

Abstract
Owing to its highly heterogeneous molecular landscape, bladder cancer (BlCa) is still characterized by non-personalized treatment and lifelong surveillance. Motivated by our previous findings on miR-143/145 value in disease prognosis, we have studied the underlying epigenetic regulation of the miR-143/145 cluster in BlCa. Expression and DNA methylation of miR-143/145 cluster were analyzed in our screening (n = 162) and The Cancer Genome Atlas Urothelial Bladder Carcinoma (TCGA-BLCA; n = 412) cohorts. Survival analysis was performed using tumor relapse and progression as clinical endpoints for non-muscle-invasive bladder cancer (NMIBC; TaT1), while disease progression and patients’ death were used for muscle-invasive bladder cancer (MIBC; T2-T4). TCGA-BLCA served as validation cohort. Bootstrap analysis was carried out for internal validation, while decision curve analysis was used to evaluate clinical benefit. TCGA-BLCA and screening cohorts highlighted MIR145 core promoter as the pivotal, epigenetic regulatory region on cluster’s expression. Lower methylation of MIR145 core promoter was associated with aggressive disease phenotype, higher risk for NMIBC short-term progression, and poor MIBC survival. MIR145 methylation-fitted multivariate models with established disease markers clearly enhanced patients’ risk stratification and prediction of treatment outcome. MIR145 core promoter methylation was identified as a potent epigenetic regulator of miR-143/145 cluster, supporting modern personalized risk stratification and management in BlCa.
Keywords: MT: Non-coding RNAs, DNA methylation, epigenetics, prognosis, tumor progression, miRNA, urothelial bladder carcinoma, bladder tumors, pyrosequencing
Graphical abstract
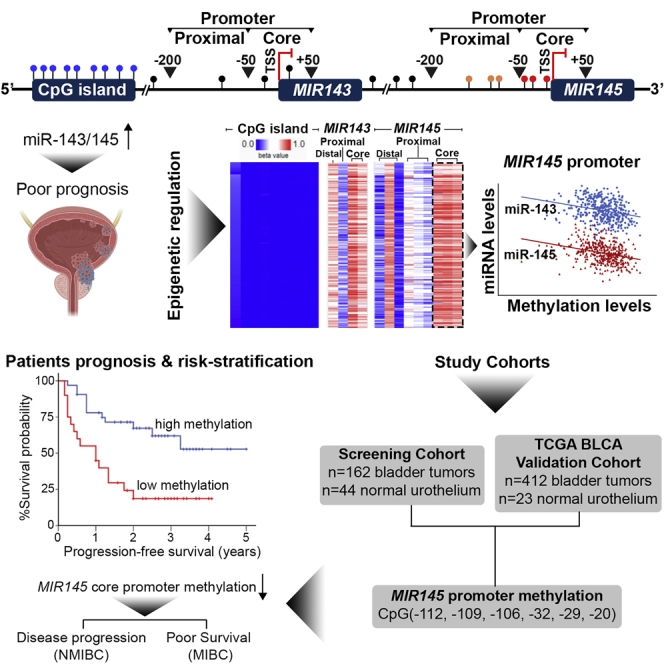
BlCa is characterized by non-personalized treatment and lifelong surveillance, aggravating patients’ quality-of-life. Herein, miR-143/145 cluster methylation was studied in BlCa, and MIR145 core promoter was highlighted as most influential on cluster’s regulation. MIR145 methylation profiling improved treatment outcome prediction, and MIR145 methylation-fitted multivariate models resulted in superior risk stratification and prognosis.
Introduction
Bladder cancer (BlCa) represents the second most common malignancy of the male genitourinary tract, succeeding prostate cancer, and the sixth most frequently diagnosed malignancy among men, worldwide.1,2 The vast majority of bladder tumors originate from the urothelium of the bladder wall, and urothelial bladder carcinomas (UBC; >90%) are further subclassified into non-muscle-invasive bladder cancer (NMIBC; Tis, Ta, T1) and muscle-invasive bladder cancer (MIBC; T2-T4), based on the invasion of the bladder’s detrusor muscle.3,4 Patients with newly diagnosed NMIBC (∼75% of primary UBC) are characterized by frequent relapses (∼50%–70%) and progression to muscle-invasive disease (∼15%),5,6 while primary MIBC (∼25% of UBC) is considered life threatening, displaying strong metastatic potential.7
Despite the marked reduction in disease-specific mortality over the last decades, owing to significant advances in disease diagnosis and therapy, there are yet to develop improvements regarding prognosis of treatment responses and personalized post-treatment management.8 Current disease prognosis relies on patients’ clinicopathological traits, mainly on pathological/clinical staging, tumor grade and multifocality, as well as the presence of carcinoma in situ (CIS). However, tumor heterogeneity—at the molecular and cellular levels—results in significantly varied disease course, even for the same risk-group patients.9, 10, 11, 12 As a result, BlCa management demands lifelong surveillance strategies, with invasive and frequent cystoscopies, affecting both patients’ quality of life and healthcare system financial costs.13 In this regard, the identification of novel molecular markers could ameliorate patients’ personalized prognosis and risk stratification and minimize unnecessary interventions, in correspondence with modern precision medicine.
MicroRNAs (miRNAs) constitute an ever growing family of endogenous small (∼22 nt) non-coding RNAs (sncRNAs), representing the most powerful post-transcriptional regulators of gene expression.14 In this regard, miRNAs finely tune numerous biological processes, including cell proliferation, apoptosis, and migration, displaying tumor-suppressive or oncogenic roles according to their effects on cellular transformation and homeostasis.15 miR-143 and miR-145 (miR-143/145) are transcribed to as bicistronic primary transcript, from the MIR143/MIR145 gene cluster (miR-143/145 cluster) on the 5q32 chromosomic region16 and are considered potent tumor suppressors via directly targeting known oncogenes, including KRAS, MYC, AKT, IGF1R, and IRS1/2.17 The expression of miR-143/145 is commonly deregulated in numerous malignancies, such as breast,18 prostate,19 clear-cell renal cell,20 colorectal,21 and head and neck squamous cell22 carcinomas, being implicated both in tumorigenesis and disease progression, as well as in supporting patient prognostication. Focusing on BlCa, our previously published findings revealed that the miR-143/145 cluster is significantly downregulated in bladder tumors compared with healthy bladder specimens, while elevated miR-143/145 levels are associated with disease aggressiveness, predicting progression of superficial tumors and high morbidity of muscle-invasive patients.23
Herein, in order to study the epigenetic regulation of miR-143/145 cluster in bladder tumors, we have analyzed DNA methylation levels of proximal and core promoter regions and evaluated their impact on miR-143/145 expression, as well as their clinical value in improving patients’ risk stratification and prediction of post-treatment disease course.
Results
MIR145 promoter epigenetically regulates miR-143/145 cluster
To identify the genomic regions of MIR143/145 cluster with epigenetic impact on miR-143/145 regulation, in silico analysis by UCSC Genome Browser (https://genome.ucsc.edu/) was performed to assign CpG sites with known Illumina CpG loci IDs (cg#) across genome. The analysis resulted in the identification of three CpG-rich regions: (1) the CpG island upstream to gene cluster (chr5:148.737.347–148.737.764), and the promoters—distal regulatory elements, proximal, and core promoter regions—of (2) MIR143 (chr5:148.808.481–148.808.586) and (3) MIR145 (chr5:148.810.209–148.810.296) genes (Figure 1A).
Figure 1.

MIR145 promoter emerges as potent epigenetic regulator of miR-143/145 cluster in bladder tumors
(A) Schematic representation of MIR143/145 locus, highlighting the CpG sites analyzed in silico within the CpG island of the cluster and the distal, proximal, and core promoters of MIR143 and MIR145 genes. (B) Spearman correlation of miR-143 and miR-145 levels in bladder tumors of the screening (left) and TCGA-BLCA (right) cohorts. (C) Boxplots representing miR-143 and miR-145 levels in bladder tumors and healthy adjacent urothelium in screening (left) and TCGA-BLCA (right) cohorts; p-values calculated by Mann-Whitney U test. (D) Heatmap illustrating the methylation levels of the CpG island and the promoters of MIR143/145 genes based on TCGA-BLCA data; visualized by XENA Browser Visualization Tool. (E–G) Spearman correlation analysis of miR-143/145 expression and methylation levels of CpG island (E), MIR143 (F), and MIR145 (G) promoters. (H, I) Methylation levels of MIR143 (H) and MIR145 (I) proximal and core promoters in bladder tumors and normal adjacent urothelium in TCGA-BLCA cohort visualized by XENA Browser Visualization Tool; p-values calculated by Welch's t-test. ∗p<0.05; ∗∗p<0.01, ∗∗∗p<0.001, ns: not significant.
The expression analysis of the cluster highlighted the strong correlation between miR-143-3p (miR-143) and miR-145-5p (miR-145) guide strands in bladder tumors of both our screening (Spearman rs = 0.934, p < 0.001) and The Cancer Genome Atlas Urothelial Bladder Carcinoma (TCGA-BLCA; Spearman rs = 0.646, p < 0.001) cohorts (Figure 1B), as well as the significantly reduced miR-143/145 expression compared with normal urothelium (Figure 1C). This potent co-regulation of miR-143/145 discloses a common and dominant regulatory mechanism in bladder tumor cells and prompted us to analyze the methylation imprinting of the identified CpG-rich regions of MIR143/145 locus using the Infinium Methylation450k data of TCGA-BLCA cohort.
The heatmap of MIR143/145 locus CpG-rich regions methylation of TCGA-BLCA cohort is depicted in Figure 1D. Within the CpG island, all CpG sites were revealed to be hypomethylated (Δβ<0.2), while their average methylation had only weak correlation with miR-143/145 levels (Figure 1E). On the contrary, CpG loci in MIR143 and MIR145 promoters displayed significantly higher imprinting, compared with CpG island (Figure 1D), while Spearman analysis highlighted the significantly stronger correlation of MIR145 promoter methylation with miR-143/145 levels compared with MIR143 promoter (Figures 1F and 1G). Accordingly, the analysis of miR-143/145 passenger strands (miR-143-5p; miR-143∗ and miR-145-3p; miR-145∗) in TCGA-BLCA cohort confirmed the higher impact of MIR145 promoter imprinting (Figure S1). Moreover, in line with the loss of miR-143/145 in bladder tumors, compared with normal urothelium, the methylation of MIR145 proximal and core promoter regions was significantly elevated in bladder tumors, which was not observed in the case of the CpGs of MIR143 promoter (Figures 1H and 1I). In the light of those findings, we decided to further study the role of MIR145 promoter imprinting on the epigenetic regulation of the miR-143/145 cluster and the clinical/treatment outcome of the BlCa patients by targeting the CpG sites −112, −109, −106 nt (proximal promoter) and −32, −29, −20 nt (core promoter) upstream of the MIR145 transcription start site.
MIR145 core promoter-mediated silencing of miR-143/145 cluster in bladder tumors
Spearman correlation analysis verified the strong negative association of miR-143/145 levels and MIR145 promoter methylation in bladder tumors (Figures 2A–2C). MIR145 core promoter region was revealed to have a superior impact on the regulation of the cluster (Figure 2A), as its hypermethylation resulted in a more robust downregulation of miR-143/145 levels (miR-143: rs = −0.401; p < 0.001; miR-145: rs = −0.425; p < 0.001) compared with the proximal promoter (miR-143: rs = −0.267; p = 0.023; miR-145: rs = −0.273; p = 0.020). Indeed, the methylation imprinting of each CpG locus analyzed was inversely correlated with miR-143 (Figure 2B) and miR-145 (Figure 2C) levels, whereas CpG (−29) and CpG (−20) of the MIR145 core promoter region revealed to hold the greatest impact on cluster’s regulation. Descriptive statistics of percent methylation levels of CpG sites (Table S1) highlighted the significantly increased methylation tendency from proximal (median percent methylation: 33.4% [−112], 32.4% [−109], 23.7% [−106]) to core (median percent methylation: 60.2% [−32], 80.8% [−29], 80.5% [−20]) promoter regions in bladder tumors, and thus the higher impact of MIR145 core promoter on cluster’s expression. Moreover, significantly elevated methylation was highlighted for all studied CpG sites in bladder tumors compared with the matched adjacent normal urothelium (Figure 2D), in line with the strong downregulation of miR-143/145 in bladder carcinoma. Representative pyrosequencing pyrograms are shown in Figure S2.
Figure 2.

MIR145 core promoter-mediated silencing of miR-143/145 cluster in bladder tumors
(A–C) Spearman correlation analysis of (A) MIR145 proximal and core promoter mean methylation, and (B, C) the individual CpG loci methylation with miR-143/145 levels. (D) Boxplots illustrating the methylation of MIR145 promoter CpG loci in bladder tumors and normal adjacent urothelium. ∗∗∗p<0.001 (E–G) Boxplots presenting the correlation of MIR145 promoter mean methylation with muscle-invasive disease (E), tumor stage (F), and tumor grade (G). p-values calculated by Mann-Whitney U test (D, E, G) and Kruskal-Wallis test (F).
Despite the tumor-suppressive functions of miR-143/145 and their loss in bladder tumors compared with the normal urothelium, our previous study revealed the significant correlation of the within-tumors miR-143/145 levels with unfavorable clinicopathological features and poor patient prognosis.23 In agreement with our previous findings and the working hypothesis of the present study, the analysis of the screening cohort highlighted the association of reduced MIR145 promoter methylation with aggressive disease phenotype (Figures 2E–2G), in terms of muscle-invasive tumors (Figure 2E), advanced tumor stage (Figure 2F), and high grade (Figure 2G). Similar to the overall methylation profile of MIR145 promoter, the core promoter CpG sites presented significantly higher methylation imprinting compared with proximal promoter, independent of the examined variable. Motivated by these observations, we decided to perform a comprehensive clinical evaluation of MIR145 core promoter methylation in BlCa patients.
Lower MIR145 core promoter methylation enhances the risk for disease progression and poor treatment outcome
Due to the different course of the disease, survival analysis was performed separately in the NMIBC and MIBC cohorts, using tumor relapse and progression (recurrence of higher/invasive stage), as well as disease progression (recurrence/metastasis or death; whichever came first) and patient’s death as clinical endpoint events, respectively. In this regard, 153 patients (NMIBC: 87; MIBC: 66) were adequately followed-up and nine patients were excluded due to insufficient monitoring data. During the median follow-up time (reverse Kaplan-Meier method) of 38.0 months (95% CI: 34.03–41.97), disease recurrence and progression were detected in 37 (42.5%) and 18 (20.7%) NMIBC patients, respectively. With respect to MIBC (T2-T4), 37 (56.1%) patients progressed, and 34 (51.5%) patients died. The mean disease-free survival (DFS) and progression-free survival (PFS) of the NMIBC patients were 45.91 months (95% CI: 38.81–53.00) and 60.80 months (95% CI: 54.98–66.62), respectively, while the overall survival (OS) of the MIBC patients was 65.81 months (95% CI: 48.92–82.69). Figure 3 presents the design and the REMARK diagram of the study.
Figure 3.

Flow and REMARK diagram of the study
For the followed-up cohort (NMIBC: 87; MIBC: 66), core/proximal promoter methylation levels were available for 85/85 NMIBC and 63/65 MIBC patients, respectively. Kaplan-Meier survival curves are presented in Figure 4, while Cox proportional regression analysis is summarized in Figure 5 and Tables S2 and S3. Kaplan-Meier survival curves clearly highlighted the markedly shorter PFS (p = 0.023; Figure 4A) interval of TaT1 patients with lower methylation of MIR145 core promoter compared with those presenting hypermethylation. Additionally, univariate Cox proportional regression corroborated the significantly higher risk for short-term disease progression (hazard ratio [HR]: 2.803; 95% confidence interval [CI]: 1.103–7.124; bootstrap p = 0.009; Figure 5A) of NMIBC patients with reduced MIR145 core promoter methylation. More importantly, multivariate Cox models strongly verified the clinical value of MIR145 core promoter hypomethylation for NMIBC progression to invasive disease stages (HR: 2.777; 95% CI: 1.071–7.198; bootstrap p = 0.029; Figure 5B) independently of tumor stage, grade, gender, and age. Regarding NMIBC relapse, both Kaplan-Meier (Figure 4B) and Cox regression (Table S2) analyses showed worse DFS of the patients with decreased methylation, although not in a statistically significant manner.
Figure 4.

Reduced methylation of MIR145 core promoter is associated with significantly higher risk for disease progression and poor treatment outcome
(A–D) Kaplan-Meier survival curves for the PFS (A) and the DFS (B) of NMIBC patients, as well as the PFS (C) and OS (D) of MIBC patients of the screening cohort according to MIR145 core promoter methylation. (E, F) Kaplan-Meier survival curves for the PFS (E) and OS (F) of the TCGA-BLCA validation cohort according to MIR145 core promoter methylation. p-values calculated by log-rank test. PFS, progression-free survival; DFS, disease-free survival; OS,overall survival; HR, hazard ratio; 95% CI, 95% confidence interval of the estimated HR.
Figure 5.

MIR145 core promoter hypomethylation represents an independent predictor of NMIBC short-term progression and poor survival of MIBC
Forest plots of univariate and multivariate Cox regression analysis for the PFS of NMIBC (A and B) and the OS of MIBC (C and D) patients. Internal validation was performed by bootstrap Cox proportional regression analysis based on 1,000 bootstrap samples. HR, hazard ratio; 95% CI, 95% confidence interval of the estimated HR intervals.
Focusing on MIBC patient’s outcome following radical cystectomy, Kaplan-Meier survival analysis unveiled the stronger risk for disease progression (p = 0.029; Figure 4C) and poor OS (p = 0.049; Figure 4D) of the T2-T4 patients with decreased methylation imprinting. Additionally, univariate Cox analysis verified the unfavorable PFS (HR: 2.060; 95% CI: 1.048–4.047; bootstrap p = 0.023; Table S3) and increased morbidity (HR: 1.983; 95% CI: 0.983–4.000; bootstrap p = 0.043; Figure 5C) of MIBC patients with hypomethylated MIR145 core promoter. Strikingly, multivariate Cox regression models, adjusted for patients’ stage, gender, and age, demonstrated the reduced methylation levels of MIR145 core promoter as an independent predictor of MIBC progression (HR: 2.962; 95% CI: 1.382–6.349; bootstrap p = 0.005; Table S3) and poor survival (HR: 2.729; 95% CI: 1.247–5.970; bootstrap p = 0.010; Figure 5D).
Consistent with our findings, the survival analysis of the TCGA-BLCA validation cohort clearly validated the inferior PFS and OS of patients with decreased methylation of the MIR145 core promoter. More precisely, Kaplan-Meier curves presented the significantly shorter PFS (p = 0.034; Figure 4E) and OS (p = 0.005; Figure 4F) of the patients with lower methylation of MIR145 core promoter CpGs, which was also confirmed by univariate Cox analysis for both PFS (HR: 1.380; 95% CI: 1.024–1.860; bootstrap p = 0.034) and OS (HR: 1.539; 95% CI: 1.140–2.077; bootstrap p = 0.003). Ultimately, the survival assessment of MIR145 proximal promoter methylation profile (Figure S3) revealed a weaker association with patients’ outcome compared with the core promoter, both in the screening and TCGA-BLCA validation cohorts, supporting the superior clinical value of core promoter CpGs in disease prognostication.
MIR145 promoter methylation analysis improves patients’ prognostication and risk stratification
Prompted by the independent prognostic value of MIR145 core promoter methylation, we thereafter analyzed its ability to improve the performance of the established disease prognostic markers. In this regard, the integration of MIR145 methylation with the European Organisation for Research and Treatment of Cancer (EORTC) risk score—a widely used clinical predictor of NMIBC progression—was shown to significantly ameliorate the risk stratification of low-risk (LR) and intermediate-risk (IR) patients for disease progression (p = 0.038; Figure 6A). Similarly, the incorporation of MIR145 methylation with tumors’ stage offered superior risk stratification of MIBC patients, enhancing their post-treatment outcome prognosis (p = 0.001; Figure 6B). Kaplan-Meier survival curves for EORTC risk score and tumor stage of the same NMIBC and MIBC cohorts, respectively, are included in Figure S4. In the light of those findings, decision curve analysis (DCA) was conducted according to Vickers et al.24 to evaluate the clinical net benefit of MIR145 methylation evaluation in disease and treatment prognostication. The DCA control model consisted of the established and clinically used markers including tumor stage, grade, and EORTC risk group for NMIBC or tumor stage for MIBC patients. Decision curves clearly highlighted the superior clinical benefit of the MIR145 core methylation-fitted multivariate model for both the PFS and OS post-treatment outcome of NMIBC (Figure 6C) and MIBC (Figure 6D), respectively, compared with the control model.
Figure 6.

MIR145 promoter methylation analysis results in superior clinical benefit of multivariate prognostic models
(A and B) Kaplan-Meier survival curves for the PFS of NMIBC (A) and the OS of MIBC (B) patients according to multivariate models of MIR145 core promoter methylation with EORTC-risk group and tumor stage, respectively. p-values calculated by log-rank test. (C and D) Decision curve analysis of the “control” and the “MIR145 methylation-fitted” multivariate prognostic models for the PFS of NMIBC (C) and OS of MIBC (D) patients.
Discussion
Despite the remarkable alleviation of BlCa-specific mortality due to recent advances in clinical treatment (evolution of imaging, improvements in surgical techniques, and new diagnostic modalities), the lack of modern precision medicine and personalized management entails inadequate prognostication and prediction of disease course.25 Meanwhile, the lifelong patient surveillance strategies, due to the high propensity for multiple recurrences and/or disease progression, classify BlCa as the most expensive per-patient-to-treat neoplastic disease, with an important financial burden for healthcare systems.26,27 In this regard, the identification of novel molecular predictors could ameliorate patients’ risk stratification and provide tailored treatment decisions, improving thus patients’ quality of life and disease management.
As highlighted by the ENCODE project, ∼70% of the human genome encodes ncRNAs,28 whereas miRNAs have emerged as the most powerful modulators of gene expression, acting at post-transcriptional and epigenetic levels.29 miRNAs are actively transcribed and orchestrate almost all aspects of biological processes, ensuring cellular homeostasis and normal physiology,30 while their aberrant regulation constitutes a hallmark of cancer onset and progression in numerous malignancies, including BlCa.31,32 Prompted by our previous study, which disclosed the potent clinical significance of miR-143/145 cluster in BlCa progression,23 we decided to further investigate the underlying epigenetic control of cluster’s expression in bladder tumors. DNA methylation constitutes a fundamental regulatory mechanism of gene expression, predominantly resulting in gene silencing.33 Nearly half of all known human miRNA genes are associated with CpG islands,34 while methylation imprinting has been found to downregulate the expression of potent onco-suppressor miRNAs, including miR-34, miR-124a, and miR-127 in different cancers.35, 36, 37 In this regard, we have analyzed the DNA methylation imprinting of the miR-143/145 gene cluster in bladder urothelium to identify its impact on the epigenetic regulation of the cluster and to assess its clinical utility in improving patients’ risk stratification and personalized prognosis.
Using in silico analysis of TCGA-BLCA cohort, the CpG island of MIR143/145 cluster was found non-methylated (“cold”), while the MIR143 promoter imprinting appeared inconsistent with miR-143/145 expression profile in malignant and normal urothelium, indicating a weak impact on cluster’s regulation. On the contrary, MIR145 promoter methylation demonstrated the most robust correlation with the miR-143/145 profile in tumors and the normal urothelium. The methylation analysis of our screening cohort confirmed the hypermethylation of the MIR145 promoter in bladder tumors compared with the matched normal urothelium, in agreement with miR-143/145 loss. Consistent with our findings, miR-143/145 cluster epigenetic silencing by MIR145 promoter hypermethylation has also been documented in prostate, lung, esophageal, and laryngeal carcinomas, resulting in apoptosis inhibition and cell proliferation enhancement.38, 39, 40, 41, 42 Strikingly, the analysis revealed a significantly increased methylation tendency from the proximal to the core MIR145 promoter, and the robust negative correlation of miR-143/145 levels with MIR145 core promoter methylation.
Focusing on miR-143/145 cluster’s clinical value for the patients, our group has previously reported the association of elevated miR-143/145 tumor levels with aggressive disease phenotype and unfavorable patient prognosis. In agreement with our previous findings, reduced MIR145 promoter methylation was significantly correlated with muscle-invasive disease and advanced tumor stage and grade. Moreover, the survival analysis highlighted the association of MIR145 core promoter hypomethylation with poor post-treatment disease outcome. In particular, the reduced MIR145 core promoter methylation resulted in significantly higher risk for the short-term progression of NMIBC (TaT1) patients to invasive disease stages following transurethral resection of bladder tumors (TURBT), as well as in worse survival outcome of MIBC (T2-T4) patients following radical cystectomy (RC), independently of patients’ clinicopathological data. Interestingly, the analysis of TCGA-BLCA validation cohort clearly confirmed the correlation of MIR145 core promoter hypomethylation with poor OS and PFS intervals. Notably, the methylation status of the MIR145 core promoter emerged as an independent and vigorous prognostic indicator, enhancing the clinical net benefit of widely used disease markers and ameliorating risk stratification of BlCa patients.
The superior risk stratification of both NMIBC and MIBC patients could be translated in the clinical practice and affect clinical decision-making either on disease treatment or post-treatment monitoring. More precisely, LR/IR NMIBC patients with significantly increased risk for progression to invasive disease stages could be candidates for High Risk-like treatment/management, including intravesical bacillus Calmette-Guérin (BCG) administration (1–3 years), more intensive monitoring, and also focusing on patients’ awareness. Those patients, in case of BCG-unresponsive tumors/short-term disease relapsing tumors, should be considered as candidates for RC. Moreover, the identification of T3/T4 patients with significantly short post-treatment survival expectancy could be considered, along patients’ age, comorbidity, and frailty, as candidates for palliative therapy to avoid unnecessary cystectomy (radical or palliative) and chemotherapy and to minimize side effects and healthcare system costs, in a shared decision-making. Definitely, future prospective studies of MIR145 promoter methylation in BlCa are of high clinical interest both to confirm our findings on disease prognosis and more importantly to highlight the clinical benefit in real-time treatment and/or monitoring decision for the patients.
The association of MIR145 promoter hypomethylation (observed in the present study) and miR-143/145 overexpression23 with poor disease prognosis seems to contradict the well-documented tumor suppressor role of the cluster.17 However, recent findings have challenged this one-way scenario of tumor suppressor function, emerging a pluripotent role of miR-143/145 in stromal and epithelial cells of epithelial origin carcinomas.43 Indeed, Dimitrova et al. have documented the pro-tumorigenic contribution of miR-143/145 in lung adenocarcinoma in vivo, where tumor-specific deletion of miR-143/145 did not affect tumorigenesis. However, stromal miR-143/145 overexpression mediated silencing of CAMK1D, an inhibitory kinase that abrogates mitotic entry, and stimulated endothelial cells proliferation and neoangiogenesis.44 This tumor-promoting role of stromal miR-143/145 is in line with the well-documented role of the cluster in facilitating the differentiation of multipotent stem cells and adult fibroblasts to vascular smooth muscle cells,45,46 as well as in maintaining the normal paracrine IGF signaling, through negative regulation of IGFBP5 by smooth muscle and myofibroblasts.47 Furthermore, miR-145 has been reported to facilitate metastasis in colorectal cancer via downstream attenuation of G1/S cell cycle checkpoint and neuregulin pathways.48 Moreover, elevated levels of miR-143/145 have been demonstrated to enhance cell invasion and epithelial-to-mesenchymal transition in breast tumors, via repressing transcriptional activators of tight junction proteins, such as CREB1, and triggering TGF-β axis by targeting TGIF, a well-known transcriptional co-repression of SMAD.49 Finally, miR-143-mediated targeting of FNDC3B documented to promote cell invasion and metastatic potential in prostate and hepatocellular cancers.50,51 Overall, these previous studies and our findings argue against a universal and cell-independent tumor-suppressor role of miR-143/145 in epithelial cancers, indicating the deregulation of the cluster’s epigenetic/transcriptional control in the tumor microenvironment as a potential tumor-promoting mode of action in human malignancies and supporting future functional studies toward this direction.
In conclusion, we have studied MIR143/145 gene cluster methylation in BlCa, highlighting that miR-143/145 cluster is epigenetically regulated in bladder tumors, while MIR145 core promoter represents the key regulatory region of cluster modulation. Reduced methylation levels of MIR145 core promoter were strongly associated with more aggressive phenotype of BlCa and higher risk for disease progression and poor treatment outcome of the patients. Notably, multivariate prognostic models including MIR145 methylation imprinting resulted in a superior risk stratification of the patients, toward personalized treatment and monitoring decisions.
Materials and methods
Screening cohort
The screening cohort of the study consisted of 162 patients diagnosed with primary UBC. Fresh-frozen bladder tumors were obtained via either TURBT for NMIBC patients (TaT1) or RC for MIBC patients (T2-T4) at “Laiko” General Hospital, Athens, Greece. Patients’ clinicopathological characteristics are summarized in Table S4. Adjacent normal bladder tissue specimens were also acquired by 96 patients of the cohort, according to pathologist’s evaluation for the absence of dysplasia and CIS. The patients received adjuvant therapy in agreement with European Association of Urology (EAU) guidelines, while none of them received any form of neoadjuvant treatment prior to surgery. Bladder tissue specimens were incubated in RNAlater Solution (Ambion), following manufacturer’s instructions, and stored at −80°C until further processing.
NMIBC patients’ risk stratification was performed according to the EORTC guidelines and post-treatment monitoring included cystoscopy and urinary cytology (for high-grade tumors) according to EAU guidelines. MIBC patients’ (T2-T4) were followed-up by renal ultrasound at 3 months and thoracoabdominal computed tomography (CT)/magnetic resonance imaging (MRI) every 6 months, while additional kidney ultrasound, thoracoabdominal CT/MRI, bone scan, and brain MRI were performed following symptoms. NMIBC patients’ disease recurrence (same or lower pathologic tumor stage) and progression (recurrence of higher/invasive stage) were confirmed by histology findings of a TURBT, which was performed after a positive follow-up cystoscopy, while MIBC patients’ recurrence was detected by a follow-up CT.
The present study was approved by the Ethics Committee of “Laiko” General Hospital, Athens, Greece, and conducted in consonance with 1975 Declaration of Helsinki ethical standards, as revised in 2008. Informed consent was obtained by all participating patients.
Validation cohort
The TCGA-BLCA cohort was utilized as validation cohort of the study. TCGA-BLCA consists of 412 patients diagnosed with UBC (n = 409), papillary adenocarcinomas (n = 1), epithelial carcinomas (n = 1), and squamous cell carcinomas (n = 1), including mainly muscle-invasive tumors (T2-T4; n = 406, 98.5%), as well as of 23 matched normal tissues. DNA methylation (available for n = 412 tumors; n = 21 normal specimens) and mRNA (available for n = 409 tumors; n = 19 normal specimens) expression profiles were generated by Illumina Infinium HumanMethylation450 platform and Illumina HiSeq 2000 RNA Sequencing platform, respectively, and their data along with patients’ clinicopathological characteristics of the TCGA-BLCA project can be retrieved by public UCSC XENA Browser (https://xenabrowser.net/datapages/).
In silico analysis
The NCBI database (https://www.ncbi.nlm.nih.gov/gene) and the UCSC Genome Browser gateway (https://genome.ucsc.edu/) were used to analyze the genome structure of the miR-143/145 cluster, exploiting GRCh37/hg19 assembly. Expression levels of miR-143/145 cluster, as well as the distribution and the methylation levels of Illumina CpG loci IDs (cg#) in MIR143/145 regulatory regions within TCGA-BLCA cohort were visualized by XENA Browser Visualization Tool (https://xenabrowser.net/).
Genomic DNA and total RNA extraction
Following pulverization of 40–100 mg of fresh-frozen tissue specimen, total RNA and genomic DNA (gDNA) were extracted using TRI-Reagent (Molecular Research Center, Cincinnati, OH, USA) according to the manufacturer’s instructions. Total RNA was dissolved in RNA Storage Solution (Invitrogen, Carlsbad, CA, USA), and genomic DNA in 8 mM NaOH, pH-adjusted by addition of 0.1 M HEPES buffer. Both RNA and DNA samples were stored at −80°C until analysis. DNA/RNA concentration and purity were determined spectrophotometrically at 260 and 280 nm, while agarose gel electrophoresis was performed to evaluate RNA integrity.
Polyadenylation of total RNA and first-strand cDNA synthesis
Polyadenylation of 1 μg of total RNA at the 3′ end was carried out in a 10-μL reaction, containing 800 μM ATP and 1 U of E. coli poly (A) polymerase (New England Biolabs, Ipswich, MA, USA), at 37°C for 60 min. Polymerase heat inactivation was performed at 65°C for 10 min.
The polyadenylated total RNA was reverse transcribed, using the oligo-dT adapter primer (Table S5) in a final reaction volume of 20 μL. The reaction mixture consisted of 50 U M-MLV Reverse Transcriptase (Invitrogen), 40 U RNaseOUT Recombinant Ribonuclease Inhibitor (Invitrogen), 500 μM dNTPs mix, and 0.25 μM oligo-dT adapter, at 37°C for 60 min. Reverse transcriptase was inactivated by heating at 70°C for 15 min.
Quantitative real-time PCR
SYBR-green fluorescent-based quantitative real-time PCR (qPCR) assays were used in order to quantify miR-143-3p and miR-145-5p levels. Specific forward primers for miR-143-3p, miR-145-5p, and the small nucleolar RNA, C/D box 48 (SNORD48), also known as RNU48, were designed based on their published sequences (NCBI Reference Sequence: NR_029684.1, NR_029686.1 and NR_002745.1, respectively) and in silico analysis. Each specific forward primer is combined with a universal reverse primer (Table S5), which is complementary to the oligo-dT adapter sequence, giving rise to 65 bp amplicons for miR-143-3p and miR-145-5p, and a 105 bp amplicon for RNU48.
The qPCR reactions were performed in the 7500 Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA), and the 10-μL reaction mixture consisted of Kapa SYBR Fast Universal 2X qPCR Master Mix (Kapa Biosystems, Woburn, MA), 200 nM of each PCR primer, and 0.2 ng of cDNA template. The thermal protocol included an initial 3-min step at 95°C for polymerase activation, followed by 40 cycles of denaturation step at 95°C for 15 s and primer annealing and extension step at 60°C for 1 min. Thereafter, dissociation curves and agarose gel electrophoresis were performed to discriminate specific amplicons from the non-specific products and/or primer dimers.
The expression analysis of miR-143 and miR-145 was carried out using the 2−ΔΔCT relative quantification method. All reactions were performed in duplicates, and the average Ct was used for the quantification analysis. RNU48 was utilized as endogenous reference control for normalization purposes.
Sodium bisulfite conversion of gDNA
Conversion of the unmethylated cytosine (C) residues to uracils (U) was performed with EpiMark Bisulfite Conversion Kit (New England Biolabs). Particularly, 1.5 μg of genomic DNA was incubated with sodium bisulfite mix under alternative cycles of thermal denaturation with incubation reactions: 95°C for 5 min, 65°C for 30 min, 95°C for 5 min, 65°C for 60 min, 95°C for 5 min, and 65°C for 90 min. Following completion of bisulfite conversion, desulfonation, sample clean up, and elution were performed via EpiMark spin columns according to manufacturer’s instructions. Bisulfite-treated DNA was stored at −80°C until analysis.
PCR amplification of bisulfite converted gDNA
Two distinctive PCR assays were developed and validated for the proximal (proximal assay) and core (core assay) promoter regions of MIR145, in which bisulfite-treated gDNA was used as template for the amplification of 127 bp and 141 bp sequencing products, respectively. Specific PCR and sequencing primers for bisulfite-treated gDNA were designed using PyroMark Assay Design 2.0 Software (Qiagen, Manchester, UK) and the published sequences (NCBI Reference Sequence: NC_000005.9) (Table S5).
Each PCR reaction was conducted in a final volume of 25 μL, containing 1.5 μL bisulfite-treated DNA, 200 μM dNTPs mix, 400 nM of forward and reverse primers, and 1 U of EpiMark Hot Start Taq DNA Polymerase (New England Biolabs). PCR cycling conditions were 95°C for 30 s, followed by 40 cycles of 95°C for 15 s, 55°C (proximal assay) or 56°C (core assay) for 30 s, and 68°C for 1 min, with a final extension at 68°C for 5 min. Non-template controls were included in each PCR reaction. Agarose gel electrophoresis was performed to evaluate the amplification of specific PCR products, as well as absence of non-specific products and/or primer dimers.
Methylation analysis by pyrosequencing
The biotinylated PCR products—in a total volume of 18 μL—were mixed with 20 μL Binding Buffer (Qiagen) and 2 μL streptavidin-sepharose high-performance beads (GE Healthcare, Chicago, USA), followed by shaking at 1,200 rpm for 20 min, in order to facilitate the immobilization. Thereafter, the immobilized PCR products were purified to single-stranded amplicons, using the PyroMark Q24 vacuum workstation (Qiagen) according to manufactures guidelines. The biotinylated ssDNA amplicons were mixed with 0.3 μM of sequencing primers (Table S5) in annealing buffer (Qiagen) and then heated for 2 min at 80°C and cooled at room temperature for 7 min for primer hybridization. Methylation analysis was carried out by pyrosequencing using the PyroMark Gold Q24 Reagents (Qiagen) in PyroMark Q24 Pyrosequencer (Qiagen), according to the manufacturer’s instructions. Quantification of percent methylation of the targeted CpGs was performed PyroMark Q24 Software 2.0 (Qiagen). Efficiency of the bisulfite conversion process was assessed by the conversion of non-CpG cytosine residues within the sequence to analyze.
Statistical analysis
Statistical analysis was performed by IBM SPSS Statistics 20 software (IBM, Armonk, NY, USA). The normal distribution of the data was evaluated by Sapiro-Wilk and Kolmogorov-Smirnov tests. Due to absence of normal distribution, the non-parametric Wilcoxon singed rank test was used to analyze miR-143/145 gene cluster expression and methylation levels between bladder tumors and normal adjacent urothelium, while Mann-Whitney U and Kruskal-Wallis tests were applied accordingly to evaluate the association of cluster’s expression and methylation with patients’ clinicopathological data.
Survival analysis was carried out by Kaplan-Meier curves, using log rank test, as well as univariate and multivariate Cox proportional regression analysis. The promoter methylation optimal cutoff values were adopted by X-tile algorithm. Internal validation was accomplished by bootstrap Cox proportional regression analysis based on 1,000 bootstrap samples. Ultimately, DCA was applied in order to evaluate MIR145 promoter methylation clinical benefit in disease prognosis and patients’ clinical outcome, in accordance with Vickers et al.,24 using STATA 13 software (StataCorp, College Station, TX, USA).
Acknowledgments
This research has been co-financed by the European Regional Development Fund of the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call RESEARCH – CREATE – INNOVATE (project code: T2EDK-02196) to the NKUA (KE17358).
Author contributions
Conceptualization and design, M.A. and A.S.; methodology, K.M.P., M.A.P., K.P., P.B., and M.A.; investigation, K.M.P., M.A.P., K.P., and P.B.; formal analysis, K.M.P., M.A.P., K.P., and M.A.; validation, K.M.P. and M.A.; visualization, K.M.P. and M.A.; resources, K.S., P.L., G.K., and A.S.; data curation, K.M.P. and M.A.; writing—original draft, K.M.P.; writing—review and editing, M.A.P., K.P., K.S., A.S., and M.A.; project administration, M.A.; funding acquisition, A.S. and M.A.; supervision, M.A. and A.S. All authors have read and agreed to the published version of the manuscript.
Declaration of interests
The authors declare no potential conflicts of interest.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.10.001.
Contributor Information
Andreas Scorilas, Email: ascorilas@biol.uoa.gr.
Margaritis Avgeris, Email: mavgeris@med.uoa.gr.
Supplemental information
Data availability
Data are available from the corresponding authors on reasonable request.
References
- 1.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2020. CA Cancer J. Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 3.Sanli O., Dobruch J., Knowles M.A., Burger M., Alemozaffar M., Nielsen M.E., Lotan Y. Bladder cancer. Nat. Rev. Dis. Primers. 2017;3:17022. doi: 10.1038/nrdp.2017.22. [DOI] [PubMed] [Google Scholar]
- 4.Prasad S.M., Decastro G.J., Steinberg G.D., Medscape Urothelial carcinoma of the bladder: definition, treatment and future efforts. Nat. Rev. Urol. 2011;8:631–642. doi: 10.1038/nrurol.2011.144. [DOI] [PubMed] [Google Scholar]
- 5.Babjuk M., Bohle A., Burger M., Capoun O., Cohen D., Comperat E.M., Hernandez V., Kaasinen E., Palou J., Roupret M., et al. EAU guidelines on non-muscle-invasive urothelial carcinoma of the bladder: update 2016. Eur. Urol. 2017;71:447–461. doi: 10.1016/j.eururo.2016.05.041. [DOI] [PubMed] [Google Scholar]
- 6.van Rhijn B.W., Burger M., Lotan Y., Solsona E., Stief C.G., Sylvester R.J., Witjes J.A., Zlotta A.R. Recurrence and progression of disease in non-muscle-invasive bladder cancer: from epidemiology to treatment strategy. Eur. Urol. 2009;56:430–442. doi: 10.1016/j.eururo.2009.06.028. [DOI] [PubMed] [Google Scholar]
- 7.Alfred Witjes J., Lebret T., Comperat E.M., Cowan N.C., De Santis M., Bruins H.M., Hernandez V., Espinos E.L., Dunn J., Rouanne M., et al. Updated 2016 EAU guidelines on muscle-invasive and metastatic bladder cancer. Eur. Urol. 2017;71:462–475. doi: 10.1016/j.eururo.2016.06.020. [DOI] [PubMed] [Google Scholar]
- 8.Antoni S., Ferlay J., Soerjomataram I., Znaor A., Jemal A., Bray F. Bladder cancer incidence and mortality: a global overview and recent trends. Eur. Urol. 2017;71:96–108. doi: 10.1016/j.eururo.2016.06.010. [DOI] [PubMed] [Google Scholar]
- 9.Jordan B., Meeks J.J. T1 bladder cancer: current considerations for diagnosis and management. Nat. Rev. Urol. 2019;16:23–34. doi: 10.1038/s41585-018-0105-y. [DOI] [PubMed] [Google Scholar]
- 10.Soukup V., Capoun O., Cohen D., Hernandez V., Burger M., Comperat E., Gontero P., Lam T., Mostafid A.H., Palou J., et al. Risk stratification tools and prognostic models in non-muscle-invasive bladder cancer: a critical assessment from the European association of Urology non-muscle-invasive bladder cancer guidelines panel. Eur. Urol. Focus. 2020;6:479–489. doi: 10.1016/j.euf.2018.11.005. [DOI] [PubMed] [Google Scholar]
- 11.Knowles M.A., Hurst C.D. Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat. Rev. Cancer. 2015;15:25–41. doi: 10.1038/nrc3817. [DOI] [PubMed] [Google Scholar]
- 12.Meeks J.J., Lerner S.P. Molecular landscape of non-muscle invasive bladder cancer. Cancer Cell. 2017;32:550–551. doi: 10.1016/j.ccell.2017.08.015. [DOI] [PubMed] [Google Scholar]
- 13.Bhanvadia S.K. Bladder cancer survivorship. Curr. Urol. Rep. 2018;19:111. doi: 10.1007/s11934-018-0860-6. [DOI] [PubMed] [Google Scholar]
- 14.Grosshans H., Filipowicz W. Molecular biology: the expanding world of small RNAs. Nature. 2008;451:414–416. doi: 10.1038/451414a. [DOI] [PubMed] [Google Scholar]
- 15.Croce C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 2009;10:704–714. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poli V., Secli L., Avalle L. The microrna-143/145 cluster in tumors: a matter of where and when. Cancers. 2020;12 doi: 10.3390/cancers12030708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Das A.V., Pillai R.M. Implications of miR cluster 143/145 as universal anti-oncomiRs and their dysregulation during tumorigenesis. Cancer Cell Int. 2015;15:92. doi: 10.1186/s12935-015-0247-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iorio M.V., Ferracin M., Liu C.G., Veronese A., Spizzo R., Sabbioni S., Magri E., Pedriali M., Fabbri M., Campiglio M., et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 19.Avgeris M., Stravodimos K., Fragoulis E.G., Scorilas A. The loss of the tumour-suppressor miR-145 results in the shorter disease-free survival of prostate cancer patients. Br. J. Cancer. 2013;108:2573–2581. doi: 10.1038/bjc.2013.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wach S., Nolte E., Theil A., Stohr C., T T.R., Hartmann A., Ekici A., Keck B., Taubert H., Wullich B. MicroRNA profiles classify papillary renal cell carcinoma subtypes. Br. J. Cancer. 2013;109:714–722. doi: 10.1038/bjc.2013.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akao Y., Nakagawa Y., Hirata I., Iio A., Itoh T., Kojima K., Nakashima R., Kitade Y., Naoe T. Role of anti-oncomirs miR-143 and -145 in human colorectal tumors. Cancer Gene Ther. 2010;17:398–408. doi: 10.1038/cgt.2009.88. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J., Sun Q., Zhang Z., Ge S., Han Z.G., Chen W.T. Loss of microRNA-143/145 disturbs cellular growth and apoptosis of human epithelial cancers by impairing the MDM2-p53 feedback loop. Oncogene. 2013;32:61–69. doi: 10.1038/onc.2012.28. [DOI] [PubMed] [Google Scholar]
- 23.Avgeris M., Mavridis K., Tokas T., Stravodimos K., Fragoulis E.G., Scorilas A. Uncovering the clinical utility of miR-143, miR-145 and miR-224 for predicting the survival of bladder cancer patients following treatment. Carcinogenesis. 2015;36:528–537. doi: 10.1093/carcin/bgv024. [DOI] [PubMed] [Google Scholar]
- 24.Vickers A.J., Elkin E.B. Decision curve analysis: a novel method for evaluating prediction models. Med. Decis. Making. 2006;26:565–574. doi: 10.1177/0272989X06295361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meeks J.J., Al-Ahmadie H., Faltas B.M., Taylor J.A., 3rd, Flaig T.W., DeGraff D.J., Christensen E., Woolbright B.L., McConkey D.J., Dyrskjot L. Genomic heterogeneity in bladder cancer: challenges and possible solutions to improve outcomes. Nat. Rev. Urol. 2020;17:259–270. doi: 10.1038/s41585-020-0304-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Svatek R.S., Hollenbeck B.K., Holmang S., Lee R., Kim S.P., Stenzl A., Lotan Y. The economics of bladder cancer: costs and considerations of caring for this disease. Eur. Urol. 2014;66:253–262. doi: 10.1016/j.eururo.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 27.Sloan F.A., Yashkin A.P., Akushevich I., Inman B.A. The cost to medicare of bladder cancer care. Eur. Urol. Oncol. 2020;3:515–522. doi: 10.1016/j.euo.2019.01.015. [DOI] [PubMed] [Google Scholar]
- 28.Consortium E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rupaimoole R., Slack F.J. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017;16:203–222. doi: 10.1038/nrd.2016.246. [DOI] [PubMed] [Google Scholar]
- 30.Ameres S.L., Zamore P.D. Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 2013;14:475–488. doi: 10.1038/nrm3611. [DOI] [PubMed] [Google Scholar]
- 31.Yoshino H., Seki N., Itesako T., Chiyomaru T., Nakagawa M., Enokida H. Aberrant expression of microRNAs in bladder cancer. Nat. Rev. Urol. 2013;10:396–404. doi: 10.1038/nrurol.2013.113. [DOI] [PubMed] [Google Scholar]
- 32.Fendler A., Stephan C., Yousef G.M., Jung K. MicroRNAs as regulators of signal transduction in urological tumors. Clin. Chem. 2011;57:954–968. doi: 10.1373/clinchem.2010.157727. [DOI] [PubMed] [Google Scholar]
- 33.Bernstein B.E., Meissner A., Lander E.S. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 34.Weber B., Stresemann C., Brueckner B., Lyko F. Methylation of human microRNA genes in normal and neoplastic cells. Cell Cycle. 2007;6:1001–1005. doi: 10.4161/cc.6.9.4209. [DOI] [PubMed] [Google Scholar]
- 35.Lujambio A., Esteller M. CpG island hypermethylation of tumor suppressor microRNAs in human cancer. Cell Cycle. 2007;6:1455–1459. [PubMed] [Google Scholar]
- 36.Wang H., Zhang T.T., Jin S., Liu H., Zhang X., Ruan C.G., Wu D.P., Han Y., Wang X.Q. Pyrosequencing quantified methylation level of miR-124 predicts shorter survival for patients with myelodysplastic syndrome. Clin Epigenetics. 2017;9:91. doi: 10.1186/s13148-017-0388-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen Z., Zhou C., Li J., Ye D., Li Q., Wang J., Cui X., Chen X., Bao T., Duan S. Promoter hypermethylation of miR-34a contributes to the risk, progression, metastasis and poor survival of laryngeal squamous cell carcinoma. Gene. 2016;593:272–276. doi: 10.1016/j.gene.2016.07.047. [DOI] [PubMed] [Google Scholar]
- 38.Zaman M.S., Chen Y., Deng G., Shahryari V., Suh S.O., Saini S., Majid S., Liu J., Khatri G., Tanaka Y., et al. The functional significance of microRNA-145 in prostate cancer. Br. J. Cancer. 2010;103:256–264. doi: 10.1038/sj.bjc.6605742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suh S.O., Chen Y., Zaman M.S., Hirata H., Yamamura S., Shahryari V., Liu J., Tabatabai Z.L., Kakar S., Deng G., et al. MicroRNA-145 is regulated by DNA methylation and p53 gene mutation in prostate cancer. Carcinogenesis. 2011;32:772–778. doi: 10.1093/carcin/bgr036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xia W., Chen Q., Wang J., Mao Q., Dong G., Shi R., Zheng Y., Xu L., Jiang F. DNA methylation mediated silencing of microRNA-145 is a potential prognostic marker in patients with lung adenocarcinoma. Sci. Rep. 2015;5:16901. doi: 10.1038/srep16901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harada K., Baba Y., Ishimoto T., Kosumi K., Tokunaga R., Izumi D., Ohuchi M., Nakamura K., Kiyozumi Y., Kurashige J., et al. Suppressor microRNA-145 is epigenetically regulated by promoter hypermethylation in esophageal squamous cell carcinoma. Anticancer Res. 2015;35:4617–4624. [PubMed] [Google Scholar]
- 42.Gao W., Zhang C., Li W., Li H., Sang J., Zhao Q., Bo Y., Luo H., Zheng X., Lu Y., et al. Promoter methylation-regulated miR-145-5p inhibits laryngeal squamous cell carcinoma progression by targeting FSCN1. Mol. Ther. 2019;27:365–379. doi: 10.1016/j.ymthe.2018.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Almeida M.I., Calin G.A. The miR-143/miR-145 cluster and the tumor microenvironment: unexpected roles. Genome Med. 2016;8:29. doi: 10.1186/s13073-016-0284-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dimitrova N., Gocheva V., Bhutkar A., Resnick R., Jong R.M., Miller K.M., Bendor J., Jacks T. Stromal expression of miR-143/145 promotes neoangiogenesis in lung cancer development. Cancer Discov. 2016;6:188–201. doi: 10.1158/2159-8290.CD-15-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cordes K.R., Sheehy N.T., White M.P., Berry E.C., Morton S.U., Muth A.N., Lee T.H., Miano J.M., Ivey K.N., Srivastava D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460:705–710. doi: 10.1038/nature08195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elia L., Quintavalle M., Zhang J., Contu R., Cossu L., Latronico M.V., Peterson K.L., Indolfi C., Catalucci D., Chen J., et al. The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: correlates with human disease. Cell Death Differ. 2009;16:1590–1598. doi: 10.1038/cdd.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chivukula R.R., Shi G., Acharya A., Mills E.W., Zeitels L.R., Anandam J.L., Abdelnaby A.A., Balch G.C., Mansour J.C., Yopp A.C., et al. An essential mesenchymal function for miR-143/145 in intestinal epithelial regeneration. Cell. 2014;157:1104–1116. doi: 10.1016/j.cell.2014.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arndt G.M., Dossey L., Cullen L.M., Lai A., Druker R., Eisbacher M., Zhang C., Tran N., Fan H., Retzlaff K., et al. Characterization of global microRNA expression reveals oncogenic potential of miR-145 in metastatic colorectal cancer. BMC Cancer. 2009;9:374. doi: 10.1186/1471-2407-9-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Avalle L., Incarnato D., Savino A., Gai M., Marino F., Pensa S., Barbieri I., Stadler M.B., Provero P., Oliviero S., et al. MicroRNAs-143 and -145 induce epithelial to mesenchymal transition and modulate the expression of junction proteins. Cell Death Differ. 2017;24:1750–1760. doi: 10.1038/cdd.2017.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fan X., Chen X., Deng W., Zhong G., Cai Q., Lin T. Up-regulated microRNA-143 in cancer stem cells differentiation promotes prostate cancer cells metastasis by modulating FNDC3B expression. BMC Cancer. 2013;13:61. doi: 10.1186/1471-2407-13-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang X., Liu S., Hu T., Liu S., He Y., Sun S. Up-regulated microRNA-143 transcribed by nuclear factor kappa B enhances hepatocarcinoma metastasis by repressing fibronectin expression. Hepatology. 2009;50:490–499. doi: 10.1002/hep.23008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available from the corresponding authors on reasonable request.