

Abstract
Idiopathic pulmonary fibrosis (IPF) is a chronic progressive disease characterized by excessive proliferation of fibroblasts and excessive accumulation of extracellular matrix (ECM). Ferroptosis is a novel form of cell death characterized by the lethal accumulation of iron and lipid peroxidation, which is associated with many diseases. Our study addressed the potential role played by ferroptosis and iron accumulation in the progression of pulmonary fibrosis. We found that the inducers of pulmonary fibrosis and injury, namely, bleomycin (BLM) and lipopolysaccharide (LPS), induced ferroptosis of lung epithelial cells. Both the ferroptosis inhibitor liproxstatin-1 (Lip-1) and the iron chelator deferoxamine (DFO) alleviated the symptoms of pulmonary fibrosis induced by bleomycin or LPS. TGF-β stimulation upregulated the expression of transferrin receptor protein 1 (TFRC) in the human lung fibroblast cell line (MRC-5) and mouse primary lung fibroblasts, resulting in increased intracellular Fe2+, which promoted the transformation of fibroblasts into myofibroblasts. Mechanistically, TGF-β enhanced the expression and nuclear localization of the transcriptional coactivator tafazzin (TAZ), which combined with the transcription factor TEA domain protein (TEAD)-4 to promote the transcription of TFRC. In addition, elevated Fe2+ failed to induce the ferroptosis of fibroblasts, which might be related to the regulation of iron export and lipid metabolism. Finally, we specifically knocked out TFRC expression in fibroblasts in mice, and compared with those in the control mice, the symptoms of pulmonary fibrosis were reduced in the knockout mice after bleomycin induction. Collectively, these findings suggest the therapeutic potential of ferroptosis inhibitors and iron chelators in treating pulmonary fibrosis.
Keywords: Pulmonary fibrosis, Ferroptosis, TGF-β, TFRC, Iron homeostasis
Graphical abstract

1. Introduction
Idiopathic pulmonary fibrosis (IPF) is a fatal fibrotic lung disease characterized by a progressive decline in lung function, with a median survival time of 2–3 years after diagnosis. Moreover, in the past decades, the prevalence and incidence of this disease have been increasing year by year [[1], [2], [3]]. The pathogenesis of IPF is now thought to be initiated by injuries of the lung alveolar epithelium with dysregulated wound healing processes. Epithelial cell apoptosis and impaired epithelial cell progenitor function are believed to be critical in fibrogenesis [[4], [5], [6]]. The accumulation of scar tissue was associated with fibroblast-to-myofibroblast transformation, fibroproliferation and excessive accumulation of extracellular matrix (ECM) proteins in the distal lung parenchyma, eventually leading to a pathological remodeling of lung architecture and the obliteration of lung tissue [1,7,8]. The most important source of myofibroblasts was the activation and transformation of fibroblasts, including not only the fibroblasts originally residing in the lungs, but also the fibroblasts recruited by inflammatory factors and chemokines, which occupied the majority of myofibroblasts [[9], [10], [11]]. Although two anti-fibrosis drugs (Nintedanib and Pirfenidone) had been approved for the clinical treatment of IPF, they only delayed pulmonary function decline and failed to stop disease progression [[12], [13], [14]]. Therefore, it is essential to characterize the underlying molecular mechanisms of epithelium injury and fibroblast activation in order to explore new treatments to improve the prognosis and survival rate of patients.
Iron is an indispensable trace element for human beings, which regulates many physiological activities of the human body [15,16]. However, disorders of iron homeostasis could lead to a number of pathological conditions including cancer, hemochromatosis, sickle cell disease, fibrotic lung disease, etc [[17], [18], [19], [20]]. Emerging evidences showed that iron metabolism disorder in the lung was closely related to the occurrence and progression of pulmonary fibrosis. Some studies observed extracellular iron and macrophage hemosiderin accumulation in histological samples from some IPF patients [[20], [21], [22]]. In addition, it had been confirmed that pulmonary fibrosis and functional decline were also related to abnormal iron overload in bleomycin-induced pulmonary fibrosis mouse model, and the similar phenomenon had also been verified in the iron overload model of homeostatic iron regulator (Hfe) gene-deficient mice [20].
Iron overload is also related to a new type of programmed cell death, ferroptosis. Membrane protein TFRC could recognize transferrin and deliver the Fe3+-transferrin complex composed of two molecules of Fe3+ and one molecule of transferrin into cells as an endosome. The reduction of Fe3+ to Fe2+ after lysosome-endosome fusion, which occurred through STEAP3 in the plasma membrane, was subsequently released it into the cytoplasm and was incorporated into the labile iron pool (LIP). Furthermore, other molecules on the cell membrane could also transport Fe2+ into the cell such as divalent metal transporter 1 (DMT1) and metal-ion transporter ZRT- and IRT-like protein 14 (ZIP14), while TFRC was relatively more important [23,24]. Besides, the amount of free iron in the cells could be regulated by other pathways. Ferritin is a complex of two basic subunits, the Ferritin heavy chain (FTH) and the Ferritin light chain (FTL1), which could bind and store intracellular free iron and thus reduce the risk of ferroptosis. In addition, the unique iron-removal channel protein called solute carrier family 40 member 1 (SLC40A1) or Ferroportin in the cell membrane could also reduce intracellular Fe2+ by transporting it out of cells. These iron regulatory elements could regulate intracellular free Fe2+ and affect the ferroptotic sensitivity of cells. Correspondingly, some iron-chelating agents such as DFO and deferiprone (DFP) were known to significantly reverse ferroptosis by reducing the intracellular iron content [16,[25], [26], [27]].
In this study, we investigate the role of iron overload and ferroptosis in the development of pulmonary fibrosis. Both lipid peroxidation accumulation and iron overload are observed during the progress of bleomycin-induced pulmonary fibrosis. In an early inflammatory phase, bleomycin and LPS directly induce ferroptosis in lung epithelial cells, while the ferroptosis inhibitor displays significant resolution for bleomycin- and LPS-induced cell death of type II alveolar epithelial (ATII) cells and lung injury. In a late fibrotic phase, TGF-β enhances intracellular labile ferrous accumulation by upregulating TFRC via the TAZ-TEAD signaling, thus promoting the fibroblast-to-myofibroblast transition. Fibroblasts also survive in response to iron-induced ferroptotic stress by regulation of the ferroptosis-defense pathway. These findings pinpoint a novel mechanism by which ferroptosis and iron accumulation lead to epithelial injury and fibroblast activation in different phases of the bleomycin-induced pulmonary fibrosis model. This study also reveals the therapeutic potential of ferroptosis-prevention therapies and iron chelator in treating lung fibrosis.
2. Materials and methods
2.1. Cell culture
BEAS-2B and IMR90 cell lines were obtained from the American type culture collection (ATCC, United States), MRC-5 was obtained from Procell Life Science&Technology Co.,Ltd. These cells had been proven to be negative for mycoplasma contamination. All cells including BEAS-2B, MRC-5, IMR90 and PLFs were maintained in RPMI 1640 with 10% FBS (Gibco, United States), 2 mM L-glutamine, 100 U/ml of penicillin, and 100 μg/ml of streptomycin at 37 °C and 5% CO2.
2.2. Isolation and culture of primary lung fibroblasts (PLFs)
Wild-type male C57 mice (less than 4 weeks) were selected. After the mice were euthanized, the lung tissues were rinsed with PBS and then infiltrated in serum-free medium. Lung tissue was cut into 1–3 mm pieces with sterile ophthalmic tweezers in a dish and added into serum-free medium with 200 U/ml penicillin, 200 μg/ml streptomycin and 1.5 mg/ml type Ⅳ collagenase. Lung tissues were incubated at 37 °C for 45 min and shook slightly every 15 min. 10% FBS was added to stop collagenase digestion and the digested lung fragments were passed through a 70 μm filter. Cells were centrifuged at 1500 rpm for 5 min. Red Cell Lysis Buffer (TIANGEN, Cat#RT122-02) was added to the cell pellets and the cells were suspended and incubated at room temperature for 5 min. Cells were then centrifuged at 1500 rpm for 5 min, and the supernatant was discarded. Cells were resuspended in complete medium for counting. 5 × 105–1 × 106 cells were added into each well of the 6-well plate, and the medium was supplemented to 2 ml per well. All procedures were performed on ice unless otherwise stated. Fibroblasts (a group of spindle cells) were obtained by repeated trypsinization and passaging.
2.3. Reagents and drugs
Reagents and Drugs used were bleomycin (Selleck, S1214), LPS (Sigma-Aldrich, L4005), recombinant Human TGF-β1 Protein (Peprotech, AF-100-21C-100), Hematoxylin (BASO, BA-4097), Eosin (BASO, BA-4098), Masson's trichrome Stain Kit (Solarbio, G1340), Hoechst 33342 (Beyotime, C1022), SYTOX™ Green (Thermo Fisher, S34860), Liproxstatin-1 (Selleck, S7699), Ferrostatin-1 (MCE, HY-100579), Deferoxamine (Sigma-Aldrich, Y0001937), Ferric ammonium citrate (FAC) (Sigma-Aldrich, F5879), SB431542 (MCE, HY-10431), Necrostatin-1 (MCE, HY-15760), Cell Counting Kit-8 (CCK-8) (Engreen, EC008).
2.4. Real-time quantitative PCR
Total RNA in cells was collected using the RNA extraction Kit (E.Z.N.A. Total RNA Kit, OMEGA, R6934) according to manufacturer's instructions. RNA concentration was measured by Nanodrop 2000C (Thermo Scientfic). cDNA synthesis was performed using a reverse transcription kit (PrimeScript RT Master Mix, TaKara, RR036A). RT-PCR was performed using SYBR Premix Ex Taq ‖ (TaKara, DRR081A). The relative expression of each target gene was normalized to GAPDH expression. The following primers were used: TFRC (human), Forward: 5′-ACCATTGTCATATACCCGGTTCA-3’; Reverse: 5′-CAATAGCCCAAGTAGCCAATCAT-3’. TFRC (mouse), Forward: 5′-GTGGAGTATCACTTCCTGTCGC-3’; Reverse: 5′-CCCCAGAAGATATGTCGGAAAGG-3’. α-SMA (human), Forward: 5′-CTATGAGGGCTATGCCTTGCC-3’; Reverse: 5′-GCTCAGCAGTAGTAACGAAGGA-3’. α-SMA (mouse), Forward: 5′-GTTCAGTGGTGCCTCTGTCA-3’; Reverse: 5′-ACTGGGACGACATGGAAAAG-3’. COL1A1 (human), Forward: 5′-GAGGGCCAAGACGAAGACATC-3’; Reverse: 5′-CAGATCACGTCATCGCACAAC-3’. COL1A1 (mouse), Forward: 5′-GTCCTCCTGGTTCTCCTGGT-3’; Reverse: 5′-GACCGTTGAGTCCGTCTTTG-3’. GAPDH (human), Forward: 5′-CAG GTG GTC TCC TCT GAC TT-3’; Reverse: 5′-CCA AAT TCG TTG TCA TAC CA-3’. GAPDH (mouse), Forward: 5′-TGACCTCAACTACATGGTCTACA-3’; Reverse: 5′-CTTCCCATTCTCGGCCTTG-3’.
2.5. Western blot and antibodies
The cultured cells were washed with PBS and then Radio-Immunoprecipitation Assay (RIPA) lysis buffer was added. Cell samples were collected with a cell scraper. Lung tissue from mice was weighed and ten times the mass of RIPA lysis buffer was added. Then the lung tissue was homogenized in a pre-cooled tissue homogenizer for 2 × 30 s at 50 Hz. Subsequently, cell or tissue samples were placed in ice, lysed for 10 min and centrifuged at 16 000 g for 15 min, and the supernatant was collected. These protein samples were separated by 10% SDS-PAGE and then were analyzed by Western blot. Primary antibodies used were anti-α-tubulin (Proteintech, 66031-1-lg), anti-GAPDH (HUABio, R1210-1), anti-α-SMA (abcam, ab7817), anti-COL1A1 (abcam, ab34710), anti-TFRC (abcam, ab214039), anti-Ferritin (abcam, ab75973), anti-SLC40A1 (abcam, ab78066) and anti-4-HNE (abcam, ab48506).
2.6. Migration and invasion assay
Transwell chamber (Millipore, PI8P01250) with or without Matrigel gel (Corning, 354 234) were used for transwell migration or invasion assay. MRC-5 cells were seeded in the upper portion of the transwell chamber in a 24-well plate suspended in 500 μl serum-free medium. Then 500 μl of RPMI 1640 with 10% FBS was added to the lower portion of the transwell chamber and the drug was simultaneously added into medium. After 48 h of incubation, the transwell chamber was washed with PBS. Cells on the upper portion of the chamber were discarded with a cotton swab. The cells were passed through an 8 μm pore membrane were fixed with 4% paraformaldehyde for 20 min and stained with 0.2% crystal violet for 15 min. The migrated or invaded MRC-5 cells could be observed and recorded using a microscope. The cells were counted by ImageJ software.
2.7. Measurement of Fe2+ in cells
The treated cells were collected by trypsin digestion. After the cells were washed with PBS, 5 μM FeRhoNox-1 (MKbio, MX4558) was added to cell suspension, and the cells were incubated at 37 °C for 1 h away from light. The FeRhoNox-1 probe entering the cell would react with Fe2+ to produce an irreversible orange-red fluorescent substance (Ab = 540 nm, FL = 575 nm). After the cells were washed with PBS again, flow cytometry (Fortessa, BD Biosciences) was able to analyze fluorescence in single cell, and the fluorescence intensity was proportional to the concentration of Fe2+ in the cells. For mouse lung tissue, it was first cut into 1–3 mm pieces, then ground and extruded, and made into a single cell suspension through a 70 μm filter. To detect Fe2+ in the α-SMA+ cells from mouse lung tissue, cells were fixed with fixation buffer (Biolegend, 420 801) at 4 °C for 30 min and the special cells were incubated with a α-SMA primary antibody (abcam, ab7817) at 4 °C for 1 h. The fluorescent secondary antibody used is Alexa Fluor 488 Donkey Anti-mouse IgG (Invitrogen, A21202). After washing with PBS, FeRhoNox-1 was incubated. In addition, depending on the probe, intracellular Fe2+ can also be reflected by fluorescence microscopy under same condition.
2.8. Measurement of ROS and lipid ROS
After the cells were digested by trypsin and collected, the cells were washed with PBS. Then the cells were resuspended in serum-free medium containing 10 μM 2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA, Invitrogen, C6827) and incubated at 37 °C for 1 h. Intracellular reactive oxygen species (ROS) could oxidize H2DCFDA to highly fluorescent 2 ′, 7 ′-dichlorofluorescein (DCF), thus flow cytometry could analyze the ROS content in a single cell. Similarly, C11 BODIPY 581/591 (Invitrogen, D3861) at a concentration of 10 μM could specifically bind to and react with the lipid reactive oxygen species (lipid ROS) in the plasma membrane. As a result, the fluorescence peak of C11 BODIPY 581/591 shifted from ∼590 nm to ∼510 nm, which could be detected by flow cytometry.
2.9. Measurement of cell death and cell viability
BEAS-2B cells were seeded in 12-well plates (5 × 104 cells/well) and were treated with bleomycin (10 μg/ml) or LPS (5 μg/ml) with or without Fer-1 (5 μM). MRC-5 cells were seeded in 12-well plates (5 × 104 cells/well) and were treated with FAC with or without DFO (50 μM). After that, cell death could be analyzed by dual staining with Hoechst33342 and SYTOX Green followed by microscopy at special node and the percentage of dead cells was counted by ImageJ software. MRC-5 cells were seeded in 96-well plates (1 × 104 cells/well) and were treated with FAC with or without DFO (50 μM) for 72 h. Then cell viability was determined by CCK8 according to manufacturer's instruction.
2.10. RNA interference
MRC-5 cells were transfected with siRNA (20 μM) through jetPRIME transfection reagent (Polyplus Transfection, PT-114-15) following the manufacturer's instructions. The human-specific siRNAs targeting TFRC were designed and synthesized by GenePharma (Shanghai). Sequences of siRNAs were as follows: siRNA1 (Sense: 5′-GGCCAGCAAAGUUGAGAAATT-3′); siRNA2 (Sense: 5′-GCUGGUCAGUUCGUGAUUATT-3′); siRNA3 (Sense: 5′-GCCCAGAUGUUCUCAGAUATT-3′); Negative Control (Sense: 5′-UUCUCCGAACGUGUCACGUTT-3′).
2.11. Lentivirus transfection
MRC-5 cells were seeded in a 6-well plate at a density of 1 × 105 cells/well. Subsequently, 5 μl of 1 × 108 TU lentivirus (Shanghai GeneChem, China) and 40 μl of 25 × HitransG A lentivirus infection regent were added to each well. Transfection efficiency was assessed by green fluorescent protein (GFP) detection using fluorescence microscopy, and cells were selected with fluorescence-activated cell sorting. The expression levels of relevant proteins (TFRC) were examined by Western blot.
2.12. Dual luciferase reporter assay
293T cells were seeded in 96-well plates at a density of 1 × 104 cells/well. When the cell density reached 60–80%, the mixture of TFRC Promoter-Luciferase Reporter plasmid (50 ng), the pCMV6-Entry-TEAD4 plasmid (0/50/100/200 ng) and Renilla plasmid (2.5 ng) was co-transfected according to jetPRIME Transfection reagent operation instructions. After 24 h, luciferase activity was measured by luminometer (Promega, Glomax-20/20) using a Dual Luciferase Reporter Assay System (Promega, E1910 and E1960) according to manufacturer's instructions. Renilla luciferase was be used as a reference for normalization of transfection efficiency.
2.13. Tissue iron content detection
Fresh mouse lung tissue of similar quality was collected. After adding 250 μl extracting solution, lung tissues were homogenized in a pre-cooled tissue homogenizer for 2 × 30 s at 50 Hz. A tissue iron assay kit was used for subsequent extraction following the manufacturer's instruction (Solarbio, BC4355). Fe3+ in tissue could be reduced to Fe2+ that could be detected colorimetrically with 2,2′-bipyridine at 520 nm which indicated the iron content in tissue.
2.14. Detection of hydroxyproline in lung
Fresh or frozen lung tissue including the middle and lower lobes of the right lung was selected for examination. According to the manufacturer's instructions (Nanjing Jiancheng, A030-2-1), the sample was added to the hydrolysate and heated at 100 °C for 20 min. The pH value of the lysate was adjusted to 6.0–6.8 and the lysate was centrifuged at 5000 rpm for 10 min. After subsequent reaction, the supernatant could be used to detect the absorbance value at 550 nm wavelength.
2.15. Immunofluorescence staining in cells
MRC-5 cells or PLFs were seeded in a 35 mm confocal dish (5 × 104 cells/dish). After treating with drug for 48 h or 72 h, the cells were washed with PBS and fixed with 4% paraformaldehyde for 10 min, and permeabilized with 0.2% Triton X-100 for 8 min. Then the cells were blocked with 2% goat serum at room temperature for 20 min. Subsequently, the cells were incubated with primary antibody at 4 °C overnight and were incubated with fluorescent secondary antibody at room temperature for 1 h after washing. Confocal microscope could analyze the expression intensity and location of molecules in cells. The concentrations of antibodies used in immunofluorescence staining of cells were anti-α-SMA (abcam, ab7817, 1:300), anti-TAZ (CST, 83669, 1:100), Donkey anti-Mouse IgG Alex Fluor 555 (Invitrogen, A-31570, 1:200) and Donkey anti-Rabbit IgG Alex Fluor 488 (Invitrogen, A-21206, 1:200). DAPI (Beyotime, C1002, 1:500) was used to label the nucleus. The cellar fluorescent images were acquired by a Nikon Eclipse Ti confocal microscopy (Nikon, Tokyo, Japan).
2.16. H&E staining and Masson's staining
After the mice were euthanized, the whole lung was taken and fixed overnight in 4% paraformaldehyde. Then gradient dehydration was carried out according to standard procedures and the tissue was paraffin embedded. Next, the tissue was sliced into 3 μm slices. After routine dewaxing and hydration, H&E staining was performed with hematoxylin (BASO, BA-4097) for 7 min and eosin (BASO, BA-4098) for 7 min. Masson's staining was performed according to the Masson's trichrome staining Kit (Solarbio, G1340). The blue collagen tissue was visible under a light microscope. H&E staining scanning was performed with PANNORAMIC 250 Flash III (3DHISTECH Ltd.), Masson's staining image was collected with an OLYMPUS IX73 microscope (Olympus, Tokyo, Japan).
2.17. Immunohistochemistry (IHC) and immunofluorescence staining for lung tissue
The drug-induced fibrotic lung tissues of mice were embedded in paraffin and sectioned in 3 μm sections, which can be used for immunohistochemistry and immunofluorescence staining. After routine dewaxing and hydration, the antigen retrieval step was performed in citrate buffer (pH6.0) (ZSGB-BIO, ZLI9065) at ∼115 °C for 2 min which could enhance the binding of primary antibody to a specific epitope. Then the section was blocked with goat serum at room temperature for 1 h. Subsequently, the section was incubated with primary antibody at 4 °C overnight. After washing, the section was incubated with HRP-labeled Goat Anti-Mouse/Rabbit IgG for 1 h at room temperature and Streptavidin/HRP for 20 min at room temperature. Finally, the DAB chromogenic kit (ZSGB-BIO, ZLI9019) was used for chromogenic reaction. The Immunohistochemistry kit (ZSGB-BIO, SP-9000) could perform specific histochemical staining. The concentrations of primary antibodies were anti-TGFβ1 (abcam, ab215715, 1:500), anti-α-SMA (abcam, ab7817, 1:300), anti-FAPα (abcam, ab207178, 1:200). Different from IHC, the section would be incubated with fluorescent secondary antibody at room temperature for 1 h after washing the primary antibody in immunofluorescence staining. The concentrations of antibodies were anti-α-SMA (abcam, ab7817, 1:3000), anti-TFRC (abcam, ab214039, 1:200), Donkey anti-Mouse IgG Alex Fluor 488 (Invitrogen, A-21202, 1:200) and Donkey anti-Rabbit IgG Alex Fluor 555(Invitrogen, A-31572, 1:200). DAPI (Beyotime, C1002, 1:500) was used to label the nucleus. The images were collected with a Nikon Eclipse Ni–U microscope (Nikon, Tokyo, Japan).
2.18. Multicolor immunofluorescence staining for lung tissue
Similarly, the lung tissues of mice were embedded in paraffin and sectioned in 3 μm sections, following gradient dewaxing and hydration. Then heat-induced antigen retrieval was performed using citrate (pH = 6) or Tris-EDTA (pH = 9) buffer. After that, the section was blocked with 5% goat serum and was incubated with primary antibody at 37 °C for 1 h, Opal Polymer HRP Mouse + Rabbit at room temperature for 10 min, and TSA Amplification at room temperature for 10 min. The Opal 7-color Manual IHC Kit (Akoya Biosciences, NEL811001KT) was purchased for the assay. Nuclei were stained with DAPI. The sample was washed by TBST for 2 min × 3 per interval. The primary antibodies used were anti-TGFβ1 (abcam, ab215715, 1:500), anti-α-SMA (abcam, ab7817, 1:3000), anti-FAPα (abcam, ab207178, 1:200), anti-Sftpc (abcam, ab90716, 1:200) and anti-FSP1 (abcam, ab197896, 1:2000). Images were collected by AKOYA BIOSCIENCES Vectra Polaris and analyzed by InForm 2.4.9 software or HALOTM software (Akoya Biosciences).
2.19. Animal experiments
For earlier experiment, wild-type male C57 mice aged 10 weeks and weighing 25 g were selected as experimental subjects, which were purchased from SPF (Beijing) Biotechnology Co.,Ltd. In the first experiment, the mice were randomly divided into two groups including control (Ctrl) and bleomycin. In the second experiment, the mice were randomly divided into four groups: control, bleomycin, bleomycin + Lip-1 and bleomycin + DFO. Finally, the mice were randomly divided into three groups: control, LPS, LPS + Lip-1. The mice were intratracheally injected with normal saline (total volume 50 μl) or bleomycin (total volume 50 μl, 5 mg/kg) or LPS (total volume 50 μl, 5 mg/kg). For the inhibitor group, mice were intraperitoneally injected with Lip-1 (total volume 150 μl, 15 mg/kg) three times a week or DFO (total volume 200 μl, 30 mg/kg) daily. When Lip-1 was injected from day 0, lung tissue samples from the mice were collected at 4, 7, 14 and 21 days. When Lip-1 was injected from day 10, samples were collected at 10, 14, 18 and 21 days.
C57BL/6J-FSP1-CreERT transgenic mice were purchased from Shanghai Model Organisms Center. C57BL/6J-TFRCflox/flox transgenic mice were purchased from Cyagen Biosciences. Crossing the TFRCflox/flox mouse with the FSP1-CreERT transgenic mice could generate FSP1-CreERT; TFRCflox/flox mice. And Tamoxifen (MCE, HY-13757A) could induce the knockout of TFRC in FSP1+ cells in FSP1-CreERT; TFRCflox/flox mice, described as TFRC−/− mice, according to the manufacturer's instructions. Bleomycin induced pulmonary fibrosis in TFRC−/− mice and TFRCflox/flox mice under similar condition.
2.20. Statistical analyses
All statistics were presented with mean ± SD, which were performed through Prism 7.0 GraphPad Software. Unpaired Student's t-test was used for comparison between two groups and one-way ANOVA was used for comparison between multiple groups, followed by Tukey test. P < 0.05 was considered as statistically significant.
3. Results
3.1. Lipid peroxidation accumulation and iron overload during bleomycin-induced pulmonary fibrosis
To explore the role and mechanism of iron and ferroptosis in IPF, we established a pulmonary fibrosis model by treating mice with bleomycin, which is commonly applied to induce pulmonary fibrosis in rodents [8,28,29]. H&E staining showed that the alveolar structure was destroyed, the lung interstitium was thickened, and the lung texture was abnormal (Fig. 1a). Bleomycin also gradually increased the deposition of collagen, which was measured by Masson's staining (Fig. 1a and b). After bleomycin administration, a significant increase in the expression of two widely accepted markers of fibroblasts, α-smooth muscle actin (α-SMA) and fibroblast activation protein-α (FAPα), was observed within 7 days, with peak levels identified at 14 days (Fig. 1a and b). TGF-β expression was also increased in bleomycin-induced lung fibrosis, occurring 4 days after bleomycin treatment, which was earlier than the upregulation of fibroblast marker expression (Fig. 1a and b). Fourteen days after the administration of bleomycin, multicolor immunofluorescence analyses showed that the expression of TGF-β and the populations of α-SMA+ fibroblasts were increased, consistent with the immunohistochemistry results (Fig. 1c). These data confirmed the establishment of pulmonary fibrosis induced with bleomycin, and a suitable time point for assessing lung fibrosis was demonstrated.
Fig. 1.

The accumulation of lipid peroxides and iron overload in lungs from bleomycin-induced pulmonary fibrosis mice. (a) Upper, lungs of mice from control (Ctrl) group and bleomycin (BLM) group on Day 21 after bleomycin administration. Bottom, representative images of H&E staining, Masson staining, and IHC staining for α-SMA, FAPα and TGF-β of lung tissue from control group and bleomycin-treated group at different days. Scale bars, 100 μm. (b) Scatter plot showing the percentage of the positive area of Masson staining and IHC staining in lung tissue. n = 3–4 mice per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (c) Left, representative images of multicolor immunofluorescence for DAPI (blue), α-SMA (green) and TGF-β (orange) in lung tissue of mice on Day 14 after bleomycin administration. Scale bars, 50 μm. Right, scatter plot showing the percentage of α-SMA+ and TGF-β+ cells, mean fluorescence intensity (MFI) of α-SMA and the positive area of TGF-β in lung tissue of mice on Day 14 after bleomycin administration. Each dot represents a microscopic image, n = 15 images per group from 3 mice. Statistical analysis was performed using the Student's t-test. *P < 0.05, ****P < 0.0001. (d) Left, immunoblotting analysis of 4-HNE in lung tissues of mice on Day 7 and Day 14 after bleomycin administration. Right, quantitative analysis of 4-HNE from three mice. Statistical analysis was performed using the Student's t-test. *P < 0.05, **P < 0.01. (e) Intracellular Fe2+ levels in lung tissue of mice were assessed by flow cytometry using FeRhoNox-1 on Day 7 and Day 14 after bleomycin administration. Upper, representative flow cytometry result for Fe2+. Bottom, scatter plot showing the results from three mice. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. **P < 0.01, ****P < 0.0001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
We then measured a putative biomarker of lipid peroxidation, 4-hydroxynonenal (4-HNE) [30,31], in the lung tissues of bleomycin-induced pulmonary fibrosis mice at different time points. Western blot analysis showed that the content of 4-HNE in the bleomycin-treated lung tissue increased compared with that in the control group tissue at 7, 14 and 21 days, suggesting the accumulation of lipid peroxides in lung tissues during bleomycin-induced pulmonary fibrosis (Fig. 1d, Supplementary Fig. 1a). In addition to elevated lipid peroxide levels, flow cytometry demonstrated the accumulation of ferrous iron (Fe2+) in cells in bleomycin-treated lung tissue at both 7 and 14 days, as indicated by Fe2+-specific fluorescent probes (Fig. 1e, Supplementary Figs. 1b and 1c). These data confirmed the accumulation of lipid peroxides and iron overload in bleomycin-induced pulmonary fibrosis. Notably, the production of lipid ROS and accumulation of ferrous iron were much lower in the lungs 14 days after bleomycin administration than 7 days after bleomycin administration. These data suggest that lipid peroxide production and iron overload tend to occur within a relatively short period after bleomycin administration, which may be subsequently partially relieved.
3.2. Bleomycin and LPS induce lipid ROS accumulation and ferroptosis in alveolar epithelial cells
Chronic injury of epithelial cells, especially ATII cells, is thought to be a key early driver of the aberrant wound-healing response and pathogenesis of IPF [32,33]. Emerging data indicate that oxidative damage might trigger the apoptosis of ATII cells, contributing to TGF-β-induced pulmonary fibrosis [34,35]. Since we demonstrated that lipid peroxides and iron accumulated in bleomycin-induced pulmonary fibrosis, we investigated whether ferroptosis was involved in bleomycin-induced lung epithelial cell damage. Human lung epithelial BEAS-2B cells were exposed to bleomycin and then evaluated for expression of ferroptosis markers. As shown in Fig. 2a and b, bleomycin significantly enhanced the production of ROS and lipid ROS in BEAS-2B cells, and the effects on lipid ROS were abolished by the lipid peroxide-trapping agent ferrostatin-1 (Fer-1). Moreover, intracellular ferrous iron was detected by FeRhoNox-1 probe staining. The accumulation of ferrous iron in bleomycin-stimulated BEAS-2B cells was inhibited by both Fer-1 and DFO (Fig. 2c). Notably, treatment with Fer-1 partially prevented the cell death triggered by bleomycin (Fig. 2d), suggesting that ferroptosis was involved in bleomycin-induced cell death. We then used LPS, another commonly used lung injury and pulmonary fibrosis inducer, to repeat the aforementioned experiment, and the results were basically consistent (Fig. 2e–h). Necroptosis inhibitor Necrostatin-1 (Necro-1) could also diminish cell death triggered by bleomycin or LPS, demonstrating that bleomycin- or LPS-induced cell death may include both ferroptosis and necroptosis (Supplementary Fig. 1d). Previous articles have proved that bleomycin could induce apoptosis of ATII cells, indicating that multiple cell death modes may contribute to injury of alveolar epithelium in bleomycin-induced pulmonary fibrosis [32,36]. Collectively, these results indicate that ferroptotic cell death of lung epithelial cells is involved in the lung injury and destruction of alveolar epithelium induced by both bleomycin and LPS.
Fig. 2.

Bleomycin and LPS induce ferroptosis in BEAS-2B cells. (a) ROS production in BEAS-2B cells was assessed by flow cytometry using H2DCFDA after 24 h of treatment with bleomycin and Fer-1. BLM: 10 μg/ml. Fer-1: 1 μM. (b) Lipid ROS production in BEAS-2B cells was assessed by flow cytometry using C11-BODIPY after 48 h of treatment with bleomycin and Fer-1. Left, representative flow cytometry result for lipid ROS. Right, scatter plot showing the results for independent samples examined. BLM, 10 μg/ml. Fer-1: 5 μM n = 3 biological replicates. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. **P < 0.01. (c) Intracellular Fe2+ levels in BEAS-2B cells were assessed by flow cytometry using FeRhoNox-1 after 48 h of treatment with bleomycin, Fer-1 and DFO. Left, representative flow cytometry result for Fe2+. Right, scatter plot showing the results for independent samples examined. BLM: 10 μg/ml. Fer-1: 5 μM. DFO: 50 μM n = 3 biological replicates. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. **P < 0.01, ****P < 0.0001. (d) Cell death of BEAS-2B cells was determined by Hoechst33342 and SYTOX Green staining coupled with fluorescent microscope after 24–84 h of treatment with bleomycin and Fer-1. BLM: 10 μg/ml. Fer-1: 5 μM n = 4–13 images per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. ****P < 0.0001. (e) ROS production in BEAS-2B cells treated with LPS and Fer-1 for 24 h was assessed by flow cytometry using H2DCFDA. LPS: 5 μg/ml. Fer-1: 1 μM. (f) Lipid ROS production in BEAS-2B cells treated with LPS and Fer-1 for 48 h was assessed by flow cytometry using C11-BODIPY. Left, representative flow cytometry result for Lipid ROS. Right, scatter plot showing the results for independent samples examined. LPS: 5 μg/ml. Fer-1: 1 μM n = 3 biological replicates. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. **P < 0.01. (g) Intracellular Fe2+ levels in BEAS-2B cells were assessed by flow cytometry using FeRhoNox-1 after 48 h of treatment with LPS, Fer-1 and DFO. Left, representative flow cytometry result for Fe2+. Right, scatter plot showing the results for independent samples examined. LPS: 5 μg/ml. Fer-1: 5 μM. DFO: 50 μM n = 3 biological replicates. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. *P < 0.05, **P < 0.01, ***P < 0.001. (h) Cell death of BEAS-2B cells was determined by Hoechst33342 and SYTOX Green staining coupled with fluorescent microscope after 24–72 h of treatment with LPS and Fer-1. LPS: 5 μg/ml. Fer-1: 5 μM n = 4–13 images per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. ****P < 0.0001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.3. Ferroptosis inhibitor and iron chelator induce significant resolution of bleomycin- and LPS-induced lung injury and fibrosis in mouse models
To examine whether blocking ferroptosis protects lung tissue from injury and fibrosis, we treated mice with the potent ferroptosis inhibitor Lip-1 three times per week following the administration of bleomycin. Consistent with our hypothesis, a decrease in the number of surfactant protein C (SPC, encoded by Sftpc)-positive type II alveolar epithelial cells was observed from 4 days through 14 days (Fig. 3a). Lip-1 treatment rescued SPC-positive type II alveolar epithelial cells after bleomycin-induced acute lung injury, indicating recovery of the alveolar epithelium (Fig. 3a). H&E staining of lung sections showed that the fibrotic symptoms characterized by alveolar septal thickening and pulmonary consolidation were relieved by Lip-1 treatment (Fig. 3b). In addition, we found that the levels of both hydroxyproline and lipid ROS in bleomycin-induced pulmonary fibrosis were significantly reduced in the Lip-1-treated group (Fig. 3c and d). Masson's trichrome staining revealed a substantial resolution in collagen deposition in the lung sections of the Lip-1-treated mice compared with that in the bleomycin-treated mice (Fig. 3e and f). Notably, the time-dependent increases in the levels of α-SMA, FAPα and TGF-β were reduced in Lip-1-treated mice compared with bleomycin-treated mice, reflecting the decreased severity of the pulmonary fibrosis (Fig. 3e and f).
Fig. 3.

Liproxstatin-1 inhibits bleomycin-induced pulmonary fibrosis. (a) Multicolor immunofluorescence showing the number of SPC+ type II alveolar epithelial cells and α-SMA+ fibroblasts in mice lung tissue from control group, bleomycin group and bleomycin + Lip-1 group at different days. Left, representative images of multicolor immunofluorescence for DAPI (blue), α-SMA (green) and SPC (yellow) in lung tissue of mice at different days after bleomycin administration. Scale bars, 50 μm. Right, scatter plot showing the percentage of SPC+ type II alveolar epithelial cells in lung tissue from control group, bleomycin group and bleomycin + Lip-1 group at different days. Each dot represents a microscopic image, n = 16–34 images per group from 4 mice. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. **P < 0.01, ****P < 0.0001. (b) Histopathological study of lung tissue in control group, bleomycin group and bleomycin + Lip-1 group at different days was performed by H&E staining. Representative photographs of lungs on Day 14 were showed. Scale bars, 1 mm. (c) Hydroxyproline (HYP) content from control group, bleomycin group and bleomycin + Lip-1 group at different days. n = 4 mice per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. *P < 0.05, **P < 0.01, ****P < 0.0001. (d) Immunoblotting analysis of 4-HNE in lung tissues of mice on Day 7 and Day 14 after bleomycin administration. Left, representative Western blot result for 4-HNE. Right, scatter plot showing the results for all samples examined. Each lane represents one mouse, n = 3 mice per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. *P < 0.05, ***P < 0.001. (e) Representative images of Masson's staining and IHC staining for α-SMA, FAPα and TGF-β of lung tissue from control group, bleomycin group and bleomycin + Lip-1 group at different days. Scale bars, 100 μm. (f) Scatter plot showing the percentage of the positive area of Masson's staining and IHC staining in lung tissue from control group, bleomycin group and bleomycin + Lip-1 group at different days. n = 4 mice per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. ***P < 0.001, ****P < 0.0001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
To determine whether Lip-1 modulates pulmonary fibrosis in the fibrotic stage, we intraperitoneally injected Lip-1 on the 10th day after bleomycin treatment. By H&E staining, we found that Lip-1 mitigated bleomycin-induced pulmonary fibrosis in the fibrotic phase (Fig. 4a). The elevated level of hydroxyproline in the lungs of the bleomycin-treated mice was also decreased by Lip-1 on Day 21 (Fig. 4b). Moreover, Lip-1 inhibited collagen deposition and α-SMA expression in the fibrotic phase (Fig. 4c–f). In addition to studying the effect of Lip-1 in pulmonary fibrosis, we tested the effect of modulating the accumulation of Fe2+ by intraperitoneal injection of the iron chelator DFO in bleomycin-induced pulmonary fibrosis mice for 14 days. Similar to Lip-1, DFO reduced collagen deposition and fibrosis levels in bleomycin-induced pulmonary fibrosis mice and inhibited the expression of α-SMA, FAPα and TGF-β in lung tissue within 14 days (Supplementary Figs. 2a and b). Moreover, DFO treatment reduced the accumulation of iron in the lungs of the bleomycin-treated mice (Supplementary Fig. 2c).
Fig. 4.

Liproxstatin-1 alleviates bleomycin-induced pulmonary fibrosis in the fibrosis stage. (a) H&E staining scanning of lung tissue from control group, bleomycin group and bleomycin + Lip-1 group at 10, 14, 18, and 21 days after bleomycin administration. Scale bars, 1 mm. (b) Hydroxyproline content from control group, bleomycin group and bleomycin + Lip-1 group at 10, 14, 18, and 21 days after bleomycin administration. n = 3–4 mice per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. *P < 0.05, **P < 0.01. (c) Representative images of Masson's staining of mice lung tissue from control group, bleomycin group and bleomycin + Lip-1 group at 10, 14, 18, and 21 days after bleomycin administration. Scale bars, 100 μm. (d) Representative images of IHC staining for α-SMA of mice lung tissue from control group, bleomycin group and bleomycin + Lip-1 group at 10, 14, 18, and 21 days after bleomycin administration. Scale bars, 100 μm. (e) Scatter plot showing the percentage of the positive area of Masson's staining in lung tissue from control group, bleomycin group and bleomycin + Lip-1 group at 10, 14, 18, and 21 days after bleomycin administration. n = 4 mice per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. *P < 0.05, ****P < 0.0001. (f) Scatter plot showing the percentage of the positive area of IHC staining in lung tissue from control group, bleomycin group and bleomycin + Lip-1 group at 10, 14, 18, and 21 days after bleomycin administration. n = 4 mice per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. **P < 0.01, ****P < 0.0001.
The aforementioned results were based on the bleomycin-induced pulmonary fibrosis mouse model, which is a classical pulmonary fibrosis model. In addition, intratracheal injection of LPS leads to pulmonary fibrosis [37,38]. Our in vitro experiments demonstrated that LPS triggered the ferroptosis of the BEAS-2B lung epithelial cell line. According to the results of H&E staining, the symptoms related to the lung tissue were aggravated and then were slowly ameliorated in the LPS-induced pulmonary fibrosis mouse model. Compared with bleomycin-induced pulmonary fibrosis, LPS-induced pulmonary fibrosis followed a shorter course of disease, with lung tissue deteriorating significantly within 4 days and peaking 7 days post-LPS treatment. H&E staining showed that Lip-1 ameliorated LPS-induced pathological changes of acute lung injury (Supplementary Fig. 3a). Masson's staining showed that Lip-1 significantly reduced LPS-induced collagen production in the lung (Supplementary Figs. 3b and f). Moreover, LPS induced the elevation of α-SMA, FAPα and TGF-β in the lung tissues, which was inhibited by Lip-1 (Supplementary Figs. 3c–f). Taken together, these results suggest that administration of a ferroptosis inhibitor can significantly protect the lung from bleomycin- and LPS-induced lung injury and pulmonary fibrosis, presumably by inhibiting the damage in alveolar epithelial cells provoked by lipid peroxidation through a burst of free radical production and ferroptosis.
3.4. TGF-β is involved in fibroblast activation by promoting intracellular Fe2+ accumulation
It is widely accepted that the bleomycin-induced lung injury model comprises two phases: an early inflammatory phase characterized by infiltration of inflammatory cells into the lungs within 7 days, followed by a late fibrotic phase characterized by increased fibroblast proliferation and differentiation to myofibroblasts, as well as synthesis of ECM during the second week [8,39,40]. Increasing evidence also suggests that alveolar epithelial injury might be the primary defect in IPF, which occurs relatively early than the inflammatory phase [41]. However, the number of α-SMA-positive fibroblasts increased from 7 days through 14 days but not within 4 days, in contrast to the control group (Fig. 3f). Flow cytometry demonstrated the accumulation of Fe2+ in α-SMA-positive fibroblasts in bleomycin-treated lung tissue on both Days 7 and 14, and the accumulation was decreased on Day 14 compared with the level on Day 7 (Fig. 5a and b). These results encouraged us to explore the reason that fibroblasts were abnormally activated under excessive iron, which is believed to be a trigger for lipid peroxidation and ferroptosis. During the initiation of the fibrotic phase, epithelial cell injury and activation of inflammation promote the production of the main profibrogenic cytokine TGF-β, which plays a critical role in the process of fibroblast differentiation into myofibroblasts and induces collagen production, causing a loss of lung elasticity and impairment of respiratory function [42,43]. To confirm the activation of fibroblasts induced by TGF-β, we stimulated two different human fibroblast cell lines (MRC-5 and IMR90 cell lines) and mouse primary lung fibroblasts (PLFs) with different concentrations of TGF-β and observed positive effects of TGF-β on the fibroblast-to-myofibroblast transition in all three types of cells. TGF-β stimulation increased both the mRNA and protein expression levels of α-SMA and COL1A1, which are classical markers of fibroblasts, in a dose-dependent manner (Supplementary Figs. 4a–d). Moreover, TGF-β significantly enhanced the migratory and invasive potential of the fibroblasts (Supplementary Fig. 4e).
Fig. 5.

TGF-β is involved in fibroblast activation by promoting the accumulation of iron in cells. (a–b) Intracellular Fe2+ levels in α-SMA+ cells in lung tissue of mice were assessed by flow cytometry using FeRhoNox-1 on Day 7 and Day 14 after bleomycin administration. Cells were first gated in α-SMA-FITC positive, and then subjected to analysis of Fe2+-PE fluorescent intensity. n = 3 mice per group. Statistical analysis was performed using the Student's t-test. ***P < 0.001. (c) Fluorescence microscopy images showing intracellular Fe2+ levels in MRC-5 stimulated with TGF-β, SB431542 (SB) and DFO using FeRhoNox-1. DAPI (blue). FeRhoNox-1 (Red). TGF-β: 10 ng/ml. SB: 1 μM. DFO: 50 μM. Scale bars, 200 μm n = 3 biological replicates. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. *P < 0.05, ***P < 0.001. (d) Fluorescence microscopy images showing intracellular Fe2+ levels in MRC-5 stimulated with FAC and DFO using FeRhoNox-1. DAPI (blue). FeRhoNox-1 (Red). FAC: 50 μM. DFO: 50 μM. Scale bars, 200 μm n = 3 biological replicates. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. ***P < 0.001. (e) Cell viability was determined by CCK8 after 72 h of treatment with FAC and DFO. FAC: 50 μM. DFO: 50 μM n = 4 biological replicates. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. *P < 0.05, **P < 0.01, ****P < 0.0001. (f) Cell viability was determined by CCK8 after 72 h of treatment with FAC and DFO. FAC: 50 μM. DFO: 50 μM n = 5 biological replicates. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. **P < 0.01, ****P < 0.0001. (g) Expression of α-SMA and COL1A1 in MRC-5 and IMR90 was assessed by Western blot after 72 h of treatment with the indicated doses of FAC and DFO. DFO: 50 μM. (h) Expression of α-SMA and COL1A1 in MRC-5 was assessed by Western blot after 72 h of treatment. TGF-β: 10 ng/ml. SB: 1 μM. DFO: 50 μM. (i) Confocal microscopy showing α-SMA in MRC-5 after 72 h of treatment with TGF-β, SB and DFO. DAPI (blue). α-SMA (red). TGF-β: 10 ng/ml. SB: 1 μM. DFO: 50 μM. Scale bars, 20 μm. (j) Representative microscopic images and the quantitation of MRC-5 cells that transferred through the transwell in the migration assay and invasion assays. Scale bars, 200 μm. TGF-β: 10 ng/ml. SB: 1 μM. DFO: 50 μM n = 6 visual fields. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. ****P < 0.0001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
We then exposed MRC-5 cells to TGF-β and detected intracellular Fe2+ with an FeRhoNox-1 probe. As shown in Fig. 5c, the fluorescence intensity of the FeRhoNox-1 probe was increased with TGF-β stimulation, while both SB431542, a blocker targeting the TGF-β receptor, and DFO attenuated TGF-β-induced intracellular labile Fe2+ accumulation. Similarly, intracellular Fe2+ was increased by FAC, and this effect was reduced by DFO (Fig. 5d). In other words, both TGF-β and exogenous iron supplementation increased intracellular labile Fe2+ accumulation. To investigate the effect of elevated Fe2+ on fibroblasts, we stimulated MRC-5 cells with different concentrations of FAC. Low concentrations of FAC within a certain range promoted cell proliferation (Fig. 5e), which was inhibited by DFO (Fig. 5f). However, when the concentration of FAC exceeded a certain level, the cell proliferation rate was decreased (Fig. 5e). This outcome was reasonable because the accumulation of intracellular Fe2+ might lead to excessive production of lipid peroxidation, triggering ferroptosis. In addition, the levels of α-SMA and COL1A1 in MRC-5 and IMR-90 cells were increased by low concentrations of FAC (Fig. 5g). This increase was prevented when FAC was applied in combination with DFO (Fig. 5g). Similarly, the increased expression of α-SMA and COL1A1 elevated by TGF-β was also diminished by DFO (Fig. 5h and i), indicating that excess iron caused by TGF-β stimulation promotes the transition of fibroblasts into myofibroblasts. In addition, the enhanced migratory and invasive abilities of MRC-5 cells promoted by TGF-β were also attenuated by treatment with SB431542 or DFO (Fig. 5j). Together, these findings suggest that TGF-β functions is a positive regulator of intracellular labile Fe2+ accumulation, which is involved in fibroblast activation.
3.5. The upregulation of TFRC expression via the TAZ-TEAD complex contributes to the function of TGF-β to promote iron accumulation and fibroblast activation
To clarify the mechanism involved in TGF-β-regulated ferrous iron accumulation in fibroblasts, we detected the expression of several iron-related proteins in MRC-5 cells stimulated with TGF-β. As shown in Fig. 6a, the expression of TFRC and ferritin was upregulated upon TGF-β stimulation. TFRC, a cell surface membrane protein, is involved in cellular iron uptake by transporting transferrin-bound iron into the cell. Iron storage protein ferritin is composed of 24 subunits, including heavy-chain ferritin (FTH) and light-chain ferritin (FTL1), which can bind and store intracellular iron [26,27,44]. Thus, the elevation of FTH implied an increase in intracellular Fe2+. SLC40A1 is the only known transmembrane exporter of iron that can directly alter the amount of iron in cells [16,24]. Correspondingly, the expression of TFRC in α-SMA-positive cells was increased in the lung tissue of bleomycin-treated pulmonary fibrosis mice compared with that of control mice, as revealed by immunofluorescence and flow cytometry (Fig. 6c and d). In addition, the upregulated transcription level of TFRC induced by TGF-β in MRC-5 and PLF cells was revealed by qPCR (Fig. 6b).
Fig. 6.

TGF-β enhances TFRC expression in fibroblasts by promoting the formation and nuclear localization of TAZ/TEAD4 complex. (a) Expression of α-SMA, COL1A1, TFRC, FTH and SLC40A1 in MRC-5 was assessed by Western blot after 72 h of treatment with the indicated doses of TGF-β. (b) Expression of TFRC in MRC-5 and PLFs were assessed by qPCR after 48 h of treatment with the indicated doses of TGF-β. n = 4 biological replicates. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. *P < 0.05. **P < 0.01, ***P < 0.001, ****P < 0.0001. (c) MFI of TFRC-PE in α-SMA+ cells in lung tissue of mice was assessed by flow cytometry on Day 14 after bleomycin administration. Left, representative flow cytometry result for MFI of TFRC-PE in α-SMA+ cells. Cells were first gated in α-SMA-FITC positive, and then subjected to analysis of TFRC-PE fluorescent intensity. Right, scatter plot showing the results for independent samples examined. n = 3 mice per group. Statistical analysis was performed using the Student's t-test. **P < 0.01. (d) Immunofluorescence showing TFRC and α-SMA in lung tissue of mice on Day 14 after bleomycin administration. DAPI (blue). TFRC (Red). α-SMA (Green). Scale bars, 10 μm. (e) Expression of α-SMA and TFRC in MRC-5 after TFRC knockdown with siRNA was assessed by Western blot after 72 h of treatment with TGF-β. TGF-β: 10 ng/ml. (f) Fluorescence microscopy images showing intracellular Fe2+ levels in MRC-5 after TFRC knockdown with siRNA using FeRhoNox-1 after 24 h of treatment with FAC. DAPI (blue). FeRhoNox-1 (Red). FAC: 2 μM. Scale bars, 200 μm. (g) Expression of α-SMA and TFRC in MRC-5 overexpressing TFRC was assessed by Western blot after 72 h of treatment with the indicated doses of FAC. (h) Fluorescence microscopy images showing intracellular Fe2+ levels in MRC-5 overexpressing TFRC using FeRhoNox-1 after 24 h of treatment with FAC. FeRhoNox-1 (Red). FAC: 2 μM. Scale bars, 200 μm. (i) Expression of TAZ in MRC-5 and PLFs was assessed by Western blot after 72 h of treatment with the indicated doses of TGF-β. (j) Expression of TAZ, TFRC and α-SMA in MRC-5 and PLFs was assessed by Western blot after 72 h of treatment with TGF-β and SB. TGF-β: 10 ng/ml. SB: 1 μM. (k) Confocal microscopy showing the expression and localization of TAZ and α-SMA in MRC-5 and PLFs after 48 h of treatment with TGF-β. DAPI (blue). TAZ (green). α-SMA (red). TGF-β: 10 ng/ml. SB: 1 μM. Scale bars, 50 μm. (l) TFRC promoter luciferase activity was determined in 293T cells transfected with pCMV6-Entry-TEAD4 plasmid using dual luciferase reporter assay. n = 4 biological replicates. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. ****P < 0.0001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
To further determine the potential role played by TFRC in TGF-β-induced fibroblast activation, we knocked down the expression of TFRC in MRC-5 cells with siRNA and then stimulated MRC-5 cells with TGF-β. SiRNA targeting TFRC effectively decreased the level of TFRC in MRC-5 cells, as determined by qPCR, Western blot and flow cytometry (Supplementary Figs. 4f–h). Knockdown of TFRC expression diminished the intracellular ferrous iron level and activation of fibroblasts induced by TGF-β, suggesting that TFRC might be the target critical for iron accumulation in TGF-β-induced fibroblast activation in pulmonary fibrosis (Fig. 6e and f). Correspondingly, overexpression of TFRC in MRC-5 cells with lentivirus also increased the intracellular ferrous iron level and promoted the activation of fibroblasts under lower concentrations of FAC (Fig. 6g and h). However, it was not clear how TGF-β upregulated TFRC expression in fibroblasts. It has been reported that TAZ expression, a transcriptional coactivator, can be upregulated, and it can be translocated into the nucleus upon TGF-β stimulation. TAZ promotes the transcription of profibrotic factors through its interaction with the TEAD family of transcription factors [[45], [46], [47]]. As shown in Fig. 6i and j, we observed increasing TAZ expression levels in TGF-β-stimulated MRC-5 and PLF cells, and SB431542 effectively reduced the effect of TGF-β on TAZ expression. Moreover, immunofluorescence showed that TGF-β stimulation resulted in increased nuclear localization of TAZ in MRC-5 cells and PLFs, which was attenuated by SB431542 (Fig. 6k and Supplementary Fig. 4i). This result was confirmed by Western blot analysis of cytoplasmic and nuclear fractions (Supplementary Fig. 4j). To validate the contribution of TEAD4 activation to TFRC expression, the transcriptional regulation of TFRC was investigated with a dual-luciferase reporter assay system. pGL3-TFRC (−3000/+250) was cotransfected with pCMV6-Entry-TEAD4 into 293T cells. As shown in Fig. 6l, transfection of the TEAD4 plasmid significantly increased the luciferase activity of the TFRC promoter region.
3.6. Regulation of the ferroptosis-defense pathway during fibroblast activation induced by iron overload
As shown in Fig. 7a and b, even low doses of FAC that induced fibroblast activation resulted in an increase in lipid peroxidation in fibroblasts, while DFO reduced the production of lipid peroxides. However, the accumulation of lipid ROS failed to trigger ferroptosis, indicating that fibroblasts might be protected from iron-induced oxidative stress by a certain ferroptosis-defense pathway (Fig. 7c). To investigate the molecular basis underlying the defense against ferroptosis, we first tested the expression of iron-related proteins in MRC-5 cells under long-term stimulation with TGF-β. Prolonged treatment of MRC-5 cells with TGF-β stimulated high levels of α-SMA, SLC40A1 and FTH, and the lipid peroxidation inhibitor Fer-1 attenuated the effect of TGF-β (Fig. 7d). The upregulation of SLC40A1 and FTH suggested increased iron export and cytosolic iron storage, thus reducing the risk of ferroptosis caused by iron accumulation.
Fig. 7.

Fibroblasts escape from ferroptotic cell death induced by iron accumulation through modulating ferroptosis-related genes. (a) Lipid ROS production in MRC-5 cells was assessed by flow cytometry using C11-BODIPY after 72 h of treatment with the indicated doses of FAC. Left, representative flow cytometry result for Lipid ROS. Right, scatter plot showing the results for independent samples examined. n = 3 biological replicates. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. ****P < 0.0001. (b) Lipid ROS production in MRC-5 cells was assessed by flow cytometry using C11-BODIPY after 72 h of treatment with FAC and DFO. Left, representative flow cytometry result for Lipid ROS. Right, scatter plot showing the results for independent samples examined. FAC: 50 μM. DFO: 50 μM n = 3 biological replicates. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. *P < 0.05, ****P < 0.0001. (c) Cell death of fibroblasts was determined by Hoechst33342 and SYTOX Green staining coupled with fluorescent microscope after 48–96 h of treatment with the indicated doses of FAC and DFO. DFO: 50 μM n = 9–12 images per group. (d) Expression of α-SMA, TFRC, FTH and SLC40A1 in MRC-5 was assessed by Western blot after 3 weeks of treatment with TGF-β and Fer-1. TGF-β: 10 ng/ml. Fer-1: 1 μM. (e) Right, the y-axis is the relative expression of genes, and the x-axis is the different group. The expression of 787 genes was up-regulated in the FAC group and down-regulated in FAC + DFO group. Statistical analysis was performed using the one-way ANOVA, P < 0.05. Left, KEGG enrichment analysis of the 787 genes ranked in the top 20. The numbers outside the brackets represent the number of genes in corresponding pathway in the figure, and the numbers in brackets represent –log10(p value). (f) KEGG pathway analysis of genes significantly upregulated in FAC group (Ctrl Vs. FAC) and significantly downregulated in FAC + DFO group (FAC Vs. FAC + DFO). The size of the bubble represents the number of differentially expressed genes, and the color of the bubble reflects the p-value. (g–h) Upper, GSEA analysis showed signal pathways enriched in FAC group. Ctrl Vs. FAC. Bottom, Heat map showed the expression of genes from corresponding GSEA analysis. (i–j) Upper, GSEA analysis showed signal pathways enriched in FAC group. FAC Vs. FAC + DFO. Bottom, Heat map showed the expression of some genes from corresponding GSEA analysis. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
To investigate the mechanism of ferroptosis defense in iron-accumulating fibroblasts, we performed RNA sequencing in parental and FAC-stimulated MRC-5 cells with or without DFO treatment (Supplementary Fig. 5a). Heatmaps comparing the overall differences in gene expression among the three groups indicated that FAC stimulation caused many changes in the transcriptome of MRC-5 cells, which were reversed by DFO (Supplementary Fig. 5a). Data analysis revealed that the expression of a total of 787 genes was upregulated in the FAC group and that their expression was recovered in the FAC + DFO group; these differences were significantly different among the three groups, as determined through one-way ANOVAs (Fig. 7e). Through a Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of these significantly different genes, we unexpectedly found that FAC stimulation significantly altered certain pathways related to lipid metabolism and cholesterol metabolism in fibroblasts, which ranked in the top 20 enriched pathways (Fig. 7e). Differentially upregulated gene expression in the FAC group and differentially downregulated gene expression in the FAC + DFO group were associated with lipid metabolism, fatty acid metabolism and unsaturated fatty acid synthesis (Fig. 7f). These differences offered us some ideas. The main characteristics of ferroptosis are iron overload and the accumulation of lipid peroxides, and lipid metabolism greatly affects the synthesis of lipid peroxides and the sensitivity of ferroptosis. Therefore, we inferred that, in general, excessive accumulation of lipid peroxides in MRC-5 cells might by inhibited through the regulation of certain pathways related to lipid metabolism when intracellular Fe2+ is moderately elevated; that is, a ferroptosis-defense pathway may be activated in these cells.
Subsequently, pairwise comparisons were performed with the control group and the FAC group and the FAC group and the FAC + DFO group, and the identified genes that might affect the accumulation of lipid peroxides and ferroptotic sensitivity are listed. For example, high expression of stearoyl-CoA desaturase (SCD) in the FAC group catalyzed the synthesis of monounsaturated fatty acids (MUFAs) in fibroblasts, which may protect cells from ferroptosis [48]. Fatty acid desaturase 2 (FADS2) is a key enzyme that regulates the biosynthesis of polyunsaturated Fatty acids (PUFAs). Some studies related to lung cancer have proven that knockdown of FADS2 can significantly reduce the expression of certain protective genes and increase the levels of lipid ROS in lung cancer cells, thus enhancing the sensitivity of cells to ferroptosis [49,50]. This outcome suggested that the upregulated expression of FADS2 in the FAC group might reduce the ferroptotic sensitivity of lung fibroblasts. In addition, acetyl-CoA acetyltransferase 2 (ACAT2), a member of the acyl-coenzyme A:cholesterol acyltransferase (ACAT) family, can catalyze the generation of cholesterol esters, and it has been confirmed that high expression of ACAT2 can promote cell proliferation [51] (Supplementary Figs. 5b and c). Furthermore, we detected the mRNA expression of several key genes in MRC-5 cells in vitro after FAC and DFO stimulation, which was consistent with the RNA sequencing results (Supplementary Fig. 5d). Moreover, a Gene Set Enrichment Analysis (GSEA) provided some relevant evidence. Compared with the control group, genes involved in glutathione metabolism and the PPAR signaling pathway were enriched in the FAC group. Acyl-CoA synthetase long-chain family member 3 (ACSL3), with upregulated expression in the FAC group, converted MUFAs into phospholipid monounsaturated fatty acids (PL-MUFAs), thereby protecting cells from ferroptosis (Fig. 7g and h). Evidence shows that overexpression of ACSL3 in melanoma cells may inhibit lipid oxidative stress through absorption of oleic acid, and high expression of ACSL3 has been associated with poor prognosis of patients with one of various tumors [[52], [53], [54]]. Compared with that in the FAC group, the expression of cell cycle and PPAR signaling pathway genes was decreased in the FAC + DFO group (Fig. 7i and j). On the one hand, this proves the effect of Fe2+ changes on cell proliferation; on the other hand, it demonstrates the effect of FAC on cell lipid metabolism.
3.7. Loss of TFRC in fibroblasts inhibits bleomycin-induced pulmonary fibrosis
Fibroblast-specific protein 1 (FSP1, also identified as S100A4) is considered as a marker of fibroblasts in different organs undergoing tissue remodeling and may serve as a sensitive and specific marker for lung fibroblasts [55]. As shown in Fig. 8a, multicolor immunofluorescence staining confirmed the colocalization of FSP1 and α-SMA in bleomycin-induced fibrotic lung tissue. To determine the role played by TFRC and TFRC-associated iron uptake in the activation of fibroblasts in pulmonary fibrosis progression, we generated mice that lack TFRC only in fibroblasts by crossing mice expressing a conditional TFRC-knockout allele (TFRCflox/flox) with mice that express Cre recombinase driven by the FSP1 promoter (FSP1-CreERT). The offspring (TFRCflox/flox, FSP1−CreERT) are referred to hereafter as TFRC−/−, and TFRC in FSP1+ cells were specifically knocked out after consecutive intraperitoneal injection of tamoxifen for 5 days. The bleomycin-induced pulmonary fibrosis model was then established with TFRC−/− mice and TFRCflox/flox mice (Fig. 8b). Western blot analysis of primary fibroblasts from TFRCflox/flox and TFRC−/− mice confirmed efficiency of conditional knockout (Fig. 8c). Comparing lung tissue 4, 7, 14, and 21 days after bleomycin administration, we found that the symptoms in TFRC−/− mice were alleviated, compared to those in the TFRCflox/flox mice, on Day 21 (Fig. 8d). H&E staining demonstrated that specific knockout of TFRC in fibroblasts alleviated the symptoms of bleomycin-induced pulmonary fibrosis, which was particularly significant in the later stage (Fig. 8e). In addition, hydroxyproline content in lung tissues decreased in TFRC−/− mice compared to TFRCflox/flox mice 21 days after bleomycin administration (Fig. 8f). Fourteen days after bleomycin administration, α-SMA, FSP1 and TFRC in the lung tissues of both groups were labeled by multicolor immunofluorescence. The expression of TFRC was significantly decreased in α-SMA+ and FSP1+ cells (Fig. 8g). Masson's staining showed that collagen deposition was relatively reduced in the lung tissues of the TFRC−/− mice (Fig. 8h and i). The expression of α-SMA and FSP1 was also decreased in the bleomycin-induced fibrotic lung tissues of the TFRC−/− mice, as indicated by IHC staining (Fig. 8h and i). These results demonstrate that TFRC expression in fibroblasts effectively promotes the activation of fibroblasts and the occurrence of pulmonary fibrosis.
Fig. 8.

Loss of TFRC in fibroblasts alleviates pulmonary fibrosis in a bleomycin model. (a) Representative images of multicolor immunofluorescence for DAPI (blue), α-SMA (green), FSP1 (orange) in lung tissue of mice on Day 14 after bleomycin administration. Scale bars, 50 μm. (b) A pattern diagram showing the progression of conditional knockout of TFRC in FSP1+ fibroblast as well as bleomycin-induced pulmonary fibrosis. (c) Expression of TFRC in primary fibroblasts from TFRCflox/flox and TFRC−/− mice was assessed by Western blot after 5 days of tamoxifen intraperitoneal injection and 7 days of waiting. (d) Images of lungs from TFRC−/− mice and TFRCflox/flox mice at 4, 7, 14, and 21 days after bleomycin administration. Scale bars, 1 cm. (e) H&E staining of lung tissue from TFRC−/− mice and TFRCflox/flox mice at 4, 7, 14, and 21 days after bleomycin administration. Scale bars, 1 mm. (f) Hydroxyproline content from TFRC−/− mice and TFRCflox/flox mice at 4, 7, 14, and 21 days after bleomycin administration. n = 3 mice per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. *P < 0.05, ***P < 0.001. (g) Left, representative images of multicolor immunofluorescence for DAPI (blue), α-SMA (green), FSP1 (orange) and TFRC (red) in lung tissue of TFRC−/− mice and TFRCflox/flox mice on Day 14 after bleomycin administration. Scale bars, 10 μm. Right, scatter plot showing the MFI of TFRC and α-SMA in FSP1+ cells and the MFI of TFRC in α-SMA+ cells in lung tissue of mice on Day 14 after bleomycin administration. Each dot represents a microscopic image, n = 18 images per group from 3 mice. Statistical analysis was performed using the Student's t-test. ****P < 0.0001. (h) Representative images of Masson's staining and IHC staining for FSP1 and α-SMA of lung tissue from TFRC−/− mice and TFRCflox/flox mice at 4, 7, 14, and 21 days after bleomycin administration. Scale bars of Masson's staining, 200 μm. Scale bars of IHC staining, 100 μm. (i) Scatter plot showing the percentage of the positive area of Masson's staining and IHC staining in lung tissue from TFRC−/− mice and TFRCflox/flox mice at 4, 7, 14, and 21 days after bleomycin administration. n = 3 mice per group. Statistical analysis was performed using the Student's t-test. *P < 0.05, **P < 0.01. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
4. Discussion
The mechanisms involved in pulmonary fibrosis are not completely understood, and the effects of drugs on pulmonary fibrosis are unsatisfactory. As a novel type of programmed cell death driven by iron-dependent lipid peroxidation, ferroptosis is implicated in various diseases related to organ damage, including ischemia/reperfusion damage, drug-induced kidney injury, rhabdomyolysis-associated renal damage, and liver damage in chronic liver diseases [[56], [57], [58]]. Iron overload induces significant increases in lipid peroxide accumulation and ferroptosis in mouse primary hepatocytes, contributing to ferroptosis-related liver damage and fibrosis in mouse liver fibrosis models established either by high dietary iron or carbon tetrachloride (CCl4) injections [[59], [60], [61]]. In this study, we found that both bleomycin and LPS induced iron overload and ferroptosis in lung epithelial cells, contributing to epithelial damage in the early inflammatory phase of pulmonary fibrosis. Interestingly, the population of fibroblasts was increased even under iron-induced oxidative stress. This phenomenon was probably due to the following reasons. The half-life of bleomycin ranges from a few hours to 21 h in vivo [62,63]. It triggers epithelial injury, which is a key event in the onset of pulmonary fibrosis. AECs occupy more than 95% of the alveolar surface area in normal lung tissues and are most likely to come into direct contact with lethal external substances [64,65]. Notably, in the early stage of bleomycin-induced pulmonary fibrosis, AEC death was not limited to ferroptosis, as demonstrated in this study, and apoptosis and necroptosis of AECs have been confirmed by other studies [32,36,66]. In previous studies, LPS is often used to induce acute lung injury models, but it has been reported that LPS could also induce symptoms of pulmonary fibrosis, which may last shorter than bleomycin-induced pulmonary fibrosis. In our study, LPS was just selected as a supplement to the bleomycin-induced pulmonary fibrosis model in order to compare the protective effects of Liproxstatin-1 in different models of pulmonary fibrosis. Our results also confirmed that LPS-induced pulmonary fibrosis models supported our conclusions.
During the following fibrotic phase in bleomycin-induced pulmonary fibrosis, the level of bleomycin remaining in lungs may not be high enough to trigger the death of activated fibroblasts. We also found that both the production of 4-HNE and the accumulation of ferrous iron decreased in the lungs of mice on Day 14 compared with that in the lungs on Day 7 after bleomycin administration. In other words, although fibroblasts might show accumulation of ferrous iron in this fibrotic phase, the iron concentration was more moderate than that in the early inflammatory phase. Notably, the increase in ferrous iron induced by low-dose FAC caused the production of lipid ROS without triggering ferroptosis. Mechanistically, our study revealed that activation of ferroptosis-defensive and iron exporting pathways protected fibroblasts from ferroptotic stress induced by moderate iron accumulation. Intracellular PUFAs are transformed into phospholipid polyunsaturated fatty acids (PL-PUFAs) by a series of catalytic processes and are eventually oxidized to lipid ROS or PL-PUFA-OOH, which ultimately leads to ferroptosis. In contrast, PL-MUFAs derived from MUFAs contribute to cell resistance to ferroptosis. Undoubtedly, ferroptosis is greatly affected by cellular lipid metabolism [54]. From RNA-sequencing data, we observed that the expression of certain genes related to fatty acid biosynthesis and lipid metabolism pathways, such as SCD, FADS2, SQLE, APOE, ACACA, and FABP4, was altered after iron level elevation. A series of studies proved that the upregulated expression of these genes promoted the survival, migration and invasion of cancer cells and was associated with poor prognosis in people of tumors [[48], [49], [50],[67], [68], [69], [70], [71]]. These upregulated genes inhibited the ferroptotic sensitivity of cells by influencing lipid metabolism and lipid peroxidation. In previous bioinformatics studies, researchers compared the gene signature in bronchoalveolar lavage fluid (BALF) or lung tissue between IPF patients and normal controls and found that certain differentially expressed ferroptosis-related genes were highly expressed in the IPF patients, which was associated with poor prognosis. Among these genes in our study, some were upregulated in FAC-stimulated fibroblasts, including MYC, AIFM2, PRKCA, RGS4 and ZFP36. This result suggests that the expression of these genes may be upregulated when fibroblasts are activated by Fe2+ in vivo, regulating ferroptotic risk and promoting pulmonary fibrosis [[72], [73], [74]]. MRC-5 cells exhibited upregulated expression of SLC40A1, which transports iron out of the cell, and FTH, which stores intracellular free iron, under long-term stimulation with TGF-β. The increase of these genes regulating iron homeostasis may protect fibroblasts against lipid oxidative stress and ferroptotic cell death by decreasing Fe2+ levels. Chen et al. [72] analyzed and compared the ferroptosis-related gene signature in BALF between IPF patients and normal controls and thus identified 19 differentially expressed genes associated with poor prognosis. Among these differentially expressed genes, we found that SLC40A1 was highly expressed in the IPF patient group, which is consistent with the results of the in vitro experiments. This finding suggests that SLC40A1 may relieve the pressure of lipid peroxidation and the risk of ferroptosis by transporting iron out of the fibroblasts during the fibrotic stage in IPF patients.
Numerous studies have confirmed that the transition of fibroblasts to myofibroblasts is a key step for the excessive accumulation of ECM in fibrotic pathological diseases [[75], [76], [77]]. As mentioned above, Fer-1 inhibited the activating effect during the long-term stimulation of MRC-5 cells by TGF-β, suggesting that lipid peroxidation might positively regulate the activation of fibroblasts. Correspondingly, Lip-1 also alleviated pulmonary fibrosis when intraperitoneal injection was performed from the 10th day after bleomycin administration, which may have been due to the inhibition of fibroblast activation by Lip-1 not the ferroptosis of the epithelial cells in vivo. In fact, lipid peroxidation has been confirmed to promote the activation and transformation of in other fibroblast cell lines into myofibroblasts, which is consistent with our results [78]. Recently, COVID-19-induced acute respiratory distress syndrome was associated with clinical, radiographic, histopathological, and ultrastructural hallmarks of pulmonary fibrosis. The accumulation of monocyte-derived macrophages with enrichment of fibrosis-associated gene signatures, such as TGF-β, in IPF was significantly similar to the macrophage populations found in severe COVID-19. The lungs of patients with severe COVID-19 also had a pronounced expansion and activation of myofibroblasts and fibroblasts, similar to that of the pulmonary fibrosis models [79,80]. Therefore, characterizing the molecular mechanisms involved in fibroblast activation may further fuel the development of potential therapies to reverse phenotype transition in fibrotic tissue and control the progression of fibrotic diseases. A series of in vitro and in vivo studies have shown that TGF-β is a critical cytokine for the accumulation of ECM and the occurrence of fibrosis. External stimulation triggered damage to AECs and subsequent inflammatory responses, resulting in the secretion of profibrotic factors represented by TGF-β, which not only recruits and activates fibroblasts to transform into myofibroblasts but also promotes the epithelial-mesenchymal transition (EMT) [81,82]. Our findings revealed a novel mechanism that was involved in TGF-β-regulated fibroblast activation. TGF-β enhanced intracellular labile ferrous accumulation by upregulating TFRC via TAZ-TEAD signaling and promoted the fibroblast-to-myofibroblast transition in the fibrotic phase. Several previous studies have reported fibrosis-enhancing effects of iron. For example, Gao et al. [83] revealed that the iron overload of hepatic stellate cells (HSCs) was able to activate HSCs and drive liver lipogenesis and fibrosis. And iron elevated collagen gene expression and increased the proliferation of rat HSCs [84,85] and promoted cirrhosis in mice [86]. Similarly, studies with murine HSCs showed transferrin-induced elevations in α-SMA, vimentin and collagen secretion [87]. Considering the positive effect of moderate iron accumulation on promoting lung fibroblast activation in our study, we presume that fibroblasts take advantage of iron and do not suffer from the oxidative stress induced by iron. Some preclinical studies have proven that inhibitors targeting TGF-β (such as P17 and Del-1) can effectively attenuate lung fibrosis in bleomycin-treated mice [88,89]. Considering the therapeutic potential of the iron chelator revealed in our study, it is possible that the combination of targeting both TGF-β and iron overload might exert a synergistic effect in lung fibrosis.
Earlier studies revealed that YAP/TAZ, as the terminal effectors of the Hippo pathway, are mainly shuttled between the nucleus and cytoplasm and interact with the TEAD family in the nucleus to promote cell proliferation and regulate gene expression [45,90,91]. Previous studies have confirmed that the YAP-TEAD complex can target the expression of ferroptosis-related genes, including TFRC. Specifically, the transcription factor TEAD4 bound to the promoter regions of the TFRC gene, and YAP overexpression significantly enhanced the transcriptional activation function of TEAD4 [47]. Our results also indicated that TGF-β boosted TFRC mRNA expression in fibroblasts. In different fibrosis models and fibrotic samples from patients, the increase in the expression and nuclear localization of YAP/TAZ has been verified, and the activation of YAP/TAZ has been shown to be expectedly regulated by TGF-β, which plays an essential role in the TGF-β-induced transformation of fibroblasts into myofibroblasts and fibrosis [46,92]. Therefore, we explored possible relationships among TGF-β, TAZ-TEAD4 and TFRC in fibroblasts. It was first found that TGF-β increased the intracellular Fe2+ level by upregulating TFRC expression via the TAZ-TEAD4 pathway and thus induced the activation of fibroblasts. This point was demonstrated by experiments in vivo and in vitro.
Understanding the course and progression of fibrosis can provide more ideas for the treatment of IPF. In this study, we demonstrated that ferroptosis is a promising therapeutic target for the prevention of pulmonary fibrosis. Liproxstatin-1 treatment effectively rescued bleomycin- and LPS-induced ferroptosis and the subsequent fibrotic course in lungs by inhibiting the accumulation of lipid peroxides. In addition, DFO inhibited the activation of fibroblasts and reduced collagen deposition in bleomycin-induced pulmonary fibrosis. This therapeutic effect was not only due to the prevention of a high ferrous iron accumulation in the early inflammatory phase but also due to the modulation of moderate iron accumulation in the fibrotic phase. Therefore, it is necessary to take ferroptosis and iron homeostasis into account when developing a therapeutic strategy of pulmonary fibrosis.
Taken together, our findings provide new insight into the type of cell death relevant to the pathogenesis of IPF. Our data suggest that ferroptosis and iron homeostasis play essential roles in the onset and progression of bleomycin- and LPS-induced pulmonary fibrosis. From a mechanistic perspective, bleomycin and LPS directly induce excessive iron overload and ferroptosis in lung epithelial cells in the early inflammatory phase. Moderate intracellular labile ferrous accumulation regulated by TFRC via the TGF-β-TAZ-TEAD signaling pathway promotes fibroblast-to-myofibroblast transition in the late fibrotic phase. These findings provide compelling evidence that therapeutic strategies for targeting ferroptosis and/or iron homeostasis disorders can be developed to treat iron overload-associated diseases, including pulmonary damage and fibrosis.
Ethics statement
The animal experiment had been reviewed and approved by the Laboratory Animal Welfare and Ethics Committee of the Air Force Medical University (approval certificate IACUC-20211102).
Author contributions
PZ, ZNC and JW conceived and designed the study. ZP, YFQ, XHF and FFY conducted the experiments. JW and ZP performed writing, review and revision of the paper. FH, PL and GZ analyzed the data. XL, SJW, HYC and JNY bred transgenic mice. PZ, ZNC, JW and ZP supervised the study. All authors reviewed and edited the manuscript.
Declaration of competing interest
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Natural Science Foundation of China 82022059 (to J.W.) and the Natural Science Basis Research Plan in Shaanxi Province of China 2019ZY-CXPT-03-01 (to P.Z.). This work was also supported by the Youth Science and Technology Nova Program of Shaanxi Province 2020KJXX-055 (to J.W.), National Key Research and Development Program of China 2019YFC1316303 (to S.W.) and National Natural Science Foundation of China 82173244 (to H.C.).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102509.
Contributor Information
Jiao Wu, Email: jiaowubio@hotmail.com.
Zhi-Nan Chen, Email: znchen@fmmu.edu.cn.
Ping Zhu, Email: zhuping@fmmu.edu.cn.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Lipid peroxidation and iron accumulation in the lungs of bleomycin-induced pulmonary fibrosis mice. (a) Immunoblotting analysis of 4-HNE in lung tissues of mice on Day 21 after bleomycin administration. Left, representative western blot result for 4-HNE. Right, scatter plot showing the results for all samples examined. Each lane represents one mouse, n = 3 mice per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. *P < 0.05. (b) The gating strategy for detecting intracellular Fe2+ levels in primary lung cells by flow cytometry using FeRhoNox-1. Upper, control group. Bottom, bleomycin group on Day 7 after bleomycin administration. (c) The gating strategy for detecting intracellular Fe2+ levels in primary lung cells by flow cytometry using FeRhoNox-1. Upper, control group. Bottom, bleomycin group on Day 14 after bleomycin administration. (d) Cell death of BEAS-2B cells was determined by Hochest33342 and SYTOX Green staining coupled with fluorescent microscope after 84 h of treatment with bleomycin, Fer-1 and Necrostatin-1 (Necro-1). BLM: 10 μg/ml. Fer-1: 5 μM. Necro-1: 50 μM n = 10 images per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. ****P < 0.0001. (e) Cell death of BEAS-2B cells was determined by Hochest33342 and SYTOX Green staining coupled with fluorescent microscope after 72 h of treatment with LPS and Fer-1. LPS: 5 μg/ml. Fer-1: 5 μM. Necro-1: 50 μM n = 9–10 images per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. ****P < 0.0001.
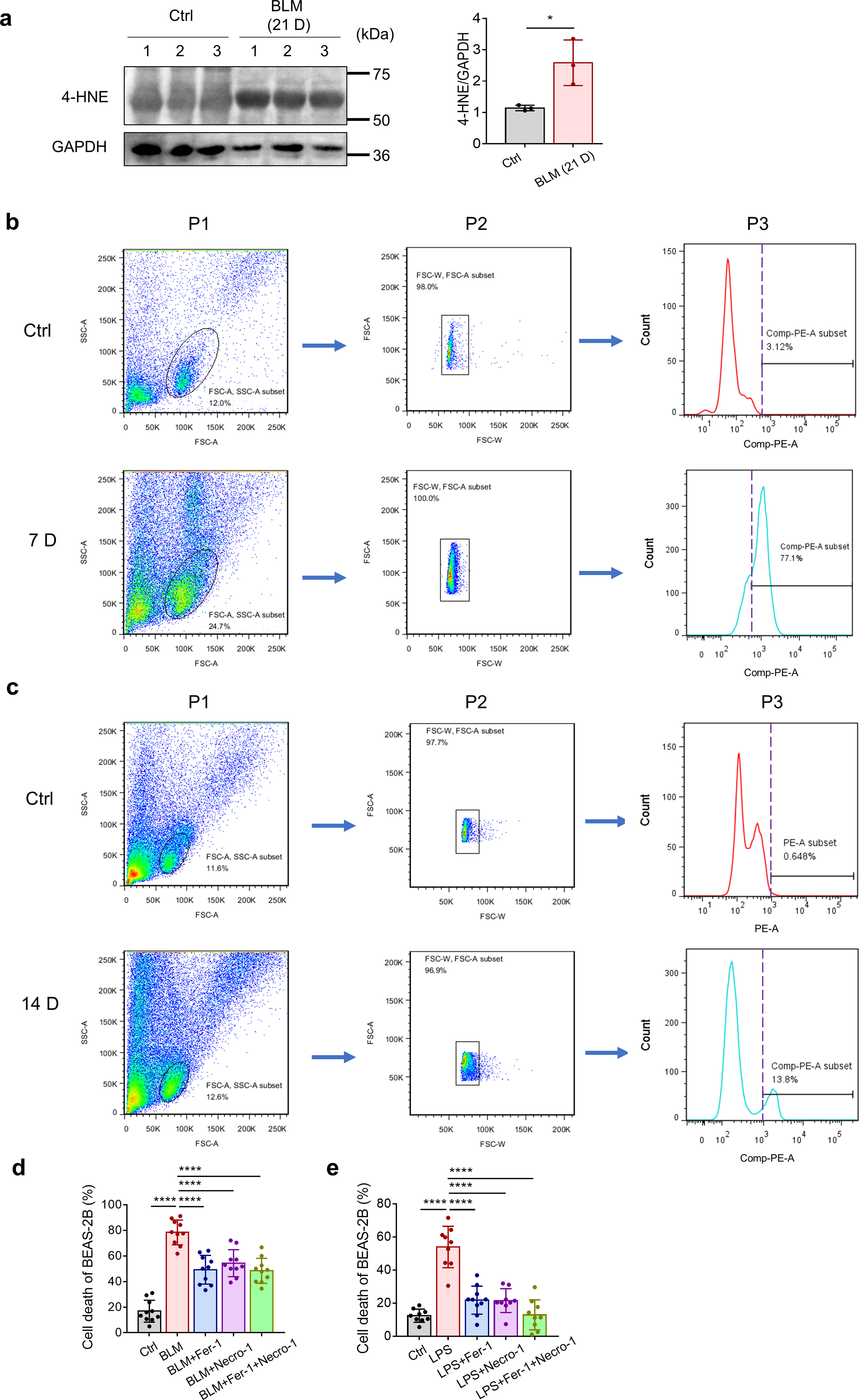{kind=link}
DFO inhibits bleomycin-induced pulmonary fibrosis. (a) Representative images of Masson's staining and IHC staining for α-SMA, FAPα and TGF-β of mice lung tissue from control, bleomycin group and bleomycin + DFO group on Day 14 after bleomycin administration. Scale bars, 100 μm. (b) Scatter plot showing the percentage of the positive area of Masson's staining and IHC staining in lung tissue from control group, bleomycin group and bleomycin + DFO group on Day 14 after bleomycin administration. n = 4–5 mice per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. *P < 0.05, ****P < 0.0001. (c) Iron levels in lung tissues of mice was determined by tissue Iron content detection Kit on Day 14 after bleomycin administration. n = 4 mice per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. **P < 0.01.
{kind=link}
Lip-1 inhibits LPS-induced lung injury and pulmonary fibrosis. (a) Upper left, The lung of mouse from LPS group and LPS + Lip-1 group on Day 7 after LPS administration. The other, H&E staining scanning of lung tissue including control group, LPS group and LPS + Lip-1 group at different days. Scale bars, 1 mm. (b) Representative images of Masson's staining of mice lung tissue from control group, LPS group and LPS + Lip-1 group at different days. Scale bars, 100 μm. (c-e) Representative images of IHC staining for α-SMA, FAPα and TGF-β of mice lung tissue from control group, LPS group and LPS + Lip-1 group at different days. Scale bars, 100 μm. (f) Scatter plot showing the percentage of the positive area of Masson's staining and IHC staining in lung tissue from control group, LPS group and LPS + Lip-1 group at different days. n = 4 mice per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
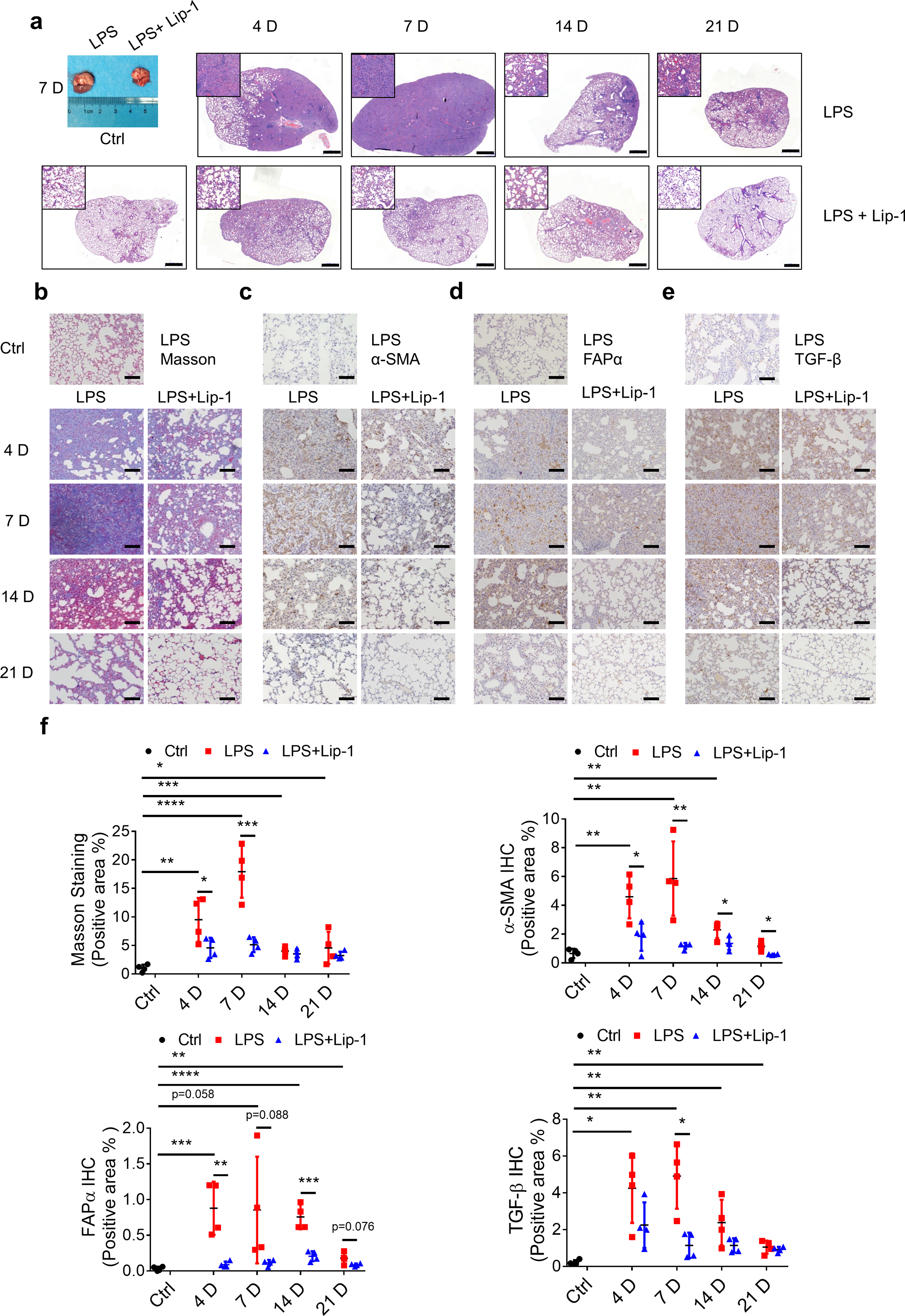{kind=link}
TGF-β promotes the activation of fibroblasts. (a-c) Expression of α-SMA and COL1A1 in MRC-5 (a), IMR90 (b) and PLFs (c) were assessed by qPCR after 24–48 h of treatment with the indicated doses of TGF-β. a: n = 3 biological replicates. b: n = 3 biological replicates. c: n = 4 biological replicates. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. *P < 0.05, ***P < 0.001, ****P < 0.0001. (d) Expression of α-SMA and COL1A1 in MRC-5, IMR90 and PLFs was assessed by western blot after 72 h of treatment with the indicated doses of TGF-β. (e) Representative microscopic images and the quantitation of MRC-5 cells that transferred through the transwell in the migration assay and invasion assays. Scale bars, 200 μm. TGF-β: 10 ng/ml n = 6 visual fields. Statistical analysis was performed using the Student's t-test. Scale bars, 200 μm **P < 0.01, ****P < 0.0001. (f) Expression of TFRC in MRC-5 after TFRC knockdown with siRNA were assessed by qPCR after 48 h of treatment. n = 4 biological replicates. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. ****P < 0.0001. (g) Expression of TFRC in MRC-5 after TFRC knockdown with siRNA was assessed by western blot after 72 h of treatment. (h) MFI of TFRC-FITC in MRC-5 after TFRC knockdown with siRNA was assessed by flow cytometry after 72 h of treatment. Scatter plot showing the results for independent samples examined. n = 3 biological replicates. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. **P < 0.01, ***P < 0.001, ****P < 0.0001. (i) Left, the expression of TAZ in nucleus was quantified by ImageJ in MRC-5 after 48 h of treatment with TGF-β and SB. TGF-β: 10 ng/ml. SB: 1 μM n = 34–37 cells from 5 images. Right, the expression of TAZ in nucleus was quantified by ImageJ in PLFs after 48 h of treatment with TGF-β and SB. TGF-β: 10 ng/ml. SB: 1 μM n = 24–32 cells from 3 images. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. **P < 0.01, ****P < 0.0001. (j) Expression of TAZ in cytoplasm and in nucleus in MRC-5 was assessed by western blot after 72 h of treatment with TGF-β and SB. TGF-β: 10 ng/ml. SB: 1 μM. Lamin B and α-tubulin were used respectively as nuclear and cytoplasmic markers.
{kind=link}
Fe2+ modulates ferroptosis-related genes in fibroblasts. (a) Heat map showed the RNA-sequence results of MRC-5 cells stimulated by FAC (50 μM) for 72 h. The red stripes represent highly expressed genes and the blue stripes represent low-expressed genes. n = 3 biological replicates. (b) Heat map showed the relative expression of some genes between control group and FAC group. The red stripes represent significantly upregulated genes and the blue stripes represent significantly downregulated genes. n = 3 biological replicates. Statistical analysis was performed using the one-way ANOVA, P < 0.05. (c) Heat map showed the relative expression of some genes between FAC group and FAC + DFO group. The red stripes represent significantly upregulated genes and the blue stripes represent significantly downregulated genes. n = 3 biological replicates. Statistical analysis was performed using the one-way ANOVA, P < 0.05. (d) Expression of some lipid metabolism and ferroptotic protective genes in MRC-5 cells was assessed by qPCR after 72 h of treatment with FAC and DFO. FAC: 50 μM. DFO: 50 μM n = 3 biological replicates. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
{kind=link}
References
- 1.Richeldi L., Collard H.R., Jones M.G. Idiopathic pulmonary fibrosis. Lancet. 2017;389(10082):1941–1952. doi: 10.1016/S0140-6736(17)30866-8. [DOI] [PubMed] [Google Scholar]
- 2.Steele M.P., Schwartz D.A. Molecular mechanisms in progressive idiopathic pulmonary fibrosis. Annu. Rev. Med. 2013;64:265–276. doi: 10.1146/annurev-med-042711-142004. [DOI] [PubMed] [Google Scholar]
- 3.Sgalla G., Biffi A., Richeldi L. Idiopathic pulmonary fibrosis: diagnosis, epidemiology and natural history. Respirology. 2016;21(3):427–437. doi: 10.1111/resp.12683. [DOI] [PubMed] [Google Scholar]
- 4.Sisson T.H., et al. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010;181(3):254–263. doi: 10.1164/rccm.200810-1615OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liang J., et al. Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nat. Med. 2016;22(11):1285–1293. doi: 10.1038/nm.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Korfei M., et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008;178(8):838–846. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barkauskas C.E., Noble P.W. Cellular mechanisms of tissue fibrosis. 7. New insights into the cellular mechanisms of pulmonary fibrosis. Am. J. Physiol. Cell Physiol. 2014;306(11):C987–C996. doi: 10.1152/ajpcell.00321.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kolb P., et al. The importance of interventional timing in the bleomycin model of pulmonary fibrosis. Eur. Respir. J. 2020;55(6) doi: 10.1183/13993003.01105-2019. [DOI] [PubMed] [Google Scholar]
- 9.Phan T.H.G., et al. Emerging cellular and molecular determinants of idiopathic pulmonary fibrosis. Cell. Mol. Life Sci. 2021;78(5):2031–2057. doi: 10.1007/s00018-020-03693-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanjore H., et al. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am. J. Respir. Crit. Care Med. 2009;180(7):657–665. doi: 10.1164/rccm.200903-0322OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hashimoto N., et al. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2010;43(2):161–172. doi: 10.1165/rcmb.2009-0031OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.King T.E., Jr., et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014;370(22):2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 13.Flaherty K.R., et al. Nintedanib in progressive fibrosing interstitial lung diseases. N. Engl. J. Med. 2019;381(18):1718–1727. doi: 10.1056/NEJMoa1908681. [DOI] [PubMed] [Google Scholar]
- 14.Gerckens M., et al. Phenotypic drug screening in a human fibrosis model identified a novel class of antifibrotic therapeutics. Sci. Adv. 2021;7(52):eabb3673. doi: 10.1126/sciadv.abb3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kerins M.J., Ooi A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxidants Redox Signal. 2018;29(17):1756–1773. doi: 10.1089/ars.2017.7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang L., et al. RNF217 regulates iron homeostasis through its E3 ubiquitin ligase activity by modulating ferroportin degradation. Blood. 2021;138(8):689–705. doi: 10.1182/blood.2020008986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bogdan A.R., et al. Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem. Sci. 2016;41(3):274–286. doi: 10.1016/j.tibs.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pietrangelo A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology. 2010;139(2):393–408. doi: 10.1053/j.gastro.2010.06.013. 408 e1-2. [DOI] [PubMed] [Google Scholar]
- 19.Menon A.V., et al. Excess heme upregulates heme oxygenase 1 and promotes cardiac ferroptosis in mice with sickle cell disease. Blood. 2022;139(6):p. 936–941.. doi: 10.1182/blood.2020008455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ali M.K., et al. Critical role for iron accumulation in the pathogenesis of fibrotic lung disease. J. Pathol. 2020;251(1):49–62. doi: 10.1002/path.5401. [DOI] [PubMed] [Google Scholar]
- 21.Ali M.K., et al. Role of iron in the pathogenesis of respiratory disease. Int. J. Biochem. Cell Biol. 2017;88:181–195. doi: 10.1016/j.biocel.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 22.Bargagli E., et al. Metabolic dysregulation in idiopathic pulmonary fibrosis. Int. J. Mol. Sci. 2020;21(16) doi: 10.3390/ijms21165663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bayir H., et al. Achieving life through death: redox biology of lipid peroxidation in ferroptosis. Cell Chem Biol. 2020;27(4):387–408. doi: 10.1016/j.chembiol.2020.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen X., et al. Iron metabolism in ferroptosis. Front. Cell Dev. Biol. 2020;8 doi: 10.3389/fcell.2020.590226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anandhan A., et al. Breakdown of an ironclad defense system: the critical role of NRF2 in mediating ferroptosis. Cell Chem Biol. 2020;27(4):436–447. doi: 10.1016/j.chembiol.2020.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Torti S.V., Torti F.M. Iron: the cancer connection. Mol. Aspect. Med. 2020 doi: 10.1016/j.mam.2020.100860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen X., et al. Broadening horizons: the role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021;18(5):280–296. doi: 10.1038/s41571-020-00462-0. [DOI] [PubMed] [Google Scholar]
- 28.Liu T., De Los Santos F.G., Phan S.H. The bleomycin model of pulmonary fibrosis. Methods Mol. Biol. 2017;1627:27–42. doi: 10.1007/978-1-4939-7113-8_2. [DOI] [PubMed] [Google Scholar]
- 29.Rangarajan S., et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat. Med. 2018;24(8):1121–1127. doi: 10.1038/s41591-018-0087-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhong H., Yin H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: focusing on mitochondria. Redox Biol. 2015;4:193–199. doi: 10.1016/j.redox.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Y., et al. Quantitative profiling of protein carbonylations in ferroptosis by an aniline-derived probe. J. Am. Chem. Soc. 2018;140(13):4712–4720. doi: 10.1021/jacs.8b01462. [DOI] [PubMed] [Google Scholar]
- 32.Parimon T., et al. Alveolar epithelial type II cells as drivers of lung fibrosis in idiopathic pulmonary fibrosis. Int. J. Mol. Sci. 2020;21(7) doi: 10.3390/ijms21072269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yao C., et al. Senescence of alveolar type 2 cells drives progressive pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2021;203(6):707–717. doi: 10.1164/rccm.202004-1274OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen H., et al. TSSK4 upregulation in alveolar epithelial type-II cells facilitates pulmonary fibrosis through HSP90-AKT signaling restriction and AT-II apoptosis. Cell Death Dis. 2021;12(10):938. doi: 10.1038/s41419-021-04232-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ptasinski V.A., et al. Targeting alveolar repair in idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2021;65(4):347–365. doi: 10.1165/rcmb.2020-0476TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X., et al. Toll interacting protein protects bronchial epithelial cells from bleomycin-induced apoptosis. Faseb. J. 2020;34(8):9884–9898. doi: 10.1096/fj.201902636RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen D., et al. Sodium propionate attenuates the lipopolysaccharide-induced epithelial-mesenchymal transition via the PI3K/Akt/mTOR signaling pathway. J. Agric. Food Chem. 2020;68(24):6554–6563. doi: 10.1021/acs.jafc.0c01302. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y.Y., et al. Telomere shortening activates TGF-beta/Smads signaling in lungs and enhances both lipopolysaccharide and bleomycin-induced pulmonary fibrosis. Acta Pharmacol. Sin. 2018;39(11):1735–1745. doi: 10.1038/s41401-018-0007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wollin L., et al. Potential of nintedanib in treatment of progressive fibrosing interstitial lung diseases. Eur. Respir. J. 2019;54(3) doi: 10.1183/13993003.00161-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang J., et al. Hyperoside attenuates bleomycin-induced pulmonary fibrosis development in mice. Front. Pharmacol. 2020;11 doi: 10.3389/fphar.2020.550955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moss B.J., Ryter S.W., Rosas I.O. Pathogenic mechanisms underlying idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2022;17:p. 515–546. doi: 10.1146/annurev-pathol-042320-030240. [DOI] [PubMed] [Google Scholar]
- 42.Hu H.H., et al. New insights into TGF-beta/Smad signaling in tissue fibrosis. Chem. Biol. Interact. 2018;292:76–83. doi: 10.1016/j.cbi.2018.07.008. [DOI] [PubMed] [Google Scholar]
- 43.Meng X.M., Nikolic-Paterson D.J., Lan H.Y. TGF-beta: the master regulator of fibrosis. Nat. Rev. Nephrol. 2016;12(6):325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 44.Brown R.A.M., et al. Altered iron metabolism and impact in cancer biology, metastasis, and immunology. Front. Oncol. 2020;10:476. doi: 10.3389/fonc.2020.00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang H., et al. Correction: TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J. Biol. Chem. 2019;294(15):5808. doi: 10.1074/jbc.AAC119.008436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Piersma B., Bank R.A., Boersema M. Signaling in fibrosis: TGF-beta, WNT, and YAP/TAZ converge. Front. Med. 2015;2:59. doi: 10.3389/fmed.2015.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu J., et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature. 2019;572(7769):402–406. doi: 10.1038/s41586-019-1426-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kikuchi K., Tsukamoto H. Stearoyl-CoA desaturase and tumorigenesis. Chem. Biol. Interact. 2020;316 doi: 10.1016/j.cbi.2019.108917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Y.L., et al. Multifaceted regulation and functions of fatty acid desaturase 2 in human cancers. Am J Cancer Res. 2020;10(12):4098–4111. [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang Y., et al. EGLN1/c-Myc induced lymphoid-specific helicase inhibits ferroptosis through lipid metabolic gene expression changes. Theranostics. 2017;7(13):3293–3305. doi: 10.7150/thno.19988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weng M., et al. ACAT2 promotes cell proliferation and associates with malignant progression in colorectal cancer. OncoTargets Ther. 2020;13:3477–3488. doi: 10.2147/OTT.S238973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quan J., Bode A.M., Luo X. ACSL family: the regulatory mechanisms and therapeutic implications in cancer. Eur. J. Pharmacol. 2021;909 doi: 10.1016/j.ejphar.2021.174397. [DOI] [PubMed] [Google Scholar]
- 53.Ubellacker J.M., et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature. 2020;585(7823):113–118. doi: 10.1038/s41586-020-2623-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hadian K., Stockwell B.R. SnapShot: ferroptosis. Cell. 2020;181(5):1188–1188 e1. doi: 10.1016/j.cell.2020.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lawson W.E., et al. Characterization of fibroblast-specific protein 1 in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2005;171(8):899–907. doi: 10.1164/rccm.200311-1535OC. [DOI] [PubMed] [Google Scholar]
- 56.Friedmann Angeli J.P., et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014;16(12):1180–1191. doi: 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gao M., et al. Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell. 2015;59(2):298–308. doi: 10.1016/j.molcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Capelletti M.M., et al. Ferroptosis in liver diseases: an overview. Int. J. Mol. Sci. 2020;21(14) doi: 10.3390/ijms21144908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang H., et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology. 2017;66(2):449–465. doi: 10.1002/hep.29117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu Y., et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood. 2020;136(6):726–739. doi: 10.1182/blood.2019002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang L., et al. Auranofin mitigates systemic iron overload and induces ferroptosis via distinct mechanisms. Signal Transduct. Targeted Ther. 2020;5(1):138. doi: 10.1038/s41392-020-00253-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Della Latta V., et al. Bleomycin in the setting of lung fibrosis induction: from biological mechanisms to counteractions. Pharmacol. Res. 2015;97:122–130. doi: 10.1016/j.phrs.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 63.Miura Y., et al. Bimodal fibrosis in a novel mouse model of bleomycin-induced usual interstitial pneumonia. Life Sci Alliance. 2022;5(1) doi: 10.26508/lsa.202101059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Katzen J., Beers M.F. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J. Clin. Invest. 2020;130(10):5088–5099. doi: 10.1172/JCI139519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Y., et al. Pulmonary alveolar type I cell population consists of two distinct subtypes that differ in cell fate. Proc. Natl. Acad. Sci. U. S. A. 2018;115(10):2407–2412. doi: 10.1073/pnas.1719474115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Minagawa S., et al. Regulated necrosis in pulmonary disease. A focus on necroptosis and ferroptosis. Am. J. Respir. Cell Mol. Biol. 2020;62(5):554–562. doi: 10.1165/rcmb.2019-0337TR. [DOI] [PubMed] [Google Scholar]
- 67.Xu H., et al. Cholesterol metabolism: new functions and therapeutic approaches in cancer. Biochim. Biophys. Acta Rev. Canc. 2020;1874(1) doi: 10.1016/j.bbcan.2020.188394. [DOI] [PubMed] [Google Scholar]
- 68.Tang W., et al. Ferroptosis regulators, especially SQLE, play an important role in prognosis, progression and immune environment of breast cancer. BMC Cancer. 2021;21(1):1160. doi: 10.1186/s12885-021-08892-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin R., et al. Identification of ferroptosis genes in immune infiltration and prognosis in thyroid papillary carcinoma using network analysis. BMC Genom. 2021;22(1):576. doi: 10.1186/s12864-021-07895-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liang Y., et al. A novel survival model based on a Ferroptosis-related gene signature for predicting overall survival in bladder cancer. BMC Cancer. 2021;21(1):943. doi: 10.1186/s12885-021-08687-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Luis G., et al. Tumor resistance to ferroptosis driven by Stearoyl-CoA Desaturase-1 (SCD1) in cancer cells and Fatty Acid Biding Protein-4 (FABP4) in tumor microenvironment promote tumor recurrence. Redox Biol. 2021;43 doi: 10.1016/j.redox.2021.102006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.He Y., et al. An 8-ferroptosis-related genes signature from Bronchoalveolar Lavage Fluid for prognosis in patients with idiopathic pulmonary fibrosis. BMC Pulm. Med. 2022;22(1):15. doi: 10.1186/s12890-021-01799-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.He J., Li X., Yu M. Bioinformatics analysis identifies potential ferroptosis key genes in the pathogenesis of pulmonary fibrosis. Front. Genet. 2021;12 doi: 10.3389/fgene.2021.788417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li M., et al. Ferroptosis-related genes in bronchoalveolar lavage fluid serves as prognostic biomarkers for idiopathic pulmonary fibrosis. Front. Med. 2021;8 doi: 10.3389/fmed.2021.693959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.King T.E., Jr., Pardo A., Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378(9807):1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 76.Gibb A.A., Lazaropoulos M.P., Elrod J.W. Myofibroblasts and fibrosis: mitochondrial and metabolic control of cellular differentiation. Circ. Res. 2020;127(3):427–447. doi: 10.1161/CIRCRESAHA.120.316958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Grande M.T., Lopez-Novoa J.M. Fibroblast activation and myofibroblast generation in obstructive nephropathy. Nat. Rev. Nephrol. 2009;5(6):319–328. doi: 10.1038/nrneph.2009.74. [DOI] [PubMed] [Google Scholar]
- 78.Gong Y., et al. Lipid peroxidation and GPX4 inhibition are common causes for myofibroblast differentiation and ferroptosis. DNA Cell Biol. 2019;38(7):725–733. doi: 10.1089/dna.2018.4541. [DOI] [PubMed] [Google Scholar]
- 79.Wendisch D., et al. SARS-CoV-2 infection triggers profibrotic macrophage responses and lung fibrosis. Cell. 2021;184(26):6243–6261 e27. doi: 10.1016/j.cell.2021.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang S., et al. A single-cell transcriptomic landscape of the lungs of patients with COVID-19. Nat. Cell Biol. 2021;23(12):1314–1328. doi: 10.1038/s41556-021-00796-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lederer D.J., Martinez F.J. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 2018;378(19):1811–1823. doi: 10.1056/NEJMra1705751. [DOI] [PubMed] [Google Scholar]
- 82.Wolters P.J., Collard H.R., Jones K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2014;9:157–179. doi: 10.1146/annurev-pathol-012513-104706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gao H., et al. Aberrant iron distribution via hepatocyte-stellate cell axis drives liver lipogenesis and fibrosis. Cell Metabol. 2022;34(8):1201–1213 e5. doi: 10.1016/j.cmet.2022.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Houglum K., Bedossa P., Chojkier M. TGF-beta and collagen-alpha 1 (I) gene expression are increased in hepatic acinar zone 1 of rats with iron overload. Am. J. Physiol. 1994;267(5 Pt 1):G908–G913. doi: 10.1152/ajpgi.1994.267.5.G908. [DOI] [PubMed] [Google Scholar]
- 85.Gardi C., et al. Effect of free iron on collagen synthesis, cell proliferation and MMP-2 expression in rat hepatic stellate cells. Biochem. Pharmacol. 2002;64(7):1139–1145. doi: 10.1016/s0006-2952(02)01257-1. [DOI] [PubMed] [Google Scholar]
- 86.Arezzini B., et al. Iron overload enhances the development of experimental liver cirrhosis in mice. Int. J. Biochem. Cell Biol. 2003;35(4):486–495. doi: 10.1016/s1357-2725(02)00298-4. [DOI] [PubMed] [Google Scholar]
- 87.Mehta K.J., et al. Iron enhances hepatic fibrogenesis and activates transforming growth factor-beta signaling in murine hepatic stellate cells. Am. J. Med. Sci. 2018;355(2):183–190. doi: 10.1016/j.amjms.2017.08.012. [DOI] [PubMed] [Google Scholar]
- 88.Arribillaga L., et al. Therapeutic effect of a peptide inhibitor of TGF-beta on pulmonary fibrosis. Cytokine. 2011;53(3):327–333. doi: 10.1016/j.cyto.2010.11.019. [DOI] [PubMed] [Google Scholar]
- 89.Kim D.Y., et al. Del-1, an endogenous inhibitor of TGF-beta activation, attenuates fibrosis. Front. Immunol. 2020;11:68. doi: 10.3389/fimmu.2020.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pocaterra A., Romani P., Dupont S. YAP/TAZ functions and their regulation at a glance. J. Cell Sci. 2020;133(2) doi: 10.1242/jcs.230425. [DOI] [PubMed] [Google Scholar]
- 91.Santucci M., et al. The Hippo pathway and YAP/TAZ-TEAD protein-protein interaction as targets for regenerative medicine and cancer treatment. J. Med. Chem. 2015;58(12):4857–4873. doi: 10.1021/jm501615v. [DOI] [PubMed] [Google Scholar]
- 92.Noguchi S., Saito A., Nagase T. YAP/TAZ signaling as a molecular link between fibrosis and cancer. Int. J. Mol. Sci. 2018;19(11) doi: 10.3390/ijms19113674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Lipid peroxidation and iron accumulation in the lungs of bleomycin-induced pulmonary fibrosis mice. (a) Immunoblotting analysis of 4-HNE in lung tissues of mice on Day 21 after bleomycin administration. Left, representative western blot result for 4-HNE. Right, scatter plot showing the results for all samples examined. Each lane represents one mouse, n = 3 mice per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. *P < 0.05. (b) The gating strategy for detecting intracellular Fe2+ levels in primary lung cells by flow cytometry using FeRhoNox-1. Upper, control group. Bottom, bleomycin group on Day 7 after bleomycin administration. (c) The gating strategy for detecting intracellular Fe2+ levels in primary lung cells by flow cytometry using FeRhoNox-1. Upper, control group. Bottom, bleomycin group on Day 14 after bleomycin administration. (d) Cell death of BEAS-2B cells was determined by Hochest33342 and SYTOX Green staining coupled with fluorescent microscope after 84 h of treatment with bleomycin, Fer-1 and Necrostatin-1 (Necro-1). BLM: 10 μg/ml. Fer-1: 5 μM. Necro-1: 50 μM n = 10 images per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. ****P < 0.0001. (e) Cell death of BEAS-2B cells was determined by Hochest33342 and SYTOX Green staining coupled with fluorescent microscope after 72 h of treatment with LPS and Fer-1. LPS: 5 μg/ml. Fer-1: 5 μM. Necro-1: 50 μM n = 9–10 images per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. ****P < 0.0001.
DFO inhibits bleomycin-induced pulmonary fibrosis. (a) Representative images of Masson's staining and IHC staining for α-SMA, FAPα and TGF-β of mice lung tissue from control, bleomycin group and bleomycin + DFO group on Day 14 after bleomycin administration. Scale bars, 100 μm. (b) Scatter plot showing the percentage of the positive area of Masson's staining and IHC staining in lung tissue from control group, bleomycin group and bleomycin + DFO group on Day 14 after bleomycin administration. n = 4–5 mice per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. *P < 0.05, ****P < 0.0001. (c) Iron levels in lung tissues of mice was determined by tissue Iron content detection Kit on Day 14 after bleomycin administration. n = 4 mice per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. **P < 0.01.
Lip-1 inhibits LPS-induced lung injury and pulmonary fibrosis. (a) Upper left, The lung of mouse from LPS group and LPS + Lip-1 group on Day 7 after LPS administration. The other, H&E staining scanning of lung tissue including control group, LPS group and LPS + Lip-1 group at different days. Scale bars, 1 mm. (b) Representative images of Masson's staining of mice lung tissue from control group, LPS group and LPS + Lip-1 group at different days. Scale bars, 100 μm. (c-e) Representative images of IHC staining for α-SMA, FAPα and TGF-β of mice lung tissue from control group, LPS group and LPS + Lip-1 group at different days. Scale bars, 100 μm. (f) Scatter plot showing the percentage of the positive area of Masson's staining and IHC staining in lung tissue from control group, LPS group and LPS + Lip-1 group at different days. n = 4 mice per group. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
TGF-β promotes the activation of fibroblasts. (a-c) Expression of α-SMA and COL1A1 in MRC-5 (a), IMR90 (b) and PLFs (c) were assessed by qPCR after 24–48 h of treatment with the indicated doses of TGF-β. a: n = 3 biological replicates. b: n = 3 biological replicates. c: n = 4 biological replicates. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. *P < 0.05, ***P < 0.001, ****P < 0.0001. (d) Expression of α-SMA and COL1A1 in MRC-5, IMR90 and PLFs was assessed by western blot after 72 h of treatment with the indicated doses of TGF-β. (e) Representative microscopic images and the quantitation of MRC-5 cells that transferred through the transwell in the migration assay and invasion assays. Scale bars, 200 μm. TGF-β: 10 ng/ml n = 6 visual fields. Statistical analysis was performed using the Student's t-test. Scale bars, 200 μm **P < 0.01, ****P < 0.0001. (f) Expression of TFRC in MRC-5 after TFRC knockdown with siRNA were assessed by qPCR after 48 h of treatment. n = 4 biological replicates. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. ****P < 0.0001. (g) Expression of TFRC in MRC-5 after TFRC knockdown with siRNA was assessed by western blot after 72 h of treatment. (h) MFI of TFRC-FITC in MRC-5 after TFRC knockdown with siRNA was assessed by flow cytometry after 72 h of treatment. Scatter plot showing the results for independent samples examined. n = 3 biological replicates. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. **P < 0.01, ***P < 0.001, ****P < 0.0001. (i) Left, the expression of TAZ in nucleus was quantified by ImageJ in MRC-5 after 48 h of treatment with TGF-β and SB. TGF-β: 10 ng/ml. SB: 1 μM n = 34–37 cells from 5 images. Right, the expression of TAZ in nucleus was quantified by ImageJ in PLFs after 48 h of treatment with TGF-β and SB. TGF-β: 10 ng/ml. SB: 1 μM n = 24–32 cells from 3 images. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. **P < 0.01, ****P < 0.0001. (j) Expression of TAZ in cytoplasm and in nucleus in MRC-5 was assessed by western blot after 72 h of treatment with TGF-β and SB. TGF-β: 10 ng/ml. SB: 1 μM. Lamin B and α-tubulin were used respectively as nuclear and cytoplasmic markers.
Fe2+ modulates ferroptosis-related genes in fibroblasts. (a) Heat map showed the RNA-sequence results of MRC-5 cells stimulated by FAC (50 μM) for 72 h. The red stripes represent highly expressed genes and the blue stripes represent low-expressed genes. n = 3 biological replicates. (b) Heat map showed the relative expression of some genes between control group and FAC group. The red stripes represent significantly upregulated genes and the blue stripes represent significantly downregulated genes. n = 3 biological replicates. Statistical analysis was performed using the one-way ANOVA, P < 0.05. (c) Heat map showed the relative expression of some genes between FAC group and FAC + DFO group. The red stripes represent significantly upregulated genes and the blue stripes represent significantly downregulated genes. n = 3 biological replicates. Statistical analysis was performed using the one-way ANOVA, P < 0.05. (d) Expression of some lipid metabolism and ferroptotic protective genes in MRC-5 cells was assessed by qPCR after 72 h of treatment with FAC and DFO. FAC: 50 μM. DFO: 50 μM n = 3 biological replicates. Statistical analysis was performed using one-way ANOVA followed by multiple comparisons. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.