 (t, J = 6.9 Hz, 1H), 4.64 (s, 1H), 4.58 (t, J = 8.4 Hz, 1H), 4.56 – 4.49 (m, 2H), 4.39 – 4.34 (m, 1H), 3.89 (d, J = 10.9 Hz, 1H), 3.80 (dd, J = 11.1, 4.0 Hz, 1H), 3.69 – 3.63 (m, 3H), 3.62 – 3.58 (m, 1H), 3.48 (t, J = 7.6 Hz, 2H), 2.92 – 2.87 (m, 4H), 2.76 – 2.74 (dt, 2H), 2.52 – 2.38 (m, 7H), 2.35 (s, 1H), 2.24 (dd, J = 13.3, 7.6 Hz, 1H), 2.11 – 2.08 (m, 2H), 1.03 (s, 9H). HRMS (m/z) for C57H67N11O6S+ [M + H]+: calcd: 1032.4913, found, 1032.4904.
(2S,4R)-1-((S)-14-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-2-(tert-butyl)-4,11-dioxo-6,9-dioxa-3,12-diazatetradecanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (35).
Compound 35 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 72c (9.4 mg, 0.02 mmol, 1.0 equiv). Compound 35 was obtained as white solid (6.2 mg, yield 37%). 1H NMR (800 MHz, CD3OD) δ 8.90 (s, 1H), 8.31 (d, J = 8.3 Hz, 1H), 8.07 – 8.02 (m, 2H), 7.97 (s, 1H), 7.90 (d, J = 7.8 Hz, 1H), 7.86 (d, J = 7.8 Hz, 1H), 7.82 (d, J = 7.9 Hz, 2H), 7.73 (d, J = 8.1 Hz, 2H), 7.39 – 7.35 (m, 5H), 7.31 (d, J = 7.5 Hz, 1H), 6.85 (t, J = 6.9 Hz, 1H), 4.73 (s, 1H), 4.61 (t, J = 8.5 Hz, 1H), 4.53 (s, 1H), 4.41 (d, J = 15.3 Hz, 1H), 4.35 (d, J = 15.4 Hz, 1H), 4.05 – 3.88 (m, 6H), 3.86 – 3.81 (m, 1H), 3.70 – 3.50 (m, 5H), 2.96 – 2.86 (m, 4H), 2.75 – 2.69 (m, 2H), 2.45 (s, 3H), 2.38 – 2.31 (m, 1H), 2.27 (dd, J = 13.0, 7.6 Hz, 1H), 2.12 – 2.04 (m, 2H), 1.04 (s, 9H). HRMS (m/z) for C57H66N11O7S+ [M + H]+: calcd: 1048.4862, found, 1048.4877.
(2S,4R)-1-((S)-16-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-2-(tert-butyl)-4,13-dioxo-7,10-dioxa-3,14-diazahexadecanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (36).
Compound 36 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 72d (9.9 mg, 0.02 mmol, 1.0 equiv). Compound 36 was obtained as white solid (9.1 mg, yield 53%). 1H NMR (800 MHz, CD3OD) δ 8.94 (s, 1H), 8.32 (d, J = 8.3 Hz, 1H), 8.05 (t, J = 7.7 Hz, 2H), 7.96 (s, 1H), 7.90 (d, J = 7.8 Hz, 1H), 7.86 (d, J = 7.6 Hz, 1H), 7.82 (d, J = 8.1 Hz, 2H), 7.74 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 7.9 Hz, 2H), 7.43 – 7.38 (m, 3H), 7.31 (d, J = 7.5 Hz, 1H), 6.85 (t, J = 7.0 Hz, 1H), 4.65 (s, 1H), 4.58 (t, J = 8.2 Hz, 1H), 4.54 (d, J = 15.4 Hz, 1H), 4.50 (s, 1H), 4.37 (d, J = 15.4 Hz, 1H), 3.89 (d, J = 10.9 Hz, 1H), 3.80 (dd, J = 11.1, 4.1 Hz, 1H), 3.72 – 3.66 (m, 4H), 3.58 – 3.51 (m, 4H), 3.48 (t, J = 7.5 Hz, 2H), 2.93 – 2.87 (m, 4H), 2.76 – 2.70 (m, 2H), 2.55 – 2.52 (m, 1H), 2.49 – 2.39 (m, 6H), 2.34 (dd, J = 11.3, 6.1 Hz, 1H), 2.23 (dd, J = 13.3, 7.6 Hz, 1H), 2.11 – 2.08 (m, 2H), 1.03 (s, 9H). HRMS (m/z) for C59H70N11O7S+ [M + H]+: calcd: 1076.5175, found, 1076.5189.
(2S,4R)-1-((S)-17-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-2-(tert-butyl)-4,14-dioxo-6,9,12-trioxa-3,15-diazaheptadecanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (37).
Compound 37 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 72e (10.1 mg, 0.02 mmol, 1.0 equiv). Compound 37 was obtained as white solid (6.8 mg, yield 39%). 1H NMR (800 MHz, CD3OD) δ 8.91 (s, 1H), 8.32 (d, J = 8.4 Hz, 1H), 8.05 (dd, J = 8.3, 4.2 Hz, 2H), 7.99 (s, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.84 (dd, J = 29.9, 7.9 Hz, 3H), 7.76 – 7.69 (m, 2H), 7.45 (d, J = 7.9 Hz, 2H), 7.44 – 7.38 (m, 3H), 7.30 (d, J = 7.6 Hz, 1H), 6.84 (s, 1H), 4.70 (s, 1H), 4.62 – 4.47 (m, 3H), 4.35 (d, J = 15.3 Hz, 1H), 4.09 – 3.84 (m, 5H), 3.80 (dd, J = 11.1, 4.0 Hz, 1H), 3.70 – 3.58 (m, 8H), 3.58 – 3.47 (m, 2H), 2.95– 2.88 (m, 4H), 2.75 – 2.69 (m, 2H), 2.47 (s, 3H), 2.38 – 2.30 (m, 1H), 2.23 (dd, J = 13.3, 7.6 Hz, 1H), 2.13 – 2.02 (m, 2H), 1.03 (s, 9H). HRMS (m/z) for C59H70N11O8S+ [M + H]+: calcd: 1092.5124, found, 1092.5135.
(2S,4R)-1-((S)-19-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)-2-(tert-butyl)-4,16-dioxo-7,10,13-trioxa-3,17-diazanonadecanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (38).
Compound 38 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 72f (10.6 mg, 0.02 mmol, 1.0 equiv). Compound 38 was obtained as white solid (8.3 mg, yield 46%). 1H NMR (800 MHz, CD3OD) δ 8.93 (s, 1H), 8.32 (d, J = 8.4 Hz, 1H), 8.05 (t, J = 8.1 Hz, 2H), 7.97 (s, 1H), 7.90 (d, J = 7.8 Hz, 1H), 7.84 (dd, J = 25.3, 7.8 Hz, 3H), 7.74 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 7.9 Hz, 2H), 7.44 – 7.37 (m, 3H), 7.31 (d, J = 7.5 Hz, 1H), 6.84 (t, J = 7.0 Hz, 1H), 4.64 (s, 1H), 4.59 – 4.47 (m, 3H), 4.37 (d, J = 15.4 Hz, 1H), 3.89 (d, J = 10.9 Hz, 1H), 3.80 (dd, J = 11.1, 4.0 Hz, 1H), 3.74 – 3.63 (m, 4H), 3.63 – 3.51 (m, 8H), 3.48 (t, J = 7.5 Hz, 2H), 2.94 – 2.86 (m, 4H), 2.76 – 2.70 (m, 2H), 2.58 – 2.51 (m, 1H), 2.49 – 2.39 (m, 6H), 2.38 – 2.31 (m, 1H), 2.23 (dd, J = 13.1, 7.7 Hz, 1H), 2.12 – 2.03 (m, 2H), 1.03 (s, 9H). HRMS (m/z) for C61H74N11O8S+ [M + H]+: calcd: 1120.5437, found, 1120.5442.
N1-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-N16-((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)-4,7,10,13-tetraoxahexadecanediamide (39).
Compound 39 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 72g (11.3 mg, 0.02 mmol, 1.0 equiv). Compound 39 was obtained as white solid (8.4 mg, yield 45%). 1H NMR (800 MHz, CD3OD) δ 8.92 (s, 1H), 8.32 (d, J = 8.4 Hz, 1H), 8.05 (dd, J = 11.9, 7.2 Hz, 2H), 7.98 (s, 1H), 7.91 (d, J = 7.8 Hz, 1H), 7.86 (d, J = 7.6 Hz, 1H), 7.82 (d, J = 8.0 Hz, 2H), 7.74 (d, J = 8.1 Hz, 2H), 7.47 (d, J = 7.8 Hz, 2H), 7.45 – 7.37 (m, 3H), 7.31 (d, J = 7.5 Hz, 1H), 6.84 (t, J = 7.0 Hz, 1H), 4.64 (s, 1H), 4.59 – 4.48 (m, 3H), 4.37 (d, J = 15.4 Hz, 1H), 3.89 (d, J = 10.9 Hz, 1H), 3.81 (dd, J = 11.1, 4.0 Hz, 1H), 3.73 – 3.63 (m, 4H), 3.63 – 3.50 (m, 12H), 3.48 (t, J = 7.5 Hz, 2H), 2.93 – 2.87 (m, 4H), 2.79 – 2.68 (m, 2H), 2.57 – 2.40 (m, 7H), 2.35 – 2.31 (m, 1H), 2.23 (dd, J = 13.3, 7.6 Hz, 1H), 2.12 – 2.04 (m, 2H), 1.03 (s, 9H). HRMS (m/z) for C63H78N11O9S+ [M + H]+: calcd: 1164.5699, found, 1164.5682.
N1-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-N17-((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)-3,6,9,12,15-pentaoxaheptadecanediamide (40).
Compound 40 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 72h (11.6 mg, 0.02 mmol, 1.0 equiv). Compound 40 was obtained as white solid (9.7 mg, yield 51%). 1H NMR (800 MHz, CD3OD) δ 8.95 (d, J = 13.9 Hz, 1H), 8.32 (d, J = 8.3 Hz, 1H), 8.11 – 7.98 (m, 3H), 7.92 (d, J = 7.8 Hz, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.82 (d, J = 8.0 Hz, 2H), 7.74 (d, J = 7.9 Hz, 2H), 7.49 – 7.36 (m, 5H), 7.31 (d, J = 7.6 Hz, 1H), 6.86 (t, J = 7.0 Hz, 1H), 4.69 (s, 1H), 4.62 – 4.50 (m, 3H), 4.37 (d, J = 15.4 Hz, 1H), 4.05 – 3.91 (m, 4H), 3.88 (d, J = 11.2 Hz, 1H), 3.82 (dd, J = 11.2, 3.9 Hz, 1H), 3.73 – 3.50 (m, 18H), 2.91 (d, J = 7.7 Hz, 4H), 2.76 – 2.71 (m, 2H), 2.48 (s, 3H), 2.37 – 2.31 (m, 1H), 2.25 (dd, J = 13.4, 7.7 Hz, 1H), 2.14 – 2.02 (m, 2H), 1.04 (s, 9H). HRMS (m/z) for C63H78N11O10S+ [M + H]+: calcd: 1180.5648, found, 1180.5672.
N1-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-N19-((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)-4,7,10,13,16-pentaoxanonadecanediamide (41).
Compound 41 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 72i (12 mg, 0.02 mmol, 1.0 equiv). Compound 41 was obtained as white solid (10.6 mg, yield 55%). 1H NMR (800 MHz, CD3OD) δ 8.96 (s, 1H), 8.33 (d, J = 8.5 Hz, 1H), 8.05 (dd, J = 12.2, 7.2 Hz, 2H), 8.00 (s, 1H), 7.91 (d, J = 7.8 Hz, 1H), 7.88 (d, J = 7.6 Hz, 1H), 7.83 (d, J = 8.1 Hz, 2H), 7.75 (d, J = 8.0 Hz, 2H), 7.48 (d, J = 7.8 Hz, 2H), 7.42 (d, J = 7.9 Hz, 2H), 7.39 (t, J = 7.7 Hz, 1H), 7.31 (d, J = 7.5 Hz, 1H), 6.85 (t, J = 6.9 Hz, 1H), 4.65 (s, 1H), 4.61 – 4.47 (m, 3H), 4.37 (d, J = 15.4 Hz, 1H), 3.89 (d, J = 10.9 Hz, 1H), 3.81 (dd, J = 11.1, 4.0 Hz, 1H), 3.70 – 3.62 (m, 4H), 3.62 – 3.51 (m, 16H), 3.49 (t, J = 7.6 Hz, 2H), 2.94 – 2.87 (m, 4H), 2.74 (q, J = 12.4, 10.6 Hz, 2H), 2.57 – 2.51 (m, 1H), 2.49 (s, 3H), 2.43 (h, J = 5.6 Hz, 3H), 2.38 – 2.30 (m, 1H), 2.24 (dd, J = 13.3, 7.7 Hz, 1H), 2.14 – 2.03 (m, 2H), 1.03 (s, 9H). HRMS (m/z) for C65H82N11O10S+ [M + H]+: calcd: 1208.5961, found, 1208.5978.
N1-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-N4-((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)succinimide (42).
Compound 42 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 73a (8.5 mg, 0.02 mmol, 1.0 equiv). Compound 42 was obtained as white solid (10.6 mg, yield 67%). 1H NMR (800 MHz, CD3OD) δ 8.97 (s, 1H), 8.32 (d, J = 8.4 Hz, 1H), 8.04 (t, J = 7.8 Hz, 2H), 7.95 (s, 1H), 7.90 (d, J = 7.8 Hz, 1H), 7.89 – 7.84 (m, 3H), 7.73 (d, J = 8.1 Hz, 2H), 7.47 (d, J = 7.9 Hz, 2H), 7.42 (d, J = 7.9 Hz, 2H), 7.41 – 7.35 (m, 1H), 7.30 (d, J = 7.6 Hz, 1H), 6.84 (t, J = 6.9 Hz, 1H), 4.64 – 4.47 (m, 4H), 4.38 (d, J = 15.4 Hz, 1H), 3.90 (d, J = 10.9 Hz, 1H), 3.85 – 3.74 (m, 1H), 3.51 – 3.41 (m, 2H), 2.96 – 2.85 (m, 4H), 2.77 – 2.69 (m, 2H), 2.60 – 2.57 (m, 1H), 2.54 – 2.41 (m, 6H), 2.39 – 2.31 (m, 1H), 2.23 (dd, J = 13.2, 7.7 Hz, 1H), 2.13 – 2.03 (m, 2H), 1.07 – 0.99 (m, 9H). HRMS (m/z) for C55H62N11O5S+ [M + H]+: calcd: 988.4651, found, 988.4659.
N1-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-N5-((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)glutaramide (43).
Compound 43 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 73b (8.7 mg, 0.02 mmol, 1.0 equiv). Compound 43 was obtained as white solid (8.3 mg, yield 52%). 1H NMR (800 MHz, CD3OD) δ 8.93 (d, J = 29.3 Hz, 1H), 8.30 (dd, J = 25.1, 8.4 Hz, 1H), 8.04 (t, J = 6.3 Hz, 2H), 7.97 (s, 1H), 7.90 – 7.85 (m, 2H), 7.82 (d, J = 8.0 Hz, 2H), 7.74 (d, J = 8.0 Hz, 2H), 7.48 (d, J = 7.8 Hz, 2H), 7.44 – 7.36 (m, 3H), 7.29 (d, J = 7.5 Hz, 1H), 6.85 (s, 1H), 4.63 – 4.46 (m, 4H), 4.37 (d, J = 15.4 Hz, 1H), 3.92 (d, J = 10.9 Hz, 1H), 3.83 (dd, J = 11.1, 4.1 Hz, 1H), 3.50 – 3.45 (m, 2H), 2.95 – 2.87 (m, 4H), 2.76 – 2.70 (m, 2H), 2.47 (s, 3H), 2.39 – 2.35 (m, 1H), 2.29 – 2.14 (m, 5H), 2.14 – 2.03 (m, 2H), 1.87 – 1.82 (m, 2H), 1.03 (s, 9H). HRMS (m/z) for C56H64N11O5S+ [M + H]+: calcd: 1002.4807, found, 1002.4811.
N1-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-N6-((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)adipamide (44).
Compound 44 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 73c (8.9 mg, 0.02 mmol, 1.0 equiv). Compound 44 was obtained as white solid (6.7 mg, yield 41%). 1H NMR (800 MHz, CD3OD) δ 8.93 (s, 1H), 8.32 (d, J = 8.4 Hz, 1H), 8.09 – 8.00 (m, 2H), 7.96 (s, 1H), 7.90 (d, J = 7.8 Hz, 1H), 7.86 (d, J = 7.7 Hz, 1H), 7.82 (d, J = 8.1 Hz, 2H), 7.74 (d, J = 8.1 Hz, 2H), 7.47 (d, J = 7.8 Hz, 2H), 7.42 (d, J = 7.9 Hz, 2H), 7.39 (q, J = 7.9 Hz, 1H), 7.30 (d, J = 7.5 Hz, 1H), 6.84 (t, J = 6.9 Hz, 1H), 4.63 (s, 1H), 4.61 – 4.50 (m, 3H), 4.37 (d, J = 15.4 Hz, 1H), 3.91 (d, J = 10.9 Hz, 1H), 3.82 (dd, J = 11.1, 4.1 Hz, 1H), 3.47 (t, J = 7.5 Hz, 2H), 2.98 – 2.83 (m, 4H), 2.77 – 2.70 (m, 2H), 2.54 – 2.45 (m, 3H), 2.39 – 2.30 (m, 1H), 2.28 – 2.12 (m, 5H), 2.13 – 2.05 (m, 2H), 1.62 – 1.51 (m, 4H), 1.04 (s, 9H). HRMS (m/z) for C57H66N11O5S+ [M + H]+: calcd: 1016.4964, found, 1016.4956.
N1-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-N7-((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)heptanediamide (45).
Compound 45 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 73d (9.2 mg, 0.02 mmol, 1.0 equiv). Compound 45 was obtained as white solid (8.6 mg, yield 52%). 1H NMR (800 MHz, CD3OD) δ 8.94 (s, 1H), 8.32 (d, J = 8.3 Hz, 1H), 8.09 – 7.99 (m, 2H), 7.96 (s, 1H), 7.90 (d, J = 7.9 Hz, 1H), 7.87 (d, J = 7.7 Hz, 1H), 7.82 (d, J = 8.1 Hz, 2H), 7.74 (d, J = 8.1 Hz, 2H), 7.48 (d, J = 7.9 Hz, 2H), 7.43 (d, J = 7.9 Hz, 2H), 7.39 (t, J = 7.7 Hz, 1H), 7.30 (d, J = 7.6 Hz, 1H), 6.85 (t, J = 6.9 Hz, 1H), 4.63 (s, 1H), 4.61 – 4.49 (m, 3H), 4.40 – 4.34 (m, 1H), 3.91 (d, J = 10.9 Hz, 1H), 3.81 (dd, J = 11.1, 4.1 Hz, 1H), 3.49 – 3.38 (m, 2H), 2.99 – 2.84 (m, 4H), 2.78 – 2.70 (m, 2H), 2.49 (s, 3H), 2.34 (dp, J = 21.6, 7.1 Hz, 1H), 2.28 – 2.19 (m, 2H), 2.20 – 2.15 (m, 3H), 2.13 – 2.04 (m, 2H), 1.61 – 1.45 (m, 4H), 1.31 – 1.27 (m, 2H), 1.04 (s, 9H). HRMS (m/z) for C58H68N11O5S+ [M + H]+: calcd: 1030.5120, found, 1030.5126.
N1-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-N8-((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)octanediamide (46).
Compound 46 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 73e (9.4 mg, 0.02 mmol, 1.0 equiv). Compound 46 was obtained as white solid (8.4 mg, yield 50%). 1H NMR (800 MHz, CD3OD) δ 8.95 (s, 1H), 8.32 (d, J = 8.3 Hz, 1H), 8.09 – 8.01 (m, 2H), 7.96 (s, 1H), 7.90 (d, J = 7.9 Hz, 1H), 7.86 (d, J = 7.5 Hz, 1H), 7.82 (d, J = 8.1 Hz, 2H), 7.77 – 7.70 (m, 2H), 7.48 (d, J = 7.8 Hz, 2H), 7.43 (d, J = 7.8 Hz, 2H), 7.41 – 7.35 (m, 1H), 7.30 (d, J = 7.5 Hz, 1H), 6.85 (t, J = 6.9 Hz, 1H), 4.64 (s, 1H), 4.62 – 4.48 (m, 3H), 4.38 (d, J = 15.5 Hz, 1H), 3.92 (d, J = 10.9 Hz, 1H), 3.82 (dd, J = 11.1, 4.1 Hz, 1H), 3.47 (t, J = 7.4 Hz, 2H), 2.98 – 2.87 (m, 4H), 2.72 (dt, J = 14.1, 8.5 Hz, 2H), 2.53 – 2.44 (m, 3H), 2.39 – 2.28 (m, 1H), 2.27 – 2.03 (m, 7H), 1.59 – 1.49 (m, 4H), 1.32 – 1.23 (m, 4H), 1.04 (s, 9H). HRMS (m/z) for C59H70N11O5S+ [M + H]+: calcd: 1044.5277, found, 1044.5289.
N1-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-N9-((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)nonanediamide (47).
Compound 47 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 73f (9.6 mg, 0.02 mmol, 1.0 equiv). Compound 47 was obtained as white solid (5.4 mg, yield 32%). 1H NMR (800 MHz, CD3OD) δ 8.93 (s, 1H), 8.32 (d, J = 7.4 Hz, 1H), 8.07 – 7.67 (m, 9H), 7.55 – 7.22 (m, 6H), 6.84 (s, 1H), 4.70 – 4.45 (m, 4H), 4.38 (dd, J = 15.3, 6.4 Hz, 1H), 3.91 (t, J = 8.7 Hz, 1H), 3.86 – 3.80 (p, J = 4.2 Hz, 1H), 3.47 (q, J = 7.6 Hz, 2H), 2.97 – 2.84 (m, 4H), 2.79 – 2.68 (m, 2H), 2.49 (s, 3H), 2.34 (s, 1H), 2.30 – 2.01 (m, 7H), 1.54 (dd, J = 13.8, 7.0 Hz, 4H), 1.32 – 1.11 (m, 6H), 1.15 – 0.94 (m, 9H). HRMS (m/z) for C60H72N11O5S+ [M + H]+: calcd: 1058.5433, found, 1058.5451.
N1-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-N10-((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)decanediamide (48).
Compound 48 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 73g (9.8 mg, 0.02 mmol, 1.0 equiv). Compound 48 was obtained as white solid (5.3 mg, yield 31%). 1H NMR (800 MHz, CD3OD) δ 8.94 (s, 1H), 8.33 (d, J = 8.4 Hz, 1H), 8.05 (dd, J = 8.4, 4.7 Hz, 2H), 7.96 (s, 1H), 7.90 (d, J = 7.7 Hz, 1H), 7.86 (d, J = 7.6 Hz, 1H), 7.82 (d, J = 8.0 Hz, 2H), 7.74 (d, J = 8.0 Hz, 2H), 7.48 (d, J = 7.8 Hz, 2H), 7.43 (d, J = 7.8 Hz, 2H), 7.38 (t, J = 7.7 Hz, 1H), 7.30 (d, J = 7.6 Hz, 1H), 6.84 (s, 1H), 4.68 – 4.62 (m, 1H), 4.63 – 4.47 (m, 3H), 4.38 (d, J = 15.4 Hz, 1H), 3.92 (d, J = 10.9 Hz, 1H), 3.82 (dd, J = 11.1, 4.1 Hz, 1H), 3.47 (q, J = 7.7 Hz, 2H), 2.94 – 2.86 (m, 4H), 2.78 – 2.68 (m, 2H), 2.49 (d, J = 4.6 Hz, 3H), 2.34 (d, J = 10.5 Hz, 1H), 2.32 – 2.24 (m, 3H), 2.19 – 2.03 (m, 4H), 1.65 – 1.56 (m, 4H), 1.23 (s, 8H), 1.05 (s, 9H). HRMS (m/z) for C61H74N11O5S+ [M + H]+: calcd: 1072.5590, found, 1072.5593.
N1-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-N11-((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)undecanediamide (49).
Compound 49 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 73h (10 mg, 0.02 mmol, 1.0 equiv). Compound 49 was obtained as white solid (8.4 mg, yield 48%). 1H NMR (800 MHz, CD3OD) δ 8.92 (s, 1H), 8.32 (d, J = 8.0 Hz, 1H), 8.05 (d, J = 8.2 Hz, 2H), 7.96 (s, 1H), 7.90 (d, J = 7.6 Hz, 1H), 7.82 (t, J = 8.4 Hz, 3H), 7.73 (d, J = 8.1 Hz, 2H), 7.55 – 7.35 (m, 5H), 7.30 (d, J = 7.4 Hz, 1H), 6.82 (s, 1H), 4.65 (s, 1H), 4.61 – 4.47 (m, 3H), 4.38 (d, J = 15.4 Hz, 1H), 3.92 (d, J = 10.7 Hz, 1H), 3.82 (dd, J = 11.1, 4.0 Hz, 1H), 3.48 (t, J = 7.4 Hz, 2H), 2.98 – 2.84 (m, 4H), 2.78 – 2.66 (m, 2H), 2.49 (s, 3H), 2.40 – 2.20 (m, 5H), 2.20 – 2.01 (m, 3H), 1.68 – 1.45 (m, 4H), 1.22 (s, 10H), 1.04 (s, 9H). HRMS (m/z) for C62H76N11O5S+ [M + H]+: calcd: 1086.5746, found, 1086.5765.
N-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)acetamide (50).
Compound 50 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 74a (5.3 mg, 0.02 mmol, 1.0 equiv). Compound 50 was obtained as yellow solid (5.3 mg, yield 42%). 1H NMR (800 MHz, CD3OD) δ 8.28 (d, J = 8.4 Hz, 1H), 8.03 (d, J = 6.0 Hz, 1H), 7.99 (d, J = 8.4 Hz, 1H), 7.88 (d, J = 5.7 Hz, 2H), 7.78 (d, J = 8.1 Hz, 2H), 7.73 (d, J = 7.7 Hz, 1H), 7.67 (d, J = 8.0 Hz, 2H), 7.35 (t, J = 7.8 Hz, 1H), 7.27 – 7.20 (m, 2H), 6.86 (d, J = 7.2 Hz, 1H), 6.77 (t, J = 6.8 Hz, 1H), 6.62 (d, J = 8.4 Hz, 1H), 5.06 (dd, J = 12.8, 5.5 Hz, 1H), 3.91 (s, 2H), 3.64 – 3.56 (m, 2H), 2.94 – 2.85 (m, 5H), 2.79 – 2.67 (m, 4H), 2.34 – 2.30 (m, 1H), 2.16 – 1.98 (m, 2H). HRMS (m/z) for C44H41N10O5+ [M + H]+: calcd: 789.3256, found, 789.3244.
N-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-3-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)propenamide (51).
Compound 51 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 74b (5.5 mg, 0.02 mmol, 1.0 equiv). Compound 51 was obtained as yellow solid (7.4 mg, yield 58%). 1H NMR (800 MHz, CD3OD) δ 8.26 (d, J = 8.4 Hz, 1H), 8.05 – 7.99 (m, 2H), 7.94 (s, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.82 – 7.78 (m, 3H), 7.68 (d, J = 8.0 Hz, 2H), 7.43 – 7.33 (m, 2H), 7.28 (d, J = 7.5 Hz, 1H), 6.97 (d, J = 7.1 Hz, 1H), 6.89 (d, J = 8.5 Hz, 1H), 6.85 – 6.76 (m, 1H), 5.01 (dd, J = 12.5, 5.3 Hz, 1H), 3.48 (q, J = 7.9 Hz, 4H), 2.94 – 2.88 (m, 2H), 2.87 (t, J = 7.3 Hz, 2H), 2.82 – 2.77 (m, 1H), 2.74 – 2.72 (m, 2H), 2.70 – 2.61 (m, 2H), 2.46 (t, J = 6.7 Hz, 2H), 2.37 – 2.30 (m, 1H), 2.06 – 2.03 (m, 2H). HRMS (m/z) for C45H43N10O5+ [M + H]+: calcd: 803.3412, found, 803.3419.
N-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-4-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)butanamide (52).
Compound 52 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 73c (5.7 mg, 0.02 mmol, 1.0 equiv). Compound 52 was obtained as yellow solid (8.5 mg, yield 65%). 1H NMR (800 MHz, CD3OD) δ 8.24 (d, J = 8.4 Hz, 1H), 8.03 (d, J = 6.2 Hz, 1H), 8.00 – 7.95 (m, 2H), 7.85 (d, J = 7.8 Hz, 1H), 7.79 (dd, J = 14.6, 7.7 Hz, 3H), 7.68 (t, J = 6.1 Hz, 2H), 7.43 – 7.37 (m, 2H), 7.31 (d, J = 7.5 Hz, 1H), 6.94 – 6.87 (m, 1H), 6.82 (t, J = 6.9 Hz, 1H), 6.77 (d, J = 8.5 Hz, 1H), 5.02 – 4.96 (m, 1H), 3.53 (p, J = 6.7 Hz, 2H), 3.07 (t, J = 7.3 Hz, 2H), 2.99 – 2.85 (m, 4H), 2.81 – 2.78 (m, 1H), 2.76 – 2.62 (m, 4H), 2.35 – 2.30 (m, 1H), 2.22 (t, J = 7.2 Hz, 2H), 2.06 (s, 2H), 1.83 – 1.77 (m, 2H). HRMS (m/z) for C46H45N10O5+ [M + H]+: calcd: 817.3569, found, 817.3576.
N-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-5-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)pentanamide (53).
Compound 53 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 74d (6 mg, 0.02 mmol, 1.0 equiv). Compound 53 was obtained as yellow solid (7.6 mg, yield 57%). 1H NMR (800 MHz, CD3OD) δ 8.25 (d, J = 8.4 Hz, 1H), 8.08 – 8.19 (m, 2H), 7.94 (s, 1H), 7.87 (d, J = 7.8 Hz, 1H), 7.79 (dd, J = 11.5, 7.6 Hz, 3H), 7.68 (d, J = 8.0 Hz, 2H), 7.43 (t, J = 7.8 Hz, 1H), 7.38 (t, J = 7.7 Hz, 1H), 7.31 (d, J = 7.6 Hz, 1H), 6.98 – 6.93 (m, 1H), 6.85 – 6.76 (m, 2H), 5.08 – 5.02 (m, 1H), 3.53 (t, J = 7.1 Hz, 2H), 3.06 (t, J = 7.1 Hz, 2H), 2.95 – 2.82 (m, 5H), 2.79 – 2.68 (m, 4H), 2.36 – 2.29 (m, 1H), 2.18 (t, J = 7.2 Hz, 2H), 2.12 (dd, J = 12.5, 5.8 Hz, 1H), 2.10 – 2.02 (m, 1H), 1.63 – 1.50 (m, 2H), 1.48 – 1.42 (m, 2H). HRMS (m/z) for C47H47N10O5+ [M + H]+: calcd: 831.3725, found, 831.3715.
N-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-6-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)hexanamide (54).
Compound 54 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 74e (6.2 mg, 0.02 mmol, 1.0 equiv). Compound 54 was obtained as yellow solid (9.2 mg, yield 68%). 1H NMR (800 MHz, CD3OD) δ 8.28 (d, J = 8.4 Hz, 1H), 8.03 (dd, J = 12.4, 7.3 Hz, 2H), 7.95 (s, 1H), 7.90 (d, J = 7.8 Hz, 1H), 7.79 (d, J = 8.0 Hz, 2H), 7.75 (d, J = 7.6 Hz, 1H), 7.68 (d, J = 8.0 Hz, 2H), 7.45 (t, J = 7.8 Hz, 1H), 7.39 (t, J = 7.7 Hz, 1H), 7.31 (d, J = 7.5 Hz, 1H), 6.97 – 6.91 (m, 1H), 6.84 – 6.78 (m, 2H), 4.99 (dd, J = 12.7, 5.4 Hz, 1H), 3.52 (t, J = 7.1 Hz, 2H), 3.07 (t, J = 7.2 Hz, 2H), 2.94 – 2.86 (m, 5H), 2.82 – 2.78 (m, 1H), 2.74 – 2.70 (m, 2H), 2.69 – 2.62 (m, 2H), 2.27 – 2.29 (m, 1H), 2.16 (t, J = 7.2 Hz, 2H), 2.10 – 2.01 (m, 1H), 1.55 (q, J = 7.5 Hz, 2H), 1.47 (q, J = 7.4 Hz, 2H), 1.27 – 1.22 (m, 2H). HRMS (m/z) for C48H49N10O5+ [M + H]+: calcd: 845.3882, found, 845.3876.
N-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-7-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)heptanamide (55).
Compound 55 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 74f (6.4 mg, 0.02 mmol, 1.0 equiv). Compound 55 was obtained as yellow solid (8.3 mg, yield 60%). 1H NMR (800 MHz, CD3OD) δ 8.29 (d, J = 8.4 Hz, 1H), 8.02 (dd, J = 12.1, 7.0 Hz, 2H), 7.94 (s, 1H), 7.89 (d, J = 7.8 Hz, 1H), 7.80 (d, J = 8.0 Hz, 2H), 7.75 (d, J = 7.7 Hz, 1H), 7.69 (d, J = 8.0 Hz, 2H), 7.47 (t, J = 7.8 Hz, 1H), 7.38 (t, J = 7.7 Hz, 1H), 7.31 (d, J = 7.6 Hz, 1H), 6.96 (d, J = 7.1 Hz, 1H), 6.90 (d, J = 8.5 Hz, 1H), 6.79 (s, 1H), 5.06 (dd, J = 12.6, 5.4 Hz, 1H), 3.52 (t, J = 7.1 Hz, 2H), 3.11 (t, J = 7.2 Hz, 2H), 2.94 – 2.83 (m, 5H), 2.78 – 2.68 (m, 4H), 2.38 – 2.30 (m, 1H), 2.17 – 2.08 (m, 3H), 2.07 – 2.05 (m, 1H), 1.53 (q, J = 7.5 Hz, 2H), 1.50 – 1.45 (m, 2H), 1.26 (q, J = 7.5 Hz, 2H), 1.21 (q, J = 7.7 Hz, 2H). HRMS (m/z) for C49H51N10O5+ [M + H]+: calcd: 859.4038, found, 859.4025.
N-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-8-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)octanamide (56).
Compound 56 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 74g (6.6 mg, 0.02 mmol, 1.0 equiv). Compound 56 was obtained as yellow solid (9.2 mg, yield 66%). 1H NMR (800 MHz, CD3OD) δ 8.31 (d, J = 8.3 Hz, 1H), 8.05 (d, J = 8.3 Hz, 1H), 8.01 (d, J = 6.2 Hz, 1H), 7.94 (s, 1H), 7.91 (d, J = 7.8 Hz, 1H), 7.79 (dd, J = 26.9, 7.8 Hz, 3H), 7.71 (d, J = 8.0 Hz, 2H), 7.50 (t, J = 7.8 Hz, 1H), 7.38 (t, J = 7.7 Hz, 1H), 7.30 (d, J = 7.6 Hz, 1H), 6.99 (t, J = 5.9 Hz, 1H), 6.94 (d, J = 8.5 Hz, 1H), 6.80 (t, J = 6.9 Hz, 1H), 5.07 – 5.01 (m, 1H), 3.51 (t, J = 7.2 Hz, 2H), 3.16 (t, J = 7.2 Hz, 2H), 3.02 – 2.93 (m, 4H), 2.89 – 2.83 (m, 1H), 2.77 – 2.66 (m, 4H), 2.38– 2.35 (m, 1H), 2.14 (t, J = 7.4 Hz, 2H), 2.12 – 2.05 (m, 2H), 1.59 – 1.46 (m, 4H), 1.26 (q, J = 7.4 Hz, 2H), 1.24 – 1.17 (m, 4H). HRMS (m/z) for C50H53N10O5+ [M + H]+: calcd: 873.4195, found, 873.4187.
N-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-3-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)propenamide (57).
Compound 57 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 75a (6.2 mg, 0.02 mmol, 1.0 equiv). Compound 57 was obtained as yellow solid (7.7 mg, yield 57%). 1H NMR (800 MHz, CD3OD) δ 8.28 (d, J = 8.3 Hz, 1H), 8.02 (dd, J = 11.4, 7.2 Hz, 2H), 7.97 (s, 1H), 7.87 (d, J = 7.7 Hz, 1H), 7.80 (dd, J = 8.0, 5.0 Hz, 3H), 7.70 (d, J = 8.0 Hz, 2H), 7.48 (t, J = 7.8 Hz, 1H), 7.39 – 7.33 (m, 1H), 7.28 (d, J = 7.5 Hz, 1H), 6.96 (t, J = 7.6 Hz, 2H), 6.82 (t, J = 6.9 Hz, 1H), 4.88 – 4.77 (m, 1H), 3.76 – 3.70 (m, 2H), 3.58 (t, J = 5.1 Hz, 2H), 3.49 (t, J = 7.5 Hz, 2H), 3.36 (t, J = 5.2 Hz, 2H), 2.95 – 2.78 (m, 5H), 2.73 – 2.69 (m, 2H), 2.62 – 2.52 (m, 2H), 2.45 (t, J = 5.8 Hz, 2H), 2.37 – 2.29 (m, 1H), 2.10 – 2.04 (m, 1H), 1.89 (d, J = 12.6 Hz, 1H). HRMS (m/z) for C47H47N10O6+ [M + H]+: calcd: 847.3675, found, 847.3673.
N-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-3-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)propenamide (58).
Compound 58 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 75b (6.9 mg, 0.02 mmol, 1.0 equiv). Compound 58 was obtained as yellow solid (8.7 mg, yield 61%). 1H NMR (800 MHz, CD3OD) δ 8.28 (d, J = 8.4 Hz, 1H), 8.02 (dd, J = 10.4, 6.3 Hz, 2H), 7.93 (s, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.81 – 7.75 (m, 3H), 7.70 (d, J = 8.0 Hz, 2H), 7.43 (t, J = 7.8 Hz, 1H), 7.36 (t, J = 7.7 Hz, 1H), 7.27 (d, J = 7.5 Hz, 1H), 6.98 – 6.92 (m, 2H), 6.79 (t, J = 6.8 Hz, 1H), 5.02 – 4.97 (m, 1H), 3.69 (t, J = 5.9 Hz, 2H), 3.63 (t, J = 5.2 Hz, 2H), 3.56 (s, 4H), 3.47 (t, J = 7.4 Hz, 2H), 3.39 (t, J = 5.2 Hz, 2H), 2.94 – 2.81 (m, 4H), 2.82 – 2.79 (m, 1H), 2.71 – 254 (m, 4H), 2.42 (t, J = 5.9 Hz, 2H), 2.36 – 2.30 (m, 1H), 2.09 – 2.01 (m, 2H). HRMS (m/z) for C49H51N10O7+ [M + H]+: calcd: 891.3937, found, 891.3945.
N-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-3-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethoxy)propenamide (59).
Compound 59 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 75c (7.6 mg, 0.02 mmol, 1.0 equiv). Compound 59 was obtained as yellow solid (7.9 mg, yield 53%). 1H NMR (800 MHz, CD3OD) δ 8.29 (d, J = 8.4 Hz, 1H), 8.03 (d, J = 8.3 Hz, 2H), 7.96 (s, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.82 – 7.78 (m, 3H), 7.71 (d, J = 8.0 Hz, 2H), 7.46 (t, J = 7.8 Hz, 1H), 7.37 (t, J = 7.7 Hz, 1H), 7.29 (d, J = 7.5 Hz, 1H), 6.98 (t, J = 7.4 Hz, 2H), 6.80 (s, 1H), 5.01 (dd, J = 12.9, 5.3 Hz, 1H), 3.67 (t, J = 6.0 Hz, 2H), 3.65 (t, J = 5.2 Hz, 2H), 3.62 (s, 4H), 3.58 (t, J = 4.5 Hz, 2H), 3.56 – 3.52 (m, 2H), 3.48 (t, J = 7.5 Hz, 2H), 3.40 (t, J = 5.2 Hz, 2H), 2.95 – 2.85 (m, 4H), 2.84 – 2.80 (m, 1H), 2.75 – 2.60 (m, 4H), 2.41 (t, J = 5.9 Hz, 2H), 2.36 – 2.30 (m, 1H), 2.11 – 2.02 (m, 2H). HRMS (m/z) for C51H55N10O8+ [M + H]+: calcd: 935.4199, found, 935.4178.
N-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-1-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3,6,9,12-tetraoxapentadecan-15-amide (60).
Compound 60 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 75d (8.3 mg, 0.02 mmol, 1.0 equiv). Compound 60 was obtained as yellow solid (8 mg, yield 51%). 1H NMR (800 MHz, CD3OD) δ 8.28 (d, J = 8.4 Hz, 1H), 8.12 – 8.01 (m, 3H), 7.92 – 7.79 (m, 2H), 7.83 (d, J = 8.0 Hz, 2H), 7.73 (d, J = 8.0 Hz, 2H), 7.44 – 7.34 (m, 2H), 7.28 (d, J = 7.5 Hz, 1H), 7.00 – 6.94 (m, 1H), 6.89 – 6.76 (m, 2H), 5.01 (dd, J = 12.9, 5.4 Hz, 1H), 3.72 – 3.69 (m, 2H), 3.67 – 3.60 (m, 6H), 3.62 – 3.56 (m, 6H), 3.58 – 3.53 (m, 2H), 3.49 (t, J = 7.7 Hz, 2H), 3.32 – 3.23 (m, 2H), 3.02 – 2.92 (m, 2H), 2.89 – 2.79 (m, 3H), 2.79 – 2.63 (m, 4H), 2.45 (t, J = 5.8 Hz, 2H), 2.39 – 2.32 (m, 1H), 2.07 (d, J = 14.0 Hz, 2H). HRMS (m/z) for C53H59N10O9+ [M + H]+: calcd: 979.4461, found, 979.4459.
N-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenethyl)-1-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3,6,9,12,15-pentaoxaoctadecan-18-amide (61).
Compound 61 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (10.8 mg, 0.02 mmol) and 75e (9 mg, 0.02 mmol, 1.0 equiv). Compound 61 was obtained as yellow solid (7.8 mg, yield 48%). 1H NMR (800 MHz, CD3OD) δ 8.30 (d, J = 8.3 Hz, 1H), 8.09 – 7.98 (m, 3H), 7.90 (d, J = 7.8 Hz, 1H), 7.84 (dd, J = 14.9, 7.9 Hz, 3H), 7.72 (d, J = 8.1 Hz, 2H), 7.50 (t, J = 7.8 Hz, 1H), 7.41 – 7.35 (m, 1H), 7.30 (d, J = 7.5 Hz, 1H), 7.02 (s, 1H), 6.98 (d, J = 8.5 Hz, 1H), 6.84 (s, 1H), 5.02 (dd, J = 12.8, 5.2 Hz, 1H), 3.70 (q, J = 5.6 Hz, 2H), 3.62 – 3.55 (m, 14H), 3.55 – 3.46 (m, 6H), 3.38 – 3.34 (m, 2H), 2.97 – 2.86 (m, 4H), 2.85 – 2.82 (m, 1H), 2.78 – 2.72 (m, 2H), 2.71 (s, 1H), 2.70 – 2.63 (m, 1H), 2.44 (t, J = 5.8 Hz, 2H), 2.37 – 2.32 (m, 1H), 2.12 – 2.03 (m, 2H). HRMS (m/z) for C55H63N10O10+ [M + H]+: calcd: 1023.4723, found, 1023.4716.
(2S,4R)-1-((S)-2-(11-(3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)propanamido)undecanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (62).
Compound 62 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (30.4 mg, 0.05 mmol) and 7831 (31.6 mg, 0.05 mmol, 1.0 equiv). Compound 62 was obtained as white solid (35.2 mg, yield 63%). 1H NMR (600 MHz, CD3OD) δ 9.02 (s, 1H), 8.30 (d, J = 8.5 Hz, 1H), 8.06 – 7.98 (m, 2H), 7.91 (d, J = 1.9 Hz, 1H), 7.90 – 7.86 (m, 1H), 7.83 (dd, J = 8.2, 6.4 Hz, 3H), 7.74 – 7.69 (m, 2H), 7.48 – 7.42 (m, 4H), 7.37 (t, J = 7.6 Hz, 1H), 7.29 (d, J = 7.5 Hz, 1H), 6.84 (dd, J = 7.6, 6.3 Hz, 1H), 5.06 – 4.98 (m, 1H), 4.64 (s, 1H), 4.59 (dd, J = 9.1, 7.6 Hz, 1H), 4.48 – 4.43 (m, 1H), 3.91 – 3.87 (m, 1H), 3.77 (dd, J = 11.0, 4.0 Hz, 1H), 3.08 (t, J = 7.0 Hz, 2H), 2.99 (t, J = 7.4 Hz, 2H), 2.94 – 2.86 (m, 2H), 2.76 – 2.68 (m, 2H), 2.54 – 2.46 (m, 5H), 2.38 – 2.16 (m, 4H), 2.12 – 2.02 (m, 1H), 2.00 – 1.94 (m, 1H), 1.63 – 1.54 (m, 2H), 1.52 (d, J = 7.0 Hz, 3H), 1.36 – 1.08 (m, 14H), 1.05 (s, 9H). 13C NMR (151 MHz, CD3OD) δ 174.63, 173.50, 171.85, 170.95, 154.31, 153.32, 151.88, 148.39, 146.91, 144.80, 144.50, 141.32, 141.26, 138.78, 137.61, 135.15, 133.07, 132.44, 129.72, 129.10 (2C), 128.98, 128.49, 128.23, 128.20 (2C), 127.67 (2C), 127.04, 126.29 (2C), 126.10, 124.73, 117.28, 112.64, 111.55, 69.58, 67.65, 59.21, 58.67, 57.64, 56.57, 48.75, 39.00, 37.55, 37.40, 36.18, 35.26, 35.09, 31.96, 31.65, 29.12, 29.10, 29.00, 28.94, 28.91, 26.47, 25.67 (3C), 25.61, 20.98, 14.10, 13.43. tR = 4.02 min, HRMS (m/z) for C64H80N11O5S+ [M + H]+: calcd: 1114.6059, found, 1114.6046.
(2R,4S)-1-((S)-2-(11-(3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)propanamido)undecanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (63).
Compound 63 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (30.4 mg, 0.05 mmol) and 8031 (31.6 mg, 0.05 mmol, 1.0 equiv). Compound 63 was obtained as white solid (37.8 mg, yield 68%). 1H NMR (600 MHz, CD3OD) δ 9.00 (s, 1H), 8.28 (d, J = 8.4 Hz, 1H), 8.03 – 7.98 (m, 2H), 7.91 (d, J = 1.8 Hz, 1H), 7.89 – 7.85 (m, 1H), 7.84 – 7.80 (m, 2H), 7.79 (dd, J = 7.7, 1.6 Hz, 1H), 7.69 – 7.65 (m, 2H), 7.54 – 7.49 (m, 2H), 7.45 – 7.40 (m, 2H), 7.35 (t, J = 7.6 Hz, 1H), 7.31 – 7.25 (m, 1H), 6.81 (dd, J = 7.7, 6.3 Hz, 1H), 5.06 – 5.01 (m, 1H), 4.60 (dd, J = 8.2, 6.7 Hz, 1H), 4.52 (s, 1H), 4.50 – 4.46 (m, 1H), 4.00 (s, 1H), 3.97 (dd, J = 10.9, 4.9 Hz, 1H), 3.73 (dd, J = 10.4, 3.3 Hz, 1H), 3.05 (t, J = 7.0 Hz, 2H), 2.99 (t, J = 7.3 Hz, 2H), 2.96 – 2.87 (m, 2H), 2.77 – 2.68 (m, 2H), 2.52 (t, J = 7.3 Hz, 2H), 2.48 (s, 3H), 2.38 – 2.19 (m, 3H), 2.17 – 2.00 (m, 3H), 1.57 – 1.48 (m, 1H), 1.47 (d, J = 7.0 Hz, 3H), 1.30 – 1.14 (m, 9H), 1.08 (s, 9H), 1.05 – 1.00 (m, 5H). 13C NMR (151 MHz, CD3OD) δ 174.89, 173.44, 172.35, 170.98, 154.30, 153.09, 151.88, 148.40, 146.75, 144.55, 144.38, 141.37, 141.20, 138.68, 137.45, 135.16, 132.98, 129.47, 129.27, 129.07, 128.96 (2C), 128.49, 128.23, 128.16 (2C), 127.72 (2C), 127.05, 126.95, 126.39 (2C), 124.71, 117.27, 112.42, 111.49, 69.14, 59.34, 58.67, 58.16, 55.61, 53.75, 48.68, 38.95, 37.64, 37.52, 35.13, 34.32, 31.96, 31.62, 29.10, 29.08, 28.98, 28.95, 28.92, 26.44, 25.66 (3C), 25.52, 25.42, 21.35, 14.19, 13.43. tR = 4.00 min, HRMS (m/z) for C64H80N11O5S+ [M + H]+: calcd: 1114.6059, found, 1114.6074.
(2R,4S)-1-((S)-2-(11-(3-(3-(3-(4-(1-Aminocyclobutyl)phenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-5-yl)phenyl)propanamido)undecanamido)-3,3-dimethylbutanoyl)-4-(benzyloxy)-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (64).
Compound 64 was synthesized following the standard procedure for preparing compound 3 from intermediates 1 (49 mg, 0.08 mmol) and 86 (58.3 mg, 0.08 mmol, 1.0 equiv). Compound 64 was obtained as white solid (52.7 mg, yield 55%). 1H NMR (600 MHz, CD3OD) δ 8.90 (s, 1H), 8.18 (d, J = 8.4 Hz, 1H), 7.93 – 7.85 (m, 2H), 7.82 – 7.74 (m, 1H), 7.73 – 7.67 (m, 3H), 7.63 – 7.56 (m, 2H), 7.37 – 7.30 (m, 4H), 7.30 – 7.09 (m, 8H), 6.71 (dd, J = 7.7, 6.2 Hz, 1H), 4.91 (q, J = 6.9 Hz, 2H), 4.60 (s, 1H), 4.51 – 4.42 (m, 2H), 4.37 (d, J = 11.5 Hz, 1H), 4.15 (d, J = 11.8 Hz, 2H), 3.59 (dd, J = 11.4, 3.8 Hz, 1H), 3.26 (s, 1H), 2.97 (t, J = 7.0 Hz, 2H), 2.88 (t, J = 7.4 Hz, 2H), 2.83 – 2.74 (m, 2H), 2.66 – 2.57 (m, 2H), 2.44 – 2.34 (m, 6H), 2.33 – 2.06 (m, 4H), 2.01 – 1.81 (m, 2H), 1.52 – 1.42 (m, 1H), 1.40 (d, J = 7.0 Hz, 3H), 1.26 – 0.97 (m, 12H), 0.95 (s, 9H). 13C NMR (151 MHz, CD3OD) δ 174.64, 173.48, 171.72, 171.04, 154.29, 153.31, 151.88, 148.39, 146.92, 144.76, 144.48, 141.32, 141.28, 138.79, 138.00, 137.57, 135.15, 133.07, 129.72, 129.10 (2C), 128.97, 128.50, 128.22, 128.19 (2C), 127.96 (2C), 127.67 (2C), 127.58 (2C), 127.38, 127.30, 127.03, 126.30 (2C), 126.12, 124.73, 117.27, 112.62, 111.53, 77.02, 70.25, 59.20, 58.67, 57.47, 53.76, 53.14, 48.75, 48.47, 39.01, 37.56, 35.26, 35.17, 34.96, 31.96, 31.66, 29.13, 29.09, 29.00, 28.95, 28.93, 28.89, 26.48, 25.65 (3C), 20.99, 14.11, 13.43. tR = 4.37 min, HRMS (m/z) for C71H86N11O5S+ [M + H]+: calcd: 1204.6529, found, 1204.6537.
(2S,4R)-4-(Benzyloxy)-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (83).
To a solution of commercially available intermediate 81 (642 mg, 2 mmol) in DMSO (10 mL) were added 8231 (436 mg, 2 mmol, 1.0 equiv), EDCI (574 mg, 3 mmol, 1.5 equiv), HOAt (408 mg, 3 mmol, 1.5 equiv), and NMM (606 mg, 6 mmol, 3.0 equiv). After being stirred at rt for 2 h, the resulting mixture was purified by reverse phase C18 column (10% – 100% methanol / 0.1% TFA in H2O) to afford the corresponding product. This product was dissolved in DCM (5 mL), and the resulting solution was treated with TFA (5 mL) for 30 min. After the solvent was evaporated, the resulting residue was purified by reverse phase C18 column (10% – 100% methanol / 0.1% TFA in H2O) to afford compound 83 as white solid (232 mg, yield 28% for two steps). 1H NMR (600 MHz, CD3OD) δ 9.00 (s, 1H), 7.51 – 7.47 (m, 2H), 7.45 (d, J = 8.3 Hz, 2H), 7.40 – 7.34 (m, 4H), 7.33 – 7.30 (m, 1H), 5.16 – 5.04 (m, 1H), 4.60 (d, J = 4.9 Hz, 2H), 4.49 (dd, J = 10.8, 7.3 Hz, 1H), 4.41 (t, J = 4.0 Hz, 1H), 3.58 – 3.50 (m, 1H), 3.44 (dd, J = 12.6, 3.9 Hz, 1H), 2.79 – 2.68 (m, 1H), 2.51 (s, 3H), 2.03 –1.93 (m, 1H), 1.55 (d, J = 7.0 Hz, 3H). HRMS (m/z) for C24H28N3O2S+ [M + H]+: calcd: 422.5665, found, 422.5678.
12-(((S)-1-((2S,4R)-4-(Benzyloxy)-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-12-oxododecanoic acid (86).
To a solution of commercially available (S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoic acid 84 (231 mg, 0.55 mmol) in DMSO (5 mL) were added 83 (232 mg, 0.55 mmol, 1.0 equiv), EDCI (158 mg, 0.83 mmol, 1.5 equiv), HOAt (113 mg, 0.83 mmol, 1.5 equiv), and NMM (168 mg, 1.66 mmol, 3.0 equiv). After being stirred at rt for 1 h, the resulting mixture was purified by reverse phase C18 column (10% – 100% methanol / 0.1% TFA in H2O) to afford the corresponding product. This product was dissolved in DCM (5 mL), and the resulting solution was treated with TFA (5 mL) for 30 min. After the solvent was evaporated, the resulting mixture was purified by reverse phase C18 column (10% – 100% methanol / 0.1% TFA in H2O) to afford compound 85 as white solid (243 mg, yield 83% for two steps). ESI-MS (m/z) [M + H]+: 535.8. To a solution of 85 (53.5 mg, 0.1 mmol) in DMSO (1 mL) were added dodecanedioic acid (77, 30.1 mg, 0.1 mmol, 1.0 equiv), EDCI (28.8 mg, 0.15 mmol, 1.5 equiv), HOAt (20.4 mg, 0.15 mmol, 1.5 equiv), and NMM (30.3 mg, 0.3 mmol, 3.0 equiv). After being stirred overnight at rt, the resulting mixture was purified by preparative HPLC (10% – 100% methanol / 0.1% TFA in H2O) to afford the corresponding product. After this product was dissolved in DCM (1 mL), the reaction mixture was treated with TFA (1 mL) for 30 min. After the solvent was evaporated, the resulting mixture was purified by preparative HPLC (10% – 100% methanol / 0.1% TFA in H2O) to afford compound 86 as white solid (58.3 mg, yield 81% for two steps). 1H NMR (600 MHz, CD3OD) δ 8.98 (s, 1H), 7.39 – 7.32 (m, 4H), 7.25 – 7.19 (m, 4H), 7.19 – 7.13 (m, 1H), 4.97 – 4.89 (m, 1H), 4.62 (s, 1H), 4.53 – 4.43 (m, 2H), 4.39 (d, J = 11.6 Hz, 1H), 4.20 – 4.12 (m, 2H), 3.60 (dd, J = 11.3, 3.6 Hz, 1H), 2.82 (t, J = 7.7 Hz, 2H), 2.41 (s, 3H), 2.34 – 2.25 (m, 1H), 2.25 – 2.07 (m, 2H), 1.92 – 1.82 (m, 1H), 1.60 – 1.47 (m, 3H), 1.41 (d, J = 7.0 Hz, 3H), 1.34 – 1.18 (m, 13H), 0.97 (s, 9H). HRMS (m/z) for C42H59N4O6S+ [M + H]+: calcd: 748.0155, found, 748.0146.
Cell Culture
All cell lines were purchased from ATCC and authenticated. Cell lines were regularly tested in the lab for mycoplasma. All cells were cultured at 37 °C under 5% CO2. Cell lines were cultured in 1X DMEM (Corning, 10–013-CV) with 10% fetal bovine serum (Atlanta Biologicals S11150) and 1X Penicillin/Streptomycin. Cells were split using 0.05% or 0.25% trypsin (Corning 25–051-Cl or 25–053-Cl, respectively) before they reached full confluence and media was changed every 3–4 days.
Western Blotting Assay
Cells were lysed in 2X sample buffer (125 mM Tris-HCl at pH 6.8, 10% βME, 2% SDS, 20% glycerol, 0.05% Bromophenol Blue, 8 M urea). Protein lysates were loaded into 4–12% Bis-Tris gels and resolved by electrophoresis. Samples were then blotted on PVDF membrane (Millipore IPVH00010) using the wet transfer technique (Invitrogen). Membranes were blocked in 5% milk-TBST for 30 min, and incubated in primary antibody in 5% milk-TBST or 5% BSA-TBST at 4 °C for 16 h. Membranes were rinsed (3 × 10 min) in TBST and incubated in horseradish peroxidase-conjugated secondary antibodies in 5% milk-TBST for at least 2 h and rinsed again in TBST (3 × 6 min). Membranes were visualized using the chemiluminescence system (Thermo 34080, 37075) on autoradiography film (Denville E3018). Primary Antibodies: β-Actin (Sigma A5316), p-AKT (Ser473 CST-9721), total AKT (CST-9272), AKT1 (2H10, CST-2947), AKT2 (CST-3063), AKT3 (CST-8018), p-S6 (Ser240/244, CST-5364), p-PRAS40 (Thr246, CST-2997). Secondary Antibodies: Mouse (Thermo 31432), Rabbit (Thermo 31460).
AKT Activity Inhibition Assay
AKT enzyme activity inhibition for ARQ-092, 62, 63 and 64 against three AKT isoforms AKT1, AKT2 and AKT3 was conducted by Reaction Biology using a HotSpot kinase assay.45 The biochemical IC50 values were determined by using a 10-concentration points mode with 3-fold dilution (the top concentration of 10 μM) in the presence of 10 μM ATP with DMSO as control.
Cell Proliferation Assay
Experiments were carried out in 96-well plates in triplicates (Corning 720089). A total of 1–3 × 103 cells per well were grown in the presence of indicated compounds, and cells were then monitored for 3–5 days using the IncuCyte live cell imaging system (Essen BioScience, Ann Arbor, MI, USA), which was placed in a cell culture incubator operated at 37 °C under 5% CO2. Cell confluence was determined using calculations derived from phase-contrast images readings on an IncuCyte ZOOM (Essen Biosciences) on live cells over time.
Cell Colony Formation Assay
Cells were cultured for 14 days in the presence of different compounds. Media with compound was replenished every two days. At the end of the experiment, media was aspirated and viable cells were stained with 0.5% crystal violet dye.
Proteomics Sample Preparation
The cell pellets were resuspended in 8 M Urea, 50 mM Tris-HCl pH 8.0, reduced with dithiothreitol (5 mM final) for 30 min at room temperature, and alkylated with iodoacetamide (15 mM final) for 45 min in the dark at room temperature. Samples were diluted 4-fold with 25 mM Tris-HCl pH 8.0, 1 mM CaCl2 and digested with trypsin at 1:100 (w/w, trypsin : protein) ratio overnight at room temperature. There were two biological samples at each condition and total 6 samples. Peptides were cleaned by homemade C18 stage tips and the concentration was determined (Peptide assay, Thermo 23275). 25 μg each was used for labeling with isobaric stable tandem mass tags (TMT10−126, 127N, 128N, 129N, 130N and 131) following manufacture instruction. The mixture of labeled peptides was fractionated into 22 fractions on C18 stage tip with buffer 10 mM Trimethylammonium bicarbonate (TMAB), pH 8.5 containing 5 to 50% acetonitrile.
Mass Spectrometry Analysis
Dried peptides were dissolved in 0.1% formic acid, 2% acetonitrile. 0.5 μg of peptides of each fraction was analyzed on a Q-Exactive HF-X coupled with an Easy nanoLC 1200 (Thermo Fisher Scientific, San Jose, CA). Peptides were loaded on to a nanoEase MZ HSS T3 Column (100Å, 1.8 μm, 75 μm x 250 mm, Waters). Analytical separation of all peptides was achieved with 110-min gradient. A linear gradient of 5 to 10% buffer B over 5 min, 10% to 31% buffer B over 70 min, 31% to 75% buffer B over 15 min was executed at a 250 nL/min flow rate followed a ramp to 100% B in 1 min and 19-min wash with 100% B, where buffer A was aqueous 0.1% formic acid, and buffer B was 80% acetonitrile and 0.1% formic acid. MS experiments were also carried out in a data-dependent mode with full MS (externally calibrated to a resolution of 60,000 at m/z 200) followed by high energy collision-activated dissociation-MS/MS of the top 15 most intense ions with a resolution of 15,000 at m/z 200. High energy collision-activated dissociation-MS/MS was used to dissociate peptides at a normalized collision energy of 32 eV in the presence of nitrogen bath gas atoms. Dynamic exclusion was 45 seconds.
Raw Proteomics Data Processing and Analysis
Peptide identification and quantification with TMT reporter ions were performed using the MaxQuant software version 1.6.10.43 (Max Planck Institute, Germany). Protein database searches were performed against the UniProt human protein sequence database (UP000005640). A false discovery rate (FDR) for both peptide-spectrum match (PSM) and protein assignment was set at 1%. Search parameters included up to two missed cleavages at Lys/Arg on the sequence, oxidation of methionine, and protein N-terminal acetylation as a dynamic modification. Carbamidomethylation of cysteine residues was considered as a static modification. Peptide identifications are reported by filtering of reverse and contaminant entries and assigning to their leading razor protein. Data processing and statistical analysis were performed on Perseus (Version 1.6.10.50). Protein quantitation was performed on biological replicates and a two-sample t-test statistics was used with a p-value of 5% to report statistically significant protein abundance fold-changes.
Mouse Pharmacokinetic Study
Compound 62 in HCl salt form was dissolved in a formulation vehicle of 5% v/v NMP, 5% v/v Solutol HS-15, and 90% v/v normal saline. Six male Swiss Albino mice were administered intraperitoneally with solution formulation of 62 at 75 mg/kg. All samples were maintained below −70 ± 10 °C until bioanalysis. Approximately 60 μL of plasma samples were collected from three mice at 0.5, 1, 2, 4, 8, and 12 h post dose. Plasma concentrations were quantified by fit-for-purpose LC-MS/MS method (LLOQ:1.02 ng/mL for plasma). Compound concentrations in plasma at each time point are average values from 3 tested mice. Error bars represent ± SEM. Experiments involving mice were performed according to the Institutional Animal Care and Use Committee (IACUC)-approved protocol.
Supplementary Material
ACKNOWLEDGEMENTS
J.J. acknowledges the support by an endowed professorship by the Icahn School of Medicine at Mount Sinai. This research was supported by the grant R35CA220491 (to R.P.) from the National Cancer Institute (NCI) at the National Institutes of Health (NIH). R.P. and J.J. are also supported by P30 CA196521 grant from the NCI at the NIH. J.X. is supported by T32 postdoc fellow training grant (T32CA078207) and Leo and Julia Forchheimer Foundation Postdoc Fellowship. This work utilized the NMR Spectrometer Systems at Mount Sinai acquired with funding from National Institutes of Health SIG grants 1S10OD025132 and 1S10OD028504.
J.J., R.P., J.L. J.X. and X.Y. are inventors of a patent application filed by the Icahn School of Medicine at Mount Sinai. J.J. is a cofounder and equity shareholder in Cullgen, Inc. and a consultant for Cullgen, Inc., EpiCypher, Inc., and Accent Therapeutics, Inc. R.P. is a shareholder and advisor of Therapten Bioscience, Inc. The Jin laboratory received research funds from Celgene Corporation, Levo Therapeutics, Inc., Cullgen, Inc. and Cullinan Oncology, Inc.
ABBREVIATIONS USED
- CRBN
cereblon
- DCM
dichloromethane
- DMSO
dimethyl sulfoxide
- EDCI
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- HOAt
1-hydroxy-7-azabenzo-triazole
- IP
intraperitoneal
- MOA
mechanism of action
- NAE
NEDD8-activating enzyme
- P-AKT
phosphorylated AKT
- PDK1
phosphoinositide-dependent kinase 1
- PEG
polyethylene glycol
- PH
pleckstrin-homology
- PK
pharmacokinetic
- POM
pomalidomide
- PROTAC
proteolysis targeting chimera
- SAR
structure-activity relationship
- T-AKT
total AKT
- TFA
trifluoroacetic acid
- TMT
tandem mass tag
- TPD
targeting protein degradation
- UPS
ubiquitin proteasome system
- VHL
von Hippel-Lindau
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Effects of compounds 20, 48–49 and 62 on colony formation inhibition in PC3 cells; effects of compounds 20, 48–49 and 62 on the degradation of AKT in SW620 and Mia PaCa-2 cells; effects of compound 62 on degradation of AKT1, AKT2 and AKT3 isoforms in SW620 cells; western blotting verification of T-AKT degradation in SW620 cell lysates for proteomic studies; 1H NMR, 13C NMR and HPLC spectra of compounds 62, 63 and 64 (PDF).
Molecular formula strings for all compounds (CSV).
REFERENCES
- 1.Cheng JQ; Lindsley CW; Cheng GZ; Yang H; Nicosia SV, The Akt/PKB pathway: molecular target for cancer drug discovery. Oncogene 2005, 24 (50), 7482–7492. [DOI] [PubMed] [Google Scholar]
- 2.Hennessy BT; Smith DL; Ram PT; Lu Y; Mills GB, Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov 2005, 4 (12), 988–1004. [DOI] [PubMed] [Google Scholar]
- 3.Yang J; Nie J; Ma X; Wei Y; Peng Y; Wei X, Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol. Cancer 2019, 18 (1), 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu P; Cheng H; Roberts TM; Zhao JJ, Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov 2009, 8 (8), 627–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoxhaj G; Manning BD, The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer 2020, 20 (2), 74–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown JS; Banerji U, Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol. Ther 2017, 172, 101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manning BD; Cantley LC, AKT/PKB signaling: navigating downstream. Cell 2007, 129 (7), 1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manning BD; Toker A, AKT/PKB Signaling: navigating the network. Cell 2017, 169 (3), 381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Testa JR; Bellacosa A, AKT plays a central role in tumorigenesis. Proc. Natl. Acad. Sci. U. S. A 2001, 98 (20), 10983–10985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martorana F; Motta G; Pavone G; Motta L; Stella S; Vitale SR; Manzella L; Vigneri P, AKT inhibitors: new weapons in the fight against breast cancer? Front Pharmacol. 2021, 12, 662232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heerding DA; Rhodes N; Leber JD; Clark TJ; Keenan RM; Lafrance LV; Li M; Safonov IG; Takata DT; Venslavsky JW; Yamashita DS; Choudhry AE; Copeland RA; Lai Z; Schaber MD; Tummino PJ; Strum SL; Wood ER; Duckett DR; Eberwein D; Knick VB; Lansing TJ; McConnell RT; Zhang S; Minthorn EA; Concha NO; Warren GL; Kumar R, Identification of 4-(2-(4-amino-1,2,5-oxadiazol-3-yl)-1-ethyl-7-{[(3S)-3-piperidinylmethyl]oxy}−1H- imidazo[4,5-c]pyridin-4-yl)-2-methyl-3-butyn-2-ol (GSK690693), a novel inhibitor of AKT kinase. J. Med. Chem 2008, 51 (18), 5663–5679. [DOI] [PubMed] [Google Scholar]
- 12.Blake JF; Xu R; Bencsik JR; Xiao D; Kallan NC; Schlachter S; Mitchell IS; Spencer KL; Banka AL; Wallace EM; Gloor SL; Martinson M; Woessner RD; Vigers GP; Brandhuber BJ; Liang J; Safina BS; Li J; Zhang B; Chabot C; Do S; Lee L; Oeh J; Sampath D; Lee BB; Lin K; Liederer BM; Skelton NJ, Discovery and preclinical pharmacology of a selective ATP-competitive Akt inhibitor (GDC-0068) for the treatment of human tumors. J. Med. Chem 2012, 55 (18), 8110–8127. [DOI] [PubMed] [Google Scholar]
- 13.Addie M; Ballard P; Buttar D; Crafter C; Currie G; Davies BR; Debreczeni J; Dry H; Dudley P; Greenwood R; Johnson PD; Kettle JG; Lane C; Lamont G; Leach A; Luke RW; Morris J; Ogilvie D; Page K; Pass M; Pearson S; Ruston L, Discovery of 4-amino-N-[(1S)-1-(4-chlorophenyl)-3-hydroxypropyl]-1-(7H-pyrrolo[2,3-d]pyrimidin −4-yl)piperidine-4-carboxamide (AZD5363), an orally bioavailable, potent inhibitor of Akt kinases. J. Med. Chem 2013, 56 (5), 2059–2073. [DOI] [PubMed] [Google Scholar]
- 14.Hirai H; Sootome H; Nakatsuru Y; Miyama K; Taguchi S; Tsujioka K; Ueno Y; Hatch H; Majumder PK; Pan BS; Kotani H, MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther 2010, 9 (7), 1956–1967. [DOI] [PubMed] [Google Scholar]
- 15.Zhao YY; Tian Y; Zhang J; Xu F; Yang YP; Huang Y; Zhao HY; Zhang JW; Xue C; Lam MH; Yan L; Hu ZH; Dinglin XX; Zhang L, Effects of an oral allosteric AKT inhibitor (MK-2206) on human nasopharyngeal cancer in vitro and in vivo. Drug Des. Devel. Ther 2014, 8, 1827–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xing Y; Lin NU; Maurer MA; Chen H; Mahvash A; Sahin A; Akcakanat A; Li Y; Abramson V; Litton J; Chavez-MacGregor M; Valero V; Piha-Paul SA; Hong D; Do KA; Tarco E; Riall D; Eterovic AK; Wulf GM; Cantley LC; Mills GB; Doyle LA; Winer E; Hortobagyi GN; Gonzalez-Angulo AM; Meric-Bernstam F, Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 2019, 21 (1), 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lapierre JM; Eathiraj S; Vensel D; Liu Y; Bull CO; Cornell-Kennon S; Iimura S; Kelleher EW; Kizer DE; Koerner S; Makhija S; Matsuda A; Moussa M; Namdev N; Savage RE; Szwaya J; Volckova E; Westlund N; Wu H; Schwartz B, Discovery of 3-(3-(4-(1-Aminocyclobutyl)phenyl)-5-phenyl-3H-imidazo[4,5-b]pyridin-2-yl)pyridin −2-amine (ARQ 092): an orally bioavailable, selective, and potent allosteric AKT inhibitor. J. Med. Chem 2016, 59 (13), 6455–6469. [DOI] [PubMed] [Google Scholar]
- 18.Mimura N; Hideshima T; Shimomura T; Suzuki R; Ohguchi H; Rizq O; Kikuchi S; Yoshida Y; Cottini F; Jakubikova J; Cirstea D; Gorgun G; Minami J; Tai YT; Richardson PG; Utsugi T; Iwama A; Anderson KC, Selective and potent Akt inhibition triggers anti-myeloma activities and enhances fatal endoplasmic reticulum stress induced by proteasome inhibition. Cancer Res. 2014, 74 (16), 4458–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yap TA; Garrett MD; Walton MI; Raynaud F; de Bono JS; Workman P, Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr. Opin. Pharmacol 2008, 8 (4), 393–412. [DOI] [PubMed] [Google Scholar]
- 20.Faes S; Dormond O, PI3K and AKT: unfaithful partners in cancer. Int. J. Mol. Sci 2015, 16 (9), 21138–21152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo K; Tang W; Zhuo H; Zhao G, Recent advance of Akt inhibitors in clinical Trials. ChemistrySelect. 2019, 4 (31), 9040–9044. [Google Scholar]
- 22.Landel I; Quambusch L; Depta L; Rauh D, Spotlight on AKT: current therapeutic challenges. ACS Med. Chem. Lett 2020, 11 (3), 225–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lai AC; Crews CM, Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discov 2017, 16 (2), 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schapira M; Calabrese MF; Bullock AN; Crews CM, Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discov 2019, 18 (12), 949–963. [DOI] [PubMed] [Google Scholar]
- 25.Dale B; Cheng M; Park KS; Kaniskan HU; Xiong Y; Jin J, Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer 2021, 21 (10), 638–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneider M; Radoux CJ; Hercules A; Ochoa D; Dunham I; Zalmas LP; Hessler G; Ruf S; Shanmugasundaram V; Hann MM; Thomas PJ; Queisser MA; Benowitz AB; Brown K; Leach AR, The PROTACtable genome. Nat. Rev. Drug Discov 2021, 20 (10), 789–797. [DOI] [PubMed] [Google Scholar]
- 27.Sun X; Gao H; Yang Y; He M; Wu Y; Song Y; Tong Y; Rao Y, PROTACs: great opportunities for academia and industry. Signal Transduct. Target Ther 2019, 4, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He M; Cao C; Ni Z; Liu Y; Song P; Hao S; He Y; Sun X; Rao Y, PROTACs: great opportunities for academia and industry (an update from 2020 to 2021). Signal Transduct. Target Ther 2022, 7 (1), 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bekes M; Langley DR; Crews CM, PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov 2022, 21 (3), 181–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.You I; Erickson EC; Donovan KA; Eleuteri NA; Fischer ES; Gray NS; Toker A, Discovery of an AKT degrader with prolonged inhibition of downstream signaling. Cell Chem. Biol 2020, 27 (1), 66–73 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu X; Xu J; Xie L; Wang L; Shen Y; Cahuzac KM; Chen X; Liu J; Parsons RE; Jin J, Design, synthesis, and evaluation of potent, selective, and bioavailable AKT kinase degraders. J. Med. Chem 2021, 64 (24), 18054–18081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu J; Yu X; Martin TC; Bansal A; Cheung K; Lubin A; Stratikopoulos E; Cahuzac KM; Wang L; Xie L; Zhou R; Shen Y; Wu X; Yao S; Qiao R; Poulikakos PI; Chen X; Liu J; Jin J; Parsons R, AKT degradation selectively inhibits the growth of PI3K/PTEN pathway-mutant cancers with wild-type KRAS and BRAF by destabilizing Aurora Kinase B. Cancer Discov. 2021, 11 (12), 3064–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu X; Xu J; Shen Y; Cahuzac KM; Park KS; Dale B; Liu J; Parsons RE; Jin J, Discovery of potent, selective, and in vivo efficacious AKT kinase protein degraders via structure-activity relationship studies. J. Med. Chem 2022, 65 (4), 3644–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu CL; Luo X; Tian T; Rao Z; Wang H; Zhou Z; Mi T; Chen D; Xu Y; Wu Y; Che J; Zhou Y; Li J; Dong X, Structure-based rational design enables efficient discovery of a new selective and potent AKT PROTAC degrader. Eur. J. Med. Chem 2022, 238, 114459. [DOI] [PubMed] [Google Scholar]
- 35.Lin K; Lin J; Wu WI; Ballard J; Lee BB; Gloor SL; Vigers GP; Morales TH; Friedman LS; Skelton N; Brandhuber BJ, An ATP-site on-off switch that restricts phosphatase accessibility of Akt. Sci. Signal 2012, 5 (223), ra37. [DOI] [PubMed] [Google Scholar]
- 36.Calleja V; Laguerre M; Larijani B, 3-D structure and dynamics of protein kinase B-new mechanism for the allosteric regulation of an AGC kinase. J. Chem. Biol 2009, 2 (1), 11–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu WI; Voegtli WC; Sturgis HL; Dizon FP; Vigers GP; Brandhuber BJ, Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS One 2010, 5 (9), e12913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lapierre JM; Eathiraj S; Vensel D; Liu Y; Bull CO; Cornell-Kennon S; Iimura S; Kelleher EW; Kizer DE; Koerner S; Makhija S; Matsuda A; Moussa M; Namdev N; Savage RE; Szwaya J; Volckova E; Westlund N; Wu H; Schwartz B, Discovery of 3-(3-(4-(1-aminocyclobutyl)phenyl)-5-phenyl-3H-imidazo[4,5-b]pyridin-2-yl)pyridin −2-amine (ARQ 092): an orally bioavailable, selective, and potent allosteric AKT inhibitor. J. Med. Chem 2016, 59 (13), 6455–6469. [DOI] [PubMed] [Google Scholar]
- 39.Han X; Wang C; Qin C; Xiang W; Fernandez-Salas E; Yang CY; Wang M; Zhao L; Xu T; Chinnaswamy K; Delproposto J; Stuckey J; Wang S, Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of Androgen Receptor (AR) for the treatment of prostate cancer. J Med Chem 2019, 62 (2), 941–964. [DOI] [PubMed] [Google Scholar]
- 40.Shen Y; Gao G; Yu X; Kim HS; Wang L; Xie L; Schwarz M; Chen X; Guccione E; Liu J; Bedford MT; Jin J, Discovery of first-in-class protein arginine methyltransferase 5 (PRMT5) degraders. J. Med. Chem 2020, 63 (17), 9977–9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu X; Li D; Kottur J; Shen Y; Kim HS; Park KS; Tsai YH; Gong W; Wang J; Suzuki K; Parker J; Herring L; Kaniskan HU; Cai L; Jain R; Liu J; Aggarwal AK; Wang GG; Jin J, A selective WDR5 degrader inhibits acute myeloid leukemia in patient-derived mouse models. Sci. Transl. Med 2021, 13 (613), eabj1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dale B; Anderson C; Park KS; Kaniskan HU; Ma A; Shen Y; Zhang C; Xie L; Chen X; Yu X; Jin J, Targeting triple-negative breast cancer by a novel proteolysis targeting chimera degrader of enhancer of zeste homolog 2. ACS Pharmacol. Transl. Sci 2022, 5 (7), 491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu Y; Savage RE; Eathiraj S; Meade J; Wick MJ; Hall T; Abbadessa G; Schwartz B, Targeting AKT1-E17K and the PI3K/AKT pathway with an allosteric AKT inhibitor, ARQ 092. PLoS One 2015, 10 (10), e0140479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soucy TA; Smith PG; Milhollen MA; Berger AJ; Gavin JM; Adhikari S; Brownell JE; Burke KE; Cardin DP; Critchley S; Cullis CA; Doucette A; Garnsey JJ; Gaulin JL; Gershman RE; Lublinsky AR; McDonald A; Mizutani H; Narayanan U; Olhava EJ; Peluso S; Rezaei M; Sintchak MD; Talreja T; Thomas MP; Traore T; Vyskocil S; Weatherhead GS; Yu J; Zhang J; Dick LR; Claiborne CF; Rolfe M; Bolen JB; Langston SP, An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458 (7239), 732–736. [DOI] [PubMed] [Google Scholar]
- 45.Anastassiadis T; Deacon SW; Devarajan K; Ma H; Peterson JR, Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol 2011, 29 (11), 1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.