

Abstract
P53 mutation is an important cause of chemoresistance in colorectal cancer (CRC). The investigation and identification of the downstream targets and underlying molecular mechanism of chemoresistance induced by P53 abnormalities are therefore of great clinical significance. In this study, we demonstrated and reported for the first time that leucine-rich pentatricopeptide repeat-containing protein (LRPPRC) is a key functional downstream factor and therapeutic target for P53 mutation-induced chemoresistance. Due to its RNA binding function, LRPPRC specifically bound to the mRNA of multidrug resistance 1 (MDR1), increasing MDR1 mRNA stability and protein expression. In normal cells, P53 induced by chemotherapy inhibited the expression of LRPPRC via miR-34a and in turn reduced the expression of MDR1. However, chemotherapy-induced P53/miR-34a/LRPPRC/MDR1 signalling pathway activation was lost when P53 was mutated. Additionally, the accumulated LRPPRC and MDR1 promoted drug resistance. Most importantly, gossypol-acetic acid (GAA) was recently reported by our team as the first specific inhibitor of LRPPRC. In CRC cells with P53 mutation, GAA effectively induced degradation of the LRPPRC protein and reduced chemoresistance. Both in vivo and in vitro experiments revealed that combination chemotherapy with GAA and 5-fluorouracil (5FU) yielded improved treatment outcomes. In this study, we reported a novel mechanism and target related to P53-induced drug resistance and provided corresponding interventional strategies for the precision treatment of CRC.
Subject terms: Tumour biomarkers, Oncogenes
Introduction
Colorectal cancer (CRC) is one of the most common malignant tumours in the world, and it caused 900,000 cancer-related deaths in 2020 [1]. Chemotherapy based on 5-fluorouracil (5FU) is the main treatment for CRC [2]. However, the effectiveness of chemotherapy needs to be improved, largely due to the recurrence and metastasis caused by drug resistance [3–6]. The recurrence rate after chemotherapy was up to 30% in patients with stage I-III disease and 65% in patients with stage IV disease [7]. Therefore, there is an urgent need to elaborate on the mechanism of chemoresistance and develop a corresponding treatment strategy.
P53 mutation frequently occurs in many cancers [8]. In CRC, P53 mutation is found in approximately 50% of all cases, which could contribute to genome instability and regulate the occurrence and progression of tumours [9–11]. The importance of P53 mutation in chemoresistance has also been reported for different cancers, such as CRC and breast cancer [12, 13]. Targeting the key factors of the P53 signalling pathway to rescue the chemoresistance caused by P53 mutation has become a research focus [14].
Leucine-rich pentatricopeptide repeat-containing protein (LRPPRC) [15] was originally studied as a mitochondrial protein that maintains the stability of the mitochondrial transcriptome and was found to be associated with the physiological processes underlying Leigh syndrome and HIV infection [16–18]. Recent studies have revealed the high expression of LRPPRC in different cancers and its association with stage, grade and poor prognosis in cancer patients [19, 20]. Mechanistic studies have revealed the effects of LRPPRC on gene transcription and mRNA stability in cancer [21]. General studies also showed that LRPPRC is a potential regulator of MDR1 [22] and participates in the regulation of chemotherapy sensitivity in the context of imatinib mesylate and cisplatin treatment [23–25]. However, the function and mechanism of LRPPRC in CRC chemoresistance are currently unclear. In addition, gossypol-acetic acid (GAA), a specific inhibitor of LRPPRC [26], was reported to inhibit the proliferation of various tumour cells, including prostate [27], breast [28] and CRC [29] cancer cells. The effect of combination treatment with GAA and 5FU is still unknown.
In this study, we identified LRPPRC as a new downstream drug-resistant protein related to P53. Wild-type (WT) P53 affected the mRNA stability of MDR1 through the negative regulation of LRPPRC by P53/miR-34a and therefore regulated the expression of MDR1. In addition, mutated (MUT) P53 failed to suppress LRPPRC after DNA damage, leading to increased MDR1 transcription, which subsequently caused chemoresistance. We also investigated the effects of a combination of 5FU and GAA. In this study, we revealed a novel potential mechanism for chemoresistance caused by P53 mutation and provided a new theoretical basis for future treatment options for patients with P53 mutation.
Materials and methods
Cell culture
The human CRC cell lines were saved in the State Key Laboratory of Molecular Oncology, National Cancer Center/National Clinical Research Center for Center/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College. All cell lines were authenticated by short tandem repeat (STR) profiling. RKO and DLD-1 cells were maintained in RPMI-1640 supplemented with 10% foetal bovine serum. The other cell lines were maintained in DMEM (Dulbecco’s Modified Eagle Medium) containing 10% foetal bovine serum. All cell lines were cultured at 37 °C in 5% CO2.
Construction of the 5FU-resistant SW480 cell line
SW480 parental cells in the logarithmic growth phase were continuously cultured in medium containing 0.5 μg/ml 5FU. When the SW480 cells exhibited stable growth, the concentration of 5FU was gradually increased to 1, 1.5, and 2 μg/ml. The cells that could survive in medium containing 2 μg/ml 5FU were used in our study.
Mass spectrometry analysis
Samples were analysed using an Orbitrap FusionTM LumosTM mass spectrometer (Thermo Fisher Scientific, USA) coupled with an Easy nLC 1000 system (Thermo Fisher Scientific, USA). Peptides were concentrated on a trap column (Acclaim PepMapTM 100, 74 μm × 2 cm (Thermo Fisher Scientific)) and eluted from an analytical column (E Acclaim PepMapTM RSLC 75 μm × 25 cm (Thermo Fisher Scientific)) with an acetonitrile gradient.
Mass spectrometry measurements were performed. Spectral data were collected in positive-ionisation mode with an electrospray voltage of 2300 V. The MS scan range was 350–1500 m/z at a resolution of 60 K. The automatic gain control target was set to 4.0 × 105, with a maximum injection time of 50 ms. The MS scan range was 350–1500 m/z at a resolution of 60 K. The automatic gain control target was set to 4.0 × 105, with a maximum injection time of 50 ms. The fragmentation mode was set to collision-induced dissociation, and the collision energy was 30%. The MS cycle time was set to 3 s, with data-dependent analysis and automated precursor peak selection. Precursors were selected for fragmentation based on the following criteria: most intense peaks, ion-intensity threshold 5.0 × 104, and charge state 2–7. Fragments were detected with an ion trap with an automatic scan range.
MS/MS spectra were searched against the UniProt bovine database (www.UniProt.org) for protein identification using Proteome Discoverer Software 2.4 (Thermo Fisher Scientific). The false discovery rates for protein and peptide identifications were set at 1%.
All samples were processed under the same conditions.
Chemicals, plasmids and antibodies
Gossypol-acetic acid (GAA) was obtained from Aladdin (Shanghai, China). Nutlin-3a was purchased from MedChemExpress. The WT P53 plasmid was saved in the State Key Laboratory of Molecular Oncology, National Cancer Center. The MUT P53 plasmid (R273H) was purchased from YouBio (Changsha, China). All plasmids were analysed by Sanger sequencing. Antibodies against LRPPRC (Santa Cruz Biotechnology Cat# sc-166178, RRID: AB_2137453), P53 (Santa Cruz Biotechnology Cat# sc-126, RRID: AB_628082) and MDR1 (Santa Cruz Biotechnology Cat# sc-13131, RRID: AB_626990) were purchased from Santa Cruz Biotech. The antibody against Bcl-2 was purchased from Proteintech (Proteintech Cat# 12789-1-AP, RRID: AB_2227948). Antibodies against Bax (Cell Signaling Technology Cat# 2772, RRID: AB_10695870) and cleaved PARP (Cell Signaling Technology Cat# 9542, RRID: AB_2160739) were purchased from Cell Signaling Technology. The antibody against β-actin was purchased from Sigma (Sigma–Aldrich Cat# A5316, RRID: AB_476743).
Animal study
All animal care and procedures were in accordance with national and institutional policies for animal health and well-being and approved by the Cancer Institute and Hospital, Chinese Academy of Medical Sciences (Beijing, China). Male BALB/c nude mice (weighing between 18 and 20 grams, aged 6 weeks) were purchased from Beijing HFK Bioscience Co., Ltd. (Beijing, China). The mice were randomly divided into groups for xenograft tumour formation.
Immunohistochemistry (IHC)
IHC assays were performed as previously described [30]. A total of 149 CRC tissues were used for IHC. According to the intensity of staining (intensity score), a pathologist who was blinded to the clinical status of the CRC tissues classified the sections into four ranks (0, negative; 1, slightly positive; 2, positive; 3, strongly positive), and the percentage of positive cells (percentage score) was classified into five ranks (0, 0%; 1, <10%; 2, 10–50%; 3, 51–80%; 4, >80%) for common types of CRC. The immunoreactivity score (IRS) was determined by the formula IRS = intensity score × percentage score. An overall score of ≤3 was defined as negative, and an overall score of ≥4 was defined as positive.
Tumour specimens
The tumour tissues and matched normal tissues were diagnosed as colorectal adenocarcinoma by pathological examination. The patient samples used for qPCR and IHC in this study were obtained from the Fourth Hospital of Hebei Medical University. The collected specimens met the following conditions: (1) all patients were not treated with chemotherapy before surgery; (2) all specimens were immediately placed in cryopreservation tubes and stored in liquid nitrogen after the specimens were removed. (3) All surgeries were radical excisions, and the upper and lower margins of the specimens were negative.
RNA extraction and qRT-PCR
RNA extraction and qRT-PCR assays were performed as previously described [31]. LRPPRC forward: GCGCAGATCACCCAGAAGAT.
LRPPRC reverse: GTTCGTCGCATTTGTCCACC.
GAPDH forward: GAGAAGGCTGGGGCTCATTT
GAPDH reverse: AGTGATGGCATGGACTGTGG
MDR1 forward: GTCGGACCACCATTGTGATAG
MDR1 reverse: CATTTCCTGCTGTCTGCATTGTG
Western blotting analysis of protein
Western Blotting assays were performed as previously described [30].
Transfection assays
The miR-34a mimic and inhibitor were purchased from GenePharma (Suzhou, China). The siRNA for LRPPRC was purchased from Invitrogen. Transfection was performed with Lipofectamine 2000 (Invitrogen, Carlsbad, California) according to the manufacturer’s instructions.
Si-LRPPRC-1: CCTCAAAGGAATGCAAGAATT
Si-LRPPRC-2: CGCAGC TTTAAGAGGTGAAAT
Si-P53-1: CGGCGCACAGAGGAAGAGAAT
Si-P53-2: CACCATCCACTACAACTACAT
Sh-LRPPRC construction and stable cell line selection
The sequence of Si-LRPPRC-1 was cloned into the psi-LvRU6rLP vector, generating the Sh-LRPPRC plasmid (GeneCopoeia, Guangzhou). Next, a plasmid mixture containing the psPAX2, pMD2.G (Addgene) and Sh-LRPPRC plasmids was cotransfected into 293 T cells to produce lentivirus. The lentiviral supernatant was collected 48 h after transfection. The purified lentivirus was obtained by passage through a 0.45 μm filter and added to medium supplemented with 8 µg/ml polybrene to transfect SW480 cells. SW480 cells infected with Sh-LRPPRC lentivirus were further selected with 2 µg/ml puromycin for 5 days to generate stable cell lines. The knockdown efficiency of LRPPRC was determined by Western blotting and qPCR.
Transwell assays
Transwell assays were performed as previously described [31].
Luciferase reporter assay
According to the sequence of the 3’-UTR of LRPPRC, microRNAs binding with LRPPRC were predicted by TargetScan (TargetScan, RRID:SCR_010845). HEK 293 T cells or RKO cells were seeded into 12-well plates at a density that would yield 60–70% confluence. The next day, HEK 293 T cells or RKO cells were transfected with pmirGLO-LRPPRC-3’UTR-WT or pmirGLO-LRPPRC-3’UTR-MUT (Generay, Shanghai, China) and miR-34a mimic or miR-NC using Lipofectamine 2000. The cells were then cultured at 37 °C in 5% CO2 for 24 h. Transfected cells were harvested and subjected to a luciferase reporter assay using the Dual-Luciferase Reporter assay system (Promega Corporation, Madison, WI, USA). Renilla luciferase was used as an internal control.
Proliferation assays and apoptosis assays
Cell proliferation assays were performed via the xCELLigence Real-Time Cell Analyser (RTCA)-MP system (Acea Biosciences/Roche), and the cells were automatically counted once every 15 min [31]. The cells were collected after treatment with drugs and then processed with an apoptosis kit (Neobioscience, China).
50% inhibitory concentration (IC50) analysis
A total of 5×103 cells were seeded in an E-Plate 96 (Roche Applied Science), which was placed in the RTCA-MP system. After the cells attached to the wall and spread out, the medium containing 5FU was replaced. For SW480 cells, the concentrations of 5FU were 0, 2.5, 5, 10, 20, and 40 μg/ml, and the IC50 at 96 h was calculated with GraphPad Software. For HCT116 cells, the concentrations of 5FU were 0, 5, 10, 20, 40, and 300 μg/ml, and the IC50 at 60 h was calculated with GraphPad Software.
RNA immunoprecipitation (RIP) assay
The EZ-Magna RIP Kit (Millipore, USA) was applied according to the manufacturer’s protocol to conduct the RIP assay. Immunoprecipitation was performed with an anti-LRPPRC antibody (Abcam, USA) [32].
Statistical analysis
The experimental results were analysed by Student’s t test (unpaired, two-tailed). P < 0.05 was considered to indicate significance. All statistical analyses were performed using Prism 5 (GraphPad Software Inc., La Jolla, CA). All assays were repeated at least three times.
Results
LRPPRC enhances the 5FU chemoresistance of CRC cells
As revealed by the GDSC database (https://www.cancerrxgene.org/), P53 mutations were associated with chemoresistance in CRC cells (Supplementary Fig. 1A). To identify P53-related proteins involved in 5FU resistance, we conducted a series of experiments designed as shown in Fig. 1A. We first established a 5FU-resistant cell line SW480 (named SW480R) (Supplementary Fig. 1B). Then, SW480R cells with MUT P53 and HCT116 cells with WT P53 were treated with 5FU for 36 h, and the remaining cells and their matched parental cell lines were analysed by protein mass spectrometry. Further analysis was performed to screen 5FU-resistant proteins regulated by P53.
Fig. 1. LRPPRC promoted 5FU chemoresistance in the context of P53 mutation.

A Schematic diagram of the experimental design. B Heatmap of differentially expressed proteins from protein quantitative mass spectrometry. C Immunoblotting of LRPPRC and P53 in SW480 and HCT116 cells treated with 5FU for 36 h. D Immunoblotting of LRPPRC and P53 in SW480 and HCT116 cells treated with DDP for 24 h. E The IC50 values of SW480 and HCT116 cells with or without LRPPRC knockdown. F The combined treatment effect of 5FU and Si-LRPPRC.
According to the results of protein mass spectrometry analysis, 2292 highly expressed proteins in SW480R cells and 1086 weakly expressed proteins in HCT116 cells were screened (fold change (FC) > 1.2 or FC < 0.8) (Fig. 1B and Supplementary Table 1). Of these, 416 candidate proteins showed highly increased expression in SW480R cells treated with 5FU and decreased expression in HCT116 cells treated with 5FU (Supplementary Fig. 1C left and Supplementary Table 2).
A total of 2744 CRC-related genes were obtained from the intersection of colon cancer-related genes and rectal cancer-related genes in the GEPIA database (FC > 1.2, P < 0.05) and CRC-related genes in the GeneCards database (https://www.genecards.org/) (Supplementary Table 3 and Supplementary Fig. 1C right). We also obtained 504 drug resistance genes from the GSE69657 dataset (FC > 1.2, P < 0.05) (Supplementary Table 4). We took the intersection of the 416 candidate proteins, 2744 CRC-related genes and 504 drug resistance genes with jvenn software (http://jvenn.toulouse.inra.fr/app/example.html) and obtained 11 genes: NOB1, DDX21, BRCC3, CPD, GTPBP4, NAT10, LRPPRC, SLC11A2, CTPS2, LARP4B, and METTL15 (Supplementary Fig. 1D). Among them, we found that LRPPRC had been reported as an oncogene in various cancers and was associated with the treatment sensitivity of imatinib mesylate and cisplatin [20, 23]. Therefore, we speculated that LRPPRC was a potential drug-resistance protein related to P53.
We subsequently verified the above results using western blotting (WB). The results showed that LRPPRC expression was increased in drug-resistant and 5FU-treated SW480 cells compared to control cells. LRPPRC expression was decreased in HCT116 cells treated with 5FU compared to control cells (Fig. 1C, Supplementary Fig. 1E). We also verified that the expression of LRPPRC was dependent on P53 in CRC cell lines, in which P53 could be induced by cisplatin and adriamycin (Fig. 1D and Supplementary Fig. 1F).
To investigate the association between LRPPRC and 5FU treatment, we designed small interfering RNA (siRNA) and the shRNA-LRPPRC lentiviral vector to knockdown LRPPRC expression (Supplementary Fig. 1G, H). We found that compared to the control (SW480 = 14.52 μg/ml, HCT116 = 18.95 μg/ml), the IC50 of 5FU was significantly decreased in cells with reduced LRPPRC expression (SW480 = 4.88 μg/ml, HCT116 = 11.7 μg/ml) (Fig. 1E). To further validate this hypothesis, apoptosis and cluster formation assays were conducted. As revealed by the results, LRPPRC knockdown significantly enhanced apoptosis induced by 5FU (Supplementary Fig. 1I, J). In RTCA analysis, the proliferation of cells transfected with Si-LRPPRC and treated with 5FU was significantly reduced compared to that of cells in the Si-NC, Si-NC + 5FU and Si-LRPPRC groups (Fig. 1F).
LRPPRC promotes CRC proliferation, migration, and invasion in vitro and in vivo
Subsequently, the function of LRPPRC in CRC was investigated. We found that LRPPRC was highly expressed in CRC cells compared to normal intestinal epithelial cells (Supplementary Fig. 2A). The proliferation ability of SW480 and HCT116 cell lines was significantly inhibited by reduced expression of LRPPRC (Supplementary Fig. 2B, C). In addition, Transwell analysis demonstrated that reduced expression of LRPPRC decreased the migration and invasion of CRC cells (Supplementary Fig. 2D). Furthermore, the malignant phenotypes of CRC cells with stable Sh-LRPPRC expression were inhibited compared to those of the control cells (Supplementary Fig. 3A–C). In BALB/c nude mice subjected to subcutaneous tumour transplantation, the tumour growth rate and volume were significantly reduced in the Sh-LRPPRC group compared with the control group (Supplementary Fig. 3D). The results of H&E staining are shown in Supplementary Fig. 3E. Overall, LRPPRC promoted the proliferation, migration and invasion of CRC in vitro and in vivo.
Clinical characteristics of LRPPRC in colorectal cancer patients
As revealed by the results in The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov/) and samples collected in this study, the mRNA of LRPPRC was highly expressed in CRC tissues compared with normal colorectal tissues (Fig. 2A). Additionally, as suggested by the Clinical Proteomic Tumour Analysis Consortium (CPTAC) database (https://proteomics.cancer.gov/programs/cptac) and WB results in this study, LRPPRC protein expression was found to be higher in CRC tissues than in normal colorectal tissues (Fig. 2B, C). Furthermore, increased protein levels of LRPPRC were highly positively correlated with lymphatic metastasis and tumour stage (Fig. 2D). Additionally, high expression of LRPPRC was negatively correlated with the overall survival and progression-free survival in patients treated with 5-FU (Fig. 2E). Further analysis suggested that patients with P53 mutation and high expression of LRPPRC had a marginally poorer five-year survival prognosis than other patients (Fig. 2F).
Fig. 2. LRPPRC was overexpressed in CRC tissues.

A The mRNA expression of LRPPRC in 58 paired clinical CRC tissues (left) and 41 paired TCGA tissues (right). The mRNA level of LRPPRC was higher in CRC tissues. B Immunoblotting for LRPPRC in ten freshly paired CRC tissues. The protein level of LRPPRC was notably elevated in CRC tissues. C The protein expression of LRPPRC in CRC was higher in the CPTAC database. D Association of LRPPRC expression assessed by IHC with lymph node metastasis (left). Association of LRPPRC expression assessed by IHC with stage (right). The tissues used for IHC were obtained from the Fourth Hospital of Hebei Medical University. E The expression of LRPPRC was negatively correlated with the overall survival and progression-free survival of patients in the GSE103479 dataset. F High expression of LRPPRC with P53 mutation had a marginally poorer five-year survival prognosis in the GSE103479 dataset. *P < 0.05; **P < 0.01; ***P < 0.001.
P53 regulates the expression of LRPPRC
As suggested by the mass spectrometry and screening results, LRPPRC is a potential downstream drug-resistance protein of P53. To study the effects of P53 on LRPPRC expression, the P53 plasmid was transfected into cells. Indeed, the overexpression of MUT P53 significantly enhanced the expression of LRPPRC in a dose-dependent manner, while WT P53 achieved the opposite result (Fig. 3A, B and Supplementary Fig. 4A left).
Fig. 3. The protein level of LRPPRC was regulated by P53, and LRPPRC was a direct target of miR-34a.

A Immunoblotting of LRPPRC and P53 in SW480 and HCT116 cells transfected with the P53 mutation plasmid or control for 36 h. B Immunoblotting of LRPPRC and P53 in SW480 and HCT116 cells transfected with WT P53 plasmid or control for 36 h. C Immunoblotting of LRPPRC and P53 in SW480 and HCT116 cells transfected with P53 siRNA. D Effects of LRPPRC and P53 in HCT116 cells after incubation with different concentrations of Nutlin-3a. E Contrasting effects on LRPPRC protein levels in HCT116 (P53−/−) cells transfected with P53 MUT and WT plasmids. F The binding site prediction for LRPPRC from four databases (TargetScan, miRWalk, mirDIP, and microRNA). G The potential binding site of miR-34a in the 3’-UTR of LRPPRC, predicted by TargetScan. H A dual-luciferase reporter assay was conducted to verify that miR-34a significantly inhibited the luciferase activity of the WT 3’-UTR but not the MUT 3’-UTR of LRPPRC. I The transfection efficiency of the miR-34a mimic in HCT116 cells (left). Immunoblotting of LRPPRC transfected with gradient mole of mimic for miR-34a in HCT116 cells (right). J The transfection efficiency of the miR-34a inhibitor in HCT116 and RKO cells (left). Immunoblotting of LRPPRC transfected with miR-34a inhibitor in HCT116 and RKO cells (right).
In addition, we also assessed the effects of silencing of endogenous P53 on LRPPRC. P53 knockdown inhibited the expression of LRPPRC in SW480 cells with P53 MUT (Fig. 3C left) but enhanced LRPPRC expression in HCT116 cells with P53 WT (Fig. 3C right). Moreover, Nutlin-3a, an inhibitor of the p53-MDM2 interaction and stabilizer of the P53 protein [33], was used to assess the effects of WT P53 on LRPPRC expression in HCT116 cells. Nutlin-3a inhibited LRPPRC expression in a dose-dependent manner (Fig. 3D). Similar results were also observed in P53-deficient cells (Fig. 3E). Here, we confirmed that P53 participated in the regulation of LRPPRC protein levels.
LRPPRC is the direct target of miR-34a
In the above results, we found that LRPPRC expression was related to P53. However, we did not find evidence indicating that the promoter of LRPPRC was transcriptionally regulated by P53. For this reason, we speculated that the regulation of LRPPRC by P53 was mediated by miRNA.
The expression of LRPPRC was potentially regulated by 11 microRNAs, namely, miR-519b-3p, miR-320c, miR-330-3p, miR-139-3p, miR-491-3p, miR-127-5p, miR-34c-3p, miR-34a-5p, miR-193a-5p, miR-129-5p and miR-107, predicted by 4 miRNA prediction databases (Fig. 3F). Among them, miR-34a was previously reported to be associated with the P53 signalling pathway [34]. Thus, we first determined whether miR-34a could be regulated in opposite directions on the WT and MUT P53 backgrounds. As we expected, overexpression of MUT P53 decreased miR-34a expression (Supplementary Fig. 4A right, Supplementary Fig. 4B), while overexpression of WT P53 enhanced miR-34a expression (Supplementary Fig. 4C). The effect of endogenous P53 on miR-34a was also evaluated. MUT P53 knockdown promoted miR-34a expression, while WT P53 achieved the opposite result (Supplementary Fig. 4D). The level of miR-34a was also enhanced in HCT116 cells after treatment with Nutlin-3a (Supplementary Fig. 4E). The opposite regulatory effect on miR-34a was also observed in P53-deficient cells transfected with MUT P53 or WT P53 (Supplementary Fig. 4F). Thus, we suspected that miR-34a mediated the regulation of LRPPRC by P53.
Using the TargetScan database (http://www.targetscan.org/vert_72/), we identified the potential binding sites of miR-34a and LRPPRC (Fig. 3G) and constructed MUT and WT control plasmids. In a luciferase assay, we found that LRPPRC was the target gene of miR-34a in CRC. miR-34a inhibited the 3’-UTR reporter gene activity of WT LRPPRC but not MUT LRPPRC (Fig. 3H). Additionally, as revealed by WB, miR-34a activation significantly reduced the protein level of LRPPRC, while inhibition of miR-34a expression increased the protein level of LRPPRC (Fig. 3I, J and Supplementary Fig. 4G). In general, miR-34a inhibited the expression of LRPPRC by directly binding to the 3’-UTR of LRPPRC.
P53 inhibits LRPPRC via miR-34a
Overall, it was proven that LRPPRC was a direct target of miR-34a. We subsequently investigated whether the inhibitory effect of P53 on LRPPRC was mediated by miR-34a.
SW480 cells with MUT P53 and HCT116 cells with WT P53 were used. As revealed by WB (upper panel) and qPCR (lower panel) in Fig. 4, abnormal expression of WT P53 in both SW480 and HCT116 cells reduced the protein level of LRPPRC by promoting the expression of miR-34a. In addition, these effects were rescued by a specific inhibitor of miR-34a (Fig. 4A, B). In HCT116 cells with high WT endogenous P53 expression induced by cisplatin treatment, LRPPRC expression was significantly suppressed, and miR-34a expression was increased. This suppression of LRPPRC expression caused by high P53 expression was rescued by the miR-34a inhibitor. In SW480 cells with MUT P53 expression, cisplatin treatment promoted the expression of LRPPRC but inhibited miR-34a expression. This increased LRPPRC expression caused by MUT P53 expression was further enhanced by the miR-34a inhibitor (Fig. 4C, D). Overall, as evidenced by our results, we found that P53 regulated the expression of LRPPRC via miR-34a (Fig. 4E).
Fig. 4. P53 mediated the expression of LRPPRC via miR-34a.

A Immunoblotting for LRPPRC and P53 after transfection with P53 plasmid or miR-34a inhibitor in SW480 cells (up). The level of miR-34a was evaluated by qPCR (bottom). B Immunoblotting for LRPPRC and P53 after transfection with P53 plasmid or miR-34a inhibitor in HCT116 cells (up). The level of miR-34a was evaluated by qPCR (bottom). C Immunoblotting for LRPPRC and P53 after transfection with miR-34a inhibitor or negative control and treatment with DDP in SW480 cells (up). The level of miR-34a was evaluated by qPCR (bottom). D Immunoblotting for LRPPRC and P53 in HCT116 cells transfected with miR-34a inhibitor or negative control and then treated with DDP (up). The level of miR-34a was evaluated by qPCR (bottom). E Model of the regulation of LRPPRC by the LRPPRC/miR-34a axis.
LRPPRC knockdown causes mRNA degradation of MDR1
According to previous results, P53 negatively regulates LRPPRC through miR-34a. LRPPRC knockdown inhibited the malignancy of CRC cells and enhanced chemotherapy sensitivity to 5FU. However, the downstream molecular mechanisms of LRPPRC in chemoresistance were still unclear. Therefore, we performed transcriptomic sequencing analysis on SW480 cells with stable low LRPPRC expression and controls (Fig. 5A and Supplementary Table 5). Forty-seven genes were enriched in the ABC_FAMILY_PROTEINS_MEDIATED_TRANSPORT pathway, and ABCB1 (encoding MDR1) was significantly downregulated (Supplementary Table 5). LRPPRC was reported to be a key regulatory factor for the expression of MDR1 [22], an important protein involved in chemoresistance in CRC. Thus, we selected MDR1 as the potential downstream target of LRPPRC.
Fig. 5. Downregulation of LRPPRC induced mRNA degradation of MDR1.

A Pathway enrichment analysis of differentially expressed genes (FC > 1.2, or FC < 0.8, P < 0.05) in Sh-LRPPRC cells (SW480) and NC cells through Reactome gene sets. Fisher’s test was used to calculate the P value for the pathway enrichment analysis. B Correlations of the mRNA expression of LRPPRC and MDR1 in the CCLE database. C Correlations of the mRNA expression of LRPPRC and MDR1 in the TCGA database. D Correlations of the protein expression of LRPPRC and MDR1 in the CPTAC database. E The protein expression of LRPPRC and MDR1 in SW480 cells and SW480R cells with or without LRPPRC knockdown (up). The mRNA expression of LRPPRC and MDR1 in SW480 cells and SW480R cells with or without LRPPRC knockdown (down). F The interaction between MDR1 and LRPPRC was confirmed by RIP and Western blotting. G The mRNA expression of MDR1 in SW480 cells with or without LRPPRC knockdown after treatment with actinomycin D (5 μg/ml). H Immunoblotting of LRPPRC, P53 and MDR1 after transfection with P53 mutation plasmid or negative control in SW480 cells and SW480R cells with or without LRPPRC knockdown. I The IC50 value of 5FU in SW480 cells transfected with P53 mutation plasmid or negative control with or without LRPPRC knockdown. J The protein expression of Bcl2, LRPPRC, and MDR1 in SW480 cells with or without LRPPRC combining treatment with 5FU.
Through the Cancer Cell Line Encyclopedia (CCLE) database (https://portals.broadinstitute.org/ccle), we found that LRPPRC was positively correlated with MDR1 in CRC cell lines (Fig. 5B). Consistent results were also obtained from the TCGA and CPTAC databases (Fig. 5C, D). Subsequently, as revealed by WB, LRPPRC knockdown reduced the mRNA and protein levels of MDR1 (Fig. 5E). LRPPRC was reported to affect the stability of mRNA. Using an RNA immunoprecipitation (RIP) assay, we also demonstrated that LRPPRC was able to bind to MDR1 mRNA, while LRPPRC knockdown promoted the degradation of MDR1 mRNA and reduced the level of the MDR1 protein (Fig. 5F, G). The above results demonstrated that LRPPRC potentially affected the expression of MDR1 by regulating the mRNA degradation of MDR1, thereby enhancing the chemoresistance of cells to 5FU treatment.
MUT P53 increases MDR1 expression via LRPPRC and enhances the 5FU chemoresistance of cells
In the next experiment, we investigated whether P53 could regulate the expression of MDR1 through LRPPRC and therefore regulate sensitivity to 5FU chemotherapy. We found that MUT P53 transfection significantly increased the levels of MDR1 and LRPPRC in cells and caused an increase in the IC50 of 5FU. Furthermore, reduced expression of LRPPRC significantly rescued the alterations in MDR1 levels and the 5FU IC50 (Fig. 5H, I). In addition, LRPPRC knockdown significantly promoted apoptosis and rescued the upregulation of MDR1 in 5FU-treated SW480 cells (Fig. 5J). This suggested that MUT P53 regulated the level of MDR1 by increasing LRPPRC expression and therefore led to 5FU chemoresistance.
GAA inhibits proliferation and enhances sensitivity to 5FU chemotherapy in CRC cells
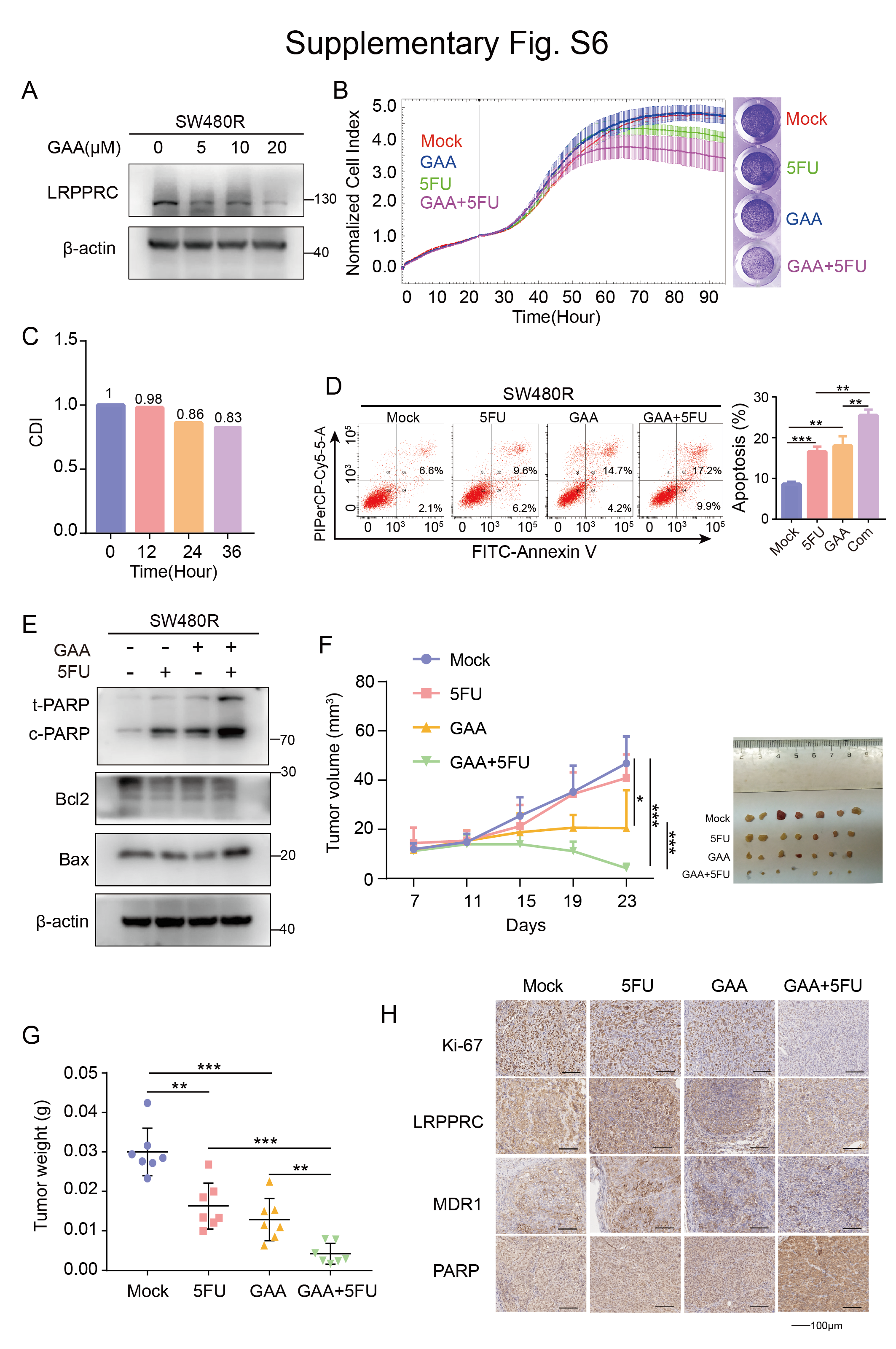
According to the above results, interventional treatment targeting LRPPRC to rescue the chemoresistance caused by P53 mutations appears promising. In a previous study, we found that GAA specifically decreased the protein level of LRPPRC [26]. As demonstrated in Fig. 6A, GAA inhibited the protein expression of LRPPRC. In the following experiment, we confirmed the effect of GAA on CRC in vitro. GAA inhibited the proliferation of CRC cells in a dose-dependent manner (Supplementary Fig. 5). We subsequently investigated the effect of combined treatment with GAA and 5FU. We divided the cells into four groups: the control, GAA, 5FU, and combination treatment groups. Both the cell proliferation test and drug sensitivity test showed that cells in the GAA combined with 5FU treatment group displayed the worst proliferation activity and the smallest cell number (Fig. 6B), and those two drugs exhibited synergistic effects (Fig. 6C). In apoptosis tests, cells in the combined treatment group also showed the highest apoptosis rate and the highest expression of apoptosis-related proteins (Fig. 6D, E). Our results demonstrated that the combination of GAA and 5FU produced significant therapeutic effects in vitro. Similar results were also obtained from the test in BALB/c nude mice. As shown in Fig. 6F, the growth rate of subcutaneous transplanted tumours in the combination treatment group was significantly lower than that in the other three groups. The volume and weight of subcutaneously transplanted tumours in the combined treatment group were also significantly lower than those in the other 3 groups (Fig. 6G, H). As revealed by immunohistochemistry, the levels of Ki-67, LRPPRC and MDR1 were highest in the 5FU treatment group. Furthermore, the level of cleaved PARP was highest in the combined treatment group (Fig. 6I). In addition, a therapeutic effect of GAA and 5FU in combination was observed in SW480R cells, which indicated that GAA sensitisation to 5FU affected tumour cell proliferation in vitro and in vivo (Supplementary Fig. 6).
Fig. 6. GAA could enhance sensitivity to 5FU chemotherapy.

A The expression of LRPPRC in SW480 and HCT116 cells treated with GAA at a range of concentrations for 24 h. B The combined therapeutic effect of GAA and 5FU in SW480 cells. C The interaction coefficient of GAA and 5FU: coefficient of drug interaction (CDI) > 1 indicates antagonism, CDI = 1 indicates addition, and CDI < 1 indicates synergism. D SW480 cells were treated with 5FU (20 μg/ml) and/or GAA (20 μM) for 48 h. Apoptosis was detected by flow cytometry (left). Statistical analysis of apoptosis (right). E SW480 cells were treated for 36 h with 5FU and/or GAA. The protein expression of Bax, Bcl2, total-PARP and cleaved-PARP was detected by WB. F Schematic diagram of combined treatment with GAA and 5FU in vivo. G The volume of subcutaneously transplanted tumours in 4 groups: mock, 5FU, GAA, and GAA + 5FU. H The weight of subcutaneously transplanted tumours in the 4 groups. I IHC of cleaved-PARP, Ki-67, LRPPRC and MDR1 in subcutaneous transplanted tumours from four groups. Scale bars, 100 μm.
Discussion
P53 mutation frequently occurs in patients with CRC, while P53 dysfunction leads to tumour progression and chemoresistance [35]. Here, we reported the potential molecular mechanism of P53 mutation-induced 5FU chemoresistance in CRC: P53 mutation caused chemoresistance through the miR-34a/LRPPRC/MDR1 signalling pathway (Fig. 7). This mechanism may be involved in the effects of chemotherapy treatment in CRC patients with P53 mutation. GSE103479 dataset analysis also suggested that among CRC patients treated with chemotherapy, patients with P53 mutations and high expression of LRPPRC (27%) demonstrated poor 5-year survival. To date, no drug targeting P53 mutation has been applied in the clinical treatment of CRC. Therefore, investigations of new treatment strategies targeting P53 mutation-related chemoresistance (especially for patients with high LRPPRC expression) are particularly important.
Fig. 7.

Proposed model of chemosensitivity modulation via the P53/miR-34a/LRPPRC/MDR1 pathway.
We first reported that LRPPRC was a new drug-resistance downstream protein of P53, participated in CRC as an oncogene, and affected sensitivity to 5FU chemotherapy in CRC cells. In P53 mutant cells, 5FU promoted the expression of MUT P53 and increased LRPPRC expression, MDR1 mRNA stability and MDR1 protein levels, which ultimately caused chemoresistance. In addition, LRPPRC knockdown rescued the increased expression of MDR1 caused by P53 mutation and enhanced sensitivity to 5FU treatment. Overall, we identified a novel mechanism of chemoresistance caused by P53 mutation via the regulation of LRPPRC.
LRPPRC was previously reported to have mRNA binding properties [36, 37]. We found that LRPPRC knockdown reduced the mRNA stability of MDR1 and therefore reduced the protein level of MDR1. We identified a new mechanism underlying the regulatory effect of LRPPRC on the mRNA stability of MDR1. This process may have affected the chemotherapy sensitivity of 5FU-treated cells.
We explained, on a molecular level, the potential effects of treatments targeting LRPPRC in P53 mutant cells. This result suggested that the combination of a small-molecule inhibitor of LRPPRC and 5FU may be effective in the treatment of patients with P53 mutation (especially those with high LRPPRC expression). We subsequently proved that combination treatment with GAA and 5FU was effective in both in vitro and in vivo experiments: we found that in cells with P53 mutation, combined use of GAA and 5FU produced improved treatment results compared with single drug use, proving the effectiveness of combination treatment with GAA and 5FU. Combination therapy with GAA and 5FU in SW480R cells was consistent with that in parental cells. Thus, for patients with P53 mutation (with high LRPPRC expression), the combined use of GAA and 5FU may be a promising strategy.
Overall, we demonstrated that MUT P53 induced chemoresistance through the miR-34a/LRPPRC/MDR1 signalling pathway. We also demonstrated the promising treatment effect of 5FU combined with GAA in the treatment of CRC. Our study provides a new potential treatment strategy for the precision treatment of CRC patients with P53 mutation.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
Thanks Professor Xiaohong Fang and Dr. Wei Zhou (University of Chinese Academy of Sciences (Zhejiang Cancer Hospital), Hangzhou Institute of Medicine, Chinese Academy of Sciences, Hangzhou, Zhejiang) for the valuable advices and help.
Author contributions
YS and GW conceived and designed the project. YY, LZ and HY performed experiments and wrote the manuscript. SG made figures and tables. SH, MT and YN performed mass spectrometry. JY performed bioinformatics analysis, data collection and data statistics. CZ and JN provided clinical samples and relevant information. YY and HY performed experiments, made figures and tables and revised the manuscript during the revision.
Funding
This work was supported by the National Natural Science Foundation of China (no. 81872398), CAMS Innovation Fund for Medical Sciences (CIFMS) (No. 2021-I2M-1-014), the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (No. 2019PT310027), and the National key research and development program of China (No. 2016YFA0500303).
Data availability
The authors declare that all data supporting the findings of this study are available within the paper in the main text or the Supplementary file.
Competing interests
The authors declare no competing interests.
Ethics approval
All animal care and procedures were in accordance with national and institutional policies for animal health and well‐being and approved by Cancer Institute and Hospital, Chinese Academy of Medical Sciences (Beijing, China).
Footnotes
Edited by M. Oren
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Yang Yang, Hongyu Yuan, Lianmei Zhao.
Contributor Information
Guiying Wang, Email: wangguiyingtgzy@163.com.
Yongmei Song, Email: symlh2006@163.com.
Supplementary information
The online version contains supplementary material available at 10.1038/s41418-022-01007-x.
References
- 1.Sung H, Ferlay J, Siegel R, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Xie Y-H, Chen Y-X, Fang J-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct Target Ther. 2020;5:22. doi: 10.1038/s41392-020-0116-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramos A, Hemann MT. Drugs, bugs, and cancer: Fusobacterium nucleatum promotes chemoresistance in colorectal. Cancer Cell. 2017;170:411–3. doi: 10.1016/j.cell.2017.07.018. [DOI] [PubMed] [Google Scholar]
- 4.Vodenkova S, Buchler T, Cervena K, Veskrnova V, Vodicka P, Vymetalkova V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: Past, present and future. Pharmacol Therapeutics. 2020;206:107447. doi: 10.1016/j.pharmthera.2019.107447. [DOI] [PubMed] [Google Scholar]
- 5.Blondy S, David V, Verdier M, Mathonnet M, Perraud A, Christou N. 5-Fluorouracil resistance mechanisms in colorectal cancer: From classical pathways to promising processes. Cancer Sci. 2020;111:3142–54.. doi: 10.1111/cas.14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Del Rio M, Molina F, Bascoul-Mollevi C, Copois V, Bibeau F, Chalbos P, et al. Gene expression signature in advanced colorectal cancer patients select drugs and response for the use of leucovorin, fluorouracil, and irinotecan. J Clin Oncol: Off J Am Soc Clin Oncol. 2007;25:773–80. doi: 10.1200/JCO.2006.07.4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Stok EP, Spaander MCW, Grünhagen DJ, Verhoef C, Kuipers EJ. Surveillance after curative treatment for colorectal cancer. Nat Rev Clin Oncol. 2017;14:297–315. doi: 10.1038/nrclinonc.2016.199. [DOI] [PubMed] [Google Scholar]
- 8.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 9.Iacopetta B. TP53 mutation in colorectal cancer. Hum Mutat. 2003;21:271–6. doi: 10.1002/humu.10175. [DOI] [PubMed] [Google Scholar]
- 10.Russo A, Bazan V, Iacopetta B, Kerr D, Soussi T, Gebbia N. The TP53 colorectal cancer international collaborative study on the prognostic and predictive significance of p53 mutation: influence of tumor site, type of mutation, and adjuvant treatment. J Clin Oncol: Off J Am Soc Clin Oncol. 2005;23:7518–28. doi: 10.1200/JCO.2005.00.471. [DOI] [PubMed] [Google Scholar]
- 11.Wang S, Zhang Y, Huang J, Wong CC, Zhai J, Li C, et al. TRIM67 activates p53 to suppress colorectal cancer initiation and progression. Cancer Res. 2019;79:4086–98.. doi: 10.1158/0008-5472.CAN-18-3614. [DOI] [PubMed] [Google Scholar]
- 12.Stiewe T, Haran T. How mutations shape p53 interactions with the genome to promote tumorigenesis and drug resistance. Drug Resist Updat. 2018;38:27–43. doi: 10.1016/j.drup.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 13.Royds JA, Iacopetta B. p53 and disease: when the guardian angel fails. Cell Death Differ. 2006;13:1017–26.. doi: 10.1038/sj.cdd.4401913. [DOI] [PubMed] [Google Scholar]
- 14.Cao X, Hou J, An Q, Assaraf YG, Wang X. Towards the overcoming of anticancer drug resistance mediated by p53 mutations. Drug Resist Updat. 2020;49:100671. doi: 10.1016/j.drup.2019.100671. [DOI] [PubMed] [Google Scholar]
- 15.Hou J, Wang F, McKeehan WL. Molecular cloning and expression of the gene for a major leucine-rich protein from human hepatoblastoma cells (HepG2). In vitro cellular & developmental biology. Animal. 1994;30a:111–4. doi: 10.1007/BF02631402. [DOI] [PubMed] [Google Scholar]
- 16.Mootha V, Lepage P, Miller K, Bunkenborg J, Reich M, Hjerrild M, et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc Natl Acad Sci USA. 2003;100:605–10. doi: 10.1073/pnas.242716699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schweitzer C, Matthews J, Madson C, Donnellan M, Cerny R, Belshan M. Knockdown of the cellular protein LRPPRC attenuates HIV-1 infection. PLoS ONE. 2012;7:e40537. doi: 10.1371/journal.pone.0040537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siira SJ, Spåhr H, Shearwood AJ, Ruzzenente B, Larsson NG, Rackham O, et al. LRPPRC-mediated folding of the mitochondrial transcriptome. Nat Commun. 2017;8:1532. doi: 10.1038/s41467-017-01221-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang X, Li X, Huang H, Jiang F, Lin Z, He H, et al. Elevated levels of mitochondrion-associated autophagy inhibitor LRPPRC are associated with poor prognosis in patients with prostate cancer. Cancer. 2014;120:1228–36. doi: 10.1002/cncr.28551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cui J, Wang L, Ren X, Zhang Y, Zhang H. LRPPRC: a multifunctional protein involved in energy metabolism and human disease. Front Physiol. 2019;10:595. doi: 10.3389/fphys.2019.00595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Z, Chen X, Xie R, Huang M, Dong W, Han J, et al. DANCR promotes metastasis and proliferation in bladder cancer cells by enhancing IL-11-STAT3 signaling and CCND1 expression. Mol Ther: J Am Soc Gene Ther. 2019;27:326–41. doi: 10.1016/j.ymthe.2018.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Labialle S, Dayan G, Gayet L, Rigal D, Gambrelle J, Baggetto L. New invMED1 element cis-activates human multidrug-related MDR1 and MVP genes, involving the LRP130 protein. Nucleic Acids Res. 2004;32:3864–76.. doi: 10.1093/nar/gkh722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu Y, Cui J, Jin L, Su Y, Zhang X. LRPPRC contributes to the cisplatin resistance of lung cancer cells by regulating MDR1 expression. Oncol Rep. 2021;45:4. doi: 10.3892/or.2021.7955. [DOI] [PubMed] [Google Scholar]
- 24.Corrêa S, Binato R, Du Rocher B, Ferreira G, Cappelletti P, Soares-Lima S, et al. ABCB1 regulation through LRPPRC is influenced by the methylation status of the GC -100 box in its promoter. Epigenetics. 2014;9:1172–83. doi: 10.4161/epi.29675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corrêa S, Pizzatti L, Du Rocher B, Mencalha A, Pinto D, Abdelhay E. A comparative proteomic study identified LRPPRC and MCM7 as putative actors in imatinib mesylate cross-resistance in Lucena cell line. Proteome Sci. 2012;10:23. doi: 10.1186/1477-5956-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou W, Sun G, Zhang Z, Zhao L, Xu L, Yuan H, et al. Proteasome-independent protein knockdown by small-molecule inhibitor for the undruggable lung adenocarcinoma. J Am Chem Soc. 2019;141:18492–9. doi: 10.1021/jacs.9b08777. [DOI] [PubMed] [Google Scholar]
- 27.McGregor N, Patel L, Craig M, Weidner S, Wang S, Pienta KJ, et al. AT-101 (R-(-)-gossypol acetic acid) enhances the effectiveness of androgen deprivation therapy in the VCaP prostate cancer model. J Cell Biochem. 2010;110:1187–94. doi: 10.1002/jcb.22633. [DOI] [PubMed] [Google Scholar]
- 28.Kisim A, Atmaca H, Cakar B, Karabulut B, Sezgin C, Uzunoglu S, et al. Pretreatment with AT-101 enhances tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis of breast cancer cells by inducing death receptors 4 and 5 protein levels. J Cancer Res Clin Oncol. 2012;138:1155–63.. doi: 10.1007/s00432-012-1187-1. [DOI] [PubMed] [Google Scholar]
- 29.Ko C, Shen S, Yang L, Lin C, Chen Y. Gossypol reduction of tumor growth through ROS-dependent mitochondria pathway in human colorectal carcinoma cells. Int J Cancer. 2007;121:1670–9. doi: 10.1002/ijc.22910. [DOI] [PubMed] [Google Scholar]
- 30.Yuan H, Zhou W, Yang Y, Xue L, Liu L, Song Y. ISG15 promotes esophageal squamous cell carcinoma tumorigenesis via c-MET/Fyn/β-catenin signaling pathway. Exp Cell Res. 2018;367:47–55. doi: 10.1016/j.yexcr.2018.03.017. [DOI] [PubMed] [Google Scholar]
- 31.Song Y, Li L, Ou Y, Gao Z, Li E, Li X, et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. 2014;509:91–5. doi: 10.1038/nature13176. [DOI] [PubMed] [Google Scholar]
- 32.Zhao Z, Li L, Du P, Ma L, Zhang W, Zheng L, et al. Transcriptional downregulation of miR-4306 serves as a new therapeutic target for triple negative breast cancer. Theranostics. 2019;9:1401–16.. doi: 10.7150/thno.30701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol. 2009;49:223–41. doi: 10.1146/annurev.pharmtox.48.113006.094723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hermeking H. MicroRNAs in the p53 network: micromanagement of tumour suppression. Nat Rev Cancer. 2012;12:613–26. doi: 10.1038/nrc3318. [DOI] [PubMed] [Google Scholar]
- 35.Capaci V, Bascetta L, Fantuz M, Beznoussenko GV, Sommaggio R, Cancila V, et al. Mutant p53 induces Golgi tubulo-vesiculation driving a prometastatic secretome. Nat Commun. 2020;11:3945. doi: 10.1038/s41467-020-17596-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Z, Chen X, Xie R, Huang M, Dong W, Han J, et al. DANCR promotes metastasis and proliferation in bladder cancer cells by enhancing IL-11-STAT3 signaling and CCND1 expression. Mol Ther. 2019;27:326–41. doi: 10.1016/j.ymthe.2018.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruzzenente B, Metodiev MD, Wredenberg A, Bratic A, Park CB, Cámara Y, et al. LRPPRC is necessary for polyadenylation and coordination of translation of mitochondrial mRNAs. EMBO J. 2012;31:443–56. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings of this study are available within the paper in the main text or the Supplementary file.