

Abstract
PI3Kδ inhibitors are active in patients with lymphoid neoplasms and a first series of them have been approved for the treatment of multiple types of B-cell lymphoid tumors, including marginal zone lymphoma (MZL). The identification of the mechanisms underlying either primary or secondary resistance is fundamental to optimize the use of novel drugs. Here we present a model of secondary resistance to PI3Kδ inhibitors obtained by prolonged exposure of a splenic MZL cell line to idelalisib. The VL51 cell line was kept under continuous exposure to idelalisib. The study included detailed characterization of the model, pharmacological screens, silencing experiments, and validation experiments on multiple cell lines and on clinical specimens. VL51 developed resistance to idelalisib, copanlisib, duvelisib, and umbralisib. An integrative analysis of transcriptome and methylation data highlighted an enrichment of upregulated transcripts and low-methylated promoters in resistant cells, including IL-6/STAT3- and PDGFRA-related genes and surface CD19 expression, alongside the repression of the let-7 family of miRNA, and miR-125, miR-130, miR-193 and miR-20. The IL-6R blocking antibody to-cilizumab, the STAT3 inhibitor stattic, the LIN28 inhibitor LIN1632, the PDGFR inhibitor masitinib and the anti-CD19 antibody drug conjugate loncastuximab tesirine were active compounds in the resistant cells as single agents and/or in combination with PI3Kδ inhibition. Findings were validated on additional in vitro lymphoma models and on clinical specimens. A novel model of resistance obtained from splenic MZL allowed the identification of therapeutic approaches able to improve the antitumor activity of PI3Kδ inhibitors in B-cell lymphoid tumors.
Introduction
The delta isoform of phosphoinositide kinase (PI3Kδ) shows prominent expression across hematopoietic tissues and has a central role in B-cell receptor (BCR) signaling.1,2 Indeed, its inhibition is being extensively explored as a therapeutic approach for patients with lymphoid neo-plasms.1-3 Idelalisib was the first-in-class specific PI3Kδ inhibitor to show clinical activity as a single agent in patients with follicular lymphoma, marginal zone lymphoma (MZL), chronic lymphocytic leukemia and mantle cell lymphoma, and in combination with rituximab in patients with chronic lymphocytic leukemia,1,2 and it received the approval from the U.S. Food and Drug Administration (FDA) for the treatment of patients with relapsed follicular lymphoma or small lymphocytic lymphoma after at least two prior systemic therapies, and, in combination with rituximab, for patients with re-lapsed chronic lymphocytic leukemia.3 Idelalisib has been successfully followed by a series of second-generation PI3KÖ inhibitors, such as parsaclisib and zandelisib, and by compounds that inhibit additional kinases, such as copanlisib (PI3Ka/PI3Kö), duvelisib (PI3Kö/PI3Ky) and umbralisib (PI3K5 and casein kinase-1e), achieving clinical responses in lymphomas, including follicular lymphoma, mantle cell lymphoma, MZL and chronic lymphocytic leukemia.13 In particular, data have so far led to the FDA approval of copanlisib for patients with relapsed/refractory follicular lymphoma, duvelisib for patients with relapsed/refractory chronic lymphocytic leukemia, small lymphocytic lymphoma or follicular lymphoma, and umbralisib for the treatment of patients with relapsed/refractory MZL or follicular lymphoma.3 The identification of the mechanisms underlying either primary or secondary resistance to PI3K5 inhibitors is fundamental to optimize the use of these drugs, and a number of studies have described mechanisms of resistance to this class of agents in lymphoid neoplasms, driven by activation of alternative signaling cascades.4-11 Here we present a model of secondary resistance to PI3KÖ inhibitors, including duvelisib, copanlisib and umbralisib, obtained by prolonged exposure of a splenic MZL cell line to idelalisib.
Methods
Development of resistant cell lines
VL51 cells were cultured according to the recommended conditions, as previously described.12 To develop resistance cell lines were exposed to a 90% inhibitory concentration (IC90) of idelalisib (Selleckchem, Houston, TX, USA) for several months until they acquired specific drug resistance (resistant cells). In parallel, cells were cultured under similar conditions in the absence of drug (parental cells). Proliferation of stable resistance was tested by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay after 2 weeks of drug-free culture. The multidrug resistance phenotype was assessed by real-time polymerase chain reaction for MDR1 and MDR2/3 genes using published primers.13 We developed biological replicates by splitting the resistant clones 1 month after the development of resistance, keeping them separate for 6 months before performing further experiments. Cell line identity was periodically authenticated by short tandem repeat DNA profiling, as previously described.14 Cells were periodically tested to confirm Mycoplasma negativity using the MycoAlert My-coplasma Detection Kit (Lonza, Visp, Switzerland).
Treatments
Response to single drugs or drug combinations was assessed after 72 h of exposure to increasing doses of the drug followed by an MTT assay. Copanlisib, duvelisib, umbralisib, everolimus, bimiralisib, vincristine, 5-azacitidine, masitinib, stattic and tocilizumab were purchased from Selleckchem, Lin28-1632 (LIN1632) from R&D Systems (Minneapolis, MN, USA), and human recombinant interleukin 6 (IL-6; CYT-213) from Prospec (Rehovot, Israel). Loncastuximab tesirine was kindly provided by ADC Therapeutics (Epalinges, Switzerland). Details are provided in the Online Supplementary Methods.
Genomics and data mining
Details of the genomics studies and data mining are provided in the Online Supplementary Methods.
Enzyme-linked immunosorbent assays
For enzyme-linked immunosorbent assays (ELISA) the conditioned medium was cultured for 72 h and collected. The medium was then filtered twice (22 µm) and centrifuged at 4000 rpm for 30 minutes in Amicon Ultra-4 tubes (Ultracel 3k, Merk Millipore) to remove cells and particles, and analyzed via cytokine array or ELISA. Cytokine arrays (Human Cytokine Array Panel A, R&D Systems) were performed according to the manufacturer’s protocols. ELISA on frozen human serum samples were performed using a Luminex Assay (R&D Systems) according to the manufacturer’s protocols. The serum samples were collected from patients treated with idelalisib and enrolled on tissue banking protocols at Dana Farber Cancer Institute; all patients signed written informed consent prior to a sample being drawn, and the protocols were approved by the Dana-Farber Harvard Cancer Center Institutional Review Board.
Flow cytometry and protein analyses
Flow cytometry was performed to determine the surface expression of platelet-derived growth factor receptor A (PDGFRA), IL-6 receptor (IL-6R), IL-6 cytokine family signal transducer (IL-6ST), CXCR4 and CD19 (Online Supplementary Table S1), levels of p-AKT, p-BTK, p-PLCG2, p-mTOR and p-ERK (Online Supplementary Table S2); and cell cycle phases. Immunoblotting was performed to determine the expression of AKT/p-AKT, ERK/p-ERK JAK/p-JAK, STAT/p-STAT and GAPDH (Online Supplementary Table S3). Details are provided in the Online Supplementary Methods.
Pharmacological combination screening
Pharmacological screening was performed by exposing idelalisib-resistant cells in parallel with parental clones to 348 compounds, as single agents (1 µM) or in combination with idelalisib (1 µM), from a custom library containing agents belonging to the following classes: “Kinase Inhibitory”, “Epigenetic Compound”, “PI3K/Akt Inhibitor”, “Apoptosis”, “Anti-cancer Compound”, and “MAPK Inhibitor” (Selleckchem). Cell viability was measured by MTT assay after 72 h of exposure to the agents.
Gene silencing
Details of the gene silencing experiments are provided in the Online Supplementary Methods.
Results
A splenic marginal zone lymphoma model of secondary resistance to PI3Kδ inhibitors
To create a novel model of resistance to the PI3Kδ inhibitors, we selected the splenic MZL-derived VL51 cell line, with transcriptome, somatic mutational status, DNA profile and methylome comparable to that of primary splenic MZL samples.15,16 VL51 harbors a series of lesions that are typical of splenic MZL, including truncating mu-
Figure 1.

Profiles of drug sensitivity differ between parental and resistant cell lines. (A-G) Acquired resistance was tested by an MTT assay (72 h) in parental (black) and resistant (red) cells of the VL51 cell line. Drug sensitivity was evaluated in resistant and parental cells for the PI3K inhibitors idelalisib (A), copanlisib (B), duvelisib (C), umbralisib (D), the dual PI3K-mTOR inhibitor bimiralisib (E), the mTOR inhibitor everolimus (F) and the chemotherapy agent and the multidrug resistance substrate vincristine (G). Error bars correspond to standard deviation of the mean. Data are derived from at least three independent experiments. P values from a Z-test, statistically significant for values <0.05. tations of NOTCH2 and BIRC3, which activate downstream NOTCH and non-canonical NF-KB, respectively, and a nuclear localization signal mutation of KLF2.15,17,18
After 6 months of continuous exposure to the FDA-approved PI3KÖ inhibitor idelalisib, VL51 developed stable resistance with an IC 25-fold higher than that of its 50 parental counterpart (Figure 1A). The resistance was confirmed to be stable by repeating the IC50 measurements after 2 weeks in medium containing no drug, and multidrug resistance was ruled out by demonstrating no changes in the expression levels of MDR1/2 genes by semiquantitative real-time polymerase chain reaction (Online Supplementary Figure S1).
The idelalisib-resistant cell line also showed resistance to other FDA-approved PI3K inhibitors, including the PI3Ka/PI3K5 inhibitor copanlisib, the PI3Kö/PI3Kg inhibitor duvelisib, and the PI3K5/CK1e inhibitor umbralisib (Figure 1B-D). Conversely, they maintained sensitivity to the mTOR inhibitor everolimus, to the dual PI3K/mTOR inhibitor bimiralisib (Figure 1E, F) and to the chemotherapy agent and MDR substrate vincristine (Figure 1G).
Resistant cells clearly differed from their parental counterparts based on RNA-sequencing (total RNA, miRNA) and methylation, as shown by unsupervised analyses and at the pharmacological profiling (Figure 1, Online Supplementary Figures S2 and S11), but no exonic nonsynonymous variants appeared to be acquired in the resistant cells. Online Supplementary Table S4 shows the single and copy number variants identified by whole-exome sequencing in resistant cells compared to parental cells.
Resistance to idelalisib is driven by IL-6 and PDGFRA
To investigate the mechanism of resistance, we integrated the gene expression (gene expression profiling, RNA-sequencing), miRNA (RNA-sequencing) and methylation (800k Illumina array) profiles obtained in resistant and parental cells (Online Supplementary Table S4). When compared to its parental counterpart, the resistant cell line was enriched in BCR/TLR/NF-κB (TLR4, CD19, SYK), IL-6/STAT3 (IL-6, CD44), chemokines (CXCL10, CXCR4, CXCR3), PDGFR (PDGFRA, PRKCE), IGF1R and RAS-RAF signaling pathways, epigenetic signatures (PRC2-complex targets and methylated genes in cancer) and genes upregulated in MZL. On the other hand, it showed decreased expression of transcripts involved in amino acid deprivation, antigen processing, drug metabolism, translation, proteosome and hypoxia (Online Supplementary Figures S2B and S3, Online Supplementary Table S5). The integrative analysis of transcriptome and methylation data highlighted an enrichment of upregulated transcripts among the low-methylated promoters in resistant cells, including IL-6 and RAS-related genes (Figure 2, Online Supplementary Figure S3). Analysis of the miRNA profiles identified repression of members of the let-7 family (let-7e, let-7g, let7d, let-7f, let-7a), along with other miRNA including miR-125, miR-130, miR-193 and miR-20. Conversely, only one member of the let-7 family (let-7c) was among the upregulated miRNA in resistant cells, together with miR-3196, miR-4492, miR-4516 and others (Online Supplementary Table S5). Among the repressed miRNA in resistant cells, miRNA potentially targeting the IL-6-PDGFRA axis were enriched. Integration of methylation and miRNA data identified fully methylated and repressed miRNA known to target upregulated genes: members of the let-7 family of miRNA (let-7d, let-7e, let-7g) that directly target IL-6,19 miR-125a that regulates IL-6, IL-6R and STAT3,20 and the negative regulators of PDGFR signaling, miR-130a,21 miR-193b, miR-20b and miR-1722,23 (Figure 2, Online Supplementary Table S5). These data indicated that an epigenetic reprogramming could sustain the observed resistance.
Figure 2.
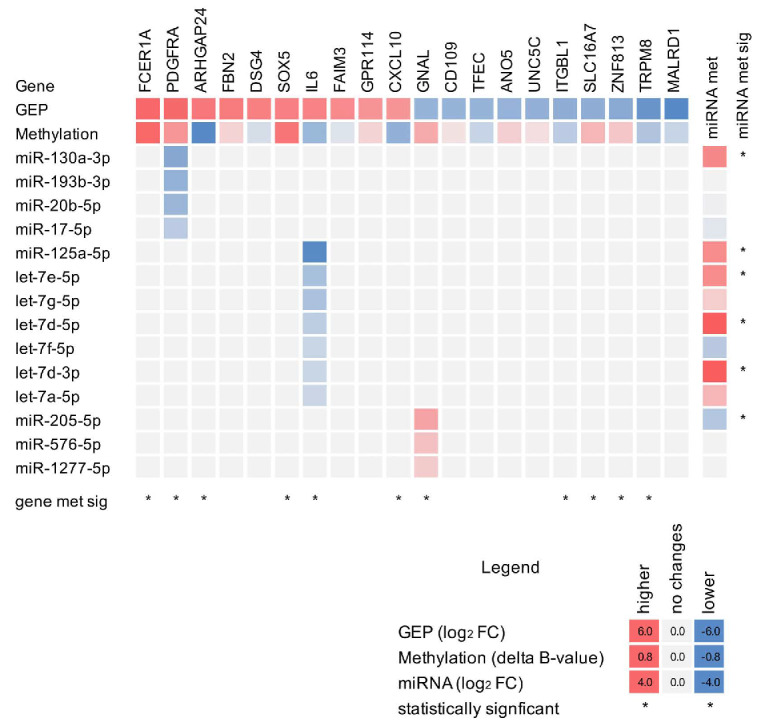
Multi-omics signature of the VL51, resistant cell line identifies activation of the IL-6-PDGFRA axis. Heatmap of RNA (gene expression profiling), methylation and miRNA profiles of resistant cells compared to parental cells. Heatmap values represent the differences between resistant and parental cells: fold change (log2 for RNA and miRNA) or D β-value (methylation), red for enrichment in resistant cells and blue for parental cells. Columns correspond to gene expression (gene expression profiling, RNA-sequencing) and methylation (MethylationEPIC BeadChip, Illumina) profiles of the top ten upregulated and top ten down-regulated genes; rows represent the differently expressed miRNA (RNA-sequencing) with values in the columns corresponding to the targeted gene. *Statistically significant differences (moderated t-test). GEP: gene expression profile; FC: fold change.
Having observed an upregulation of the secreted factor IL-6 at the RNA level in resistant cells, we evaluated whether the transfer of conditioned media to parental cells was able to transfer the resistance to PI3K inhibitors. The media from resistant cells (taken at 48 h), but not media taken from other cells, including parental cells, conferred resistance (Figure 3A). The secretion of IL-6 was confirmed by ELISA (Online Supplementary Figure S4), and the overexpression of surface PDGFRA, CXCR4 and CD19 was shown by flow cytometry (Figure 3B, Online Supplementary Figure S5). Increased levels of p-ERK (immunob-lot and phosphoFlow) and p-STAT (immunoblot) were observed in resistant cells (Figure 3C, Online Supplementary Figures S6 and S7).
Figure 3.

Proteome profiles of resistant cells differ from those of parental cells. (A) Cell viability, determined by the MTT assay, of parental cells (black line) exposed to idelalisib in the presence or not of conditioned medium from VL51 idelalisib-resistant cells (red line), cultured for 48 h, and parental + RES-cond RPMI (dotted black line). Data were derived from the average of three independent experiments. (B) Expression of surface PDGFRA (top), CXCR4 (center) and CD19 (bottom) by fluorescence activated cell sorting in parental (gray) and resistant (red) cell lines. The dotted black line represents the negative control (neg-cnt). Data were derived from two independent experiments. (C) Levels of protein phosphorylation by immunoblot in resistant cells. Values correspond to average fold-change of resistant compared to parental cells in two independent experiments. Data were normalized to GAPDH levels. Error bars represent standard error of the mean. *Statistically significant differences (t-test). MFI: mean fluorescence intensity; FACS: fluorescence activated cell sorting.
While silencing of individual genes had only partial effects, the concomitant silencing of both IL-6 and PDGFRA by siRNA reverted the resistance (Figure 4A, Online Supplementary Figure S8). Consistent with the effect of IL-6 silencing, exposure of parental cells to recombinant IL-6 induced resistance (Figure 4B).
Targeting IL-6/STAT3, LIN28, or PDGFRA reverts the resistance to idelalisib
We then explored pharmacological approaches to overcome the acquired resistance. Based on IL-6 involvement we tested tocilizumab, an IL-6R blocking antibody approved by the FDA for the treatment of various autoimmune disorders and cytokine release syndrome.24 The addition of tocilizumab to idelalisib overcame the resistance increasing the potency (i.e., the minimal active concentration) of the small molecule in the resistant cells (Figure 4B). No advantage was given by adding the antibody in parental cells. Tocilizumab as a single agent showed limited cytotoxicity in both parental and resistant cells (Online Supplementary Figure S9). To investigate the signaling activation in resistant cells downstream of IL-6, we combined idelalisib with the STAT3 inhibitor stattic. Consistent with the involvement of IL-6/STAT3 activation in resistant cells, and similarly to tocilizumab, addition of stattic decreased cell viability and enhanced sensitivity to idelalisib in resistant cells, with minimal effects in parental cells (Online Supplementary Figure S10).
Since members of the let-7 family were downregulated in the resistant cells, we evaluated LIN1632, an inhibitor of LIN28, an RNA binding protein involved in the downregulation of such miRNA. While the compound had no activity as a single agent, addition of LIN1632 to idelalisib improved the activity of the PI3Kδ inhibitor (1 µM) (Figure 5A), and increased the expression of let-7 in resistant cells but not in parental ones (Online Supplementary Figure S11). While a low dose of LIN1632 (1 µM) was effective only in resistant cells, higher doses of LIN1632 (50 µM) increased expression of let-7 in parental cells as well, confirming the activity of the LIN28/let-7 axis in VL51 cells (Online Supplementary Figure S11B).
Screening with 348 anticancer agents and compounds targeting important biological pathways identified acquired sensitivity of the resistant cells to the PDGFR inhibitor masitinib (Online Supplementary Figure S12). The efficacy of idelalisib was enhanced by the presence of masitinib (500 nM) only in resistant cells and not in parental ones (Figure 5B, Online Supplementary Figure S13).
CD19 upregulation in resistant cells determines increased sensitivity to CD19-targeting agents
Due to the increased CD19 (RNA and surface) expression in the idelalisib-resistant VL51 cell line compared to its parental counterpart (Figures 6A and 3B bottom, Online Supplementary Figure S5E), we evaluated a CD19-targeting treatment, namely the CD19-directed antibody-drug conjugate loncastuximab tesirine (ADCT-402), recently approved by the FDA for the treatment of patients with relapsed/refractory large B-cell lymphoma.25 The idelalisib-resistant cells were much more sensitive than their parental counterpart (Figure 6B).
Figure 4.
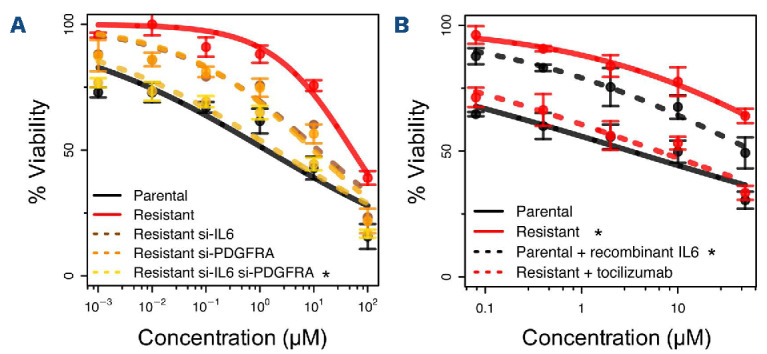
Interfering with IL-6 or PDFGRA overcomes resistance in the VL51 model. (A) Small interfering RNA were used for gene expression silencing of IL6 alone (brown dotted line), PDGFRA alone (orange dotted line) or concomitant silencing of IL6 and PDGFRA (yellow dotted line). Black and red lines for parental and resistant controls. *Statistically significant differences when compared to resistant control (red line, Z-test P<0.05). (B) Stimulation with recombinant IL-6 (30 ng/mL) conferred resistance to idelalisib in the parental cells (dotted black line) and blocking of IL-6 signaling with the monoclonal antibody tocilizumab (25 µg/mL) overcame resistance to idelalisib (red dotted line). Black and red continuous lines represent parental and resistant controls. *Statistically significant differences when compared to parental control (black continuous line, Z-test P<0.05). Sensitivity to all treatments was tested by an MTT assay at 72 h. Data were derived from three independent experiments, error bars represent standard deviation of the mean.
Figure 5.
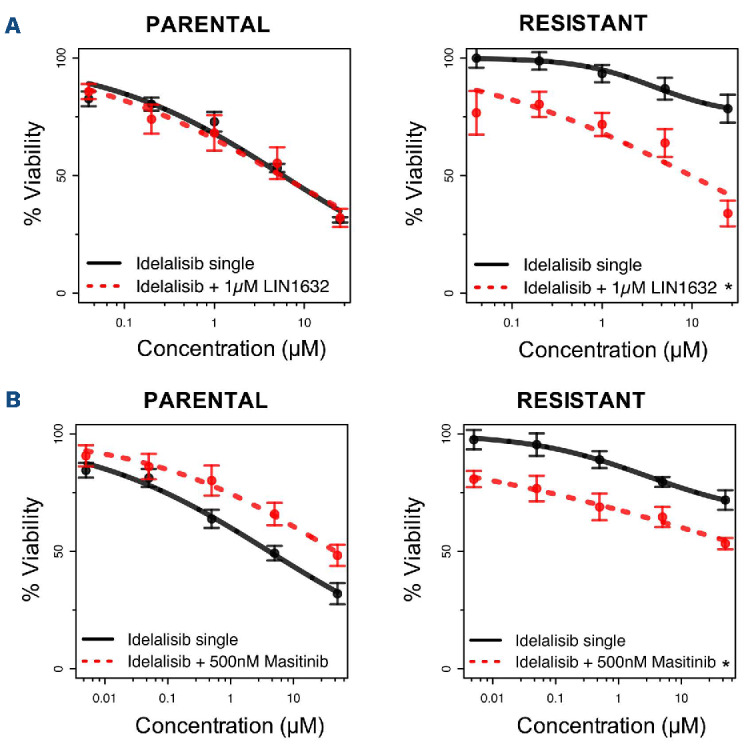
Combination with LIN28 or PDFGRA inhibitors restores sensitivity to idelalisib. (A) Addition of 1 µM of the LIN28 inhibitor LIN1632 restored sensitivity to idelalisib in the resistant cell line with no effect in the parental cells and very limited sensitivity as a single agent for both parental and resistant cell lines (Online Supplementary Figure S11B). (B) The combination of idelalisib with 500 nM of the PDGFR inhibitor masitinib increased sensitivity to idelalisib in the resistant cells with limited benefit in the parental cells. Treatment with masitinib as a single agent was beneficial only in resistant cells but not in parental ones (Online Supplementary Figure S13B). Sensitivity to all treatments was tested by an MTT assay at 72 h. Data were derived from three independent experiments, error bars represent standard deviation of the mean. *Statistically significant differences when compared to treatment with idelalisib as a single agent (black continuous line, Z-test P<0.05).
The detected mechanisms of resistance are not limited to idelalisib or the VL51 model
We then investigated whether the mechanism of resistance identified might affect sensitivity to other PI3K inhibitors, such as duvelisib, umbralisib and copanlisib. Addition of the IL-6R blocking antibody tocilizumab, the STAT3 inhibitor stattic, the PDGFR inhibitor masitinib or the LIN28 inhibitor LIN1632 improved the anti-lymphoma activity not only of idelalisib but also of the other clinically relevant PI3K inhibitors in resistant cells but not in the parental VL51 cell line (Online Supplementary Figure S14A, B). Conversely, as observed for idelalisib in parental cells, stimulation with recombinant IL-6 decreased sensitivity to duvelisib, umbralisib and copanlisib, which was restored by the addition of tocilizumab (Online Supplementary Figure S14C, D).
To further extend the significance of our findings beyond the VL51 model, we first took advantage of a large series of lymphoma cell lines we had previously characterized at transcriptome level and for their sensitivity to idelalisib.26 IL6, PDGFRA and LIN28 expression levels were inversely correlated with idelalisib sensitivity, while the latter was positively correlated with let-7 and miR-125 levels also in these additional B-cell lymphoma models (P<0.05) (Figure 7). Second, we selected two B-cell lymphoma models, another splenic MZL SSK41 and the diffuse large B-cell lymphoma-derived RCK8, based on their low sensitivity to idelalisib and their expression of IL6R, PDGFRA and LIN28. We tested the response to idelalisib, duvelisib, umbralisib and copanlisib in combination with the corresponding inhibitors tocilizumab (anti-IL-6R), masitinib (PDGFR inhibitor) and LIN1632 (LIN28 inhibitor). In line with what was observed in the VL51 model, both SSK41 and RCK8 benefited from the addition of tocilizumab, masitinib and LIN1632 (Online Supplementary Figure S15A, B). Third, the effect of IL-6 stimulation on the sensitivity to idelalisib and to the additional PI3K inhibitors duvelisib, umbralisib and copanlisib was evaluated in primary idelalisib-sensitive B-cell lymphoma cell lines with expression of IL6R, including the mantle cell lymphoma models Granta519 and JVM2. Similarly to VL51 parental cells, stimulation with recombinant IL-6 decreased sensitivity to all PI3K inhibitors, and addition of tocilizumab restored response to the drugs (Online Supplementary Figure S15C, D).
Figure 6.
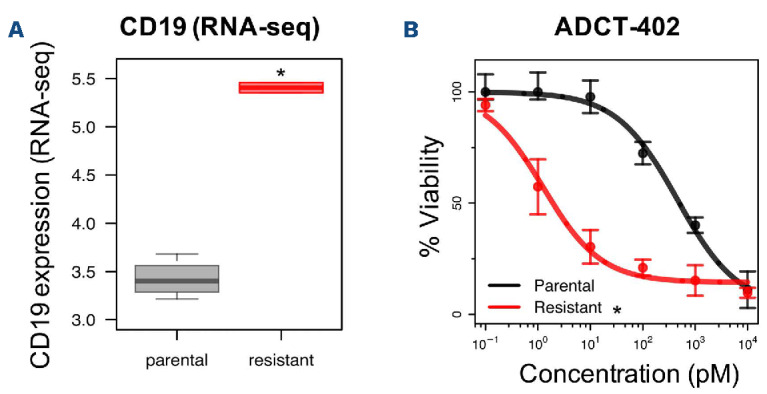
Idelalisib-resistant cells exhibited increased sensitivity to an anti-CD19 treatment. (A) CD19 expression was increased in resistant (red) compared to parental (grey) cells, both at the mRNA level (shown by RNA-sequencing) and with regards to surface expression (shown by fluorescence activated cell sorting, Figure 3B bottom panel). (B) The response to loncastuximab tesirine (ADCT-402) was tested by an MTT assay at 72 h of exposure. Data were derived from three independent experiments, error bars represent standard deviation of the mean. *Statistically significant differences between parental (black) and resistant (red) cell lines (Z-test, P<0.05). RNA-seq: RNA sequencing
Exposure to epigenetic drugs improves sensitivity to idelalisib
Based on the different epigenetic profiles observed in parental and resistant cells, we evaluated whether resistance may be reverted using epigenetic drugs. Due to the enrichment in resistant cells for targets of the PRC2-complex and methylated genes in cancer (Online Supplementary Figure S2B), we tested the combination of idelalisib with the EZH2 inhibitor tazemetostat and the demethylating agent 5-azacitidine. Resistant and parental cells were exposed to 5-azacitidine or to tazemetostat given concomitantly with idelalisib or 5 days before the PI3K inhibitor. The concomitant combination exhibited very limited effects in either parental or resistant cells (Online Supplementary Figure S16A). Nevertheless, and consistent with a mechanistic role of methylation in the resistance to idelalisib, pre-treatment with either 5-azacitidine (200 nM) or tazemetostat (1 µM) was beneficial in resistant cells and not in parental ones (Online Supplementary Figure S16B).
The factors associated with resistance in cell lines are also relevant in clinical specimens
To extrapolate our findings to the clinical context, we studied both available expression datasets and a series of serum samples. PDGFRA, IL-6 and IL-6 receptor appeared expressed in two series of retrospective collections of MZL clinical specimens and in a large series of diffuse large B-cell lymphomas27-29 (Online Supplementary Figure S17A-D). Taking advantage of the previously reported gene expression profile of splenic MZL clinical specimens,28 we determined the top 200 genes positively correlated (Pearson correlation) with the expression of either IL6 or PDGFRA, defining IL6 and PDFGRA signatures. When applied to our resistant model, the two signatures were enriched among the transcripts more expressed in the resistant cells than in the parental VL51 cells (Online Supplementary Figure S17E), highlighting the similarities between our model and the clinical setting.
Finally, secreted levels of IL-6 were evaluated in the serum of patients treated with idelalisib, comparing chronic lymphocytic leukemia patients with primary or acquired resistance to the PI3Kδ inhibitor to patients responding to the drug and paired for similar clinical features (Online Supplementary Table S6). In agreement with the in vitro data, all samples but one secreting IL-6 were non-responders to idelalisib (Figure 8A). Longitudinal analyses, comparing responders and non-responders to idelalisib were carried out for those patients with IL-6 secretion and available data along time. Non-responders showed increasing IL-6 levels upon treatment, while the paired responders remained with no IL-6 expression (Figure 8B).
These data suggest that the secreted factors identified in vitro can be present in the tumor microenvironment and that lymphoma cells might express the corresponding receptors to take advantage of available chemokines. Thus, although different mechanisms may drive resistance to PI3K inhibitors in clinical cases, the mechanism identified here might apply to some of these patients.
Figure 7.

Expression levels of crucial factors are correlated with resistance to idelalisib. IL6, PDGFRA, and LIN28 expression levels are inversely correlated with idelalisib sensitivity. Conversely, expression of miR-125a and members of the let-7 family of microRNA is associated with sensitivity to idelalisib. Expression and sensitivity data were analyzed from a previous publication by our group in a panel of 34 B-cell lymphoma cell lines.26 Cell lines were split into two groups based on higher (positive, black bars) or lower (negative, grey bars) values than the median expression of the corresponding gene or miRNA. Means of the idelalisib IC50 were calculated for these two groups and compared by a t-test. *P<0.05. IC50: half maximum inhibitory concentration.
Discussion
We developed and characterized a model of secondary resistance to PI3K inhibitors in splenic MZL. Our results indicate that: (i) epigenetic reprogramming can drive resistance to PI3K inhibitors by promoting the secretion of cytokines; and (ii) the resistance can be overcome using various drugs, which could be tested in novel clinical trials.
Idelalisib was the first-in-class PI3Kδ inhibitor and several additional PI3K inhibitors with different selectivity, including duvelisib, copanlisib and umbralisib, have entered the clinical setting as single agents and in combinations.1-3 The model of secondary resistance we developed via prolonged in vitro exposure to idelalisib presented decreased sensitivity to other PI3K inhibitors, such as copanlisib, duvelisib and umbralisib. The role of the identified factors in the resistance to PI3K inhibitors has been further validated in additional B-cell lymphoma in vitro models and across a large series of cell lines derived from different types of lymphoma, and their expression demonstrated in various clinical specimens, including the serum of idelalisib-resistant patients.
Targeting epigenetic vulnerabilities has been recently proposed as a valuable strategy to overcome resistance to therapy in cancer, including resistance to PI3K inhibitors.30-33 Epigenetic cell plasticity, which might be pharmacologically reverted, allows the development of drug-tolerant subpopulations even in the absence of genetic lesions or can provide a first permissible environment for the emergence, later on, of cells carrying DNA changes.34 A relevant role for methylation has been suggested in splenic MZL, in which a hypermethylation phenotype, elevated expression of EZH2, and enrichment of PRC2-complex targets are associated with a more aggressive clinical outcome.16
The resistant cell line secreted interleukins and chemokines, such as IL-6, and showed upregulation of prosurvival networks, including PDGFRA, JAK-STAT and NF-κB pathways. Our findings are in line with the notion that secreted factors can give resistance to PI3K inhibitors,35,36 and with the background of the cell line we have used, representative of the recently described NNK splenic MZL subgroup, driven by mutations in genes involved in NF-κB/NOTCH/KLF2.37 IL-6 can protect cancer cells from apoptosis and DNA damage induced by drugs and can decrease sensitivity to tyrosine kinase inhibitors by activating different signaling pathways, such as the JAK-STAT, AKT-mTOR and NF-κB signaling pathways.38 Release of IL-6 mediates resistance to the BTK inhibitor ibrutinib in Waldenström macroglobulinemia.39 to duvelisib and copanlisib in diffuse large B-cell lymphoma and T-cell lymphoma cells,35 and, in head and neck squamous cell carcinoma cell lines, blockade of IL-6 signaling overcomes resistance to the pan-PI3K inhibitor buparlisib.36 In our resistant cell line, upregulation of IL-6 was associated with downregulation of let-7 and miR-125 microRNA, which directly target the interleukin,19 and with the loss of methylation in the IL6 gene promoter. The latter was methylated in the parental cell line but not in resistant lines, and methylation-based repression of the IL-6-targeting miRNA, miR-125 and members of let-7 family, might contribute to the increased IL-6 expression. LIN28 is an RNA binding protein that functions as an oncogene inhibiting the expression of let-7 family microRNA, dysregulating the normal balance between differentiation and cell growth.40 Since IL-6 induces transcription of NF-κB targets, a positive feedback loop leads to B-cell activation under the control of LIN28 and let-7.19 The miRNA mir-125 is located at 19q13 and frequently downregulated or deleted in cancer, and it can also regulate cell proliferation by targeting p53.41 Loss of miR-125 leads to high expression of STAT3, IL-6 itself and of a subunit of IL-6 receptor complex (IL-6R).20 Hence, in the context of NF-κB and JAKSTAT activation, miR-125 might work concomitantly with let-7. Alongside IL-6, PDGFRA too was upregulated in the resistant cell line. Active PDGFRA signaling, as a consequence of genomic aberrations affecting PDGFRA, is associated with tumor development and progression in solid cancer,42 it mediates the activation of PI3K-AKT, JAK-STAT and RAS-ERK signaling,43 and it confers resistance to the tyrosine kinase inhibitor imatinib.44 PDGFR signaling is known to increase IL-6/IL-6R axis expression,45 and, furthermore, PDGFR is also linked with CXCR4,46 upregulated in the resistant cells. CXCR4 is upregulated after exposure to BCR inhibitors, including PI3K and BTK inhibitors26 and, in diffuse large B-cell lymphoma models, it has been suggested as a potential mechanism of resistance to the tyrosine kinase inhibitors themselves.5,6
Figure 8.

Levels of serum IL-6 are higher in idelalisib-resistant chronic lymphocytic leukemia patients than in idelalisib-sensitive ones. (A) IL-6 secretion was evaluated in serum samples from patients with chronic lymphocytic leukemia at the end of the treatment by enzyme-linked immunosorbent assay (Luminex, R&D Systems). Patients who responded to idelalisib (responders, blue) or did not (non-responders, red) were compared by a t-test. P represents adjusted P-value. (B) Longitudinal analyses were performed on clinically-paired patients with secretion of IL-6. Secretion of IL-6 was compared between patients not responding to idelalisib (red) and their corresponding matched controls (responding, blue). MFI: mean fluorescence intensity. ELISA: enzymelinked immunosorbent assay.
Since a multitude of experiments, including genetic silencing and use of IL-6 recombinant protein, indicated that the mechanism of resistance to PI3K inhibitors in the VL51 model was driven by activation of the IL-6/STAT3 and PDFGRA signaling cascades and was dependent on the action of LIN28, we explored possible therapeutic interventions. Tocilizumab is an IL-6R blocking antibody in clinical use to treat different autoimmune disorders and more recently for the cytokine release syndrome that can be seen with chimeric antigen receptor T-cell therapy or in patients with coronavirus disease 2019.24 Masitinib, a small molecule that inhibits PDGFR signaling and targets the innate immune system, is under clinical development for various indications, including systemic mastocytosis, solid tumors, and amyotrophic lateral sclerosis.47 The combination with these two already clinically available drugs, and with the LIN28 inhibitor LIN1632 or with the STAT3 inhibitor stattic, two compounds in the preclinical phase of investigation, restored sensitivity to PI3K inhibition. Moreover, the resistant cells had higher CD19 expression on their cell surface, which gave much higher activity to the CD19 targeting antibody drug conjugate loncastuximab tesirine, recently approved by the FDA for the treatment of patients with relapsed or refractory large B-cell lymphoma.25 In the clinical routine, CD19 could be easily investigated (by immunohistochemistry or flow cytometry) and its expression provides a rationale for testing CD19-targeting antibody drug conjugates, naked antibodies, and cellular therapies in patients exposed to PI3K inhibitors.
In conclusion, a model of secondary resistance to PI3K inhibitors, derived from splenic MZL, has revealed mechanisms of resistance to these drugs and allowed the identification of a first series of active therapeutic approaches that could be explored further.
Supplementary Material
Funding Statement
Funding: This study was supported in part by the Swiss National Science Foundation (SNSF 31003A_163232/1) with funds to EZ, DR and FB. JRB was supported by National Institutes of Health grant RO1 CA 213442 (PI: Jennifer Brown).
References
- 1.Phillips TJ, Michot JM, Ribrag V. Can next-generation PI3K inhibitors unlock the full potential of the class in patients with B-cell lymphoma? Clin Lymphoma Myeloma Leuk. 2021;21(1):8-20.e3. [DOI] [PubMed] [Google Scholar]
- 2.Kienle DL, Stilgenbauer S. Approved and emerging PI3K inhibitors for the treatment of chronic lymphocytic leukemia and non-Hodgkin lymphoma. Expert Opin Pharmacother. 2020;21(8):917-929. [DOI] [PubMed] [Google Scholar]
- 3.Tarantelli C, Argnani L, Zinzani PL, et al. PI3Kδ inhibitors as immunomodulatory agents for the treatment of lymphoma patients. Cancers (Basel). 2021;13(21):5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scheffold A, Jebaraj BMC, Tausch E, et al. IGF1R as druggable target mediating PI3K-δ inhibitor resistance in a murine model of chronic lymphocytic leukemia. Blood. 2019;134(6):534-547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen L, Ouyang J, Wienand K, et al. CXCR4 upregulation is an indicator of sensitivity to B-cell receptor/PI3K blockade and a potential resistance mechanism in B-cell receptor-dependent diffuse large B-cell lymphomas. Haematologica. 2020;105(5):1361-1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim JH, Kim WS, Ryu KJ, et al. CXCR4 can induce PI3Kδ inhibitor resistance in ABC DLBCL. Blood Cancer J. 2018;8(2):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Faia K, White K, Murphy E, et al. The phosphoinositide-3 kinase (PI3K)-δ,γ inhibitor, duvelisib shows preclinical synergy with multiple targeted therapies in hematologic malignancies. PLoS One. 2018;13(8):e0200725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yahiaoui A, Meadows SA, Sorensen RA, et al. PI3Kδ inhibitor idelalisib in combination with BTK inhibitor ONO/GS-4059 in diffuse large B cell lymphoma with acquired resistance to PI3Kδ and BTK inhibitors. PLoS One. 2017;12(2):e0171221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iyengar S, Clear A, Bodor C, et al. P110alpha-mediated constitutive PI3K signaling limits the efficacy of p110delta-selective inhibition in mantle cell lymphoma, particularly with multiple relapse. Blood. 2013;121(12):2274-2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murali I, Kasar S, Naeem A, et al. Activation of the MAPK pathway mediates resistance to PI3K inhibitors in chronic lymphocytic leukemia. Blood. 2021;138(1):44-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matera E-L, Fouret J, Baulu E, et al. Enhanced sensitivity of idelalisib and ibrutinib-resistant cell lines to anti-CD38 antibodies. J Cancer Sci Clin Ther. 2020;4:71-77. [Google Scholar]
- 12.Spriano F, Chung EYL, Gaudio E, et al. The ETS inhibitors YK-4-279 and TK-216 are novel antilymphoma agents. Clin Cancer Res. 2019;25(16):5167-5176. [DOI] [PubMed] [Google Scholar]
- 13.Hoellein A, Decker T, Bogner C, et al. Expression of multidrug resistance-associated ABC transporters in B-CLL is independent of ZAP70 status. J Cancer Res Clin Oncol. 2010;136(3):403-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaudio E, Tarantelli C, Spriano F, et al. Targeting CD205 with the antibody drug conjugate MEN1309/OBT076 is an active new therapeutic strategy in lymphoma models. Haematologica. 2020;105(11):2584-2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piva R, Deaglio S, Fama R, et al. The Kruppel-like factor 2 transcription factor gene is recurrently mutated in splenic marginal zone lymphoma. Leukemia. 2015;29(2):503-507. [DOI] [PubMed] [Google Scholar]
- 16.Arribas AJ, Rinaldi A, Mensah AA, et al. DNA methylation profiling identifies two splenic marginal zone lymphoma subgroups with different clinical and genetic features. Blood. 2015;125(12):1922-1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rossi D, Deaglio S, Dominguez-Sola D, et al. Alteration of BIRC3 and multiple other NF-kappaB pathway genes in splenic marginal zone lymphoma. Blood. 2011;118(18):4930-4934. [DOI] [PubMed] [Google Scholar]
- 18.Rossi D, Trifonov V, Fangazio M, et al. The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med. 2012;209(9):1537-1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 microRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139(4):693-706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li D, Kong C, Tsun A, et al. MiR-125a-5p decreases the sensitivity of Treg cells toward IL-6-mediated conversion by inhibiting IL-6R and STAT3 expression. Sci Rep. 2015;5:14615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh BN, Kawakami Y, Akiyama R, et al. The Etv2-miR-130a network regulates mesodermal specification. Cell Rep. 2015;13(5):915-923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mazzu YZ, Hu Y, Shen Y, et al. miR-193b regulates tumorigenesis in liposarcoma cells via PDGFR, TGFbeta, and Wnt signaling. Sci Rep. 2019;9(1):3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slattery ML, Mullany LE, Sakoda LC, et al. The MAPK-signaling pathway in colorectal cancer: dysregulated genes and their association with microRNAs. Cancer Inform. 2018;17:1176935118766522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Actemra (tocilizumab): highlights of prescribing information. Revised: 03/2021. Genentech, Inc. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm. 2021. [Google Scholar]
- 25.Zynlonta (loncastuximab tesirine): highlights of prescribing information. Revised: 04/2021. ADC Therapeutics, Inc. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm. 2021. [Google Scholar]
- 26.Tarantelli C, Gaudio E, Arribas AJ, et al. PQR309 is a novel dual PI3K/mTOR inhibitor with preclinical antitumor activity in lymphomas as a single agent and in combination therapy. Clin Cancer Res. 2018;24(1):120-129. [DOI] [PubMed] [Google Scholar]
- 27.Arribas AJ, Campos-Martín Y, Gómez-Abad C, et al. Nodal marginal zone lymphoma: gene expression and miRNA profiling identify diagnostic markers and potential therapeutic targets. Blood. 2012;119(3):e9-e21. [DOI] [PubMed] [Google Scholar]
- 28.Arribas AJ, Gomez-Abad C, Sanchez-Beato M, et al. Splenic marginal zone lymphoma: comprehensive analysis of gene expression and miRNA profiling. Mod Pathol. 2013;26(7):889-901. [DOI] [PubMed] [Google Scholar]
- 29.Lenz G, Wright G, Dave SS, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med. 2008;359(22):2313-2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu D, Yan Y, Wei T, et al. A targetable epigenetic vulnerability in PI3K/AKT inhibitor resistant cancers. bioRxiv 2020.08.27.269613; doi: 10.1101/2020.08.27.269613. [preprint, not peer-reviewed] [DOI] [Google Scholar]
- 31.Quagliano A, Gopalakrishnapillai A, Barwe SP. Understanding the mechanisms by which epigenetic modifiers avert therapy resistance in cancer. Front Oncol. 2020;10:992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Romero-Garcia S, Prado-Garcia H, Carlos-Reyes A. Role of DNA methylation in the resistance to therapy in solid tumors. Front Oncol. 2020;10:1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia-Martinez L, Zhang Y, Nakata Y, et al. Epigenetic mechanisms in breast cancer therapy and resistance. Nat Commun. 2021;12(1):1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wright SCE, Vasilevski N, Serra V, et al. Mechanisms of resistance to PI3K inhibitors in cancer: adaptive responses, drug tolerance and cellular plasticity. Cancers (Basel). 2021;13(7):1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim JH, Kim WS, Park C. Interleukin-6 mediates resistance to PI3K-pathway-targeted therapy in lymphoma. BMC Cancer. 2019;19(1):936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yun MR, Choi HM, Kang HN, et al. ERK-dependent IL-6 autocrine signaling mediates adaptive resistance to pan-PI3K inhibitor BKM120 in head and neck squamous cell carcinoma. Oncogene. 2018;37(3):377-388. [DOI] [PubMed] [Google Scholar]
- 37.Bonfiglio F, Bruscaggin A, Guidetti F, et al. Genetic and phenotypic attributes of splenic marginal zone lymphoma. Blood 2022;139(5):732-747. [DOI] [PubMed] [Google Scholar]
- 38.Kumari N, Dwarakanath BS, Das A, et al. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016;37(9):11553-11572. [DOI] [PubMed] [Google Scholar]
- 39.Chen JG, Liu X, Munshi M, et al. BTK Cys481Ser drives ibrutinib resistance via ERK1/2 and protects BTK wild-type MYD88-mutated cells by a paracrine mechanism. Blood. 2018;131(18):2047-2059. [DOI] [PubMed] [Google Scholar]
- 40.Piskounova E, Polytarchou C, Thornton JE, et al. Lin28A and Lin28B inhibit let-7 microRNA biogenesis by distinct mechanisms. Cell. 2011;147(5):1066-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen J, Ouyang H, An X, et al. miR-125a is upregulated in cancer stem-like cells derived from TW01 and is responsible for maintaining stemness by inhibiting p53. Oncol Lett. 2019;17(1):87-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ip CKM, Ng PKS, Jeong KJ, et al. Neomorphic PDGFRA extracellular domain driver mutations are resistant to PDGFRA targeted therapies. Nat Commun. 2018;9(1):4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li GZ, Raut CP. Targeted therapy and personalized medicine in gastrointestinal stromal tumors: drug resistance, mechanisms, and treatment strategies. Onco Targets Ther. 2019;12:5123-5133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lim SE, Esain V, Kwan W, et al. HIF1α-induced PDGFRβ signaling promotes developmental HSC production via IL-6 activation. Exp Hematol. 2017;46:83-95.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bernat-Peguera A, Simón-Extremera P, da Silva-Diz V, et al. PDGFR-induced autocrine SDF-1 signaling in cancer cells promotes metastasis in advanced skin carcinoma. Oncogene. 2019;38(25):5021-5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laforgia M, Marech I, Nardulli P, et al. An evaluation of masitinib for treating systemic mastocytosis. Expert Opin Pharmacother. 2019;20(13):1539-1550. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.