

Abstract
Long-chain polyunsaturated fatty acids (LCPUFAs) are critical for fetal brain development. Infants born to preeclamptic mothers or those born growth restricted due to placental insufficiency have reduced LCPUFA and are at higher risk for developing neurodevelopmental disorders. Since plasma levels of testosterone (T) and fatty acid-binding protein 4 (FABP4) are elevated in preeclampsia, we hypothesized that elevated T induces the expression of FABP4 in the placenta leading to compromised transplacental transport of LCPUFAs. Increased maternal T in pregnant rats significantly decreased n-3 and n-6 LCPUFA levels in maternal and fetal circulation, but increased their placental accumulation. Dietary LCPUFAs supplementation in T dams increased LCPUFA levels in the maternal circulation and further augmented placental storage, while failing to increase fetal levels. The placenta in T dams exhibited increased FABP4 mRNA and protein levels. In vitro, T dose-dependently upregulated FABP4 transcription in trophoblasts. Testosterone stimulated androgen receptor (AR) recruitment to the androgen response element and trans-activated FABP4 promoter activity, both of which were abolished by AR antagonist. Testosterone in pregnant rats and cultured trophoblasts significantly reduced transplacental transport of C14-docosahexaenoic acid (DHA) and increased C14-DHA accumulation in the placenta. Importantly, FABP4 overexpression by itself in pregnant rats and trophoblasts increased transplacental transport of C14-DHA with no significant placental accumulation. Testosterone exposure, in contrast, inhibited this FABP4-mediated effect by promoting C14-DHA placental accumulation.
Keywords: pregnancy, androgens, placenta, preeclampsia, fatty acids, FABP4, fetal growth
In summary, our studies show that maternal hyperandrogenism increases placental FABP4 expression via transcriptional upregulation and preferentially routes LCPUFAs toward cellular storage in the placenta leading to offspring lipid deficiency.
Graphical Abstract
Graphical Abstract.
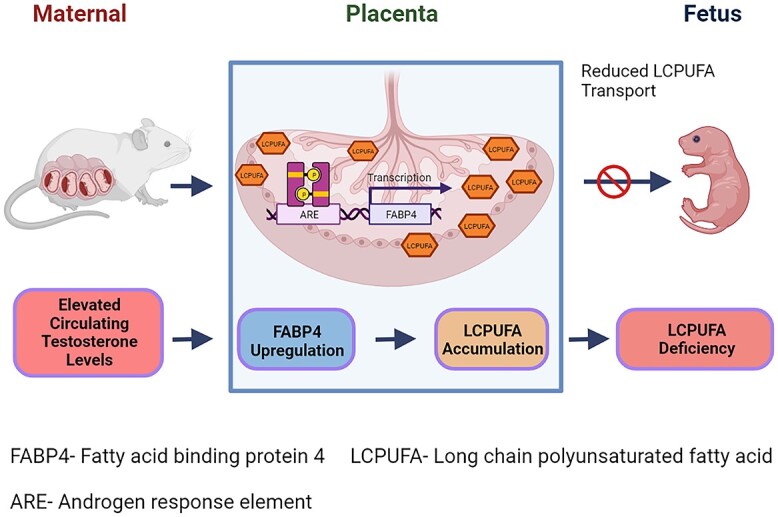
Introduction
The developing fetus requires saturated and unsaturated fatty acids as a source of energy and structural components of cell membranes. They are also precursors of bioactive molecules, including steroid hormones, prostaglandins, prostacyclins, thromboxanes, and leukotrienes, involved in brain development, blood pressure regulation, smooth muscle contraction, and blood clotting [1]. The essential long-chain polyunsaturated fatty acids (LCPUFAs) linoleic (LA; 18: 2 n-6) and alpha-linolenic (ALA; 18:3 n-3) acids and their respective long-chain derivatives, mainly arachidonic (ARA; 20:4 n-6) and docosahexaenoic (DHA; 22:6 n-3) acids, are crucial for fetal tissue growth and development [2–4]. Both the human [5] and rat fetuses [6] have limited ability to synthesize LCPUFAs, including AA and DHA [7–11]. Fetal LCPUFA demand must, therefore, be met from the maternal circulation and LCPUFA transfer across the syncytiotrophoblast, the transport epithelium of the placenta, and the primary barrier between maternal and fetal circulations [4]. The transplacental transport of LCPUFAs is first enabled by lipoprotein and endothelial lipases in the syncytiotrophoblast microvillous plasma membrane. Placental enzymatic hydrolysis of circulating maternal triglycerides and phospholipids enables the release of free fatty acids [12]. Liberated free fatty acids are then transferred from the maternal circulation across the microvillous plasma membrane by simple diffusion or via membrane-bound fatty acid transport proteins (FATPs) and fatty acid translocase/cluster of differentiation 36 (FAT/CD36). Once inside syncytiotrophoblasts, free fatty acids bind with cytosolic fatty acid-binding proteins (FABPs) and are transferred through intracellular aqueous cytoplasm to either target organelles or the fetal-facing syncytiotrophoblast basal plasma membrane for transport to the fetal circulation [13]. Long-chain polyunsaturated fatty acids are preferentially transferred across the syncytiotrophoblast, and this explains the higher content of LCPUFA, in particular of DHA, in fetal circulation compared with the maternal compartment [14, 15].
The requirement for fatty acids increases exponentially with fetal age, especially during the last weeks of intrauterine life [2, 16]. For example, human fetal brain DHA accretion occurs at a rate of ∼14.5 mg/week during the last trimester [17]. In rodents, brain DHA content increases by 10-fold (from 0.64 to 6.6 mg/brain, at ∼2 mg/week) during the first 3 weeks of neonatal life [18]. This sharp increase in brain DHA accretion coincides with human fetal and rodent prepubertal growth spurts when extensive axonal and dendritic outgrowth and development of synaptic connections are made [16, 19]. This exemplifies the importance of having an optimal amount of DHA in the fetal circulation during the perinatal period in both humans and rodents. Unfortunately, infants born to mothers with preeclampsia (PE) or those born growth restricted due to placental insufficiency have reduced circulatory LCPUFAs, and fat stores compared with normal babies [20–23], and are at increased risk for developing neurological [24, 25] and cardiovascular disorders [26, 27]. The reasons for the reduced transplacental transport and lower fetal levels of LCPUFA in PE and fetal growth restriction are not clear but may be related to compromised transplacental transfer of LCPUFAs, as reflected by lower cord vein and fetal AA and DHA concentrations [21, 28, 29] accompanying increased placental accumulation [30]. Intriguingly, even maternal LCPUFA supplementation appears to be of little or no beneficial effects in mothers with PE [31–33]. Strikingly, pregnant women with PE are consistently reported to have higher circulatory and placental levels of fatty acid-binding protein 4 (FABP4) [34–43]. FABP4 is known to have a higher binding affinity to LCPUFAs [44] and regulates trophoblastic lipid transport and accumulation during placental development [45]. It has also been reported that maternal FABP4 levels are elevated in PE, even before the clinical onset of the disease [39]. However, the functional role of FABP4 in the transplacental transport of LCPUFAs and the mechanisms contributing to its placental upregulation remain unknown.
Epidemiological studies show that testosterone (T) levels are elevated 1.5- to 2.4-fold in women with PE compared to normotensive pregnant women [46] and correlate with the delivery of small-for-gestational-age babies [47]. Elevated T, as observed in polycystic ovary syndrome (PCOS), is associated with increased FABP4 mRNA in granulosa cells [48]. Experimental studies also show that elevated T increases FABP4 expression in the rat placenta [49] with an associated decrease in placental weight in rats [50], an increase in placental differentiation in sheep [51], and a decrease in placental amino acid transport capacity in rats [50]. Despite the well-documented effects of elevated maternal T on fetoplacental growth, together with the available circumstantial evidence of its association with increased FABP4 expression, the direct effect of T on transplacental transport of LCPUFAs and placental FABP4 expression has not been studied. In the present study, we hypothesized that elevated T induces the expression of FABP4 in the placenta leading to compromised transplacental transport of LCPUFAs. Thus, the objectives are to determine (1) the effect of elevated T on maternal, fetal, and placental LCPUFA concentrations, (2) whether dietary LCPUFA supplementation rescues T-induced changes in fatty acid levels, (3) whether T alters placental expression of genes involved in fatty acid transport, (4) whether T regulates FABP4 transcription in the placenta, and (5) the (patho)physiological role of FABP4 on transplacental fatty acid transport.
Methods
Animals and treatment
All procedures were approved by the Animal Care and Use Committee at the University of Wisconsin-Madison and were in accordance with those guidelines published by the US National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996). Timed pregnant Sprague–Dawley rats (gestational day [GD] 12; the presence of copulation plug is day 1), purchased from Envigo (Indianapolis, IN), were used in this study. After acclimatization, on GD 14, dams were randomly divided into control and treatment groups. Dams in the treatment group were subcutaneously injected with T propionate (0.5 mg/kg/day, n = 12) for 5 days from GD 15–20. The control group received vehicle (sesame oil, n = 12). This dose and duration of T exposure are commonly used to mimic plasma T levels observed in PE pregnancies, and this model consistently exhibited PE features [46, 49, 52]. The rats were fed with standard rodent (Teklad global rodent diet # TD.94045, Envigo). A subset of rats in these two groups were fed an LCPUFA-enriched diet (AIN-93G, # 112325, Dyets, Inc. Bethlehem, PA) during the course of T exposure. Food and water were provided ad libitum, and all groups were housed in a room with controlled temperature and a 12-h light–dark cycle.
Serum and placenta collection
Between 9 and 10 a.m. on GD 21, rats were anesthetized using isoflurane, and maternal laparotomy was done to expose individual fetal units. Fetal blood was collected by nicking the left ventricle of the heart using 26G BD precision slide needle and 1 ml BD tuberculin slip tip syringe. Maternal blood was collected by cardiac puncture. Collected samples from six to eight fetuses of individual dams were pooled, serum separated, and processed in parallel with the corresponding maternal samples. Placentas were collected, rinsed thoroughly in ice-chilled saline, and blotted with Whatman filter paper. Maternal tissues, including liver and adipose tissue, were also collected. All samples were snap-frozen in liquid nitrogen and stored at −80 °C until further processing.
Liquid chromatography-Mass spectrometry (LC–MS/MS) quantification of LCPUFAs
Total fatty acids were extracted from maternal serum, using chloroform:methanol 2:1 (v/v) containing butylated hydroxytoluene (BHT) to prevent oxidation during sample preparation [53]. Five microliters of plasma was mixed with 1.5 ml of chloroform:methanol (2: 1 v/v). To this mixture, 300 μl of water was added to facilitate phase separation. The bottom phase was removed and mixed with 500 μl of chloroform:methanol:water (3/48/47; v/v/v), vortexed, and centrifuged for 5 min at 16 000g at 4 °C. The lower phase containing lipids was dried down in a speed vac and reconstituted with 200 μl of methanol:dichloromethane (1:1 v/v) containing 2 mM ammonium acetate prior to mass spectrometric analysis. For placental tissues, fatty acid extraction was performed according to the modification of the method of Brown et al. [30]. In brief, 40 mg of frozen placental tissue was homogenized in 400 μl methanol containing 50 μg/ml of BHT using a bead homogenizer. The homogenate was vortexed for 2 min and left on ice for 2 min (repeated three times). Insoluble material was cleared by centrifugation for 5 min at 16 000g at 4 °C. The supernatant (25 μl) was added to 250 μl MTBE and vortexed overnight at 4 °C. Ammonium acetate (150 μl of 100 mM) was added to induce phase separation. The upper organic layer was removed to a new vial, dried in a speed vac, resuspended in 125 μl of methanol:dichloromethane (1:1 v/v) containing 2 mM ammonium acetate prior to mass spectrometric analysis. All samples subjected to ESI-MS were at a concentration below 20 μM; conditions at which ion-suppression effects are minimal [54].
Mass spectra were acquired using an ABSciex TripleTOF 5600 (Sciex, Foster City, CA) equipped with an electrospray interface with a 25 μm iD capillary and coupled to an Eksigent μUHPLC (Eksigent, Redwood City, CA) according to the methods of Tran et al. [55]. An Analyst TF 1.7 software was utilized to cycle all experiments and for data processing and acquisition. The sample was analyzed in MS/MS all acquisition mode by direct infusion, in both positive and negative ionization mode. Methanol:dichloromethane (1:1 v/v) containing 2 mM ammonium acetate was used as mobile phase. Data were analyzed with LipidView (ABSciex) software version 1.2, including smoothing, identification, removal of isotope contribution from lower mass species, and correction for isotope distribution.
Quantitative real-time polymerase chain reaction for expression of genes related to fatty acid synthesis and transport
Total RNA was extracted using the RNeasy mini kit (QIAGEN, Valencia, CA) according to the manufacturer’s instructions. RNA concentration and integrity were determined using a DS-11 spectrophotometer (DeNovix, Wilmington, DE). One microgram of total RNA was reverse transcribed using an iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). After dilution, cDNA corresponding to 100 ng of RNA was amplified by quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR) using FAM (Invitrogen; Thermo Scientific, Grand Island, NY) as the fluorophore in a CFX96 real-time thermal cycler (Bio-Rad). Polymerase chain reaction conditions for TaqMan Gene Expression Assay were 2 min at 50 °C and 10 min at 95 °C for 1 cycle, then 15 s at 95 °C and 1 min at 60 °C for 50 cycles. Results were calculated using the 2–ΔΔCT method and expressed as fold change of the gene of interest in treated vs control (vehicle) samples. All reactions were performed in duplicate, and β-actin was used as an internal control. All gene-specific primers used in this study are provided in Supplementary Table S1.
Western blotting
Tissues were homogenized in ice-cold RIPA buffer (Cell Signaling Technology, Danver, MA) containing a protease inhibitor tablet and phosphatase inhibitor cocktail-2 and -3 (Sigma-Aldrich, St. Louis, MO). Tissue lysates were centrifuged (14 000g for 10 min at 4 °C), and the protein content was measured using the BCA protein assay kit (Pierce; Thermo Scientific). The supernatant was resuspended in NuPAGE lithium dodecyl sulfate sample buffer and reducing agent (Invitrogen; Thermo Scientific). Proteins (30 μg) alongside Precision Plus Standard (Kaleidoscope; Bio-Rad, Hercules, CA), and negative controls were resolved on 4–12% gradient NuPAGE Bis-Tris gels (Invitrogen) at 100 V for 2 h at room temperature and then transferred onto Immobilon-P membranes (Millipore, Billerica, MA) at 100 V for 1 h. The membranes were blocked with 5% nonfat dry milk for 1 h and then incubated overnight at 4 °C with respective primary antibodies (Human anti-FABP4, # HPA002188, Sigma-Aldrich; Rat anti-FABP4, # PA5-79231, Invitrogen; Thermo Scientific) and β-actin (#4070, Cell Signaling Technologies). After washing, the membranes were incubated with secondary antibodies (anti-rabbit conjugated with horseradish peroxidase) and detected with the Pierce ECL detection kits (Thermo Scientific, Waltham, MA). The densitometric measurement was done using ImageJ software. Results were normalized to vehicle results and expressed as ratios of the intensity of a specific band to that of β-actin.
Cell culture and treatments
BeWo cells (CCL-98; American Type Culture Collection, Manassas, VA) were cultured at 37 °C in a 5% CO2 atmosphere in Ham F12 K medium (Wako) with 10% FBS (Hyclone), as described previously [56]. HepG2 cells (American Type Culture Collection) were maintained in Dulbecco modification of Eagle medium (DMEM; Cellgro, Manassas, VA) containing 1% l-glutamine, 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin in 5% CO2 at 37 °C. 3T3-L1 cells were maintained in Dulbecco modified Eagle medium with 10% fetal bovine serum in 5% CO2. BeWo, HepG2, and 3T3-L1 were treated with respective medium containing increasing concentrations of T (0.1–100 nM, Sigma) for 24 h, in the absence or presence of androgen receptor (AR) antagonist (hydroxyflutamide; 100 nM).
In silico analysis of FABP4 promoter
Androgen receptor binding sites on the human FABP4 gene (NCBI ID:2167) promoter were identified by LASAGNA motif search tool [57]. Briefly, the analysis was performed on the entire upstream (2 kb) sequences from the transcriptional start site (TSS) using human reference core matrices (MA0007.2) from JASPAR transcription factor database [58] with the cutoff P-value of 0.01. The identified AR binding sites were utilized for the downstream chromatin immunoprecipitation (ChIP) assay to assess the binding efficiency of the AR to the respective motifs, followed by the luciferase assay to determine whether AR binding to these motifs positively activates the FABP4 transcriptional machinery.
ChIP and quantitative PCR
Chromatin immunoprecipitation assay was performed using a simple ChIP kit (#9005; Cell Signaling Technologies) to validate androgen response element (ARE) binding sites on human FABP4 gene promoter as we described previously [59]. Briefly, BeWo cells were washed and cross-linked with formaldehyde treatment. Cells/tissues were lysed, and chromatin was fragmented by enzymatic digestion using micrococcal nuclease for 20 min at 37 °C with frequent mixing. The nuclei were separated, sonicated for 20 s twice with 40% power to break the nuclear membrane and clarified by centrifugation at 10 000 rpm for 10 min at 4 °C. DNA samples from the digested chromatin were separated using spin columns, and the quality was analyzed by agarose gel electrophoresis. Chromatin (5 μg of DNA in 500 μl volume for each) was then subjected to immunoprecipitation with an AR antibody (25 μl per reaction) along with positive (10 μl anti-histone antibody; Cell Signaling Technologies #4620) and negative (1 μl of rabbit IgG, Cell Signaling Technologies #2727) controls. ChIP-grade Protein G magnetic beads were used to capture the antigen–antibody complex along with specific DNA target sites. Cross-linking was then reversed by 5 mM NaCl and followed by proteinase K treatment to remove the protein contamination. Sample DNA fragments containing specific AR binding sites were purified using spin columns, quantified, and stored for further qPCR analysis. Using custom-designed primers (Supplementary Table S2) that flank the predicted putative AREs, qPCR was performed to identify the ligand-dependent AR binding to AREs in FABP4 gene promoter, and the results were analyzed as we described previously [59].
FABP4 promoter constructs and reporter assay
Promoter sequences corresponding to the human FABP4 transcript ENST00000256104.4 were obtained from Eukaryotic Promoter Database [60]. The ChIP detected AR binding motifs located within 1 kb (AREs 1, 2, and 3) FABP4 promoter fragments containing Mlu I and Xho I flanks were custom synthesized through IDT and cloned into respective multiple cloning sites of pGL3 basic backbone, as described previously [61]. For the reporter assay, these individual promoter constructs were transiently transfected into ~6 × 108 BeWo cells using Fugene HD transfection reagent (Promega, Madison, WI) along with pmaxGFP vector to monitor the transfection efficiency. After 24 h, cells were washed and incubated in the presence of vehicle (ethanol), T (10 nM), T+ hydroxyflutamide (100 nM), and hydroxyflutamide alone for 24 h at 37 °C with 5% CO2 in F-12K media containing 10% FBS and 1% antibiotics (Invitrogen; Thermo Scientific). Then, the cells were washed with ice-cold PBS twice and were lysed by adding 250 μl of passive lysis buffer per well (Promega). A luciferase assay was performed using the Luciferase reporter assay system (E1500; Promega), following the manufacturer’s instructions. Cell lysates (20 μl) were transferred onto a 96-well plate, and GFP fluorescence was measured using FLUOstar Omega (BMG Labtech, Ortenberg, Germany). After that, 100 μl of luciferase assay reagent was dispensed in the same plate, and luminescence was measured using Fluorstar Omega. The luciferase activity was normalized with GFP fluorescence, and the results were expressed as relative luminescence.
Lentiviral particles and FABP4 overexpression
A Lenti ORF clone of human FABP4 (PS100092, OriGene, Rockville, MD) was obtained, and the lentivirus was produced by using a Lenti-vpak Packaging Kit (TR30037, OriGene) following the manufacturer’s instructions. Briefly, 5 × 106 of 293T cells were plated in a 15-cm tissue culture dish and incubated for 1 day. For transfection, 5 μg lentiviral vector containing the cytomegalovirus promoter-driven FABP4 ORF and 6 μg of lentiviral packaging plasmids were mixed with 1.5 ml Opti-MEM and 33 μl turbofectin transfection reagent. After 15 min incubation at room temperature, the mixture was added to T293 cells in culture. After incubation at 37 °C for 18 h, fresh culture media was added, and viral supernatant was collected every 24 h for 3 days. The cell debris from collected viral supernatant was removed by filtration through a 0.45 μm filter. Viral titer was determined using a one-wash lentivirus titer kit (TR30038, Origene). The FABP4 lentiviral particles were concentrated by using a Lenti-X Concentrator (Cat. No. 631231, Clontech, San Jose, CA). The lentiviral particles were then injected into pregnant rats and transduced into BeWo cells. Pregnant rats were injected with lentiviral FABP4 particles intravenously via the tail vein on GD 12 and 16 at a dose of 8 × 1010 viral particles/kg. The controls were injected with the same dose of viral particles lacking FABP4. For transduction in BeWo, the cells were plated at 2.5 × 105 cells per well and, after overnight incubation, transduced with lentiviral FABP4 particles at a multiplicity of infection of 100. Polybrene (5 μg/ml) (Sigma) was added to increase the infection efficiency. After overnight culture at 37 °C, the virus-containing media was replaced with fresh media. The selection was carried out for a week with 2 μg/ml puromycin (Sigma), which was changed every 2 days. The selection was terminated when the uninfected control cells died. The FABP4 expression was confirmed by Western blotting (Supplementary Figure S1).
Placental transport of 14C-DHA in pregnant rats
On GD 21, rats were administered with 0.3 ml of 2.5 μmol/kg (2 μCi/kg) 14C-DHA (Moravek Biochemicals, Brea, CA) via tail vein injection [62]. Freshly prepared, 1:1 molar ratio of 14C-DHA diluted in saline containing BSA (pH = 6.0–6.5) was used for the injection. Maternal and fetal blood and placenta were collected as described above after 3 h of radioisotope injection; studies show that there is minimal 14C-DHA back flux at this time [63]. Serum and placental samples were lysed in biosol biodegradable tissue solubilizer (National Diagnostics) for 2 h at room temperature. The bioscint liquid scintillation fluid (National Diagnostics) was used for β-counting (Tri-Carb 4910TR liquid scintillation counter, PerkinElmer). Transport data were presented as placental disintegrations per minute (dpm) per gram placenta (representing placental uptake/accumulation of isotope) and fetal dpm per milliliter fetal serum (representing the amount of isotope transported to the fetus). Transport data were expressed for the T group relative to the controls.
Transcellular 14C-DHA transport using the transwell system
The transcellular fatty acid transport studies were performed as described previously [56]. Briefly, the Corning Snapwell Transwell polyester 12-well transwell plates containing permeable membranes were coated with human placental collagen (0.01% w/v). Cells were then seeded at 100 000 cells per cm2. The cells formed a monolayer at day 3–4 post-seeding, and all experiments were carried out on day 4 after syncytialization, which was confirmed by hPL and hCG mRNA expression and secretion of hCG, as we described previously [64]. Trypan blue was added to the apical reservoir, and no color seeping through to the basal reservoir indicated cell confluency. The cells were treated with vehicle or T (0.1–100 nM) for 24 h, and then serum-free F12 medium containing 1% fatty acid-free albumin and 10 μCi of 14C-DHA was replaced in the apical reservoir of the transwell. Serum-free F12 medium containing 1% fatty acid-free albumin was added to the basal reservoir of the transwell. The dose- and time-dependent changes in transcellular transport of DHA across the BeWo monolayer were measured as the rate of appearance of DHA in the lower (“fetal side”) reservoir following transfer from the upper (“maternal side”) reservoir. After incubation (every 4 h for up to 24 h), aliquots of cell media from the basal reservoir were collected, and transport of 14C-DHA through the BeWo cell monolayer was determined by liquid scintillation counting. After the final aliquot, cells were thoroughly washed with cold PBS to remove any surface-bound FAs, then lysed and harvested in liquid scintillation fluid and prepared for scintillation counting to estimate the intracellular accumulation of 14C-DHA. The results were expressed as disintegrations per minute per microgram of protein.
Statistical analysis
All data are presented as mean ± S.E.M. The normality and homogeneity of the variances were analyzed using the one-sample Kolmogorov–Smirnov test and Anderson–Darling test, respectively. One- or two-way ANOVA followed by Bonferroni post hoc test was performed on multiple observations and unpaired Student t-test for comparison of single observations between control and T propionate groups. Repeated measures ANOVA was used to compare the time-dependent effect. Data analysis was done using GraphPad Prism for Windows (GraphPad Software, San Diego, CA). Since observations in individual pups and placentas of the same litter were not independent, an average was obtained for each litter. Therefore, n = 1 represents averaged values in one litter. Differences were considered statistically significant at P < 0.05.
Results
Effect of T and dietary supplementation on placental and fetal weights
Testosterone administration during pregnancy in rats caused Intrauterine growth restriction (IUGR). At GD 21, fetal weights in T dams were diminished by 11% (control: 5.52 ± 0.21 g; T: 4.84 ± 0.27 g; n = 6 litters in each group; P < 0.05) and placental weights were reduced by 12% (control: 553 ± 14.4 mg; T: 488 ± 11.9 mg; n = 6 dams in each group; P < 0.05) in comparison to controls. Feeding a diet supplemented with LCPUFAs did not alter fetal weights in control and also did not abrogate lower fetal weight in the T group (control-supplemented: 5.65 ± 0.23; T-supplemented: 4.72 ± 0.21 g; n = 6 dams in each group; P < 0.05). Placental efficiency (fetal to placental weight ratio) in T-treated animals was not statistically different from controls. LCPUFA supplementation did not alter placental weight in control, however, did abrogate lower placental weight in the T group (control-supplemented: 584 ± 19 mg; T-supplemented: 557 ± 21 mg; n = 6 dams in each group). Placental efficiency was significantly lower in the T-supplemented group compared with the control-supplemented group. No significant differences were noted in daily feed intake and mean litter size between control and T dams with or without LCPUFA supplementation. Overall, elevated maternal T caused IUGR, and LCPUFA supplementation restored placental, but not fetal, weights in T dams.
Effect of T and dietary supplementation on LCPUFA concentrations in maternal and fetal circulations
Elevated T significantly decreased total n-3 and n-6 fatty acids by 43 and 23% in maternal (Figure 1A), and 40% and 33% in fetal (Figure 1B) circulations, respectively, compared with controls (n = 6 dams; P < 0.05). Notably, maternal and fetal concentrations of ALA (maternal: ↓46%; fetal: ↓40%), DHA (maternal: ↓34%; fetal: ↓40%), linoleic acid; LNA (maternal: ↓15%; fetal: ↓32%), and ARA (maternal: ↓34%; fetal: ↓27%) were significantly decreased in the T group compared with controls (Tables 1 and 2).
Figure 1.
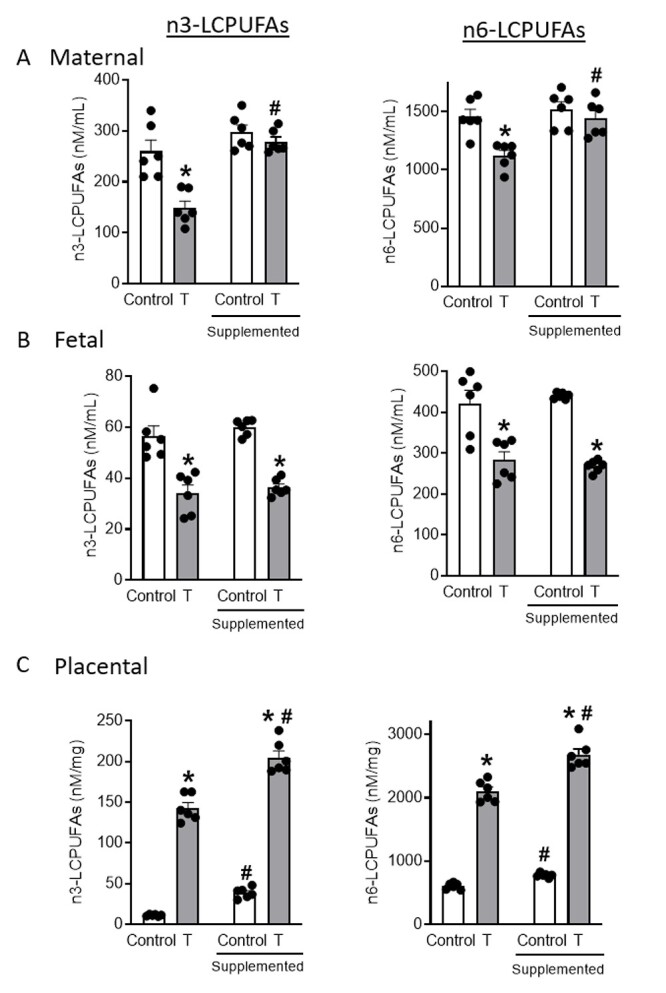
Absolute amounts of total n3 and n6 fatty acids in (A) maternal serum, (B) fetal serum, and (C) placenta of control and T dams with and without dietary LCPUFA supplementation. Fatty acids were quantified using LC–MS/MS. Data presented as mean ± SEM of six rats in each group. *P < 0.05 vs respective control group, #P < 0.05 vs respective group without LCPUFA supplementation.
Table 1.
n3- and n6-LCPUFA levels in maternal serum (nM/ml)
| Fatty acid | Fatty acid chain | Control | T | Control-supplemented | T-Supplemented |
|---|---|---|---|---|---|
| Omega-3 fatty acids (n-3 LCPUFAs) | |||||
| Stearidonic acid | 18:4 | 4.99 ± 0.77 | 2.55 ± 0.82* | 5.63 ± 0.3 | 2.6 ± 0.6* |
| Alpha-linolenic acid | 18:3 | 180.77 ± 23.35 | 98.16 ± 23.79* | 208.72 ± 11.1 | 190.39 ± 12.6# |
| Eicosapentaenoic acid | 20:5 | 1.71 ± 0.25 | 1.05 ± 0.27* | 2.31 ± 0.17 | 2.34 ± 0.2# |
| Docosahexaenoic acid | 22:6 | 60.64 ± 2.45 | 40.2 ± 4.39* | 66.42 ± 8.15 | 62.53 ± 6.71# |
| Docosapentaenoic acid (n-3) | 22:5 | 11.84 ± 1.29 | 6.97 ± 1.28* | 13.99 ± 0.72 | 10.79 ± 1.07# |
| Docosatrienoic acid | 22:4 | 0.35 ± 0.05 | 0.21 ± 0.06* | 0.37 ± 0.04 | 0.33 ± 0.04# |
| Total n-3 LCPUFAs | 260.29 ± 26.37 | 149.14 ± 13.61* | 297.45 ± 14.4 | 278.99 ± 9.14# | |
| Omega-6 fatty acids (n-6 LCPUFAs) | |||||
| Gamma-linolenic acid | 18:3 | 296.26 ± 36.2 | 166.85 ± 35.82* | 284.72 ± 24.94 | 279.89 ± 26.81# |
| Linoleic acid | 18:2 | 1060.94 ± 24.26 | 903.73 ± 35.64* | 1124.99 ± 110.72 | 1068.03 ± 130.34# |
| Arachidonic acid | 20:4 | 55.37 ± 3.63 | 36.75 ± 4.69* | 60.46 ± 3.8b | 53.33 ± 3.42# |
| Dihomo-gamma-linolenic acid | 20:3 | 2.65 ± 0.38 | 1.45 ± 0.4* | 2.73 ± 0.23 | 2.45 ± 0.23# |
| Eicosadienoic acid | 20:2 | 4.48 ± 0.51 | 2.51 ± 0.59* | 4.11 ± 0.38 | 3.3 ± 0.34# |
| Docosapentaenoic acid (n-6) | 22:5 | 30.05 ± 1.87 | 8.37 ± 4.23* | 37.17 ± 2b | 27.99 ± 2.65*# |
| Docosatetraenoic acid | 22:4 | 8.19 ± 0.98 | 3.26 ± 1.29* | 7.82 ± 0.24 | 7.62 ± 0.21# |
| Docosadienoic acid | 22:2 | 0.09 ± 0.02 | 0.05 ± 0.02* | 0.09 ± 0.02 | 0.08 ± 0.02# |
| Total Omega 6 fatty acids | 1458.04 ± 62 | 1122.99 ± 44.42* | 1522.1 ± 101.94 | 1442.68 ± 63.99# | |
Values are expressed as mean ± SEM. n = 6 in each group. *P < 0.05 vs respective control group, #P < 0.05 vs respective group without supplementation.
Table 2.
n3- and n6-LCPUFA levels in fetal serum (nM/ml)
| Fatty acid | Fatty acid chain | Control | T | Control-supplemented | T-Supplemented |
|---|---|---|---|---|---|
| Omega-3 fatty acids (n-3 LCPUFAs) | |||||
| Stearidonic acid | 18:4 | 1.44 ± 0.20 | 1.04 ± 0.18* | 1.54 ± 0.06 | 1.09 ± 0.22* |
| Alpha-linolenic acid | 18:3 | 22.92 ± 2.37 | 13.57 ± 3.46* | 24.42 ± 0.03 | 13.56 ± 1.07* |
| Eicosapentaenoic acid | 20:5 | 0.53 ± 0.04 | 0.36 ± 0.06* | 0.69 ± 0.14 | 0.23 ± 0.07* |
| Docosahexaenoic acid | 22:6 | 30.04 ± 1.55 | 17.94 ± 3.74* | 31.61 ± 0.83 | 20.09 ± 0.49* |
| Docosapentaenoic acid (n-3) | 22:5 | 1.39 ± 0.14 | 1.20 ± 0.09 | 1.64 ± 0.03 | 1.54 ± 0.17 |
| Docosatrienoic acid | 22:4 | 0.15 ± 0.01 | 0.07 ± 0.03* | 0.15 ± 0.01 | 0.05 ± 0.01* |
| Total n-3 LCPUFAs | 56.47 ± 4.08 | 34.17 ± 3.15* | 60.04 ± 1.27 | 36.34 ± 0.81* | |
| Omega-6 fatty acids (n-6 LCPUFAs) | |||||
| Gamma-linolenic acid | 18:3 | 39.87 ± 3.89 | 23.43 ± 5.87* | 41.32 ± 1.65 | 19.74 ± 2.09* |
| Linoleic acid | 18:2 | 350.25 ± 25.73 | 239.75 ± 42.05* | 360.28 ± 4.58 | 230.72 ± 4.36* |
| Arachidonic acid | 20:4 | 19.70 ± 1.48 | 14.46 ± 2.34* | 23.15 ± 0.47 | 12.39 ± 2.07* |
| Dihomo-gamma-linolenic acid | 20:3 | 1.06 ± 0.07 | 0.53 ± 0.16* | 1.61 ± 0.13# | 0.55 ± 0.11* |
| Eicosadienoic acid | 20:2 | 1.10 ± 0.09 | 0.62 ± 0.16* | 1.46 ± 0.11 | 0.48 ± 0.10* |
| Docosapentaenoic acid (n-6) | 22:5 | 8.71 ± 0.83 | 3.49 ± 1.49* | 9.13 ± 0.29 | 3.76 ± 0.51* |
| Docosatetraenoic acid | 22:4 | 1.15 ± 0.09 | 0.89 ± 0.12 | 2.89 ± 0.44# | 0.56 ± 0.11*# |
| Docosadienoic acid | 22:2 | 0.06 ± 0.01 | 0.04 ± 0.009* | 0.36 ± 0.14 | 0.06 ± 0.01* |
| Total Omega 6 fatty acids | 421.88 ± 31.78 | 283.21 ± 20.51* | 440.21 ± 3.09 | 268.2 ± 5.91* | |
Values are expressed as mean ± SEM. n = 6 in each group. *P < 0.05 vs respective control group, #P < 0.05 vs respective group without supplementation.
Dietary LCPUFA supplementation negated the marked decrease observed in total fatty acids in the maternal circulation of T dams by increasing n-3 by 67% and n-6 by 20%, but was without significant effect in the controls (Figure 1A; n = 6 dams; P < 0.05). Long-chain polyunsaturated fatty acid supplementation, therefore, normalized total n-3 and n-6 fatty acid concentrations in the maternal circulation of T dams by restoring levels of ALA, DHA, LNA, and ARA to those found in control-supplemented dams (Tables 1 and 2).
When compared to the respective counterparts on a standard diet, dietary LCPUFA supplementation in T dams failed to abrogate deficiencies in total n-3 and n-6 fatty acids in the T fetal circulation (n-3: ↓40%; n-6: ↓39%) and was without significant effects in control fetuses (Figure 1B; n = 6 dams; P < 0.05). Fetuses in the LUPUFA-supplemented T group thus maintained lower n-3 and n-6 fatty acids by 40 and 39%, respectively, as well diminished fetal concentrations of ALA (↓45%), DHA (↓36%), LNA (↓36%), and ARA (↓47%) compared with control-supplemented fetuses (Tables 1 and 2).
Effect of T and dietary supplementation on LCPUFA concentrations in the placenta
Elevated T significantly increased total n-3 and n-6 fatty acids in the placentae of T dams compared to controls (n-3: ↑1166%; n-6: ↑245%; Figure 1C; n = 6 dams; P < 0.05). Specifically, placental concentrations of ALA (↑252%), DHA (↑1218%), LNA (↑89%), and ARA (↑218%) were increased in the placentae of T dams (Table 3).
Table 3.
n3- and n6-LCPUFA levels in placenta (nM/mg)
| Fatty acid | Fatty acid chain | Control | T | Control-supplemented | T-Supplemented |
|---|---|---|---|---|---|
| Omega-3 fatty acids (n-3 LCPUFAs) | |||||
| Stearidonic acid | 18:4 | 1.14 ± 0.57 | 57.48 ± 4.85* | 19.62 ± 4.18# | 77.85 ± 7.83*# |
| Alpha-linolenic acid | 18:3 | 13.25 ± 1.31 | 46.59 ± 6.56* | 18.98 ± 4.3 | 81.69 ± 4.65*# |
| Eicosapentaenoic acid | 20:5 | 0.4 ± 0.06 | 67.25 ± 5.92* | 3.66 ± 0.43# | 92.58 ± 3.07*# |
| Docosahexaenoic acid | 22:6 | 46.81 ± 2.94 | 617.11 ± 35.71* | 146.11 ± 8.91# | 903.55 ± 31.92*# |
| Docosapentaenoic acid (n-3) | 22:5 | 5.39 ± 0.39 | 67.98 ± 5.92* | 44.34 ± 16.68# | 72.23 ± 7.4* |
| Docosatrienoic acid | 22:4 | 0.78 ± 0.06 | 1.34 ± 0.23* | 0.86 ± 0.09 | 1.17 ± 0.17* |
| Total n-3 LCPUFAs | 11.29 ± 0.42 | 142.96 ± 6.49* | 38.93 ± 2.65# | 204.85 ± 8.31*# | |
| Omega-6 fatty acids (n-6 LCPUFAs) | |||||
| Gamma-linolenic acid | 18:3 | 11.25 ± 0.82 | 27.15 ± 0.98* | 20.38 ± 0.76# | 16.25 ± 2.88# |
| Linoleic acid | 18:2 | 358.44 ± 13.73 | 677.93 ± 11.88* | 268.48 ± 10.56# | 843.92 ± 50.02*# |
| Arachidonic acid | 20:4 | 174.33 ± 10.26 | 554.62 ± 14.46* | 229 ± 9.13 | 597.12 ± 29.44* |
| Dihomo-gamma-linolenic acid | 20:3 | 6.92 ± 0.46 | 17.99 ± 1.19* | 8.62 ± 0.67 | 26.83 ± 1.29*# |
| Eicosadienoic acid | 20:2 | 13.37 ± 0.36 | 491.54 ± 41.1* | 87.73 ± 4.62# | 748.64 ± 53.45*# |
| Docosapentaenoic acid (n-6) | 22:5 | 26.12 ± 1.31 | 305.21 ± 36.08* | 133.36 ± 5.76# | 402.11 ± 14.49* |
| Docosatetraenoic acid | 22:4 | 17.36 ± 1.99 | 24.65 ± 1.9* | 31.03 ± 2.84# | 41.01 ± 2.88*# |
| Docosadienoic acid | 22:2 | 1.41 ± 0.11 | 2.06 ± 0.26* | 1.37 ± 0.12 | 4.69 ± 0.48*# |
| Total Omega 6 fatty acids | 609.19 ± 20.37 | 2101.17 ± 67.51* | 779.96 ± 13.73# | 2680.59 ± 90.79*# | |
Values are expressed as mean ± SEM. n = 6 in each group. *P < 0.05 vs respective control group, #P < 0.05 vs respective group without supplementation.
Dietary LCPUFA supplementation augmented total n-3 and n-6 fatty acid levels in the placentae of T dams (n-3: ↑43%; n-6: ↑27%) as well as control dams (n-3: ↑245%; n-6: ↑28%) compared with their respective counterparts on the standard diet. Thus, placental n-3 and n-6 fatty acids were significantly higher in T-supplemented dams (n-3: ↑426%; n-6: ↑244%) compared with control-supplemented dams. Notably, increased placental levels of ALA (↑330%), DHA (↑518%), LNA (↑214%), and ARA (↑161%) were observed in the placenta of T-supplemented dams compared with control-supplemented dams (Table 3). Total fatty acid levels in fetal liver and adipose tissue, nevertheless, were comparable in control and T dams (Supplementary Figure S2).
Effect of T on the expression of fatty acid synthesis and transport genes in the placenta
To determine whether fetal deficiency and placental accumulation of LCPUFAs in T dams correlated with alteration fatty acid synthesis/transport in the placenta, placental mRNA levels of genes involved in the fatty acid synthesis, FATP, and FABP were determined with qRT-PCR. As shown in Figure 2A and B, while there were no significant changes in mRNA expression placental fatty acid synthesis genes, FATPs and translocase (CD36), lipoprotein lipase mRNA expression was increased by 2-fold in T placentae compared with controls (n = 6 in each group; P < 0.05). Strikingly, placental FABP4 was the only FABP that significantly increased (4-fold) in the placentae of T dams compared with controls (Figure 2C; n = 6 in each group; P < 0.05). Western blotting showed that FABP4 protein levels were significantly increased by 3.3-fold in T placentae compared with controls (Figure 2D; n = 6 in each; P < 0.05). Increased FABP4 expression, however, was limited to placentae as no differences in FABP4 mRNA levels were observed in fetal liver and adipose tissue of T compared with control groups (Figure 2E; n = 6 in each).
Figure 2.

Effect of elevated T on mRNA expression of genes involved in (A) fatty acid synthesis, (B) FATP, and (C) FABPs in the placenta. (D) Effect of elevated T on FABP4 protein expression in the placenta. Representative Western blots for FABP4 and β-actin are shown at top; blot density obtained from densitometric scanning of FABP4 normalized to β-actin is shown at bottom. (E) FABP4 mRNA expression in the liver and adipose tissue of control and T dams. mRNA expression was normalized to β-actin. Data presented as mean ± SEM of six rats per group. *P < 0.05 vs control. Acac, Acetyl-CoA Carboxylase Alpha; Fasn, Fatty Acid Synthase; Lipe, Lipase E, Hormone Sensitive Type; Fads2, Delta-6-Desaturase; Fads1, Delta-5 Desaturase; Elovl5, ELOVL Fatty Acid Elongase 5; Lpl, Lipoprotein Lipase; Lipg, Lipase G, Gastric Type.
Effect of T on FABP4 mRNA expression in cultured trophoblast cells in vitro
We next tested whether T directly regulates the expression of FABP4 by examining T effects in cultured human trophoblast (BeWo) cells. Testosterone, at a clinically relevant concentration range (0.1–10 nM), induced dose-dependent increases in FABP4 mRNA expression up to 3.7-fold in the trophoblasts, and this effect was prevented by the selective AR antagonist hydroxyflutamide (100 nM) (Figure 3A; n = 4; P < 0.05). Also, T at 10 nM induced a 6-fold increase in FABP4 protein levels compared to vehicle (Figure 3B; n = 4; P < 0.05). Consistent with the effects observed in vivo, T did not affect FABP4 mRNA expression in cultured hepatocytes (hepG2) and adipocytes (3T3-L1) (Figure 3C; n = 4 in each). Testosterone, therefore, selectively upregulated FABP4 transcription in human trophoblast cells but not in hepatocytes and adipocytes.
Figure 3.

Effect of T on FABP4 (A) mRNA and (B) protein expression in cultured trophoblast cells (BeWo) in vitro. (C) Effect of T on FABP4 mRNA expression in cultured hepatocytes (HepG2) and adipocytes (3T3-L1). Representative Western blots for FABP4 and β-actin are shown at top; blot density obtained from densitometric scanning of FABP4 normalized to β-actin is shown at bottom. mRNA expression was normalized with β-actin. Data presented as mean ± SEM from four independent experiments. *P < 0.05 vs vehicle control, #P < 0.05 vs 10 nM T treatment. HF, hydroxyflutamide (100 nM).
AR binding sites on FABP4 gene promoter
We probed FABP4 for promoter analysis. Bioinformatic analysis of FABP4 promoter using the LASAGNA motif search tool identified 5 putative AREs within the −2 kb upstream region from the FABP4 TSS (Figure 4A). We further probed FABP4 with ChIP assay to identify if any of these AREs interacts with AR in the presence of T in trophoblast cells. Our results show that ARE1, ARE2, and ARE3 interacted with AR in a ligand-dependent fashion showing 1.8-, 2.7-, and 2.8-fold enrichment, respectively, when treated with T compared with controls (Figure 4B; n = 4; P < 0.05). Other predicted AREs (ARE4 and ARE5) did not show significant ligand-dependent enrichment.
Figure 4.

Bioinformatic analysis of FABP4 gene promoter and functional validation of androgen receptor binding motifs. (A) Schematics showing the location of AREs on FABP4 promoter, (B) ChIP assay showing T-induced AR interaction with a putative ARE (C) reporter assay showing luciferase activity in BeWo cells transfected with the reporter plasmid containing ARE 1, 2 and 3 in the presence of T (10 nM) with and without hydroxylflutamide (HF, 100 nM). Data presented as mean ± SEM from four independent experiments. *P < 0.05 vs Vehicle, #P < 0.05 vs T treatment.
Functional effect of AR–ARE interaction on FABP4 gene expression
Since AR interacted with ARE1, 2, and 3, we further probed whether the AR-ARE binding could result in the activation of FABP4 promoter. We cloned 1 kb promoter region of FABP4 and performed a reporter assay. Results showed that AR was able to induce transcription in a ligand-dependent manner. Testosterone increased luciferase activity by ~3-fold and was significantly higher than vehicle (Figure 4C; n = 4; P < 0.05). Hydroxyflutamide treatment abolished T-mediated luciferase activity (Figure 4C; n = 4; P < 0.05). Our data thus suggest that the functional AR binding site in the −1 kb promoter region of the FABP4 gene has a promoter engaging function.
Effect of T and FABP4 overexpression on placental DHA transport in pregnant rats in vivo
To understand the functional effect of T and the role of FABP4 overexpression on transplacental transport LCPUFAs, we performed radiotracer transport studies using C14-DHA. In normal (wild-type) pregnant rats, elevated T inhibited transplacental C14-DHA transport to the fetus by 46% accompanied by a 2-fold increase in C14-DHA accumulation in placentae compared to controls (Figure 5; n = 6; P < 0.05). Overexpression of FABP4 in pregnant rats augmented transplacental transport of C14-DHA to the fetus by 2.2-fold without significant accumulation in placentae compared with wild-type pregnant rats (Figure 5; n = 6; P < 0.05). Testosterone treatment inhibited the FABP4 overexpression-induced increase in transport of DHA to the fetus by 66% with an accompanying 2-fold increase in C14-DHA accumulation in placentae (Figure 5; n = 6; P < 0.05).
Figure 5.
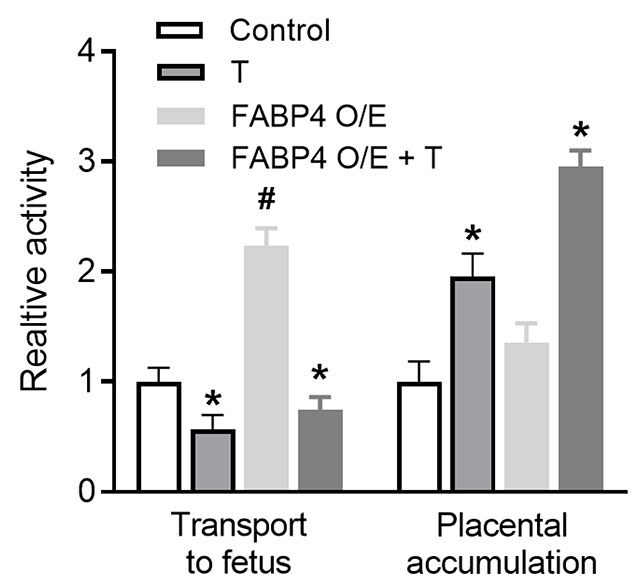
Effect of T and FABP4 overexpression on transplacental DHA transport in pregnant rats in vivo. Radiotracer transport studies using C14-DHA were performed and placental transport to the fetus (fetal dpm per milliliter serum) and placental accumulation (placental dpm per gram placenta) were measured. Data presented as mean ± SEM from n = 6 dams in each group. *P < 0.05 vs respective control, #P < 0.05 vs wild type. FABP O/E, FABP4 overexpressed.
Effect of T and FABP4 overexpression on transcellular DHA transport in cultured trophoblast cells in vitro
In normal BeWo trophoblast cells, T dose- and time-dependently decreased the rate of C14-DHA transfer across the monolayer (Figure 6A; n = 4; P < 0.05), and this was associated with an increase in the intracellular accumulation of C14-DHA at 24 h time point (Figure 6B; n = 4; P < 0.05). FABP4 overexpression in BeWo cells induced a time-dependent increase in transcellular transport of C14-DHA, and T treatment inhibited the FABP4 overexpression-induced increase in transcellular C14-DHA transport (Figure 6C; n = 4; P < 0.05). FABP4 overexpression in BeWo cells did not alter intracellular C14-DHA levels compared to normal trophoblasts, but T treatment induced significant accumulation of C14-DHA in FABP4-overexpressed cells (Figure 6D; n = 4; P < 0.05).
Figure 6.

Effect of T and FABP4 overexpression on transcellular DHA transport in cultured BeWo cells in vitro. (A) T-induced dose- and time-dependent changes in transcellular C14-DHA transport from apical to the basal reservoir in normal trophoblasts, (B) dose-dependent changes in intracellular C14-DHA accumulation in normal trophoblasts after 24 h of T treatment, (C) Time-dependent changes in transcellular C14-DHA transport in normal and FABP4-overexpressed trophoblasts in the presence and absence of T (10 nM) treatment. (D) Changes in intracellular C14-DHA accumulation in normal and FABP4-overexpressed trophoblasts in the presence and absence of 24 h T (10 nM) treatment. Data presented as mean ± SEM of four independent experiments. *P < 0.05 vs control. FABP O/E, FABP4 overexpressed.
Discussion
The major finding of this study is that, at clinically relevant concentrations, elevated maternal T levels during pregnancy led to increased accumulation of LCPUFAs in the placenta with reduced concentrations in both maternal and fetal circulations. In T-treated dams, dietary LCPUFA supplementation normalized maternal LCPUFA levels and augmented placental LCPUFA accumulation, but failed to increase fetal LCPUFA levels. Testosterone directly upregulated placental FABP4 mRNA transcripts by positively regulating FABP4 transcription through a functional ARE in the FABP4 gene promoter; yet, the transplacental transport of LCPUFAs was reduced. FABP4 overexpression by itself increased transplacental transport of DHA, but elevated maternal T prevented this FABP4 overexpression effect by promoting LCPUFA accumulation in the placenta. Therefore, we suggest that maternal hyperandrogenism during pregnancy induces placental FABP4 expression via transcriptional upregulation and preferentially routes LCPUFAs toward placental intracellular storage leading to offspring lipid deficiency.
Although PE is one of the major causes of maternal and perinatal mortality and morbidity, the pathophysiology of this disease has yet to be completely understood. Placental AR expression is increased in women with PE, coincident with increased placental T production [65]. We have previously shown that elevated maternal T in pregnant rats, at levels similar to that in women with PE, decreased transplacental transport of amino acid, but not glucose, to the fetus coincident with decreased offspring birth weight [50]. In this study, we not only confirm that elevated maternal T decreases fetal growth, but we also provide novel findings demonstrating decreased absolute amounts of n-3 and n-6 LCPUFA in both maternal and fetal circulations, especially essential fatty acids such as ARA and DHA. The decreased maternal LCPUFA concentrations were not due to inhibition of its synthesis, as the expression of Δ5- and Δ6-desaturase in placentae, liver, and adipose tissue (Supplementary Figure S3), which are major sites of synthesis, is not altered in T dams. Our findings that T did not alter regulatory enzymes of fatty acid synthesis are not consistent with the reports of Kelly et al. [66], which showed that T suppressed fatty acid synthesis. This discrepancy could be related to the T status and sex differences. Kelly et al. [66] examined the effects of T replacement in orchiectomized males, while our study examined the impact of elevated T in pregnant females. The increased uptake and storage of LCPUFAs in the placentae of T dams in this study suggests that such ectopic lipid accumulation may contribute to decreased LCPUFA concentrations in both maternal and fetal circulations, similar to deficiencies that reported for women with PE [21]. Whether T decreases intestinal absorption of fatty acids, however, needs to be examined in the future.
Increased placental storage of fatty acids along with low fetal circulatory LCPUFA concentrations suggests that the transfer of fatty acids across the placenta may be impaired in T rats. This idea is supported by dietary supplementation of LCPUFA enhancing maternal and placental concentrations of LCPUFAs, while failing to abrogate fetal LCPUFA deficiencies in T rats. This premise is further strengthened by our radioactive tracer experiment that shows elevated T increasing placental accumulation of DHA, while at the same time decreasing transplacental transport of DHA to the fetus. Our in vitro studies of T dose-dependently increasing intracellular DHA accumulation in parallel with decreased transcellular DHA transport in BeWo cells further confirm that T stimulates increased storage of fatty acids in placental trophoblast cells while decreasing their transport to the fetus. Although previous studies have shown that lipid storage occurs in placentae of the rat L-NAME model of PE [67], reminiscent of lipid accumulation in human PE placentae [21], we provide the first evidence that maternal hyperandrogenism acting via placental AR stimulates ectopic storage of LCPUFAs in the placenta while limiting the transfer of these critical nutrients to the fetus.
The transport of fatty acids through the placenta is orchestrated by several proteins, including lipoprotein lipase, fatty acid transfer proteins such as FATP-1-6, CD36, and cytoplasmic FABP-1-9 [13]. Placentae from our T groups, however, demonstrated increased expression of lipoprotein lipase and FABP4. Lipoprotein lipase is the key enzyme abundantly expressed on the maternal side of the placenta [12] involved in hydrolyzing circulating triglycerides to increase the availability of “free” fatty acids for uptake into syncytiotrophoblasts. Increased expression of lipoprotein lipase in T placenta could, therefore, be responsible for increased uptake of fatty acids into the placenta, similar to its role in promoting fatty liver [68]. The mechanism leading to increased expression of lipoprotein lipase in the T placenta, however, needs to be clarified.
Once inside the syncytiotrophoblast, fatty acids bind to FABPs and are shuttled from the apical (maternal facing) to the basolateral (fetal facing) plasma membrane for delivery to the fetus. There are nine family members of FABPs, but our studies only demonstrated the expression of FABP-1, -2, -3, -4, -5, -7, and -9 in rat placentae. Out of these FABPs expressed, only FABP4 expression was increased in T placentae. Importantly, this is also the only FABP4 increased in the placentae of women with PE [34–43]. Placental upregulation of FABP4, however, is not a compensatory response to T-induced decreases in fetal LCPUFA concentrations, because T-exposed cultured trophoblasts upregulate FABP4 mRNA, suggesting that FABP4 is a physiological target for T and that androgens normally upregulate placental FABP4 expression. Testosterone induction of FABP4 expression was indeed placenta-specific since T was consistently unable to stimulate FABP4 expression in hepatocyte and adipocyte cultures. Using the AR antagonist hydroxyflutamide, we demonstrated AR-mediated T stimulation of FABP4 in trophoblast. This effect is expected as the trophoblast cells express AR, consistent with previous human studies [65, 69]. In silico analysis of the entire upstream sequences of the FABP4 transcription start codon revealed multiple AREs within human FABP4 promoter, which can bind to AR. ChIP-PCR analysis using specific AR antibodies showed that AR was recruited to AREs in the human FABP4 promoter in trophoblast cells. These data provide the first line of evidence regarding the role of AR in mediating androgen stimulation of FABP4 expression in trophoblast cells. Indeed, this premise is supported by AR-ARE3-mediated, T-induced trans-activation of human FABP4 promoter-driven luciferase reporter gene in trophoblast cells in vitro. Since AR interactions with co-activators or co-repressors can regulate ARE-regulated genes [69], future studies should discern whether co-regulators additionally contribute to T-induced trophoblast-specific augmentation of FABP4 expression.
Although FABP4 is shown to have a higher binding affinity to LCPUFAs [70], there is no direct evidence regarding the function of FABP4 in placental fatty acid transport. This study, however, is the first, to our knowledge, showing that FABP4 is involved in transplacental DHA transport. Overexpression of FABP4 in pregnant rats and BeWo trophoblast cells increased transplacental transport of DHA with no significant accumulation of DHA in the placenta. In contrast, the presence of T dramatically decreased the FABP4 overexpression-induced increase in DHA transport in pregnant rats with a simultaneous increase in placental DHA accumulation. The in vitro studies in BeWo cells thus confirm that T directly promotes disruption of cellular DHA trafficking. It is intriguing, therefore, as to the mechanism by which upregulated placental FABP4 fails to adequately maintain the transfer of fatty acids across the placenta in T rats and women with PE. Mass spectrometry-based screens have independently identified numerous posttranslational modifications on FABP4, including phosphorylation, acetylation, and carbonylation [71–73], but only phosphorylation has been validated in in vitro systems, and the functional relevance of such modification(s) remains to be addressed [73]. It thus remains to be elucidated whether posttranslational modifications contribute to FABP4 dysfunction in the placenta of T rats and women with PE. Although we focused on DHA transport in this study, it would be interstring to confirm if similar regulatory effects are operational in the transplacental transport of other LCPUFAs. Further, it is also necessary to examine if T and FABP4 effects on placental fatty acid handling vary depending on fetal sex.
In conclusion, this study indicates that while elevated maternal T increases placental FABP4 expression by an AR-mediated mechanism, it concomitantly impair transport of fatty acids across the placenta leading to offspring lipid deficiency. These results suggest a role for AR-mediated maternal T action as a possible dysregulator of placental lipid trafficking. It is, therefore, interesting to speculate that in human pregnancies with PE, or in women with hyperandrogenic PCOS in whom the incidence of PE is increased [74], ectopic lipid storage in the placenta decreased maternal and fetal LCPUFA levels and inconsistent beneficial effects of DHA supplementation during PE may all indeed be AR-mediated. In a nonhuman primate model for PCOS, increasing maternal T during early-to-mid gestation diminished fetal circulating levels of total free fatty acids later in gestation [74]. The ability of elevated maternal T to influence the transfer of critical nutrients to the fetus may contribute to some of the adverse effects of T on fetal growth and development observed in both women with PE and with PCOS. Strategies that target excessive AR-mediated dysregulation of LCPUFA trafficking in the placenta could lead to new approaches in preventing the dyslipidemic effects in pregnancies complicated by PE and fetal growth restriction.
Conflict of interest
The authors have no conflicts of interest to declare.
Data availability
The data underlying this article are available in the article and in its online supplementary material.
Supplementary Material
Footnotes
† Grant Support: Financial support from the National Institute of Health (NIH) through grant HL134779, awarded to S.K., is greatly appreciated. The content is solely the responsibility of authors and does not necessarily represent the official views of NIH. The funding agency was not involved in the design, analysis, or interpretation of the data reported.
Contributor Information
Kathirvel Gopalakrishnan, Department of Comparative Biosciences, School of Veterinary Medicine, University of Wisconsin-Madison, Madison, WI, USA.
Jay S Mishra, Department of Comparative Biosciences, School of Veterinary Medicine, University of Wisconsin-Madison, Madison, WI, USA.
Jordan R Ross, Department of Comparative Biosciences, School of Veterinary Medicine, University of Wisconsin-Madison, Madison, WI, USA.
David H Abbott, Department of Obstetrics and Gynecology, School of Medicine and Public Health, University of Wisconsin-Madison, Madison, WI, USA; Endocrinology-Reproductive Physiology Program, University of Wisconsin-Madison, Madison, WI, USA; Wisconsin National Primate Research Center, University of Wisconsin-Madison, Madison, WI, USA.
Sathish Kumar, Department of Comparative Biosciences, School of Veterinary Medicine, University of Wisconsin-Madison, Madison, WI, USA; Department of Obstetrics and Gynecology, School of Medicine and Public Health, University of Wisconsin-Madison, Madison, WI, USA; Endocrinology-Reproductive Physiology Program, University of Wisconsin-Madison, Madison, WI, USA.
References
- 1. Kremmyda LS, Tvrzicka E, Stankova B, Zak A. Fatty acids as biocompounds: their role in human metabolism, health and disease: a review. Part 2: fatty acid physiological roles and applications in human health and disease. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2011; 155:195–218. [DOI] [PubMed] [Google Scholar]
- 2. Innis SM. Perinatal biochemistry and physiology of long-chain polyunsaturated fatty acids. J Pediatr 2003; 143:S1–S8. [DOI] [PubMed] [Google Scholar]
- 3. Haggarty P. Fatty acid supply to the human fetus. Annu Rev Nutr 2010; 30:237–255. [DOI] [PubMed] [Google Scholar]
- 4. Haggarty P. Placental regulation of fatty acid delivery and its effect on fetal growth—a review. Placenta 2002; 23:S28–S38. [DOI] [PubMed] [Google Scholar]
- 5. Dunlop M, Court JM. Lipogenesis in developing human adipose tissue. Early Hum Dev 1978; 2:123–130. [DOI] [PubMed] [Google Scholar]
- 6. Lorenzo M, Caldes T, Benito M, Medina JM. Lipogenesis in vivo in maternal and foetal tissues during late gestation in the rat. Biochem J 1981; 198:425–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Y, Botolin D, Christian B, Busik J, Xu J, Jump DB. Tissue-specific, nutritional, and developmental regulation of rat fatty acid elongases. J Lipid Res 2005; 46:706–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rodriguez A, Sarda P, Nessmann C, Boulot P, Leger CL, Descomps B. Delta6- and delta5-desaturase activities in the human fetal liver: kinetic aspects. J Lipid Res 1998; 39:1825–1832. [PubMed] [Google Scholar]
- 9. Mercuri O, Elena de Tomas M, Itarte H. Prenatal protein depletion and delta 9, delta 6 and delta 5 desaturases in the rat. Lipids 1979; 14:822–825. [PubMed] [Google Scholar]
- 10. Wadhwani NS, Dangat KD, Joshi AA, Joshi SR. Maternal micronutrients and omega 3 fatty acids affect placental fatty acid desaturases and transport proteins in Wistar rats. Prostaglandins Leukot Essent Fatty Acids 2013; 88:235–242. [DOI] [PubMed] [Google Scholar]
- 11. Campbell FM, Clohessy AM, Gordon MJ, Page KR, Dutta-Roy AK. Uptake of long chain fatty acids by human placental choriocarcinoma (BeWo) cells: role of plasma membrane fatty acid-binding protein. J Lipid Res 1997; 38:2558–2568. [PubMed] [Google Scholar]
- 12. Lager S, Powell TL. Regulation of nutrient transport across the placenta. J Pregnancy 2012; 2012:179827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gil-Sanchez A, Koletzko B, Larque E. Current understanding of placental fatty acid transport. Curr Opin Clin Nutr Metab Care 2012; 15:265–272. [DOI] [PubMed] [Google Scholar]
- 14. Innis SM. Essential fatty acid transfer and fetal development. Placenta 2005; 26:S70–S75. [DOI] [PubMed] [Google Scholar]
- 15. Gil-Sanchez A, Larque E, Demmelmair H, Acien MI, Faber FL, Parrilla JJ, Koletzko B. Maternal-fetal in vivo transfer of [13C]docosahexaenoic and other fatty acids across the human placenta 12 h after maternal oral intake. Am J Clin Nutr 2010; 92:115–122. [DOI] [PubMed] [Google Scholar]
- 16. Ballabriga A, Martinez M. A chemical study on the development of the human forebrain and cerebellum during the brain ‘growth spurt’ period. II. Phosphoglyceride fatty acids. Brain Res 1978; 159:363–370. [DOI] [PubMed] [Google Scholar]
- 17. Clandinin MT, Chappell JE, Leong S, Heim T, Swyer PR, Chance GW. Intrauterine fatty acid accretion rates in human brain: implications for fatty acid requirements. Early Hum Dev 1980; 4:121–129. [DOI] [PubMed] [Google Scholar]
- 18. Sinclair AJ, Crawford MA. The accumulation of arachidonate and docosahexaenoate in the developing rat brain. J Neurochem 1972; 19:1753–1758. [DOI] [PubMed] [Google Scholar]
- 19. Kolb B, Whishaw IQ. Plasticity in the neocortex: mechanisms underlying recovery from early brain damage. Prog Neurobiol 1989; 32:235–276. [DOI] [PubMed] [Google Scholar]
- 20. Cetin I, Alvino G. Intrauterine growth restriction: implications for placental metabolism and transport. A review. Placenta 2009; 30:S77–S82. [DOI] [PubMed] [Google Scholar]
- 21. Mackay VA, Huda SS, Stewart FM, Tham K, McKenna LA, Martin I, Jordan F, Brown EA, Hodson L, Greer IA, Meyer BJ, Freeman DJ. Preeclampsia is associated with compromised maternal synthesis of long-chain polyunsaturated fatty acids, leading to offspring deficiency. Hypertension 2012; 60:1078–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alvino G, Cozzi V, Radaelli T, Ortega H, Herrera E, Cetin I. Maternal and fetal fatty acid profile in normal and intrauterine growth restriction pregnancies with and without preeclampsia. Pediatr Res 2008; 64:615–620. [DOI] [PubMed] [Google Scholar]
- 23. Wadhwani N, Patil V, Pisal H, Joshi A, Mehendale S, Gupte S, Wagh G, Joshi S. Altered maternal proportions of long chain polyunsaturated fatty acids and their transport leads to disturbed fetal stores in preeclampsia. Prostaglandins Leukot Essent Fatty Acids 2014; 91:21–30. [DOI] [PubMed] [Google Scholar]
- 24. Nomura Y, John RM, Janssen AB, Davey C, Finik J, Buthmann J, Glover V, Lambertini L. Neurodevelopmental consequences in offspring of mothers with preeclampsia during pregnancy: underlying biological mechanism via imprinting genes. Arch Gynecol Obstet 2017; 295:1319–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dachew BA, Scott JG, Mamun A, Alati R. Pre-eclampsia and the risk of attention-deficit/hyperactivity disorder in offspring: findings from the ALSPAC birth cohort study. Psychiatry Res 2019; 272:392–397. [DOI] [PubMed] [Google Scholar]
- 26. Auger N, Fraser WD, Healy-Profitos J, Arbour L. Association between preeclampsia and congenital heart defects. JAMA 2015; 314:1588–1598. [DOI] [PubMed] [Google Scholar]
- 27. Fugelseth D, Ramstad HB, Kvehaugen AS, Nestaas E, Stoylen A, Staff AC. Myocardial function in offspring 5-8 years after pregnancy complicated by preeclampsia. Early Hum Dev 2011; 87:531–535. [DOI] [PubMed] [Google Scholar]
- 28. Rodie VA, Caslake MJ, Stewart F, Sattar N, Ramsay JE, Greer IA, Freeman DJ. Fetal cord plasma lipoprotein status in uncomplicated human pregnancies and in pregnancies complicated by pre-eclampsia and intrauterine growth restriction. Atherosclerosis 2004; 176:181–187. [DOI] [PubMed] [Google Scholar]
- 29. Kulkarni AV, Mehendale SS, Yadav HR, Kilari AS, Taralekar VS, Joshi SR. Circulating angiogenic factors and their association with birth outcomes in preeclampsia. Hypertens Res 2010; 33:561–567. [DOI] [PubMed] [Google Scholar]
- 30. Brown SH, Eather SR, Freeman DJ, Meyer BJ, Mitchell TW. A lipidomic analysis of placenta in preeclampsia: evidence for lipid storage. PLoS One 2016; 11:e0163972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Burchakov DI, Kuznetsova IV, Uspenskaya YB. Omega-3 long-chain polyunsaturated fatty acids and preeclampsia: trials say “no,” but is it the final word? Nutrients 2017; 9(12):1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. De Giuseppe R, Roggi C, Cena H. n-3 LC-PUFA supplementation: effects on infant and maternal outcomes. Eur J Nutr 2014; 53:1147–1154. [DOI] [PubMed] [Google Scholar]
- 33. Makrides M, Gibson RA, AJ MP, Yelland L, Quinlivan J, Ryan P, DOMInO Investigative Team . Effect of DHA supplementation during pregnancy on maternal depression and neurodevelopment of young children: a randomized controlled trial. JAMA 2010; 304:1675–1683. [DOI] [PubMed] [Google Scholar]
- 34. Fasshauer M, Seeger J, Waldeyer T, Schrey S, Ebert T, Kratzsch J, Lossner U, Bluher M, Stumvoll M, Faber R, Stepan H. Serum levels of the adipokine adipocyte fatty acid-binding protein are increased in preeclampsia. Am J Hypertens 2008; 21:582–586. [DOI] [PubMed] [Google Scholar]
- 35. Kelly CB, Hookham MB, Yu JY, Lockhart SM, Du M, Jenkins AJ, Nankervis A, Hanssen KF, Henriksen T, Garg SK, Hammad SM, Scardo JAet al. Circulating adipokines are associated with pre-eclampsia in women with type 1 diabetes. Diabetologia 2017; 60:2514–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li B, Yang H, Zhang W, Shi Y, Qin S, Wei Y, He Y, Yang W, Jiang S, Jin H. Fatty acid-binding protein 4 predicts gestational hypertension and preeclampsia in women with gestational diabetes mellitus. PLoS One 2018; 13:e0192347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lin YP, Xu CL, Lin KS, Gu HB, Chen L, Wang Y, Weng BC, Huang HQ, Li YP, Zou YL, Li ZS. Study on the correlation between adipocyte fatty-acid binding protein, glucolipid metabolism, and pre-eclampsia. J Obstet Gynaecol Res 2018; 44:655–662. [DOI] [PubMed] [Google Scholar]
- 38. Qiao GH, Sun XZ. Increased plasma fatty acid binding protein 4 concentration at the first prenatal visit and its relevance to preeclampsia. Hypertens Res 2018; 41:763–769. [DOI] [PubMed] [Google Scholar]
- 39. Scifres CM, Catov JM, Simhan H. Maternal serum fatty acid binding protein 4 (FABP4) and the development of preeclampsia. J Clin Endocrinol Metab 2012; 97:E349–E356. [DOI] [PubMed] [Google Scholar]
- 40. Tuuri AL, Jauhiainen MS, Tikkanen MJ, Kaaja RJ. Systolic blood pressure and fatty acid-binding protein 4 predict pregnancy-induced hypertension in overweight nulliparous women. Placenta 2014; 35:797–801. [DOI] [PubMed] [Google Scholar]
- 41. Wotherspoon AC, Young IS, McCance DR, Patterson CC, Maresh MJ, Pearson DW, Walker JD, Holmes VA, Diabetes and Pre-eclampsia Intervention Trial (DAPIT) Study Group . Serum fatty acid binding protein 4 (FABP4) predicts pre-eclampsia in women with type 1 diabetes. Diabetes Care 2016; 39:1827–1829. [DOI] [PubMed] [Google Scholar]
- 42. Yan JY, Wang XJ. Expression and significance of adipocyte fatty acid-binding protein in placenta, serum and umbilical cord blood in preeclampsia. Zhonghua Fu Chan Ke Za Zhi 2010; 45:885–890. [PubMed] [Google Scholar]
- 43. Yan Y, Peng H, Wang P, Wang H, Dong M. Increased expression of fatty acid binding protein 4 in preeclamptic placenta and its relevance to preeclampsia. Placenta 2016; 39:94–100. [DOI] [PubMed] [Google Scholar]
- 44. Haggarty P, Ashton J, Joynson M, Abramovich DR, Page K. Effect of maternal polyunsaturated fatty acid concentration on transport by the human placenta. Biol Neonate 1999; 75:350–359. [DOI] [PubMed] [Google Scholar]
- 45. Scifres CM, Chen B, Nelson DM, Sadovsky Y. Fatty acid binding protein 4 regulates intracellular lipid accumulation in human trophoblasts. J Clin Endocrinol Metab 2011; 96:E1083–E1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kumar S, Gordon GH, Abbott DH, Mishra JS. Androgens in maternal vascular and placental function: implications for preeclampsia pathogenesis. Reproduction 2018; 156:R155–R167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Carlsen SM, Jacobsen G, Romundstad P. Maternal testosterone levels during pregnancy are associated with offspring size at birth. Eur J Endocrinol 2006; 155:365–370. [DOI] [PubMed] [Google Scholar]
- 48. Hu W, Qiao J. Expression and regulation of adipocyte fatty acid binding protein in granulosa cells and its relation with clinical characteristics of polycystic ovary syndrome. Endocrine 2011; 40:196–202. [DOI] [PubMed] [Google Scholar]
- 49. Gopalakrishnan K, Mishra JS, Chinnathambi V, Vincent KL, Patrikeev I, Motamedi M, Saade GR, Hankins GD, Sathishkumar K. Elevated testosterone reduces uterine blood flow, spiral artery elongation, and placental oxygenation in pregnant rats. Hypertension 2016; 67:630–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sathishkumar K, Elkins R, Chinnathambi V, Gao H, Hankins GD, Yallampalli C. Prenatal testosterone-induced fetal growth restriction is associated with down-regulation of rat placental amino acid transport. Reprod Biol Endocrinol 2011; 9:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Beckett EM, Astapova O, Steckler TL, Veiga-Lopez A, Padmanabhan V. Developmental programming: impact of testosterone on placental differentiation. Reproduction 2014; 148:199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chinnathambi V, Blesson CS, Vincent KL, Saade GR, Hankins GD, Yallampalli C, Sathishkumar K. Elevated testosterone levels during rat pregnancy cause hypersensitivity to angiotensin II and attenuation of endothelium-dependent vasodilation in uterine arteries. Hypertension 2014; 64:405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ulmer CZ, Patterson RE, Koelmel JP, Garrett TJ, Yost RA. A robust lipidomics workflow for mammalian cells, plasma, and tissue using liquid-chromatography high-resolution tandem mass spectrometry. Methods Mol Biol 2017; 1609:91–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stahlman M, Ejsing CS, Tarasov K, Perman J, Boren J, Ekroos K. High-throughput shotgun lipidomics by quadrupole time-of-flight mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2009; 877:2664–2672. [DOI] [PubMed] [Google Scholar]
- 55. Tran PN, Brown SH, Mitchell TW, Matuschewski K, McMillan PJ, Kirk K, Dixon MW, Maier AG. A female gametocyte-specific ABC transporter plays a role in lipid metabolism in the malaria parasite. Nat Commun 2014; 5:4773. [DOI] [PubMed] [Google Scholar]
- 56. Tobin KA, Johnsen GM, Staff AC, Duttaroy AK. Long-chain polyunsaturated fatty acid transport across human placental choriocarcinoma (BeWo) cells. Placenta 2009; 30:41–47. [DOI] [PubMed] [Google Scholar]
- 57. Lee C, Huang CH. LASAGNA-Search: an integrated web tool for transcription factor binding site search and visualization. Biotechniques 2013; 54:141–153. [DOI] [PubMed] [Google Scholar]
- 58. Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, Lee R, Bessy A, Cheneby J, Kulkarni SR, Tan G, Baranasic D, Arenillas DJet al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res 2018; 46:D1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Blesson CS, Chinnathambi V, Hankins GD, Yallampalli C, Sathishkumar K. Prenatal testosterone exposure induces hypertension in adult females via androgen receptor-dependent protein kinase Cdelta-mediated mechanism. Hypertension 2015; 65:683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dreos R, Ambrosini G, Groux R, Cavin Perier R, Bucher P. The Eukaryotic Promoter Database in its 30th year: focus on non-vertebrate organisms. Nucleic Acids Res 2017; 45:D51–D55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hsu SC, Huang SM, Lin SH, Ka SM, Chen A, Shih MF, Hsu YJ. Testosterone increases renal anti-aging klotho gene expression via the androgen receptor-mediated pathway. Biochem J 2014; 464:221–229. [DOI] [PubMed] [Google Scholar]
- 62. Xu Y, Agrawal S, Cook TJ, Knipp GT. Maternal di-(2-ethylhexyl)-phthalate exposure influences essential fatty acid homeostasis in rat placenta. Placenta 2008; 29:962–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Metherel AH, Kitson AP, Domenichiello AF, Lacombe RJS, Hopperton KE, Trepanier MO, Alashmali SM, Lin L, Bazinet RP. Docosahexaenoic acid (DHA) accretion in the placenta but not the fetus is matched by plasma unesterified DHA uptake rates in pregnant Long Evans rats. Placenta 2017; 58:90–97. [DOI] [PubMed] [Google Scholar]
- 64. Mishra JS, Zhao H, Hattis S, Kumar S. Elevated glucose and insulin levels decrease DHA transfer across human trophoblasts via SIRT1-dependent mechanism. Nutrients 2020; 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sathishkumar K, Balakrishnan M, Chinnathambi V, Chauhan M, Hankins GD, Yallampalli C. Fetal sex-related dysregulation in testosterone production and their receptor expression in the human placenta with preeclampsia. J Perinatol 2012; 32:328–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kelly DM, Nettleship JE, Akhtar S, Muraleedharan V, Sellers DJ, Brooke JC, McLaren DS, Channer KS, Jones TH. Testosterone suppresses the expression of regulatory enzymes of fatty acid synthesis and protects against hepatic steatosis in cholesterol-fed androgen deficient mice. Life Sci 2014; 109:95–103. [DOI] [PubMed] [Google Scholar]
- 67. Sun MN, Yang Z, Ma RQ. Effect of high-fat diet on liver and placenta fatty infiltration in early onset preeclampsia-like mouse model. Chin Med J (Engl) 2012; 125:3532–3538. [PubMed] [Google Scholar]
- 68. Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology 2010; 51:679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Heinlein CA, Chang C. Androgen receptor (AR) coregulators: an overview. Endocr Rev 2002; 23:175–200. [DOI] [PubMed] [Google Scholar]
- 70. Richieri GV, Ogata RT, Kleinfeld AM. Equilibrium constants for the binding of fatty acids with fatty acid-binding proteins from adipocyte, intestine, heart, and liver measured with the fluorescent probe ADIFAB. J Biol Chem 1994; 269:23918–23930. [PubMed] [Google Scholar]
- 71. Xu Z, Ande SR, Mishra S. Temporal analysis of protein lysine acetylation during adipocyte differentiation. Adipocyte 2013; 2:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hellberg K, Grimsrud PA, Kruse AC, Banaszak LJ, Ohlendorf DH, Bernlohr DA. X-ray crystallographic analysis of adipocyte fatty acid binding protein (aP2) modified with 4-hydroxy-2-nonenal. Protein Sci 2010; 19:1480–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hresko RC, Hoffman RD, Flores-Riveros JR, Lane MD. Insulin receptor tyrosine kinase-catalyzed phosphorylation of 422(aP2) protein. Substrate activation by long-chain fatty acid. J Biol Chem 1990; 265:21075–21085. [PubMed] [Google Scholar]
- 74. Abbott DH, Bruns CR, Barnett DK, Dunaif A, Goodfriend TL, Dumesic DA, Tarantal AF. Experimentally induced gestational androgen excess disrupts glucoregulation in rhesus monkey dams and their female offspring. Am J Physiol Endocrinol Metab 2010; 299:E741–E751. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material.